Article Text

Abstract
Introduction Systemic sclerosis (SSc) is a severe and costly multiorgan autoimmune connective tissue disease characterised by vasculopathy and fibrosis. One of the major causes of SSc-related death is pulmonary arterial hypertension (PAH), which develops in 12–15% of patients with SSc and accounts for 30–40% of deaths. In situ thrombosis in the small calibre peripheral pulmonary vessels resulting from endothelial dysfunction and an imbalance of anticoagulant and prothrombotic mediators has been implicated in the complex pathophysiology of SSc-related PAH (SSc-PAH), with international clinical guidelines recommending the use of anticoagulants for some types of PAH, such as idiopathic PAH. However, anticoagulation has not become part of standard clinical care for patients with SSc-PAH as only observational evidence exists to support its use. Therefore, we present the rationale and methodology of a phase III randomised controlled trial (RCT) to evaluate the efficacy, safety and cost-effectiveness of anticoagulation in SSc-PAH.
Methods and analysis This Australian multicentre RCT will compare 2.5 mg apixaban with placebo, in parallel treatment groups randomised in a 1:1 ratio, both administered twice daily for 3 years as adjunct therapy to stable oral PAH therapy. The composite primary outcome measure will be the time to death or clinical worsening of PAH. Secondary outcomes will include functional capacity, health-related quality of life measures and adverse events. A cost-effectiveness analysis of anticoagulation versus placebo will also be undertaken.
Ethics and dissemination Ethical approval for this RCT has been granted by the Human Research Ethics Committees of all participating centres. An independent data safety monitoring board will review safety and tolerability data for the duration of the trial. The findings of this RCT are to be published in open access journals.
Trial registration number ACTRN12614000418673, Pre-results.
- Systemic sclerosis
- Pulmonary arterial hypertension
- Apixaban
- Randomised controlled trial
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
This is the first clinical trial ever to evaluate the efficacy, safety and cost-effectiveness of anticoagulation as adjunct treatment in systemic sclerosis-related pulmonary arterial hypertension (PAH).
The blinded randomised placebo-controlled design of this trial is intended to minimise bias.
The choice of apixaban 2.5 mg two times a day as the anticoagulant treatment is based on consideration of the risk to benefit ratio in systemic sclerosis-related PAH. However, this study is not intended to specifically evaluate the efficacy, safety and costeffectiveness of other anticoagulant doses or drugs in this condition.
The use of a composite clinical worsening primary end point and health-related quality of life as a secondary end point is in line with the most recent expert taskforce recommendations.
Among the limitations of this study is the inclusion of patients with PAH of varying durations and not exclusively incident cases, and the use of self-reported health service usage in cost-effectiveness analysis. In addition, indirect costs are not quantified.
Introduction
