Article Text

Abstract
Objectives Newborn screening for cystic fibrosis (CF) facilitates early diagnosis and genetic counselling for parents of affected infants. Many parents elect to use prenatal testing for subsequent pregnancies, and this may affect the prevalence of CF. The aim of this study was to assess the evidence for changes in the live-birth prevalence of CF since the introduction of newborn screening for CF.
Methods The authors reviewed the records of the Victorian newborn screening programme and the clinical records of the three centres caring for patients with CF in Victoria, Australia, in order to determine the live-birth prevalence of patients with CF; before (1979–1988) and after (1989–2006) the introduction of newborn screening. The authors reviewed the records of the Victorian Clinical Genetics Service to ascertain the number and outcome of prenatal tests for CF (1979–2006). Live births in Victoria were obtained from the state birth register.
Findings Between 1979 and 1988, the live-birth prevalence of CF was 3.96 (95% CI 3.48 to 4.49) per 10 000 live births. Following the introduction of newborn screening (1989–2006) the live-birth prevalence of CF was 3.28 (95% CI 2.97 to 3.63) per 10 000 live births, representing a reduction of 17% (95% CI 2% to 29%, p=0.025). In the prescreening period, there were 10 prenatal tests, which identified three affected pregnancies, all of which were terminated. In the later period, there were 304 prenatal tests (mean 17/year), of which 76 were affected, and 70 of these pregnancies were terminated.
Conclusion The authors observed a modest reduction in the live-birth prevalence of CF since the introduction of newborn screening. This is principally due to at-risk couples detected by newborn screening electing to use prenatal testing on subsequent pregnancies.
Statistics from Altmetric.com
Background
Newborn screening for cystic fibrosis (CF) was introduced in Victoria, Australia in 1989 to facilitate early diagnosis and genetic counselling for parents of affected infants.1 All infants have a heel prick test on day 2–4 of life, and the initial screen is for elevated serum levels of trypsinogen by immunoreactive assay (IRT). In the first 2 years of the screening programme (1989–1990), a second IRT was requested at 4–6 weeks if the initial value was elevated (>99th centile of values), and the diagnosis was confirmed by a sweat test. From 1991, the gene-mutation analysis was incorporated into the screening programme, testing for the common CF transmembrane conductance regulator (CFTR) gene mutation, p.508del (1991–2006).2 In addition to detecting infants with CF, newborn screening also detects a number of healthy carriers. These infants have an elevated IRT, one CFTR gene mutation and a normal sweat chloride value. The families of these infants are also offered genetic counselling and cascade family testing.3
What is already known on this topic
▶. Newborn screening for cystic fibrosis (CF) identifies most babies with CF in the first few weeks of life.
▶. Newborn screening for CF can also identify some babies as heterozygotes (healthy carriers).
▶. Parents of babies with CF (or carriers) can access genetic counselling and have reproductive choices available before the birth of their next child.
What this study adds
▶. Cystic fibrosis carrier parents identified through newborn screening use prenatal testing on subsequent pregnancies.
▶. The detection of carrier parents following newborn screening for cystic fibrosis has resulted in a small decline in the live-birth prevalence of cystic fibrosis.
Although the inherited nature of CF had been recognised for a long time, prior to 1989 there were limited options with regard to testing of carrier parents and virtually no ability to offer prenatal testing to detect affected pregnancies. The identification of the CFTR gene and mutations responsible for CF allowed the inclusion of gene mutation testing as part of the newborn screening paradigm and created a clear understanding among parents at the time of diagnosis that CF is a genetic condition and that testing of subsequent pregnancies was possible.4
We hypothesised that the live-birth prevalence of CF would decrease after the introduction of newborn screening, as a result of carrier parents utilising prenatal testing on subsequent pregnancies. The primary aim of this study was to determine the live-birth prevalence of CF in the years before and after the introduction of newborn screening for CF.
