Abstract
Sodium transition metal oxides (NaMO2) with a P2 structure exhibit good Na+ ion conductivity and are promising sodium-ion battery cathode materials. Manganese-based compounds have a high working potential vs. Na+/Na, and high capacity. Yet, the layered nature of these materials means that they are prone to structural rearrangements at high voltage/low Na contents, the phase transformations and Na+ ion/vacancy ordering transitions resulting in capacity fade and poor reversibility. This review discusses the latest advances in the field and focuses mainly on recent work on NayMn1-xMxO2 (x, y ≤ 1, M = Ni, Mg, Li) compounds. We compare the different lithium and sodium transition metal layered oxides (P2, O3, etc.) and describe the structures and mechanisms observed on alkali (de)intercalation. The strategies used to enhance the electrochemical properties and stabilize the structural framework of sodium transition metal oxides are reviewed. We show how X-ray diffraction and 7Li/23Na solid-state Nuclear Magnetic Resonance can be combined to provide a detailed insight into the structural and electronic processes occurring upon electrochemical cycling of these materials.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Why Are We Interested in Na-Ion Battery Cathodes?
Together, the increasing demand for energy and the threat from Global Warming make electrical energy storage (EES) a world wide strategic priority. EES is expected to play a key role in the decarbonization of electric power sources. The two billion people worldwide not currently served by a reliable electricity supply are likely to be connected via local grids, for which EES is essential. While Li-ion batteries (LIBs) will play a role, a key challenge is to develop lower cost batteries that deliver safe, reliable storage with high cycle and calendar life. In this regard, Na-ion batteries (NIBs) are potentially important. NIBs operate in a similar fashion to LIBs, offering both advantages and disadvantages. Concerning the former, the ability to use Al instead of Cu as a current collector at the anode could substantially reduce cost. Na is also far more abundant in the Earth's crust than Li, and more widely distributed geographically. Regarding the disadvantages, the standard operating potential of Na metal is 300 mV more positive than Li metal. This generally translates into lower operating potentials for Na-ion systems, although the potential of a full cell depends on the difference in the chemical potentials of Na in the anode and cathode materials. There are considerable opportunities for scientists to develop new combinations of anode, electrolyte and cathode that are optimized for a variety of applications with different criteria such as high energy density, high power, and high operating potential. This has encouraged a rapid growth of interest in research into NIB components. Hard carbons show some promise as anodes,1–3 but more must be done to improve performance. The cathode remains a major challenge and it is to this that we direct the current review. A comparative plot showing the performance of various Li/Na-ion cathodes, in terms of average working potential, volumetric capacity, and volumetric energy density, is given in Figure 1. Although the highest energy density is observed for Li-ion cathodes, several Na-ion cathodes exhibit values that make them potential candidates. The figure also highlights the challenge of finding better Na-ion cathodes. Transition metal oxides typically exhibit high reversible capacities. Their compact structural framework makes them ideally suited for applications for which a high volumetric density is required. On the other hand, the inductive effect of the polyanion group in phosphates, fluorophosphates, sulfates, etc., generally leads to higher voltages. Some polyanionic frameworks are also excellent candidates for high power applications, due to their open frameworks that provide facile diffusion pathways for Na+ ions and enable fast ionic transport. However, the low density of the polyanionic structures means that they score poorly when it comes to volumetric energy density.
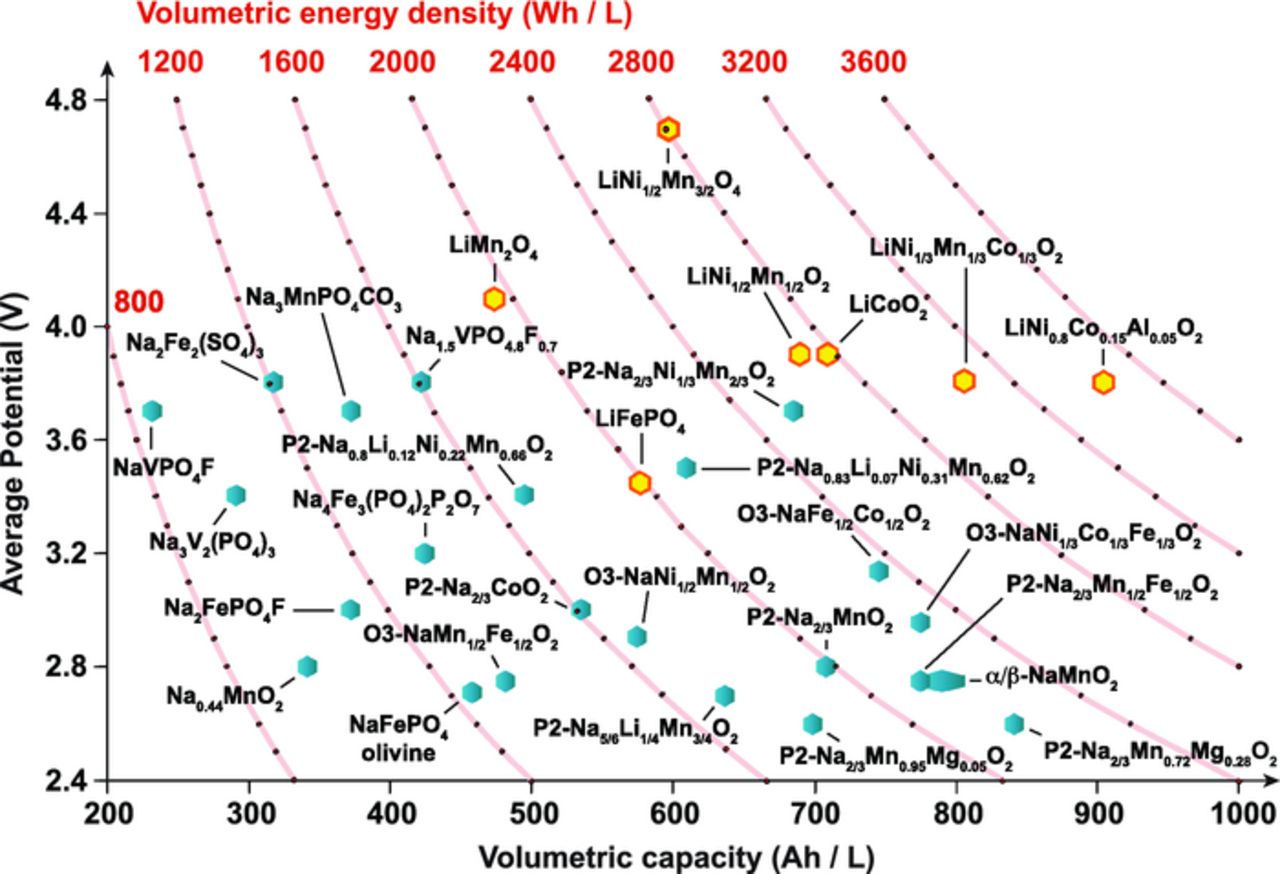
Figure 1. Average discharge potential (V vs. Na+/Na or vs. Li+/Li for the blue and yellow hexagons, respectively) and volumetric energy density (Wh/L) vs. volumetric capacity (Ah/L) for selected positive electrode materials for NIBs and LIBs. The mass and volume of the active electrode material have been taken into account to calculate the capacity and energy density. The data, collected in Table S1 of the Supplementary Information, were obtained from references.4–34
Outline of the Review
In this review, we will focus on recent advances in the understanding of manganese-based sodium transition metal oxides, NayMn1-xMxO2 (x, y ≤ 1, M = Ni, Mg, Li, etc.), used as NIB cathode materials. These compounds are promising materials for high performance cathodes due to their high potentials vs. Na+/Na, relatively good cycling ability, good volumetric capacity relative to other sodium and lithium cathodes, as shown in Figure 1, and easy synthesis. The layered structure of these materials is challenging to stabilize at high voltage/low Na contents. This review will focus mainly on our own work in the field and related studies. We shall begin by comparing lithium and sodium transition metal oxides used as intercalation materials in rechargeable batteries and consider the specific challenges faced by transition metal oxides upon alkali (de)intercalation. We will then discuss the strategies employed to enhance the electrochemical properties and stabilize the structural framework of sodium transition metal oxides.
In the second half of the review, we will briefly review the use of NMR spectroscopy to investigate structural transformations and electronic processes occurring upon cycling of electrode materials. We will highlight the different pieces of information that can be obtained from diffraction and ssNMR techniques, as applied to Li-based and Na-based systems. Finally, we will illustrate, with examples taken from our own work, how solid-state NMR, in combination with diffraction techniques and ab initio calculations, gives considerable insight into the phase transformations occurring upon Na extraction from/reinsertion into NayMn1-xMxO2 (x, y ≤ 1, M = Ni, Mg, Li) compounds.
Manganese-Based Sodium Transition Metal Oxide Cathode Materials for NIBs
Layered intercalation materials for Li/Na-ion rechargeable battery electrodes
Layered materials are attractive intercalation hosts, as they generally exhibit weak interlayer interactions and an empty 2D space for guest ion diffusion. When prepared in the discharged state (alkali guest already present), on extracting the guest alkali species from the interlayer space the structure initially undergoes an anisotropic expansion along the axis perpendicular to the layers, by convention the c axis, due to the increased repulsion between the negatively charged MO2 layers. The interlayer expansion is usually below 0.5 Å for Li extraction, but is close to 1.5 Å for Na+ deintercalation, in part due to the larger ionic radius of Na+ (1.02 Å) vs. Li+ (0.76 Å), but also due to the different sites occupied by these ions. Large structural expansions induce strain between domains with different degrees of intercalation in the crystal structure, resulting in cracks, particle fragmentation,35 and amorphization after repeated cycling.36 Large expansion coefficients along c may also contribute to the staging often observed at small x values (low intercalation), wherein the guest cations are unevenly distributed among the interlayer spacings.37
The different crystal chemistries of intercalated Li+ and Na+ ions in transition metal oxides
In the 1970s, Fouassier et al. conducted a systematic study of the structure and physical properties of AxMO2 compounds (A = Na, K; M = Cr, Mn, Co). Lamellar AxMO2 structures, stabilized for x > 0.5, are composed of brucite-type layers of edge-sharing MO6 octahedra with the alkali ions filling interlayer spaces.38 In the early 1980s, Delmas et al. devised a specific nomenclature to describe the variety of MO2 layer stackings in AxMO2 phases, where A is an alkali ion and M a transition metal ion. Each structure (polytype) is denoted by Xn, where X refers to the alkali coordination (O: Octahedral, P: trigonal Prismatic, T: Tetrahedral), and n to the number of MO2 slabs in the hexagonal cell.39 A prime indicates a distortion of an ideal polytype. Na+ and Li+ ions have different preferred coordination environments, and different crystal structures are observed for the intercalated transition metal oxides. While the larger Na+ ions have a strong preference for 6-coordinate octahedral or prismatic sites, Li+ ions are found in octahedral and tetrahedral sites. Na-intercalated transition metal oxides are found as O- and P-type structures, the most common polytypes being P2, O3 and P3, depicted in Figure 2. Li-intercalated oxides, on the other hand, crystallize as T and O structures.
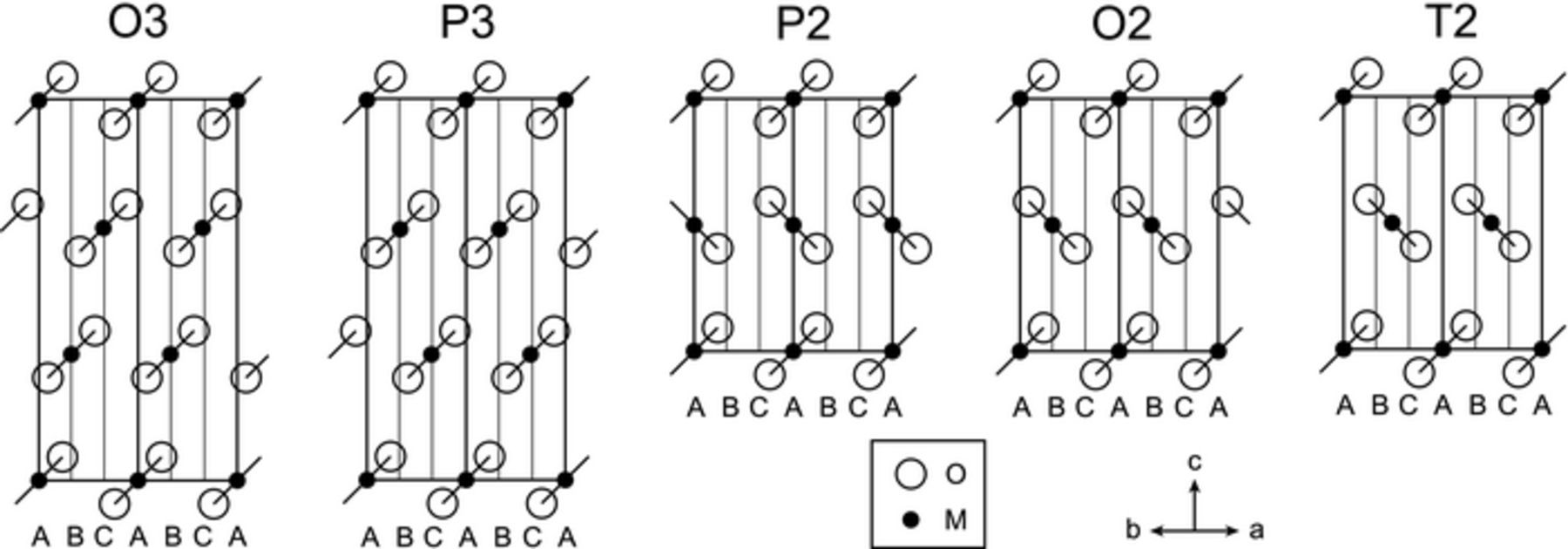
Figure 2. Diagram of different layered structures consisting of rigid MO2 sheets, viewed from the [110] direction. The alkali metal ions are omitted. Adapted with permission from Paulsen et al.40 Copyright 2000 American Chemical Society.
The different preferred coordination environments for Na+ and Li+ account for the changes in MO2 layer stacking upon Na/Li ion-exchange reactions: P-type sodium phases become O- or T-type lithium phases. In addition, the facile interconversion and low energy separation between the different structural polytypes often leads to stacking faults in the ion-exchanged material. For example, ion-exchange of P2-Na5/6[Li1/4Mn3/4]O2 produces a mixture of O2-Lix[Li1/4Mn3/4]O2, with O2'- and O6-type impurity phases.13
LixMO2 vs. NaxMO2 as positive electrode materials
The electrochemical performance of Na-intercalated transition metal oxides can be strikingly different from that of their Li counterparts. For instance, even though both LixCrO2 and NaxCrO2 crystallize in the same structural polytype, the former has been found to be electrochemically inactive,41 while the latter has demonstrated a reversible capacity of 120 mAh/g when cycled in a Na cell,42 and a recent study showed that the capacity retention of carbon-coated NaxCrO2 is excellent.43 The metastable layered phase, LiFeO2, can be prepared from ion-exchanged α-NaFeO2. However, it is electrochemically inactive,44 in part due to ready Fe3+ migration on cycling; Fe4+ has also been proposed to be unstable in these materials.27 On the other hand, Mössbauer spectroscopy measurements have indicated that Fe4+ is stable in O3-type NaFeO244, and approximately 80 mAh/g of reversible capacity could be delivered in a Na cell when the cut-off voltage was limited to 3.4 V45. Although Fe migration processes are more frequent in Li cells,46 due to the similar ionic radii of Fe3+ and Li+, the presence of Fe in the alkali layer leads to serious deterioration of the electrode performance in both Na and Li systems. For instance, poor electrode reversibility was observed when O3-type NaFeO2 was cycled above 3.5 V.45 The different electrochemical behaviors of Li- and Na-intercalated transition metal oxides suggest that novel, high performance Na-based energy storage systems remain to be discovered, with unique characteristics. The different destabilization mechanisms which affect the performance of LixMO2 and NaxMO2 cathodes upon electrochemical (de)intercalation of the alkali ion are discussed in the next sections.