Scleroderma or systemic sclerosis (SSc) is a multiorgan autoimmune connective tissue disease (CTD) characterised by vasculopathy and fibrosis, which is estimated to affect over 2 million people worldwide, with some studies indicating a rising incidence.1–3 Owing to the multiorgan nature and chronicity of the disease, SSc is associated with significant morbidity and is one of the most costly rheumatic diseases.4–7 SSc is also a life-threatening condition that carries the greatest burden of case-based mortality among the rheumatic diseases, reducing life expectancy by an average of 16 years per male patient and 34.1 years per female patient.8 It is now well established that pulmonary arterial hypertension (PAH), a condition of increased resistance in the pulmonary vasculature, is one of the leading causes of death in SSc, accounting for 30–40% of deaths in this disease.9–13 Untreated, SSc-related PAH (SSc-PAH) may follow a rapidly fatal course, with death resulting from right ventricular failure and arrhythmias.9
So-called ‘advanced’ PAH therapies target mediators of the complex pathophysiology underlying PAH (figure 1), predominantly molecules responsible for vascular remodelling, that result in an imbalance between endogenous pulmonary vasoconstriction and vasodilation.14 ,15 In SSc-PAH, these advanced PAH therapies demonstrate improved survival, exercise capacity as measured by 6 min walk distance (6MWD) and health-related quality of life (HRQoL) outcomes, compared with placebo.14–16 Prior to the advent of advanced PAH therapies in the early 2000s, the 1-year survival of patients with SSc-PAH was 45%.17 Subsequently, a systematic review of all randomised controlled trials (RCTs) of advanced PAH therapies, including patients with primary ‘idiopathic’ PAH (iPAH) and PAH secondary to CTD (CTD-PAH), reported an absolute reduction in mortality of 39% (p=0.04) with specific PAH treatment compared with placebo.18 Further, two Australian observational studies have shown improved survival with combination PAH therapy compared with monotherapy in patients with iPAH and CTD-PAH (3-year survival 85% with combination therapy vs 60% with monotherapy in CTD-PAH).19 ,20 Thus, survival has improved dramatically since the introduction of advanced therapies. However, PAH still carries a high burden of morbidity and mortality.10 ,15 Importantly, SSc-PAH continues to display the poorest prognosis compared with iPAH and other CTD-PAH subgroups.21 ,22
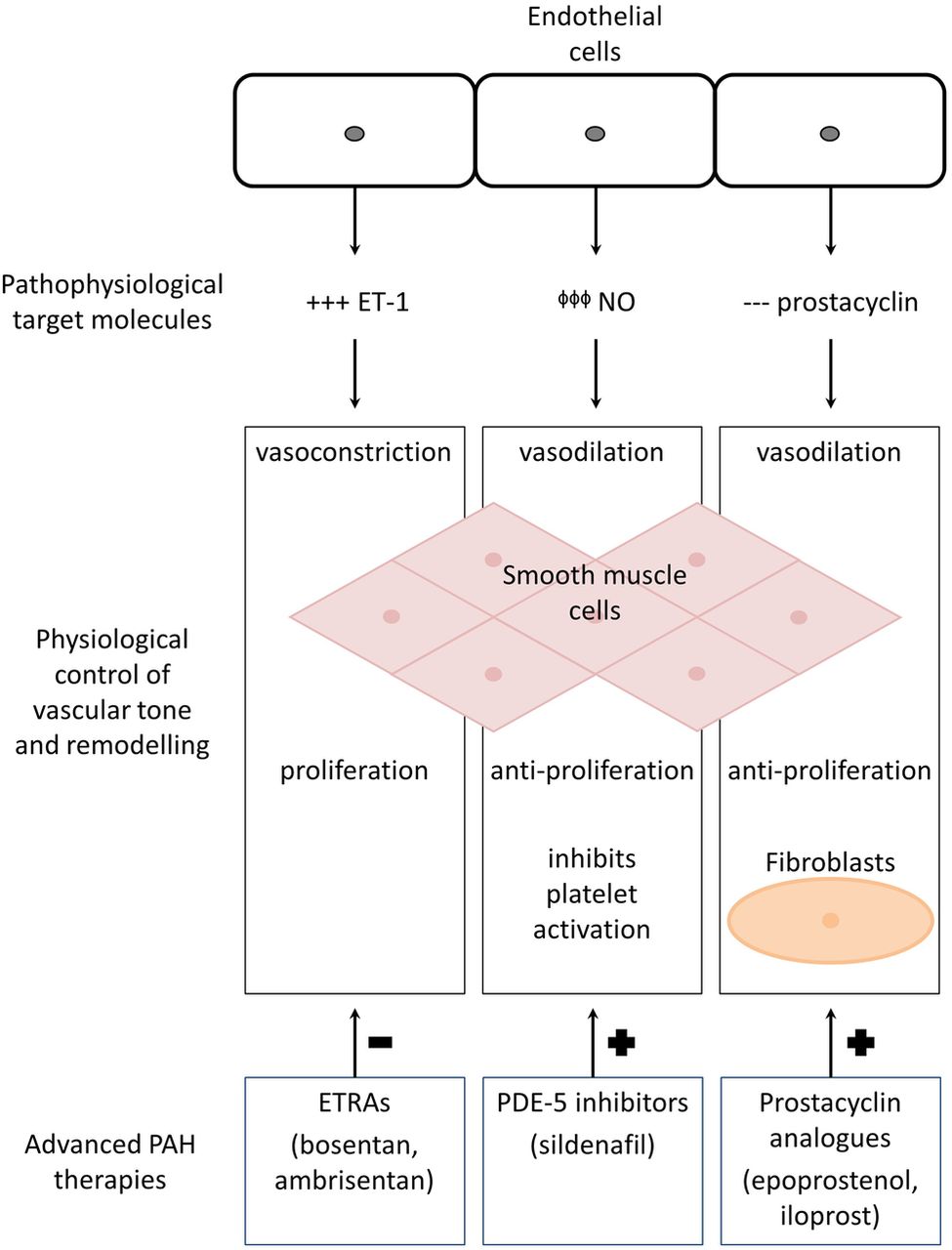
The pathophysiological targets of advanced PAH therapies. Pulmonary artery endothelial cell dysfunction impacts on vascular smooth muscle cell tone and remodelling in the following ways, targeted by the three main classes of advanced PAH therapy to prevent (–) or promote (+) the physiological mechanisms described in the centre of the diagram: (+++) overexpression of ET-1 has a potent vasoconstrictor effect. Thus, ETRAs such as bosentan and ambrisentan block vasoconstriction of pulmonary artery smooth muscle cells. (ϕϕϕ) Impaired production of NO is remedied by PDE-5 inhibitors such as sildenafil that enhance NO-mediated vasodilation. (---) Prostacyclin is a vasodilator with antiproliferative effects that is deficient in the setting of PAH. Prostacyclin analogues such as epoprostenol, treprostinil and iloprost therefore promote vasodilation in pulmonary smooth muscle cells and prevent vascular remodelling which may involve numerous cells, including platelets and fibroblasts. ET-1, endothelin-1; ETRA, ET-1 receptor antagonist; NO, nitric oxide; PAH, pulmonary arterial hypertension; PDE-5, phosphodiesterase type-5.
