


Abstract
We show, in a series of established experimental breast adenocarcinomas and fibrosarcomas induced by carcinogen de novo in mice, that the therapeutic efficacy of doxorubicin treatment is dependent on CD8 T cells and IFN-γ production. Doxorubicin treatment enhances tumor antigen–specific proliferation of CD8 T cells in tumor-draining lymph nodes and promotes tumor infiltration of activated, IFN-γ–producing CD8 T cells. Optimal doxorubicin treatment outcome also requires both interleukin (IL)-1β and IL-17 cytokines, as blockade of IL-1β/IL-1R or IL-17A/IL-17Rα signaling abrogated the therapeutic effect. IL-23p19 had no observed role. The presence of γδ T cells, but not Jα18+ natural killer T cells, at the time of doxorubicin treatment was also important. In tumor samples taken from breast cancer patients prior to treatment with anthracycline chemotherapy, a correlation between CD8α, CD8β, and IFN-γ gene expression levels and clinical response was observed, supporting their role in the therapeutic efficacy of anthracyclines in humans. Overall, these data strongly support the pivotal contribution of both innate and adaptive immunity in treatment outcomes of anthracycline chemotherapy. Cancer Res; 71(14); 4809–20. ©2011 AACR.
Introduction
Chemotherapy treatment in cancer has long been associated with development of systemic immunosuppression (1, 2). However, more recent studies have reported augmentation of antitumor immune responses by chemotherapy (reviewed in ref. 3). The immunostimulatory properties of chemotherapy have been associated with increased expression of “stress-like” molecules by damaged or dying tumor cells, such as killer cell lectin-like receptor sub-family K ligands, Fas, and tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) death receptors and HSPs, that render cells receptive to direct attack by cells of the innate immune system; including natural killer (NK) cells, natural killer T cells (NKT cells), and γδ T cells (reviewed in ref. 4). Until recently, the mechanisms by which chemotherapy treatment facilitated an adaptive, tumor-specific immune response were largely uncharacterized. Certain alarmins or danger-associated molecular patterns are expressed or released during tumor cell death induced by immunogenic chemotherapeutic agents, including anthracyclines. These include translocation of the endoplasmic reticulum resident calreticulin complex to the plasma membrane, providing an “eat-me” signal (5–7) and release of the nuclear alarmin HMGB1 to engage TLR-4 on dendritic cells (DC; refs. 8, 9). In addition, we showed recently that dying tumor cells release ATP, which then acts on P2X7 purinergic receptors from DCs and triggers the NOD-like receptor family, pyrin domain–containing-3 protein (NLRP3)-dependent caspase-1 activation complex (inflammasome), allowing for the secretion of proinflammatory interleukin (IL)-1β. Accordingly, anticancer chemotherapy turned out to be inefficient against tumors established in purinergic receptor P2rx7− or Nlrp3− or Casp1-deficient hosts (10).
Anthracyclines have been used in anticancer treatment for more than 40 years and now have a major role in the management of leukemia, lymphoma, uterine, ovarian, sarcoma, and breast malignancies. Doxorubicin became the most widely used anthracycline because of lower toxicity and potent antitumor activity against solid tumors. Although there were earlier reports evidencing the immune system contribution to doxorubicin-mediated antitumor effects (11, 12), Maccubbin and colleagues were the first to show that doxorubicin was an effective immunomodulator capable of boosting CTL responses (13). A multitude of studies investigating combination treatment of doxorubicin and immunotherapies followed; however, the underlying mechanisms of how immunogenic cell death caused by doxorubicin links to induction of a cytotoxic CD8 T-cell response were largely unknown.
Using experimental and carcinogen-induced mouse models of breast cancer and fibrosarcoma, respectively, here we show key cytokines and immune cell populations required for the antitumor therapeutic efficacy of doxorubicin. We show that CD8 T cells and IFN-γ are critical effectors of IL-1β- and IL-17–dependent signaling in response to doxorubicin-treated breast tumors and sarcomas, and corroborating this, we also report that CD8α, CD8β, and IFN-γ gene expression levels in breast cancer patients treated with anthracycline chemotherapy correlate with treatment response.
Materials and Methods
Mice
Inbred wild-type (WT) C57BL/6 and BALB/c mice, and OT-I mice carrying a MHC class I-restricted transgenic T-cell receptor (TCR) for the ovalbumin (OVA257–264; SIINFEKL) peptide were obtained from Walter and Eliza Hall Institute (WEHI; Parkville, Australia). C57BL/6 gene–targeted knockout IFN-γ−/−, IL-1R−/−, IL-17A−/−, TCRδ−/−, Jα18−/−, IL-23p19−/−, and IL-12p35−/− mice were bred and maintained at the Peter MacCallum Cancer Center (Peter Mac; East Melbourne, Australia) as previously described (14, 15). TCRδ−/− mice were obtained from I. Frazer (University of Queensland, Australia) and IL-17A−/− mice were kindly provided by Y. Iwakura (University of Tokyo, Japan). Mice of 6 to 14 weeks of age were used in all experiments, and all procedures were approved by the Peter Mac Animal Ethics Committee.
Tumor models
Experimental.
C57BL/6-derived AT3 (kindly provided by Trina Stewart, Peter Mac) and EO771 (16) and BALB/c-derived H2N100 mammary adenocarcinoma (17) lines, and BALB/c-derived MCA2 (derived in-house from mice with an MCA-induced tumor) and C57BL/6-derived MCA205 (provided by L.Z.) fibrosarcoma lines, were all maintained in RPMI 1640 supplemented with 10% fetal calf serum (FCS). The OVA-expressing AT3 line (AT3OVA) was generated by retrovirally infecting the AT3 parental line with pMIG/MSCV-IRES-eGFP plasmid encoding membrane-bound OVA. To examine s.c. tumor growth, WT or gene-targeted mice were inoculated s.c. on the hind flank with the indicated number of cells and tumor size monitored. Once tumors were established (0.2–0.5 cm2) mice received a single intratumoral (i.t.) or i.v. treatment of doxorubicin hydrochloride (DOX; Sandoz) or equivalent volume of PBS. Some mice received control Ig (cIg; Mac-4 or agp3), anti-IFN-γ (R4-6A2), anti-CD8α (53.6.72), anti-CD8β (53.5.8), anti-IL-1β (B122.10), anti-IL-17A (M210), anti-IL-17Rα (M751), or anti-IL-23p19 (16E5) monoclonal antibody (mAb) at the time of PBS/doxorubicin treatment, as indicated in the legends to neutralize or deplete cell subsets. Anti-agp3, anti-IL-23p19, anti-IL-17A, and anti-IL-17Rα mAb were kindly provided by AMGEN Inc.
