Abstract
The lithium-ion battery is an ideal candidate for a wide variety of applications due to its high energy/power density and operating voltage. Some limitations of existing lithium-ion battery technology include underutilization, stress-induced material damage, capacity fade, and the potential for thermal runaway. This paper reviews efforts in the modeling and simulation of lithium-ion batteries and their use in the design of better batteries. Likely future directions in battery modeling and design including promising research opportunities are outlined.
Export citation and abstract BibTeX RIS
Lithium-ion (Li-ion) batteries are becoming increasingly popular for energy storage in portable electronic devices. Compared to alternative battery technologies, Li-ion batteries provide one of the best energy-to-weight ratios, exhibit no memory effect, and experience low self-discharge when not in use. These beneficial properties, as well as decreasing costs, have established Li-ion batteries as a leading candidate for the next generation of automotive and aerospace applications.1,2 Li-ion batteries are also a promising candidate for green technology. Electrochemical power sources have had significant improvements in design, economy, and operating range and are expected to play a vital role in the future in automobiles, power storage, military, mobile-station, and space applications. Lithium-ion chemistry has been identified as a good candidate for high-power/high-energy secondary batteries and commercial batteries of up to 100 Ah have been manufactured. Applications for batteries range from implantable cardiovascular defibrillators operating at 10 μA, to hybrid vehicles requiring pulses of up to 100 A. Today the design of these systems have been primarily based on (1) matching the capacity of anode and cathode materials, (2) trial-and-error investigation of thicknesses, porosity, active material and additive loading, (3) manufacturing convenience and cost, (4) ideal expected thermal behavior at the system level to handle high currents, etc., and (5) detailed microscopic models to understand, optimize, and design these systems by changing one or few parameters at a time. The term 'lithium-ion battery' is now used to represent a wide variety of chemistries and cell designs. As a result, there is a lot of misinformation about the failure modes for this device as cells of different chemistries follow different paths of degradation. Also, cells of the same chemistry designed by various manufacturers often do not provide comparable performance, and quite often the performance observed at the component or cell level does not translate to that observed at the system level.
Problems that persist with existing lithium-ion battery technology include underutilization, stress-induced material damage, capacity fade, and the potential for thermal runaway.3 Current issues with lithium-ion batteries can be broadly classified at three different levels as shown schematically in Fig. 1: market level, system level, and single cell sandwich level (a sandwich refers to the smallest entity consisting of two electrodes and a separator). At the market level, where the end-users or the consumers are the major target, the basic issues include cost, safety, and life. When a battery is examined at the system level, researchers and industries face issues such as underutilization, capacity fade, thermal runaways, and low energy density. These issues can be understood further at the sandwich level, at the electrodes, electrolyte, separator, and their interfaces. Battery researchers attribute these shortcomings to major issues associated with Solid-Electrolyte Interface (SEI)-layer growth, unwanted side reactions, mechanical degradation, loss of active materials, and the increase of various internal resistances such as ohmic and mass transfer resistance. This paper discusses the application of modeling, simulation, and systems engineering to address the issues at the sandwich level for improved performance at the system level resulting in improved commercial marketability.

Figure 1. Current issues with Li-ion batteries at the market level and the related performance failures observed at the system level, which are affected by multiple physical and chemical phenomena at the sandwich level.
"Systems engineering can be defined as a robust approach to the design, creation, and operation of systems. The approach consists of the identification and quantification of system goals, creation of alternative system design concepts, analysis of design tradeoffs, selection and implementation of the best design, verification that the design is properly manufactured and integrated, and post-implementation assessment of how well the system meets (or met) the goals."4 Process systems engineering has been successfully employed for designing, operating, and controlling various engineering processes and many efforts are currently being attempted for Li-ion batteries. The development of new materials (including choice of molecular constituents and material nano- and macro-scale structure), electrolytes, binders, and electrode architecture are likely to contribute toward improving the performance of batteries. For a given chemistry, the systems engineering approach can be used to optimize the electrode architecture, operational strategies, cycle life, and device performance by maximizing the efficiency and minimizing the potential problems mentioned above.
The schematic in Fig. 2 shows four systems engineering tasks and the interactions between these tasks. Ideally, the eventual goal of the systems engineering approach applied to Li-ion batteries would develop a detailed multiscale and multiphysics model formulated so that its equations can be simulated in the most efficient manner and platform, which would be employed in robust optimal design. The first-principles model would be developed iteratively with the model predictions compared with experimental data at each iteration, which would be used to refine the detailed model until its predictions became highly accurate when validated against experimental data not used in the generation of the model. The following sections describe each of these systems engineering tasks in more detail.
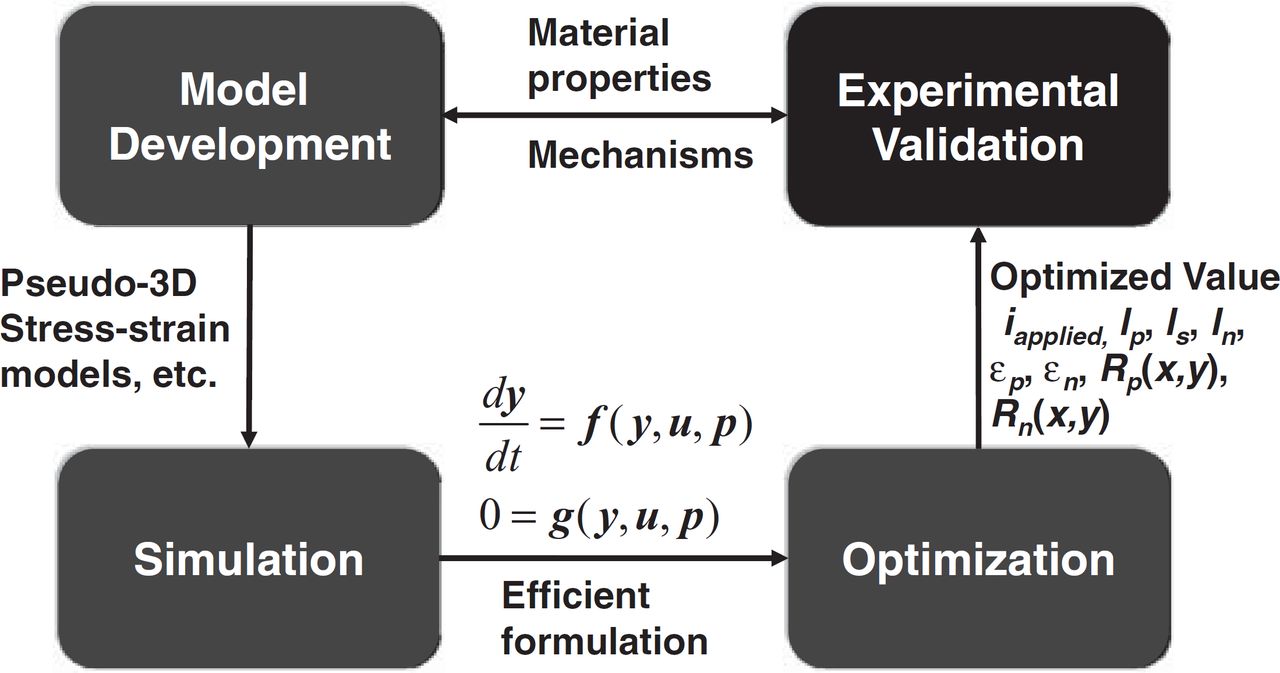
Figure 2. Schematic of systems engineering tasks and the interplay between them: In the figure, u, y, and p are vectors of algebraic variables, differential variables, and design parameters, respectively.
Systems engineering approaches have been used in the battery literature in the past, but not necessarily with all of the tasks and their interactions in Fig. 2 implemented to the highest level of fidelity. Such a systems engineering approach can address a wide range of issues in batteries, such as
- (1)Identification of base transport and kinetic parameters
- (2)Capacity fade modeling (continuous or discontinuous)
- (3)Identification of unknown mechanisms
- (4)Improved life by changing operating conditions
- (5)Improved life by changing material properties
- (6)Improved energy density by manipulating design parameters
- (7)Improved energy density by changing operating protocols
- (8)Electrolyte design for improved performance
- (9)State estimation in packs
- (10)Model predictive control that incorporates real-time estimation of State-of-Charge (SOC) and State-of-Health (SOH).
- (11)Improved protocols for optimum formation times.
The next section reviews the status of the literature in terms of modeling, simulation, and optimization of lithium-ion batteries, which is followed by a discussion of the critical issues in the field, and methods for addressing these issues and expected future directions in the conclusions section.
Background
In Fig. 2, model development forms the core of the systems engineering approach for the optimal design of lithium-ion batteries. Generally, the cost of developing a detailed multiscale and multiphysics model with high predictive ability is very expensive, so model development efforts begin with a simple model and then add more physics until the model predictions are sufficiently accurate. That is, the simplest fundamentally strong model is developed that produces accurate enough predictions to address the objectives. The best possible physics-based model can depend on the type of issue being addressed, the systems engineering objective, and on the available computational resources. This section describes various types of models available in the literature, the modeling efforts being undertaken so far, and the difficulties in using the most comprehensive models in all scenarios.