In situ thrombosis is a likely contributor to the pathophysiology of SSc-PAH, with pulmonary vascular (venous and arterial) thrombosis in the small calibre peripheral pulmonary vessels appearing as a common histological feature in both iPAH and CTD-PAH tissue specimens (figure 2).23–25 While several observational studies, including the Australian Scleroderma Cohort Study, have suggested a survival benefit with anticoagulation in PAH, other observational studies have not supported this finding.19 ,26–31 However, many of the patients included in these studies were not on advanced PAH therapy, and the majority had iPAH.28 ,31 In contrast, the Australian Scleroderma Cohort Study data revealed a substantial survival benefit with anticoagulation when administered in conjunction with advanced PAH therapy.19 In this CTD-PAH cohort (95% of whom were patients with SSc-PAH), exhibiting a median survival of only 5 years, an estimated 5-fold reduction in mortality was observed with warfarin treatment, prescribed at physician discretion, over an average 2.6±1.8 years follow-up.19 Furthermore, in contrast to the support for anticoagulation in European and American guidelines for treatment of iPAH, owing to the absence of RCT data, recommendations for anticoagulation in SSc-PAH are based on weak evidence and reflect a state of clinical equipoise among experts.32–36 Although pulmonary vascular pathobiology may be similar to that seen in iPAH, patients with SSc-PAH have other clinical features which may impact the risk to benefit ratio of anticoagulation. Hence, there is great variability in beliefs and prescribing habits regarding anticoagulation as adjunct therapy in SSc-PAH.26 ,37 The weight of preliminary evidence, societal costs and high morbidity of SSc-PAH demand an urgent resolution of this contentious issue through an RCT.

The pathogenic triad of systemic sclerosis-related PAH. Vasoconstriction, vascular remodelling and thrombosis constitute the pathogenic ‘triad’ of PAH in SSc (SSc-PAH). The ETRAs, PDE-5 inhibitors and prostacyclin promote vasodilation and prevent vascular remodelling, while anticoagulants may have a beneficial effect in SSc-PAH by preventing thrombosis. ETRA, endothelin receptor antagonist; PAH, pulmonary arterial hypertension; PDE-5, phosphodiesterase type-5; SSc, systemic sclerosis.
In the design of this RCT, several considerations favour the use of novel oral anticoagulants as safer, more effective and more convenient than warfarin for patients with SSc-PAH. Factor Xa is a pivotal component of the coagulation cascade, and oral factor Xa inhibitors such as apixaban and rivaroxaban, which are hypothesised to have antiplatelet and endothelial effects, may target multiple pathways critical to SSc-PAH pathogenesis.38–41 Oral factor Xa inhibitors may offer more stable blood levels than warfarin, assuming full compliance. These agents are administered at fixed doses, have fewer diet or drug interactions, are eliminated through multiple pathways and do not require routine international normalised ratio (INR) monitoring.38 ,39 The reliable bioavailability of the factor Xa inhibitors is particularly advantageous in patients with SSc, many of whom have gut hypomotility and bacterial overgrowth, which may affect warfarin and vitamin K absorption, resulting in unstable INRs.42 With up to 6% of patients with SSc exhibiting intestinal telangiectasiae or gastric antral vascular ectasiae which may bleed,43 ,44 the lower risk of gastrointestinal bleeding with apixaban, observed in large clinical trials of other patient groups, is reassuring.45–53 Finally, patients with SSc often have difficult venous access due to skin fibrosis and subcutaneous joint contractures.26 Such patients are typically reluctant to have the multiple venesections required for INR monitoring. Since oral factor Xa inhibitors do not require monitoring of blood levels and dose adjustment,38 ,39 there is potential to blind treatment assignment for RCTs and participant retention in clinical trials could possibly increase.
Objective