3-Methylcholanthrene-induced carcinogenesis.
Groups of male BALB/c WT mice were inoculated s.c. in the hind flank with 400 μg of 3-methylcholanthrene (MCA; Sigma-Aldrich in 0.1 mL of corn oil as described; ref. 18). When sarcomas had established (palpable tumor 0.2–0.45 cm2) mice received i.t. doxorubicin or PBS once a week for 2 weeks. Some mice received cIg, anti-CD8α, anti-IFN-γ, anti-IL-1β, anti-IL-23p19, or anti-IL-17Rα on days −1, 0, and weekly thereafter for 6 weeks relative to initial PBS/doxorubicin treatment. Development of fibrosarcomas was monitored weekly over the course of 250 days.
In vivo CD8 T-cell assays and flow cytometry.
OT-I T cells were purified from spleens and lymph nodes of OT-I transgenic mice, with positive selection using a CD8 T-cell isolation kit (Miltenyi Biotec). CD8 T-cell purity was always found to be greater than 90%. Purified OT-I cells were labeled with 2.5 μmol/L carboxyfluorescein succinimidyl ester (CFSE) and injected i.v. (2 × 106) into the tail vein of C57BL/6 mice harboring established, PBS/doxorubicin-treated AT3OVA or AT3-empty vector (AT3EV) tumors. Five days later, tumor-draining lymph nodes (TDLN) were harvested and CFSE dilution in CD8+KbOVA257–264-tetramer+ cells was assessed by flow cytometry. Indices of proliferation were generated using FlowJo software (Tree Star). For tumor-infiltrating lymphocyte (TIL) analysis, AT3-OVA tumors were excised and single-cell suspensions created by a combination of collagenase type 4 (Worthington Biochemical Corp.) and DNase I (Roche) enzymatic digestion at 37°C, and mechanical disruption. OT-I T cells were identified by antibody staining with anti-CD45.2 (104; eBioscience), anti-CD8α (Ly-2; eBioscience), and KbOVA257–264-tetramer (provided by A. Brooks). Dead cells were excluded by addition of 1 μg/mL of fluorogold (Sigma-Aldrich) in the final wash. Cells were acquired on an LSR-II (BD) or Canto II (BD) flow cytometer and analysis was done using FlowJo software. For intracellular IFN-γ analysis, tumor cell suspensions were cultured overnight in RPMI 1640 supplemented with 10% FCS in the presence of 0.1 μmol/L OVA257–264 (SIINFEKL) peptide (Auspep Pty) and 5 ng/mL recombinant mouse IL-2 (BD Biosciences). Monensin (BioLegend) was added to the cells in the final 4 hours of culture to inhibit cytokine release from the Golgi/endoplasmic reticulum complex. Permeabilization and fixation of cells were conducted using the BD Cytofix/Cytoperm kit according to the manufacturers' instructions (BD Biosciences) before staining with anti-IFN-γ mAb (XMG1.2; eBioscience).
Gene expression analysis
Two cohorts of breast cancer gene expression data sets were used for these analyses. Complete clinical data are included in the Supplementary Information (Supplementary Table S1). The first comprised early-stage breast cancer patients who had received no systemic treatment to determine the association of the genes with clinical outcome without anthracycline (i.e., association with prognosis). The 4 data sets used (NKI; ref. 19; VDX; ref. 20; MAINZ; ref. 21; and TRANSBIG; ref. 22) have been previously described. Clinical endpoint used for the analyses was the first distant metastatic event. Tumor biopsy samples taken for gene expression profiling were collected at surgery. Gene expression data sets were retrieved from public databases or authors' Web sites. We used normalized data (log2 intensity in single-channel platforms or log2 ratio in dual-channel platforms). Hybridization probes were mapped to the Entrez GeneID as in Shi and colleagues, 23, using RefSeq and Entrez database version 2007.01.21. When multiple probes were mapped to the same GeneID, the one with the highest variance in a particular data set was selected to represent the GeneID. Each gene analyzed was then scaled such that quartiles 2.5% and 97.5% are equal to −1 and +1, respectively. This scaling is robust to outliers and hence allows combination of the microarray data from each data set. This meta-analytic technique has been previously described (24–26). Univariate Cox regression analysis was used to determine prognostic significance with clinical outcome of the genes as a continuous measurement and were calculated stratified by data set.
The second cohort of breast cancer patients was treated with anthracycline chemotherapy to determine the association of immune genes with clinical response to doxorubicin. These women received epirubicin as single-agent chemotherapy (100 mg/m2) for 4 cycles prior to surgery in the setting of a neoadjuvant clinical trial (clinical trials.gov NCT00162812; ref. 27). Epirubicin is an anthracycline chemotherapy favored in clinical practice due to its better toxicity profile than doxorubicin. Tumor biopsy samples were taken using a core biopsy 14- to 16-G needle prior to chemotherapy. Clinical endpoint used was pathologic complete response (pCR) documented at surgery, which is an accepted surrogate for disease-free and overall survival in breast cancer. Genes were correlated with pCR as a continuous variable (i.e., to determine whether higher expression correlated with a higher chance of obtained pCR), using a receiver operating characteristic (ROC), and measured using an area under the curve (AUC). Microarray analysis was done using Affymetrix GeneChips.
Statistical analysis
Mouse.
Statistical analyses were done using GraphPad Prism software. Significant differences between groups were assessed by a 2-tailed t test or Mann–Whitney U test, as indicated. Values of P < 0.05 were considered significant.