An important task is to experimentally validate the chosen model to ensure that the model predicts the experimental data to the required precision with a reasonable confidence. This task is typically performed in part for experiments designed to evaluate the descriptions of physicochemical phenomena in the model whose validity is less well established. However, in a materials system such as a lithium-ion battery, most variables in the system are not directly measurable during charge-discharge cycles, and hence are not available for comparison to the corresponding variables in the model to fully verify the accuracy of all of the physicochemical assumptions made in the derivation of the model. Also, model parameters that cannot be directly measured experimentally typically have to be obtained by comparing the experimental data with the model predictions.
A trial-and-error determination of battery design parameters and operating conditions is inefficient, which has motivated the use of battery models to numerically optimize battery designs. This numerical optimization can be made more efficient by use of reformulated or reduced order models.5–10 Simulation time plays a role in determining the use of these models in various applications, and high simulation times have limited the application of battery optimization based on physics-based models. Efficient ways of simulating battery models is an active area of research and many researchers have published various mathematical techniques and methods to simulate physics-based battery models faster.5,6,9,10 This has enabled greater use of optimization and systems engineering based on physics-based models.11–13
Once an efficient method of simulating the battery models is devised, the next step is to formulate optimization problems to address the real-world challenges described in the previous section. The objective function can be chosen based on the required performance objectives at the system level. Optimization of operating conditions, control variables, and material design (architecture) can be performed based on specific performance objectives described in more detail in a later section. After obtaining either an optimal operating protocol or electrode architecture for a specific performance objective, the results should be verified using experiments.
Mathematical models for lithium-ion batteries vary widely in terms of complexity, computational requirements, and reliability of their predictions (see Fig. 3). Including more detailed physicochemical phenomena in a battery model can improve its predictions but at a cost of increased computational requirements. Therefore simplified battery models continue to be applied in the literature when appropriate for the particular needs of the application. This section summarizes the literature on model development for lithium-ion batteries, and the application of these models in systems engineering. Models for the prediction of battery performance can be roughly grouped into four categories: empirical models, electrochemical engineering models, multiphysics models, and molecular/atomistic models.
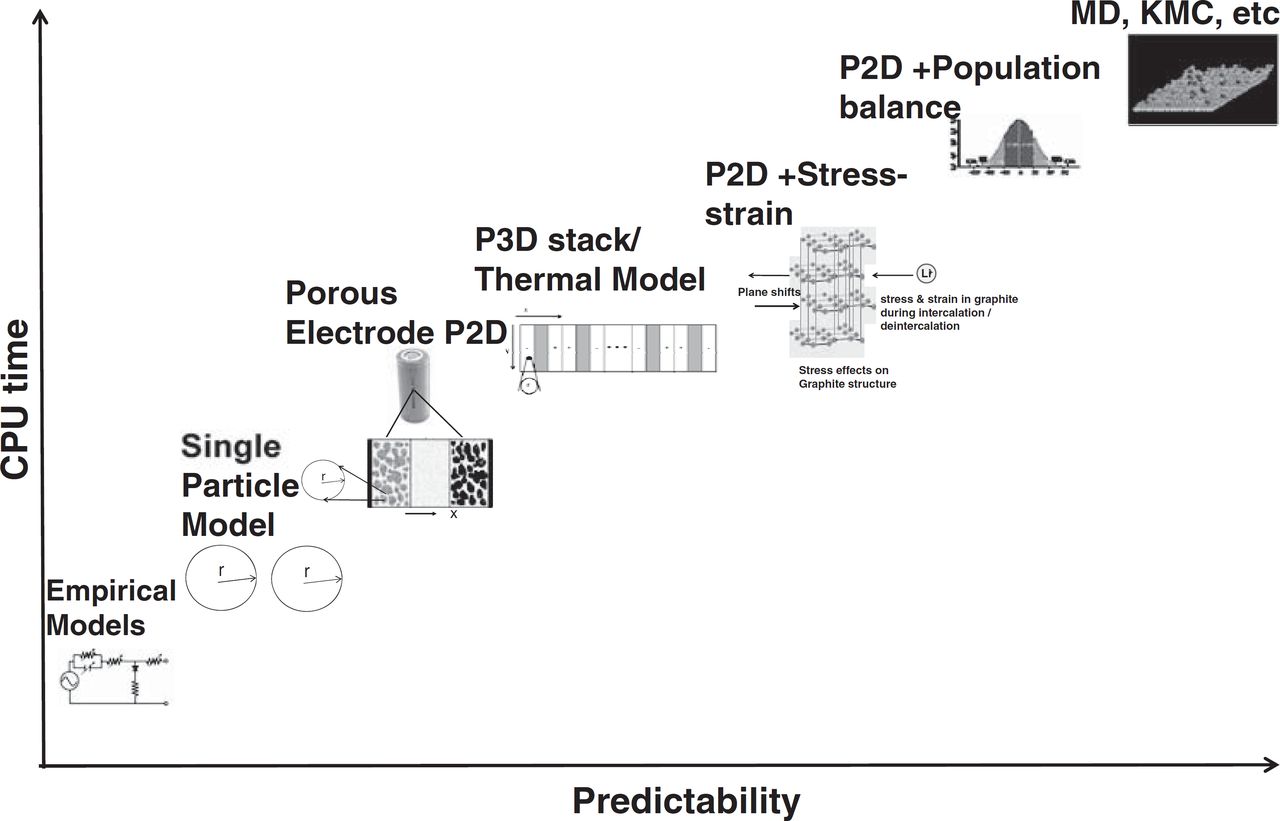
Figure 3. Wide range of physical phenomena dictates different computational demands.
Empirical models
Empirical models employ past experimental data to predict the future behavior of lithium-ion batteries without consideration of physicochemical principles. Polynomial, exponential, power law, logarithmic, and trigonometric functions are commonly used as empirical models. The computational simplicity of empirical models enables very fast computations, but since these models are based on fitting experimental data for a specific set of operating conditions, predictions can be very poor for other battery operating conditions. Such battery models are also useless for the design of new battery chemistries or materials.
Electrochemical engineering models
The electrochemical engineering field has long employed continuum models that incorporate chemical/ electrochemical kinetics and transport phenomena to produce more accurate predictions than empirical models. Electrochemical engineering models of lithium-ion batteries have appeared in the literature for more than twenty years.14 Below is a summary of electrochemical engineering models, presented in order of increasing complexity.
Single-particle model
The single-particle model (SPM) incorporates the effects of transport phenomena in a simple manner. Zhang et al.15 developed a model of diffusion and intercalation within a single electrode particle, which was expanded to a sandwich by considering the anode and cathode each as a single particle with the same surface area as the electrode.16 In this model, diffusion and intercalation are considered within the particle, but the concentration and potential effects in the solution phase between the particles are neglected.16,17 The following typical reactions are considered in each of the particle in the SPM (MO refers to metal oxide):

Due to these simplifications, this model can be quickly simulated, but is only valid for limited conditions, such as low rates and thin electrodes.17 Greater efficiency can be obtained by including a parabolic profile approximation for the lithium concentration within the particle.16,18
Ohmic porous-electrode models
The next level of complexity is a porous-electrode model that accounts for the solid- and electrolyte-phase potentials and current while neglecting the spatial variation in the concentrations. The model assumes either linear, Tafel, or exponential kinetics for the electrochemical reactions and incorporates some additional phenomena, such as the dependency of conductivities as a function of porosity. Optimization studies have been performed using this model to design the separator and electrode thicknesses19–21 and ideal spatial variations of porosity within electrodes.13
Pseudo-two-dimensional models
The pseudo-two-dimensional (P2D) model expands on the ohmic porous-electrode model by including diffusion in the electrolyte and solid phases, as well as Butler-Volmer kinetics (see Fig. 4). Doyle et al.14 developed a P2D model based on concentrated solution theory to describe the internal behavior of a lithium-ion sandwich consisting of positive and negative porous electrodes, a separator, and a current collector. This model was generic enough to incorporate further advancements in battery systems understanding, leading to the development of a number of similar models.14,22–32 This physics-based model is by far the most used by battery researchers, and solves for the electrolyte concentration, electrolyte potential, solid-state potential, and solid-state concentration within the porous electrodes and the electrolyte concentration and electrolyte potential within the separator. This model based on the principles of transport phenomena, electrochemistry, and thermodynamics is represented by coupled nonlinear partial differential equations (PDEs) in x, r, and t that can take seconds to minutes to simulate. The inclusion of many internal variables allow for improved predictive capability, although at a greater computational cost than the aforementioned models.
