Article Text

Abstract
Introduction Preeclampsia is a leading cause of maternal and perinatal morbidity and mortality. There is a need for adjuvant, targeted therapies to improve outcomes. Broccoli sprout extract, rich in the antioxidant sulforaphane, reduces oxidative stress and placental secretion of the antiangiogenic factors that contribute to vascular dysfunction in preeclampsia. We propose a phase III trial investigating broccoli sprout extract. We will assess broccoli sprout extract in women with early onset (<34 weeks) preeclampsia, investigating (1) the interval between enrolment and delivery (days), (2) biomarkers of placental and endothelial function and (3) maternal and fetal outcomes.
Methods A double-blind, placebo-controlled randomised trial will be conducted at Monash Health, Melbourne, Australia. One hundred and eighty women (45 each arm of each stratum) with early onset preeclampsia (defined as per Society for Obstetric Medicine of Australia and New Zealand guidelines) will be recruited. Consenting women will be randomised to receive an oral dose of either broccoli sprout extract (24 mg of activated sulforaphane) or identical placebo, twice daily until delivery. Maternal blood will be collected antenatally for measurement of biomarkers of preeclampsia, including soluble fms-like tyrosine kinase 1 (sFlt-1), placental growth factor (PlGF), soluble endoglin (sEng) and activin A, as well as circulating sulforaphane metabolites. Maternal and perinatal outcomes will be monitored throughout. All clinical care decisions, including the timing of delivery, will be made by the treating team, blinded to treatment allocation. Participation in this trial will not affect routine care. At delivery, maternal and cord blood and placentae will be collected to measure sulforaphane metabolites and sFlt-1, PlGF, sEng and activin A.
Ethics and dissemination Approval to conduct the trial has been granted by Monash Health Human Research and Ethics Committee (RES-18-0000-109A). Deidentified data will be published in peer-reviewed journals and presented at learnt society conferences, both nationally and internationally. This study has not yet commenced and is pre-results.
Trial registration number
- Preeclampsia
- broccoli sprout
- sulforaphane
- antioxidant
- clinical trial
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Study design is a double-blind, randomised, placebo-controlled trial.
Intervention is a naturally occurring nutritional supplement with an excellent safety profile.
Sample size not adequate for secondary outcomes.
Study participants restricted to women with early onset preeclampsia.
Introduction
Preeclampsia is defined as new onset hypertension after 20 weeks’ gestation with associated maternal organ dysfunction and/or fetal growth restriction.1 It complicates 5%–8% of pregnancies and is a leading cause of maternal and perinatal morbidity and mortality worldwide.1 Even in high-resource settings the risk of neonatal mortality is fivefold greater in those born to a mother with preeclampsia compared with those born to a normotensive mother. This increased mortality is largely due to associated fetal growth restriction and the need for premature delivery. Indeed, preeclampsia is the leading cause of iatrogenic premature delivery, implicated in 20% of all premature births.1 Unfortunately, the incidence of preeclampsia has not changed over the last century and, beyond controlling maternal blood pressure, we continue to lack effective targeted therapies for this serious disorder.1 2
Though much remains unknown about the pathological progression of preeclampsia, it is broadly accepted that a placenta, chronically injured by ischaemic-reperfusion insult, releases excessive vasoactive and inflammatory factors into the maternal circulation. In turn, these factors induce systemic maternal endothelial dysfunction.3 The resulting vasoconstriction and increased vessel permeability cause hypertension, oedema, renal endotheliosis and secondary organ ischaemic injury. For the last 50 years, the pharmacological management of preeclampsia has aimed solely to correct the maternal hypertension, allowing safer continuation of the pregnancy in the interests of improving fetal maturity. While the focus on controlling hypertension has improved maternal and perinatal outcomes it has neglected the underlying pathological processes of the disease and limited the potential gains in mitigating fetal risk, particularly in the setting of early onset disease.1 Seeking to prolong the pregnancy further by targeting the oxidative stress-induced endothelial dysfunction is an additional approach worth exploring.