Human.
Statistical analysis using gene expression data was done with R version 2.5.1 and BioConductor version 1.8. R code used is available as “genefu” from the comprehensive R archive network. The remaining statistical analyses were conducted on SPSS 18.0 (SPSS, Inc.). Results were not corrected for multiple testing, as the analyses were considered hypothesis generating.
Results
The efficacy of doxorubicin therapy in established s.c. tumors requires CD8+ cells and IFN-γ
Both CD8 T cells and IFN-γ are important in immunosurveillance of a range of malignancies (28–31). More recently, adaptive immunity and IFN-γ have been implicated in the immunogenic effects of chemotherapy (10, 32), but CD8+ T cells have not been previously directly shown to contribute to the antitumor activity of doxorubicin. Despite the fact that anthracyclines are commonly used in the clinical management of human breast cancer, very little data concerning the mechanism of action of doxorubicin against mammary carcinomas has been obtained in experimental models or from clinical samples. We assessed the requirement for CD8+ T cells and IFN-γ in the effectiveness of doxorubicin chemotherapy treatment against a variety of established breast cancers and fibrosarcomas. In WT mice, a localized (i.t.) treatment of doxorubicin was sufficient to suppress s.c. growth of AT3, H2N100, and EO771 mammary tumors and MCA205 fibrosarcomas (Fig. 1A–D). Depletion of CD8α+ cells or neutralization of IFN-γ with mAb administration immediately prior to doxorubicin treatment severely abrogated the antitumor effect of the drug in the established tumor setting with significant loss of tumor growth suppression (Fig. 1A–D). Abrogation of doxorubicin efficacy was also observed in IFN-γ−/− mice (Fig. 1C). To confirm that the loss of therapeutic effect of doxorubicin following anti-CD8α mAb treatment was specifically due to depletion of CD8+ T cells, we assessed an additional group of mice receiving anti-CD8β mAb prior to doxorubicin treatment of established MCA2 fibrosarcoma tumors. Depletion of CD8 T cells alone was sufficient to abrogate the antitumor effect of doxorubicin (Fig. 1E). In addition, mice challenged with MCA2 tumors were treated with i.v. delivery of doxorubicin to establish that systemic administration of doxorubicin was comparable with localized treatment at inducing CD8 T cell and IFN-γ–mediated antitumor activity. For the first time, these studies validate across a series of experimental tumors the critical role of CD8+ T cells and IFN-γ in vivo in the mechanism of antitumor activity of doxorubicin.
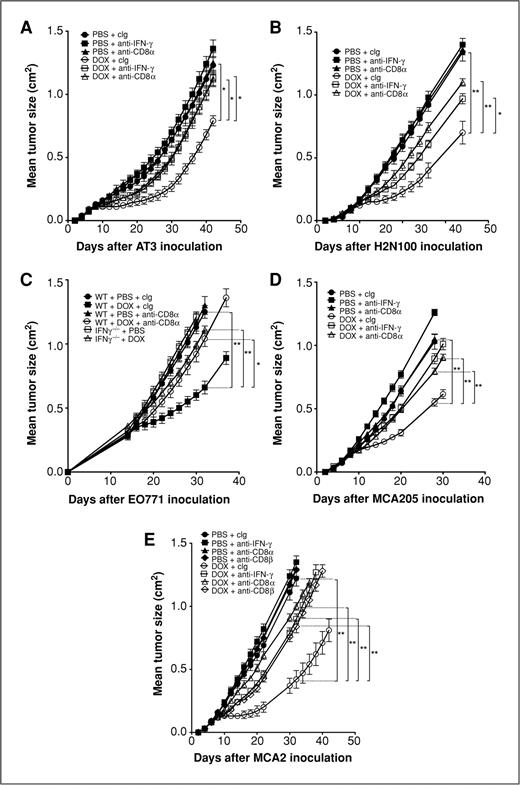
Doxorubicin therapy requires CD8+ T cells and IFN-γ. Groups of 5 syngeneic WT or gene-targeted mice as indicated were injected s.c. with 5 × 105 AT3 mammary adenocarcinoma cells (A), 5 × 105 H2N100 mammary adenocarcinoma cells (B), 5 × 105 EO771 mammary adenocarcinoma cells (C), 8 × 105 MCA205 fibrosarcoma cells (D), or 1 × 105 MCA2 fibrosarcoma cells (E). Mice then received either i.t. PBS or doxorubicin (50 μL, 2 mmol/L) on day 7 (A), day 8 (B), day 14 (C), and day 7 (D) after tumor inoculation or PBS or doxorubicin (2 mg/kg) i.v. on days 7 and 14 (E) after tumor inoculation. Some mice received control Ig (cIg), anti-CD8α, anti-CD8β, or anti-IFN-γ (100 μg i.p.) on day −1, 0, and weekly thereafter relative to initial PBS/doxorubicin treatment. Tumor size was measured as indicated. Data show means ± standard errors of 5 mice per group. Data for AT3, H2N100, EO771 (A–C), and MCA2 (E) are representative of 2 independent experiments. Statistical analyses were conducted at the time point indicated on the figure using Mann–Whitney test (*, P < 0.05; **, P < 0.01).
Doxorubicin therapy requires CD8+ T cells and IFN-γ. Groups of 5 syngeneic WT or gene-targeted mice as indicated were injected s.c. with 5 × 105 AT3 mammary adenocarcinoma cells (A), 5 × 105 H2N100 mammary adenocarcinoma cells (B), 5 × 105 EO771 mammary adenocarcinoma cells (C), 8 × 105 MCA205 fibrosarcoma cells (D), or 1 × 105 MCA2 fibrosarcoma cells (E). Mice then received either i.t. PBS or doxorubicin (50 μL, 2 mmol/L) on day 7 (A), day 8 (B), day 14 (C), and day 7 (D) after tumor inoculation or PBS or doxorubicin (2 mg/kg) i.v. on days 7 and 14 (E) after tumor inoculation. Some mice received control Ig (cIg), anti-CD8α, anti-CD8β, or anti-IFN-γ (100 μg i.p.) on day −1, 0, and weekly thereafter relative to initial PBS/doxorubicin treatment. Tumor size was measured as indicated. Data show means ± standard errors of 5 mice per group. Data for AT3, H2N100, EO771 (A–C), and MCA2 (E) are representative of 2 independent experiments. Statistical analyses were conducted at the time point indicated on the figure using Mann–Whitney test (*, P < 0.05; **, P < 0.01).