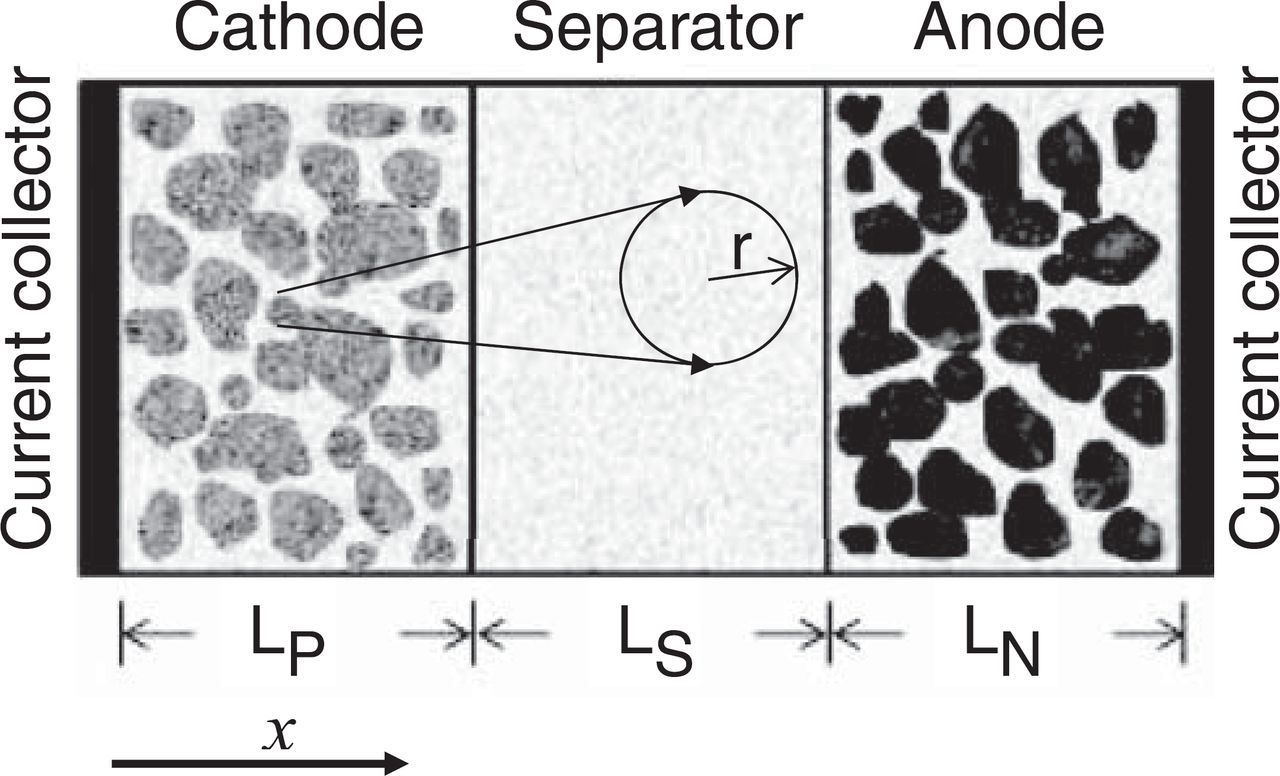
Figure 4. P2D model with schematic of the cell sandwich with the cathode, anode, and separator also showing the spherical particles in the pseudo-second dimension.
Multiphysics models
Multiscale, multidimensional, and multiphysics electrochemical-thermal coupled models are necessary to accurately describe all of the important phenomena that occur during the operation of lithium-ion batteries for high power/energy applications such as in electric/hybrid vehicles.
Thermal models
Including temperature effects into the P2D model adds to the complexity, but also to the validity, of the model, especially in high power/energy applications. Due to the added computational load required to perform thermal calculations, many researchers have decoupled the thermal equations from the electrochemical equations by using a global energy balance, which makes it impossible to monitor the effects on the performance of the cells due to temperature changes.33–37 Other researchers have similarly decoupled the thermal simulation of the battery stack from the thermal/electrochemical simulation of a single cell sandwich.38,39 Other thermal models have been reported that are coupled with first-principles electrochemical models both for single cells and cell stacks.40–42 The global energy balance is only valid when the reaction distribution is uniform all over the cell; for accurate estimation of heat generation in a cell, the local variations in the reaction current and SOC must be incorporated.43 Recently, Guo et al.17 published a simplified thermal model applied to a single particle. Some papers have presented 2D thermal-electrochemical coupled models for lithium-ion cells that take into account the effects of local heat generation.44,45 Similar studies predict battery discharging performance at different operating temperatures.46 Additionally, the coupling of a 1D electrochemical model with a lumped thermal model by means of an Arrhenius form of temperature dependence for the physicochemical properties has been reported.47–49 Recently, researchers have begun considering 3D thermal models to better understand the dynamic operation and control of lithium-ion batteries for large-scale applications. Since such models are quite computationally expensive, several approximations are made, resulting in various shortcomings. Some models cannot monitor the thermal effect of electrochemical parameters,35,50 while other models require empirical input from experiments or other simulations,51,52 (or use volume-averaged equations for the solid-phase intercalation). Another approach assumes a linear current-potential relationship and neglects spatial concentration variations and is therefore only valid for low power operations.53 A Multi-Scale Multi-Dimensional (MSMD) model54 and a model derived from a grid of 1D electrochemical/thermal models55 have also been implemented for 3D thermal simulation of batteries.
Stress-strain and particle size/shape distributions
Intercalation of lithium causes an expansion of the active material, such as graphite or manganese oxide, while lithium extraction leads to contraction. The diffusion of lithium in graphite is not well understood, but some work has been done to model the diffusion and intercalation of lithium into the electrode material.27,56,57 Since lithium diffuses within the particle, the expansion and contraction of the material will not happen uniformly across the particle (i.e., the outer regions of the particle will expand first due to lithium intercalating there first). This spatial nonuniformity causes stress to be induced in the particle and may lead to fracture and loss of active material,58,59 which is one of the mechanisms for capacity fade. Various models have been developed to examine the volume change and stress induced by lithium-ion intercalation for single particles.60–62 A two-dimensional microstructure model was developed63 to extend the stress-strain analysis from single particles and was eventually incorporated into the full P2D model.64 These models show that high rates of charging result in increased stress and increased chance of fracture, which can be somewhat mitigated by using smaller particles, or ellipsoidal particles. Additionally during battery cycling, some particles are lost or agglomerate to form larger sized particles, which results in performance degradation. In addition, porous materials rarely have uniform particle size and shape. Some continuum models have accounted for the distribution of particle sizes and its effect on the battery performance,65,66 for example, through the equation65
![Equation ([1])](https://content.cld.iop.org/journals/1945-7111/159/3/R31/revision1/jes_159_3_R31eqn1.jpg)
where  is the fraction of total current flowing in solution, N (r) is the number of particles per unit volume of composite electrode with a radius between size r and r + dr in the porous electrode, Y(r) is a function that relates the outward normal current density per unit surface area of a particle to the potential difference, and
is the fraction of total current flowing in solution, N (r) is the number of particles per unit volume of composite electrode with a radius between size r and r + dr in the porous electrode, Y(r) is a function that relates the outward normal current density per unit surface area of a particle to the potential difference, and  is the potential difference between the solid particle and the adjacent solution. A promising future direction would be to extend such models to include variations in particle size and shape distribution by (1) writing N in terms of the multiple independent particle coordinates that define the particle shape (typically 3), and (2) replacing the single integral with a more complicated volume integral. The time-dependent change in the particle size distribution due to breakage and agglomeration can be modeled by a spatially-varying multi-coordinate population balance equation:
is the potential difference between the solid particle and the adjacent solution. A promising future direction would be to extend such models to include variations in particle size and shape distribution by (1) writing N in terms of the multiple independent particle coordinates that define the particle shape (typically 3), and (2) replacing the single integral with a more complicated volume integral. The time-dependent change in the particle size distribution due to breakage and agglomeration can be modeled by a spatially-varying multi-coordinate population balance equation:
![Equation ([2])](https://content.cld.iop.org/journals/1945-7111/159/3/R31/revision1/jes_159_3_R31eqn2.jpg)
where f (l, x, t) is the particle size and shape distribution function, x is the spatial coordinate, li is the ith independent size coordinate, l is the vector with elements li (typically of dimension three), Gi (l,t) = dli/dt is the growth rate along the ith independent size coordinate (which is negative for shrinkage), h(l, x, t, f) is the generation/disappearance rate of particle formation (e.g., due to breakage and agglomeration), and t is time.67–70 The expression for h(l, x, t, f) for breakage and agglomeration contains integrals over the f (l, x, t), and the h and Gi have dependencies on additional states such as local lithium-ion concentrations. This model to capture the effects of morphology within a material, called a mesoscale model,71,72 would enable the material degradation due to spatially-varying and time-varying changes in the particle size and shape distribution to be explicitly addressed.