The aim of this study is to evaluate the efficacy, safety and cost-effectiveness of treatment over 3 years with the novel oral anticoagulant apixaban (a factor Xa inhibitor) in SSc-PAH, by undertaking a multicentre, double-blind, placebo-controlled RCT. The intervention will occur on a background of advanced PAH therapy prescribed as standard of care for participants assigned to treatment and placebo arms.
Methods and analysis
Study design
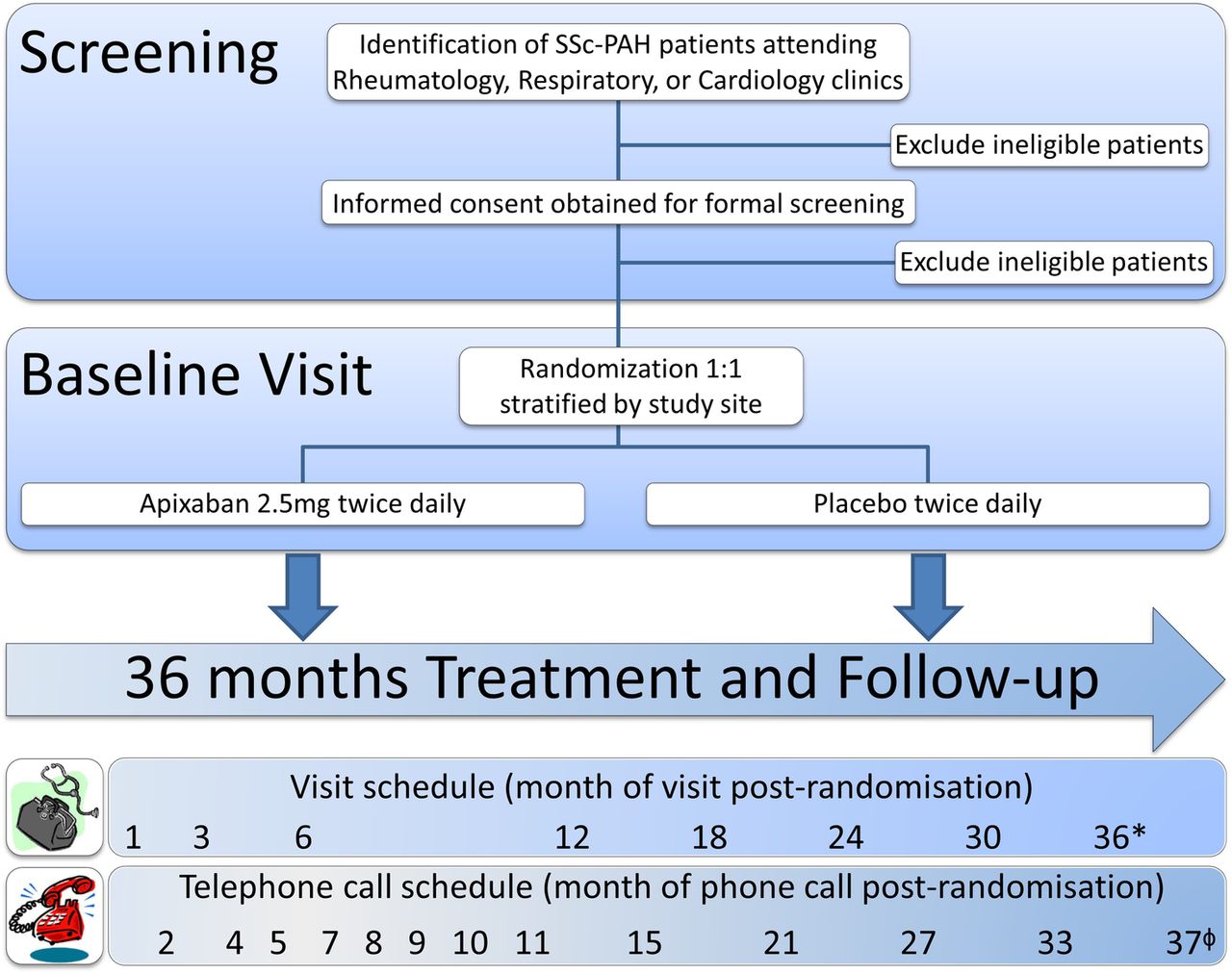
The study is designed as a multicentre, participant-blinded and investigator-blinded, placebo-controlled phase III clinical trial to compare the efficacy, safety and cost-effectiveness of apixaban 2.5 mg twice daily versus placebo, randomised in a 1:1 ratio, over a treatment period of 3 years, as additional therapy in patients with SSc-PAH who are already on advanced pulmonary vasodilators. The study design and assessment timeline is illustrated in figure 3.

{kind=link}
{kind=link}
{kind=link}
Study design and assessment timeline. During the initial stages of screening, patients with SSc-PAH will be identified via review of medical records at the multidisciplinary study sites. Formal screening assessments to confirm eligibility for the study will occur after the patient has provided informed consent. Patients who meet all inclusion criteria and none of the exclusion criteria will be randomised in a 1:1 ratio, stratified by study site, to receive double-blinded treatment with either 2.5mg apixaban or placebo, twice daily for 36 months. Over the course of study treatment, participants will visit study sites at the following times post-randomisation: 1, 3, 6, 12, 18, 24, 30 and 36 months (*=end of study visit, performed on the day of permanent cessation of the study drug, sooner than 36 months in exceptional circumstances). Telephone follow-up will occur monthly in the first 12 months, then third-monthly thereafter, between scheduled visits until 30 days after the end of study visit (ϕ=37 months post-randomisation at the latest), to ensure no adverse events have occurred and to capture all healthcare usage, including changes to concomitant medication. SSc-PAH, scleroderma-related pulmonary arterial hypertension.
Study population
Study participants will be identified by cardiologists, rheumatologists and respirologists during the course of routine care at 13 Australian PAH treatment centres across six states (New South Wales, Queensland, South Australia, Tasmania, Victoria and Western Australia). Recruitment will take place over 24 months or until sample size requirements are met and participants will be treated for 36 months. Participants will be adult men and women with symptomatic SSc-PAH as defined by the American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) 2013 classification criteria for SSc54 and international guidelines for diagnosis of PAH.35 ,54 Inclusion and exclusion criteria are listed in tables 1 and 2, respectively. Many of the exclusion criteria focus on reducing the risk of adverse bleeding events in the study population.44 ,50 All eligible participants will sign informed consent prior to study enrolment, following adequate explanation of the aims, methods, objectives and potential hazards of the trial by the responsible investigator.
The SPHInX study inclusion criteria
The SPHInX study exclusion criteria
Randomisation and allocation concealment
Ethical considerations mandate background treatment with advanced PAH therapies as standard of care in all study participants.35 ,36 Since sites differ in rates of use of PAH-specific therapies, including combination therapy, randomisation will be stratified according to site, with the effect of various PAH therapies subsequently adjusted for in multiple regression analysis.
Randomisation to placebo or study drug in a 1:1 ratio will be performed by a statistician who is not associated with any study site, using computer-generated block randomisation, stratified according to study site. After the investigator obtains informed consent and confirms eligibility, patients who meet all inclusion criteria and none of the exclusion criteria will be assigned to study treatment by the site pharmacist at baseline visit, according to the site randomisation schedule.