In particular, inducers of the nuclear factor E2-like related factor 2 (Nrf2) antioxidant pathway offer an attractive approach. Inducing Nrf2 would be expected to have anti-inflammatory and antioxidant effects in both the placenta and in the maternal vasculature. Nuclear factor E2-related factor 2 is an endogenous inducer of cellular antioxidants.4 5 Under physiological conditions, bioavailable levels of Nrf2 are regulated by cytosolic binding to kelch-like ECH-associated protein 1 (KEAP-1), preventing rapid proteasome degradation.5 Exposure to oxidative stress induces cysteine modifications to KEAP-1, loss of binding to Nrf2 and translocation of Nrf2 to the nucleus.4 Within the nucleus, by combining with small maf-proteins in the promoter region of antioxidant ‘safeguarding’ genes, Nrf2 stimulates antioxidant response elements resulting in the transcription of mRNA for a number of cellular antioxidants and phase II enzymes.4 Numerous studies have shown therapeutic benefits from Nrf2 stimulation both in maintaining endothelial health and in treating vasculopathies.6
The Nrf2 inducer sulforaphane is a naturally occurring organosulphur abundant in broccoli sprout extract7–9 that has attracted attention in cardiovascular and cancer medicine.7 8 It stabilises Nrf2 by impairing ubiquitination and increasing Nrf2 phosphorylation, thereby preventing proteasomal degradation and causing cytosolic accumulation.5 Sulforaphane also induces cytosolic transcription and nuclear translocation of Nrf2. As such, sulforaphane uses the Nrf2 pathways to enhance production of phase II and antioxidant enzymes, improving cellular resilience to oxidative stress.4 10
Rationale
Preeclampsia remains a leading cause of maternal and perinatal morbidity and mortality worldwide.1 While the introduction of antihypertensives 60 years ago represented a major advance in the care of women with preeclampsia, further progress has all but stalled. Future benefits in maternal and/or perinatal outcomes are likely to come from improved screening and prevention11 or from more effective treatment, beyond simply managing maternal hypertension.12 13 In particular, therapies that target the maternal endothelial dysfunction that underlies the hypertension offer promise in further improving maternal and perinatal outcomes. The antioxidant and anti-inflammatory sulforaphane may be one such therapy. Preliminary data from our group support a role for sulforaphane in reducing placental production of the antiangiogenic factors soluble fms-like tyrosine kinas 1 (sFlt-1) and activin A. We have further shown that sulforaphane improves endothelial cell health and function after activation with tumour necrosis factor alpha and serum from pre-eclamptic women. Whether sulforaphane has beneficial in vivo effects on placental and/or endothelial function in women with early onset preeclampsia remains unexplored. We aim to examine this possibility in our clinical trial, Prolong.
Aims and hypothesis
We hypothesise that administration of Broccomax will significantly increase duration of pregnancy, specifically the interval between diagnosis of preeclampsia and delivery.
The overarching aim of this trial is to assess the utility of a commercial broccoli sprout extract (BroccoMax) as an adjuvant therapy in the management of women with early onset (<34 weeks) preeclampsia.
Aim 1. To assess whether broccoli sprout extract can safely prolong the interval between enrolment and delivery (recorded in days) in women with early onset (<34 weeks) preeclampsia.
Aim 2. To assess the effects of a broccoli sprout supplement on production of maternal circulating biomarkers of placental and endothelial health in women with early onset (<34 weeks) preeclampsia.
Aim 3. To assess effects of a broccoli sprout extract on maternal and perinatal outcomes (safety and tolerance) in women with early onset (<34 weeks) preeclampsia.
Methods and analysis
Study design
Double-blind, randomised, placebo-controlled superiority trial (figure 1).