Destabilizing mechanisms for LixMO2 structures
The similar ionic sizes of the Li+ and Mn+ species, as shown in Figure 3, leads to the destabilization of layered LixMO2 compounds via two related mechanisms: (i) antisite disorder (when a transition metal ion in the transition metal layer, M, exchanges with Li+ in the alkali metal layer) and (ii) the formation of a spinel phase (see Figure 4) by migration of 25% of the M ions into the alkali metal layers, with Li+ occupying the tetrahedral sites,47 a phenomenon that occurs in partially delithiated materials when the spinel is the thermodynamic ground state structure for x = 0.5.
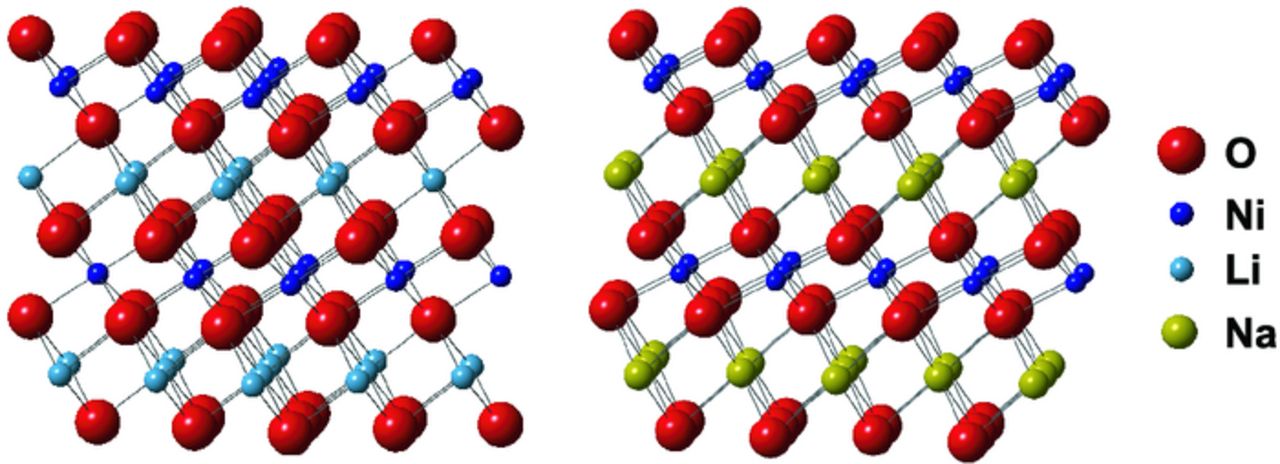
Figure 3. Diagrams of the O3-LiNiO2 (R-3m) and O3'-NaNiO2 (C2/m) structures, showing the similar sizes of Li+ and Ni2+ (with ionic radii of 0.76 and 0.69 Å, respectively) and the more dissimilar size of Na+ (with an ionic radius of 1.02 Å). A significant amount of Li/Ni antisite disorder is observed in O3-LiNiO2 (not shown here),48 but there is no evidence for Na/Ni site exchange in O3'-NaNiO2.49,50
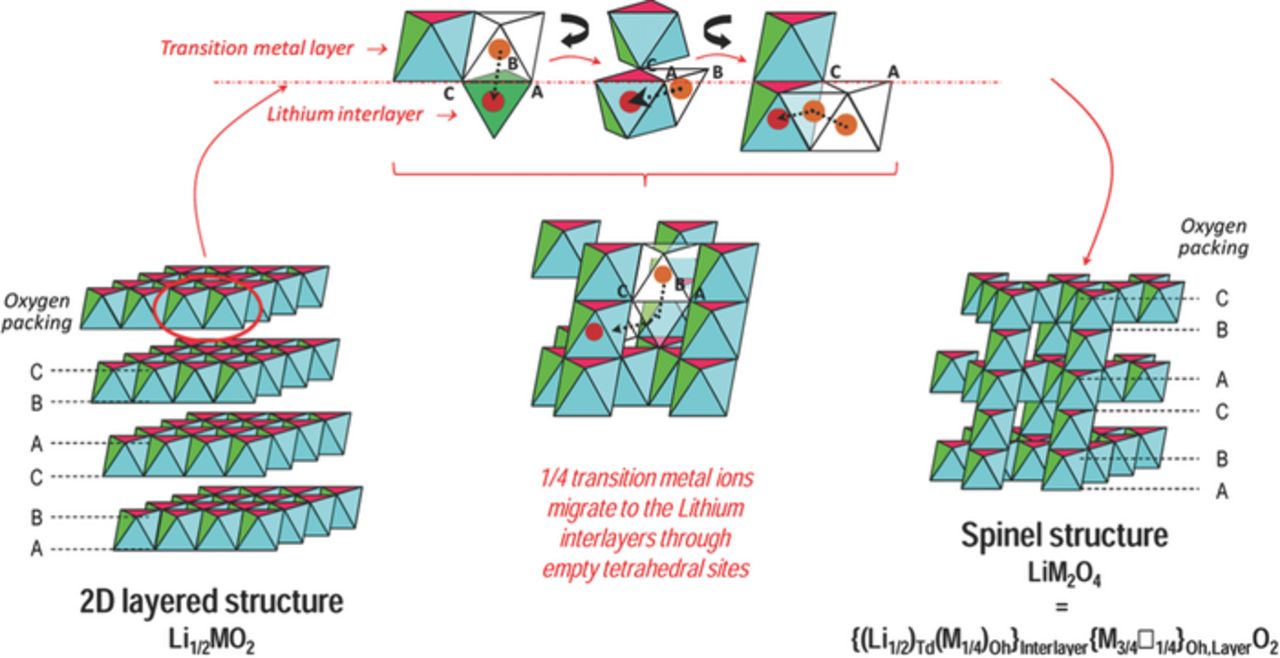
Figure 4. Scheme of the layered-to-spinel transformation observed upon deintercalation of layered oxides LixMO2 at high temperature. Td and Oh stand for tetrahedral and octahedral sites, respectively. Lithium ions are not represented for the sake of simplicity. Adapted with permission from Guilmard et al.51,52 Copyright 2003 American Chemical Society.
In a recent first principles study of O3-type AxMO2 (A = Li, Na) cathodes, Ceder and coworkers showed that, unlike lithiated phases, Na-intercalated phases do not have a thermodynamic driving force to transform to the spinel structure. They further argued that transition metal ion mobility, a requirement for the layered-to-spinel transformation process, is much lower in Na compounds than in their Li counterparts, the activation energy barrier to form the NaTd-MTd dumbbell transition state along the M migration path, in which both Na and M species occupy tetrahedral sites, being high.53 Indeed, ab initio calculations on spinel NaMn2O4 predicted that this compound is unstable by more than 50 meV/atom, and decomposes into NaMnO2, Na2Mn3O7, and Mn2O3.54 This claim was confirmed by an experimental study by Komaba and coworkers,55 and more recently by Liu et al.,56 who cycled spinel-type LiMn2O4 samples in a Na cell. In the latter work, sodium was inserted into the cathode material after initial electrochemical extraction of lithium, but the poor stability of the Na-containing spinel structure led to the transformation to a layered structure after several cycles.55,56
Destabilizing mechanisms for NaxMO2 structures
The mechanisms responsible for capacity loss and poor reversibility in NaxMO2 cathodes are different from those observed in lithium systems. Layered NaxMO2 materials have a tendency to undergo phase transformations induced by oxygen layer glides at low Na contents, and transitions between the different Na+ ion/vacancy ordered patterns formed at particular Na stoichiometries.
Oxygen layer glides and phase transformations upon charge
MO2 sheets are held together by the Na+ ions present in the interlayer space. As the cell potential is increased Na+ ions are deintercalated from the cathode material and fewer atoms maintain the initial stacking of the layers. Labile MO2 layer stacking leads to oxygen layer glides upon Na extraction. These phase transitions appear as extended regions of constant potential in the electrochemical curve. Phase transitions from P2 to O2,4,10,11 OP4,6,58 or between P3 and O3 polytypes,17,59–61 are associated with a low energy cost and take place at room temperature, as they do not require the breaking of M-O bonds. Conversely, the P2 and P3 (or O3) polytypes not only differ by shifts of the MO2 slabs, but also by a 60° rotation of all MO6 octahedra. P2 to P3 (or O3) phase transitions require the breaking of M-O bonds and only occur at elevated temperatures.
Phase transitions from structures containing prismatic Na sites to structures with octahedral Na environments are electrostatically driven upon Na extraction. At the end of charge, the number of Na+ ions in the interslab space is small, and the screening of the repulsive O2−−O2− interactions by Na+ ions becomes less effective. The increase in the repulsive interactions between adjacent MO2 layers is effectively mitigated by oxygen layer glides perpendicular to the c axis, typically accompanied by a drastic shrinkage of the interslab distance. In other words, Na+ vacancies are less stable in prismatic sites than in octahedral sites,39 and structures containing octahedral interslab sites form as the Na layers are depleted.
A variety of O-like structures, with or without a long-range ordered stacking sequence of the MO2 sheets, have been reported to form upon sodium electrochemical deintercalation from P2 structures. In many cases, the determination of the long-range order of layer stacking from X-ray diffraction patterns is particularly tricky. The diffraction pattern of the cathode in the high voltage region can either not be fully indexed due to extensive broadening of the diffraction peaks, or the goodness of fit is similar with different structural models (O2, O6, OP4), as was highlighted in a recent study of P2-NaxMn1/2Fe1/2O2 by Mortemard de Boisse et al.62
Lu and Dahn, in their study of the P2-NaxNi1/3Mn2/3O2 material, described two possible directions for oxygen layer shifts to form an octahedral close-packed oxygen framework, starting from the prismatic framework.10 The authors explained that, because these two choices are selected at random (see Figure 5), the resulting structure necessarily develops stacking faults. Only if all central layers glided according to choice 1 or all choice 2 would the perfect O2 structure form.10
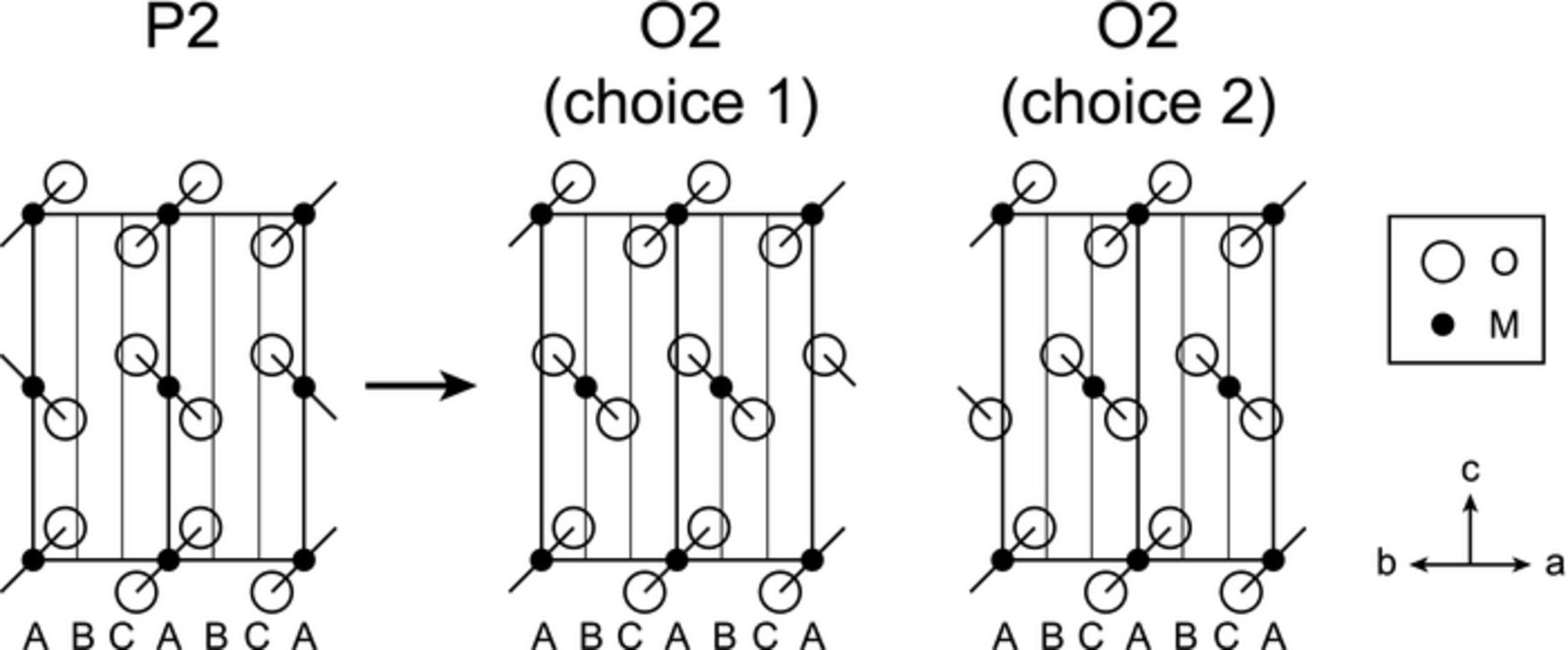
Figure 5. [110] sections of the P2 and O2 structures. When Na is removed from the P2 structure, a sliding of adjacent MO2 layers occurs to create Na octahedra (from trigonal prisms). This can be accomplished by sliding the MO2 slabs so that the transition metals from the middle layer in the left panel move from A to B (choice 1) or from A to C (choice 2), thus allowing for the possibility of stacking faults. Reprinted with permission from Lu and Dahn.10 Copyright 2001, The Electrochemical Society, replotted for clarity.
Starting from a P2 material, the formation of an OP4 phase, rather than a complete transformation to an O2 or O6 phase, can be rationalized in terms of staging phenomena occurring upon sodium extraction. The OP4 phase obtained upon partial layer shearing is composed of an alternate stacking of octahedral (O2) and prismatic (P2) layers along the c-axis. The Na+ vacancies are located in O-type layers, while the Na+ ions remain in the P-type layers. The transition to the OP4 phase, intermediate between the P2 and O2 layer stackings, is less structurally damaging, hence more reversible, than the P2-O2 phase transition observed upon Na extraction from e.g. P2-NaxNi1/3Mn2/3O2.11 The OP4 phase was recently reported upon Na deintercalation from P2-NaxFe1/2Mn1/2O26 and P2-NaxMn1-yMgyO2 (0 < y < 0.2).12 O2 and O6 phases are expected to occur as more Na is removed from the cathode material, and when the OP4 structure is destabilized by creation of Na+ vacancies in P-type layers.