Participants, healthcare providers, investigators, data collectors and outcome assessors will be blinded to treatment assignment. To ensure allocation concealment, the appearance of the investigational drug (apixaban, BMS-562247, Bristol-Myers Squibb Limited (BMS)) and its packaging will be indistinguishable from the matching placebo, both manufactured by BMS. The labelling and packaging of apixaban and matching placebo will be conducted according to Good Manufacturing Practice, Good Clinical Practice and national regulatory requirements, coordinated by the study lead pharmacy.
A password-protected restricted access electronic database of all randomisation codes will be kept for emergency unblinding purposes. If any participant experiences a medical emergency where management would be improved by knowledge of the blinded treatment assignment, unblinding will be available 24 hours per day. A set of tamper-proof sealed envelopes containing the blinding code for each participant will be kept at each site in case contact with the database server fails. The integrity of these sealed envelopes will be periodically checked. A log of every access to the unblinding codes will be kept and all requests for unblinding must be clearly justified.
Treatment exposure and compliance
The study drug will be administered orally, twice daily as 2.5 mg tablets of apixaban or matching placebo, with a dose interval of ∼12 hours. Participants will be asked to return all the unused study drug at follow-up visits and to self-report any missed doses of therapy. Study drug adherence will be assessed by recording quantities of the returned study drug at each follow-up visit. Participants will cease using the study drug 36 months after initiation at the baseline visit.
The study design mandates the concomitant use of at least one advanced pulmonary vasodilator, such as an endothelin-1 receptor antagonist (ETRA) and/or a phosphodiesterase type-5 (PDE-5) inhibitor. However, these therapies must be at a stable dose for at least 2 months prior to baseline. Permissible concomitant medication includes diuretic therapy, provided that a stable dose was maintained for at least 1 month prior to baseline; one antiplatelet agent will be allowed at the physician's discretion. However, the combination of clopidogrel or ticagrelor and aspirin is not allowed due to increased risk of bleeding.51 Prohibited concomitant medications from 1 month prior to baseline until study drug cessation include any investigational drug other than the study drug; oral or subcutaneous anticoagulation with warfarin, apixaban, rivaroxaban, dabigatran, enoxaparin, dalteparin or heparin. Participants must not be receiving continuous intravenous infusion of epoprostenol or iloprost for PAH at baseline or be planned to initiate this therapy within the next 3 months. However, the following exceptions may apply following study initiation: (1) the study drug may be temporarily suspended to receive prophylactic anticoagulation during a therapeutic or surgical procedure if this is deemed in the participant's best interest; and (2) addition of intravenous epoprostenol to oral advanced PAH therapy for participants in the modified New York Heart Association/WHO (NYHA/WHO) functional class (FC) IV failing ETRAs and PDE-5 inhibitors.35 Short-term treatment with IV prostacyclin for severe Raynaud's phenomenon or digital ulcers may be administered at any time during the study without constituting a clinical worsening event (CWE).
Concomitant medications will be monitored closely from 1 month following the baseline visit. Participants will be required to self-report all changes to therapy throughout the study treatment period using a healthcare usage diary. Initiation of any new PAH-specific treatment or a dose increase of such a drug without adjudicated clinical worsening of PAH is strongly discouraged during the study period. If continued administration of the study drug is believed to be contrary to the best interests of the participant (ie, adverse event, diagnostic or therapeutic procedure, laboratory abnormalities, pregnancy, unblinding or withdrawal of consent), interruption or permanent discontinuation of the study drug is mandated. Participants will resume the study drug as long as the investigator feels it is safe for them to do so and no more than 8 weeks of study treatment has been missed. Participants who prematurely discontinue the study drug for any reason will not be replaced and, unless they withdraw consent, will continue to be followed up six-monthly until 36 months from baseline.
Study assessments
The study assessment schedule is illustrated in figure 2, starting with screening and ending with follow-up 30 days after permanent cessation of the study drug. Additional visits may also take place at any time during the treatment period in case of a suspected CWE. Screening assessments to confirm study eligibility may occur at any time prior to randomisation, or be completed on the same day as the baseline visit. Adverse event surveillance is prioritised at follow-up assessments. With reference to their healthcare usage diary, participants will be required to self-report all healthcare usage (ie, visits to healthcare/allied health practitioners and hospitalisations), side effects and pregnancy test results if applicable. Data collection requirements over the duration of the study are described in table 3. All data collected will be entered de-identified into a customised electronic case report form, created on the REDCap platform, that is password protected and stored securely on the central server at St Vincent's Hospital Melbourne. Hard copies of source documents will be retained for 5 years following the end of the study.
Data collection requirements over the duration of the study
The 6 min walk test (6MWT) will be performed in a standardised, non-encouraged fashion, measuring the walking distance covered by the patient during a 6 min period followed immediately by the Borg dyspnoea index, which rates dyspnoea severity on a visual analogue scale from ‘0’ to ‘10’.55 The following validated HRQoL questionnaires will be completed by the patient: the Medical Outcomes Study 36-Item Short Form Health Survey (SF-36),56 the scleroderma-modified Stanford Health Assessment Questionnaire (sHAQ)57 ,58 and the Cambridge Pulmonary Hypertension Outcome Review (CAMPHOR).59 The 6MWT and HRQoL questionnaires will be omitted from visit 2 (1 month post-randomisation), which will serve as an abridged safety assessment only, unless there is a suspected CWE.