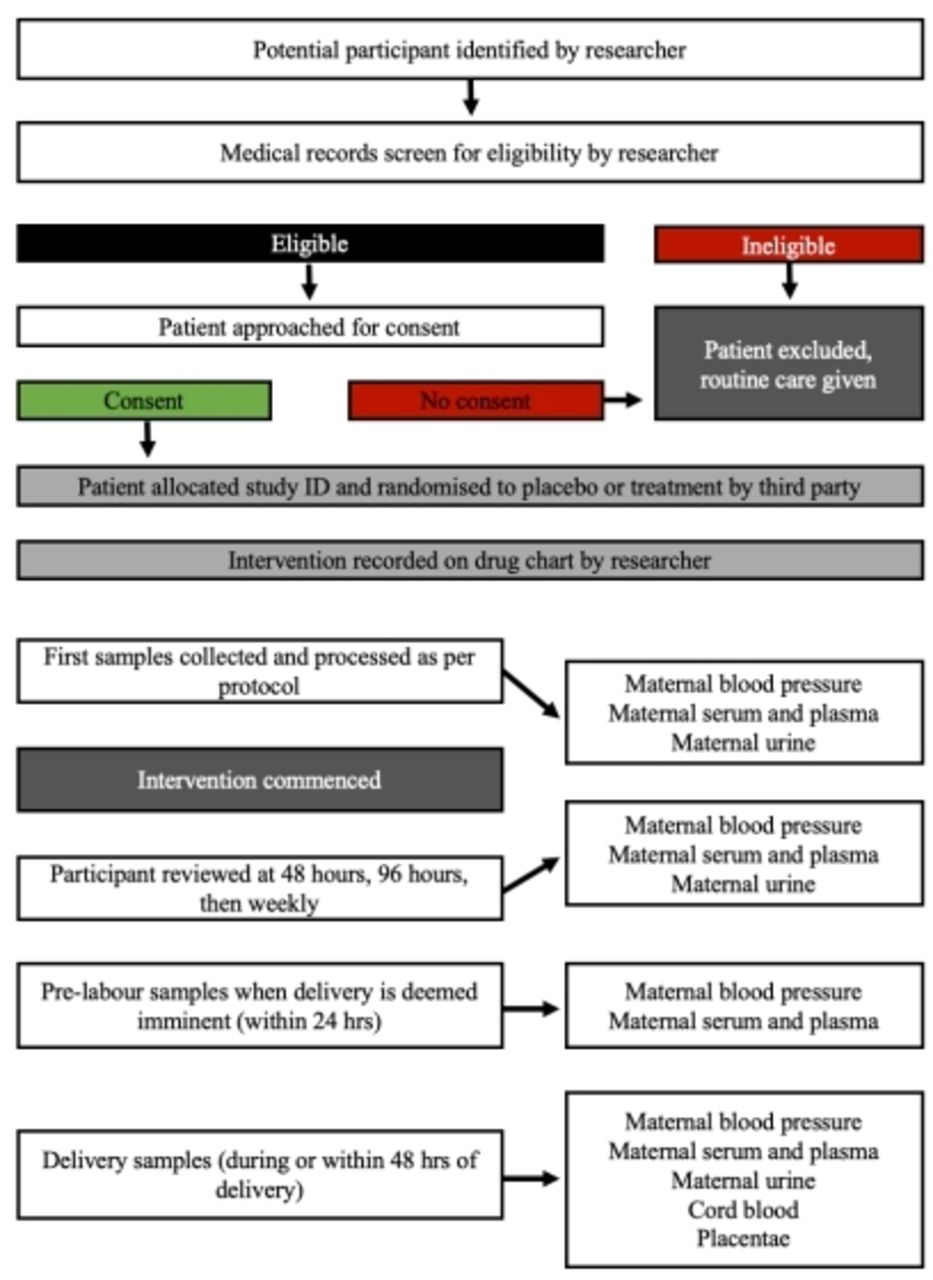
Flowchart indicating participant recruitment, enrolment and sample collection. Potential participants will be identified from the labour ward and clinic and will be screened for eligibility by the research team. Eligible women will be approached for consent to participate. Where a woman is not eligible or declines to participate, no change will be made to her routine care and she will not be approached again. Consenting participants will be randomised to receive either broccoli sprout extract or placebo which will be written on the participant drug chart and given as per hospital protocol. Samples will be collected throughout the participant stay in hospital. Initial samples will include maternal blood pressure, maternal bloods (10 mL for serum and plasma) and maternal urine (50 mL). At 48, 96 hours, then weekly until delivery, maternal bloods and urine will be collected and blood pressure recorded. Immediately prior to labour maternal blood will be collected. After delivery, placentae will be collected along with cord blood (5 mL). Maternal urine sample will also be collected.
