Article Text

Abstract
BACKGROUND Current guidelines on the treatment of childhood asthma recommend the introduction of an anti-inflammatory drug in children who have persistent symptoms and require regular treatment with a bronchodilator. The efficacy and safety of inhaled nedocromil sodium (Tilade Mint aerosol) administered using a Fisonair spacer at a dose of 4 mg three times daily was compared with placebo in the treatment of asthmatic children aged 6–12 years who are symptomatic and recovering from an acute exacerbation of asthma.
METHODS A group comparative, double blind, placebo controlled trial was performed in children who were recovering from an acute episode of asthma following treatment in the emergency department of the hospital or in children referred from their general practitioner following a wheezing episode and documented evidence of at least two previous episodes of wheezing. A two week baseline period on existing bronchodilator treatment was followed by a 12 week treatment period on either nedocromil sodium (2 mg/puff) or placebo. Both treatments were administered using a Fisonair spacer at a dose of two puffs three times daily. Changes from baseline values in daytime asthma and night time asthma symptom scores, usage of rescue bronchodilators, mean peak expiratory flow (PEF) recorded twice daily on diary cards, patients’ opinion of treatment, and withdrawals due to treatment failure were measured during the primary treatment period (last six weeks of treatment).
RESULTS One hundred and forty two children aged 6–12 years entered the baseline period. Sixty three were withdrawn due to failure to meet the entry criteria (18) or the criteria for asthma symptom severity (15) or reversibility (9), because they developed uncontrolled asthma (2), because they took disallowed treatment (2), or for other non-trial related reasons (17). Seventy nine patients (46 boys) of mean age 8.8 years entered the treatment period. There were significant differences in the changes from baseline values during the last six weeks of treatment in favour of nedocromil sodium compared with placebo in the primary variables of daytime asthma and night time asthma, morning and evening PEF, and the usage of rescue inhaled bronchodilators; 53% of patients reported nedocromil sodium to be very or moderately effective compared with 44% placebo. Improvement in asthma symptoms, PEF, and reduction in use of rescue bronchodilators did not reach statistical significance until after six weeks of treatment. Twenty two patients were withdrawn or dropped out during the treatment phase, 12 due to uncontrolled asthma or persistence of asthma symptoms, four due to suspected adverse drug reactions (nedocromil sodium 3 (headaches 2, angio-oedema/urticaria 1), placebo 1(persistent cough)), and six due to non-treatment related reasons. Seventy one adverse events were reported by 27 patients in the nedocromil group and 75 by 30 patients in the placebo group.
CONCLUSIONS Asthma symptoms, use of bronchodilators, and lung function can be improved significantly in children recovering from an acute exacerbation of asthma or wheeze and currently receiving treatment with bronchodilators alone by the addition of inhaled nedocromil sodium at a dose of 4 mg three times daily administered using a Fisonair holding chamber.
- asthma
- nedocromil sodium
- childhood asthma
- spacer
Statistics from Altmetric.com
The current international guidelines on the management of asthma in children1 recommend a stepwise approach in which children with infrequent episodic asthma commence treatment with intermittent short acting β2 adrenergic drugs. Once these are used more than three times a week, or exacerbations occur more frequently than every 4–6 weeks, prophylactic treatment is indicated. Inhaled sodium cromoglycate is the first prophylactic compound to be introduced and inhaled corticosteroids should be substituted if this fails to control symptoms. In the UK either inhaled sodium cromoglycate or inhaled corticosteroids are recommended as the first line prophylactic treatment.
Nedocromil sodium is a chromone. It is the disodium salt of a pyranoquinoline dicarboxylic acid developed as an anti-inflammatory treatment for asthma2 and is administered from a metered dose inhaler in unit doses of 2 mg. It has been compared with sodium cromoglycate in in vitro and in vivo models of asthma and has been shown to be up to 10 times more potent.3-5 A number of clinical trials have been conducted in adult patients6-9and a meta-analysis of all placebo controlled trials has shown that the drug is an effective treatment for adult asthma.10However, there have been very few trials of this drug to date in children with asthma.
Many children who attend hospital accident and emergency departments with acute episodes of asthma are not receiving anti-inflammatory therapy. In addition, there are many asthmatic children under the care of their general practitioners who have not been started on anti-inflammatory treatment despite having recurrent episodes of wheezing. We therefore decided to evaluate the efficacy and safety of nedocromil sodium in the treatment of children who had recently attended a hospital emergency department with an acute episode of asthma, and symptomatic children referred by their general practitioners who had had a recent episode of wheezing and were currently receiving bronchodilator treatment alone.