Stack models
In order to simulate battery operation more accurately, battery models are improved by considering multiple cells arranged in a stack configuration. Simulation of the entire stack is important when thermal or other effects cause the individual cells to operate differently from each other. Since it is often not practical or possible to measure each cell individually in a stack, these differences can lead to potentially dangerous or damaging conditions such as overcharging or deep-discharging certain cells within the battery, which can cause thermal runaway or explosions. The ability to efficiently simulate battery stacks would facilitate the health monitoring of individual cell behavior during charging and discharging operations and thereby increasing safety by reducing the chances of temperature buildup causing thermal runaway. The significant increase in computational requirements to simulate a stack model has slowed its development and most examples of stack modeling perform some approximation or decoupling to facilitate efficient simulation.36,39,73 Researchers have also published simplified coupled thermal electrochemical models applied to a single particle for stacks in parallel and series configurations.74 Fully coupled battery stack simulations have been performed for a limited number of cells by using reformulation techniques to expedite simulation.75
Molecular/atomistic models.— Kinetic Monte Carlo method
The Kinetic Monte Carlo (KMC) method is a stochastic approach that has been used to model the discharge behavior of lithium ions during intercalation. Such models76–79 have been used to simulate diffusion of lithium from site to site within an active particle to aid in understanding on how different crystal structures affect lithium mobility75 and how the activation barrier varies with lithium-ion concentration.78,79 Additionally, Monte Carlo methods have been used to predict thermodynamic properties.80 KMC has also been applied to simulate the growth of the passive SEI-layer across the surface of the electrode particle, to simulate one of the mechanisms for capacity fade.81
Molecular dynamics
Molecular dynamics has been used to gain insight into several molecular-scale phenomena that arise during the operation of lithium-ion batteries. One of the applications has been to the simulation of the initial growth of the passivating SEI film at the interface of the solvent and graphite anode. The application of a large negative potential during initial charging decomposes ethylene carbonate (EC) in the solvent, to produce the passivating SEI film containing lithium ethylene dicarbonate and salt decomposition products. Although molecular dynamics is computationally very expensive for simulation of more than tens of picoseconds of battery operation, the method was demonstrated to be fast enough for simulation of the initial stage of SEI layer formation.82 The simulations were able to predict the formation of carbon monoxide, which has been detected in experiments, and predicted that the initial SEI layer formation occurs is initiated at highly oxidized graphite edge regions of the anode.
Another application of molecular dynamics to lithium-ion batteries has been the simulation of the initial transport of lithium ions through a polycrystalline cathode.83 Between each crystal grain is an amorphous intergranular film (IGF), and the motivation for the study was the conjecture that lithium ions diffuse much faster through the IGF than through the crystal grains. Although the simulations employed a particular lithium silicate glass as a solid electrolyte and vanadia with an amorphous V2O5 IGF separating the crystal grains, the results are expected to have more general applicability. The simulations were feasible with molecular dynamics because the conclusions only required that the lithium ion diffuse far enough into the cathode to quantify the differences in diffusion rates through the IGF and crystal grains. The simulation of effective diffusivities is one of the most common applications of molecular dynamics.84
Density functional theory
Density functional theory (DFT) calculations can be used to provide predictive insight into the structure and function of candidate electrode materials. The ground-state energy is given as a unique functional of the electron density, which can be calculated by self-consistently solving for the atomic orbitals. Geometry optimizations are used to determine structures, energetics, and reaction mechanisms. In the area of sustainable energy storage, DFT calculations have been used to predict and rationalize the structural changes that occur upon cycling of electrode materials, for example, in the calculation of activation barriers and thermodynamic driving forces for Ni ions in layered lithium nickel manganese oxides. Similar calculations have been used to determine the lattice properties and electronic structure of graphite and LiC6.85 Additionally, DFT calculations can be used to examine the effect of lithium intercalation on the mechanical properties of a graphite electrode, including Young's modulus, expansion of the unit cell, and the resulting stress effects,86 as well as to compare the stability of LiPF6 (a common electrolyte) in various solvents.87 DFT calculations have also been used to examine the mechanisms affecting the stability and function of the organic electrolytes separating the electrode materials, as in the reductive decompositions of organic propylene carbonate and ethylene carbonate to build up a solid-electrolyte interface that affects cycle-life, lifetime, power capability, and safety of lithium-ion batteries.
Simulation
Multiple numerical methods are available for the simulation of any particular battery model. For empirical models, analytical solutions are usually possible and can be easily solved in Microsoft Excel or Matlab.88 Analytical solutions can be implemented in a symbolic language such as Mathematica,89 or Maple,90 or Mathcad,91 or in a compiled language such as FORTRAN or C++. Analytical solutions based on linear model equations often involve eigenvalues, which might have to be determined numerically. For nonlinear model equations, sometimes analytical series solutions using perturbation methods92 or other symbolic techniques93 can be derived. Numerical simulation methods are more flexible, with multiple methods available for any particular battery model. The best numerical methods tend to be more sophisticated when moving toward the upper right of the battery models shown in Fig. 3.
For SPMs for a single electrode, analytical solutions have been derived for constant-current operation and cannot be obtained directly for the constant-potential operation, due to the fact that the boundary flux is implicitly determined by the nonlinear Butler-Volmer equation particularly when the open circuit voltage changes with state of charge. At this scale, especially for AC impedance data, analytical solutions are easily obtained and have been heavily used even for estimating unknown diffusion coefficients. A numeric symbolic solution was also derived for the AC impedance response that showed similar results to the analytical solution.94–96
When two electrodes are included in an SPM, an analytical solution is available for constant-current operation but not for constant-potential operation, for reasons as stated above, or when film formation for the SEI layer is modeled. Beyond SPM and porous electrode ohmic resistance models, analytical solutions are not possible for simulating charge-discharge curves. A SPM with two electrodes consists of a single partial differential equation for each electrode. Conversely, a finite-difference scheme discretized with 50 node points in the radial direction generates 50 × 2 + 50 × 2 = 200 differential algebraic equations (DAEs). Recall that the SPM is computationally efficient but is not accurate, especially for high rates. For P2D models14 typically the finite-difference approach has been used. A P2D model with polynomial approximation18 for the solid phase, when discretized with 50 node points in the spatial direction for each variable, results in a system of 250 DAEs for each electrode and 100 DAEs for the separator. Thus, the total number of DAEs to be solved for the P2D model across the entire cell is 250 + 250 + 100 = 600 DAEs. The addition of temperature effects to this model results in 750 DAEs to be solved simultaneously. Stack models are much more computationally expensive, as the number of DAEs is equal to the number of cells in the stack (N) times the number of equations coming from each sandwich. Using the finite-difference discretization of spatial variables in x, y, and r with 50 node points along each direction in a pseudo-3D thermal-electrochemical coupled model would generate 15,000 + 7500 + 15,000 = 37,500 DAEs to be solved simultaneously for a single sandwich.
The speed and accuracy of a numerical method depends upon the complexity of the model equations, including operating and boundary conditions, and the numerical algorithm. The most common numerical methods for simulation of lithium-ion batteries are the finite-difference method (FDM), finite-volume method (FVM, or sometimes called the control volume formulation), and finite-element method (FEM). The main continuum simulation methods reported in the literature for the simulation of batteries can be classified as
- (1)DUALFOIL.26 This software employs Newman's BAND subroutine,97 which is a finite-difference method used to simulate electrochemical systems for more than four decades. Symbolic software such as Mathematica89 and Maple90 can be used for determining analytical expressions for the Jacobians and for generating the associated FORTRAN code for use with the BANDJ subroutine.23
- (2)FVM with various time-discretization schemes,98 which has been applied to P2D models.
- (3)
- (4)Finite-difference or reformulation schemes in spatial coordinates with adaptive solvers such as DASSL in time.23
Each approach has its advantages and disadvantages. DUALFOIL is a freely available FORTRAN code. The FDM has been used extensively in battery simulation23 as it is easy to implement and modify. The FVM is closely related to the FDM but more easily handles irregular geometries. The FEM handles both irregular geometries and heterogeneous compositions, but is much harder to implement by hand, and so is usually only applied to batteries using commercial FEM software such as COMSOL. An advantage of commercial software like COMSOL is ease of use and that the numerical implementation is invisible to the user and results from COMSOL can be directly integrated to MATLAB environment, which is a widely used tool for control and optimization. However, a disadvantage is that COMSOL's numerical implementations cannot be modified by the user to (1) increase computational efficiency by exploiting additional mathematical structure in the model equations or (2) integrate such efficient simulation results into advanced systems engineering algorithms for optimal design, operation, or control in a computationally efficient manner.
When optimization fails while using COMSOL-like codes, detective work is required to determine whether the numerical simulation was robust enough to provide accurate numerical Jacobians. Also, as of today, global optimization methods are readily available only for algebraic equations. Algebraic optimizations can be formulated by discretization of all the variables and parameters including the control variables,101,102 but these optimization schemes typically have too high complexity to be solvable using existing global optimization software. Many groups are working on the development of optimization software that is more computationally efficient at computing local optima for dynamic optimizations or on ensuring convergence to a global optimum.103,104
BATTERY DESIGN STUDIO100 has a module for the simulation of P2D lithium-ion battery models. Adaptive solvers provide advantages in speed compared to fixed time-discretization schemes. Researchers have used DASSL for solving battery models.23 DASSL/DASPK use backward differentiation schemes in time, which are numerically stable and efficient. For the same set of equations, these adaptive schemes can provide an order of magnitude savings in time. Battery models more advanced than the P2D model are usually solved offline in the literature (an exception is the P2D thermal model from Gu et al.44,48 and the stress-strain model from Renganathan et al.63).