Sample size
The size effect on the primary outcome was based on the results of a trial of melatonin as an adjuvant therapy in women with early onset preeclampsia.14 15 In that trial, melatonin prolonged the enrolment-to-delivery interval by 6 days, from a mean (SD) of 10.4 (8.3) to 16.4 (11).15 Using these data, we calculated that 42 women in each treatment group (1:1 ratio) would be sufficient to detect a 6-day difference in mean (two-sided comparison) enrolment-to-delivery interval with 80% power. To allow for a 5% attrition rate, we elected a sample size of 45 in each arm, equating to a total of 90 participants. Randomisation for this study will be stratified within two gestation brackets: 24+0–30+0, 30+0–33+6. Because the power analysis was performed based on a study with a single stratum, we elected to have 90 participants in each stratum, requiring a total of 180 participants. This study is powered on the primary outcome of interval between enrolment and delivery, rather than secondary outcomes.
Trial sites
Women will be recruited from Monash Medical Centre and Jessie McPherson Private Hospital, Clayton, Victoria, Australia. Both sites are level 6 maternity services, as per Victorian government Maternity Capability Framework.16
Participant inclusion criteria
A woman will be eligible for inclusion in the trial only if the following criteria are met:
Aged 18–45 years.
Singleton pregnancy.
Diagnosis of preeclampsia, as defined by the Society for Obstetric Medicine of Australia and New Zealand guidelines.17
Gestation between 24+0 and 33+6 weeks.
Live fetus.
Able to safely continue pregnancy for at least 48 hours, as determined by the treating obstetrician.
No known significant fetal anomaly.
Able to give written, informed consent.
Participant exclusion criteria
A woman will not be eligible for inclusion in this trial if any of the following criteria apply:
Eclampsia.
Current use of broccoli sprout extract supplement.
Contraindications to use of broccoli sprout extract supplement (eg, intolerance of broccoli sprout).
Unknown gestation.
Unwillingness or inability to follow the procedures outlined in the participant information and consent form.
Mentally, cognitively or legally incapacitated or ineligible to provide informed consent.
Corecruitment/participation in another clinical trial where there is a pharmaceutical, herbal or nutritional intervention (such trial interventions would also include complementary and alternative medicines).
Participant recruitment
Potential participants will be identified by the research team from the antenatal clinic, pregnancy assessment unit, in-patient wards and birth suite at Monash Medical Centre. Following discussion with the attending clinical team caring for the woman, eligible women will be approached by a member of the research team who has no involvement in the provision of patient care and provided with the participant information and consent form for the trial. The research team member will provide a verbal explanation of the trial, including a description of the trial processes, the voluntary nature of the trial, and that a decision to participate, or not, will not affect normal clinical care. No trial-related procedures will be performed on any individual without their prior written, informed consent.
Women who provide written and informed consent to participate will be randomised to receive either broccoli sprout extract (BroccoMax, Jarrow Formulas, Los Angeles, California, USA) or an identical placebo (Jarrow Formulas). Allocation will be determined by a computer-generated sequence. After recruitment, each participant will be provided with a unique code so as to maintain participant confidentiality.
