Article Text

Abstract
Introduction Otitis media (OM) starts within weeks of birth in almost all Indigenous infants living in remote areas of the Northern Territory (NT). OM and associated hearing loss persist from infancy throughout childhood and often into adulthood. Educational and social opportunities are greatly compromised. Pneumococcus and non-typeable Haemophilus influenzae (NTHi) are major OM pathogens that densely colonise the nasopharynx and infect the middle ear from very early in life. Our hypothesis is that compared to current single vaccine schedules, a combination of vaccines starting at 1 month of age, may provide earlier, broadened protection.
Methods and analyses This randomised outcome assessor, blinded controlled trial will recruit 425 infants between 28 and 38 days of age and randomly allocate them (1:1:1) to one of three pneumococcal conjugate vaccine (PCV) schedules: Synflorix at 2, 4, 6 months of age, Prevenar13 at 2, 4 and 6 months of age, or an investigational schedule of Synflorix at 1, 2 and 4 months plus Prevenar13 at 6 months of age. The blinded primary outcomes at 7 months of age are immunogenicity of specific vaccine antigens (geometric mean concentration (GMC) and proportion of participants with above threshold GMC of 0.35 µg/L). Secondary outcomes at all timepoints are additional immunogenicity measures and proportion of participants with nasopharyngeal carriage of vaccine-type pneumococci and NTHi, and any OM, including any tympanic membrane perforation. Parental interviews will provide data on common risk factors for OM.
Ethics and dissemination Ethical approval has been obtained from NT Department of Health and Menzies HREC (EC00153), Central Australian HREC (EC00155) and West Australian Aboriginal Health Ethics Committee (WAAHEC- 377-12/2011). Final trial results, data analyses, interpretation and conclusions will be presented in appropriate written and oral formats to parents and guardians, participating communities, local, national and international conferences, and published in peer-reviewed open access journals.
Trial registration numbers ACTRN12610000544077 and NCT01174849.
- IMMUNOLOGY
- MICROBIOLOGY
- PUBLIC HEALTH
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
This study addresses an important health and social issue for Australian Indigenous children who live in the only western country named by the WHO as being in the highest category (>6%) of chronic suppurative otitis media (CSOM) prevalence.
This study is a randomised controlled trial of two licensed pneumococcal conjugate vaccines (PCVs) and a novel early combination schedule of these vaccines. Funding is independent of vaccine manufacturers.
Primary outcomes are accepted global immunogenicity criteria for licensing and varying PCV schedules. Secondary nasopharyngeal carriage outcomes are measured according to WHO recommendations. Secondary clinical outcomes are prepublished standardised assessments of middle ear status.
Limitations of the study include small sample size for secondary outcomes and slow recruitment.
Introduction
Background and rationale
In the Northern Territory of Australia, otitis media (OM) is a major public health crisis. By 3 months of age all infants have had a diagnosis of OM, and the episode persists for months.1 ,2 Our studies have identified communities where as many as 60% of children under 2.5 years of age have tympanic membrane perforations (TMP). Less than 20% children have normal aerated middle ears.3 The conductive hearing loss associated with persistent middle ear effusion (around 25 dB) or TMP (35–60 dB) has detrimental effects on auditory processing, speech and language development, behavioural and communication skills, and educational achievement. In randomised controlled trials, compared to placebo, long-term amoxicillin has been shown to halve the risk of TMP4 and long-term topical antibiotics will resolve the ear discharge (but not the perforation) in only 30% children.5 Health services struggle to deal with the diagnosis of acute OM (AOM), and with the management of perforation or persistent hearing loss. Surgical options are rarely available. Strategies to prevent early ear disease or reduce the incidence, duration and severity of disease are urgently needed.
Onset of first OM is preceded by acquisition of bacterial pathogens that colonise the nasopharynx soon after birth.1 The primary pathogens recovered from ear discharge of Aboriginal children with AOM with perforation (AOMwiP) in the first 18 months of life are pneumococcus (Spn), and non-typeable Haemophilus influenzae (NTHi). Pre-PCVs (pneumococcal conjugate vaccines) Pneumococcus was recovered from 38% middle ear discharge samples and NTHi from 55%.6
Our research question is to determine the potential for the two licensed PVCs PHiD-CV10 and PCV13 to be used early and in combination to broaden protection from the OM pathogens. The combination schedule chosen (PHiD-CV10 at 1, 2 and 4 months plus PCV13 at 6 months) was based on (1) data showing improved HiD immunogenicity following a third dose of PHiD-CV10 compared to two doses (data provided by GSK), (2) the opportunity to start vaccination earlier, at 1 month rather than 2 months of age, (3) the Northern Territory's relatively low prevalence of invasive pneumococcal disease caused by PCV13-only serotypes 3, 6A and 19A (compared to other serotypes) and (4) data showing reasonable immune response to a single dose of PCV.7
If superior, the COMBO vaccine schedule is likely to be recommended for Indigenous infants living in high-risk populations.
