
Abstract
 materials have recently shown promise as high capacity stable electrodes for advanced rechargeable lithium batteries. Using first principles quantum mechanical energy computations we demonstrate that the stability of these materials is due to the particular valence distribution on the transition metals in this material. Spin density calculations indicate that the Mn ion has oxidation state
materials have recently shown promise as high capacity stable electrodes for advanced rechargeable lithium batteries. Using first principles quantum mechanical energy computations we demonstrate that the stability of these materials is due to the particular valence distribution on the transition metals in this material. Spin density calculations indicate that the Mn ion has oxidation state  independently of the Li content in the material, while Ni is oxidized from
independently of the Li content in the material, while Ni is oxidized from  to
to  upon removing Li. The high insertion voltage for the
upon removing Li. The high insertion voltage for the  can be partly attributed to the change in Mn-Ni interaction upon Li cycling. © 2002 The Electrochemical Society. All rights reserved.
can be partly attributed to the change in Mn-Ni interaction upon Li cycling. © 2002 The Electrochemical Society. All rights reserved.
Export citation and abstract BibTeX RIS
Layered lithium-manganese oxides are of interest for use in rechargeable lithium batteries because of their potential for very high capacity, relative safety, and affordability. Their inherent safety is derived from the fact that both  (present at the end of discharge) and
(present at the end of discharge) and  (at the end of charge) are quite stable valence states. While manganese oxides with the spinel structure and stoichiometry
(at the end of charge) are quite stable valence states. While manganese oxides with the spinel structure and stoichiometry  have been used in batteries, their capacity is limited, and severe stability issues exist with the material. Layered lithium manganese oxides, on the other hand, have a theoretical capacity of 288 mAh/g. Unfortunately, the layered
have been used in batteries, their capacity is limited, and severe stability issues exist with the material. Layered lithium manganese oxides, on the other hand, have a theoretical capacity of 288 mAh/g. Unfortunately, the layered  structure is not the ground state for
structure is not the ground state for  1 and one has to resort to either metastable processing routes2
3 starting from a
1 and one has to resort to either metastable processing routes2
3 starting from a  or to compositional modifications to increase the stability of the layered phase over the other possible polytopes.4
5
6
7 Almost all of the pure or lightly doped layered manganese oxides have shown a rapid transformation to a spinel upon cycling.8
9 While this spinel can in some cases maintain capacity,10 it has a less favorable voltage profile and the remaining disorder in the structure limits its current density.
or to compositional modifications to increase the stability of the layered phase over the other possible polytopes.4
5
6
7 Almost all of the pure or lightly doped layered manganese oxides have shown a rapid transformation to a spinel upon cycling.8
9 While this spinel can in some cases maintain capacity,10 it has a less favorable voltage profile and the remaining disorder in the structure limits its current density.