Methods
Subjects
We utilised the records of the newborn screening programme for CF run by Victorian Clinical Genetics Service to determine the number of babies detected by newborn screening since its introduction on 1 January 1989 until 31 December 2006. We verified the diagnoses of CF against clinic-held records of the Royal Children's Hospital, Monash Medical Centre and the Alfred Hospital, the three providers of CF care in the state of Victoria, Australia. This was to capture infants born with meconium ileus or a family history of CF, or who may have been missed by screening. A patient was considered to have CF if they met the criteria outlined by the US CF Foundation consensus documents.5 6 We stopped at 2006, to allow any children who were missed by screening to have presented clinically and because population-based carrier screening was introduced in Victoria in 2006.
To identify infants with CF born prior to the introduction of newborn screening, between 1979 and 1988, we used the CF clinic records of the Royal Children's Hospital, Melbourne and the Alfred Hospital, Melbourne as described above. These two hospitals provided all CF care in Victoria at the time.
The number of live births for the state of Victoria was taken from the state register of births, the Perinatal Data Collection Unit managed by the Department of Human Services, Victoria. It is a mandatory registration system for all Victorian births of at least 20 weeks' gestation.
The records of the Molecular Genetics laboratory of Victorian Clinical Genetics Service were reviewed to determine the number of prenatal tests for CF, the number of affected fetuses and the outcomes of the pregnancies. Victorian Clinical Genetics Service is the sole provider of CF gene mutation testing in Victoria. The Molecular Genetics laboratory and the Newborn Screening laboratory are accredited to ISO/IEC 15189 (Medical Laboratories standard) by the National Association of Testing Authorities. All molecular genetics test requests and results are recorded in the laboratory information system.
Statistical analysis
Birth prevalence was calculated by dividing the number of cases of CF by the number of live births, and standard methods based on the Poisson distribution were used to obtain 95% CIs for prevalence rates and their ratio.
Ethical approval
The study was approved by the Ethics in Human Research Committee of the Royal Children's Hospital, Melbourne as a clinical audit.
Results
Figure 1 displays the annual birth prevalence of CF for the period of study, along with the prevalence of all known CF in pregnancies. A gradual downturn in live-birth prevalence is apparent after the introduction of screening in 1989. More specifically, between 1979 and 1988 there were 238 live births of patients with CF (from 601 745 live births in Victoria) giving a prevalence of 3.96 (95% CI 3.48 to 4.49) per 10 000 live births, while in the period following the introduction of newborn screening (1989–2006) there were 377 live births of patients with CF (from 1 148 223 live births) giving a prevalence of 3.28 (95% CI 2.97 to 3.63) per 10 000 live births. Formal comparison between the two periods in the form of a prevalence rate ratio gave RR=0.83 (95% CI 0.71 to 0.98; p=0.025), representing a 17% (95% CI 2% to 29%) reduction in prevalence of live births with CF since the introduction of newborn screening.