Serum and platelet-free plasma samples collected at baseline, 6-month and 24-month follow-up visits will be stored at −80° C for the N-terminal prohormone of the B-type natriuretic peptide (NT-proBNP) assay and exploratory biomarker testing.60 ,61 Factor Xa levels in platelet-free plasma specimens will also be compared between baseline and 6 months to reflect bioavailability of apixaban in the treatment group.62 Anti-factor Xa assays and biomarker assays will be performed for all samples in triplicate, in a single laboratory, at the conclusion of the study.
Outcome measures
In line with the Task Force on Endpoints and Clinical Trial design recommendation for phase III trials at the Fourth World Symposium on pulmonary hypertension in Dana Point, California,63 a composite primary end point will be employed, providing measurable parameters to support an independent adjudication of ‘time to clinical worsening’. The primary end point will be time from randomisation up to 36 months to the first adjudicated CWE from the composite parameters listed in table 4. CWEs will be adjudicated in a blinded fashion by an end point adjudication committee consisting of four of the investigators (MN, WS, DP, SMP) who will adjudicate each event independently and then meet to discuss any that were not unanimously agreed on. The study drug will be continued in a blinded fashion after a CWE is adjudicated, to enable quantification of the total number of CWEs during the study period as a secondary end point.
Definition of measurable composite primary end point parameters
Selection of secondary end points was informed by Expert Panel recommendations for a ‘core set’ of outcome measures to be used in clinical trials of new therapies in SSc-PAH.64 Secondary efficacy end points include all-cause mortality; absence of worsening in the NYHA/WHO functional class; change in 6MWD and the Borg dyspnoea index; change in the SF-36, sHAQ and CAMPHOR questionnaire subscales. Secondary end point comparisons will be evaluated from baseline to each of the 12-month, 24-month and 36-month follow-up time points, adjusted for time since diagnosis of PAH. The last valid post-baseline value will be carried forward to compensate for any missing values at each time point.
Safety and tolerability end points will comprise treatment-emergent adverse events (serious and non-serious) including marked laboratory abnormalities up to 7 days after the last study drug intake, adverse events leading to premature discontinuation of study drug, change from baseline to end of study in vital signs. Health economic end points will include number per year, and associated costs, of all-cause and PAH-related hospitalisations and in-patient hospital days, general practitioner, specialist visits, allied health service usage and initiation of new medications.
In participants who discontinue the study, where possible, CWE will be captured every 6 months to the end of 36 weeks from enrolment.
Sample size estimation
Sample size was calculated based on a comparison of two survival curves for the primary outcome of clinical worsening over 3 years, applying the method of Rubinstein et al.65 This method uses median survival rather than event rate. The following variables were used to determine sample size: (1) α=0.05, two-sided; (2) β=0.2 (power 80%); (3) difference to be detected expressed as an HR of placebo:treatment=2.0, based on previous Australian observational data, but reduced by 60% to provide a more conservative estimate for the purposes of an RCT;19 (4) control group median survival=45.6 months also based on previous Australian observations;19 (5) ratio of participants randomised to control and experimental groups=1:1; (6) block randomisation stratified according to 13 centres; (7) duration of recruitment=24 months; (8) duration of follow-up=36 months; (9) expected attrition=10%. However, substantial loss to follow-up is unlikely as trial participants are required to attend for regular review to continue receiving PAH therapy subsidised under the Pharmaceutical Benefit Scheme (PBS). Based on these assumptions, it is expected that 65 events will be observed in this study and a total sample size of 170 participants (85 per arm) is required.
Statistical analyses
The hypothesis to be tested is: null hypothesis (H0)=the distribution of the primary end point is the same in the treatment groups; alternative hypothesis (H1)=the distribution of the primary end point in the placebo group differs from the distribution in the active group. The ratio of the hazards of a CWE in the two groups is not expected to change over time. Therefore, the use of methods requiring proportional hazards is considered appropriate. The main analyses for the primary and secondary end points will test the null hypothesis by means of the log-rank and Wilcoxon tests, performed in the intention-to-treat population, which includes all participants randomised. No adjustment for covariates is planned for the primary analysis. However, in order to evaluate the robustness of results, the primary end point will also be analysed on the per-protocol set, with 80% used as the cut-off to define an adherent patient. Supportive analyses will be conducted using appropriate covariates (eg, the date PAH was first diagnosed by right heart catheterisation (RHC), the start date of concomitant PAH medications and combination PAH therapy) in a Cox regression model.
The time to occurrence of the first CWE up to 30 days after the last study drug intake will be described by the Kaplan-Meier survival curves. The HR of placebo:treatment with two-sided 95% CIs of the event-free proportion estimates at relevant time points will be presented for each treatment group in graphical and tabular form, in addition to descriptive statistics to summarise patient and disease characteristics. No imputation method will be used for the primary end point and if there is a missing assessment (eg, no confirmatory 6MWT or NYHA/WHO FC) for a CWE; the end point adjudication committee will be responsible for qualifying or disqualifying such events before primary end point analysis. Patients without a CWE permanently discontinuing treatment will be censored 30 days after study treatment discontinuation or date of last contact.