Using higher doping levels, it has been possible to stabilize the layered structure against transformation to spinel. In particular, co-doping of Li and Cr5 11 has been particularly successful. Ni-doped Mn materials were first synthesized several years ago,12 13 but this approach has gained renewed interest now that good cycling behavior for these materials has been demonstrated.5 14
Some uncertainty exists with regards to the valence states in these mixed-metal compounds. In  the observed capacity could only be explained by the cycling of
the observed capacity could only be explained by the cycling of  to
to  5 a fact later confirmed with X-ray absorption spectroscopy.15 In
5 a fact later confirmed with X-ray absorption spectroscopy.15 In  it has been speculated5
14 that the Ni and Mn ions, respectively, have valence
it has been speculated5
14 that the Ni and Mn ions, respectively, have valence  and
and  though earlier work by Spahr13 assumed both Ni and Mn to have valence
though earlier work by Spahr13 assumed both Ni and Mn to have valence  in the starting material. Because the capacity of
in the starting material. Because the capacity of  is
is  the assumption of
the assumption of  and
and  in the starting material requires that the nickel ion cycles between
in the starting material requires that the nickel ion cycles between  and
and  The purpose of this paper is to clarify the valence states in
The purpose of this paper is to clarify the valence states in  and characterize the electronic and structural changes that occur upon delithiation. The origin for the high potential of this material is also discussed. The energies, intercalation potentials, geometries, and electronic structure of the
and characterize the electronic and structural changes that occur upon delithiation. The origin for the high potential of this material is also discussed. The energies, intercalation potentials, geometries, and electronic structure of the  materials are obtained using first principles quantum mechanical computations in the generalized gradient approximation to density functional theory. Ultrasoft pseudopotentials and the Perdew-Wang exchange correlation function were used, as implemented in VASP.16 All calculations were performed with spin polarization, previously demonstrated to be crucial in manganese oxides.1 Reasonable intercalation potentials and geometrical information can be obtained with first principles methods, as has been amply demonstrated.17
18
19 To describe the
materials are obtained using first principles quantum mechanical computations in the generalized gradient approximation to density functional theory. Ultrasoft pseudopotentials and the Perdew-Wang exchange correlation function were used, as implemented in VASP.16 All calculations were performed with spin polarization, previously demonstrated to be crucial in manganese oxides.1 Reasonable intercalation potentials and geometrical information can be obtained with first principles methods, as has been amply demonstrated.17
18
19 To describe the  system, a supercell with two formula units was used. Because this computational approach requires the use of periodic cells (as do most computational methods), the Mn and Ni are long-range ordered in rows on the triangular lattice of transition metal sites. In all cells, the symmetry was lowered enough (lower than that associated with Li/vacancy or Ni/Mn ordering alone) so that Jahn-Teller distortions could take place if energetically favorable. In practice, this means that the symmetry is always a subgroup of the C2/m group of the monoclinic layered
system, a supercell with two formula units was used. Because this computational approach requires the use of periodic cells (as do most computational methods), the Mn and Ni are long-range ordered in rows on the triangular lattice of transition metal sites. In all cells, the symmetry was lowered enough (lower than that associated with Li/vacancy or Ni/Mn ordering alone) so that Jahn-Teller distortions could take place if energetically favorable. In practice, this means that the symmetry is always a subgroup of the C2/m group of the monoclinic layered 
The valence state of a high-spin transition metal ion can best be determined by integrating the spin-polarization density in a sphere around the ion. Integrating spin density is much more effective than integrating the charge density, as the former filters out the electronic contribution from the oxygen p -states which usually carry very little net electron spin. For the relevant ions, 
 and
and  we expect a total electron spin count of, respectively, 3, 4, and 5 (in units of
we expect a total electron spin count of, respectively, 3, 4, and 5 (in units of  ). For
). For 
 and
and  we expect 0, 1, and 2, respectively, electron spins as the
we expect 0, 1, and 2, respectively, electron spins as the  has a core of nonspin polarized, full
has a core of nonspin polarized, full  levels. Figure 1a shows the integrated spin as function of integration radius around Ni and Mn in the
levels. Figure 1a shows the integrated spin as function of integration radius around Ni and Mn in the  structure. The integrated moment increases steeply as we integrate through the d-states of the metal ion, but then reaches a plateau value because the charge density of the oxygen ions does not contribute to the spin density. After this plateau the integrated value increases again as spin from neighboring transition metals is picked up. The Mn ion in