Carcinogen-induced tumors respond to doxorubicin in a CD8 T-cell- and IFN-γ–dependent manner
The activity and mechanism of action of most cancer therapies, including doxorubicin, have rarely if ever been assessed in mouse models of de novo tumorigenesis. This type of model is more practical in assessing any agent's action in the context of a developing and progressing tumor, as opposed to a transplant setting. Recently, we have employed established fibrosarcomas induced by MCA in mice as a therapeutic model for various immune targets (33, 34) and to determine the importance of host IL-1β in the mechanism of action of doxorubicin (10). To date, however, the importance of many other host immune elements in doxorubicin antitumor activity has not been examined in a mouse model in which the tumor has arisen de novo in the host. Depletion of CD8 T cells following establishment of palpable sarcomas (0.20–0.45 cm2) did not significantly alter the growth kinetics of the established tumor (Fig. 2, left; Supplementary Fig. S4). Doxorubicin treatment (i.t.) of established MCA sarcomas achieved modest tumor growth suppression in most mice, and 4 of 30 (13%) showed a significant period of retarded tumor growth but eventually succumbed (Fig. 2A; Supplementary Fig. S4). This variable response was consistent with the vast heterogeneity between individual MCA-induced fibrosarcomas. Importantly, the significant therapeutic effect of doxorubicin in inhibiting fibrosarcoma growth across the whole cohort of control mAb-treated mice (P < 0.0001; Supplementary Fig. S4) was abolished if mice received anti-CD8α mAb or anti-IFN-γ mAb during doxorubicin therapy (Fig. 2B and C; Supplementary Fig. S4), indicating that both CD8+ T cells and IFN-γ were critical for the antitumor effects of doxorubicin.

Tumors induced de novo by carcinogen respond to doxorubicin in a CD8+ T cell– and IFN-γ–dependent manner. Groups of 15 to 30 male BALB/c WT mice were injected s.c. on the flank with 400 μg MCA on day 0. When sarcomas had established (the second week of palpable tumor 0.20–0.45 cm2), BALB/c mice received either i.t. PBS (left) or doxorubicin (50 μL, 2 mmol/L; right) once a week for 2 weeks. Some mice received (A) control Ig (anti-agp3), (B) anti-CD8α, or (C) anti-IFN-γ (100–500 μg i.p.) on days −1, 0, and weekly thereafter for 6 weeks relative to initial PBS/doxorubicin treatment. Mice were then monitored for tumor development over 250 days and recorded as the growth curves (tumor size in cm2) of individual mice with tumor in each group.
Tumors induced de novo by carcinogen respond to doxorubicin in a CD8+ T cell– and IFN-γ–dependent manner. Groups of 15 to 30 male BALB/c WT mice were injected s.c. on the flank with 400 μg MCA on day 0. When sarcomas had established (the second week of palpable tumor 0.20–0.45 cm2), BALB/c mice received either i.t. PBS (left) or doxorubicin (50 μL, 2 mmol/L; right) once a week for 2 weeks. Some mice received (A) control Ig (anti-agp3), (B) anti-CD8α, or (C) anti-IFN-γ (100–500 μg i.p.) on days −1, 0, and weekly thereafter for 6 weeks relative to initial PBS/doxorubicin treatment. Mice were then monitored for tumor development over 250 days and recorded as the growth curves (tumor size in cm2) of individual mice with tumor in each group.
Doxorubicin treatment enhances antigen-specific CD8 T-cell proliferation in vivo
We next investigated in more detail the antigen-driven responses of CD8 T cells following chemotherapy in breast cancer. Immunocompetent B6 mice showed considerable resistance to challenge with s.c. AT3OVA tumors in comparison with AT3-empty vector (AT3EV) control tumors (Supplementary Fig. S1). Next, we adoptively transferred naive OVA-specific CD8 T cells (OT-I cells) 1 day after mice harboring established AT3OVA or AT3EV were treated with doxorubicin. Not surprisingly, the proportion of OT-I cells recovered from TDLN after 5 days was significantly higher in mice with AT3OVA tumors than in those inoculated with AT3EV tumors. More importantly, the proportion of OT-I cells in TDLN was enhanced following doxorubicin treatment, however, only in the context of cognate antigen recognition (AT3OVA setting; Fig. 3A). Furthermore, in vivo proliferation of OT-I cells in the presence of OVA, as measured by CFSE dilution, was significantly increased after doxorubicin treatment (Fig. 3B). Overall, localized doxorubicin treatment of established tumors enhances the accumulation and proliferation of CD8 T cells responding to cognate tumor antigen in secondary lymphoid organs.

Doxorubicin treatment enhances CD8 T-cell proliferation to cognate tumor antigen. C57BL/6 mice were inoculated s.c. with either AT3EV (1 × 106) or AT3OVA (1 × 106) tumors. After 30 days, groups of mice (n = 5) were i.t. treated with doxorubicin (1 mmol/L) or PBS, and the following day all mice received i.v. transfer of purified, CFSE-labeled CD8+ OT-I T cells (2 × 106). Five days after OT-I cell transfer, TDLNs were excised and fluorescence-activated cell-sorting (FACS) analyses on KbOVA-tetramer–reactive CD8 T cells were conducted. A, representative FACS plots of CD8+KbOVA-tetramer+ (CD8+KbOVAtet+) cells in the DLN of mice harboring AT3EV or AT3OVA tumors. Data shown are the percentage of OT-I cells within the lymphocyte population in individual mice with the indicated tumors and treatment. *, P < 0.02, t test; ns, not significant. B, FACS histograms showing representative CFSE dilution within the gated CD8+KbOVA-tetramer+ transferred OT-I cell population in DLN of mice harboring AT3EV or AT3OVA tumors after prior treatment with PBS (top) or doxorubicin (bottom). OT-I cell proliferation indices with means are shown in the underlying graph. ***, P < 0.0001, t test; ns, not significant. All graphs are representative of 3 independent experiments.