Explanation for choice of comparators
Three PCVs were licensed or soon to be licensed in Australia at the time of writing this proposal in 2009. PCV7 had been effective in eradicating almost all OM episodes caused by the seven vaccine serotypes. However, this eradication had not been translated to a clinical benefit6 due to replacement disease by non-PCV7 serotypes8 and a significant role of NTHi as a primary pathogen in TMP (Pneumococcus was recovered from around 31% middle ear discharge samples from PCV7-vaccinated children less than 18 months of age and NTHi from 58%).6 We also believe persistent high rates of pneumonia in the Northern Territory9 are not necessarily related to low efficacy of PCV7, but to serotype replacement and additional pathogens including NTHi, as shown in Papua New Guinea.10 Extended coverage vaccines are urgently needed. One strategy with potential to extend serotype coverage has been addressed by another study of ours (NHMRC 490320 ‘PneuMum’),11 which has evaluated maternal immunisation with Pneumovax (23-valent pneumococcal polysaccharide vaccine, 23PPV) to provide early and expanded serotype protection in Indigenous infants. In 2006, results of an RCT of 11-valent PCV (pneumococcal H. influenzae protein D conjugate vaccine, 11Pn-PD) showed efficacy for vaccine-type pneumococcal AOM and NTHi-AOM.12 The 10-valent vaccine, PHiD-CV10, was licensed on the basis of immunogenicity of the pneumococcal antigens and in 2009 was under consideration by Therapeutic Goods Administration for recommendation in Australia. In October 2009, the Northern Territory was the only Australian jurisdiction to change recommendations for PCV from PCV7 at 2, 4 and 6 months (followed by PPV23 at 18 months for Indigenous children), to PHiD-CV10 at 2, 4, 6 and 18 months (for all children). In 2011, the 13-valent CRM197-conjugated vaccine, Prevenar13, was approved in Australia as a 2, 4 and 6 month schedule (followed by PCV13 at 18 months for Indigenous children). In October 2011, Prevenar13 replaced PHiD-CV10 in the Northern Territory.
Objectives
Specific objectives or hypotheses.
Overall objective
To determine if the COMBO vaccination schedule is superior to the other two vaccination schedules with regard to overall protection from OM pathogens.
Primary hypotheses:
The COMBO group will have greater immunogenicity for HiD* compared to the PCV13 group at 7 months;
The COMBO group will have greater immunogenicity to pneumococcal serotype 19A** compared to the PHiD-CV10 group at 7 months;
The COMBO group will have greater immunogenicity to pneumococcal serotype 3** compared to the PHiD-CV10 group at 7 months;
The COMBO group will have greater immunogenicity to pneumococcal serotype 6A** compared to the PHiD-CV10 group at 7 months.
Key secondary hypotheses
The COMBO group will have less carriage of NTHi* compared to the PCV13 group at 7 months;
The COMBO group will have less carriage of pneumococcal serotype 19A** compared to the PHiD-CV10 group at 7 months;
The COMBO group will have less carriage of pneumococcal serotype 3** compared to the PHiD-CV10 group at 7 months;
The COMBO group will have less carriage of pneumococcal serotype 6A** compared to the PHiD-CV10 group at 7 months;
The COMBO group will have less OM compared to the PCV13 group at 7 months;
The COMBO group will have less OM compared to the PHiD-CV10 group at 7 months.
*HiD is not in PCV13, but is in PHiD-CV10.
**Serotypes in PCV13 but not in PHiD-CV10 are: 3, 6A and 19A.
Methods
Participants, interventions and outcomes
Trial design
The PREVIX_COMBO trial is an open label, allocation concealed, primary outcome assessor blinded, parallel group and randomised controlled trial with three groups (1:1:1), to determine the superiority of combination schedule over single vaccines.
Study setting
The trial will take place in remote Aboriginal and Torres Strait Islander communities (see figure 1). Traditional Owners (elders) have provided signed agreement for their community to participate in this research.

PREV-IX_COMBO study sites in the Northern Territory and Western Australia.
Eligibility criteria
Participants: Inclusion criteria—Aboriginal or Torres Strait Islander male and female infants living in a participating remote community (whose Traditional Owners have provided signed Expressions of Interest and confirmation of participation in PREV-IX_COMBO trial) in the Northern Territory or Western Australia, 28–38 days of age, healthy and eligible for National Immunisation Programme routine vaccines.
Exclusion criteria: Prior adverse reaction to PCVs according to the Australian Immunisation Handbook; gestational age <32 weeks. Not the eldest of the multiple births. Parents (usually the mother) are asked whether they expect to be resident in that community until their baby is 7 months of age. Those planning to move to a non-participating community are not eligible to be in the study.
Interventions and dosing schedules
Infants are randomly allocated in a 1:1:1 ratio to (1) Synflorix (PHiD-CV10) at 2, 4 and 6 months of age, (2) Prevenar13 (PCV13) at 2, 4 and 6 months of age or (3) the combination schedule of PHiD-CV10 at 1, 2 and 4 months, and PCV13 at 6 months of age. Infants allocated the combination schedule received a single dose of PHiD-CV10 at the time of randomisation (table 1). Study vaccines are administered into the left anteriolateral thigh. Immunisations are performed by nurses who had completed the About Giving Vaccines course.