To understand the importance of capacity fade in a lithium-ion secondary battery system, significant efforts have been devoted to the development of mathematical models that describe the discharge behavior and formation of the active and passive SEI layers. The majority of these models are empirical or semi-empirical.105,106 Other works have attempted to simulate capacity fade by considering the lithium deposition as a side reaction and the resulting increased resistance.31,107–111 Others have simulated capacity fade by modeling the active material loss, or change of internal parameter with cycling.31,108–113 Other researchers have used KMC methods to examine the SEI layer formation at the microscale level.81 Such a model, however, is computationally expensive, which makes online simulation difficult. Further work is needed to couple such fundamental models to the popular continuum models in use.
Optimization applied to Li-ion batteries
Several researchers have applied optimization to design more efficient electrochemical power sources. Newman and co-workers obtained optimal values of battery design parameters such as electrode thickness and porosity.21,24,26,114–117 To simplify the optimization, many of these papers employed models with analytical solutions, which are only available in limiting cases. Battery design optimization using a full order model has been demonstrated by several researchers.11,24,26,115,116 Newman and co-workers report the use of Ragone plots for studies regarding the optimization of design parameters, changing one design parameter at a time, such as electrode thickness, while keeping other parameters constant, Ragone plots for different configurations can be obtained. Hundreds of simulations are required when applied current is varied to generate a single curve in a Ragone plot, which is tedious and computationally expensive. An alternative is to simultaneously optimize the battery design parameters and operating conditions such as the charging profile.11 Parameters have been simultaneously optimized for different models and goodness of fits compared based on statistical analysis.118 Parameter estimation has also been used in a discrete approach to analyze and predict capacity fade using a full-order P2D model.110,111 Golmon et al.119 attempted a multiscale design optimization for improving electrochemical and mechanical performance of the battery by manipulating both micro- and macro-scale design variables such as local porosities, particle radii, and electrode thickness to maximize the capacity of the battery. A surrogate-based framework using global sensitivity analysis has been used to optimize electrode properties.120 Simulation results from P2D models have been used to generate approximate reduced-order models for use in global sensitivity analysis and optimization. Rahimian et al.12 used a single-particle model when computing the optimum charging profile for maximizing the life of battery during cycling. The following section describes the systems engineering tasks of (1) parameter estimation, (2) model-based optimal design, and (3) state estimation that have been applied to lithium-ion batteries.
Parameter estimation is typically formulated as the minimization of the sum-of-squared differences between the model outputs and their experimentally measured values for each cycle i, for example,121–123
![Equation ([3])](https://content.cld.iop.org/journals/1945-7111/159/3/R31/revision1/jes_159_3_R31eqn3.jpg)
where yi(tj) is the measured voltage at time tj for cycle i, ymodel, i(tj; θi) is the voltage computed from the battery model at time tj for cycle i for the vector of model parameters θi (the parameters being estimated from the experimental data), and ni is the number of time points in cycle i. Solving the optimization [3] is known in the literature as least-squares estimation.121–123 Many numerical algorithms are available for solving the nonlinear optimization [3], such as the steepest descent, Gauss-Newton, and Levenberg-Marquardt methods.122 These iterative methods reduce the sum-of-squared differences between the model outputs and the experimental data points until the error is no longer significantly reduced. More sophisticated Bayesian estimation methods employ the same numerical algorithms but use optimization objectives that take into account prior information on the model parameters.124
Battery design parameters such as cell thickness and electrode porosity and operating profiles can be optimized using the same numerical algorithms, for objectives such as maximization of performance (e.g., energy density, life) or minimization of capacity fade and mechanical degradation. These optimizations are solved subject to the model equations and any physical constraints. The optimal estimation of unmeasured states in lithium-ion batteries can also be formulated in terms of a constrained model-based optimization. The optimization objectives, models, and constraints differ for different systems engineering tasks, but can all be written in terms of one general formulation:125
![Equation ([4])](https://content.cld.iop.org/journals/1945-7111/159/3/R31/revision1/jes_159_3_R31eqn4.jpg)
![Equation ([5])](https://content.cld.iop.org/journals/1945-7111/159/3/R31/revision1/jes_159_3_R31eqn5.jpg)
![Equation ([6])](https://content.cld.iop.org/journals/1945-7111/159/3/R31/revision1/jes_159_3_R31eqn6.jpg)
![Equation ([7])](https://content.cld.iop.org/journals/1945-7111/159/3/R31/revision1/jes_159_3_R31eqn7.jpg)
where Ψ is the optimization objective,126 z(x) is the vector of differential state variables, y(x) is the vector of algebraic variables, u(x) is the vector of control variables, and p is the vector of design parameters. Although there are many numerical methods for solving constrained optimization problems,127–129 this paper summarizes only control vector parameterization (CVP) as this is the method that is easiest to implement and most commonly used in industrial applications. The CVP method parameterizes the optimization variables, by employing basis functions or discretization, in terms of a finite number of parameters to produce a nonlinear program that can be solved using standard software. First-principles models for lithium-ion batteries tend to be highly stiff, requiring adaptive time-stepping for reasonable computational efficiency.104 CVP is well suited for optimizations over such models, as CVP incorporates the model equations by calling a user-specified subroutine for simulating the model equations. Any speedup obtained by an adaptive time-stepping for the model equations directly translates into a speedup on the CVP calculations.
More specifically, the control variable u(x) in CVP is parameterized by a finite number of parameters, typically as a polynomial or piecewise-linear function or by partitioning its values over space, and the resulting nonlinear program is solved numerically. Most numerical optimization algorithms utilize an analytically or numerically determined gradient of the optimization objective and constraints to march toward improved values for the optimization variables in the search space. In CVP, as the number of intervals increases, the number of equations increases and makes optimization more computationally expensive. Hence the fastest and most efficient battery model and code for the desired level of accuracy is recommended when applying CVP or any alternative optimization methods.
A discussion of simulating lithium-ion batteries at the systems-level is incomplete without addressing issues pertaining to the estimation of state-of-charge and health of the battery. Designing a tool to predict the life or performance of a battery is an interesting optimization problem with implications on material modifications during the initial battery formulation for a particular application, allowance for making a specific maintenance plan during the course of the life of the battery, and, most importantly, on the cost of the battery. Precise estimations of SOC and SOH are also essential to ensure the safe operation of batteries, that is, preventing the battery from overcharging and thermal runaway.
Some commonly used methods in the industry to monitor the SOC of the battery include monitoring of the cell impedance,130–133 equivalent circuit analyses,134,135 techniques based on fuzzy logic,136,137 or pattern recognition.138 Optical and eddy current methods139,140 are being devised to monitor available capacity in battery systems with flat response surfaces. Based on the algorithm used for estimation, the models used to estimate SOC and SOH can be classified broadly into two categories. Some utilities such as the battery packs used in on-board satellites during the lack of solar energy or cells used in watches follow a routine or pre-programmed load. In such instances, it is possible to develop a degradation model based on a priori testing, knowing the operating conditions and the design parameters of the cell. Such a model does not require frequent updates for the parameters, unless there is a significant change in the operating conditions. In some other applications, such as battery packs used in vehicles, the battery is subjected to a dynamic load that changes as frequently as every few milliseconds. In these cases, the degradation mechanism and hence state of charge or the state of health of the power system depends on the load conditions imposed in the immediate past and it is necessary to monitor the cell on a regular basis. There are some differences between the algorithms used to make life-estimates for the case with the known operating parameters compared to the dynamic-load case. The latter situation is less forgiving in terms of the calculation time, for example. SOC and SOH estimators have been an integral part of battery controllers; however, the estimations have been primarily based on empirical circuit-based models that can fail under abusive or non-ideal operating conditions. Precise estimations of SOC and SOH are very essential for the safe operation of the batteries, in order to prevent them from overcharging and thermal runaway. Santhanagopalan et al.141 reviewed past efforts on the monitoring and estimation of SOC in the literature, and reported an online Kalman filter-based SOC estimation for lithium-ion batteries based on a single-particle model. Klein et al.9 recently published state estimation using a reduced order model for a lithium-ion battery. Smith et al.'s10 analysis of a 1D electrochemical model for a lithium-ion battery indicated that the electrode surface concentration was more easily estimated from the real-time measurements than the electrode bulk concentration. Domenico et al.142 designed an extended Kalman filter for SOC estimation based on an electrochemical model coupling the average solid active material concentration with the average values of the chemical potentials, electrolyte concentration, and the current density.
Critical Issues in the Field
This section describes the challenges that arise when building predictive models for lithium-ion batteries and employing these models for systems engineering.
Sparsity of manipulated variables
Once the battery is manufactured and closed in a sealed case, the battery is discharged (used) according to the requirements of the application. The only variables that can be manipulated during battery operation to make best use of the battery is the charging current profile and operating temperature, which can affect transport and electrochemical rates resulting in modified performance.
Before the battery is sealed, the design variables such as the electrode dimensions, the type of materials, and materials properties such as porosity, active surface area, and microstructure can be selected so as to provide the best possible performance. The resulting battery design can be verified at small scale (e.g., few milli- or micro-Ah batteries) relatively easily in the laboratory, but scaling up to the large-scale batteries required for some industrial applications is challenging.