Methods
PATIENTS
Patients with asthma of either sex aged 6–12 years were eligible for selection. Those with a history of renal, hepatic, or cardiovascular disease, or chronic respiratory disease other than asthma were excluded. Patients had either had a recent episode of asthma treated at hospital or were referred by their general practitioner with a current episode of wheeze and with documented evidence of at least two previous episodes of wheeze in the previous six months. Asthma was defined as current wheeze with airways obstruction that is reversible by at least 15% or an increase in forced expiratory volume in one second (FEV1) of ⩾140 ml following the inhalation of two puffs of a bronchodilator administered by a metered dose inhaler. All patients had to have a minimum score of asthma symptoms (22) on a daily diary card during a two week baseline period. Patients and their parents had to be co-operative, to keep a daily diary card for the duration of the trial, and to be able to use a pressurised metered dose aerosol, a peak flow meter, and a spacer (Fisonair). Patients could have received a short course of treatment with oral corticosteroids for their recent asthma attack, but could not be receiving current treatment with either inhaled sodium cromoglycate, oral or inhaled corticosteroids. Patients were also excluded if they had been treated with inhaled sodium cromoglycate during the previous month or with inhaled corticosteroids during the previous three months.
Patients and/or their parents gave written informed consent and the protocol was approved by the local ethics review committee.
TRIAL DESIGN
The trial was of a double blind, group comparative design. After a two week baseline period during which they had to have a minimum asthma symptom score (see below), the children were randomised to receive treatment with either inhaled nedocromil sodium or inhaled placebo for a period of 12 weeks. Patients were seen by the investigator at the beginning and end of the baseline period and every two weeks during the trial.
The trial was conducted according to the principles established by the declaration of Helsinki (as modified in Tokyo in 1975, Venice in 1983, and Hong Kong in 1989). As the trial was conducted in three countries, both the clinical investigators and the nurses responsible for patient recruitment and monitoring met on at least one occasion in order to ensure a common understanding of the trial protocol.
TEST MEDICATIONS
The test medications were administered from a metered dose inhaler using a 750 ml spacer (Fisonair). As the active drug was yellow in colour, the spacers were made of yellow material in order to maintain blindness. The test medications were either inhaled nedocromil sodium (2 mg per puff) or placebo (containing liquefied gas propellants and excipients only), administered at a dose of two puffs three times daily.
Patients were allowed to use inhaled bronchodilators as needed to control acute attacks of wheezing or bronchospasm. The number of doses they required over each 24 hour period was recorded on the daily diary card. The inhaled bronchodilators could be administered via a metered dose inhaler, a dry powder delivery system, or an aqueous nebuliser. Oral bronchodilators were not allowed. Patients were allowed to use topical nasal and/or ophthalmic preparations or antihistamines as required for the relief of nasal and ophthalmic symptoms, but were not allowed to use any form of corticosteroid (apart from topical corticosteroids to the skin) or any other anti-asthma treatment.
MEASUREMENTS
Patient diary cards
Throughout the trial, including the baseline period, parents kept a daily diary card on which they recorded the severity of daytime and night time asthma symptoms, the morning and evening peak expiratory flow (PEF), and the number of doses of rescue bronchodilators used during the 24 hours. Details of the scoring system used to record the severity of symptoms are presented in Appendix .
The PEF reading was the best of three measurements taken standing before any medication was administered.
Investigator assessments
At each visit the investigator asked the parent or guardian to grade the severity of the day and night asthma symptoms over the preceding 14 days using a five point scale. The scoring system used was similar to that used on the patient diary card but covered a two week period. Pulmonary function tests were carried out at each clinic visit and included FEV1, forced vital capacity (FVC), and PEF.
Global assessments
At the end of the trial the parent/guardian and the investigator recorded the overall efficacy of the test treatment administered using a five point scale where 1 = very effective, 2 = moderately effective, 3 = slightly effective, 4 = no effect, and 5 = made condition worse.
Safety and tolerability
At each clinic visit the investigator asked the parent/guardian if the child had had any unusual event since the previous visit. An unusual event was defined as any symptom, sign, illness, or experience that developed or increased in severity during the course of the study. It included any major alteration in laboratory values and any apparently unrelated illness, accident, or unanticipated surgery. Any suspected adverse drug reactions related to the test treatment were also recorded.