Differences in baseline characteristics of patients in the apixaban and control arms will be compared using univariate methods (χ2, t-tests and Mann-Whitney tests). Univariate and multivariable methods (logistic and linear regression) will be used to compare differences in echocardiographic parameters, 6MWD, NYHA/WHO FC, NT-proBNP level and HRQoL in the apixaban and control arms at 1, 2 and 3 years. Covariates included in multivariable analyses will include specific PAH therapy, cardiovascular medications and immunosuppressives. Sensitivity analyses will be performed to evaluate the effect of poor treatment adherence and loss to follow-up in patients whose fate is unknown at the end of the study. No interim efficacy analyses are planned at this stage. However, a planned re-estimation of the sample size may be performed prior to the expected closure of recruitment, based on the observed blinded event rate in the composite end point.
Predefined subgroup analyses include a comparison of efficacy and safety in incident versus prevalent PAH, limited versus diffuse SSc disease subtypes and according to the autoantibody profile (anti-centromere anti-nuclear antibody vs anti-topoisomerase antibody). All statistical analyses will be performed by a biostatistician using STATA software.
Cost-effectiveness analysis
On completion of the RCT, a health economic analysis will be undertaken to determine the incremental cost-effectiveness ratio, in terms of ‘net costs’ per unit of ‘health gain’. Net costs will comprise the costs of treatment with apixaban and advanced PAH therapies for the duration of life-years gained, minus costs saved from hospitalisation and health service usage in the same 3-year time period. In order to enable this type of analysis, we will collect detailed usage data for medications, primary care, outpatient consultations, emergency department and elective hospitalisations, through participant health service usage diaries, questionnaires administered at study contact and source databases of the participating hospitals. Since actual costs of health service usage are not recorded, in cost-effectiveness analysis, we are making the assumption that the unit cost assigned to each service in the Medicare Benefits Schedule (MBS) is an accurate estimate of true costs.
Collection of time-to-event data and HRQoL data will enable calculation of quality-adjusted life years (QALYs) gained by the inclusion of anticoagulation therapy. Depending on the findings of the initial cost-effectiveness analysis, further economic modelling beyond 3 years, using the Markov approach, may be required.
We will be applying a 5% annual discount rate to projected future costs and benefits. We will undertake sensitivity analyses to test the robustness of our cost-effectiveness results, and to identify key input parameters to which the results are most sensitive. Both ‘one-way’ sensitivity analyses and ‘multi-way’ probabilistic sensitivity analyses will be undertaken.
Ethics, safety monitoring, auditing and access to data
Ethical approval of this trial has been acknowledged by the Governance offices of all hospitals involved in the trial (Fiona Stanley Hospital, Gold Coast University Hospital, Liverpool Hospital, Monash Health, Royal Adelaide Hospital, Royal Hobart Hospital, Royal Prince Alfred Hospital, The Alfred Hospital and The Queen Elizabeth Hospital). The findings of this RCT are to be published in open access journals, with none of the participants identifiable.
An independent Data and Safety Monitoring Board (DSMB), comprising a rheumatologist, haematologist, cardiologist and gastroenterologist, will review unblinded safety and tolerability data at three-monthly intervals to ensure safety of participants for the duration of the study. Members of the DSMB are independent of the study investigators and are free of competing interests. A formal DSMB charter has been produced for this study.
The randomisation code will not be broken and made available to investigators, including the study statistician, until after data analysis is complete.
The project coordinator based at St Vincent's Hospital Melbourne will audit the trial conduct and data entry every 3–4 months and will undertake site visits. At this stage, no independent audit of trial conduct is planned but would occur at the request of the DSMB or regulatory bodies.
Only the lead chief investigator, trial coordinator and biostatistician will have access to the final unblinded trial data set for the purpose of analysis and dissemination of the findings from this study. None of these team members will have access to unblinded trial data prior to the completion of the study.
Communication with investigators and dissemination of findings
Any protocol modifications such as changes to eligibility criteria and analysis plans will be communicated by the lead chief investigator (MN) to the principal and associated investigators, and affected trial participants, through personal communication including emails and teleconferences and circulation of written documents including an amended study protocol. Authorship of papers arising from this study will be based on contribution to the study including intellectual content.
Study limitations
Limitations of this study include the inclusion of patients with PAH of varying durations and not exclusively incident cases, as well as the use of self-reported health service usage in cost-effectiveness analysis. In addition, in this study, indirect costs are not quantified.
Discussion
The design of this clinical trial was not without its challenges. Numerous studies have demonstrated that survival in SSc-PAH declines precipitously over the first 3 years following diagnosis and thereafter plateaus, with an overall median survival of 5 years.19 ,21 ,22 ,42 ,66 Furthermore, registry studies have shown that prevalent cohorts of patients with PAH have better overall survival than incident cohorts, suggesting that there may be survivor bias in patients with long-standing PAH.19 Therefore, in an RCT of a novel therapy for SSc-PAH where the end point is a combination of mortality and clinical worsening, it would be ideal to limit enrolment to those with <3 years' duration since diagnosis of PAH on RHC. However, given the low disease prevalence, this restriction could limit enrolment of an appropriate sample size in a timely manner. Despite these more generous inclusion criteria, the recruitment of a sufficient number of patients to power this clinical trial remains the biggest challenge to its timely completion. The investigators are currently in the process of enlisting more recruitment sites to meet sample size requirements.
