Abstract
Sodium ion batteries offer a low-cost, sustainable, and environment-friendly solution to large-scale electrochemical energy storage systems. Layered oxides represent a family of promising cathode materials with a potential to improve the energy and power densities while reducing the material cost of sodium ion batteries. However, due to the chemical and structural instability of layered oxides in an aqueous solution, the current battery electrode manufacturing requires expensive and hazardous organic solvents, which impedes the full benefit of the low-cost, sustainable, and eco-friendly advantages. We need an effective technology that empowers a cathode with water processable properties. In this study, we set a representative example, P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2, to explore its performance under water-processing conditions. This material achieves a discharge capacity of 180 mAh/g and a discharge energy of 544 Wh/kg at 22°C. The aging experiments indicate its superior stability against water, having negligible bulk structural or chemical changes. The surface sensitive soft X-ray absorption spectroscopy shows that the P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 has stable surface chemistry in the aqueous solution. Moreover, the cells with water-processed cathodes delivered stable cycling performance with minor voltage decay, originating from the decreased cell impedance. Therefore, the present study sets a refined example to establish a low-cost, sustainable, and eco-friendly solution by developing water-processable electrode materials for sodium ion batteries.
Export citation and abstract BibTeX RIS
With the continuous concern for limited lithium and cobalt resources, there is much doubt that lithium ion batteries are able to meet the increasing demands for electric vehicles and renewable energy storage. Therefore, low-cost sodium ion batteries have gained broad attention and rocketed in number of publications.1–3 Layered oxide compounds, denoted as NaxTMO2 (TM=transition metal), are among the most promising cathode materials for sodium metal/ion batteries.4 They can be mainly classified into O3, P2, P3 phases, where the letters denote the sodium coordination environment (octahedral: O and prismatic: P) and the digits represent the number of repeated oxygen layers in one unit cell.5 P2 materials with a relatively lower initial sodium content (0.5≤x≤0.8) deliver a highly reversible capacity in a wide voltage range. O3 phase materials normally contain a higher initial sodium content (0.8≤x≤1), but the utilization of available sodium ions is lower than that of P2 materials, which ultimately leads to a relatively lower reversible capacity.4 Unique to sodium containing layered oxides, many TMs, such as Ni, Co, Mn, Cu, Fe, Cr, V, Ti, Mo, and Nb, can be combined or solely incorporated to form various cathode materials with different layered structures,6 which largely extends the layered oxide family. In addition, the diversity of the electronic configurations in layered oxides containing different TMs helps to expand the knowledge on TM redox behaviors in layered oxides.
Water-soluble binders attract great attention, because they are low-cost, human- and environment-friendly, and are prone to form stable interfaces.7 Thus far, most anodes and many non-layered cathodes can be manufactured using various water-soluble binders (e.g., polyacrylic acid, carboxymethyl cellulose, alginate) to replace the conventional polyvinylidene fluoride (PVDF).7–10 However, alkali layered oxides, an important family of practical cathodes, encounter significant challenges when used with water-soluble binder due to their highly reactive surfaces. Most sodium containing layered oxides suffer from structural and chemical instability, particularly when stored in moist air and/or during electrochemical cycling in wide voltage ranges.11,12 The water co-intercalation in the structure13,14 and structural deterioration by Mn3+ oxidation in reactions with H2O/CO2/O215 are considered the main reasons for sensitivity with air. The air-sensitive cathode materials not only deteriorate the battery performance, but also increase the cost of storage/transport by having to avoid exposure to air. Thus, the shelf life is considered an important evaluative prerequisite for sodium layered cathode materials.16–18 Furthermore, due to the interfacial chemistry and structural instability of cathode materials in the liquid solution,19–21 the current sodium battery cathode manufacturing requires expensive and hazardous organic solvents (e.g., N-Methyl-2-pyrrolidone), which impedes the full use of the low-cost, sustainable, and environment-friendly advantages. Therefore, the field is in urgent need for an effective solution that can enable the water processing of cathodes.
Herein, we develop an air-stable P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 material that can also be processed with a water-based binder for electrode manufacturing. The cathode material delivers a high reversible capacity of 180 mAh/g and a discharge energy density of 544 Wh/kg in the voltage range of 1.0–4.5 V. The Cu/Ti co-substitution smoothens charge/discharge profiles by partially suppressing the Na+/vacancy ordering. The cells containing the water-soaked material, as well as the ones with water-processed cathodes, deliver comparable reversible capacity and cycle life. The cells with the water-processed cathodes show negligible voltage decay, which is associated with the stabilized surfaces introduced by the aqueous manufacturing. The electrochemical impedance spectroscopy and synchrotron soft X-ray absorption spectroscopy reveal that the decreased impedance and mitigated surface degradation might be the origins for the stabilized electrochemical performance.