Children could be withdrawn from the trial at any stage at the request of the parents/guardian and the investigator could also withdraw patients. The reason for withdrawal was recorded.
STATISTICAL ANALYSIS
The primary variables for defining the efficacy of the test treatments were the diary card symptom scores, the morning and evening PEF, and the usage of rescue inhaled bronchodilators during the last six weeks of the treatment period. The global opinion of treatment efficacy as assessed by the patients and withdrawals due to treatment failure were also primary variables.
The secondary variables were the assessment of symptom severity carried out at the clinic visits, the pulmonary function tests performed at the clinic visits, and the investigator’s global opinion of treatment efficacy.
Before the trial it was estimated that 80 patients per treatment group would be required to detect a true difference of 0.27 in the change from baseline for the symptom scores recorded on the daily diary cards. This calculation was made using a two sided statistical test with alpha = 0.05 and 80% power assuming a standard deviation of 0.6.
The analysis was performed on an “intention to treat” basis. Patients who did not complete the study for both treatment related and non-treatment related reasons were included in the analysis as far as possible. Patients who were withdrawn because of lack of efficacy of the test treatment were included in all analyses of efficacy using imputed data. For the diary card variables the imputed data were the mean of the three days before withdrawal. For clinic assessments (with the exception of pulmonary function) they were included as extreme cases following withdrawal for that treatment period. For patient opinions they were included as treatment failures. Patients were included provided that the test medication had been taken for at least seven days. Patients who dropped out for reasons not related to treatment efficacy were included in the analyses up to the time of withdrawal. Patients who failed to take the test treatment for five or more days in any 14 day period were excluded from the analysis of that period.
The mean values of all diary card variables were calculated both for the primary period (weeks 6–12) and for each two week period of the trial. The results were compared between treatment groups as changes from baseline. The severity of symptoms and pulmonary function recorded at each clinic visit was analysed as changes from those recorded at the end of the baseline and the treatment groups compared. In addition, median values, the change from baseline, and the difference between treatment groups for diary card symptom scores and bronchodilator usage were calculated together with the 95% confidence intervals for the differences.
Pulmonary function data at clinic visits and recorded on the daily diary card were analysed using two sample ttests. All other data were analysed using the Wilcoxon rank sum test. All tests were two tailed and treatment effects were based on a p value of 0.05.
A secondary analysis was carried out for the primary variables during the primary period which stratified the patients according to whether they had received a course of oral corticosteroids during their acute attack which preceded participation in the study. Analysis of variance was used for this analysis with steroid use and treatment group used as factors.
Results
PATIENT CHARACTERISTICS
One hundred and forty two patients were entered into the study at seven centres, four in Australia, one in New Zealand, and two in South Africa. Sixty three patients were withdrawn from the study before being randomised to test treatment. Eighteen patients did not satisfy the entry criteria, 15 did not meet the required level of symptom severity during the baseline period, nine did not meet the requirements for reversibility, and 15 were unable to comply with the requirements of the trial protocol, The remaining six subjects either developed uncontrolled asthma (2), took corticosteroids during the baseline (2), or were excluded for other non-specified reasons (2). None of these patients was included in the analysis.
Seventy nine patients (46 boys) of mean age 8.8 years (range 6–12) were randomised to receive test treatment, 38 to treatment with nedocromil sodium and 41 to placebo. The characteristics of these patients are summarised in table 1. Information concerning the acute attack that resulted in admission to the trial was available in 67 cases. Details of the suspected cause of the acute attack, its duration, and the treatment used are shown in table 2.
Patient characteristics
Details of acute episodes
Twenty two children were withdrawn during the treatment phase of the trial, 12 (four from the nedocromil sodium group and eight from the placebo group) because of the onset of uncontrolled asthma (11 patients) or due to persistent asthma symptoms not controlled by test treatment (one patient). These 12 patients were classified as treatment failures and were included in all analyses of efficacy using imputed data. Ten patients dropped out or were withdrawn for reasons not related to efficacy. Four or these were withdrawn because of suspected adverse drug reactions, three from the nedocromil sodium group (two with headaches and one with angio-oedema and urticaria) and one from the placebo group with persistent cough. Four patients in the placebo group dropped out because they were unable to comply with the requirements of the protocol. Two patients in the nedocromil sodium group were withdrawn at the third visit because they had not satisfied the entry criteria. One was taking sustained release theophylline and in the other reversibility had not been demonstrated at the baseline visits. These 10 cases were included in the analyses up to the time of withdrawal.