Schedule of enrolment, interventions, and assessments
Criteria for discontinuing
Infants may be withdrawn from the study by their parent(s), guardian, the local healthcare provider or study staff.
Strategies to improve adherence
Parents are advised of their baby's scheduled visit dates. Where the parent provides a contact phone number, calls are made in advance to advise of a scheduled study visit. A dedicated study vehicle is available to transport parents and infants from their homes to the local clinic to receive vaccinations and healthcare by research staff. Parents receive a small gift following each of the two longer study visits which include a blood draw.
Relevant concomitant care
Research nurses also provide all vaccinations according to the current National Immunisation Programme for the Indigenous population. Infants receive treatment or referral for all concomitant conditions, particularly ear, skin, respiratory and growth problems, according to local guidelines.13 No concomitant medications were prohibited.
Outcomes
Primary, secondary and other outcomes.
Immunogenicity
Serotype-specific IgG are measured using a modified third-generation ELISA based on WHO recommendations.14 Sample dilutions are analysed in duplicate and three controls (low, medium and high) are included on each plate. Serotype-specific IgG is measured onto 13 vaccine serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 15B, 18C, 19F and 23F. IgG antibodies to the protein D of H. influenzae is measured by a classic ELISA, with non-lipidated protein D as coating material and expressed in ELISA units (EL.U/mL). Data are analysed using GraphPad Prism V.5 (GraphPad Software Inc, USA). Pneumococcal serotype-specific antibody concentrations and HiD antibody concentration are log10 transformed, and the geometric mean concentration (GMC) and 95% CIs are determined. Serotype-specific and HiD IgG GMC comparisons are made using unpaired t tests of the logged data.15
Nasopharyngeal carriage and ear discharge microbiology
NP swabs are collected, transported and stored as previously described,5 and in accordance with WHO recommendations for pneumococcal NP carriage studies.16 Swab quality is also recorded as good (swab inserted to the NP and held for 5 s), fair (swab inserted partially and briefly), poor (swab of outer nares only) or very poor (outer nares and no discharge visible). Any swab with visible secretions is coded as ‘good’. Swabs of ED are collected after cleaning the external canal and collecting discharge from as close as possible to the TMP.5 NP and ED swabs are cultured on selective and non-selective media, and semiquantitative colony counts recorded as previously described.5 Bacterial and viral DNA are extracted for detection by pathogen-specific quantitative PCR. For ED swabs with swarming species that preclude selection of single NTHi colonies, a millipore filtration step is used.5 At least two presumptive pneumococcal colonies and two presumptive NTHi colonies are selected from each specimen for confirmation. Colonies of minority colony morphology are chosen, if present. Pneumococci are identified by colony morphology, optochin sensitivity and positive reaction with typing sera (Statens Serum Institut, Denmark); serotype is determined by Quellung reaction. NTHi are identified by colony morphology, dependence on X and V growth factors, and lack of reaction with capsular antisera using the Phadebact Haemophilus coagglutination test. PCR discrimination of Haemophilus haemolyticus is not uniformly undertaken after confirmation that less than 0.2% presumptive NTHi isolated from NP swabs in this population are misidentified.17 Antimicrobial susceptibility is determined by the calibrated dichotomous susceptibility (CDS) disc diffusion method.18 ,19 Minimum inhibitory concentrations (MICs) are determined for macrolide and β-lactam antibiotic resistance in Streptococcus pneumoniae isolates and azithromycin resistance in NTHi isolates using Etest strips (AB bioMérieux, Sweden). β-Lactamase production by NTHi is determined using nitrocephin (Oxoid, Australia). Resistance is defined using European Committee on Antimicrobial Susceptibility Testing (EUCAST) breakpoints (http://www.eucast.org). Penicillin non-susceptibility of Spn is defined as MIC >0.06 mg/L and azithromycin resistance as MIC >0.5 mg/L. Azithromycin resistance in NTHi is defined as MIC >4 mg/L; intermediate resistance as MIC >0.12 and ≤4 mg/L, and susceptibility as MIC ≤0.12 mg/L.
Otitis media
Ear examinations and general health assessments: All clinical assessments are made by ear health research nurses with extensive training in the diagnosis and management of OM in this population. Otoscopic findings are recorded on a standardised form. Assessments are made using a tympanometer (Grason Stadler GSI 38), a LumiView (Welch Allyn), and a video-otoscope (Welch Allyn macroview or MedRx video-otoscopes).