Results and Discussion
Due to the voltage plateaus in the P2-Na0.67Ni0.33Mn0.67O2 cathode material, efforts have been made to incorporate electrochemically inactive elements (Li+, Mg2+, Zn2+, Ti4+) to mitigate the Na+/vacancy ordering and/or irreversible P2-O2 phase transformation at high voltages (e.g., 4.5 V).12,22–25 However, incorporating inactive elements decreases the Ni concentration and ultimately reduces the capacity.12,23,25 Multiple transition metals in one layered oxide are usually beneficial to battery performance.26–28 Therefore, we incorporated Cu16,17 to substitute Ni and Ti to substitute Mn to form the resulting material of P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2. The X-ray diffraction pattern (XRD, Figure 1a) shows the typical P2-type structure with minor impurities. The primary particle (layered plates stacking to form a step-like morphology) size was approximately 1–5 μm (Figure 1b). Furthermore, we discovered that the particles had smooth surfaces free of tiny particles. The well crystalline nature of the P2 material with a typical layered structure was demonstrated by the transmission electron microscopy (TEM) imaging (Figure 1c). The diffraction dots of the Fast Fourier Transform (FFT) presented two different periodic contrasts (Figure 1c inserted), which might be due to the cation ordering of two Na+ sites in P2 phase (Figure 1a inserted).29,30 The Ti4+, Mn4+, Cu2+ and Ni2+ were verified using the soft X-ray absorption spectroscopy (XAS) in the TEY mode,31 which is in agreement with the charge neutrality of the material (Figure 1d). We performed XRD on the same cathode material after it was either stirred in water or stored in an ambient environment. Many sodium containing layered oxides cannot maintain their original crystal structure when storing in an environment where water is present, let alone being soaked in water with continuous stirring for a long period.11,13,16,18,32 However, there were no other impurity peaks for these water treated samples (Figure 2a). The supernatant solution remained colorless after 6 days of stirring in water, indicating that the Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 cathode material was resilient against dissolution. For the one stored in the ambient environment for 6 months, we observed that the (002) peak slightly shifted to a lower angle, indicating expanded c parameter due to the formation of carbonate species on the surfaces (i.e., loss of Na+ from the layered lattice). We did not observe any major morphology changes other than the formation of tiny particles roughening the surfaces for the latter three samples (Figures 2b–2d). The atomic ratios detected by inductively coupled plasma atomic emission spectroscopy (ICP-AES) are presented in Table I, showing that the atomic ratio in all of the samples stayed close to the nominal ratio of Na0.67Ni0.22Cu0.11Mn0.56Ti0.11. These results provided direct evidence for the bulk structural stability of the P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2. More characterizations are needed to explore the interfacial stability against water since the interfacial properties also influence the battery performance.20,33 The water/air stability of layered cathode materials is related to the TM-TM and TM-O coordination, electronic configuration of various TMs, and chemical and physical properties of sodium containing layered oxide compounds.
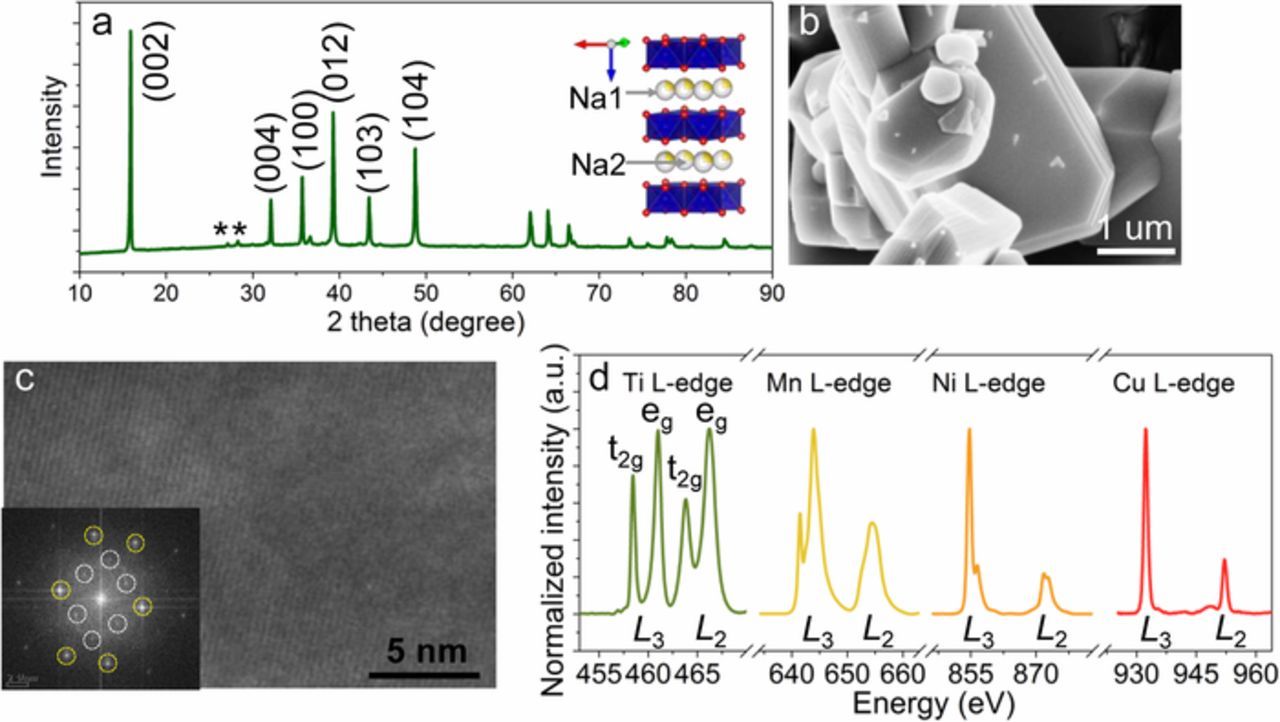
Figure 1. Characterization of the pristine P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 material. (a) XRD pattern, where inserted shows the atomic configuration of a P2 material with two different Na+ sites; the asterisks (*) denote the minor impurity. (b) SEM image; (c) TEM image with the fast Fourier transformation (FFT) inserted; the yellow and white dash circles represent the dots with lighter and darker contract, respectively; (d) Ti, Mn, Ni, and Cu L-edge XAS spectra in the total electron yield (TEY) mode.