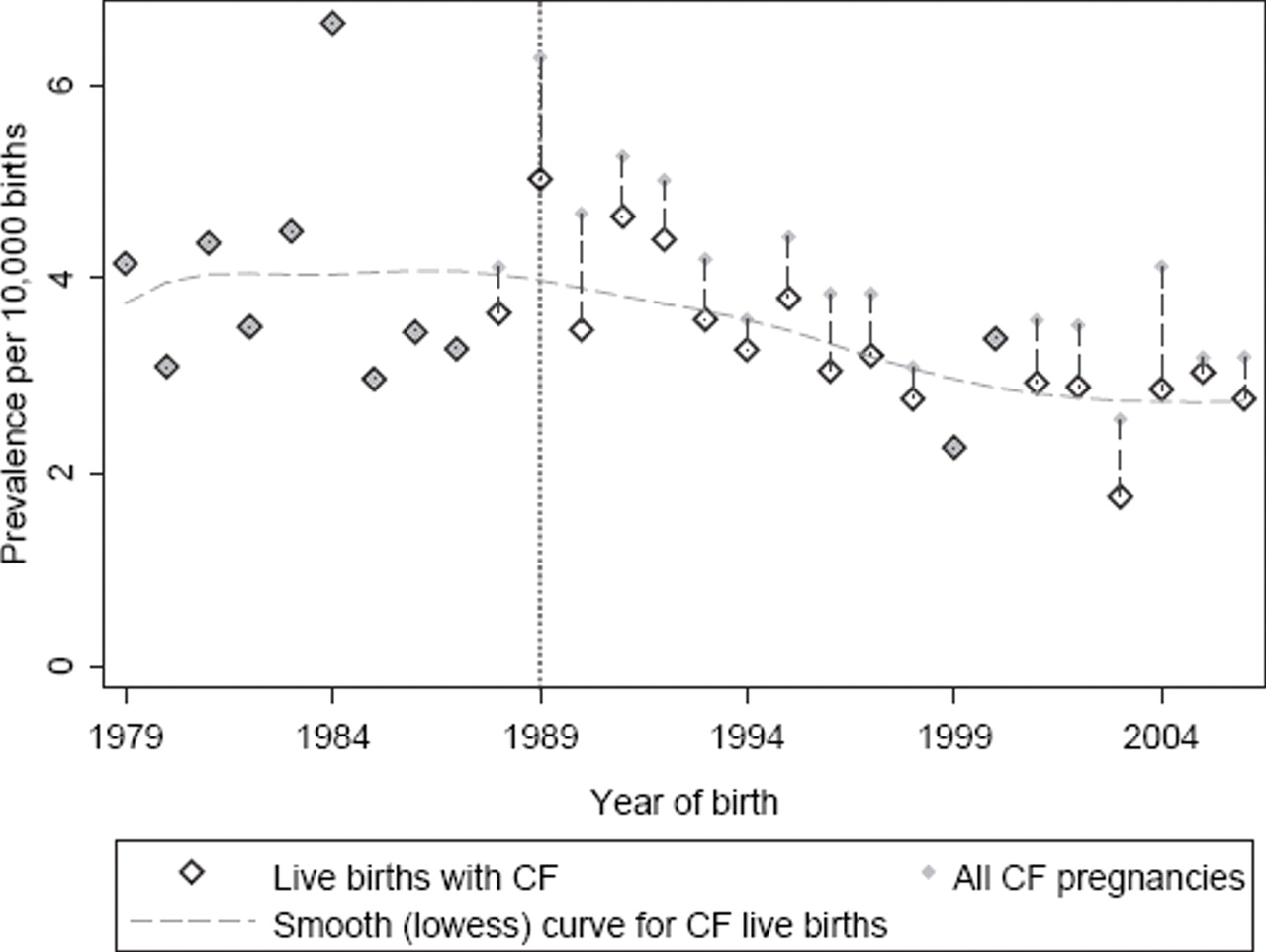
{kind=link}
Prevalence of cystic fibrosis at birth and prevalence of known cystic fibrosis pregnancies, by year of pregnancy outcome for the state of Victoria (1979–2006).
In the period before universal prenatal screening, 10 prenatal tests were recorded (one in 1987 and nine in 1988), identifying three affected fetuses (all in 1988), all of which were terminated. Since universal screening, there were 304 prenatal tests (mean 17/year) of which 76 were affected, and 70 of these were terminated (representing a prevalence, relative to all pregnancies, of 0.6 per 10 000 live births).
Discussion
We have found evidence of a modest reduction in the live-birth prevalence of CF since the introduction of newborn screening in Victoria, Australia in 1989. In the absence of a population-based carrier screening programme for CF, the reduction in live births appears to be due to the identification of at-risk couples through newborn screening. Most at-risk couples who had an affected pregnancy identified by prenatal testing elected to terminate the pregnancy.