structure. The integrated moment increases steeply as we integrate through the d-states of the metal ion, but then reaches a plateau value because the charge density of the oxygen ions does not contribute to the spin density. After this plateau the integrated value increases again as spin from neighboring transition metals is picked up. The Mn ion in  clearly carries three electrons, corresponding to a valence of
clearly carries three electrons, corresponding to a valence of  The moment around Ni is slightly below what is expected for
The moment around Ni is slightly below what is expected for  The remainder of the moment is probably on the oxygen ions as is typical for nickel oxides. Some evidence for this lies in the fact that the point where the integrated spin density rises from the plateau value is at a shorter radius than for the
The remainder of the moment is probably on the oxygen ions as is typical for nickel oxides. Some evidence for this lies in the fact that the point where the integrated spin density rises from the plateau value is at a shorter radius than for the  ion. The spin integration in Fig. 1a indicates that the formal valence states are
ion. The spin integration in Fig. 1a indicates that the formal valence states are  Further evidence can be found from the changes in spin density upon lithium removal. Figure 1b shows similar spin integrations for the delithiated material
Further evidence can be found from the changes in spin density upon lithium removal. Figure 1b shows similar spin integrations for the delithiated material  The spin on Mn is barely different from what it is in the fully lithiated material, while Ni has lost most of its moment, consistent with the electron configuration for
The spin on Mn is barely different from what it is in the fully lithiated material, while Ni has lost most of its moment, consistent with the electron configuration for  in
in  Figure 1a and b offer strong evidence that in
Figure 1a and b offer strong evidence that in  the correct valence assignment is
the correct valence assignment is  and
and  Upon Li removal
Upon Li removal  is oxidized to
is oxidized to  while the
while the  ion remains unchanged.
ion remains unchanged.

Figure 1. (a) Integrated spin as a function of integration radius (Å) around Ni and Mn in  (b) Integrated spin as a function of integration radius (Å) around Ni and Mn in
(b) Integrated spin as a function of integration radius (Å) around Ni and Mn in 
The selective oxidation of Ni in these materials is also consistent with the metal-oxygen bond length variations predicted from the computations (Fig. 2). Ni-O and Mn-O are separately shown in 
 and
and  The vertical bars on the result indicate the variation in bond length in the structure because the symmetry of our supercell (lower than
The vertical bars on the result indicate the variation in bond length in the structure because the symmetry of our supercell (lower than  ) allows for slightly different bond lengths in a given octahedron. The Mn-O bond length hardly varies with Li composition, confirming that little or no change to its valence state occurs. In contrast, the Ni-O bond length changes dramatically with Li composition. For the delithiated material, the Ni-O bond length is around 1.90 Å, very typical of
) allows for slightly different bond lengths in a given octahedron. The Mn-O bond length hardly varies with Li composition, confirming that little or no change to its valence state occurs. In contrast, the Ni-O bond length changes dramatically with Li composition. For the delithiated material, the Ni-O bond length is around 1.90 Å, very typical of  20
21 The very large spread in Ni-O distances at
20
21 The very large spread in Ni-O distances at  is due to the Jahn-Teller distortion around
is due to the Jahn-Teller distortion around  ion. The Jahn-Teller distortion is of the positive Q3 type,22 so that there are four short and two long bonds. In a real material, the spread in bond lengths may be somewhat less due to the disorder of the Ni/Mn ions. Table I shows the calculated and experimentally measured lattice parameters. The calculated numbers are somewhat larger than those measured, as is often the case for computations in the generalized gradient approximation.
ion. The Jahn-Teller distortion is of the positive Q3 type,22 so that there are four short and two long bonds. In a real material, the spread in bond lengths may be somewhat less due to the disorder of the Ni/Mn ions. Table I shows the calculated and experimentally measured lattice parameters. The calculated numbers are somewhat larger than those measured, as is often the case for computations in the generalized gradient approximation.

Figure 2. Plot of Ni-O (-⋅-) and Mn-O (⎯) bond lengths as a function of lithium content. Calculations performed at compositions of 
 and
and  The data points correspond to the average bond lengths while the bars at each point indicate the spread between the maximum and minimum bond lengths. Note that at 1/2 lithium content the minimum Ni-O bond length is less than the minimum Mn-O bond length, while the maximum Ni-O bond length is greater than the maximum Mn-O bond length. This is due to the
The data points correspond to the average bond lengths while the bars at each point indicate the spread between the maximum and minimum bond lengths. Note that at 1/2 lithium content the minimum Ni-O bond length is less than the minimum Mn-O bond length, while the maximum Ni-O bond length is greater than the maximum Mn-O bond length. This is due to the  undergoing a Jahn-Teller distortion.
undergoing a Jahn-Teller distortion.