EFFICACY
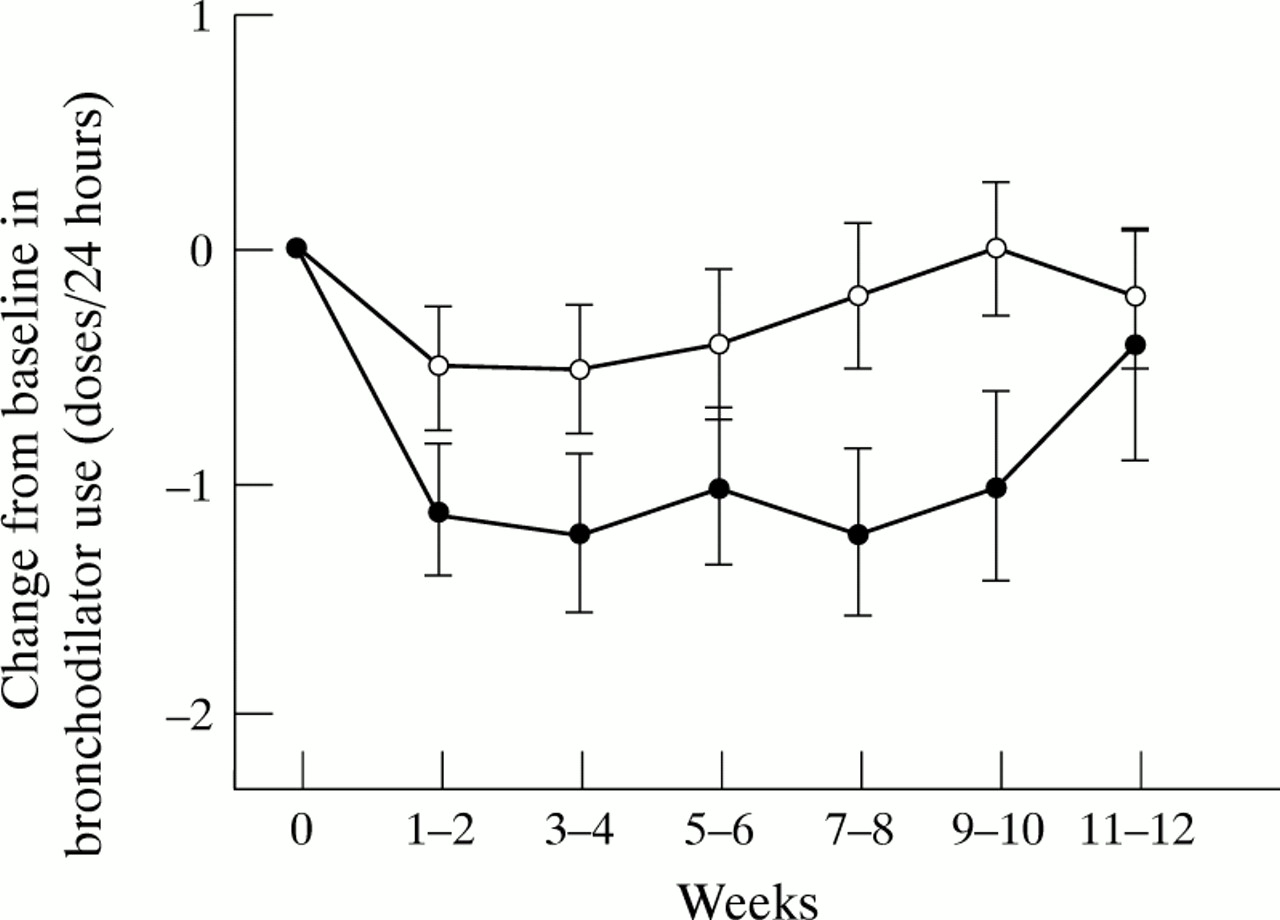
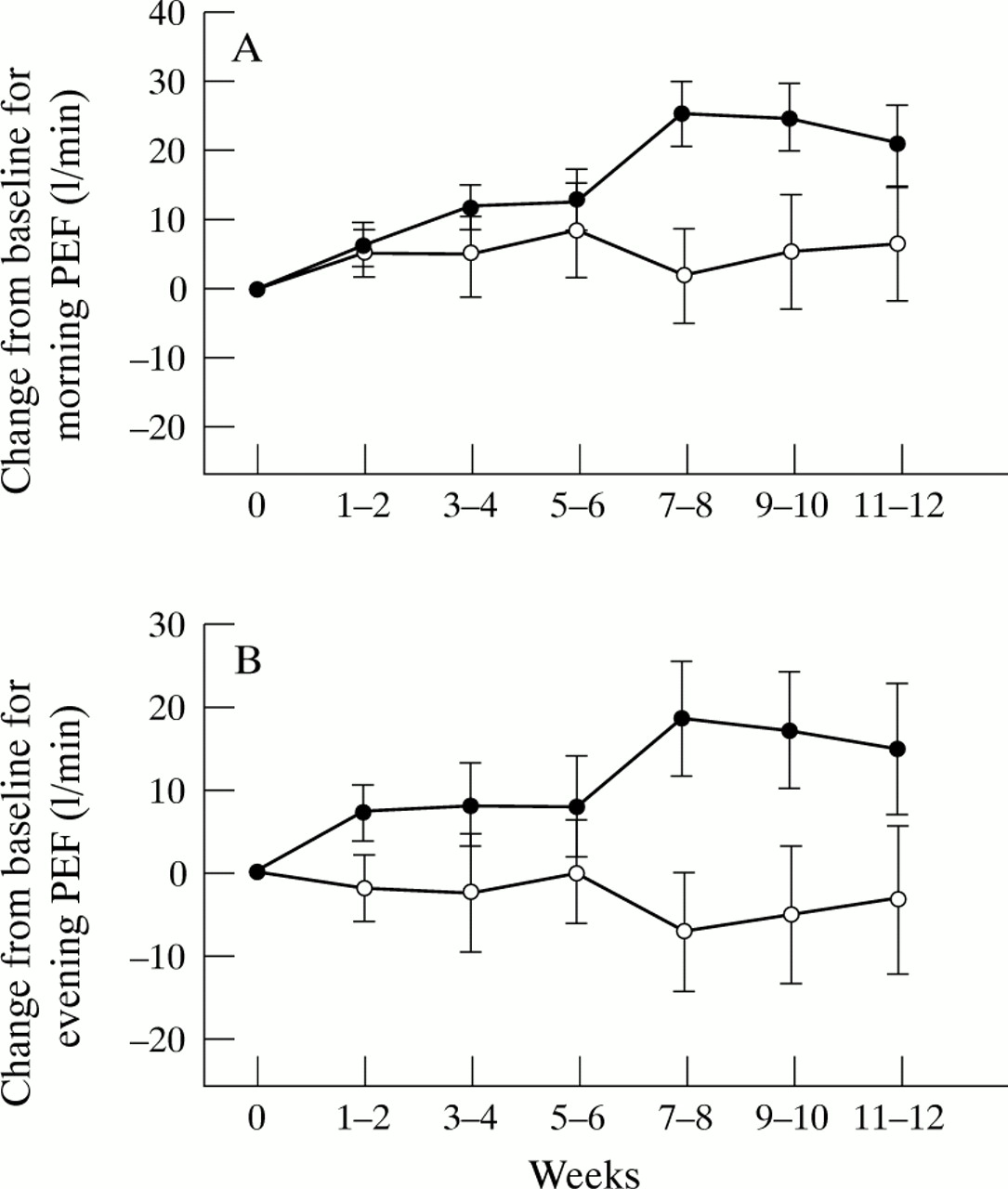
The mean baseline values and differences in the changes from mean baseline values to mean values during the primary period for all primary variables are shown in table 3. The baseline values for these variables were not significantly different apart from night time asthma. For this variable the baseline value for the nedocromil sodium group was 1.6 (0.63) and for the placebo group 1.3 (0.46) (p = 0.037). There were significant differences in the changes from baseline values to the last six weeks of treatment in favour of nedocromil sodium compared with placebo in the primary variables of daytime asthma (p = 0.03) and night time asthma (p = 0.001), morning PEF (p = 0.036) and evening PEF (p = 0.033), and the use of rescue inhaled bronchodilators (p = 0.011) The size of the differences in treatment effects between nedocromil sodium and placebo are shown in table 4 which details the changes from baseline of median values (diary card symptoms and bronchodilator usage) and mean values (PEF) together with the differences between treatments and the 95% confidence intervals of the differences. The mean changes in the two weekly values are illustrated in figs 1, 2, and 3. The distribution of categories of patients’ opinions of treatment, as shown in table 3, just failed to reach significance (p = 0.059); 53% of nedocromil sodium treated patients considered the test treatment to be very or moderately effective compared with 44% of those treated with placebo.