Ordering transitions upon Na insertion and extraction
In P2-type phases, the relative occupation of the face centered (P(2b)) and edge centered (P(2d)) prismatic Na crystallographic sites results from the interplay between the repulsive Na+-Na+ and Na+-Mn+ interactions, and from Mn+/M(n+1)+ charge ordering on the transition metal lattice. Alkali ion/vacancy ordering is more prevalent in NaxMO2, compared with LixMO2 compounds (although Li+ ion/vacancy ordering is seen in e.g. LixCoO228,65,66), because Na is more ionic and because the larger Na+ ions result in strong Na+-Na+ in-plane repulsions. The formation of ordered superstructures is also highly dependent on the synthetic conditions. Berthelot et al. claimed that the interslab cationic distribution in P2-NaxCoO2 results from the energy minimization between in-plane electrostatic repulsions that tend to maximize Na+-Na+ distances, occupation of face centered Na sites, with a higher Na+-Co3+ repulsive term due to the common face shared between the NaO6 and CoO6 polyhedra, and electron–electron interactions in the cobalt layer. The balance between all of these interactions is highly sensitive to sodium content, and various cationic distributions are observed for each composition along the electrochemical cycle.67,68 Na+ ion/vacancy ordered patterns have also been observed at particular Na stoichiometries in NaxVO2,69 NaxMnO2 compounds,6,12,36,70,71 NaxNi1/3Mn2/3O2,10,11 and NaxCo2/3Mn1/3O2.72
Na+ ion/vacancy ordering transitions have detrimental effects on the electrochemical properties of P2 type cathode materials. Transitions between ordered superstructures upon Na removal, indicated by regions of constant voltage on the electrochemical curve, may lower the Na+ diffusion coefficient by up to three orders of magnitude,73 and reduce the dimensionality of Na+ transport.74 The various plateaus on the electrochemical curve of P2-type NaxMnO2, shown in Figure 6, have been assigned to structural transformations and to transitions between single-phase domains with different Na+ ion/vacancy patterns.12
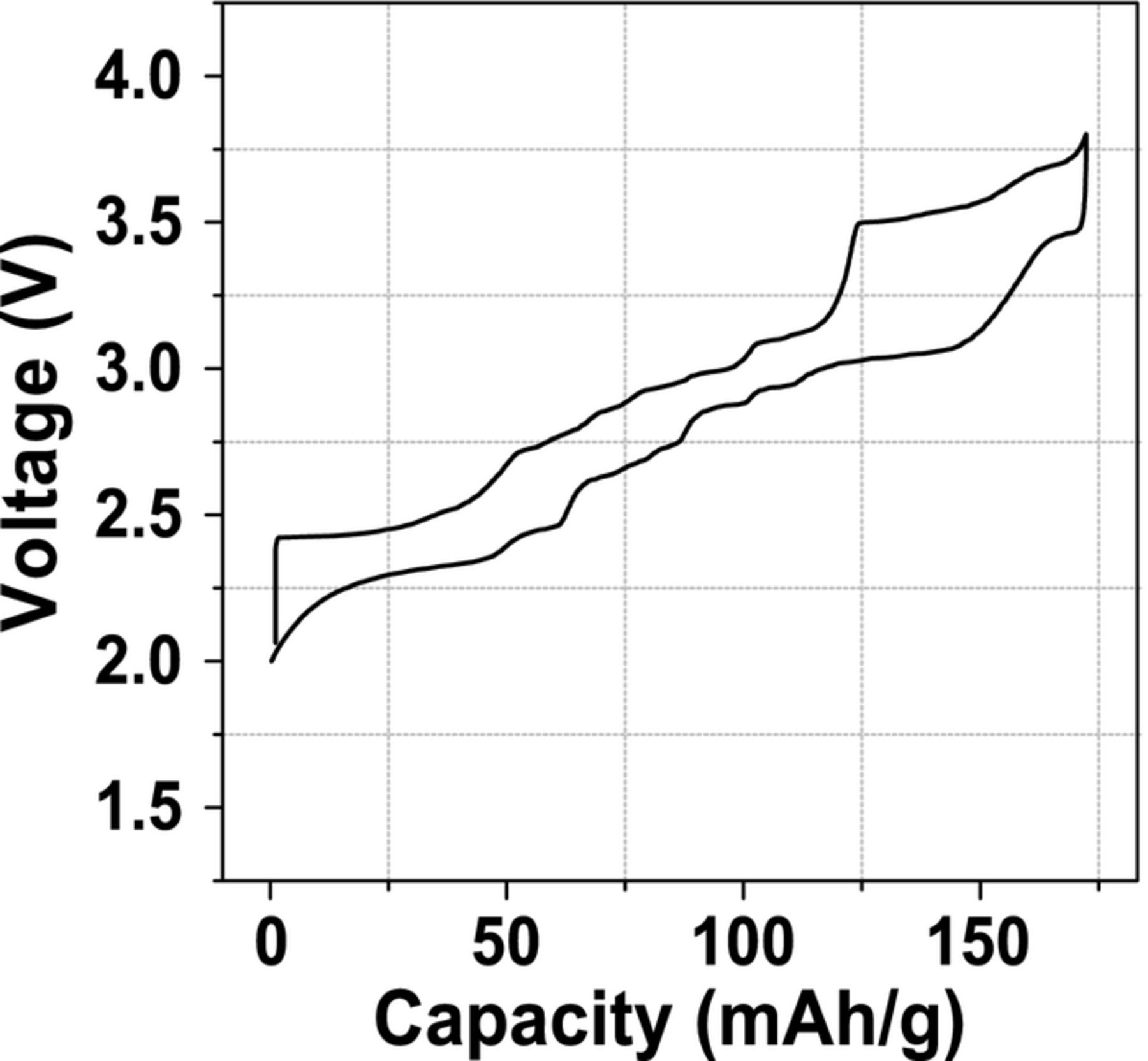
Figure 6. Electrochemical profile of quenched P2-NaxMnO2 cycled at 12 mA/g between 2.0 and 3.8 V vs. Na+/Na. Adapted from Billaud et al.12 (http://dx.doi.org/10.1039/c4ee00465e) with permission of The Royal Society of Chemistry.
The complex phase stability of NaxMO2 compounds: the case of M = Co
The complex phase stability diagrams of NaxMO2 materials result from the close energetics of the different structural polytypes, as illustrated by Wang et al.'s first principles study of the phase stability of NaxCoO2.64 This study used a combination of ab initio Density functional Theory (DFT) calculations within the Generalized Gradient Approximation (GGA) and Monte Carlo simulations to determine the ground state Na+ ion/vacancy arrangements, as a function of x, for a variety of structural polytypes (O1, O2, O3, P2, and P3). The resulting 0 K phase stability diagram and the formation energies obtained for the lowest energy Na+ ion/vacancy ordered arrangements, plotted as a function of x, are presented in Figure 7a.64 A similar combination of DFT and Monte Carlo methodologies was subsequently used by Meng et al. to investigate the lowest energy Na+ ion/vacancy orderings for P2-NaxCoO2.129 The authors compared the results obtained from DFT calculations within the GGA and GGA+U (i.e., with a Hubbard U correction) approaches and demonstrated that, at certain Na concentrations, the most stable Na+ ion/vacancy patterning is influenced by the charge localization on the Co layer. Meng et al. obtained a global energy minimum for the P2 phase for x = 0.75,129 while Wang et al. found a global energy minimum at ca. 0.33,64 presumably due to the different sizes of the supercells used, hence the different Na+ ion/vacancy arrangements explored in the two studies (and the different level of theory). On the other hand, an experimental study by Lei et al. explored the NaxCoO2 synthesis phase diagram. NaxMO2 compounds are typically prepared via solid-state synthesis,8,57 or a solution-state co-precipitation method,9,12 with calcination temperatures ranging from 500 to 1000°C. The results shown in Figure 7b indicate that the thermodynamic stability of different NaxCoO2 layered structural polytypes depends on both composition and synthesis temperature: the P2-NaxCoO2 phase is obtained for 0.55 < x < 0.88, but only at a high synthesis temperature (650–900°C).
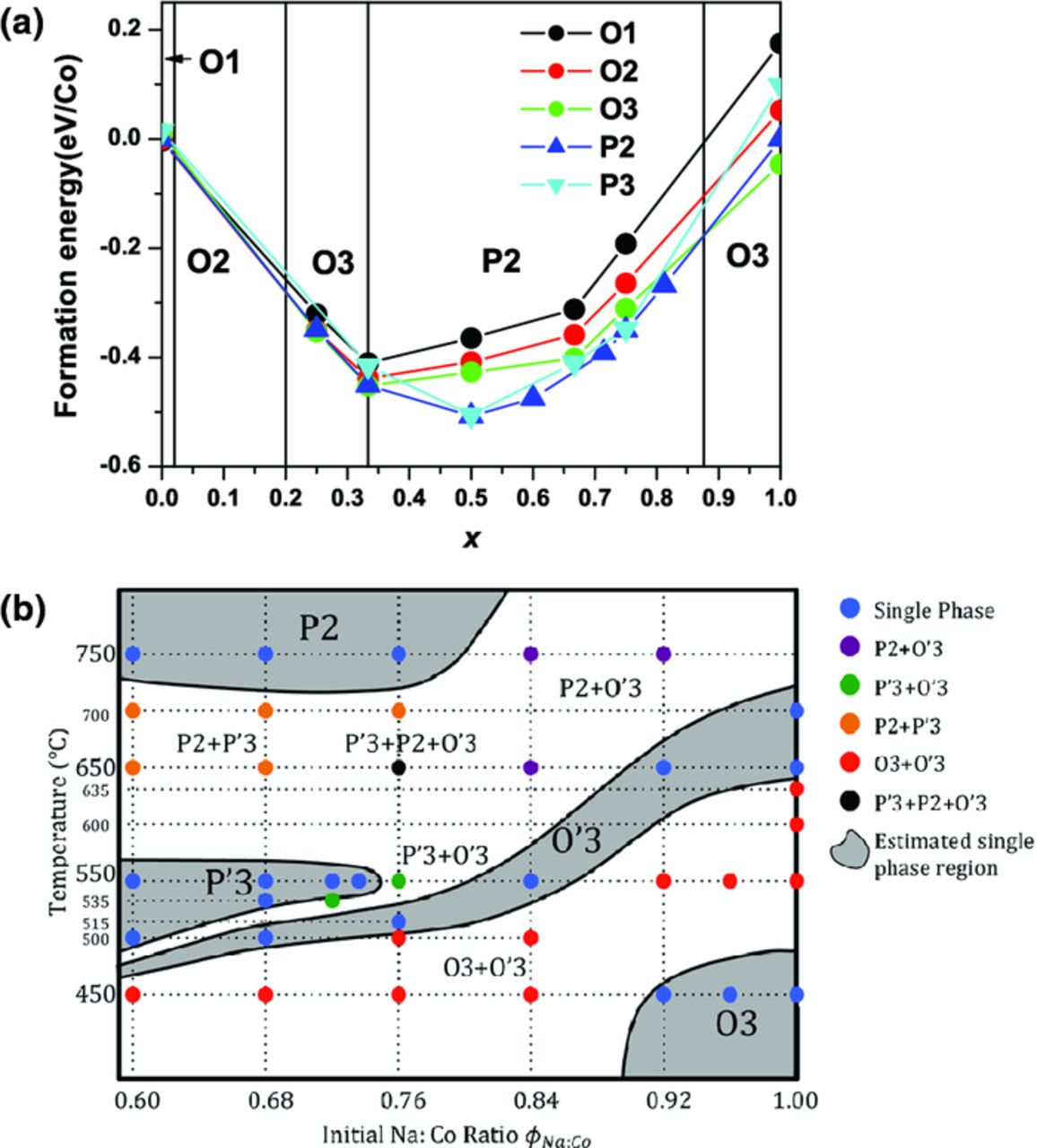
Figure 7. (a) First principles derived ground state phase diagram of NaxCoO2 as a function of x. The formation energies presented here for the different structural polytypes (O1, O2, O3, P2, P3) were obtained on the ground state Na+ ion/vacancy arrangement (for a given MO2 layer stacking and Na content x). The phase name between the phase boundaries denotes the stable phase in the region. Reproduced with permission from Wang et al.64 Copyright IOP Publishing. All rights reserved. (b) Synthesis phase diagram of NaxCoO2 as a function of the precursor Na:Co ratio φNa:Co (X axis) and the sintering temperature (Y axis). Reprinted from Lei et al.57 according to the ACS Author Choice Usage agreement.
A recent study by Levi et al.63 demonstrated that the phase stability of AxMO2 (A = Li, Na) materials upon synthesis (and during electrochemical cycling), can be predicted from the size of the lattice strains present in the material. The lattice strains for O3- and P2-NaxCoO2 were evaluated using bond valence parameters obtained from a correlation of experimental M-O bond lengths obtained from high quality structural data on a variety of Ni, Mn, and Co oxides to the M metal valence. It was found that for x > 0.5 the lattice strains were caused by steric mismatch between the Na and Co cations, resulting in elongated M-O bonds and compressed Na-O bonds. On the other hand, for small x values (x < 0.5) the lattice strains were caused by competing effects: shorter M-O bonds due to the higher M oxidation states, and increased repulsion between the highly charged M cations. For x < 0.5, stretching of the M-O bonds must be compensated by a compression of the M-M bonds.63 The global instability index, a measure of the valence violations for the whole material, was plotted as a function of x, for O3- and P2-NaxCoO2 and was found to be in good agreement with the formation energies obtained by Wang et al. for these phases (shown in Figure 7a).64 This study highlights the importance of lattice strains in explaining the highly unstable NaxMO2 phases obtained at low Na content.63
Challenges for Mn3+/Mn4+ systems
Manganese-based sodium-ion battery cathode materials fulfill the requirements for low cost, sustainable, and environmentally friendly energy storage technologies. Layered and tunnel NaxMnO2 compounds are by far the most investigated, with only very few reports of other types (e.g. spinel56) of manganese-based oxide cathode materials. An early study by Parant et al. showed that the structures of NaxMnO2 phases split into two main groups: tunnel-like structures at low x (e.g. Na0.2MnO2, Na0.4MnO2, Na0.44MnO2), and structures consisting of slabs of edge-sharing MO6 octahedra at high x (e.g. Na0.7MnO2, NaMnO2).75 Mn3+, with a (t2g)3(eg)1 electronic configuration, is Jahn-Teller active. Cooperative Jahn-Teller distortions, corresponding to long-range ordered arrangements of tetragonally distorted Mn3+O6 octahedra,76 are observed in AxMnO2 (A = Li, Na) type compounds, and result in a reduction of the unit cell symmetry. The case of P2-NaxMnO2 is a good example of the complexity of Mn3+-containing layered structures. The distortion of the ideal P2 structure obtained for NaxMnO2 is highly dependent on the synthesis conditions. More oxidizing conditions (lower temperatures) stabilize a higher average Mn oxidation state, resulting in a small distortion of the ideal hexagonal P2 structure. Parant et al. have reported two polymorphs for P2-type NaxMnO2, with different Na contents. The α form crystallizes in the ideal P2 structure, and is stable below 600°C, while the β form is an orthorhombic distorted P2' structure, and is believed to be stable above 600°C and to coexist with the O3' α-NaMnO2 phase.75 The high temperature form of Na2/3MnO2 reported by Paulsen and Dahn, β-Na0.7MnO2, exhibits a monoclinic distortion, while the low temperature α form is essentially undistorted.77 Stoyanova et al. have reported an orthorhombic distortion (Cmcm) when quenching Na2/3MnO2 from 1000°C.78 We obtained a biphasic Na2/3MnO2 material, made of an orthorhombic phase (Cmcm) and of a monoclinic (C2/m) phase upon quenching from 1000°C.12 The lifting and restoration of Jahn-Teller distortions, as the material is charged and discharged and the Mn oxidation state fluctuates between +3 and +4, induces a complicated sequence of electronic and structural processes evidenced by the large number electrochemical plateaus on the charge/discharge curve, as shown in Figure 6.