Definitions of OM: We categorise middle ear states as follows: (1) normal; (2) OM with effusion (OME); (3) acute OM without perforation (AOMwoP); (4) AOM with perforation (AOMwiP); (5) dry perforation and (6) chronic suppurative OM (CSOM). The final middle ear diagnosis reflects the child's most severely affected ear (highest category). Criteria for diagnosis are based on recommendations for clinical practice in this population20: (1) OME—intact and non-bulging tympanic membrane (TM) and type B tympanogram; (2) AOMwoP—any bulging of the TM and type B tympanogram; (3) AOMwiP—middle ear discharge observed and TM perforation recently healed or present for less than 6 weeks or covering less than 2% of the pars tensa of the TM; (4) dry perforation—TM perforation without any discharge observed and (5) CSOM—middle ear discharge observed and perforation present for longer than 6 weeks and covering at least 2% of the pars tensa of the TM. Combination categories of any suppurative OM (any AOM, AOMwiP or CSOM) and any TMP (any AOMwiP, dry perforation or CSOM) are included. Where duration of discharge is not known, size of perforation is used to distinguish AOMwiP and CSOM. Where otosocopy is not successful the infant's tympanometry result is used and the infant's status is defined as OME if either ear had a type B tympanogram. We ask the mother if she thought her baby had ear pain that day or during the previous evening. These a priori diagnostic criteria have been applied in all our surveillance and clinical trials conducted in this population since 2001.3
Other outcomes
General health measures: Common conditions of childhood are recorded at the time of ear assessment by direct observation; axillary temperature, the infant's skin (head, arms, legs and trunk) is examined for the presence of scabies, tinea, skin sores or other skin condition; presence of nasal discharge (visible at a distance of 1 m) and any cough (spontaneous or cough on request, either wet or dry). We categorise ‘not sure’ as absent. Antibiotics and other treatments or referrals are provided to participants according to local guidelines.
Risk factor questionnaires: The parent or guardian (usually the mother) is asked a standardised set of questions about common risk factors for OM, including the number of children she has, the number of people and children (less than 5 years of age) living in the infant's house, whether the mother or infant's siblings ever had TMP (‘runny ears’), the mother's highest level of education, whether the mother smokes, smoked during pregnancy or if she or other household members smoke inside the house, and whether the infant was exposed to campfire smoke during the previous week, had ever used a pacifier, or was ever breastfed or bottle fed.
Sample size
Immunogenicity: Target enrolment is 425 participants to provide 339 evaluable infants (113 per group) at 7 months (we assume 20% withdrawal and/or loss to follow-up over the total study period).
Immunogenicity: If 20% of mothers refuse blood collection, bloods from 270 infants at 7 months will be available, 90 from each group. This will provide 99% power to detect an increase in the proportion of infants with immunogenicity above threshold for 3, 6A or 19A from 20%, 37% or 57%, respectively (data from non-vaccinated controls21) in the PHiD-CV10 group to 87% for 6A and >90% for 19A in the COMBO group (data from single toddler dose7), and for anti-HiD, there will be 90% power to detect a 21% increase from 67% in the PCV13 group to 88% in the COMBO group (data from adults receiving two doses HiD22).
Carriage: We will have over 90% power to detect reductions in NTHi and 19A carriage at 7 months of age, from 80% to 50% (for NTHi) and 20% to 5% (for 19A), respectively, between the COMBO group and the PCV13 group (for NTHi), and the COMBO group and PHiD-CV10 group (for 19A).
OM: We will have 55% power to detect a doubling in the proportion of children free from OM at 7 months in the COMBO group compared to another group (20% vs 10%).
Four hundred and twenty-five is likely to be plenty for the primary outcomes and insufficient for the secondary outcomes. The PREV-IX_BOOST study (see online supplementary appendix for a brief description) depends on this trial achieving 7-month follow-up on ∼300 infants.
Progress to date: Randomisations started in September 2011 and will be completed in late 2016.
Recruitment and participant selection
Each local health service provider is regularly briefed about the study and the midwives asked to notify us of pregnant women who may have an interest in participating. The research staff provide potential participants with verbal, written and pictorial study information with assistance in local language from community members. Written informed consent or assent (from mothers younger than 16 years of age) is obtained when the baby is 28–38 days of age and after eligibility has been confirmed.
Assignment of interventions
Allocation
Sequence generation: The allocation sequence is generated by the NHMRC Clinical Trial Centre (NHMRC_CTC) randomisation service. Minimisation is used to guarantee balance by community.
Allocation concealment mechanism: An independent centralised telephone service is employed to determine allocation at the time of the call from the study research nurse.
Implementation: The research nurse calls the automated NHMRC_CTC 24/7 randomisation service with information regarding the eligibility of the participant, and community. The allocation to one of the three study interventions is received immediately and confirmed. The allocation is also immediately provided to the study coordinator by confidential fax.
Blinding (masking): The primary outcome (immunogenicity) assessors are blinded to the vaccine group allocation. Samples are labelled with a sample random number before being transported to Melbourne for testing. Only the Menzies database manager has the code for linking the participant to the immunogenicity data. The secondary outcomes (nasopharyngeal carriage with OM pathogens and OM) are not blinded. Unblinding a participant's allocated intervention during the trial will be permissible only if requested in writing by the iDSMB following a unanimous decision by the iDSMB to do so and in accordance with the Terms of Reference.