Mean (SD) baseline values and changes from baseline in diary card primary variables during primary period (last six weeks of treatment)
Comparison of change from baseline to last six weeks of treatment between nedocromil sodium and placebo for median values (symptom scores, bronchodilator use) and mean values (PEF)

Mean change from baseline values for (A) daytime and (B) night time asthma symptom scores with nedocromil sodium (•) and placebo (○). Data were analysed every two weeks. Error bars show standard errors of means. Baseline values (scale 0–5) for daytime asthma: 1.6 (0.72) for nedocromil sodium group, 1.4 (0.62) for placebo group; night time asthma 1.6 (0.63) for nedocromil sodium group, 1.3 (0.46) for placebo group.

Mean change from baseline values for (A) morning and (B) evening peak expiratory flow (PEF) with nedocromil sodium (•) and placebo (○). Data were analysed every two weeks. Error bars show standard errors of means. Baseline values for morning PEF: 222.7 (72.12) l/min for nedocromil sodium group, 227.2 (74.33) l/min for placebo group; evening PEF 231.9 (71.72) l/min for nedocromil sodium group, 241.2 (75) l/min for placebo group.
{kind=link}
{kind=link}
{kind=link}
Mean change from baseline values for mean daily usage of rescue bronchodilators with nedocromil sodium (•) and placebo (○). Data were analysed every two weeks. Error bars show standard errors of means. Baseline values 2.3 (1.8) doses/24 hours for nedocromil sodium group, 1.9 (2.00) doses/24 hours for placebo group.
The secondary variables at baseline and after 12 weeks of treatment are presented in table 5. There was no difference between nedocromil sodium and placebo for these variables at this time point. No differences were seen in the treatment effects according to whether corticosteroids had been used in the treatment for the acute attack, but the relative numbers in the groups were small.
Mean (SD) baseline values and changes from baseline in assessments and lung function recorded at final clinic visit (after 12 weeks of treatment)
SAFETY AND TOLERABILITY
Fifty one patients found the test treatment to be acceptable (nedocromil sodium 25, placebo 26) and four patients (nedocromil sodium 3, placebo 1) found it to be unacceptable. The acceptability was not recorded in 24 cases. Of the three patients who found nedocromil sodium to be unacceptable, one reported that the treatment dried out his throat, one did not like the taste, and one did not like having to take the inhaler three times a day. The placebo treated patient did not like the taste.
During the course of the treatment 27 patients treated with nedocromil sodium reported 75 adverse events and 30 patients treated with placebo reported 71 adverse events. Adverse events were generally reported as being of minor clinical significance. Headache was reported on eight occasions by patients treated with nedocromil sodium and on four occasions by patients treated with placebo. Apart from events related to the respiratory tract which were reported more frequently by placebo treated patients, all reports were either in equal numbers or one or two reports only. In four cases patients were withdrawn from the trial because of adverse events (three on nedocromil sodium (headaches in two cases and angio-oedema/urticaria in one) and one on placebo who developed a persistent cough).
Discussion
Nedocromil sodium is an anti-inflammatory drug developed as a prophylactic treatment of asthma. A meta-analysis of trials has shown its efficacy in adult asthma,10 but to date few therapeutic trials have been carried out in children. This report demonstrates the efficacy of nedocromil sodium in paediatric patients with asthma.
Businco et al 11 compared nedocromil sodium and placebo in 31 patients aged 4–21 years with grass pollen asthma. The dose used was 4 mg four times daily and the treatment period was four weeks. There were significant differences in favour of nedocromil sodium for morning tightness and daily mean morning PEF, the use of inhaled bronchodilators, and pulmonary function tests. Armenio et al 12 compared nedocromil sodium and placebo in 209 asthmatic children aged 6–17 years. Test treatments were given in addition to existing therapy. Statistically significant differences in favour of nedocromil sodium were seen for clinician assessment of asthma severity, diary card symptoms, pulmonary function, and the use of inhaled β2bronchodilators. In this trial the total symptom score was reduced by 50% during the last four weeks of treatment compared with baseline scores. This is very similar to the result of our study. The trials reported by Businco et al and by Armenioet al used a dose of 4 mg four times daily and neither used a holding chamber to assist delivery. In our trial we used a dose of 4 mg three times daily delivered by a spacer. Fooet al 13 used a spacer and a dose of 4 mg three times daily in 120 children aged 6–19 years. However, the patients in their trial had stable asthma with low symptom scores. The main objective of their trial was to examine the effect of nedocromil sodium on histamine responsiveness. No effect on histamine responsiveness nor symptom scores was seen but there was a significant increase in pulmonary function. The results of our trial are in line with those of other paediatric studies but provide more consistent evidence of drug efficacy. We have also demonstrated the effectiveness of nedocromil sodium in the circumstances in which it was used.