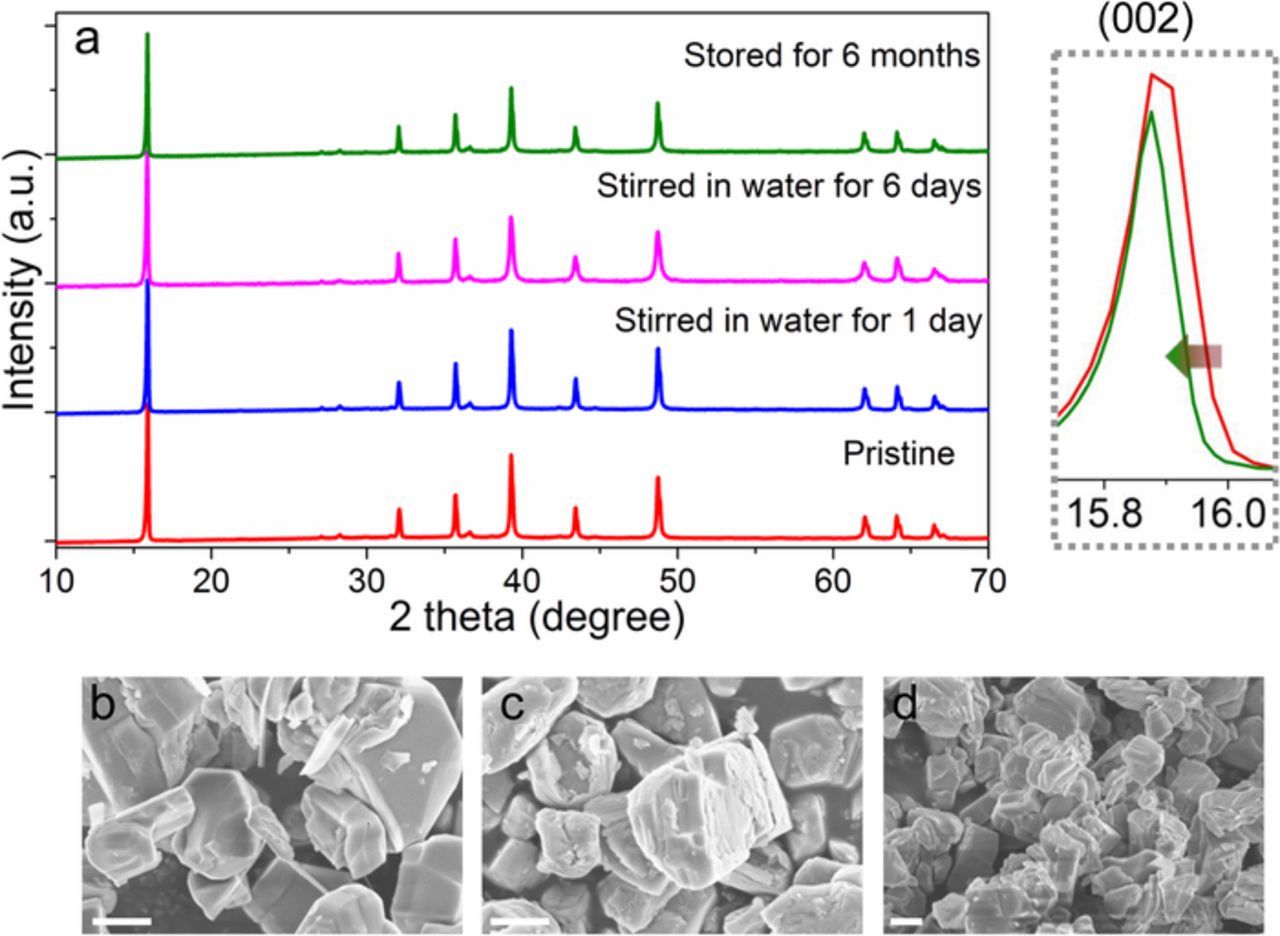
Figure 2. (a) XRD patterns of the samples: pristine P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2, P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 powder stirred in DI (deionized) water for one day, P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 powder stirred in DI water for six days, P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 powder stored in the ambient environment for 6 months; the figure on the right shows the (002) peak of the pristine and sample stored in the ambient environment for 6 months; (b-d) morphology of the three samples, from left to right is the sample stirred in water for 1 day, 6 days, and stored in the ambient environment for 6 months, the scale bars are 1 μm.
Table I. Atomic ratios of the pristine, samples stirred in DI water for 1 day and 6 days.