Need for better fundamental models to understand SEI-layer, structure
The physicochemical understanding is incomplete for much of the phenomena that occur inside a battery, such as capacity fade, stress-strain effects, mechanical degradation, and mechanisms for failure due to shocks, defects, and shorts. Much progress has been made in the last twenty years on failure mechanisms, stress-strain models, capacity fade mechanisms involving side reactions, SEI-layer formation, and other phenomena, and studies have been published with the objective of understanding battery operation at the molecular scale, using Kinetic Monte Carlo simulation, molecular dynamics, and density functional theory calculations, and at the mesoscale using population balance models. The molecular-scale models are simulated off-line (that is, not in real-time) and their predictions have been fed to continuum-scale models. A potential future application of molecular- and mesoscale models would be in the real-time prediction of the states of the battery at the small length scales for use in more accurate prediction of the whole battery performance in real time.
Robustness and computational cost in simulation and optimization
Battery models result in multiple DAEs to be simulated with unknown initial conditions while operating for multiple cycles of charge and discharge. For these models adaptive time steppers are usually more than an order of magnitude faster than uniform time-discretization. Several adaptive solvers are available for solutions of DAE models.143–146 Recently, many easy-to-use ODE solvers have been made available (ode15s, ode15i, etc.) from MATLAB,88 "NDsolve" from MATHEMATICA,89 and "dsolve" from MAPLE90 to solve non-stiff, stiff and moderately stiff DAE models of index-1.
In spite of recent advancements, many of these DAE solvers and initialization routines can fail due to numerical convergence problems during Newton iteration to solve nonlinear equations and singular/ill-conditioned Jacobian matrices resulting from small integration steps. The complexity in battery model simulation is increased by steep variations of the dependent variables (concentrations and potentials) between charging and discharging.
Battery simulations for extended operations, such as during switching from constant-current to constant-potential operations, typically require some form of event detection. The DAEs for battery models increase in complexity and also in number as the accuracy and predictability of models increase. Simulation times for battery models range from milliseconds for empirical circuit-based models to minutes for P2D/P3D models and even days for a multiscale model such as a P2D model coupled with KMC simulation, limiting the options for real-time simulations.
Uncertainties in physicochemical mechanisms
Although much literature exists for capacity fade, SEI-layer formation, and other phenomena, no existing model simulates all of the mechanisms for capacity fade or battery failure. More detailed information is required to sufficiently specify a hypothesized mechanism for a phenomenon before it can implemented in a simulation model, such as
- Which chemical species are formed and consumed in each phase and at the interface between phases?
- What is the physical configuration of each chemical species at the interface between phases (e.g., is a molecule on an electrode surface sticking out into the electrolyte or flat against the surface)?
- How many sites does each molecule on a solid surface cover?
Substantial experimental design efforts are required to answer such questions so the answers can be incorporated into first-principles lithium-ion battery models. Also, most applications using batteries for long-term requirements depend on projections made from model predictions coupled with limited test data; however, the relationship between failure modes during the test conditions and those during actual operating scenarios have not been clearly established – necessitating the tools used in SOC and SOH predictions to be independent of the operating or manufacturing conditions. Quite often in such scenarios, the use of look-up tables limits the confidence in the predictive capabilities of the models.
Conventional degradation models based on extensive testing of batteries under various operating conditions and loads have in general attributed the degradation of battery performance to loss of the active material and loss of lithium that can be cycled. Several detailed models to quantify the signature of these parameters on the aging profile of lithium ion batteries have been presented.31,147 Other approaches include the use of arbitrary empirical parameters obtained by regressing test data. These models usually interpolate the SOC and the health of the battery based on pre-stored database of information. Such models are widely employed in the industry when sufficient information on the physics of the materials in the batteries is not available – this problem is commonplace among module and pack manufacturers, who assemble the units from cells manufactured by a third-party. It is standard industrial practice to calibrate such models148,149 since monitoring the evolution of all of the physical parameters such as transport coefficients and the reaction rates within each cell inside the pack is expensive, if not impossible. Network models have also been used to address non-uniform degradation in large format cells.150
Addressing the Critical Issues, Opportunities, and Future Work
This section describes some approaches for addressing the critical issues raised in the previous section, looking toward likely future research directions in the modeling and systems engineering of lithium-ion batteries.
Sparsity of manipulated variables
Currently, batteries are charged at constant current until a cutoff potential is reached or a time limit followed by charging at constant potential. However, these charging protocols may result in thermal runaway, leading to under-utilization and possibly even explosions. Given the limited variables that are available for manipulation, it is especially important to make the best utilization of these variables during battery operations. A first-principles battery model can be employed in a dynamic optimization framework to compute a time-varying charging profile that maximizes life, minimizes capacity fade, and improves battery performance.
The determination of an optimized charging profile requires a first-principles model that has high predictive accuracy for a wide range of operating conditions, since charge transfer, reaction kinetics, and diffusion rates may be quite different than in the experiments used in the model development. A first-principles model that describes the battery behavior at the meso- and microscale models would be able to take these effects into account during the dynamic optimization. The application of dynamic optimization to compute an optimal charging profile is illustrated here for a P2D model11 for lithium-ion batteries. The dynamic optimization for a cell was formulated as:
![Equation ([8])](https://content.cld.iop.org/journals/1945-7111/159/3/R31/revision1/jes_159_3_R31eqn8.jpg)
where the optimization objective E is the total energy stored in the cell, V is the voltage obtained from the cell as computed from the first-principles model, iapplied is the applied current to the cell, the charging time tf was restricted to 1 hr, the maximum allowed voltage was 4.05 V, and the value for V as a function of time. The implementation of dynamic optimization is facilitated by the use of a reformulated model6 to compute the optimization objective. The time profiles for the electrolyte concentration at the cathode/current collector interface in Fig. 6 are for three different charging scenarios: (1) conventional charging at constant-current followed by constant-potential charging, (2) constant-current charging at an optimized value obtained by solving the dynamic optimization for a fixed value, and (3) the time-varying charging profile given by Eq. 5. The electrolyte concentration at X = 0 (the cathode/current collector interface) has the highest peak value during dynamically optimized charging, due to its higher initial current. For the chosen chemistry, mass transfer limitations in the electrolyte occur at higher currents. This protocol indicates that to increase the energy density, more energy should be stored at shorter time, albeit causing mass transfer limitations in the electrolyte, and allow the concentration to equilibrate at longer times to ensure longer operability of the battery. During dynamically optimized charging, the electrolyte concentration decreases in the latter part of charging, as lithium-ion transfer slows while more lithium ions are packed into the carbon matrix in the negative electrode. In contrast, after the first 10 minutes the electrolyte concentration is nearly constant during optimized constant-current charging. When a meaningful global objective function was chosen at the system level and robust optimization tool and meaningful models are used, improvements in 'local' battery behavior are observed.

Figure 6. Dynamic analysis of electrolyte concentration at the positive electrode for the three charging protocols. The solid line at C = 1 represents the equilibrium concentration.
The above approach can be considered as a top-down approach, where operating conditions or charging protocols are determined at the system level (battery as a whole), and the system-level behavior is affected by the local mass/charge transfer and reaction effects (Fig. 1) and indirectly manipulates non-measurable internal variables such as the electrolyte concentration or potential or also the solid-phase concentrations as shown schematically in Fig. 6. Physics-based models are required in the dynamic optimization to correctly relate the local effects to the system-level behavior as quantified by the optimization objective. The more detailed and accurate the model, the more optimal 'local' behavior can be determined using the few manipulated variables at the system level.
Note that the SPM model lacks sufficient information on the behavior in the cell to be of much usefulness in the above optimizations. If the first-principles model employed in the optimization includes a high fidelity thermal model, then the localized temperatures in the cell can be included as a constraint in the optimization. A more detailed multiscale model that includes more of the physicochemical phenomena would be needed for optimization of battery operations for very quick charging generally involving rates of 2C or higher.
Another approach that can be used to address the sparsity of manipulated variables is to have the limited number of material properties (manipulated variables) vary spatially. If the electrode architecture is designed to minimize and address every possible local nonideality at the sandwich level, then the system level performance will improve. This can be viewed as the bottom-up approach, where the material properties or electrode architecture, etc. are determined at the electrode level (micro-scale), to produce improved performance at the system level (Fig. 1). Physics-based models are required in the optimization framework to correctly relate the local effects to the system-level behavior as quantified by the optimization objective. For example, consider the minimization of the ohmic resistance at the sandwich level (Fig. 1). Optimization of spatially-uniform porosity reduced the ohmic resistance by 20%, whereas optimization for a spatially-varying profile results in a reduction of 33% (Fig. 7).13 Physics-based models are required in the optimization framework to correctly relate the local effects to the system-level behavior as quantified by the optimization objective. Note that improved performance for both solid-phase potential and current are obtained locally, which leads to reduced ohmic resistance across the sandwich, which then relates to improved performance for charge-discharge curves at the system level.