The newborn screening programme for CF in Victoria, Australia is coordinated through the Victorian Clinical Genetics Service, a single provider of genetic services in our state. All parents of babies with CF identified by newborn screening undergo genetic counselling at the time of diagnosis and are offered prenatal testing for subsequent pregnancies. Previous data from our group indicate that 67% of families use prenatal testing subsequently.4 We know that some families have used prenatal testing on multiple occasions, but we were unable to quantify this in this study because of constraints on the linkage of anonymised data. At the time of the study (to 2006) all parents wishing to avoid having a second child with CF had to use prenatal testing. Subsequently, preimplantation genetic diagnosis has become available through in vitro fertilisation providers and has been used by some families.
The sources of error in our data collection are likely to be small. Newborn screening and CF testing are undertaken by a single provider. Cases missed by screening are in the order of 5% and usually detected by 6 months of age.1 Our dataset includes babies missed by screen, and we have waited until 2008 to complete the analysis, so that it is unlikely that the reduction in live-birth prevalence is due to children with CF yet to be diagnosed. Interstate movement between birth and notification of the screening result at 4 weeks is rare, and in general, diagnoses of CF made on a child in another state of Australia are notified to the Victorian newborn screening programme. The unusually high number of cases of CF in 1984 (prescreening) may be influential in the comparison between the two periods but is not particularly extreme under the assumption of Poisson variation from year to year. It is clear from examining the figure that the reduction in live-birth prevalence may not have occurred until some years after 1989.
We do not have data to clearly separate whether the prenatal tests were performed on at-risk couples identified with a baby with CF or at-risk couples identified with a carrier baby. Although the number of carrier babies detected by newborn screening is higher than expected (approximately 1.8 times the population frequency of carriers), in most cases only one parent is a carrier (only 1/25 of their partners will also be carriers).2 3 This makes it likely that the reduction in the live-birth prevalence is mostly due to parents of an affected baby using prenatal testing on subsequent pregnancies to avoid having a second child with CF. In either case, newborn screening, through detection of affected babies or carriers, was clearly associated with a decrease in the live-birth prevalence of CF.
Other groups have reported reductions in the incidence of CF, although little exploration of the reasons for this is explored.7,–,10 In Canada, using CF registry data, a 25% reduction in the incidence of CF was reported following the identification of CFTR (in 1989) examining the years 1971–2000.7 This is in the absence of a newborn screening programme, and no mention of a population-based carrier screening is made. In north-western France, a reduction of 30.5% was reported with a mixed contribution from parents of children diagnosed clinically, through newborn screening and those found to have echogenic bowel on fetal malformation screening.9
Given that CF is an inherited condition, it might seem that our result is modest, and that a greater decline in the live-birth prevalence could have been anticipated. The answer partly lies in the poor uptake of cascade family testing, a process whereby family members (up to third degree relatives) of an affected baby or carrier baby are offered free genetic testing (and counselling). We are currently investigating the uptake of cascade family testing in Victoria. Modelling of cascade testing in the UK anticipated that even testing all siblings and first cousins of carriers would detect only 15% of new cases of CF.11 This is consistent with our experience that over 95% of parents with a baby identified by newborn screening have no family history of CF. This indicates that cascade family testing, while appropriate for the families identified by newborn screening, does not identify many carriers in the community. Only a population-based carrier screening programme is likely to substantially reduce the live-birth prevalence of CF.12 13
We have demonstrated a reduction in the live-birth prevalence of CF since the introduction of newborn screening. The impact on the live-birth prevalence is modest, and is principally due to at-risk couples electing to use prenatal testing on subsequent pregnancies.
Acknowledgments
The authors wish to thank S Ranganathan for reviewing the manuscript.
References
Footnotes
-
Competing interests None.
-
Ethics approval Ethics approval was provided by the Ethics in Human Research, Royal Children's Hospital.
-
Provenance and peer review Not commissioned; externally peer reviewed.