| Samples | Na | Cu | Ni | Mn | Ti |
|---|---|---|---|---|---|
| Pristine | 0.683 | 0.228 | 0.112 | 0.566 | 0.104 |
| Stirred in water for 1 day | 0.682 | 0.227 | 0.113 | 0.563 | 0.096 |
| Stirred in water for 6 days | 0.654 | 0.234 | 0.112 | 0.559 | 0.094 |
Multiple voltage plateaus (labeled from I to III) for the P2-Na0.67Ni0.33Mn0.67O2 cathode material are regarded as a drawback due to the periodic Na+/vacancy ordering and disordering at the stages I and II (Figure 3a) and the undesired phase transformation from P2 to O2 at the stage III.12,30,34 The cells containing the P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 cathode material presented much smoother charge/discharge curves compared to the P2-Na0.67Ni0.33Mn0.67O2 cathode material, suggesting the Cu/Ti substitution can effectively mitigate the Na+/vacancy ordered structure35 and partially hinder the unwanted, irreversible structural change (Figure 3a).12 In addition, the initial charge and discharge capacity of 130 and 180 mAh/g were achieved at C/5, respectively. Based on the capacity and charge/discharge profiles, the half-cell can deliver a discharge energy density of 544 Wh/kg.6,36 The cells containing the water-stirred cathode material and the one with the directly water-processed cathode delivered a lower initial discharge capacity of 170 mAh/g (Figures 3c–3d). The water-stirring treatment was not detrimental to the cycling performance. The initial charge and discharge capacity of 130 and 162 mAh/g were obtained at C/5 in the voltage range of 1.0–4.5 V for the cell containing the water-processed cathode (Figure 3d). The charge/discharge profiles for the initial ten cycles of the water-processed cathode overlapped better than the ones in Figures 3b–3c. We discovered minor charge/discharge voltage fading for the water-processed cathode, while major voltage decay was observed in the initial 10 cycles for the former two electrodes. We performed cyclic voltammetry (CV) on the cells containing the pristine and the one containing water-processed cathode. For the cell containing the pristine cathode (Figure 3e), there were 5 distinct oxidation peaks and two reduction peaks in the voltage range of 2.8–4.5 V, and only one redox couple in the voltage range of 1.0–2.8 V for the first cycle. Two drastic changes were discovered in the following cycles. First, with increasing cycle number, the current decreased considerably for the redox peaks at in the 4.0–4.5 V and 1.5–2.3 V. Second, the oxidation peak around 2.1 V shifted to a higher voltage, while the corresponding reduction peak around 1.5 V migrated to a lower voltage. The CV features indicated the irreversible phase transformation that subsequently induced rapid voltage and capacity fading, which is consistent with the charge/discharge profiles (Figure 3b).37 In contrast, there are only two main redox couples in the cell containing the water-processed cathode in the range of 2.8–4.5 V (Figure 3f). In addition, except for the minor changes to the first cycle, the shape of the curves and the peak positions overlapped well, indicating good structural reversibility during the electrochemical cycling. The results are indirect evidence that the water-processing for the P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 cathode material can be beneficial for the battery performance.
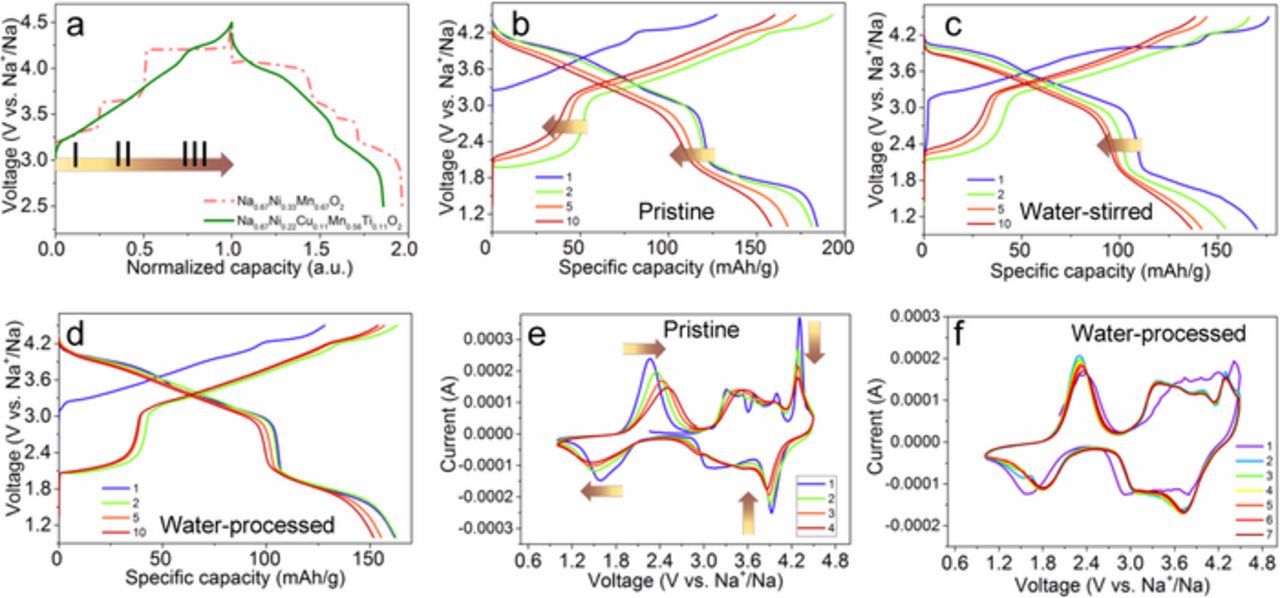
Figure 3. Electrochemical performance of sodium half cells at 22°C. (a) Second charge-discharge curves of the cells containing the P2 Na0.67Ni0.33Mn0.67O2 and Na0.67Ni0.22Cu0.11Mn0.55Ti0.11O2 cathode materials at C/5 in the range of 2.5–4.5 V vs. Na+/Na; (b-c) Initial 10 cycles of charge-discharge curves of the cells containing the pristine P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 cathode material and sample after being stirred in water for 1day cycled at C/5 in the voltage range of 1.0–4.5 V vs. Na+/Na; (d) the cathode manufactured with sodium alginate binder (SA) and water as the solvent; these cells were cycled at C/5 in the voltage range of 1.0–4.5 V vs. Na+/Na. Cyclic voltammetry (CV) of the electrodes containing (e) the pristine material and (f) the water-processed cathode at a scan rate of 0.5 mV/s in the voltage range of 1.0–4.5 V vs. Na+/Na.
We also compared the battery performance of the cells cycled in a narrow voltage range (2.5–4.5 V vs. Na+/Na) (Figure 4). For the pristine cathode, despite the well overlapped charge curves, the discharge voltage profiles faded continuously from a starting voltage of 4.25 V to one lower than 3.9 V in the first 20 cycles (Figure 4a). The voltage fading might be ascribed to the irreversible phase transformation and crack formation.12,37 In the following cycles, the charge/discharge voltage profiles overlapped well and a capacity retention of 83% was achieved after 50 cycles. For the cell containing the water-processed cathode (Figure 4b), the irreversible capacity at about 4.0 V can only be found in the initial charge cycle and vanished in the subsequent cycles. The speed of the discharge voltage fading is much slower (Figure 4b). The capacity retention for the water-processed cathode was 84% after 50 cycles, which is close to that of one containing the pristine cathode (Figure 4c). However, significant differences were observed concerning the discharged average voltage profiles (Figure 4d). For the pristine cathode, the initial average voltage of 3.64 V quickly decayed to 3.35 V with a retention of only 91% after 50 cycles. For the water-processed cathode, the initial average voltage was slightly lower (3.6 V) but maintained a higher retention of 97% after 50 cycles. The results give direct evidence that the water-processed cathode was more stable.