Doxorubicin treatment enhances CD8 T-cell proliferation to cognate tumor antigen. C57BL/6 mice were inoculated s.c. with either AT3EV (1 × 106) or AT3OVA (1 × 106) tumors. After 30 days, groups of mice (n = 5) were i.t. treated with doxorubicin (1 mmol/L) or PBS, and the following day all mice received i.v. transfer of purified, CFSE-labeled CD8+ OT-I T cells (2 × 106). Five days after OT-I cell transfer, TDLNs were excised and fluorescence-activated cell-sorting (FACS) analyses on KbOVA-tetramer–reactive CD8 T cells were conducted. A, representative FACS plots of CD8+KbOVA-tetramer+ (CD8+KbOVAtet+) cells in the DLN of mice harboring AT3EV or AT3OVA tumors. Data shown are the percentage of OT-I cells within the lymphocyte population in individual mice with the indicated tumors and treatment. *, P < 0.02, t test; ns, not significant. B, FACS histograms showing representative CFSE dilution within the gated CD8+KbOVA-tetramer+ transferred OT-I cell population in DLN of mice harboring AT3EV or AT3OVA tumors after prior treatment with PBS (top) or doxorubicin (bottom). OT-I cell proliferation indices with means are shown in the underlying graph. ***, P < 0.0001, t test; ns, not significant. All graphs are representative of 3 independent experiments.
Tumor-infiltrating CD8 T cells and IFN-γ production are enhanced by doxorubicin treatment
We harvested AT3OVA tumors from mice that had received i.t. doxorubicin or PBS treatment and naive OT-I T cell adoptive transfer to determine whether localized doxorubicin administration increased the proportion and function of CD8 TILs. A significant increase in the proportion of total CD8 T cells amongst AT3OVA TILs was observed following doxorubicin treatment (Fig. 4A), and a similar increase was observed for infiltrating Ag-specific OT-I cells (Fig. 4B). Doxorubicin treatment also induced Ag-specific IFN-γ production from CD8 TILs, observed after in vitro restimulation with cognate antigen (Fig. 4C). No cytokine was detected from CD8 TILs in the absence of restimulation (data not shown). These experiments are amongst the first to directly isolate and show the increase in the proportion of tumor-localized and functional CD8+ T cells following treatment with doxorubicin.

Local doxorubicin treatment enhances numbers of CD8 TILs and IFN-γ production. C57BL/6 mice were inoculated s.c. with AT3OVA (1 × 106) tumors. After 42 days, groups of mice (n = 5) were treated i.t. with doxorubicin (1 mmol/L) or PBS, and the following day received i.v. transfer of purified, CD8+ OT-I T cells (2 × 106). Seven days after OT-I cell transfer, tumors were excised and FACS analyses on CD8+ TILs were conducted. CD8 T cells (A) and KbOVA-tetramer–reactive CD8 T cells (B) as a percentage of total TILs for individual mice treated with PBS or doxorubicin are shown. C, antigen-specific IFN-γ cytokine production from CD8 TILs measured by intracellular staining after overnight in vitro stimulation of AT3OVA tumor cell suspensions with OVA peptide + IL-2. **, P < 0.01; ***, P < 0.0001, t test. Data are representative of 3 independent experiments.
Local doxorubicin treatment enhances numbers of CD8 TILs and IFN-γ production. C57BL/6 mice were inoculated s.c. with AT3OVA (1 × 106) tumors. After 42 days, groups of mice (n = 5) were treated i.t. with doxorubicin (1 mmol/L) or PBS, and the following day received i.v. transfer of purified, CD8+ OT-I T cells (2 × 106). Seven days after OT-I cell transfer, tumors were excised and FACS analyses on CD8+ TILs were conducted. CD8 T cells (A) and KbOVA-tetramer–reactive CD8 T cells (B) as a percentage of total TILs for individual mice treated with PBS or doxorubicin are shown. C, antigen-specific IFN-γ cytokine production from CD8 TILs measured by intracellular staining after overnight in vitro stimulation of AT3OVA tumor cell suspensions with OVA peptide + IL-2. **, P < 0.01; ***, P < 0.0001, t test. Data are representative of 3 independent experiments.
Doxorubicin treatment requires IL-1β and IL-17 but not IL-23p19
We have recently shown that IL-1β production is pivotal for a robust antitumor response to chemotherapy, by facilitating the priming of tumor-specific CD8 T cells (10). We now have extended these findings in a range of mammary adenocarcinomas to show that IL-1β/IL-1R signaling is also critical to the therapeutic outcome of doxorubicin treatment (Fig. 5). Because IL-1β is known to regulate IL-17 responses (35–37), we utilized both IL-17A−/− mice and an anti-IL-17Rα mAb to also show a critical role for IL-17 in doxorubicin treatment of transplantable mammary tumors (Fig. 5). In concert with the mammary carcinoma data, a critical role for IL-1β and IL-17A in doxorubicin antitumor activity was also illustrated using fibrosarcomas generated de novo by MCA (Fig. 6B and D; Supplementary Fig. S4). Interestingly, the absence of IL-1 or IL-17 signaling had little or no direct effect on the growth of untreated established tumors. In contrast IL-23, another immunoregulatory cytokine known to cooperate with IL-1β in the regulation of IL-17 responses (35–37), was dispensable in these models, as no alteration in doxorubicin efficacy was observed in IL-23p19−/− mice or by neutralizing IL-23 in WT mice using an anti-IL-23 mAb (Fig. 6C; Supplementary Figs. S2 and S4). In addition, by comparison, the antitumor efficacy of doxorubicin in transplanted tumors was partially reduced in the absence of the IL-12p35 subunit (Supplementary Fig. S2).