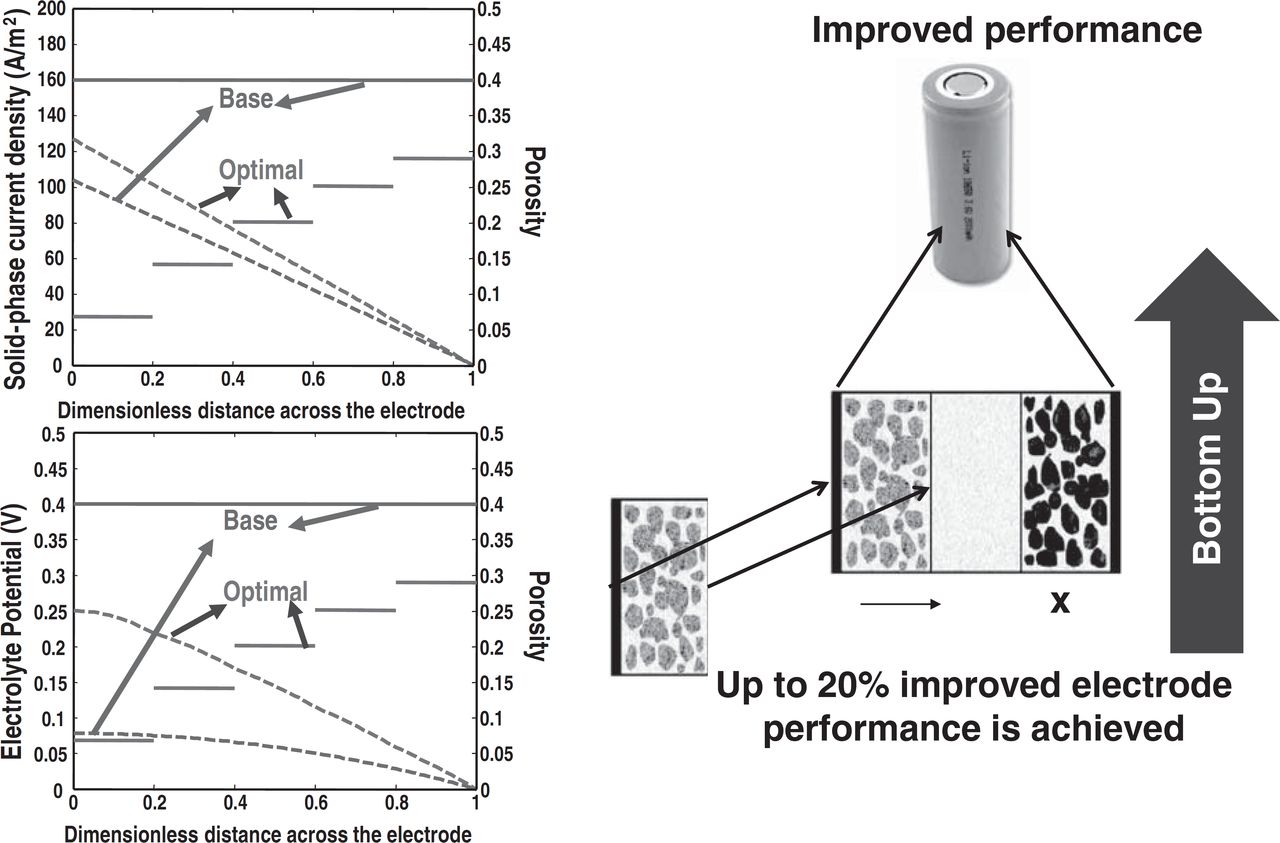
Figure 7. Model-based optimal battery design based on a porous electrode model. Solid lines are for porosity, and dashed lines represent solid-phase current density (A/m2)/ Electrolyte potential (V).
To address all the issues in Fig. 1, a more detailed model is required (i.e., moving right along the diagonal in Fig. 3). Possible material properties that can be varied as a function of distance are given in Fig. 2. Note that for particle radius, optimization with the P2D model would yield only the smallest possible radius, but stress-strain models would suggest a different size for mechanical stability.119
The more sophisticated the battery model, more computationally intensive the simulations and optimization. While the value of adding more physicochemical phenomena into battery models is clear, and discussed in more detail below, there is also a need to improve the computational efficiency in the simulation of these models by reformulation or order reduction.
Need for better fundamental models to understand SEI-layer, structure
Different simulation methods are effective at different scales (see Fig. 5), which has motivated efforts to combine multiple methods to simulate multiscale systems. Battery models that dynamically couple the molecular- through macro-scale phenomena could have a big impact in understanding and designing lithium-ion batteries. The above continuum models could be coupled with stress-strain models and population balance models to describe the time evolution of the size and shape distribution of particles. Probably the first step would be to couple molecular models with P2D models, to thoroughly validate the coupled simulation algorithms before moving to more computationally expensive 3D continuum models. KMC methods could be combined with P2D models to analyze surface phenomena such as growth of the SEI layer in a detailed manner, similarly as has been applied to other electrochemical systems.72,151–160 For a 125 × 125 mesh, 2D KMC coupled with P2D model with time steps ranging from nanoseconds to seconds would require simulation times ranging from minutes to hours and even days for a single cycle. Another multiscale coupling that could be useful is to occasionally employ molecular dynamics to update transport parameters in a P2D or 3D model. Molecular dynamics can provide information that cannot be predicted using a P2D or 3D continuum model, but long times cannot be simulated using molecular dynamics, so the combination of the two approaches has the potential to increase fidelity while being computationally feasible.

Figure 5. Approximate ranking of methods appropriate for the simulation of different time and length scales.
The current literature review suggests that typically researchers have expertise and skills in one or two of the models/methods reported in Fig. 6. If researchers with expertise in different fields collaborate, the task of multiscale model development becomes easier and faster progress can be expected. While black-box approaches are available for some of the methods in Fig. 5, it is strongly recommended that, at least for case studies, hard-coded direct numerical simulation is carried out to enable better understanding of coupling between models at different length and time scales.
Robustness and computational cost in simulation and optimization
The complexities of battery systems have made efficient simulation challenging. The most popular model, the P2D model, is often used because it is derived from well understood kinetic and transport phenomena, but the model results in a large number of highly nonlinear partial differential equations that must be solved numerically. For this reason, researchers have worked to simplify the model though reformulation or reduced order methods to facilitate effective simulation. One method of simplification is to eliminate the radial dependence of the solid phase concentration using a polynomial profile approximation,18 by representing it using the particle surface concentration and the particle average concentration, both of which are functions of the linear spatial coordinate and time only. This type of volume-averaging161,162 combined with the polynomial approximation163,164 has been shown to be accurate for low to medium rates of discharge.18,165–168 At larger discharge rates, other approaches have been developed to eliminate the radial dependence while maintaining accuracy.106,165–168 Approximate solution methods have also been developed for phase-change electrodes, for solid phase diffusion.169 Recently, discretization in space alone has been used by researchers to reduce the model to a system of DAEs with time as the sole independent variable in order to take advantage of the speed gained by time-adaptive solvers such as DASSL/DASPK.5,6,144 Such solvers also have the advantage of being capable of detecting events, such as a specific potential cutoff, and running the simulation only to that point.
Complications arise when determining consistent initial conditions for the algebraic equations. Consequently, many good solvers fail to solve DAE models resulting from simulation of battery models.170 As a result, it is necessary to develop initialization techniques to simulate battery models. Many such methods can be found in the literature for a large number of engineering problems. Methods and solvers specifically focusing on initialization of battery models are also available in the literature.170,171 Recently, a perturbation approach has been used to efficiently solve for consistent initial conditions for battery models.172 An alternative continuum representation of the discrete events in the charge/discharge cycle of a battery that does not require initialization between the discrete events of a given cycle or between any two cycles was also proposed.173
Proper orthogonal decomposition (POD) has been used to reduce the computational cost in various sets of model equations, by fitting a reduced set of eigenvalues and nodes to obtain a reduced number of equations.5 Alternatively, model reformulation techniques have been used to analytically eliminate a number of equations before solving the system.6 Other researchers have used orthogonal collocation and finite elements, rather than finite differences, in order to streamline simulations.75,174,175
For stack and/or thermal modeling of certain battery systems, many attempts have decoupled equations within the developed model.33–42 This approach breaks up a single large system into multiple, more manageable systems that can be solved independently. This allows the model to be solved quickly, but at the expense of accuracy. For this reason, efficient models that maintain the dynamic online coupling between the thermal and electrochemical behavior, as well as between individual cells in the stack are preferred.
Numerical algorithms for optimization can get stuck in local optima, which can be nontrivial to troubleshoot when the number of optimization parameters is large. This problem can at least be partly addressed using a sequential step-by-step approach (see Fig. 8). For illustration purposes, consider the maximization of the energy density with lp, ln, ls, ɛp, and ɛn, where l is the thickness of each region and ɛ the porosity (p – positive electrode, s – separator, and n – negative electrode).
- (1)Choose a battery model that can predict the optimization objective and is sensitive to the manipulated variables (e.g., a P2D model).