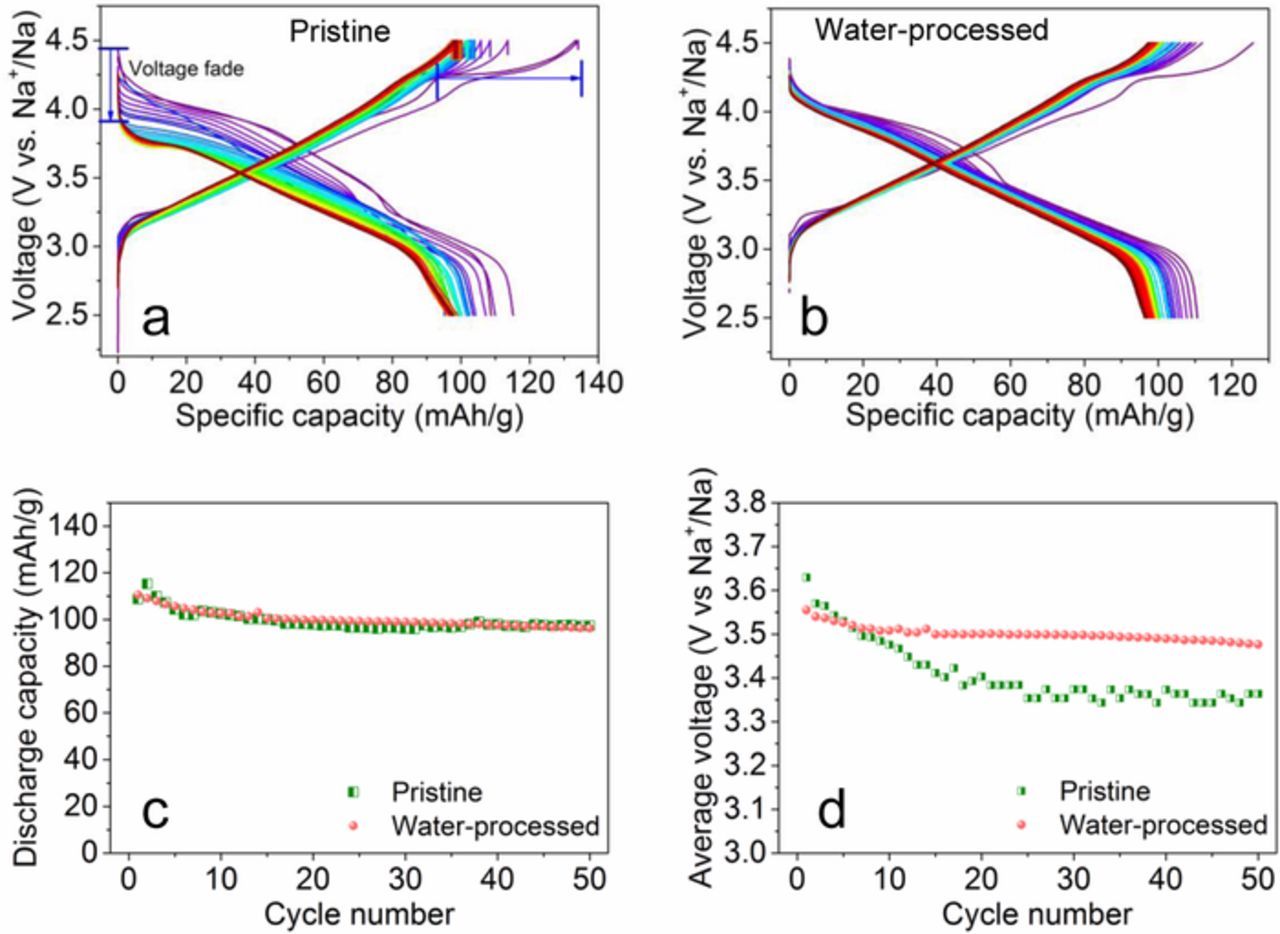
Figure 4. Charge-discharge curves of the cells (at 22°C) containing (a) the P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 cathode material and (b) the water-processed cathode; (c) Discharge capacity, (d) average discharged voltage as a function of the cycle number; these cells were cycled at C/5 in the voltage range of 2.5–4.5 V; the average discharged voltage is defined as the total discharge energy divided by the discharge capacity.
The bulk P2 structure did not change after being stirred in water for 6 days, even after being stored in the ambient environment for 6 months. However, at the surface, the crystal structure, electronic configuration, and valence states of transition metals are usually transformed by the solvent treatment, which eventually governs the performance of batteries.19,20 To understand the surface change of the P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 material, the global surface sensitive soft X-ray absorption spectroscopy (XAS) was performed at the O K-edge and Mn L-edge in the total electron yield (TEY) mode31 (Figure 5). The TEY mode probes for the chemical information of a metal oxide material with a depth sensitivity of 5−10 nm.21 For the pristine P2- Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 material, the peak located at 534 eV is associated with the surface carbonate species (Figure 5a).19,20,38 The peaks lower than 533 eV are ascribed to the hybridization of TM3d and O2p orbitals. The carbonate species disappeared after the samples were stirred in water or processed into electrodes (either with PVDF or sodium alginate binder). Negligible change was found for the powders after being stirred in water for 1 day and 6 days. The result indicates the superior stability of the cathode surface against water. However, a major difference was discovered for the electrode processed using sodium alginate (SA) in water. One possible reason is that the O K-edge information mainly comes from the oxygen in sodium alginate. The Mn oxidation state at the surface is indicative of the stability of the TM3d–O2p hybridization in Mn-containing sodium layered oxides.20,38,39 Mn4+ remained on the surface of Mn for all the powders and electrodes (Figure 5b), providing another direct evidence for the surface stability. Therefore, the surface chemistry of the P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 cathode material is highly stable against water, thus allowing for a low-cost water manufacturing process.