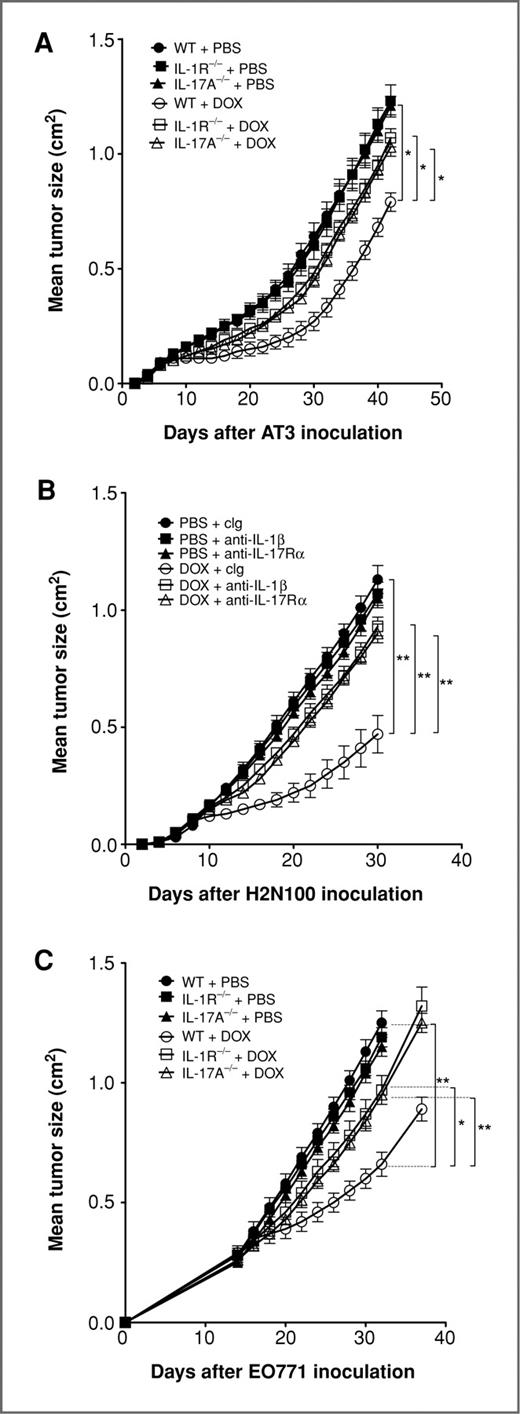
Doxorubicin therapy requires IL-1R and IL-17A. Groups of 5 syngeneic WT or gene-targeted mice as indicated were injected s.c. with 5 × 105 AT3 mammary adenocarcinoma cells (A), 5 × 105 H2N100 mammary adenocarcinoma cells (B), or 5 × 105 EO771 mammary adenocarcinoma cells (C). Mice then received either i.t. PBS or doxorubicin (50 μL, 2 mmol/L) on day 7 (A), day 8 (B), and day 14 (C) after tumor inoculation. Some mice received control Ig (cIg), anti-IL-1β, or anti-IL-17Rα (500 μg i.p.) on days 7, 8, 15, and 22 relative to tumor inoculation. Tumor size was measured as indicated. Data show means ± standard errors of 5 mice per group, representative of 2 independent experiments. Statistical analyses were conducted at the time point indicated on the figure by using Mann–Whitney test (*, P < 0.05; **, P < 0.01).
Doxorubicin therapy requires IL-1R and IL-17A. Groups of 5 syngeneic WT or gene-targeted mice as indicated were injected s.c. with 5 × 105 AT3 mammary adenocarcinoma cells (A), 5 × 105 H2N100 mammary adenocarcinoma cells (B), or 5 × 105 EO771 mammary adenocarcinoma cells (C). Mice then received either i.t. PBS or doxorubicin (50 μL, 2 mmol/L) on day 7 (A), day 8 (B), and day 14 (C) after tumor inoculation. Some mice received control Ig (cIg), anti-IL-1β, or anti-IL-17Rα (500 μg i.p.) on days 7, 8, 15, and 22 relative to tumor inoculation. Tumor size was measured as indicated. Data show means ± standard errors of 5 mice per group, representative of 2 independent experiments. Statistical analyses were conducted at the time point indicated on the figure by using Mann–Whitney test (*, P < 0.05; **, P < 0.01).
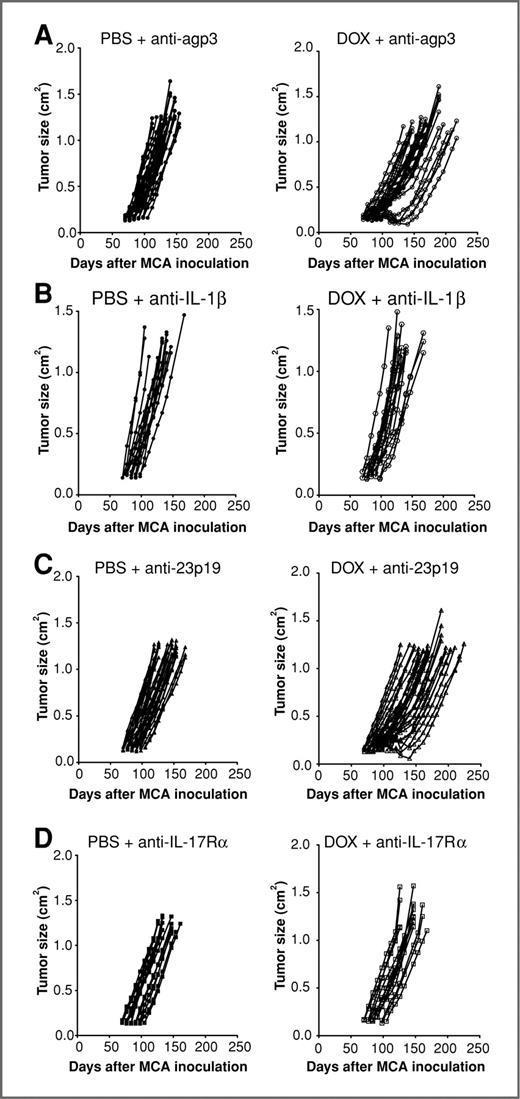
Tumors induced de novo by carcinogen respond to doxorubicin in an IL-1β– and IL-17–dependent manner. Groups of 15 to 30 male BALB/c WT mice were injected s.c. on the flank with 400 μg MCA on day 0. When sarcomas had established (the second week of palpable tumor 0.20–0.45 cm2) BALB/c mice received either i.t. PBS (left) or doxorubicin (50 μL, 2 mmol/L; right) once a week for 2 weeks. Some mice received (A) control Ig (anti-agp3), (B) anti-IL-1β, (C) anti-IL-23p19, or (D) anti-IL- 17Rα (100–500 μg i.p.) on days −1, 0, and weekly thereafter for 6 weeks relative to initial PBS/doxorubicin treatment. Mice were then monitored for tumor development over 250 days and recorded as the growth curves (tumor size in cm2) of individual mice with tumor in each group. Control Ig graphs in (A) are reproduced from Fig. 2A.