One cause of poor cycling performance of mixed valence Mn3+/Mn4+ materials is anisotropic structural expansion upon charge and discharge, as has been studied in some depth for the tunnel-like NaxMnO2 (x = 0.19–0.44) cathode material. The structure of this material first expands along the b axis as the Na content x is increased from 0.22 to 0.66, then it extends along the a and c axes from x = 0.44 to x = 0.66, as shown in Figure 8b. These anisotropic changes in the lattice parameters are attributed to Jahn-Teller effects and to the sequential reduction of Mn ions at different crystallographic positions, shown in Figure 8a, and result in the loss of half of the initial capacity by the 50th cycle at C/20.18
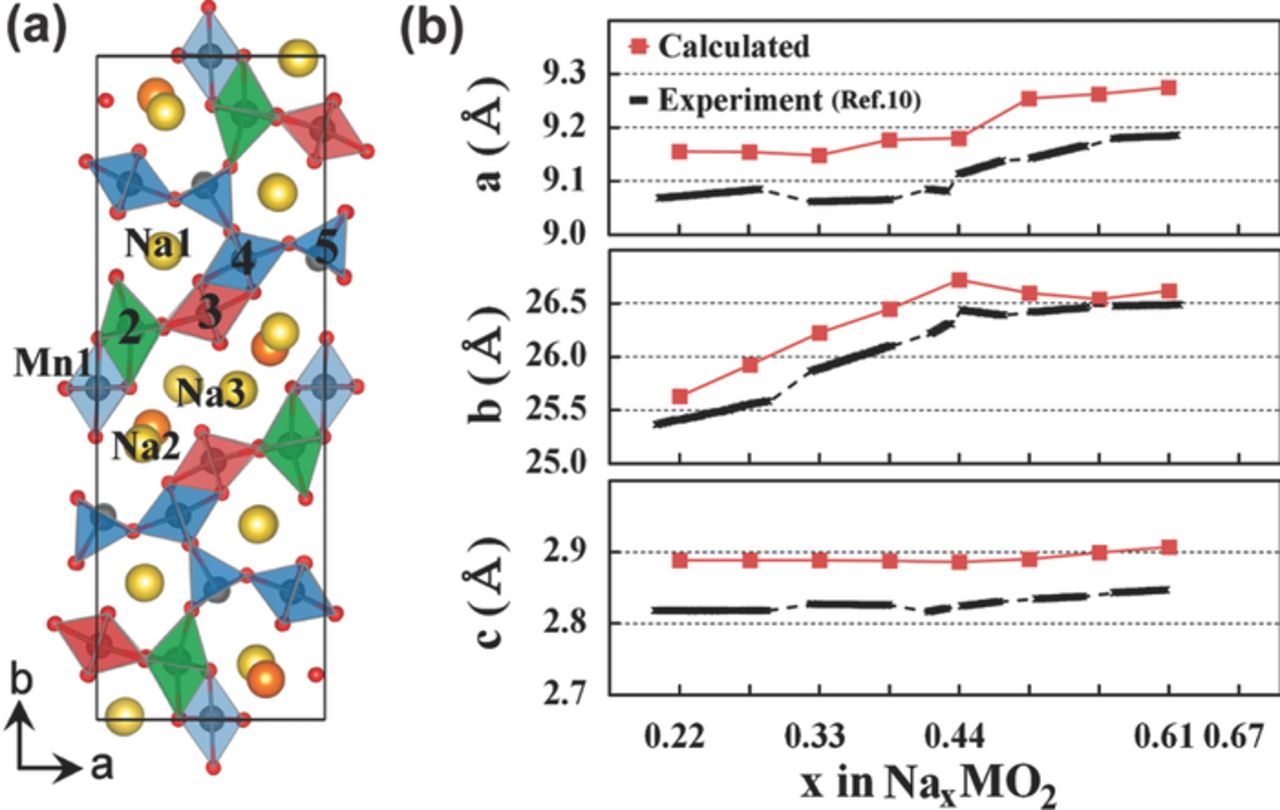
Figure 8. (a) Crystal structure of the tunnel-like Na0.44MnO2 cathode material, with five crystallographic sites for Mn, and three for Na. (b) Experimental and calculated values of the lattice parameters, and their evolution as a function of total Na content. At x = 0.22, all Mn ions are present as Mn4+, apart from those in Mn5 positions. Between x = 0.22 and x = 0.44, Mn ions in Mn2 positions are reduced, resulting in an expansion along their Jahn-Teller elongated axis (the b axis). And between x = 0.44 and x = 0.66, Mn ions in Mn3 positions are reduced, resulting in an expansion along their Jahn-Teller elongated axis (the a axis). This particular sequence of reduction events was found to be in good agreement with the calculated local density of states (LDOS) at each Mn site: the reduced Mn site, at a particular Na content, is the one with the largest density at the LUMO states. Reprinted with permission from Kim et al.79 Copyright 2012 American Chemical Society.
The intricate interactions present in Mn3+-containing oxides account for their complex electrochemical properties and have general implication for Li- and Na-ion batteries.80 A recent experimental and DFT study of O3-Na5/8MnO2 revealed that its superstructure, shown in Figure 9a, results from strong coupling between cooperative Jahn-Teller distortions, Mn3+/Mn4+ charge ordering, and an unusual Na+ ion/vacancy ordering not directed by electrostatic interactions, unlike in P2-NaxCoO267,68 or in P2-NaxVO2.69 Previous work on the LixNiO2 system (which contains Jahn-Teller active low spin Ni3+ ions with a (t2g)6(eg)1 configuration) demonstrated that the presence of two Li at the extensions of the two 180° O–Ni3+–O bonds significantly lowers the energy of the Ni3+ e*g orbital, through hybridization of the Ni d orbital, O p orbital, and Li s orbital. This mechanism leads to enhanced electron localization and Jahn-Teller activity of the metal species,81 even for partially delithiated materials. The periodic arrangement of full Na stripes, half-full Na stripes, and of Mn3+/Mn4+ stripes in O3-Na5/8MnO2, shown in Figure 9a, is believed to arise from a similar mechanism. For those stripes with not enough Na+ ions to create only Na–O–Mn3+–O–Na interactions, rather than create VNa–O–Mn3+ –O–Na configurations, the Na+ ions relax into highly distorted octahedral sites where they share the symmetric attraction of the two neighboring Jahn–Teller distorted –O–Mn3+–O–Na configurations (see Figure 9d).80
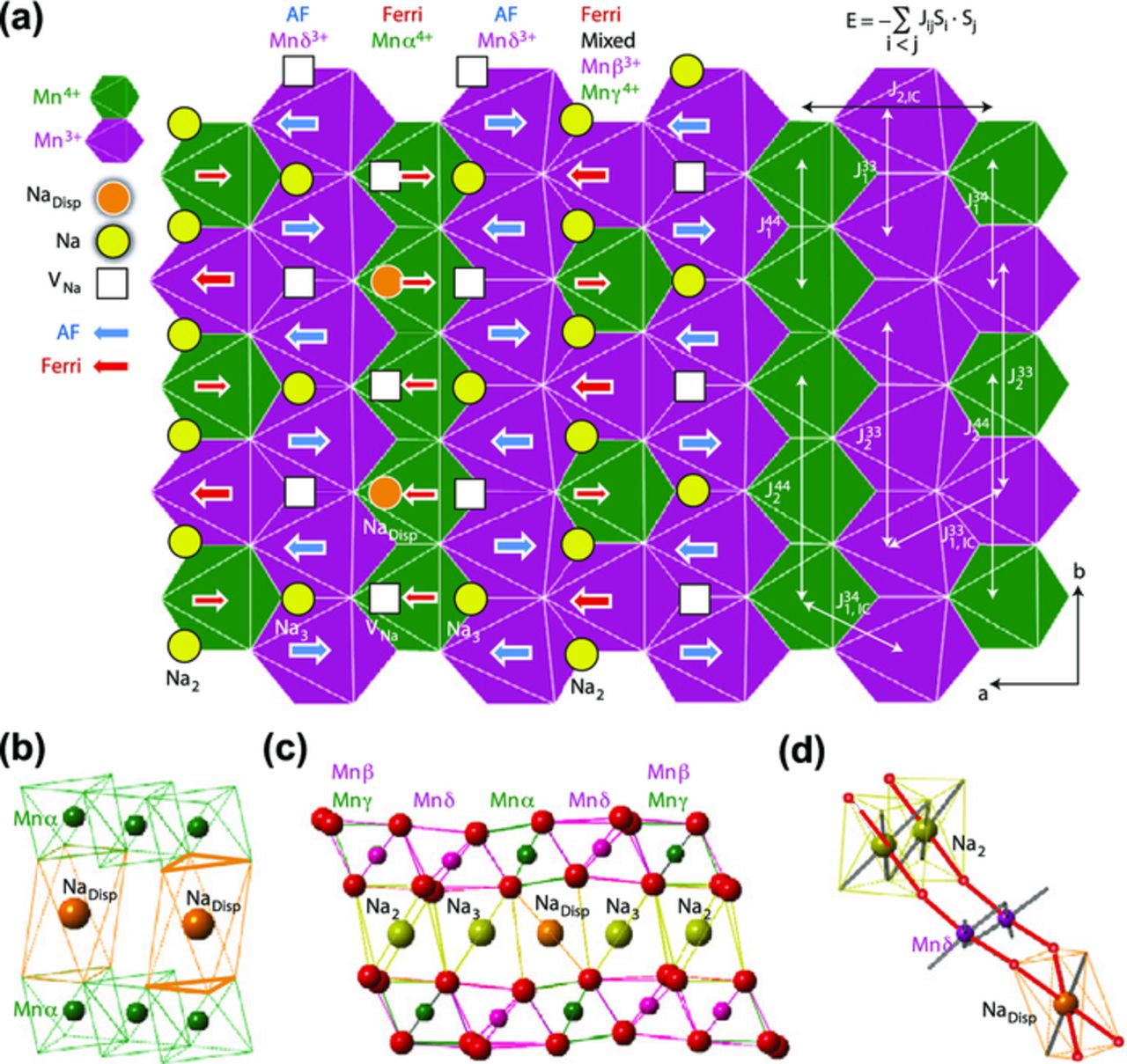
Figure 9. Diagram of the O3-Na5/8MnO2 superstructure resulting from Na+ ion/vacancy, Mn3+/Mn4+ charge, and magnetic stripe orderings. (a) The Na ordering in the ab layer includes Na+ ions (yellow circles), displaced Na+ ions (orange circles labeled with NaDisp) and Na vacancies (open squares). The transition metal layer charge ordering includes pure Mn3+O6 (purple hexagon) stripes, pure Mn4+O6 (green hexagon) stripes and the mixed-valence Mn stripes. The electronic and Na+ ion/vacancy ordering leads to a new magnetic ordering at low temperature, with antiferromagnetically coupled Mn3+ stripes (blue arrows), ferrimagnetically coupled Mn4+ and mixed Mn3+/Mn4+ stripes (red arrows). (b) The local environment of two displaced Na+ ions face-sharing with Mn4+O6 octahedra. The two triangles that share faces with one Na site are labelled with thick lines. (c) The structure viewed from the b direction shows oxygen O3 stacking; and (d) the basic unit of the spd hybridization interaction that drives the Na displacement to the distorted octahedral site. The spd hybridized bonds, in red, connect Na+, O2−, and Mn3+ ions. Reprinted by permission from Macmillan Publishers Ltd: Nature Materials (http://www.nature.com/nmat/index.html), Li et al.,80 copyright 2014.
Devising high performance NaxMO2 intercalation cathodes
The good performance of NaxMO2 materials as NIB positive electrodes depends critically on their ability to reversibly (de)intercalate sodium over a large composition range, and to operate at a sufficiently high voltage in order to achieve high energy density. High power applications also require high rate capability, i.e. high ionic and electronic conductivity. In addition, a stable structure is key for long cycle life.
High capacity cathodes: maximizing the Na content
The capacity of P2-type phases is limited by the compositional range over which the structural framework is thermodynamically stable. P2 structures are usually found for xNa ≤ 2/3, i.e. when 1/3 of the Na sites are vacant.13 It is highly unfavorable for the Na content to exceed 0.9, as this necessarily results in simultaneous occupation of nearest-neighbor Na sites.67,69 Instead, O3-type structures are typically obtained at high Na contents. As discussed in Yabuuchi et al.'s recent paper, the Na deficiency is readily compensated for in half cells, when the cathode is cycled against excess metallic sodium, but becomes a problem in a full cell, when a conventional carbon-based anode is used.82 In the case of P2-NaxFe1/2Mn1/2O2, it was found that about 50 mAh/g (corresponding to 0.2 Na per formula unit), given by the difference between the first charge and discharge capacities, has to be compensated in advance for the full cell to reach the 190 mAh/g capacity observed in a half cell.6,82 An idea that has been put forward to mitigate irreversible capacity losses in full cells is the addition of sacrificial salts to the cathode mix, which act as a source of sodium upon first discharge. For example, irreversible capacity loss on the first cycle of P2-NaxFe1/2Mn1/2O2 was halved upon addition of 5%wt. NaN3 (NaN3 → Na + 3/2 N2), but led to concomitant nitrogen evolution in the half cell configuration.83
In an attempt to enhance the capacity of P2-type NaxMnO2, doping of the MnO2 layers has been investigated. As shown in Figure 10, transition metal substitution is an effective way to increase the Na content in the as-synthesized P2-type phases. P2 structures with a Na content as high as 0.85 can be stabilized by partial substitution of Mn for Ni, Li, or a combination of both. The upper cutoff voltage is limited to about 4.2 V to prevent structural changes that would affect the reversible reinsertion of Na into the structure upon discharge. This in turn leads to only moderate reversible capacities (ranging from 63 to 140 mAh/g) for materials in which some of the Mn has been replaced by Ni and Li.9,84,85
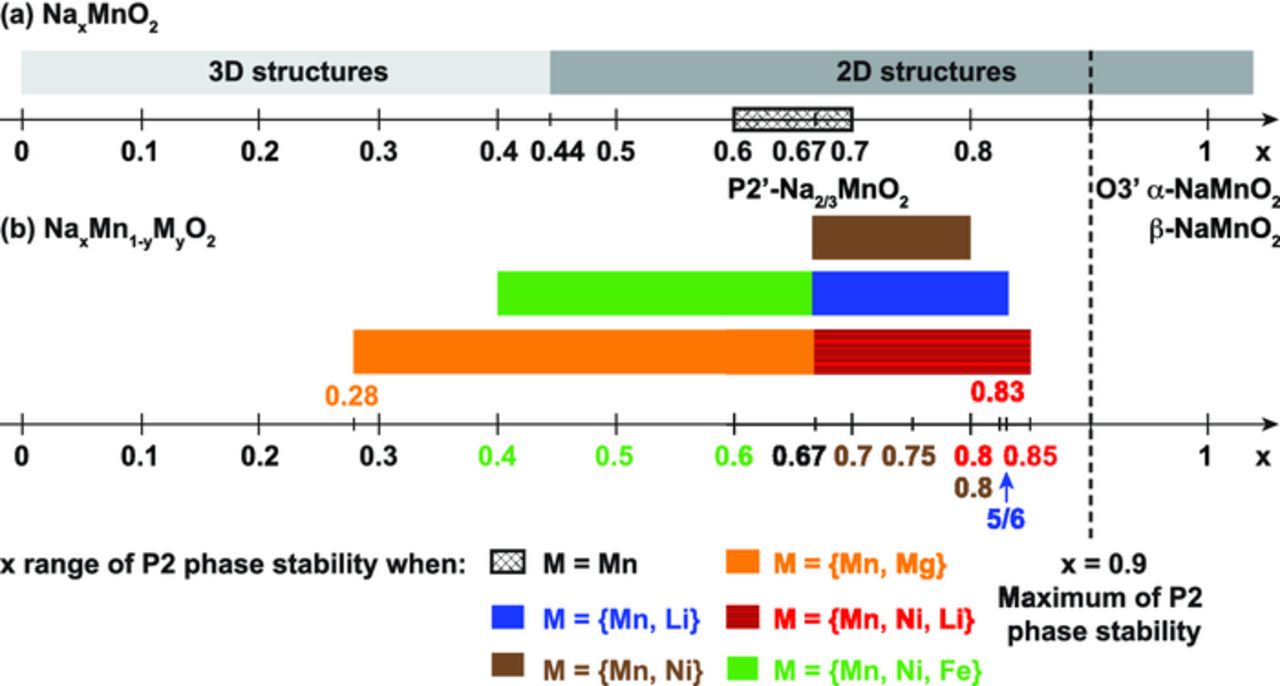
Figure 10. (a) Domain of stability of the different structural types observed for as-synthesized NaxMnO2 compounds;70,75 (b) doping on the Mn lattice leads to an extension of the range of Na content over which the P2-NaxMO2 structure is stable, as demonstrated by examples taken from the literature: M = [Mn, Ni],86 [Mn, Li],13 [Mn, Mg],5 [Mn, Ni, Li],9,84,85 [Mn, Ni, Fe].87 The upper limit in Na content (x = 0.9) for P2 phase stability arises from unfavorable occupation of nearest neighbor sites.
Several research groups have also looked into composite layered transition metal oxide cathode materials. Johnson et al. recently showed that the incorporation of Li into NaNi0.5Mn0.5O2 leads to topotactic intergrowth of P2 and O3 domains in the structure. The presence of multiple phases perhaps surprisingly results in an enhancement of the specific capacity and rate performance. Once the O3 phase transforms to a P3 phase at the beginning of the first charge, the structure is stabilized by interlocking of the P2 and P3 phases which prevents any further layer shifts upon extended cycling.88 These composite electrodes open up the possibility of reaching high capacities, since the O3 phase provides a larger sodium-ion reservoir, as well as high rate performance, since the P2 layered spacing is beneficial for easy diffusion of Na+ ions. Guo et al. obtained similar findings when they integrated a minor O3 component into a Li-doped P2-type majority phase. The P2+O3 Na0.66Li0.18Mn0.71Ni0.21Co0.08O2+δ composite displayed the highest energy density, 640 Wh/kg, of all reported cathode materials for Na systems, when cycled between 1.5 and 4.5 V at a rate of 20 mA/g (0.1C).89
Raising the electrochemical potential of the positive electrode material
Transition metal mixing has been extensively explored to improve the electrochemical performance of AxMO2 (A = Li, Na) type materials. One of the important characteristics that can be tuned upon metal substitution is the voltage at which the material (de)intercalates Li+/Na+.