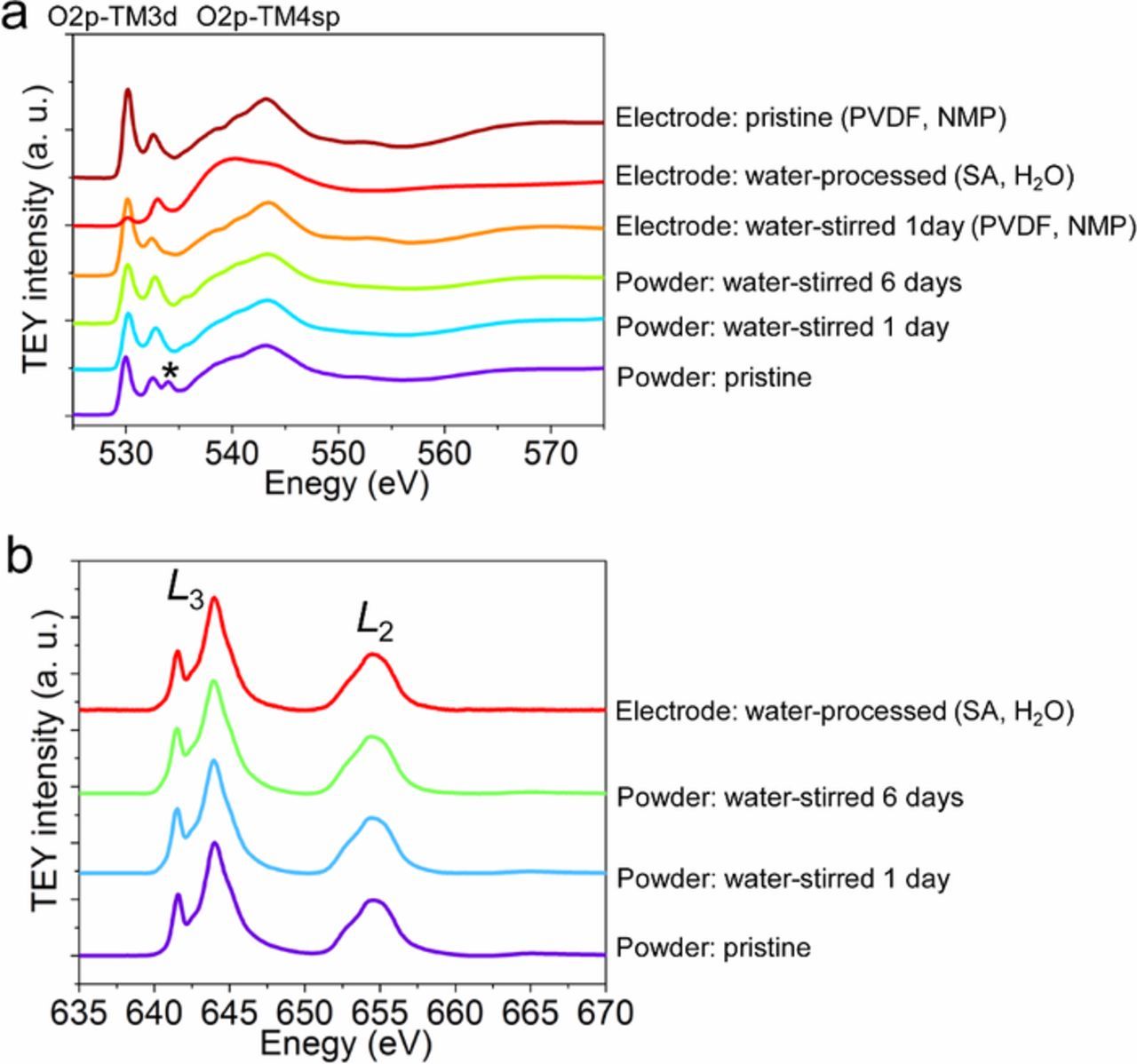
Figure 5. (a) O K-edge XAS spectra in the TEY mode of the corresponding materials and cathodes. The asterisk (*) denotes the peak associated with the carbonate group. (b) Mn L-edge XAS spectra in the TEY mode of the corresponding materials and cathode. The results showed that the TM3d–O2p hybridization and Mn oxidation state were stable upon stirring in water and electrode processing.