Perhaps the more important question to answer is whether nedocromil sodium can be regarded as an alternative first line anti-inflammatory treatment to sodium cromoglycate or inhaled corticosteroids in asthmatic children. There are no direct comparative therapeutic trials in childhood asthma between sodium cromoglycate and nedocromil sodium. However, there have been comparative studies of nedocromil sodium and sodium cromoglycate in exercise induced asthma. Comiset al 14 compared 10 mg sodium cromoglycate with 4 mg nedocromil sodium, with and without a Fisonair. There was no difference in efficacy between the two compounds but both were better than placebo. The use of the Fisonair did not confer any advantage. Novembre et al 15also compared the effects of 10 mg sodium cromoglycate, 4 mg nedocromil sodium, and placebo in exercise induced asthma in 19 children aged 6–15 years. Nedocromil sodium gave a better overall performance with complete protection in 14 patients compared with nine patients pre-treated with sodium cromoglycate and two patients with placebo. A similar comparison in 17 children has been carried out by de Benedictis et al 16 who also compared the duration of action.17 Both drugs were shown to provide significantly better protection than placebo but with no difference between them. Both provided significant protection when given 20 minutes before exercise but not at 140 minutes. It is difficult to extrapolate from the results of challenge studies, but the results of these trials in exercise induced asthma do suggest that nedocromil sodium is the more potent compound and is at least as effective as, and may be more effective than, sodium cromoglycate.
There have been no direct comparisons of nedocromil sodium and inhaled corticosteroids in children so it is not possible to predict the comparative usefulness. Recent trials of sodium cromoglycate or inhaled corticosteroids compared with placebo have not used similar designs to our own and comparisons between such trials are not therefore valid.
A comparison of the three treatments was made in 1996 using an open retrospective design by Korppi and Renes18 who reviewed the lung function of 297 school-aged children who had been treated since 1989 according to international guidelines. Sixty children were not using any preventive drugs, 169 were using chromones (97 sodium cromoglycate, 72 nedocromil sodium), and 68 inhaled corticosteroids (three beclomethasone and 65 budesonide). The mean values for PEF, FVC, and FEV1 were over 95% of the height related reference values in all treatment groups and the lower limits of the 95% confidence interval were at the level of more than 90% of those predicted. Only minor differences between the different therapeutic groups were observed and the authors concluded that, in their population, up to 70% of asthmatic children requiring maintenance therapy for asthma can be treated with chromones.
Clinical trials of new drugs in children with asthma attending hospital clinics do present problems. Most children are either well maintained on currently available effective treatments for childhood asthma such as sodium cromoglycate and inhaled corticosteroids or have severe asthma which makes them unsuitable for placebo controlled trials. Patients whose asthma is well controlled are usually free of symptoms and have normal or near normal lung function apart from when they have an acute exacerbation. In such patients there is minimal room for improvement in the primary measures of asthma severity. It is unethical to withdraw treatment to induce symptoms or reduce lung function and then to substitute a placebo or an untried treatment. The pre-trial estimate of the need for 160 patients (80/treatment group) was not achieved as 63 were rejected because of low symptom scores. However, this planned sample size was based on an estimated difference in symptom scores of 0.27. In the actual trial the difference was 0.4 for daytime asthma and 0.7 for night time asthma and significant differences were obtained with fewer numbers.
Patients are most likely to be symptomatic when recovering from an acute episode of asthma or wheezing and this trial with nedocromil sodium illustrates the importance and value of adding an anti-inflammatory drug to the treatment of such patients, in this case, children. The trial also demonstrates the importance of clearly defining the degree of severity of asthma required in order to ensure that a treatment effect would be seen if the test drug is therapeutically active. Although this meant the rejection of a number of patients as they did not meet the entry criteria, it did ensure that a homogeneous population entered the treatment phase.