Data collection, management and analysis
Data collection methods
Data quality: Standardised assessment and data collection forms are used. For the primary outcomes (immunogenicity), the collection method (heel prick or venepuncture) and volume of whole blood is recorded. For the nasopharyngeal swabs, the WHO recommendations are followed and swab quality is recorded according to a standardised category (good, fair, poor). For OM, a standardised form is used to record detailed clinical features of the diagnosis. Features include presence of wax, pus or foreign bodies in the canal, TM colour and translucency, position and degree of bulging, integrity and perforation size, amount of discharge. The final diagnosis for each ear is determined by the research nurse for purposes of disease management. Tympanometry is performed and results printed. Diagnostic algorithms are checked using the clinical feature scores and tympanogram. A video recording of the TM is used to assist the diagnostic decisions.
Retention: Participant retention till 7 months of age is promoted through the eligibility criteria (intention of the family and infant to remain in the community) and encouragement at each interaction with the study staff.
Data management
Data collected during each study visit are recorded on standardised forms in participant workbooks. Paper forms are returned to the Menzies office and data entered electronically by the research nurse into a MySQL database with Microsoft Access front-end, within 1 week of data collection. A protocol for changing data on paper according to Good Clinical Practice is used and all changes made electronically are tracked on the database. Microbiology data are entered directly onto the same MySQL database by the laboratory scientist. All data are checked using a series of logical checks and 10% of all electronic data are cross-checked with paper forms. An overall error rate of 1:200 precipitates a 100% check of targeted data fields and a further 10% check of all data. Immunology results are sent in an Excel spreadsheet via email to the senior database manager at Menzies to be linked with the main database at the time of unblinding.
Statistical methods
The description below describes the main analyses of the trial that will be reported.
Analyses will be by intention to treat (ITT) and according to protocol (ATP). All available outcome data that are collected at each timepoint (ITT) (table 1), and data collected within the study visit window for each timepoint (ATP) will be used. There will be no imputation of outcomes where data are missing (figure 2). It is expected that compliance will be high and will not vary according to treatment group.

{kind=link}
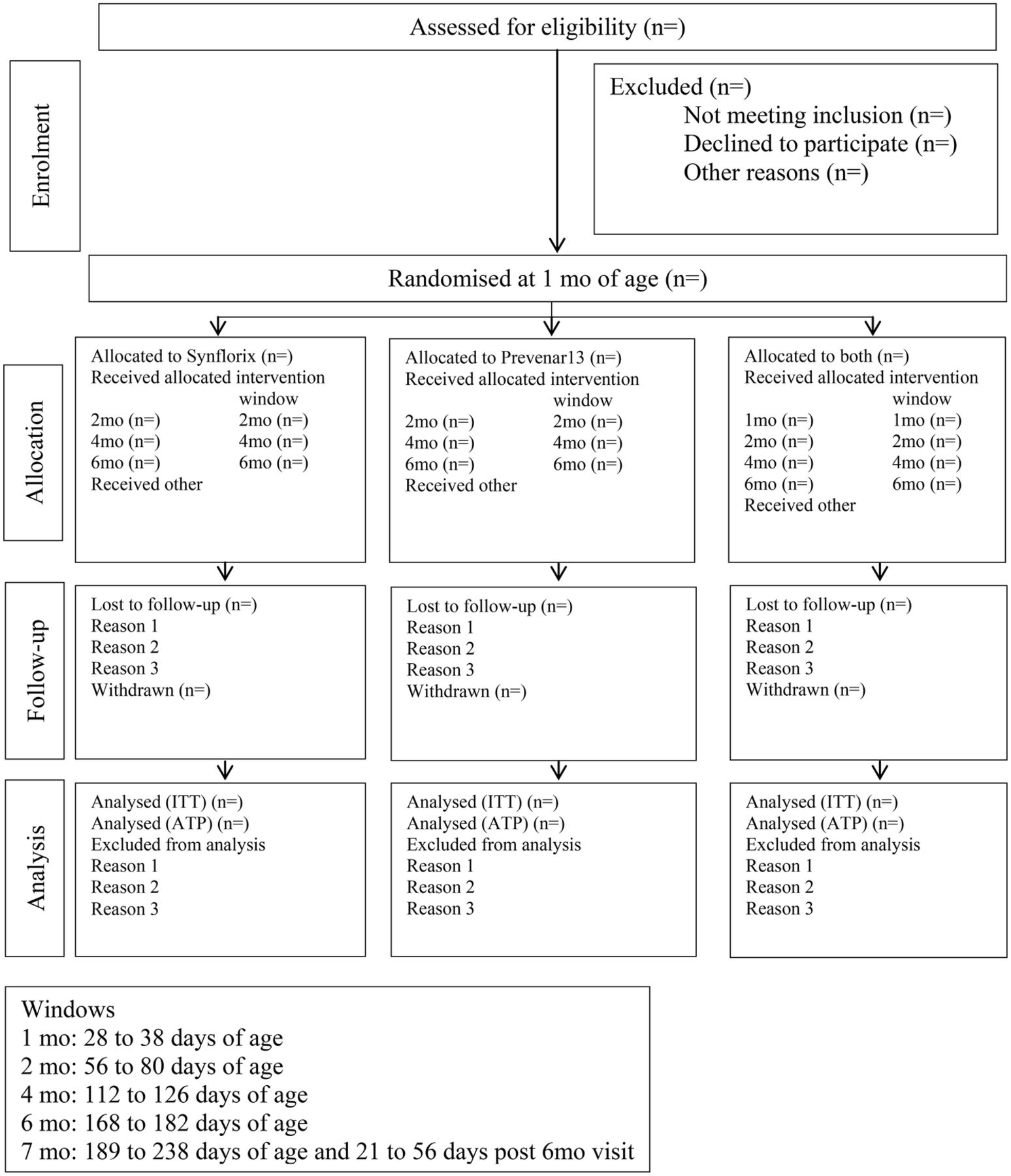{kind=link}
Participant flow chart.