Tumors induced de novo by carcinogen respond to doxorubicin in an IL-1β– and IL-17–dependent manner. Groups of 15 to 30 male BALB/c WT mice were injected s.c. on the flank with 400 μg MCA on day 0. When sarcomas had established (the second week of palpable tumor 0.20–0.45 cm2) BALB/c mice received either i.t. PBS (left) or doxorubicin (50 μL, 2 mmol/L; right) once a week for 2 weeks. Some mice received (A) control Ig (anti-agp3), (B) anti-IL-1β, (C) anti-IL-23p19, or (D) anti-IL- 17Rα (100–500 μg i.p.) on days −1, 0, and weekly thereafter for 6 weeks relative to initial PBS/doxorubicin treatment. Mice were then monitored for tumor development over 250 days and recorded as the growth curves (tumor size in cm2) of individual mice with tumor in each group. Control Ig graphs in (A) are reproduced from Fig. 2A.
Doxorubicin therapy requires γδ T cells but not type I NKT cells
Both NKT cells and γδ T cells have been implicated in innate immunosurveillance of tumors (38, 39) and are also a potent early source of IL-17 in response to IL-1β cytokine stimulation (35, 36). We assessed the requirement for these innate lymphocytes in doxorubicin therapy of established AT3 mammary tumors and MCA205 fibrosarcomas (Supplementary Fig. S3). The absence of type I Jα18+ NKT cells in TCRJα18−/− mice did not perturb the antitumor effects of doxorubicin; however, deficiency of γδ T cells in TCRδ−/− mice resulted in significant inhibition of doxorubicin activity in both AT3 and MCA205 tumors. The loss of NKT cells or γδ T cells did not alter the outgrowth of these tumors in the absence of therapy (Supplementary Fig. S3).
Clinical relevance using gene expression data from human breast cancer patients
We went on to explore whether these genes were important for therapeutic efficacy of anthracycline treatment in breast cancer patients, using publicly available gene expression data sets. The first cohort consisted of patients diagnosed at multiple different hospitals who had their tumor biopsy samples taken at surgery. The samples examined here are representative of the global breast cancer population, as there was a mixture of different tumor sizes, nodal status, and estrogen receptor (ER) expression (Supplementary Table S1). As these women received no systemic treatment, the true prognostic effects of these genes could be examined without treatment confounders. Therefore, we could examine the prognostic significance of CD8A, CD8B, IFN-γ, IL1B, IL17A, and IL23p19 genes independent of anthracycline therapy in patients who had received no systemic treatment (refs. 19–22; Table 1). As observed, only high IFN-γ levels were associated with a good prognosis in these women, that is, high levels were associated with fewer long-term breast cancer relapses.
Association of genes with clinical outcome in breast cancer patients treated with and without anthracycline chemotherapya
| Genes | Cohort 1b HR (95% CI) | Cohort 2c AUC (95% CI) |
|---|---|---|
| IFN-γ | 0.8 (0.65–0.95), P = 0.031 | 0.69 (0.56–0.81), P = 0.016 |
| CD8A | 0.82 (0.5–1.3), P = 0.4 | 0.72 (0.59–0.84), P = 0.005 |
| CD8B | 0.87 (0.59–1.3), P = 0.21 | 0.65 (0.52–0.78), P = 0.049 |
| IL17A | 0.82 (0.67–1.1), P = 0.051 | 0.5, P = 0.44 |
| IL1B | 0.87 (0.7–1.06), P = 0.18 | 0.39, P = 0.19 |
| IL23 | 0.88 (0.75–1.03), P = 0.13 | 0.56, P = 0.42 |
| Genes | Cohort 1b HR (95% CI) | Cohort 2c AUC (95% CI) |
|---|---|---|
| IFN-γ | 0.8 (0.65–0.95), P = 0.031 | 0.69 (0.56–0.81), P = 0.016 |
| CD8A | 0.82 (0.5–1.3), P = 0.4 | 0.72 (0.59–0.84), P = 0.005 |
| CD8B | 0.87 (0.59–1.3), P = 0.21 | 0.65 (0.52–0.78), P = 0.049 |
| IL17A | 0.82 (0.67–1.1), P = 0.051 | 0.5, P = 0.44 |
| IL1B | 0.87 (0.7–1.06), P = 0.18 | 0.39, P = 0.19 |
| IL23 | 0.88 (0.75–1.03), P = 0.13 | 0.56, P = 0.42 |
Abbreviation: pCR, pathologic complete response.
aAssociation of genes with clinical outcome in breast cancer patients treated with and without anthracycline chemotherapy. Cohort 1 consisted of 1,062 patients from 4 gene expression data sets who received no systemic treatment after their surgery to determine the clinical outcome association independent of anthracycline. Seventy percent of tumors were ER-positive, 85% were node negative at diagnosis. Univariate HRs presented are stratified by data set. Genes are correlated as continuous variables. Here, increasing expression of IFN-γ is correlated with a good prognosis or a longer time free from distant metastases. Cohort 2 consisted of 114 patients treated in a neoadjuvant trial of the anthracycline epirubicin given as a sole therapy prior to surgery. All patients were ER-negative with 86% of tumors greater than 2 cm and 54% with positive lymph nodes at study entry. Biopsies were taken prior to therapy. Here, increasing expression of IFN-γ, CD8A, and CD8B is associated with a higher chance of a complete response from chemotherapy at surgery. In this second cohort, a pCR was strongly correlated with a better survival from breast cancer. (Full clinical information for cohorts 1 and 2 can be found in the Supplementary Table S1.)
bEndpoint: distant metastases; number of patients = 1,062; treatment = none.
cEndpoint: pCR (complete disappearance of invasive tumor at surgery after 4 cycles of epirubicin chemotherapy given at 100 mg/m2); number of patients = 114; treatment = epirubicin monotherapy.