Randomisation
A randomisation sequence will be generated by a perinatal statistician not involved in the clinical trial, using a computer-generated code. Because the gestation at diagnosis of preeclampsia may influence the duration of the interval between diagnosis and delivery, randomisation will be stratified within two gestation brackets: 24+0–30+0, 30+0–33+6. Randomisation will be done through block sequence to ensure equivalent sample sizes are allocated to each treatment group (BroccoMax or placebo).18
The randomisation sequence will be provided to the pharmacist who will allocate capsules (BroccoMax or placebo) to each participant and will dispense the allocated intervention into bottles accordingly. The pharmacist will maintain a record of participant trial identification number and treatment group.
Intervention
Each participant will take three Broccomax capsules, each containing 8 mg of activated sulforaphane (total of 24 mg), twice daily (BD) or three identical placebo capsules twice daily (BD). Both participants and the research team will be blinded to group allocation. Capsules (BroccoMax or placebo) will be dispensed by the pharmacy in individualised bottles containing sufficient capsules for 5 days, with additional capsules (amount known only by the research team), and provided to the midwives in charge of ward care. Dosing will be recorded on the patient drug chart and administered as per hospital protocol.
Where participants are discharged home they will record taking the capsules in a patient self administration diary and return the capsule bottle, including any residual capsules, after 5 days, or sooner if delivered earlier. After delivery, residual capsules will be collected and discarded; they will not be reissued to a participant.
Outcomes
Primary outcome
The interval between enrolment and delivery, recorded in days.
Secondary outcomes
The secondary outcomes will be collected principally as measures of safety and tolerability.
Preeclampsia severity, as assessed by: escalation of antihypertensive therapy, systolic and diastolic blood pressures, severe renal involvement (serum or plasma creatinine >90 µmol/L, oliguria <80 mL/4 hour), haematological involvement (haemolysis (schistocytesor red cell fragments on blood film, raised bilirubin, raised lactate dehydrogenase >600 IU/L, decreased haptoglobin), platelets <104/µL, disseminated intravascular coagulation) liver transanimases >500 IU.
Indication for delivery (maternal or fetal compromise).
Mode of labour and birth (prelabour caesarean section, intrapartum caesarean section, induced or spontaneous labour, spontaneous vaginal birth, assisted vaginal birth).
Composite maternal outcome including maternal death, eclampsia, HELLP (haemolysis, elevated liver enzymes, low platelets) syndrome (haemolysis (lactate dehydrogenase ≥600 u/L, platelet count <100×109/L, aspartate aminotransferase >60 /L, haemolysis on peripheral blood smear or a raised haptoglobin level), pulmonary oedema (clinical signs and symptoms warranting treatment in the presence of oxygen saturations <90%), thromboembolic event (significant deep vein thrombosis or pulmonary embolus), placental abruption (retroplacental clot of >15% of maternal surface), major postpartum haemorrhage (>1000 mL of blood loss), severe renal impairment (creatinine >125 umol/L or need for dialysis), liver haematoma or rupture.
Intrauterine fetal death (stillbirth).
Changes in fetal surveillance (fetal Doppler studies—umbilical or middle cerebral artery pulsitility index, or abnormal ductus venosus—amniotic fluid volume <5 cm, abnormal fetal heart rate on CTG (cardiotocography).
Birth weight <5th percentile.
Gestation at birth.
Composite neonatal outcomes, including neonatal death before hospital discharge, 5 min Apgar score <7, umbilical lactate >5.0 at birth, admission to the neonatal intensive care unit (NICU), diagnosis of respiratory distress syndrome, bronchopulmonary dysplasia (need for oxygen after 28 days of life), sepsis, necrotising enterocolitis, intraventricular haemorrhage (grade III or IV), stage 4 or 5 retinopathy of prematurity, as determined by the treating clinician.
Duration of NICU care (days).
Maternal serum and placental angiogenic markers sFlt-1, soluble endoglin, placental growth factor (PlGF) and activin A.
Maternal TSH (thyroid stimulating hormone) and free and total T3/T4 (measured at baseline and after delivery).
Maternal demographics will be sourced from patient medical records. These will include maternal BMI (body mass index), smoking status, drug and alcohol use, age, parity, maternal comorbidities (thyroid dysfunction, diabetes (gestational type I or type II)) and maternal medications.