The distribution of IgG for each of the pneumococcal serotypes 3, 6A and 19A as well as for HiD will be presented for the three groups at the three timepoints using (1) boxplots and (2) the proportion of infants above a threshold (pneumococcal serotype-specific IgG >0.35 μg/mL and HiD IgG >100 EL.U/mL).
Carriage data, for each of the pneumococcal serotypes 3, 6A and 19A as well as for NTHi, will be presented for the three groups (as proportions) at the four timepoints.
OM data will be presented for the three groups (as proportions) at the four timepoints.
Two-sided p values (unadjusted for multiplicity) from Mann-Whitney tests on IgG data and Fisher's exact tests comparing proportions with carriage and proportions with OM will be reported for (1) COMBO group versus PHiD-CV10 group at infant age 7 months and (2) COMBO group versus PCV13 group at infant age 7 months.
Monitoring
Data monitoring
Data monitoring committee: An independent Data Safety and Monitoring Board (iDSMB) and Terms of Reference were established before the first randomisation. The iDSMB met three monthly via teleconferences in the first year, followed by six monthly meetings. Participant recruitment and retention, protocol deviations and violations, reactogenicity, adverse events and serious adverse events are regularly reviewed. Aggregate clinical and NP carriage outcomes, and adequacy of blood collection for immunogenicity outcomes are reported. A closed session is scheduled for the end of each iDSMB meeting. Further details can be found in the PREVIX_COMBO Trial Master File held on the Menzies secure server.
Interim analyses and stopping: There are no interim analyses planned. Stopping guidelines are based on safety alone and will be a decision made by the iDSMB according to the Terms of Reference. The iDSMB will hold an extraordinary meeting to discuss modification, suspension or termination of the trial on the grounds of safety. Interim independent analyses may be requested to establish whether the trial result has been attained or will not be able to be attained. Should it be felt necessary, the iDSMB may request interim independent analyses.
Harms: Standardised data collection forms are used to extract data from the medical records on adverse events (anticipated reactogenicity and unanticipated events), including hospitalisation. Adverse reactions are directly observed during postvaccination period of 15 min. The iDSMB is informed about population trends in hospitalised cases of the disease potentially related to interventions or the probable-related adverse events. The NT Department of Health, Disease Control provides monthly data on cases of Invasive Pneumococcal Disease (total cases and cases of serotype 3, 6A and 19A by ethnicity, age and location). Notifications of hospitalised cases of bronchiolitis at Royal Darwin Hospital are also provided, as bronchiolitis is the most common cause of hospitalisation in the age group of this vaccine trial. An independent safety monitor assesses relatedness of adverse events to study intervention.
Auditing: An independent trial monitoring consultant (Clinical Network Services) has been appointed to monitor the PREVIX_COMBO trial processes and documentation at key milestones including after the first five randomisations to the COMBO group, after 50% of all randomisations and after 75% of all randomisations. The processes reviewed include study progress, identification of unreported SAEs, review of informed consent for all participants, performing source document verification of completed Case Report Forms (according to visit schedule, eg, 100% visit 1 COMBO group participants, 10% other visits), review investigational product accountability and handling, review biological sample handling and storage, review investigator site file maintenance, ensure applicability to HREC and regulatory approvals/reporting. Document and seek corrective action for all discrepancies and findings. The consultant will provide the sponsor (Menzies School of Health Research) with the report within 15 working days.
Ethics and dissemination
Research ethics approval
The PREVIX_COMBO trial has ethical approval from all relevant human research ethics committees in the Northern Territory and in Western Australia, namely the Human Research Ethics Committee of the Northern Territory Department of Health & Families and the Menzies School of Health Research (HREC—EC00153), the Central Australian Human Research Ethics Committee (CAHREC—EC00155), and the West Australian Aboriginal Health Information Ethics Committee (WAAHIEC—377-12/2011). Progress reports are provided according to each EC reporting templates.