As shown in Figure 6, there was no visible difference between the as-made electrode and the cycled electrode (20 cycles) for both the NMP/PVDF-processed (Figures 6a–6b) and water/SA-processed electrodes (Figures 6d–6e). The water-processed cathode seemed to display a higher packing density of cathode particles. For the cross-section after 20 cycles (Figure 6c), we observed the electrode was slightly detached from the current collector in the NMP/PVDF-processed electrode. This could have been caused by the repeated expansion/contraction of the electrode during electrochemical cycling. The water-processed electrode remained in good contact with the current collector (Figure 6f). The difference could be partially responsible for the improved stability in the water-processed cathode. It has been reported that the SA is an effective binder to suppress the irreversible structural transformation and improv the capacity retention.8 In addition, the inhibited voltage fading in the water-processed cathode might be associated with the properties of the cathode−electrolyte interface that is not detectable by the SEM imaging. The rich carboxylic groups in the SA binder might improve the electrochemical stability of cathode particles with the electrolyte.10

Figure 6. Morphology evolution of the electrodes after electrochemical cycling. NMP/PVDF-processed P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 cathode (a) before cycling, and (b, c) after 20 cycles; Water-processed P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 cathode (d) before cycling, and (e, f) after 20 cycles. All the cells were cycled at C/5 in the voltage range of 2.5−4.5 V, the scale bars in a, b, d, e are 2 μm, the scale bars in c and f are 10 μm.
Finally, we measured the electrochemical impedance spectroscopy (EIS) of the cells after 1, 5, 20, and 60 cycles (Figure 7). For both cells, all of the spectra showed two semi-circles: one at the high frequency region (HF, left semi-circle) that is associated with the resistance (Ri) at the electrode–electrolyte interface (EEI, including CEI and SEI), and the other (Rct) at the middle frequency region (MF, right semi-circle) that is ascribed to the charge transfer process.40–42 For the cell containing the NMP/PVDF-processed cathode, the radius of the semi-circles gradually decreased with the increase of cycle number (Figure 7a), suggesting the reduction of both resistances originating from interfaces and charge transfer. For the cell containing the water-processed cathode, there was a significant reduction of the resistances from the 1st cycle to the 5th cycle and then a negligible change was noticed until 60 cycles (Figure 7b). The significant impedance decreasing of the cell containing the pristine cathode material most likely originated from the sodium anode. In addition, the improved interface of the water-processed cathode might also help deliver the overall less resistive and more stable interfaces. In order to make a direct comparison between the two cells, we used the same modeling circuit (Figure 7) to fit the Ri and Rct values (Table II). After the initial cycle, the cell containing the water-processed cathode showed consistently smaller Ri and Rct. The results indicated that the water-processed cathode allowed for a stable and less resistive interface, which provides partial explanation for the improved electrochemical performance observed in the cells containing the water-processed cathode.
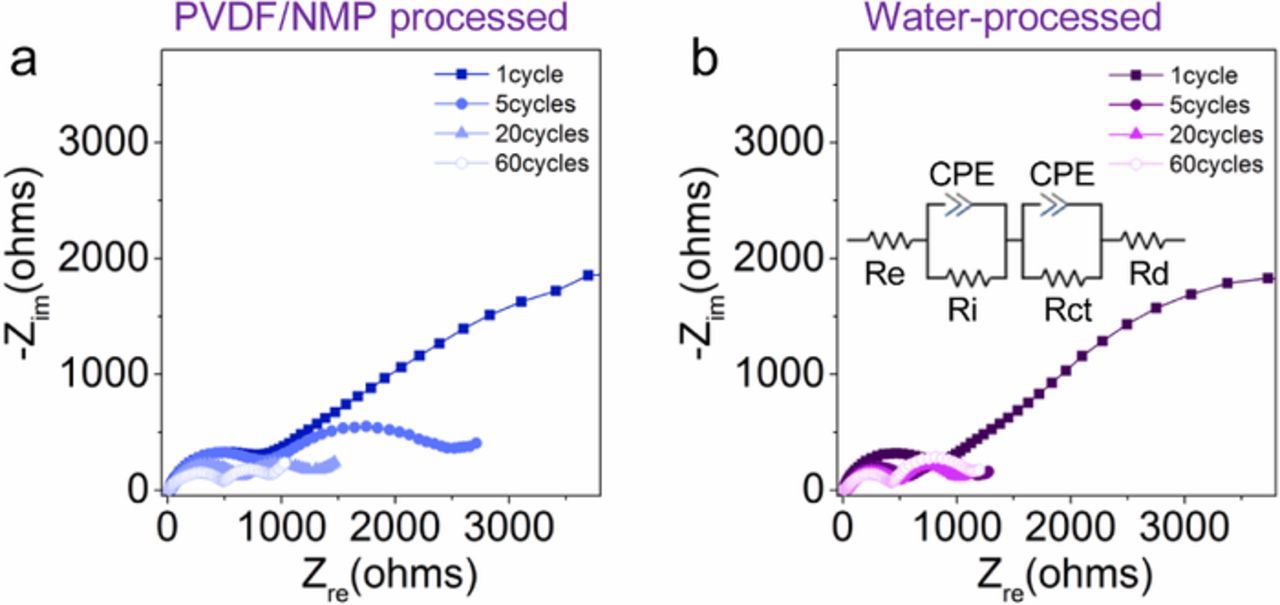
Figure 7. Electrochemical impedance spectroscopy (EIS) of the cells at 22°C containing (a) the PVDF/ NMP-processed cathode, and (b) the water-processed cathode after 1, 5, 20, and 60 cycles in the frequency of 10 kHz–10 mHz; the inserted diagram in b is the modeling circuit to fit the Nyquist plots, the Re, Ri, Rct, Rd, and CPE represent resistances from the electrolyte, interfaces, charge transfer, Warburg impedance reflecting the Na-ion diffusion in the solid, and the constant phase element, respectively.40–42
Table II. Summary of the fitted parameters for the Nyquist plots in Figure 7. Resistance evolution in the high frequency (HF) and middle frequency (MF) of the cells containing the NMP/PVDF-processed and water-processed P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 cathodes; Ri and Rct represent the resistance from the interface and charge transfer, respectively. The fitting was performed using the ZView software based on the equivalent circuit (Figure 7 insert).