Two recent reports have shown that P2-NaxMnO2 exhibits a high reversible capacity, comprised between 175 mAh/g and 190 mAh/g,12,13 but a low average operating voltage of approximately 2.8 V vs. Na+/Na. On the other hand, the much higher redox potential of O3-NaFeO2 (3.3 V vs. Na+/Na)45 has motivated research into P2-type Fe-only and Fe/Mn systems. Yabuuchi et al. investigated the partial substitution of Mn3+ for Fe3+ to stabilize the P2 phase, and showed that both Mn3+/Mn4+ and Fe3+/Fe4+ redox couples are active in P2-NaxFe1/2Mn1/2O2. This Fe/Mn bimetallic oxide exhibits 190 mAh/g of reversible capacity when cycled between 1.5 and 4.2 V at a rate of 12 mA/g, but it does not show the expected increase in average operating voltage (2.75 V) compared to P2-NaxMnO26. Lu and Dahn have examined the effect of Ni-substitution on the electrochemical properties of P2-NaxMnO2, and prepared the P2-Na2/3Ni1/3Mn2/3O2 electrode.10 Several reports have demonstrated that P2-Na2/3Ni1/3Mn2/3O2 and Li-doped P2-NaxMO2 (M = Mn, Ni) compounds operate on the Ni2+/Ni4+ redox couple, and exhibit a higher potential, of ca. 3.7 V vs. Na+/Na, compared to cathodes operating on the Mn3+/Mn4+ redox reaction.9–11,84,85 Although "Li rich" (1-x)LiMO2.xLi2MnO3 (M = Mn, Ni, Co) compounds90 suffer from voltage decay (or "droop") upon cycling, which has to date prevented their implementation in practical cells, Na/M antisite disorder is uncommon in sodium transition metal oxides, and recent reports on Mg2+- and Li+-doped P2-NaxMO2 (M = Mn, Ni) have revealed no significant voltage drop upon the introduction of spectator ions in MO2 layers.4,5,9,12,13,85
The role that oxide anions play in the redox reaction is still a subject of much debate.47 Covalency effects control the redox couples in a well established manner, for example, replacing an oxide anion by a polyanion (such as a phosphate) raises the redox couple of the metal ion, due to an increase in the ionicity of the metal (the so-called inductive effect).27 More recently, there has been considerable discussion concerning the direct role of oxygen ions in the redox processes that occur in the lithium-rich LIB layered materials,91–93 and the part they play in providing excess capacity beyond that estimated based on the redox-active transition metal ions.94–97 Although the exact mechanisms that occur in the LIB electrode remain controversial, there is no reason to expect that similar mechanisms would not operate in NIBs for appropriate compositions and Li-substitution.
Reaching high ionic and electronic conduction
A good electrode material should be a good electronic and ionic conductor. In layered materials, electronic conductivity and Na+ ion mobility are two important factors that affect intercalation.98 The electronic conductivity in AxMO2 (A = Li, Na) compounds is highly dependent on the nature of the transition metal species. In an early study, Delmas et al. rationalized the electronic properties of AxMO2 (x < 1) type compounds in terms of the spatial expansion of the metal t2g orbitals. Extended t2g orbitals and short M-M distances lead to strong M-M interactions and delocalization of the d electrons across the solid, with formation of partially filled bands, as in the case of Co systems. On the other hand, when the electrons are localized, such as in Mn and Cr systems, the oxide is semiconducting39 and electron conduction takes place via a hopping mechanism.70
When it comes to Na+ ion mobility, P2-type phases perform better than O2-type phases, due to low energy conduction pathways in the latter structures. In an O2-type phase, a Na+-ion jump from one octahedral environment to an adjacent equivalent environment occurs via the occupation of an intervening tetrahedral site. This involves crossing through an octahedral-tetrahedral face, as shown in Figure 11, giving rise to a relatively high activation barrier. The strong Coulombic repulsions between ions in the tetrahedral sites and the metal ions in the MO2 layers serve to raise the energy of the intermediate state.99 Conversely, Na+ ions in P2-type layered phases are distributed over a number of trigonal prisms (the Na sublattice is only partially filled) sharing their rectangular faces and thus providing wide passages for Na+-ion transport, and the activation energy barriers for cation hopping between adjacent sites are low.100 The presence of Jahn-Teller active cations (such as Mn3+) or Na+ ion/vacancy ordering101 will affect the activation energy barrier for Na+-ion hops between adjacent sites, hence Na+-ion conduction. As discussed previously, a stable P2 structure can be difficult to achieve in practice, due to the strongly disfavored vacant prismatic sites in partially charged systems, and to spontaneous transformation to an O-like structure.39
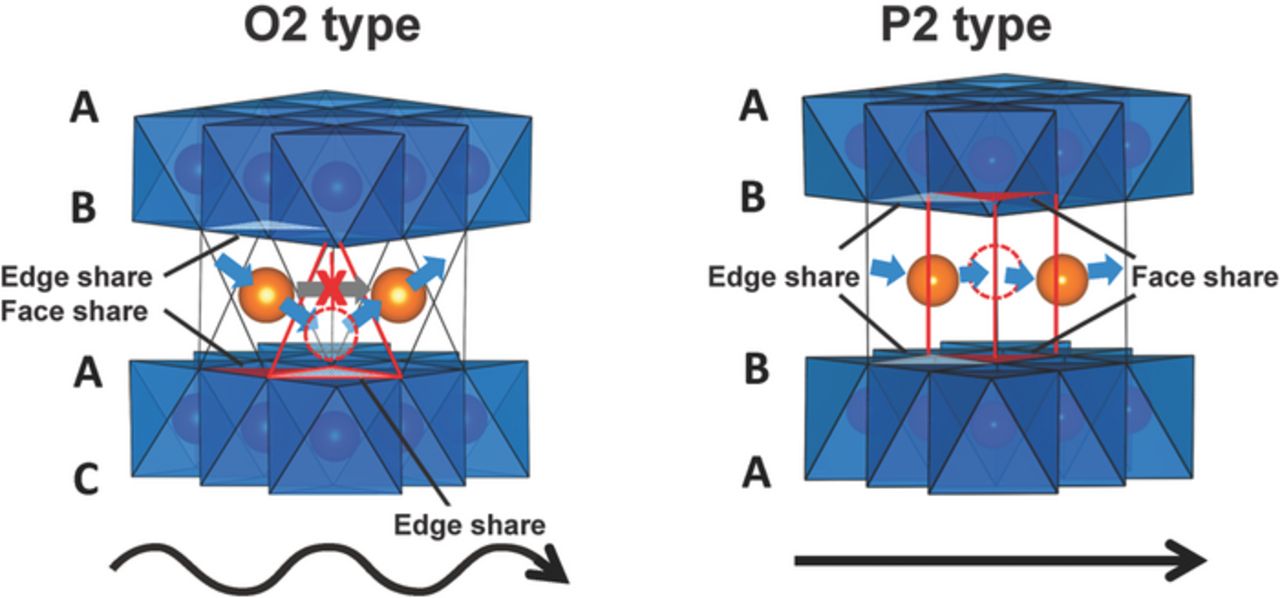
Figure 11. Na+ ion migration paths in P2- and O2-type structures. Adapted with permission from Yabuuchi et al.82 Copyright 2014 American Chemical Society.
Preventing Na+ ion/vacancy ordering transitions in NaxMO2 cathodes
Transition metal substitution for electrochemically active or inactive species leads to a more disordered structure. The composition of the metal lattice can be tuned so as to avoid ordering. Three types of ordering are common in NaxMO2 type compounds containing more than one metal M species. A bimetallic phase composed of M1 and M2 transition metal species may exhibit M1/M2 cation ordering, charge ordering on the transition metal lattice, and Na+ ion/vacancy ordering in the Na layer. It has been argued that M1/M2 cation ordering occurs when there is a simple compositional ratio between the M1 and M2 species (e.g. 1:1, 1:2, 1:3, etc.), and when their Fermi levels (ca. their oxidation states) and ionic radii are sufficiently different.102,103
Contradictory results, in terms of the role the M cation disorder plays on Na+ ion/vacancy disorder, have been obtained for Te-containing layered materials and for conventional NaxMO2 compounds based on first row transition metals. On one hand, the formation of a Na+ ion/vacancy ordered superstructure was found to be relatively independent on the degree of cation ordering in Na2M2TeO6 (M = Ni, Co, Zn, Mg) type compounds. Despite an ordered arrangement of the Te and M cations in the MO2 layers, the Na+ ions in the interlayer gaps are disordered, and a high Na+ ion mobility is observed.102 On the other hand, M1/M2 cation disorder is a necessary, but not sufficient, condition for Na+ ion/vacancy disorder in NaxMO2 (M = Mn, Ni, Co, Fe) phases, the latter being intimately linked to charge ordering on the MO2 lattice. As observed in NaxCoO2, charge order in the MO2 slabs only occurs if it can match the existing Na+ ion/vacancy order on the prismatic lattice, to satisfy charge neutrality at the local level.67,68,104 When x = 0.5, for instance, the lock-in of Na+ ions in well ordered superstructures, and strong coupling between the electronic charge carriers and Na+ ions, leads to charge localization.104
In a recent study on the origin of Na+ ion/vacancy ordering phenomena in NaxMO2 materials, Wang et al. suggested that the presence of transition metals with similar ionic radii and very different Fermi levels should limit electron delocalization, hence charge and Na+ ion/vacancy ordering. The authors investigated a Cr3+- and Ti4+-containing P2 material, intentionally choosing transition metals with very similar ionic radii and substantially different redox potentials vs. Na. A symmetric full cell was prepared with P2-NaxCr0.6Ti0.4O2 as both the positive and negative electrode. The material exhibited full disorder on both metal and Na lattices over the whole range of Na contents studied (x = 0.33–1.0), and an exceptional rate performance, with 75% capacity retention at a very high rate of 12C.103 Spectator ionic dopants, such as Mg2+, Li+, Ti4+, Zn2+, with no d electrons, are expected to effectively remove the ordering observed in mixed valence Mn3+/Mn4+ cathode materials. The lack of any plateau in the electrochemical profiles of Li-doped P2-NaxMO2 (M = Ni, Mn) compounds,9,85,88 and the smoothing of the electrochemistry upon increasing the Mg doping level in P2-NaxMn1-yMgyO2 (0 < y < 0.2) phases (see Figure 15),12 confirms this.
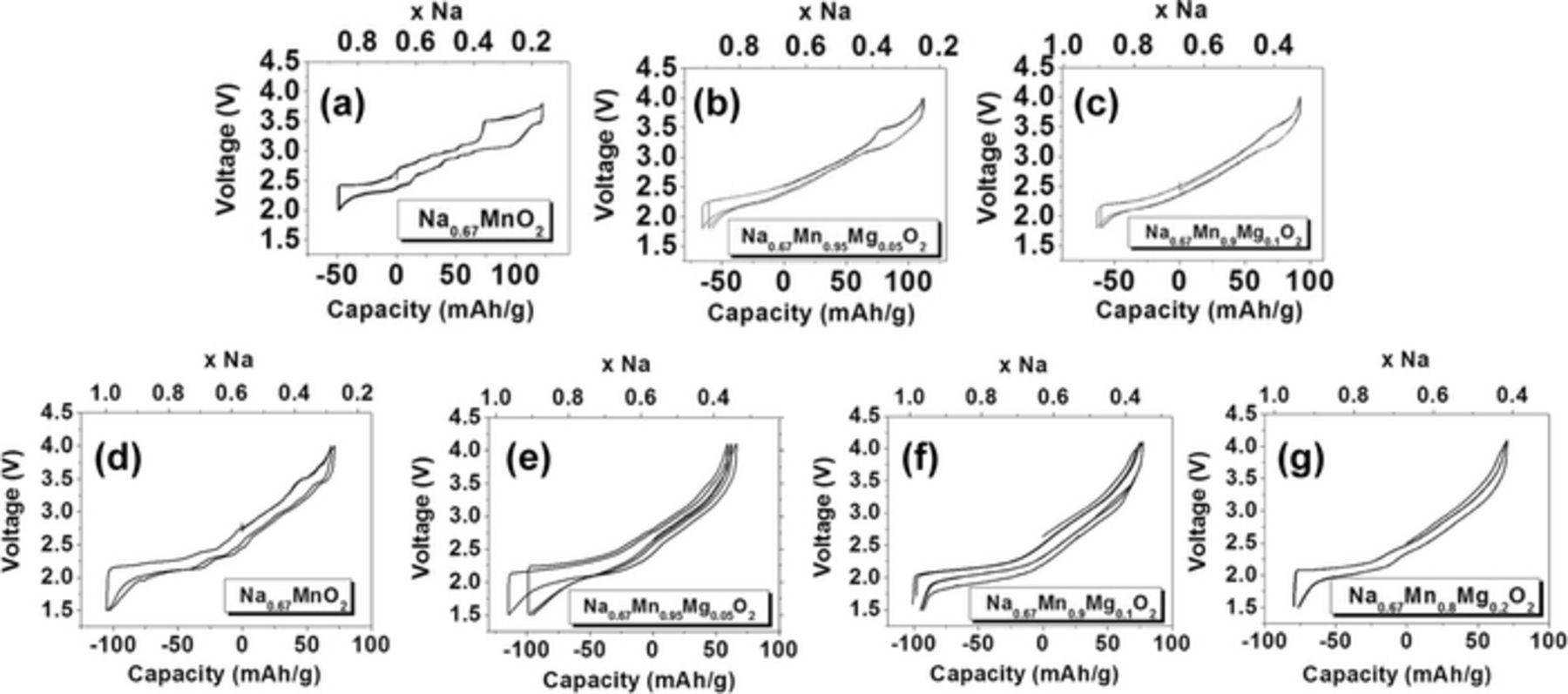
Figure 15. Electrochemical charge/discharge curves for quenched (a) Na2/3MnO2, (b) Na0.67Mn0.95Mg0.05O2, (c) Na0.67Mn0.9Mg0.1O2, and for slow-cooled (d) Na2/3MnO2, (e) Na0.67Mn0.95Mg0.05O2, (f) Na0.67Mn0.9Mg0.1O2, and (g) Na0.67Mn0.8Mg0.2O2. Adapted from Billaud et al.12 (http://dx.doi.org/10.1039/c4ee00465e) with permission of The Royal Society of Chemistry.
Structurally stable cathode materials
Preventing phase transformations and cation rearrangements in NaxMO2 cathodes
Since P2 phases are important for high rate Na cells, research efforts have focused on maintaining a single-phase behavior and preventing cation ordering transitions upon electrochemical cycling of P2-type sodium transition metal oxides. Monophasic reactions reduce the overpotential and capacity drop (resulting from a combination of low reversible capacity and poor capacity retention) of the material upon extended cycling. Single-phase behavior can be achieved by careful choice of the synthetic conditions (precursors, preparation temperature, slow cooling vs. quenching) and composition. The introduction of dopants on the transition metal lattice is also a common strategy to prevent oxygen layer glides upon Na deintercalation, and to increase the range of Na content over which the P2 structure is stable, as shown in Figure 10 for substituted NaxMnO2 phases. The amount of Na that must remain in the NaxMO2 structure to prevent structural transformations at high voltage is still under debate,84 but Li-doped P2-NaxMO2 (M = Ni, Mn) compounds, for instance, have been found to be stable up to a potential of 4.2 V, corresponding to a Na content of 0.35.9,85
Ensuring the stability of structures containing Jahn-Teller ions
Structural stability is a key requirement for the reversible behavior and long cycle life of an intercalation electrode. For Mn3+-containing materials, the lifting and restoration of the Jahn-Teller distortions during charge and discharge induces large stresses on the structure: large overpotentials, low reversibility, the accumulation of extended structural defects, and amorphization, have been observed upon extended cycling.36 Dilution of the Mn3+ species in NaxMnO2 type phases can effectively break the cooperative effect of the Jahn-Teller ions. Paulsen and Dahn demonstrated that the monoclinic distortion observed in β-Na0.7MnO2 becomes orthorhombic for weakly substituted Na2/3Mn1-xMxO2 (M = Co, Li, Ni), and an ideal P2 structure is observed for heavily substituted samples.77 Substitution of Mn3+ for a lower valence element both lowers the concentration of Mn ions in the material, and results in an increase in the average Mn oxidation state (from +3 to +4), which in turn reduces the extent of the Jahn-Teller distortions in the material.