Table I.
| Comparison of experimental with calculated lattice parameters. | ||
|---|---|---|
| Experimental and calculated lattice parameters | ||
| Comp | Structure | Lattice parameters |
| (exp or calc) | ||
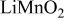 | C2/m layered | |
| exp. |
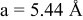 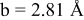 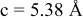 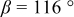 | |
| calc. |
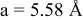 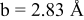 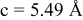 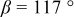 | |
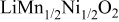 | rhombohedral layered | |
| exp.(1) |
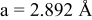 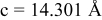 | |
| exp.(2) |
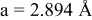 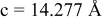 | |
| calc. |
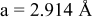 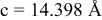 | |
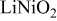 | rhombohedral layered | |
| exp.(1) |
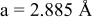 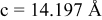 | |
| exp.(2) |
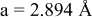 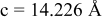 | |
| calc. |
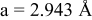 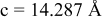
| |
We have also calculated the average discharge potential for the system  These are compared to experimental data in Table II. The calculated potentials are below the experimental values, as is typical with standard first principles energy methods.17 Hence, to make a better prediction possible, we have estimated a correction based on the difference between measured and calculated potentials for
These are compared to experimental data in Table II. The calculated potentials are below the experimental values, as is typical with standard first principles energy methods.17 Hence, to make a better prediction possible, we have estimated a correction based on the difference between measured and calculated potentials for  This correction
This correction  is added to the calculated potential to give the result in the last column. We emphasize that this adjustment is purely phenomenological and for the purpose of facilitating the direct comparison with experiments for
is added to the calculated potential to give the result in the last column. We emphasize that this adjustment is purely phenomenological and for the purpose of facilitating the direct comparison with experiments for  The data in this column agrees well with the measured values for
The data in this column agrees well with the measured values for  In the last row of the table the potential is broken down into the average for the interval
In the last row of the table the potential is broken down into the average for the interval  to
to  and
and  to
to  Some indication of the variation of potential upon charge can be derived from this.
Some indication of the variation of potential upon charge can be derived from this.
Table II.
Comparison between calculated and measured average discharge potentials for the 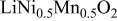 and and 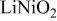 systems. The last column includes a correction to the computed voltage based on the difference between calculation and experiment for pure systems. The last column includes a correction to the computed voltage based on the difference between calculation and experiment for pure 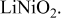 The last two rows of the table show the predicted average potentials in the first and last half of the discharge. The last two rows of the table show the predicted average potentials in the first and last half of the discharge. | |||||
|---|---|---|---|---|---|
| Calculated | Experimental | Adjusted | |||
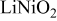 | 3.17 | 3.9 [28] | 3.9 | ||
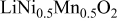 | 3.22 | 3.9 [14] | 3.95 | ||
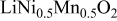 |
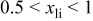 |
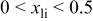 |
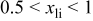 |
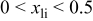 | |
| 2.94 | 3.51 | 3.67 | 4.24 | ||
Table II highlights the fact that the potential of  is actually very close to that of
is actually very close to that of  This is surprising because our results indicate that different redox couples are active in both materials. In
This is surprising because our results indicate that different redox couples are active in both materials. In  only
only  is active, while both
is active, while both  and
and  occur in
occur in  Hence the average potential for
Hence the average potential for  should be lower than for
should be lower than for  Even if one believed that Mn participates in the redox process the higher potential is difficult to explain, since the
Even if one believed that Mn participates in the redox process the higher potential is difficult to explain, since the  couple is below that of