Additional covariates will include baseline sulforaphane and circulating sFlt-1 and PlGF levels. Adjustment will be made in statistical modelling for any significant difference in these covariates between treatment arms.
Sample collection and storage
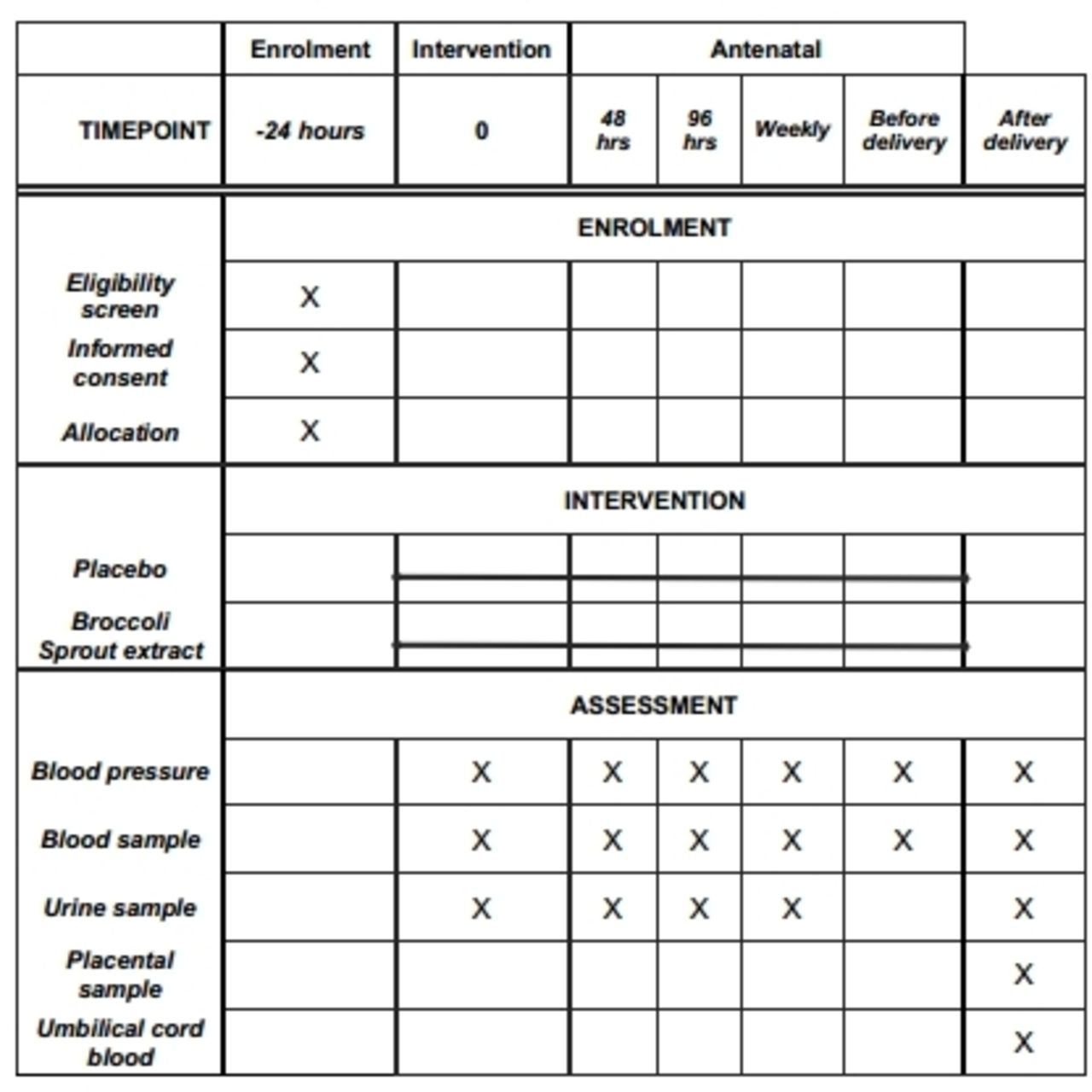
Samples will be collected at a number of time points (figure 2). All blood (10 mL for serum and plasma and 5 mL of cord blood) and urine samples (50 mL) will be centrifuged at 4°C and stored on-site at −80°C. Placental cotyledons will be removed, washed free of blood and either fixed in 10% buffered formalin or frozen in RNAlater (Sigma-Aldrich) until analysis. All biomarker investigations will be performed using ELISA and run in triplicates. Sulforaphane and its metabolites will be measured in plasma by liquid chromatography mass spectrometry using an established in-house methodology.