Doping opens up the possibility of having a more flexible structure, with little volume change upon cycling. In addition, the P2 phase can be successfully stabilized over a large voltage window by the introduction of low valence spectator ions (e.g. Li+). Al doping increases the safety of LixMO2 cathode materials,47,105 such as LiNi0.80Co0.15Al0.05O2 (or NCA), used for example as a positive electrode in the Panasonic cells currently powering the Tesla electric vehicles. Although currently safety and thermal stability studies of NaxMO2-type phases are in their infancy, the lower upper voltage cutoffs required for reversible operation of the NaxMO2 electrode systems likely reduce safety concerns in comparison to their Li analogues (but other factors need also be accounted for, e.g. the reactivity of the Na cathode materials towards the Na electrolytes, the kinetics of the unwanted reactions, etc.).
Studying Phase Transformations and Electronic Phenomena Occurring Upon Electrochemical Cycling of NaxMO2 Compounds
We now provide three case studies from our work and those of coworkers, highlighting the use of diffraction-based and solid-state Nuclear Magnetic Resonance (ssNMR) techniques to investigate layered NaxMO2 materials. The choice of these methods is motivated by their relative ease of access, and because diffraction techniques probe long-range structural order, while ssNMR gives insight into the local structure.
Diffraction-based techniques
Diffraction data allow for the identification of new phases, and for solid-solution vs. two-phase reaction mechanisms to be distinguished. Diffraction-based techniques can be used in situ, i.e. as the cell is charged and discharged, providing information on the nature and dynamics of phase transition processes as alkali ions are extracted from and reinserted into the layered material. However, the requirement for long-range order is a serious drawback for the study of NaxMO2 compounds, as these materials often become poorly crystalline at low Na contents. The quantification of each crystalline phase present in a multiphasic sample is possible in theory. In practice, if the material is composed of multiple phases with similar lattice parameters (e.g. a mixture of an ideal NaxMO2 P2 phase, and of another NaxMO2 phase with a higher Mn3+ content and a slight Jahn-Teller distortion), Rietveld refinement of the mixture is difficult due to the overlap of the main reflections. Rietveld refinements are also complicated by hkl-dependent peak broadening caused by the small size of the single-phase domains, or by the high concentration of stacking faults. Owing to the low scattering power of Na and Li, the study of intrinsic disorder on the alkali sublattice with X-ray diffraction (XRD) is challenging. XRD is usually sufficient to obtain average structural information on Na systems, but Li systems require neutron diffraction. When more than one metal species is present in the sample, the complexity introduced at the local level is difficult to capture. For instance, the similar XRD scattering powers of Ni and Mn make it impossible to gain any insight into the atomic level ordering in Ni- and Mn-containing materials with this method.86 Transition metal ordering can have a great impact on the magnetic and electronic properties of layered NaMO2 compounds and may change with Na content in the as-synthesized material. It is therefore important to identify the nature of these orderings by using local probe techniques.
ssNMR
ssNMR, as a local-probe, is well suited for the investigation of amorphous or disordered systems, such as partially-deintercalated electrode materials. It is also an ideal technique with which to investigate local distortions and variations in the electronic structure and in oxidation states associated with both transition metal substitution and alkali deintercalation.106 Charge ordering transitions in the MO2 layers, and migration of electrochemically active or inactive species, can be monitored. ssNMR is a site-specific, non-invasive and quantitative technique which allows sequences of events to be followed, by studying samples stopped at different stages of charge and discharge ex situ. ssNMR can be used in situ to study dynamic processes occurring on the NMR timescale.106,107 To date, in situ NMR is difficult to combine with sample rotation, i.e. Magic Angle Spinning (MAS), which is crucial for chemical (and hyperfine) shift resolution enhancement of paramagnetic materials in ssNMR. The vast majority of ssNMR studies of paramagnetic materials are therefore performed ex situ. The key NMR properties of Li and Na are summarized in Table I. The dominant NMR interactions for 23Na and 7Li in the NaxMO2 cathodes of interest are the quadrupolar and hyperfine terms.
Table I. NMR properties of relevant nuclei. Taken from the Encyclopedia of Magnetic Resonance article, Nuclear spin properties and conventions for chemical shifts (IUPAC Recommendations 2001).108
| Nucleus | I | Quadrupole moment Q / fm2 | Gyromagnetic ratio g / 107 rad T−1 s−1 | Natural Abundance/ % |
|---|---|---|---|---|
| 6Li | 1 | −0.0808 | 3.937 | 7.59 |
| 7Li | 3/2 | −4.01 | 10.398 | 92.41 |
| 23Na | 3/2 | 10.40 | 7.080 | 100 |
Quadrupolar interactions
The magnitude of the quadrupolar interaction depends on the quadrupole moment of the nucleus, and on the electric field gradient (EFG) at the nucleus. The latter increases with the anisotropy of the charge distribution of the crystal environment. The nuclear quadrupole coupling constant is defined as:
![Equation ([1])](https://content.cld.iop.org/journals/1945-7111/162/14/A2589/revision1/jes_162_14_A2589eqn1.jpg)
where h is Planck's constant and q is the charge of the quadrupolar nucleus. Q is the quadrupole moment, which is negligible for 6Li (see Table II). 7Li quadrupolar interactions are typically considered negligible compared with the much larger hyperfine interactions present in paramagnetic compounds.107 On the other hand, 23Na quadrupolar interactions can be significant, and first- and second-order quadrupolar terms need to be accounted for in order to correctly interpret the spectra. First and second-order isotropic linebroadening effects are at least partially removed by MAS, but the anisotropic part of the second order term cannot be averaged out and broadens the observed central transition line.106 In the absence of other sources of linebroadening, the first order quadrupolar lineshape can be fitted to determine the quadrupolar NMR parameters. The second-order term leads to a small quadrupole induced shift, δqis. The overall experimental shift, δexp, is the sum of the field-independent isotropic shift δiso, and of the field-dependent δqis:
![Equation ([2])](https://content.cld.iop.org/journals/1945-7111/162/14/A2589/revision1/jes_162_14_A2589eqn2.jpg)
23Na NMR can, in theory, give access to a larger range of structural information than 6,7Li NMR, including information on the EFG experienced by the Na electron cloud, but the interpretation of paramagnetic Na spectra is typically more complicated than that of paramagnetic Li spectra, due to the complex interference between paramagnetic and quadrupolar interactions in the former case. Paramagnetic interactions still dominate in Mn-based NaxMO2 compounds, and paramagnetic linebroadening often masks the finer details of the quadrupolar lineshape of the NMR peaks, preventing information on the EFG tensor to be obtained from fits of the peak lineshape.
Table II. ab initio hyperfine and quadrupolar NMR parameters calculated for the Mn4+-containing P2-Na2/3Ni1/3Mn2/3O2 material using the CRYSTAL09 code124,125 and the B3LYP hybrid exchange correlation functional, and their corresponding structural parameters. M stands for metal ions nearest neighbor to the central Na, and M' for metal ions next-nearest neighbor to the central Na. The range of M-O-Na bond angles and M...Na distances present in the ab initio optimized P2-Na2/3Ni1/3Mn2/3O2 structure, and corresponding to the calculated M-O-Na bond pathway contributions (BPCs), are quoted, and are in good agreement with those obtained experimentally by Cabana et al.86 The face centered (P(2b)) site in the ideal P2 phase results in very different M-O-Na BPCs than the edge centered environment P(2d). The temperature dependent hyperfine shifts were calculated at 320 K. Further experimental details are presented in the Supplementary Information.
| Site | CQ / MHz | η | BPC (Mn4+) / ppm | M-O-Na / ° | d(M...Na) / Å |
|---|---|---|---|---|---|
| P(2b) (F+F) | 3.5 (±0.3) | 0.1 | M = −36 | 81–82 | 2.81–2.82 |
| M' = 104 | 129–138 | 3.96–3.99 | |||
| P(2d) (E+E) | 5.1 | 0.83 | M = 456 | 94–101 | 3.13–3.29 |
| M' = −55 | 161–167 | 4.06–4.38 |
Paramagnetic interactions
Hyperfine interactions result from the coupling between the nuclear moment of the NMR probe and the time-average of the local field due to unpaired d electrons present on neighboring paramagnetic transition metals, Sz. Hyperfine interactions can induce large shifts and shift anisotropies in the order of 100–10000 ppm in the NMR spectra109,110 and, when using high magnetic field strengths, necessitate broadband excitation NMR techniques.111 Hyperfine interactions can either be through-space (dipolar coupling) or through-bond (Fermi contact) interactions.
The Fermi contact shift, δFC, is proportional to the electron spin density ρ(r = 0) transferred from the transition metal (M) d orbital to the s orbital of the species of interest (A), either directly or indirectly via bridging oxygen p orbitals and so-called M-O-A bond pathways:
![Equation ([3])](https://content.cld.iop.org/journals/1945-7111/162/14/A2589/revision1/jes_162_14_A2589eqn3.jpg)
![Equation ([4])](https://content.cld.iop.org/journals/1945-7111/162/14/A2589/revision1/jes_162_14_A2589eqn4.jpg)
Ac is the hyperfine coupling constant, the size and sign of which determine the magnitude and direction, respectively, of the chemical shift observed. ge is the electron g-factor; μB the Bohr magneton; μ0 the permeativity in free space; γN, S and ω0 are the gyromagnetic ratio, spin and Larmor frequency of the NMR probe. A number of ssNMR studies on paramagnetic materials have shown that, when the Fermi contact shift contribution is dominant, the observed chemical shift is given by the sum of the individual M-O-A bond pathway shift contributions, where M is a transition metal in the first coordination shell of A.107,111–114 Examples of the relevant M coordination shell around Na, for various NaxMO2 polytypes, are shown in Figure 12. The sign and magnitude of individual shift contributions depend on the geometry (bond lengths and bond angles) and covalency of the M-O-A bond pathways, as well as on the magnetic susceptibility of the material, and they are sensitive to both orbital occupation and transition metal oxidation state.107,115–118 The limiting 90° and 180° M-O-A spin transfer mechanisms can be readily rationalized by the Goodenough-Kanamori rules, originally designed to predict the sign of the coupling between d electrons in transition metal oxides.106 It is clear that careful assignment of the features in paramagnetic ssNMR spectra can provide a wealth of information, ranging from the local environments experienced by the NMR probe to the extent of electron (de)localization.119 In practice, individual M-O-Na shift contributions are first obtained on model compounds with well-known structures, which are then used to understand the shifts of more complex materials. ab initio calculations of NMR parameters play an increasing role in understanding the experimental data.111,120,121 The reader is referred to recent papers by Carlier et al.,115 Middlemiss et al.,118 and Zhang et al.,122 which describe the current state-of-the-art in the field of DFT calculations of paramagnetic cathode materials.
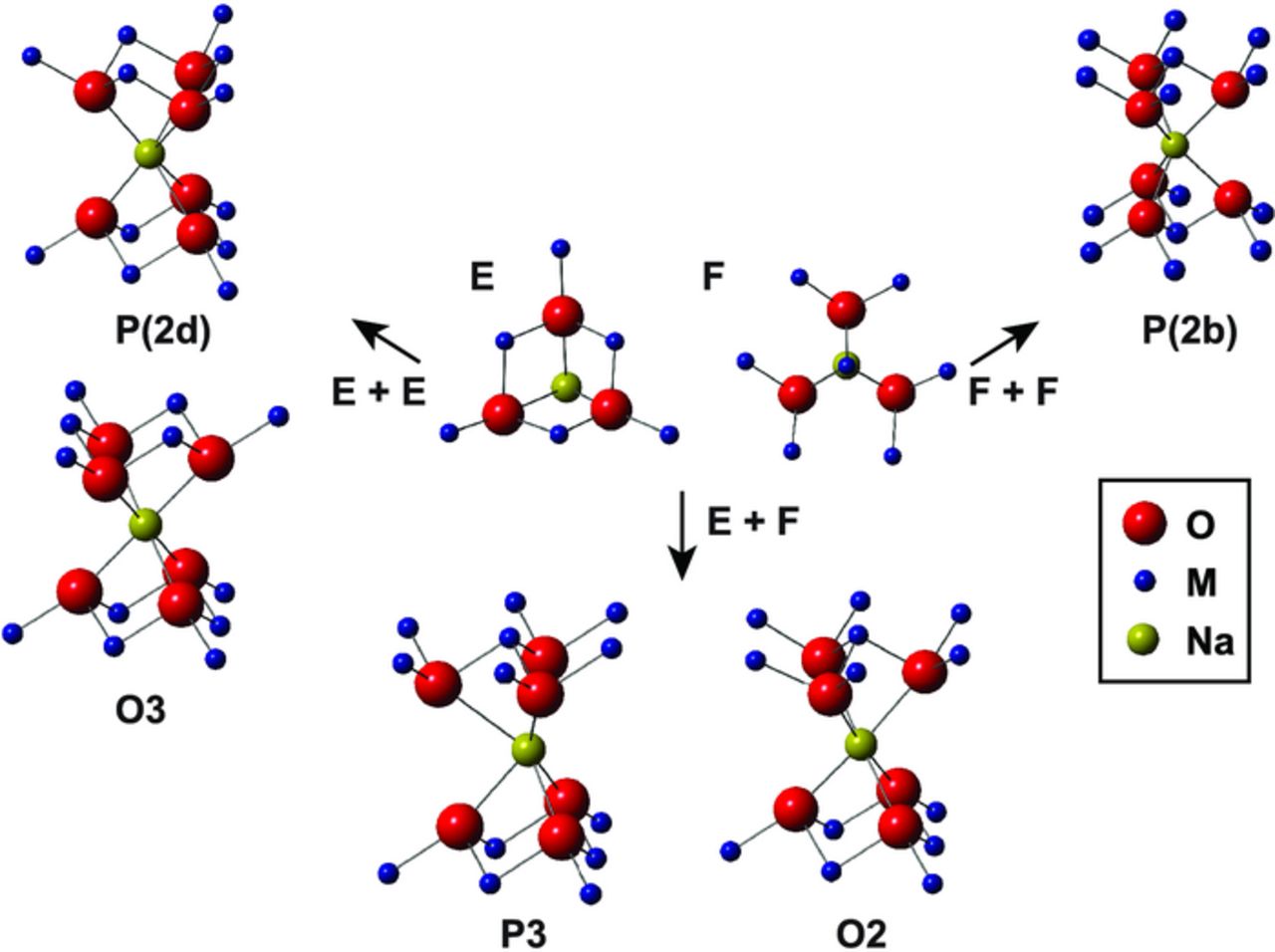
Figure 12. Local Na environments observed in P and O NaxMO2 polytypes. E and F stand for edge centered and face centered coordination to the MO2 slab above or below. Different combinations of E and F coordination to the two neighboring MO2 layers lead to different polytypes. P(2b) to O3, and P3 to O2 transitions require a 60° rotation of the top half of the Na coordination shell, and are not observed at room temperature. Different NMR parameters are expected for the various Na environments, as discussed in Analysis of the 23Na environments present in different NaxMO2 structural polytypes section.
The strong interactions between the unpaired d metal electrons and the NMR probe in paramagnetic materials, such as NaxMO2 compounds, limit the range of experiments that can be used to extract information on the system. The relaxation of the spin of interest in ssNMR is often too rapid to allow for experiments probing ionic diffusion and exchange to be performed (the NMR signal decays completely during the "mixing period"). In addition, the interference between the paramagnetic and quadrupolar effects in the 23Na NMR of NaxMO2 materials has so far precluded the use of pulse sequences designed for quadrupolar nuclei (e.g. MQMAS). And the broad 23Na NMR resonances severely limit the resolution of static in situ NMR experiments (when the sample is not spinning at the magic angle).