- (2)Reformulate or reduce the order of the model for efficient simulation. This step has to be judiciously made to ensure that the reduced order model is valid in the range of manipulated variables for optimization.
- (3)Maximize energy density with lp,
- (4)Using the solution from Step 3 as an initial guess, find optimal values for the two parameters (lp, ɛp).
- (5)Add parameters one by one, in the same manner as in Step 4.
- (6)Arrive at optimal performance with multiple parameters.
- (7)If needed before Step 3, find results with a simpler and less accurate model for a good initial guess.
- (8)After convergence, feed in more sophisticated models (for example, including stress effects) to make sure mechanical stability is not compromised.

Figure 8. Sequential approach for robust optimization of battery models with multiple design parameters.
A similar approach can be used for CVP for dynamic optimization with the total time interval divided as 2, 4, 8, etc. for subsequent optimizations until convergence.
The above algorithm will tend to have better convergence if the parameters in Steps 3–5 are rank ordered from having the largest to the lowest effect on the optimization objective. While advances have been made in the computation of global optima for dynamic optimizations,104,176 it will be at least a decade before such methods are computationally efficient enough for application to the optimal design of lithium-ion batteries using nontrivial physics-based models.
Fig. 9 shows improved performance at each step of an optimization while successively adding manipulated variables. Capacity matching was used a constraint for the thickness of the negative electrode.

Figure 9. Optimization of the energy density for a lithium-ion battery, showing the effect of electrode thickness and porosities.
Uncertainties in physicochemical mechanisms
Uncertainty quantification methods have been applied to hundreds of different kinds of systems to assess the progress of the development of first-principles models and to assess the confidence in model predictions.124,177,178 The Monte Carlo method and its many variants for uncertainty quantification are computationally expensive and have become less used over time compared to power series and polynomial chaos expansions. These expansion-based approaches avoid the high computational cost associated with applying the Monte Carlo method or parameter gridding by first computing an approximation to the full simulation model, followed by application of robustness analysis to the approximate model. These expansion-based methods are computationally efficient enough for application to lithium-ion batteries.
For example, consider the discrete estimation of model parameters as a way to track the effects of capacity fade. As of today, capacity fade is attributed to many reasons. This depends upon the chemistry, mode of operation, and size. A wide range of reasons can be linked to transport and kinetic parameters as published elsewhere.110,111,179 Five effective transport and kinetic parameters were estimated by applying least-squares estimation to the 250 mAh button cells experimental voltage-discharge data. The estimated parameters were the effective diffusion coefficient of lithium ion in the solution phase (D1), effective diffusion coefficient of lithium in the solid phase for the negative and positive electrodes (Dsn and Dsp), and electrochemical reaction rate constants for the negative and positive electrodes (kn and kp).
The effective negative-electrode solid-phase diffusion coefficient and reaction rate constant (Dsn and kn) were found to decrease monotonically with cycle #, whereas the other three parameters did not follow any particular trend. This suggested that the voltage-discharge curves may not contain sufficient information to accurately estimate the effective values of D1, Dsp, and kp, and that the change in the voltage-discharge curves with cycle # could be captured by estimation of only the effective solid-phase diffusion coefficient Dsn and reaction rate constant kn for the negative electrode. A more detailed analysis suggested that the voltage-discharge curves were very sensitive to the value of the effective solid-phase diffusion coefficient Dsn but weakly sensitive to deviations in the model parameters D1, Dsp, kp, and kn from their nominal values, resulting in large uncertainties in their values when fit to experimental voltage-discharge curves. That the voltage-discharge curves were much sensitive to a negative-electrode parameter (Dsn) suggests that mechanisms for capacity fade in the negative electrode, rather than the electrolyte or positive electrode, were the most important for this battery under these operating conditions.111
The overall trend in the variation of model parameters is more reliably assessed by plotting nominal estimates over many cycles. A discrete approach was adopted for the prediction of capacity fade by tracking the change in effective transport and kinetic parameters with cycle number (N). The model parameters Dsn and kn fit to the experimental data for cycles 25, 100, 200, 300, 400, and 500 were used to predict the remaining battery life based on voltage-discharge curves measured in past cycles. To characterize the degradation in the model parameters, a power law was fit to the estimated parameter values from cycles 25 to 500 as shown in Fig. 10a. By implicitly assuming that the changes in the parameter values are the result of the same mechanism in later cycles, the parameter values for the subsequent cycles were predicted using the power-law expressions. The voltage-discharge curve predicted by this model was in very good agreement with the experimental data at cycle 1000, indicating that the model was able to predict capacity fade as shown in Fig. 10b. Each red dot is an experimental data point and the blue line is the model prediction. It is likely that when more detailed multiscale models become available, there will not be a need to perform fitting and tracking of transport and kinetic parameters with cycles.

Figure 10. Parameter estimation, uncertainty analysis, and capacity fade prediction for a lithium-ion battery.
A rapid update of the parameters usually involves some form of a moving horizon algorithm that estimates the parameters used in the model using an initial set of data points (for example between from the start of the experiment to some interval of time t). These values for the parameters (θt) are then used to predict the cell performance for the next few data points (e.g., between times t and t + Δt). The error between the model predictions and the actual data points collected between t and t + Δt is then used to calculate the updated set of parameters θt+Δt. This process is repeated at periodic intervals of time or the load. Filtering techniques are commonly employed for on-line estimation;141,180,181 in most of these algorithms, the measured change in the response is divided between the actual battery response and system noise, based on pre-determined weights assigned to the functions calculating the noise and the battery models. The predicted response for the voltage is compared at the next time step and a correction term is introduced to the weights. More elaborate moving horizon estimates include the influence from several sets of parameters from the past on the current estimates. One example is the use of exponential forgetting functions.182,183 In this example, the effect of the parameter values θt, θt+Δt, θt+2Δt, etc. on the current estimate θt+kΔt is assumed to decay exponentially. The steps are summarized below:
- Step 1: Choose a subset of data points N0 that end when the parameters need to be updated. Calculate the initial value for the SOC.
- Step 2: Calculate the value of the exponential forgetting function at the end of N0.
- Step 3: Use the next set of data points N0+1 to N1, to calculate the updated values for the parameters in the model equations.
- Step 4: Update the SOC for the next set of data points using the parameter values from the previous step.
- Step 5: Update the exponential forgetting function, based on the data points N0+1 to N1, new values for the parameters, and the current value of the SOC.
- Step 6: Repeat Steps 1 to 5 until the end of the data set. This procedure produces a set of values for the SOC updated whenever the error between the model and the experimental data is significant.
The use of such online-tracking algorithms, together with reliable models requiring modest computational effort, greatly reduces the uncertainty associated with assessing the failure mode of the batteries, and can be implemented for a variety of operating conditions. The states of interest are tracked as and when the system operates; the advantages offered by this approach are two-fold: (1) any mitigation scheme can be implemented fairly quickly since the operator does not wait until performing the scheduled capacity checks on the batteries and (2) the proposed methodology does not rely solely on a characterization chart made under lab-scale testing environment, and captures the wear-and-tear imposed by the system on the batteries.
Conclusions
Advances in nanostructured multifunctional materials and new electrolytes will potentially improve the performance of lithium-ion batteries in the next two decades. Meanwhile, fundamental understanding is currently lagging behind the technological advancement in lithium-ion batteries as seen by the manufacturing of batteries for vehicles and other applications. New in-situ methods are currently being studied to experimentally understand intercalation and processes inside the lithium-ion battery in real-time.184–186 Whatever understanding that is gained will be incorporated into first-principles models and used in optimization to maximize the battery performance obtainable using the chemistry and materials of today. Although the details of the chemistry may be different, the approaches established for the optimization of today's battery designs are based on first principles that will be valid for tomorrow's materials and systems.
The main objective of this paper is to discuss recent developments and challenges in model development and simulation of lithium-ion batteries at different length and time scales from empirical models to atomistic models. The improved predictability of detailed multiscale models will enable precise manipulation of non-measurable variables at the micro and nanoscale. Numerical methods for the simulation of these more sophisticated models are expected to continue to improve in the future, to enable more details physicochemical phenomena to be included in battery design optimization.
The suggested directions for future research in this area are
- Development of multiscale models with improved fidelity over the full range of battery operations of interest.
- Development of robust and accurate reduced-order and reformulated models at different scales to enable efficient simulation for optimization.
- Development and implementation of robust and efficient numerical simulation and optimization schemes and software platforms to couple models of different kinds (continuum and non-continuum).
- Validation of improved performance at the system level and development of design procedures more capable of scale-up from small-scale batteries to large-scale batteries.
Acknowledgments
The authors are thankful for the financial support by the National Science Foundation under contract numbers CBET-0828002, CBET-0828123, and CBET-1008692, the International Center for Advanced Renewable Energy and Sustainability at Washington University in St. Louis (ICARES), Institute for Advanced Computing Applications and Technologies at University of Illinois, Urbana-Champaign, and the U.S. government. One of the authors (SS) gratefully acknowledges David Howell, Brian Cunningham, and the U.S. DOE Office of Vehicle Technologies Energy Storage Program for funding and support.