{kind=link}
{kind=link}
Timeline for sample collection. After eligibility screening by the research team, eligible participants will be consented within 24 hours. Consenting participants will be randomised to receive either broccoli sprout extract or placebo which will be written on the participant drug chart and given as per hospital protocol. This will be classified as time point 0. Samples will be collected throughout the participant stay in hospital at the beginning of treatment, 48 and 96 hours later and then weekly until and including delivery.
Information regarding participant demographics, blood pressure, fetal biometry and results from routine investigations will be collected from patient records. All information will be deidentified and stored on password-protected devices within the institution. Only the research team will have access to the dataset.
Proposed analysis
This is a superiority trial. Participant data will be analysed using intention to treat. All continuous measures will be assessed for normality of distribution. Differences in the primary outcome, time from enrolment to delivery in days, and secondary outcomes (safety data) will be compared between the two treatment groups. Continuous variables will be compared with a t-test (normally distributed variables) or Mann-Whitney U (non-normally distributed data). Categorical data will be assessed using a χ2. If possibly, non-parametric data will be transformed to allow parametric comparisons. The interaction between gestation at diagnosis and treatment group will also be assessed and regression approaches (using either an interaction term or gestation as a covariate) will be used to assess the relationship between treatment arm and time to delivery after assessing assumptions. Survival analysis will also be performed (after assessing assumptions) to account for censoring and survival/failure will be graphed with Kaplan-Meier curves. Linear mixed models regression will be used to compare differences in maternal angiogenic markers, TSH and T3/T4 over time between the two treatment groups. If there is a non-constant interaction between time and the outcome of interest, we will include this parameter in the model and investigate biochemical samples at specific pregnancy time points.
In the initial analysis, correction will only be made for baseline characteristics. Where appropriate, adjustment will be made using regression using a multivariate model.
A p value <0.05 (two-tailed) will be considered statistically significant.
Adverse events
While not expected, there may be unexpected adverse reactions associated with broccoli sprout supplements when used in pregnancy. To date, clinical studies have not demonstrated any serious adverse reactions to broccoli sprout supplements. However, metabolic changes during pregnancy may alter the pharmacological properties in unanticipated ways. A senior obstetrician on the treating team will monitor participants for the duration of their inpatient admission. The investigator will be contactable by phone at all times. Adverse event (AE) assessment and reporting will be undertaken in line with the requirements of the Sponsor, Monash Health and the National Health Medical Research Council.19 All observed or volunteered AE and serious AE (SAE) will be recorded and reported in detail in participant medical records, to the Monash Health Human Ethics Committee and the Sponsor, Monash Health within 24 hours.
Written summaries of the trial status will be submitted to the sponsor, annually or more frequently, if requested. All participant information and trial records will be securely stored to allow retrieval for audit or review purposes.
Data safety monitoring board reporting
A data safety monitoring board (DSMB) has been established to ensure the safe continuation of this trial by reviewing data on the following:
Maternal admission to intensive care unit or coronary care unit.
Apgar score <7 at 5 min of age requiring active resuscitation (±subsequent admission to the NICU).
Fetal surveillance outcomes (Doppler studies, CTG, biophysical profile).
Maternal or perinatal death.
All SAE/AEs submitted to the Sponsor, Monash Health.