Analysis of the 23Na environments present in different NaxMO2 structural polytypes
The Na environments present in a variety of NaxMO2 structural polytypes, depicted in Figure 12, differ in the number and geometry of the M-O-Na spin density transfer pathways. Coordination of Na to the adjacent MO2 slabs above and below can either be face centered (F), or edge centered (E). Face centered coordination corresponds to one metal (M) sitting directly above Na, transferring spin density onto the Na nucleus via three approximately 90° M-O-Na bond pathways, and 6 metal ions in equivalent next-nearest neighbor positions (M'), interacting with Na via a bond pathway at an angle of ca. 170°. Edge centered coordination leads to three nearest-neighbor metal ions (M) transferring spin density via two approximately 90° M-O-Na bond pathways, and three next-nearest neighbor metal ions (M') interacting via a bond pathway at an angle of ca. 170°. In the P2 structure, face centered coordination to the MO2 slabs above and below leads to a P(2b) Na environment (F+F), and edge centered coordination to the MO2 slabs above and below to a P(2d) site (E+E). ab initio calculations of individual Mn-O-Na Fermi contact shift contributions were performed within the Mn4+-only P2-Na2/3Ni1/3Mn2/3O2 material, as shown in Table II, and confirm that very different shifts result from the different bond pathway contributions (BPCs). The Na environment in P3-type compounds is half face centered, half edge centered coordinated to the adjacent MO2 layers (E+F). This is also the case for the octahedral Na site in O2-type (and O6-type) structures. The Na site present in O3-type structures is edge centered coordinated to the MO2 slabs above and below the Na plane (E+E). OP4 and OPP9 structures are composed of alternating P2 and O2 or O3 layers,123 and Na environments similar to those found in such structures are present. In addition to causing large variations in hyperfine shift, the different coordination geometries around the central Na are also expected to cause differences in the EFG experienced by the Na electron cloud, hence in the quadrupolar NMR parameters (see Table II).
The results presented in Table II illustrate that, in principle, experimental 23Na ssNMR and ab initio calculations should be sensitive to the various NaxMO2 structural polytypes, the sites within the polymorphs, and the presence of different cations in the 1st and 2nd cation coordination shells, on the basis of their 23Na NMR parameters. In practice, local structural disorder, and the presence of a range of highly distorted Na sites in the structures obtained in the high voltage region (end of charge), leads to significant broadening of the spectra. In addition, Na+ ion mobility can lead to NMR signal averaging, as discussed below.
What have we learnt from diffraction and ssNMR studies on NaxMn1-yMyO2 electrode materials?
We will now present a few case studies taken from our recent work on NaxMn1-yMyO2 electrode materials.
Unraveling the nature and evolution of stacking faults upon cycling of the β-NaMnO2 cathode
We have been interested in the electrochemical and structural properties of the β-NaMnO2 positive electrode material. This material was prepared directly via solid-state synthesis from Na2CO3 and Mn2O3 powders by heating at 950°C under oxygen, and then quenching to room temperature.8 This cathode exhibits a high reversible capacity of 190 mAh/g at a rate of C/20 (see Figure 13c), and a good rate capability, demonstrated by the 142 mAh/g reversible capacity maintained at a higher rate of C/2. In addition, 70% capacity retention is observed after 100 cycles at a rate of 2C. These promising electrochemical performances are surprising given the complexity of the structure revealed by powder XRD, HRTEM, and NMR. Structural complexity arises from the polymorphic nature of NaMnO2. The two forms are α-NaMnO2, a Jahn-Teller distorted O3' structure with monoclinic (C2/m) symmetry, and β-NaMnO2, with orthorhombic symmetry (Pmnm) and corrugated (zig-zag) layers. Although α-NaMnO2 is the thermodynamically stable form at ambient pressure and temperature, the small energy separation between the two forms leads to facile α/β intergrowth, as indicated by the significant number of stacking faults (twin planes) observed with HRTEM.126 The β-NaMnO2 corrugated structure and an intergrowth model between α- and β-NaMnO2 are presented in Figures 13a and 13b.8
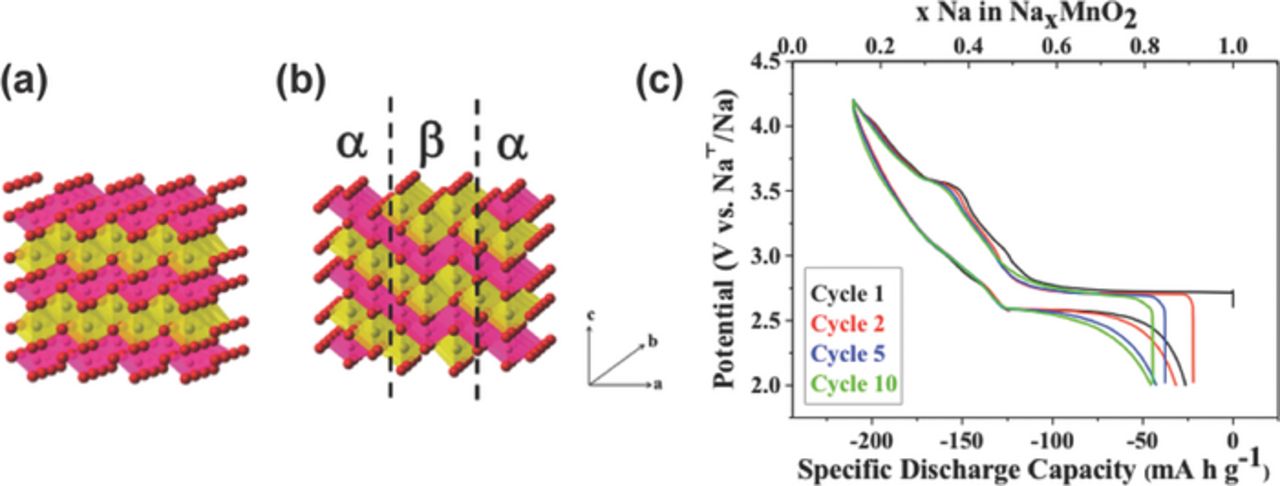
Figure 13. (a) Schematic representation of β-NaMnO2 in the Pmnm space group; and (b) intergrowth model between α and β-NaMnO2. MnO6 octahedra are pink, NaO6 octahedra are yellow and O atoms are red. (c) Charge/discharge curves for β-NaMnO2 cycled at a rate of C/20 (10 mA/g). The 1st, 2nd, 5th and 10th Na extraction/reinsertion cycles are represented in black, red, blue and green, respectively. Reprinted with permission from Billaud et al.8 Copyright 2014 American Chemical Society.
The broadening of the (011) peak in the powder XRD pattern of the pristine β-NaMnO2 phase was successfully simulated using the DIFFaX simulation program with 25% of random stacking faults in the structure, corresponding to the insertion of a monoclinic α-NaMnO2 cell in between two blocks of orthorhombic β-NaMnO2. The presence of structural defects was even more obvious in the 23Na NMR, which exhibited three resonances at 237, 433, and 656 ppm (see Figure 14) instead of the unique Na crystallographic site expected for the ideal β structure. The more intense peak at 237 ppm and the weak peak at 656 ppm were assigned to the Na site in the ideal β structure and to an α-type Na environment, respectively. The difference in chemical shift between the α and β site can be rationalized by the two additional ca. 170° Mn3+-O-Na+ interactions present in the α environment, each of which gives rise to a positive Fermi contact contribution, the positive shift coming from the occupied eg orbital that points along the Mn3+-O-Na pathway, similarly to the LiMnO2 case.127 The peak with an intermediate shift at 433 ppm was believed to arise from Na sites at the boundary between domains of monoclinic and orthorhombic symmetry, with only one 170° Mn3+-O-Na+ interaction. TEM images of as-synthesized β-NaMnO2 presented clear structural disorder along the c axis. Powder XRD data indicated a two-phase region as Na was deintercalated from β-NaxMnO2, in the range x = 1 to x = 0.57. An increase in the number of stacking faults, followed by loss of long-range and short-range order at the end of charge was clearly observed in the XRD, TEM, and NMR data. Structural disorder was found to be reversible over both lengthscales as Na was reinserted into the material, ensuring good cyclability of the compound. An increase in the number of planar defects was noted after five cycles, as demonstrated by the growth of the 656 ppm α peak relative to the spectrum obtained for the as-synthesized material (see Figure 14).8
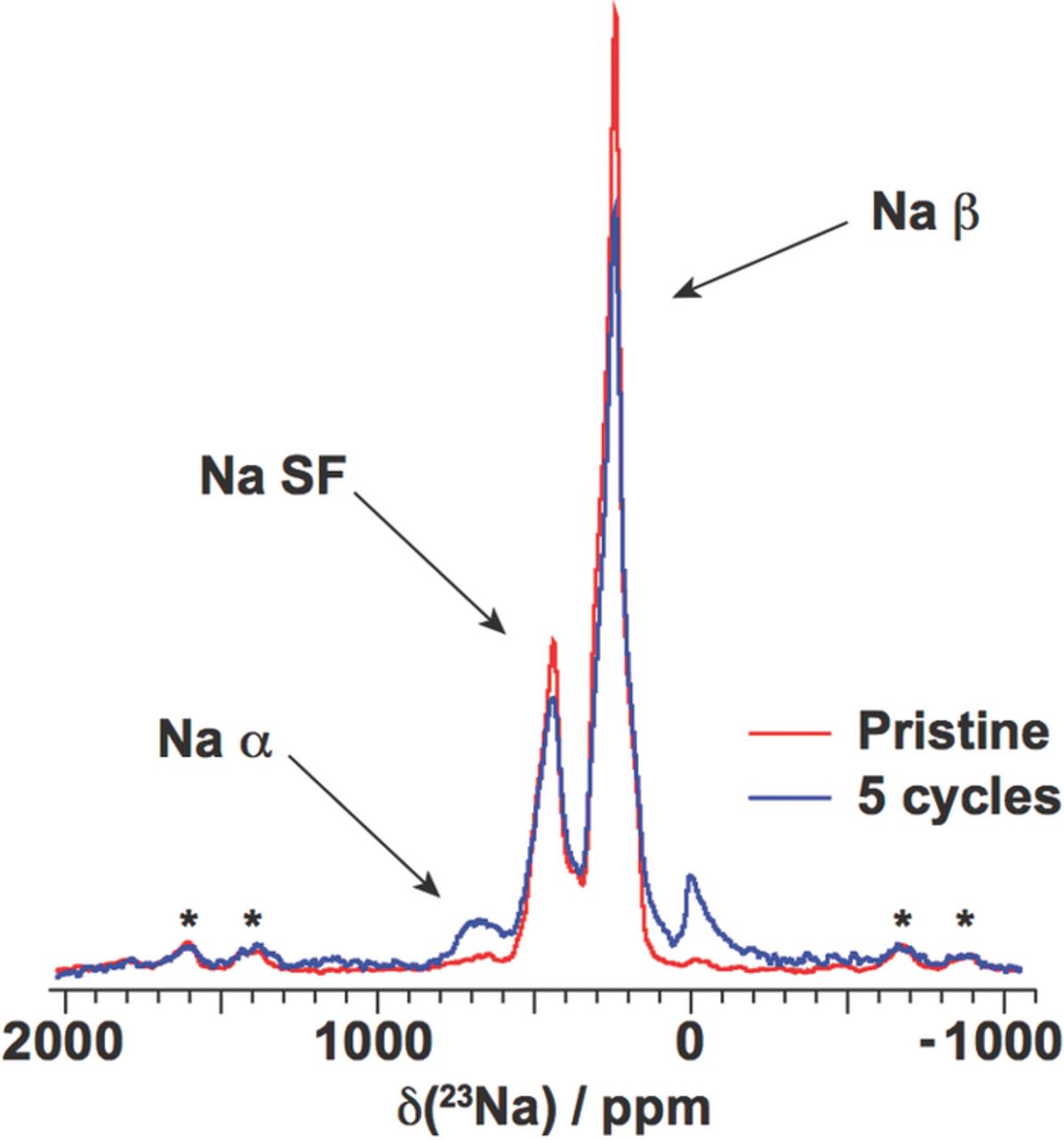
Figure 14. Comparison of the 23Na NMR spectra acquired on the β-NaMnO2 pristine phase and after the 5th discharge. Spinning sidebands are denoted with (*). Three types of Na environments are present in the structure: Na atoms in a pure α environment (Na α), in a pure β environment (Na β), and in the vicinity of a stacking fault (Na SF). The peak near 0 ppm is due to Na+ in a diamagnetic environment, most probably from residual electrolyte or its decomposition products formed during cycling. Adapted with permission from Billaud et al.8 Copyright 2014 American Chemical Society.
Contrary to the common expectation that a stable and reproducible charge/discharge behavior requires a stable structural framework with minimal changes upon Na (de)intercalation, this study shows that β-NaMnO2 exhibits surprisingly good cycling performance despite loss of long- and short-range order at low Na content.8 An in-depth DFT and ssNMR investigation of the exact nature of the stacking faults formed in the as-synthesized material, and those that develop upon cycling, is in progress.
Understanding the effect of Mg doping into P2-Na2/3MnO2
The series Na2/3Mn1-xMgxO2 (x = 0, 0.05, 0.1, 0.2) was prepared to explore how Mg substitution affects the structural phase transitions and Na-ordering. The phases were prepared via a co-precipitation technique, whereby an aqueous solution of Na2CO3 was added dropwise to an aqueous solution of Mn(CH3COO)2 and Mg(CH3COO)2.4H2O. The resulting precipitates were fired at a final temperature of 800°C and then either quenched or cooled slowly to room temperature. The charge/discharge profile of the undoped P2-Na2/3MnO2 cathode material exhibits many voltage steps due to various structural and electronic transitions, since 2/3rd of the manganese is present as the Jahn-Teller active Mn3+ ion (see Figure 15a).
However, slow-cooling and Mg-doping led to a higher average Mn oxidation state and the suppression of the Jahn-Teller driven orthorhombic distortion (with space group Cmcm) of the ideal P2 structure present in Na2/3MnO2. A smoother electrochemical profile resulted from only 5% Mg substitution for Mn (Figures 15d–15g).12 For the quenched samples, the orthorhombic distortion was still present in the 5 and 10% Mg-doped phases, but the 20% Mg-doped phase crystallized in the ideal P2 structure. The extended voltage plateau at 3.5 V vs. Na+/Na, evident in the electrochemical curves of the quenched 0 and 5% Mg-doped phases, was ascribed to the P2 to OP4 phase transformation previously reported in P2-Na2/3Fe1/2Mn1/2O26. A Mg content higher than 10% and slow cooling proved to reduce the 3.5 V plateau (with no clear evidence of a long-range P2 to OP4 transition in the XRD pattern), cell polarization, and capacity fade upon extended cycling, at the cost of a smaller initial charge capacity due to the introduction of spectator Mg2+ ions.12 This study, and work by others,4,5 demonstrates the effectiveness of the divalent Mg ion in preventing structural, electronic, and ordering transitions in P2-NayMnO2, in agreement with Wang et al.'s predictions.103
Unraveling the role of Li in the electrochemical and structural properties of P2-Na0.8Li0.12Ni0.22Mn0.66O2
Quadrupolar interactions are essentially negligible in Li NMR. In addition, the ranges spanned by Li chemical and hyperfine shifts are better established than those observed in 23Na NMR. These two effects make the 6,7Li NMR data of Li-doped P2-NaxMO2 phases simpler to interpret than the 23Na NMR spectra. Several studies on the P2-Na2/3Ni1/3Mn2/3O2 material have reported an initial reversible capacity between 134 and 161 mAh/g (see Figure 16a), but only about 64% of it is retained after 10 cycles,9,11 presumably due to the P2 to O2 phase transformation occurring upon charge to 4.2 V. Ni2+ and Mn4+ cations are proposed to form a two-dimensional honeycomb superstructure on the transition metal lattice, described by a  expansion of the primitive hexagonal cell (space group P63). Li-doped P2-Na0.8Li0.12Ni0.22Mn0.66O2 exhibits improved electrochemical properties, with a 115 mAh/g initial reversible capacity when cycled between 2 and 4.4 V, as shown in Figure 16b, and a 91% capacity retention after 50 cycles.9
expansion of the primitive hexagonal cell (space group P63). Li-doped P2-Na0.8Li0.12Ni0.22Mn0.66O2 exhibits improved electrochemical properties, with a 115 mAh/g initial reversible capacity when cycled between 2 and 4.4 V, as shown in Figure 16b, and a 91% capacity retention after 50 cycles.9
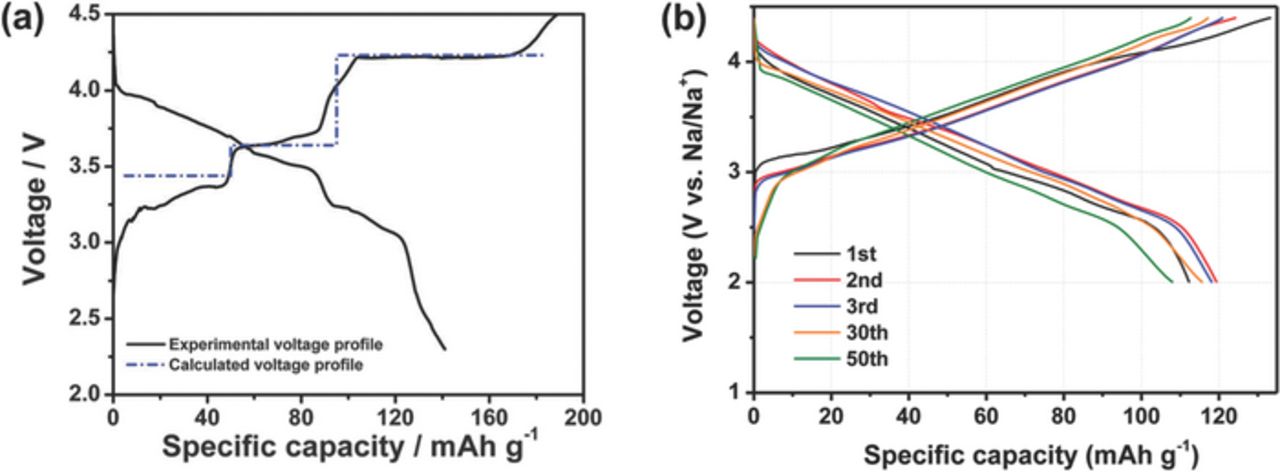
Figure 16. Electrochemical charge/discharge profiles of (a) P2-Na2/3Ni1/3Mn2/3O2 cycled at a rate of C/100 between 2.3 and 4.5 V, and (b) P2-Na0.8Li0.12Ni0.22Mn0.66O2 cycled at a rate of C/10 between 2.0 and 4.4 V. Figure 16a reproduced from Lee et al.11 with permission from the PCCP Owner Societies. Figure 16b reprinted with permission from Xu et al.9 Copyright 2014 American Chemical Society.