Protocol amendments
All protocol modifications are reported to the relevant HREC in the Northern Territory and in Western Australia. Trial registries are regularly updated. Investigators, the iDSMB and other stakeholders, including participating community health services, are advised of important protocol amendments, such as those that may impact on participant safety, scientific validity, scope or ethical rigour. Substantive protocol amendments are agreed by the PREVIX_COMBO investigator team and are approved by HREC before implementation. Minor administrative amendments are documented in notes to file.
Consent or assent
Who obtains consent: Only staff who have been appropriately trained are conducting informed consent and assent. Community midwives ask pregnant women if they would like to discuss enrolling their baby in a study of baby needles for ear health. If the mother agrees, the midwife notifies the research nurses of the mothers’ contact details. Information is provided to these parents (usually the mother) in written, verbal and pictorial formats (see online supplementary appendix), including verbal translation where requested. Consultation commences during pregnancy to allow sufficient time for families to be broadly consulted. Signed consent is not required until the infant is between 28 and 38 days of age, when the eligibility criteria are confirmed. The consent process includes explanations of all elements of consent according to Good Clinical Practice, the Declaration of Helsinki, NHMRC requirements and according to local requests to ensure cultural safety (as recommended by the Indigenous Reference Group).
Ancillary studies: Additional consent is sought from parents or guardians to use participant data and biological specimens for future research relating to OM. Options to refuse each or all requests are provided, including whether permission is granted to send specimens to other laboratories and for the disposal of biological specimens.
Confidentiality
Personal information is collected on standardised paper forms. Data are collected from participants’ medical records and by face-to-face interview. Paper-based information is stored in locked cabinets with limited access at Menzies School of Health Research and electronic data is stored on a secure password protected server. Participant ID codes are used. Names are removed for analysis purposes. Electronic data are reidentifiable if required by regulatory authorities or by the HREC or iDSMB. Specimens are labelled with participant ID code and will be destroyed at the end of the study according to the participants’ requests made during the consent process.
Access to data
The final trial dataset will be under the custody of the trial sponsor, the Menzies School of Health Research. The Menzies senior data manager will have access to the full anonymised final dataset. Access to the final dataset or identifiable data by others will require written requests to be approved by the HREC, iDSMB, all study Investigators and the Director, Menzies.
Ancillary and post-trial care
All participants have access to ancillary care from their usual health care provider (local community health centre). Should the trial show benefit or harm of the any vaccine schedule, recommended vaccination schedules may change accordingly. This will not be likely to affect trial participants who will not be age- eligible for primary course vaccination by the time this information will be available.
Compensation for trial participants for trial- related harms are provided through the trial insurance and indemnity by the Menzies School of Health Research.
Dissemination policy
Plans: Trial results will be communicated in aggregate to participant families and their communities via written and oral presentations. Results will be provided for individual communities if requested (randomisation is stratified by community). Trial results will be published in peer-reviewed international journals, will be presented at relevant national and international conferences, and will be reported to local policy makers (Australian Technical Advisory Group on Immunisation, Therapeutic Goods Administration and the Pharmaceutical Benefits Advisory Committee) with the responsibility for immunisation in Australia. Additional stakeholders, including the trial funders (NHMRC and Financial Markets for Children) and vaccine manufacturers, will be informed of the trial findings. Results will be disseminated regardless of the magnitude or direction of effect. There are no publication restrictions.
Authorship eligibility
A Publication Subcommittee will be appointed to review and classify all proposed publications according to the PREVIX publication guidelines. Publications will be classified according to outcome reported (A, primary; B, secondary; C, ancillary), target audience (i, international; ii, national; iii, local) and medium (1, journal; 2, oral; 3, poster). Authorship will be proposed by lead authors and approved by the Publications Subcommittee according to journal or other guidelines. If no clear journal or other guidelines are provided, the Publications Subcommittee will refer to established recommendations of peer-reviewed international journals. The role of each author will be published. The goal is to acknowledge substantive contributions to design, conduct, interpretation and reporting of the trial through authorship, and to enhance transparency and avoid disputes or misunderstanding after trial completion. Protocol authors will be acknowledged in subsequent publications for their role in design. All NHMRC grant application chief investigators and associate investigators will be acknowledged as group authors for their role in funding, in addition to any additional individually-named authorship on the basis of criteria and approved by the Publication Subcommittee. All study staff who participate for at least one full-time year or equivalent will be named as a group author and acknowledged for their role. Where journals do not allow such groups to be acknowledged, this publication or the PREVIX_COMBO protocol will be cited as reference for the full authorship group.