couple is below that of  These experimental and theoretical results are further evidence that strong interactions exist between the redox couples of metals when they are mixed.23 In general alloy theory,24 a measure of the effective Ni-Mn interactions can be obtained by comparing the energy of
These experimental and theoretical results are further evidence that strong interactions exist between the redox couples of metals when they are mixed.23 In general alloy theory,24 a measure of the effective Ni-Mn interactions can be obtained by comparing the energy of  to the average energy of
to the average energy of  and
and  If
If  is negative, Ni and Mn have an effective attractive interaction and the system will be either randomly mixed or ordered, depending on the strength of the interaction and the preparation temperature. If
is negative, Ni and Mn have an effective attractive interaction and the system will be either randomly mixed or ordered, depending on the strength of the interaction and the preparation temperature. If  is positive, local phase separation into Mn and Ni rich regions is energetically preferred, though random mixing may be achieved if the synthesis temperature is high enough. From calculating the relevant energy numbers in the above equation we find that for
is positive, local phase separation into Mn and Ni rich regions is energetically preferred, though random mixing may be achieved if the synthesis temperature is high enough. From calculating the relevant energy numbers in the above equation we find that for 
 is
is  per formula unit, indicating a strong ordering (attractive) tendency between Ni and Mn. Similarly, for the delithiated material
per formula unit, indicating a strong ordering (attractive) tendency between Ni and Mn. Similarly, for the delithiated material  is computed to be
is computed to be  indicating repulsive Ni-Mn interactions. These results give some insight as to why the voltage is higher than may be expected for this system. Ni-Mn interactions go from being attractive in
indicating repulsive Ni-Mn interactions. These results give some insight as to why the voltage is higher than may be expected for this system. Ni-Mn interactions go from being attractive in  to being repulsive in
to being repulsive in  Hence, to remove lithium one not only has to supply the binding energy for the Li ion and electron, but also the strong energy increase in the system due to the Mn-Ni bonds becoming unfavorable (as that interaction turns from attractive to repulsive). It can be easily deduced that the effect of changes in the metal-metal interactions upon the average discharge potential is given by
Hence, to remove lithium one not only has to supply the binding energy for the Li ion and electron, but also the strong energy increase in the system due to the Mn-Ni bonds becoming unfavorable (as that interaction turns from attractive to repulsive). It can be easily deduced that the effect of changes in the metal-metal interactions upon the average discharge potential is given by
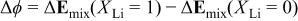
where ϕ is the equilibrium potential over the range  For the
For the  system, this result indicates that the potential is raised by about 266 mV over what would be expected if no Ni-Mn interactions were present (e.g., if
system, this result indicates that the potential is raised by about 266 mV over what would be expected if no Ni-Mn interactions were present (e.g., if  acted in a pure host, without the presence of Mn). These numbers are derived from calculations on an ordered supercell of Ni and Mn. We have estimated that if the Ni and Mn ions were fully randomized (rather than ordered in the supercell that we used for the calculations) the increase in potential would be slightly less and about 200 mV.
acted in a pure host, without the presence of Mn). These numbers are derived from calculations on an ordered supercell of Ni and Mn. We have estimated that if the Ni and Mn ions were fully randomized (rather than ordered in the supercell that we used for the calculations) the increase in potential would be slightly less and about 200 mV.
The effect of Li on the Ni-Mn interactions can be easily understood, using what is known about the miscibility of oxides.25 The effective interaction, for studying phase stability and mixing is not the bare ionic interaction but the energy difference between the average of identical pairs (i.e., Ni-Ni and Mn-Mn) and different pairs (i.e., Ni-Mn). Hence the simplest way to sample this difference is to consider the difference in energy between  (where Ni-Mn bonds are present) and
(where Ni-Mn bonds are present) and  (with Ni-Ni bonds) and
(with Ni-Ni bonds) and  (with Mn-Mn bonds). In the delithiated state, Ni and Mn have the same
(with Mn-Mn bonds). In the delithiated state, Ni and Mn have the same  valence and there is no net electrostatic interaction for exchanging their positions. It can be shown that for such iso-valent ions, the net interaction is due to size effects, and is always repulsive.26 This agrees with our result of a positive mixing energy in the delithiated state. On the other hand, in the lithiated material the different valence of Ni and Mn leads to a strong effective attractive interaction and hence explains the ordering or mixing tendency.