The DSMB may request unbinding and will advocate for cessation or re-evaluation of the trial if either arm has a statistically significant or a 50% above baseline increase in any of these outcomes.
Trial discontinuation or modification
The trial will prematurely, permanently or temporarily cease recruitment if the investigator, or the sponsor believes that there are important issues pertaining to maternal and/or fetal welfare. Given the progressive nature of preeclampsia, worsening disease will not be considered an indication for discontinuation.
The trial will conclude when:
All participants (n=180) have been studied, delivered and discharged from Monash Health.
Data collection and entry is complete and database lock has occurred.
All data analysis has been performed.
All necessary reporting has been completed.
There will be no allowance for modification of the trial intervention or protocol after recruitment has commenced unless directed by the DSMB or the HREC (Human Research Ethics Committee) .
Unblinding
Unblinding in the trial may occur in the following circumstances:
To make clinical treatment decisions or when an unexpected SAE occurs and the intervention must be made known. This is called emergency unblinding.
During an unmasked analysis in accordance with the trial analysis plan.
At the request of the DSMB.
At the conclusion of the trial to determine the effect of the intervention.
When all participants (n=180) have completed the trial, all data entry and processing are complete and the database has been locked, the CPI (clinical principal investigator) will contact the Clinical Trials Pharmacy and request that unblinding take place, prior to statistical analysis.
Ethics and dissemination
This trial will be conducted in compliance with all stipulations of this protocol, the conditions of Monash Health HREC approval, and all other relevant local national and international guidelines. Any amendments to the trial conduct, except those necessary to remove an apparent, immediate hazard to the participant, will be submitted, in writing to the Monash Health HREC, for their review and approval, before they are implemented
Data will be published in peer-reviewed journals and presented at conferences, both nationally and internationally. All patient information will be deidentified for the purpose of publication.
Patients and public involvement
Patients were not involved in the design of this trial, establishing the research question or development of recruitment procedures. Participants will be provided with the opportunity to receive the study findings ahead of publication or presentation at learnt meetings.
Discussion
Prolong is a pragmatic superiority trial designed to increase the interval between enrolment and delivery for women with preeclampsia. Here, we propose the use of a novel antioxidant to target the oxidative stress underlying preeclampsia. Through this trial, we aim to add to the collective knowledge about novel therapeutics for preeclampsia and, if successful, ultimately establish a new medical intervention that improves outcomes for women with preeclampsia and their babies.
If effective, we believe that adjuvant use of a broccoli sprout extract, or a similar sulforaphane source, will significantly reduce the serious disease burden attributed to preeclampsia. Cheaply and simply reducing the morbidity and mortality associated with disease for both mother and child will have application in both high-resource and low-resource settings. However, sample size limitations are inevitable in a phase III trial and we acknowledge that there is a risk of under power and type II error. Therefore, this study was designed to power for only our primary outcome. Future investigation with larger populations and further assessment of short-term and long-term infant outcomes will be necessary. Similarly, the single centre nature of this trial and subsequent issues in population bias are a limitation of this study that will be addressed in future investigations. Larger trials of the efficacy and clinical application of broccoli sprout extract will be necessary if prolong produces positive results. We hope that this initial trial will provide sufficient evidence to support and inform future such trials.
Acknowledgments
We would like to acknowledge Joanne Mockler whose input has been paramount to the application for ethical approval of this trial. We thank the NHMRC for providing funding for this project.
References
Footnotes
Twitter @@A_LangstonCox, @euan_wallace
Contributors The original concept for this study came from EW. The trial design was established through discussions between EW and AGC, with considerable input from SAM and KRP. The manuscript was written by AGC with drafting from SAM, KRP and EW. All authors have read and approved the final manuscript.
Funding This work is funded by an NHMRC Program grant to EMW, ID: 111 3902. The funding body had no role in trial design or the writing of the manuscript.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The Monash Health Ethics Committee approved this trial (RES-18-0000-109A) on 2 March 2018.
Provenance and peer review Not commissioned; externally peer reviewed.