The P2-Na0.8Li0.12Ni0.22Mn0.66O2 phase was synthesized via a co-precipitation technique, using a final calcination step at 900°C in air.9 in situ synchrotron XRD data obtained on the P2-Na0.8Li0.12Ni0.22Mn0.66O2 cathode material indicated the formation of O2-like stacking faults upon charge to 4.4 V, with a characteristic broadening of the (10l) peaks (space group P63/mmc), but a complete P2 to O2 phase transformation was not observed. Li substitution delays, rather than completely prevents, the phase transition from P2 to O2, which inevitably happens upon removal of all Na+ ions in the structure. In this material, the stabilized P2 structure greatly enhances the capacity retention of the material, although the initial reversible capacity is lowered upon introduction of electrochemically inactive Li+ dopant ions.9 Rietveld refinement of the XRD pattern obtained on P2-Na0.8Li0.12Ni0.22Mn0.66O2 showed the presence of a  superstructure but the intensities of the superstructure peaks could not be fitted satisfactorily. Figure 17 shows the 7Li NMR data obtained on the as-synthesized material and on three samples stopped at different stages along the first electrochemical cycle. 7Li NMR revealed that, in the as-prepared P2-Na0.8Li0.12Ni0.22Mn0.66O2 phase, Li+ ions are not only found in octahedral sites in the transition metal lattice surrounded by 6 Mn4+ nearest-neighbors, as would be expected from a simple substitution for similar sized Ni2+ ions in an ordered honeycomb metal lattice, and based on Coulombic arguments, but that at least six additional Li local environments are present. The Li+ ions preferentially occupy octahedral sites in the MO2 layers, with 85% of the integrated NMR signal intensity obtained over the corresponding (1100–1800 ppm) range of Li shifts. As expected, these Li+ ions have a high number of Mn4+ nearest neighbors: 73.5% of them have 6 Mn atoms in their first metal coordination shell, and 11% have 5 Mn and 1 Ni. The 1465 ppm resonance is assigned to Li2MnO3 (see below). Surprisingly, 15% Li+ ions are found in octahedral and lower coordination (tetrahedral) sites in the interlayer space occupied by Na+ ions, with shifts in the range 500–750 ppm and <500 ppm, respectively. The evolution of the 7Li NMR spectra during the first charge/discharge cycle reveals that on charging to 4.4 V, when only 0.35 Na is left in the structure, the total Li content decreases to 45% of its initial value and some of the Li+ moves to occupy both octahedral and tetrahedral interlayer sites, created upon oxygen layer glides. Li+ ions are not static, as originally thought, but migrate from the transition metal to the Na layer upon charge.9
superstructure but the intensities of the superstructure peaks could not be fitted satisfactorily. Figure 17 shows the 7Li NMR data obtained on the as-synthesized material and on three samples stopped at different stages along the first electrochemical cycle. 7Li NMR revealed that, in the as-prepared P2-Na0.8Li0.12Ni0.22Mn0.66O2 phase, Li+ ions are not only found in octahedral sites in the transition metal lattice surrounded by 6 Mn4+ nearest-neighbors, as would be expected from a simple substitution for similar sized Ni2+ ions in an ordered honeycomb metal lattice, and based on Coulombic arguments, but that at least six additional Li local environments are present. The Li+ ions preferentially occupy octahedral sites in the MO2 layers, with 85% of the integrated NMR signal intensity obtained over the corresponding (1100–1800 ppm) range of Li shifts. As expected, these Li+ ions have a high number of Mn4+ nearest neighbors: 73.5% of them have 6 Mn atoms in their first metal coordination shell, and 11% have 5 Mn and 1 Ni. The 1465 ppm resonance is assigned to Li2MnO3 (see below). Surprisingly, 15% Li+ ions are found in octahedral and lower coordination (tetrahedral) sites in the interlayer space occupied by Na+ ions, with shifts in the range 500–750 ppm and <500 ppm, respectively. The evolution of the 7Li NMR spectra during the first charge/discharge cycle reveals that on charging to 4.4 V, when only 0.35 Na is left in the structure, the total Li content decreases to 45% of its initial value and some of the Li+ moves to occupy both octahedral and tetrahedral interlayer sites, created upon oxygen layer glides. Li+ ions are not static, as originally thought, but migrate from the transition metal to the Na layer upon charge.9
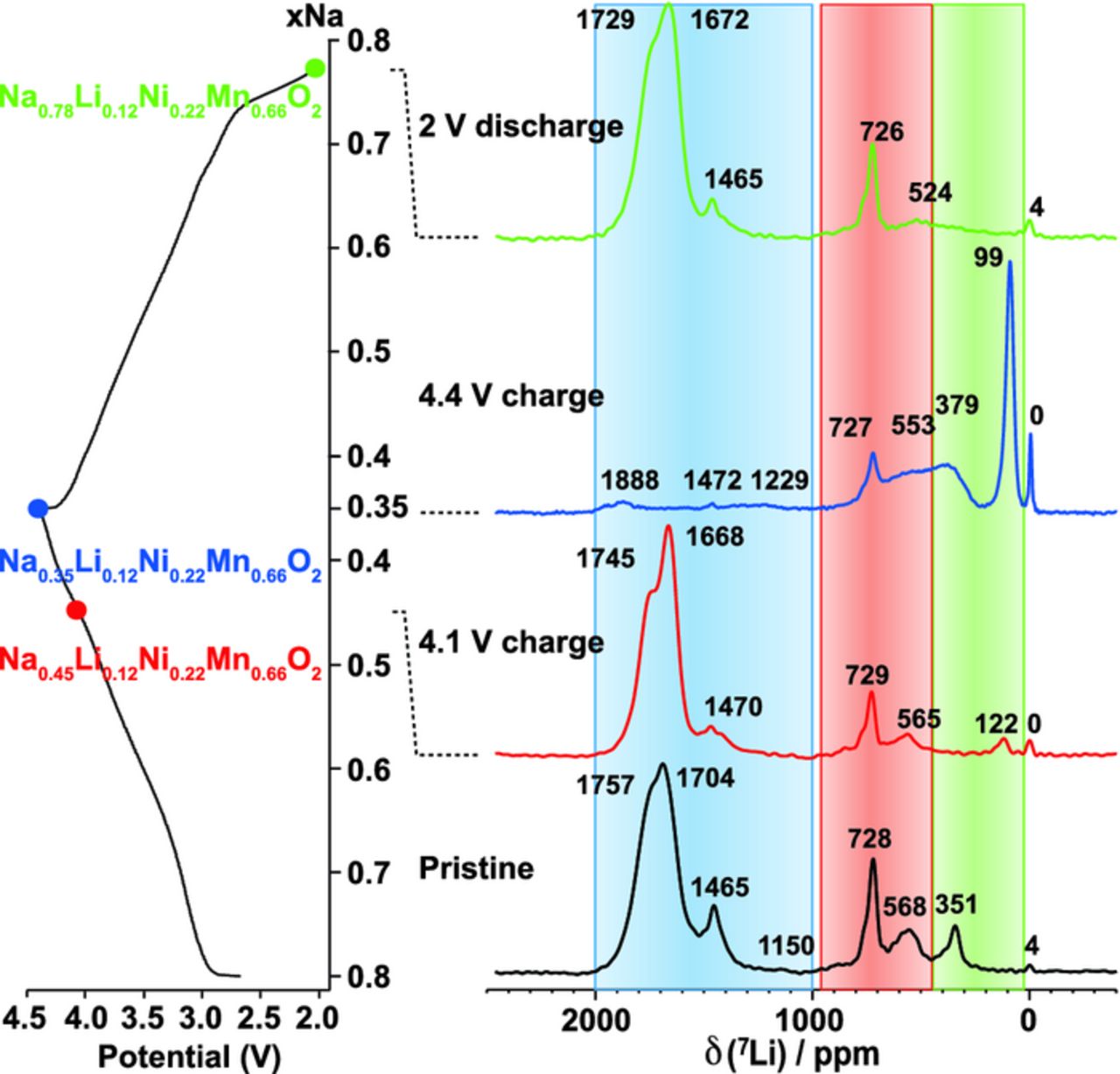
Figure 17. Isotropic slice of pj-MATPASS128 7Li NMR spectra acquired at 200 MHz on as-synthesized P2-Na0.8Li0.12Ni0.22Mn0.66O2 and at three different stages along the first electrochemical cycle. The shaded regions correspond to the ranges of shifts for Li in octahedral sites in the transition metal layer (blue), octahedral sites in the Na layer in (red), and low coordination sites (tetrahedral; green) in the Na layer. The spectra have not been scaled to represent the total Li content in the sample at each stage of the cycle. Adapted with permission from Xu et al.9 Copyright 2014 American Chemical Society.
The excellent rate capability of the material was ascribed to minimal Ni/Na antisite disorder.85 We also believe that Li+ ions stabilize the structure by reducing the in-plane electrostatic repulsion between the Mn4+ and Ni2+ cations. Perhaps most importantly, substitution of Ni2+ by a lower valence species (Li+) decreases the average ionic charge on the transition metal lattice, and more Na+ ions need to be injected in the interlayer regions for charge balance. Therefore, more Na+ ions hold the MO2 layers together up to the end of charge, and the P2 structure is stabilized over a greater potential range.9 The introduction of Li into the MO2 layers disrupts the Ni/Mn ordering on the transition metal lattice, and prevents Na+ ion/vacancy ordering transitions at the origin of the many voltage plateaus on the electrochemical curve of P2-Na2/3Ni1/3Mn2/3O2. The removal of Na+ ion/vacancy order may be due to electron localization as a result of the significant difference between the Fermi levels of the metals in the MO2 lattice.103 The presence of Li in the interlayer space should also contribute to the disruption of any order on the Na lattice.9,85,88
This study emphasizes the importance of using both diffraction and NMR techniques to obtain a full picture of Li migration processes upon cycling. NMR data indicate the presence of phases not resolved with XRD. For instance, 7Li NMR revealed that Li occupies octahedral and tetrahedral interlayer sites, which suggests the presence of structural domains with O-type layer stacking. These O-like domains likely results from stacking faults in the pristine material and/or minor (O3) impurity phases. In recent work by Karan et al., high-resolution XRD on the as-synthesized P2-Na0.85Li0.17Ni0.21Mn0.64O2 material revealed the presence of a minor monoclinic (C2/m) Li2MnO3 phase integrated into the major P2-type Na structure. This proposal is consistent with our NMR spectra (Figure 17), Li2MnO3 giving two characteristic sets of resonances at approximately 1450–1500 ppm and 700–780 ppm.84 The authors claimed that peak asymmetry and anisotropic linebroadening of the XRD pattern could be due to the presence of stacking faults, or to the presence of Li-rich regions with significant deviation from the average lattice parameters. They suggested that the presence of Li2MnO3-like domains in which the Li is immobile ensured structural stability and prevented layer gliding at low Na contents.84
Conclusions
The different applications of rechargeable batteries have different requirements in terms of cost, capacity, weight, voltage, and cell volume. Compact battery packs, with a high volumetric energy density, are required for domestic and commercial load-leveling applications, whereas portable electronics require both light and low volume battery packs, that is, high gravimetric and volumetric energy densities. Other applications may require high power, in which case a high operating voltage and high ionic and electronic conductivities are key. Transition metal oxides typically demonstrate high reversible capacities and exhibit a compact structural framework, which makes them ideally suited for applications for which a high volumetric density is required.
Work on NaxMO2 compounds has outlined a few key design principles for high performance layered NIB cathode materials. For instance, the Na content in the typically Na-deficient P2 phases should be maximized, in order to achieve high density. For P2-Nax[LiyNizMn1-y-z]O2 (0 < x, y, z < 1) compounds, the Ni2+ content should also be maximized. Because in bi- and trimetallic oxides, the ratio of the different (transition) metal species on the transition metal lattice determines the structural polytype, in mixed Ni/Mn cathodes, the P2 phase is obtained if the Ni:Mn ratio is less than 1:29.
One of the questions that remain unanswered, for many P2-type NaxMO2 systems studied until now, concerns the structural changes taking place at the end of charge. The instability of the P2 framework at low Na contents leads to oxygen layer glides at high voltage, but the exact nature of the resulting structure is usually difficult to ascertain because of the extensive disorder, e.g. the formation of stacking faults, which broadens the peaks in the diffraction patterns. Although NMR is, in theory, able to distinguish between the OP4 and O2/O6 polytypes, the Na+ ions tend to only occupy Oh sites (i.e. they are present in O-type layers) at the end of charge, it can be used to explore the role that O vs. P substitution plays in stabilizing the different layer stackings.
The strong interactions between the unpaired d metal electrons, and the NMR-active nuclei (e.g., 6,7Li and 23Na) give rise to characteristic shifts that can be used to identify local structure. For instance, 23Na NMR revealed the presence of ca. 25% stacking faults in β−NaMnO2 and the existence of new Na environments intermediate between "pure α" and "pure β" sites; these environments are located in the vicinity of a twin plane boundary between domains of α-like and β-like structural order. The evolution of the 23Na NMR signal and of the XRD data as a function of charge and discharge of β−NaMnO2 indicated the loss of long- and short-range order at low Na contents, but the reversibility of the process leads to good cycling performance. 7Li NMR on P2-Na0.8Li0.12Ni0.22Mn0.66O2 revealed that Li+ ions are mobile upon Na (de)intercalation. The role of Li in the stabilization of the P2 structure up to 4.4 V is linked to its ability to migrate from the MO2 layer to the Na interlayer space upon charge, and to return to the MO2 layer upon discharge, as clearly evidenced by the changes in the relative population of the Li sites in the ex situ 7Li NMR spectra obtained at different points along the electrochemical cycle. The various case studies outlined in this review demonstrate the power of using complementary structural techniques, such as diffraction and ssNMR, to study structural changes and often cation dynamics in battery materials. When coupled with synthesis and electrochemical studies, these methods play an important role in the development and optimization of new electrode materials.
Acknowledgments
CPG and RJC thank the Assistant Secretary for Energy Efficiency and Renewable Energy, Office of Vehicle Technologies of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231, under the Batteries for Advanced Transportation Technologies (BATT) Program subcontract #7057154 for partial support of this work. RJC also thanks the ECS for the F.M. Becket Summer Fellowship for 2015.
Via our membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202), the ab initio calculations of NMR parameters presented in this work used the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk). The first principles calculations were also carried out in part at the Center for Functional Nanomaterials, Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-SC0012704.