| NMP/PVDF- | Water- | |||
|---|---|---|---|---|
| processed | processed | |||
| Sample | Ri | Rct | Ri | Rct |
| 1cycle | 754.6 | 7934 | 734.8 | 9938 |
| 5cycles | 853.6 | 1774 | 645.2 | 580.8 |
| 20cycles | 531.7 | 669.4 | 489.6 | 502.6 |
| 60cycles | 498.8 | 483 | 498 | 488.6 |
Materials Synthesis
All the materials were synthesized through a solid-state reaction method. The stoichiometric starting materials of Na2CO3, CuO, NiO, MnO2, TiO2 were mixed by ball milling in a ZrO2 jar at 400 rpm for 8 h. Then, the uniformly mixed materials were transferred into an alumina crucible and heated at 900°C for 12 h in a box furnace to obtain the final product (P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2). The P2-Na0.67Ni0.33Mn0.67O2 was synthesized without adding TiO2 or CuO. The aging samples (samples soaked in water for 1 day and 6 days) were collected directly from the pristine powder and stirred in DI water for 1 day and 6 days at 1000 rpm. Then, the powders were centrifuged from water and dried in a vacuum oven at 100°C overnight.
Materials Characterization
The morphologies of the materials and electrodes were investigated using a scanning electron microscope (SEM) (LEO FESEM) at an accelerated voltage of 5 KV. The overall elemental composition of the material was evaluated using inductively coupled plasma atomic emission spectroscopy (ICP-AES). The ICP-AES instrument is a Spectro ARCOS SOP (side-on or radial view of the plasma interface) made by Spectro Analytical Instruments, Inc. For sequential multiple elemental measurement, the relative standard deviation and the coefficient of variation are less than 2% for concentration higher than the background equivalent concentrations. The X-ray powder diffraction (XRD) of the materials was obtained on a PANalytical X-ray diffractometer with the Cu Kα (λ = 1.54 Å) radiation. Transmission electron microscopy (TEM) was performed on a JOEL 2100 S/TEM operated at 200 keV. Soft XAS measurements were performed on the 31-pole wiggler beamline 10–1 at Stanford Synchrotron Radiation Lightsource (SSRL) using a ring current of 350 mA and a 1000 l·mm−1 spherical grating monochromator with 20 μm entrance and exit slits, providing ∼1011 ph·s−1 at 0.2 eV resolution in a 1 mm2 beam spot. Data were acquired under ultrahigh vacuum (10−9 Torr) in a single load at room temperature using total electron yield (TEY). All spectra were normalized by the current from freshly evaporated gold on a fine grid positioned upstream of the main chamber. XAS samples were mounted on an aluminum sample holder with double-sided carbon tape in a Ar-filled glove box and transferred to the load-lock chamber in a double-contained container, using a glove bag purged with argon for the transfer.
Electrochemical Characterization
The composite cathodes were prepared by spreading the slurry (N-Methyl-2-pyrrolidone as the solvent, or water as the solvent) with active materials (90 wt%), acetylene carbon (5 wt%) and binder (5 wt%, polyvinylidene fluoride or sodium alginate) and casting them on carbon-coated aluminum foils. The electrodes were then dried overnight at 120°C in a vacuum oven and transferred into a Ar filled glove box for future use. The active mass loading for all the electrodes was ∼ 4 mg/cm2. CR2032 coin cells (half cells) were assembled in a Ar-filled glove box (O2<0.5 ppm, H2O<0.5 ppm) using the composite cathode, sodium foil as the anode, Whatman glass fiber (1827-047 934-AH) as the separator, and saturated NaPF6 dissolved in propylene carbonate (PC) as the electrolyte. All the coin cells were cycled with an electrochemical workstation (Wuhan Land Company) at 22°C. The average voltage and energy density were calculated using the LANDdt software. 1C was defined as fully charging a cathode in 1 h, corresponding to a specific current density of 120 mA/g. The electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) measurements were performed on a Princeton Applied Research of VersaSTAT 4 at 22°C.
Conclusions
In this study, we proposed a quaternary P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 cathode material that achieved a high energy density (discharge capacity: 180 mAh/g, discharge energy: 544 Wh/kg) at 22°C. The Cu/Ti substitution mitigated the Na+/vacancy ordering. The aging experiments showed that the P2 material had a superior structural and chemical stability in water and in the ambient environment for 6 months. The aging process had a negligible effect on the battery performance. Furthermore, the cells containing the water-processed cathodes delivered improved cycle life, more stable voltage profiles, and decreased cell impedance. These results show the unique properties of the P2-Na0.67Ni0.22Cu0.11Mn0.56Ti0.11O2 cathode material that can further reduce the cost of material production, storage, and transportation, ultimately lowering the price of sodium ion batteries. Therefore, we anticipate that this work will enable more efforts in developing air-stable cathode materials and promoting the practical applications of sodium ion batteries. Finally, there are still some open questions regarding how water molecules interact with the cathode particles, thus we expect to perform more studies to further understand the interaction mechanism at the molecular level.
Acknowledgment
The work was supported by the Department of Chemistry Startup Funds and ICTAS Junior Faculty Award at Virginia Tech. The Stanford Synchrotron Radiation Light Source, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility is operated for the US Department of Energy Office of Science by Stanford University. Use of the Stanford Synchrotron Radiation Light source, SLAC National Accelerator Laboratory, is supported by the US Department of Energy, Office of Science (SC), Office of Basic Energy Sciences (BES) under Contract No.DE-AC02-76SF00515. Z. Y. and Y. D. acknowledge the support by the U.S. DOE SC Early Career Research Program. A portion of the work was performed using EMSL, a DOE SC User Facility sponsored by the Office of Biological and Environmental Research. The authors declare no competing financial interest.
ORCID
Feng Lin 0000-0002-3729-3148