Committees
The Child Health Division of the Menzies School of Health Research has convened an Indigenous Reference Group (CHD_IRG), chaired by Professor Ngiare Brown. The IRG is comprised of Indigenous members with expertise in science, ethics and Indigenous affairs. Role of the CHD_IRG is to review and advise on cultural relevance of research, from inception to implementation. Prof Leach regularly reports to the CHD_IRG and seeks their advice and feedback. CHD_IRG members: Chair: N Brown. Members: Pirrawayingi (M Puruntatameri), M Mayo, L Murakami-Gold, D Campbell, B Lee, E Hawthorn, I Raye, L Quall (secretariat).
An independent Data Safety and Monitoring Board (iDSMB) review participant recruitment and retention, protocol deviations and violations, reactogenicity, adverse events and serious adverse events. Aggregate clinical and NP carriage outcomes, and adequacy of blood collection for immunogenicity outcomes are reported. PREVIX iDSMB members: Chair: D Isaacs. Members: T Nolan, H Gunasekera, P Forder, N Woods, H D'Antoine. An independent Study Monitor (N Woods) assesses relatedness of adverse events to study intervention.
Data management group. The PI, Prof Leach, is responsible for data quality, security and regulatory requirements. The Menzies Data Management Group, led by Mrs Robyn Liddle and currently, Mr Steve Buchanan, manage the database and Ms Jemima Beissbarth manages the data dictionary and data cleaning.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Collaborators Additional investigators: PREVIX staff (current or at least one year): Coordinators: T Grierson, N Wilson. Research nurses: N Birt, Z Hayes, N Wienert, B Nankervis, K Whykes, S Sprenger, B Arrowsmith, V Coomber, R Sharp. Laboratory scientists: D Woltring, J Beissbarth. Statistician: M Chatfield. Admin: C Gage-Pearson. Causal staff: L Bell, N Glasson, M Downie, R Lennox, E Nosworthy.
Contributors NHMRC 605 810 grant application investigators: Chief Investigators (CIs): AJL (PI), EKM, MS, PJT, NJB, PM, HS-Vaughan, SS, AB; Associate Investigators (AIs): RA, JC, JM, VK, PSM. CIA Leach (Principal Investigator, PI) conceived the study, led funding applications, obtained HREC approval and other regulatory approvals, undertook consultations, reporting and had overseen day to day management and implementation of the trial, and wrote the manuscript. CIB EKM advised on study design, assisted with funding application, participated in investigator meetings, advised on risk management and read the final version of the manuscript. CIC MS advised on study design, assisted with funding application, participated in investigator meetings, advised on risk management and read the final version of the manuscript. CID PJT advised on study design, assisted with funding application, participated in investigator meetings, advised on risk management and read the final version of the manuscript. CIE NJB advised on study design, assisted with funding application, participated in investigator meetings and advised on risk management, particularly in relation to engagement of Aboriginal Medical Services and cultural matters, and read the final version of the manuscript. CIF PM advised on study design, assisted with funding application, participated in investigator meetings, advised on risk management and read the final version of the manuscript. CIG HS-V advised on study design, assisted with funding application, participated in investigator meetings, advised on laboratory protocols, particularly microbiology, and reviewed the final version of the manuscript. CIH SS advised on study design, assisted with funding application and read the final version of the manuscript. CII AB advised on study design, assisted with funding application, participated in investigator meetings and advised on laboratory protocols, particularly immunogenicity, and read the final version of the manuscript. AI RA advised on study design, assisted with funding application and read the final version of the manuscript. AI JC advised on study design, assisted with funding application and read the final version of the manuscript. AI JM advised on study design, assisted with funding application and advised with statistical matters, and read the final version of the manuscript. AI VK advised on study design, assisted with funding application and advised on immunisation policy implications, and read the final version of the manuscript. AI PSM advised on study design, assisted with funding application, participated in investigator meetings, advised on risk management and provided day to day supervision of clinical training, and read the final version of the manuscript.
Funding The PREVIX_COMBO trial is funded by the Australian National Health and Medical Research Council, NHMRC (Project Grant 605 810). The trial sponsor is the Menzies School of Health Research, Northern Territory, Australia.
Competing interests In the past 5 years, AJL has received research funds for surveillance of otitis media and OM pathogen nasopharyngeal carriage from GlaxoSmithKline (manufacturers of Synflorix) and Pfizer (manufacturers of Prevenar13). AJL has had costs of conference attendance reimbursed by GSK and Pfizer. AJL has no paid consultancies with either company. KM has served on Advisory Boards for GSK and Pfizer. GSK is providing in kind support for the Vietnam Pneumococcal trial, of which he is the PI. HSV, RMA and PSM have received research funds for surveillance of otitis media and OM pathogen nasopharyngeal carriage from GlaxoSmithKline (manufacturers of Synflorix).
Ethics approval Human Research Ethics Committee of the Northern Territory Department of Health & Families and the Menzies School of Health Research (HREC EC00153), the Central Australian Human Research Ethics Committee (CAHREC EC00155), and the West Australian Aboriginal Health Information Ethics Committee (WAAHIEC 377-12/2011).
Provenance and peer review Not commissioned; internally peer reviewed.
Data sharing statement Public Access There are no plans to grant public access to the full protocol, participant-level data or statistical code.