Since the novel oral anticoagulants are unable to be readily reversed, safety considerations were of utmost importance to study design. The overall drug safety profile indicates that apixaban is generally well tolerated with an elimination half-life of 12 hours.39 For stroke prevention in atrial fibrillation, apixaban administered in a ‘full dose’ (5 mg two times a day) has been demonstrated to be superior to warfarin for stroke prevention (p=0.01), with lower risk of bleeding (p<0.001).47 Similarly, there is emerging evidence that apixaban administered in ‘low dose’ (2.5 mg two times a day) may yield comparable efficacy to full-dose apixaban in certain clinical settings, such as thromboprophylaxis postarthroplasty or treatment of venous thromboembolism, with no increased risk of bleeding.45 ,46 ,52 Furthermore, in acute coronary syndromes, full-dose apixaban demonstrated a 2.45-fold increased risk of bleeding compared with placebo (p=0.005), whereas an increased risk of bleeding was not observed with low-dose treatment (p=0.09).53 Therefore, in our study, treatment comprising ‘low-dose’ apixaban should offer safety comparable to placebo, without compromising efficacy.
While a comparison with warfarin would have been interesting, from a practical and safety point of view, this was not possible. First, SSc-PAH is an infrequent condition and the addition of a third arm to the study would have increased sample size requirements, posing a serious threat to the feasibility of this study. Furthermore, two recently published studies have cast doubt over the safety of warfarin relative to its potential efficacy in SSc-PAH, suggesting that this treatment may in fact be harmful in this group of patients.29 ,30 Possible reasons for this include the presence of SSc disease features such as gastrointestinal tract hypomotility and bacterial overgrowth affecting the absorption of warfarin, and the presence of gastric antral vascular ectasia and intestinal telangiectasia, which place patients at risk of gastrointestinal bleeding. For these reasons, as well as those discussed earlier, we have not included a warfarin arm in this trial.
Non-anticoagulant effects of heparins have been described.67 The potential of such actions is currently being investigated for the novel anticoagulants, further supporting our choice of anticoagulant in this study.68
The clinical impact of this study is likely to be realised in the near term and the scope for cost-savings from reduced need for hospitalisations is considerable. Owing to the high cost of pharmacotherapy in SSc-PAH, it was important to build a health economic analysis into this study to determine cost-effectiveness of adjunct anticoagulation. If anticoagulant therapy is successful at prolonging life in SSc-PAH, patients will spend a greater period of time on costly advanced PAH-specific therapies (typically approaching $A40 000 per drug, per patient year).69 ,70 Therefore, HRQoL outcomes must be balanced against these costs. In Australia, there is no official threshold for incremental cost-effectiveness ratios, although $A50 000 per QALY saved is commonly used. The WHO recommends the use of gross domestic product per capita as a starting point to consider cost-effectiveness thresholds for a country, which for Australia at present is ∼$A88 000.71
This study seeks to determine the efficacy of a novel therapy with the goal of improving survival in a disease with very high short-term mortality. Until now, there have been no published RCTs of anticoagulation in SSc-PAH and there are no other trials currently registered in the WHO trials portal. Blinding of treatment assignment is an innovative feature of our study design as the majority of oral anticoagulation studies have been open label. Positive findings in this study may provide a rationale for further studies of factor Xa inhibition in other pulmonary vascular diseases, including iPAH. Thus, positive findings may have far-reaching implications beyond SSc. If the findings are negative, patients will be spared the potential risk, inconvenience and cost of anticoagulation. Since 30% of patients with SSc-PAH are being anticoagulated at present in clinical practice,19 this presents a unique situation where a negative study may be as important in terms of changing practice as a positive study. Regardless of the outcome, our study has the potential to redefine the standard of care in a disease entity where there is much uncertainty.
References
Footnotes
Contributors AC participated in the design of the study and coordinated the drafting of the manuscript. WS, DP, HN, EG, SMP, TW, DC, JS, PKKW, VT, ND, JW, WC, MS and RB made substantial contributions to the conception and design of the study. MN conceived of the study, coordinated its design and drafted the manuscript. All authors read and were involved in critically reviewing and revising important intellectual content of the manuscript. They also approved the final manuscript prior to submission.
Funding This study is financially supported by a peer-reviewed 5-year Project Grant (APP1062638) from the National Health and Medical Research Council of Australia (NHMRC). MN holds an NHMRC Research Fellowship (APP1071735). RB holds an NHMRC Senior Research Fellowship (APP1082138). The study drug and matching placebo are being supplied at no cost by Bristol-Myers Squibb Pty. Bristol-Myers Squibb were permitted to review the manuscript and make suggestions, but the final decision on content was exclusively retained by the authors. MN has received research support from Actelion, GlaxoSmithKline and Pfizer.
Disclaimer The contents of the published material are solely the responsibility of the individual authors and do not reflect the views of the NHMRC.
Competing interests None declared.
Ethics approval Ethical approval for this trial has been granted by the Human Research Ethics Committees of St Vincent's Hospital (Melbourne), the Royal Perth Hospital, the University of Western Australia, the Menzies Research Institute of Tasmania.
Provenance and peer review Not commissioned; externally peer reviewed.