All of the key measures of asthma severity used in the present study improved both significantly compared with a placebo treatment and clinically in terms of asthma control. The absence of a significant effect in the secondary variables illustrates the importance of using outcome measures that reflect the day by day variation in asthma severity rather than clinic assessments at wide intervals when the patient will reflect the condition on that day rather than over a period.
In paediatric practice acute attacks of asthma are frequently treated appropriately with bronchodilators and a short course of corticosteroids but consideration is not always given at that stage to the longer term needs. In a review of 422 children with acute asthma who attended the emergency department of the Royal Children’s Hospital in Parkville, Victoria, Australia between 1 January and 31 December 1989, Barnett and Oberklaid19 found that 39% had been taking no medication at all and only 24% had been taking regular anti-inflammatory treatment. In their study upper respiratory tract infections were the precipitating cause of the acute attack in 82% of cases in which it was recorded compared with 54% in our study. On discharge from their study 33% of patients had been prescribed anti-inflammatory treatment and in 45% of cases no arrangements for follow up were documented. The relative lack of improvement of the placebo treated patients in our trial would suggest that children who have had a recent episode of wheezing requiring acute treatment and who have some degree of persistent symptoms would benefit from the addition of an anti-inflammatory drug such as nedocromil sodium. This conclusion is supported by a retrospective review of the effect of drug treatment on the long term outcome of childhood asthma. König and Shaffer20 have concluded that treatment with anti-inflammatory drugs (cromolyn sodium or inhaled corticosteroids), but not as needed bronchodilators alone, improves the long term prognosis of asthma.
It is uncertain whether the use of the Fisonair spacer increased the efficacy of nedocromil sodium. Barry et al 21 in an in vitro study measuring particle size showed that the use of a Fisonair spacer increased the amount of drug below 5 μm delivered from 0.417 mg for a standard metered dose inhaler to 0.498 mg for the Fisonair. The equivalent amounts for particles below 3 μm were 0.206 mg and 0.294 mg. This was dependent upon only one actuation being put into the holding chamber at a time and there being no delay between actuation and inhalation. This was the technique used in our trial. As well as having the advantage of the lack of need to co-ordinate actuation with inhalation, the Fisonair, as with other spacers, reduces the amount of ineffective drug being deposited in the oropharynx and results in an increase in the amount reaching the lungs.
This trial has shown that nedocromil sodium at a dose of 4 mg three times daily delivered by a Fisonair holding chamber provides significant and clinically useful improvements in asthma control in children recovering from an acute episode of asthma. We conclude that this treatment regimen can be considered as a possible first line prophylactic treatment in children with mild to moderate episodic asthma.
Acknowledgments
This study was funded by Fisons Pharmaceutical Division who also supplied the medications and Fisonair holding chambers. We wish to extend our thanks to M T Stevens of EMStat Ltd who checked the statistical analysis and to Sue Bryan, Ian Murphy, and Carolyn Newton who contributed to the organisation.
Scoring system used on daily diary cards
NIGHT TIME ASTHMA
The night time asthma symptoms were based on the severity of cough, wheeze, chest tightness, and shortness of breath that resulted in disturbed sleep using the following 0–5 scale: 0 = No symptoms during the night or on waking in the morning. 1 = No symptoms during the night, but symptoms on waking at the usual time. 2 = Symptoms during the night but not causing the child to wake. Awake at the usual time. 3 = Symptoms during the night causing the child to wake (including waking early).4 = Symptoms causing the child to be awake for most of the night. 5 = Symptoms so severe that the child did not sleep at all.
DAYTIME ASTHMA
The daytime symptoms were based on the severity of cough, wheeze, and breathlessness on exertion experienced during the day using the following 0–5 scale: 0 = No symptoms during the day. 1 = Symptoms for one short period during the day. 2 = Symptoms for two or more short periods during the day. 3 = Symptoms for most of the day which did not affect the child’s normal daily activities. 4 = Symptoms for most of the day which did affect the child’s normal daily activities. 5 = Symptoms so severe that the child could not go to school or perform normal daily activities.
References
Footnotes
Conflicts of interest: A M Edwards and J Lyons were employees of the sponsors, Fisons Pharmaceuticals, at the time this trial was undertaken. Current positions: AME is Honorary Clinical Assistant, University Medicine, Southampton General Hospital, Southampton, UK and JL is Clinical Project Manager, Astra Pharmaceuticals Pty Ltd, North Ryde, NSW, Australia.