valence and there is no net electrostatic interaction for exchanging their positions. It can be shown that for such iso-valent ions, the net interaction is due to size effects, and is always repulsive.26 This agrees with our result of a positive mixing energy in the delithiated state. On the other hand, in the lithiated material the different valence of Ni and Mn leads to a strong effective attractive interaction and hence explains the ordering or mixing tendency.
Our results conclusively indicate that in  (and hence in the related systems
(and hence in the related systems  )14 Ni is the electrochemically active ion and cycles between
)14 Ni is the electrochemically active ion and cycles between  and
and  The material remains kinetically stable against transformation to spinel because Mn is not present in oxidation states lower than
The material remains kinetically stable against transformation to spinel because Mn is not present in oxidation states lower than  We recently showed that the very rapid transformation of layered
We recently showed that the very rapid transformation of layered  to spinel is due to the ease with which
to spinel is due to the ease with which  disproportionates to
disproportionates to  and
and  27 This allows Mn to rapidly migrate through tetrahedral sites as
27 This allows Mn to rapidly migrate through tetrahedral sites as 
 on the other hand, was shown to have a very high activation barrier for diffusion through the tetrahedral site. Hence, layered oxides with only manganese in the
on the other hand, was shown to have a very high activation barrier for diffusion through the tetrahedral site. Hence, layered oxides with only manganese in the  oxidation state are expected to be quite stable. In cycling between
oxidation state are expected to be quite stable. In cycling between  and
and  the Ni-Mn arrangement remains fixed due to the lack of any transition metal mobility at room temperature, but the interactions between the ions change considerably. In this system, this change in Ni-Mn interactions causes the voltage to increase for the
the Ni-Mn arrangement remains fixed due to the lack of any transition metal mobility at room temperature, but the interactions between the ions change considerably. In this system, this change in Ni-Mn interactions causes the voltage to increase for the  couple over what it would be in a noninteracting matrix.
couple over what it would be in a noninteracting matrix.
The class of materials in which the valence of Ni is  and Mn is
and Mn is  seems to possess many desirable features for a cathode material. They also point at new and interesting directions for cathode research. The combination of experimental data on highly doped systems, and our understanding of the role of
seems to possess many desirable features for a cathode material. They also point at new and interesting directions for cathode research. The combination of experimental data on highly doped systems, and our understanding of the role of  in the problems of many Mn-oxides, clearly indicates that stable layered Mn oxides, containing only
in the problems of many Mn-oxides, clearly indicates that stable layered Mn oxides, containing only  can be made. In these materials Mn has given up its role as an electrochemically active center and is present only as "filler." Hence, other elements that can take on the
can be made. In these materials Mn has given up its role as an electrochemically active center and is present only as "filler." Hence, other elements that can take on the  oxidation state in the layered oxide environment, could be selected on the basis of cost, weight, processability, environmental behavior, etc., as a substitute for Mn.
oxidation state in the layered oxide environment, could be selected on the basis of cost, weight, processability, environmental behavior, etc., as a substitute for Mn.
Acknowledgments
The authors acknowledge the support of the National Science Foundation (MRSEC Program) under contract no. DMR 98-08941. Discussions with Dane Morgan, Elena Arroyo, and Anton Van der Ven were helpful in developing the ideas in this paper. Computing resources from NPACI the National Partnership for Advanced Computing Infrastructure (NSF) are gratefully acknowledged.
The Massachusetts Institute of Technology assisted in meeting the publication costs of this article.