We then went on to examine these genes in the context of treatment with anthracycline chemotherapy. The second cohort consisted of women enrolled into a breast cancer clinical trial specifically designed to investigate biomarkers to anthracycline chemotherapy. As a consequence, all of the women in this study were negative for expression of ER (as this is the more chemoresponsive breast cancer population) and all received single-agent epirubicin, an anthracycline-type chemotherapy commonly used in clinical practice. Tumor biopsy samples were taken with core biopsy needles and were subject to gene expression profiling prior to chemotherapy (27). In these breast cancer patients, increasing levels of IFN-γ, as well as CD8α and CD8β were associated with a better response to therapy, as measured by the amount of tumor left at surgical resection after 4 cycles of therapy. Notably, the complete disappearance of invasive tumor after neoadjuvant chemotherapy was associated with an excellent survival from breast cancer and is an accepted surrogate for long-term survival from breast cancer (40). There was no significance for the other genes in these cohorts. Overall, these data support further the role of IFN-γ, CD8α, and CD8β in the clinical outcomes in breast cancer patients who have received anthracycline-type chemotherapy.
Discussion
Anthracyclines are front-line chemotherapeutic agents in the treatment of breast cancers and sarcomas. In this study, we have provided an extensive amount of new data exploring the mechanism of action of doxorubicin in a series of transplantable mammary carcinomas and fibrosarcomas generated de novo by MCA. The new data in AT3, H2N100, and EO771 mammary tumor models showed the critical role of host CD8+ T cells and IFN-γ, additionally supported by novel human clinical data in 2 different cohorts of women with breast cancer. Our study is the first to describe the specific importance of CD8+ T cells in vivo, as opposed to broadly assessing adaptive immunity by employing RAG-deficient or nude mice that are deficient in many immune components including all T-cell subsets and B cells. Furthermore, analysis included CD8α- and CD8β-specific depletions to specifically define the importance of CD8+ T cells. Doxorubicin treatment enhanced the proliferation of CD8 T cells in the tumor DLN in a cognate antigen-specific manner. Moreover, localized doxorubicin treatment increased the proportions of CD8 T cells infiltrating the tumor and enhanced tumor antigen–specific IFN-γ production from these CD8 TILs. Combined, these data are amongst the strongest to indicate that tumor cell death associated with doxorubicin treatment enhances the generation and functional activation of tumor-reactive CD8 T cells that are required for the antitumor activity of doxorubicin.
We also completed the most extensive characterization of mechanism of action of doxorubicin in tumors established de novo by carcinogen, including a comparative evaluation of the role of CD8+ T cells, IFN-γ, IL-1β, IL-17A, and IL-23. These types of established tumors generated de novo are arguably more relevant than any short-term transplanted tumors that have been used to previously establish the principles that chemotherapy may be immunogenic. Our study has clearly shown that CD8+ T cells, IFN-γ, IL-1β, and IL-17A but not IL-23, were critical in the antitumor activity of doxorubicin. We further illustrated the key role of IL-1β and IL-17A, but not IL-23, in doxorubicin control of a series of mammary carcinomas. Our data extend our recently published data that illustrated the importance of IL-17A in the antitumor activity of doxorubicin against transplanted MCA205 tumors and tracked the generation of IL-17–producing γδ+ T cells following doxorubicin treatment (41). We confirmed the role of γδ+ T cells, but not type I NKT cells, in the mechanism of action of doxorubicin, using transplantable mammary carcinomas.
In human breast cancer patients, although IFN-γ levels seemed relevant for patient clinical outcome independent of anthracycline, the importance of both CD8 T cells and IFN-γ in therapeutic efficacy was supported by the observation that increasing levels of CD8α, CD8β, and IFN-γ gene expression correlated with a better response to anthracycline chemotherapy. Notably, here the tumor biopsy samples were taken prior to chemotherapy, supporting the concept that an intact immune system aids therapeutic response to anthracyclines. However, it would be most interesting to also see the relative changes in gene expression before and shortly after administration of therapy. Although the analyses in human breast cancer patients presented here are limited by the use of retrospective data sets, nonrandomized treatment cohorts, and patients treated with epirubicin rather than doxorubicin, which is a different type of anthracycline chemotherapy, it supports our laboratory observations and a recent study (42) of the clinical relevance of antitumor immunity for better clinical outcomes from anthracycline therapy in breast cancer.
It is likely that the context in which IL-17A is produced, particularly the constituents of the tumor microenvironment, will determine the outcome of IL-17 secretion. IL-17 production, particularly from innate T lymphocytes such as NKT cells and γδ T cells, is dependent on IL-1R signaling (36, 43) and has been shown to boost CD8 T-cell IFN-γ responses (44). Given the requirement for IL-1β and CD8 T cells in the antitumor immune response following doxorubicin treatment, we assessed the role for IL-17 as an intermediary molecule in this pathway. IL-17A or IL-17Rα blockade resulted in loss of doxorubicin efficacy, very similar to that of IL-1β/IL-1R blockade, indicating that IL-17 is probably a major downstream response cytokine to IL-1β secretion. Interestingly, IL-23, itself a modulator of antitumor immunity (45–48), but also an important cofactor along with IL-1β in the stimulation of IL-17 production from memory T cells (37), NKT cells (35), and γδ T cells (36), was dispensable in doxorubicin treatment of breast cancers and fibrosarcomas, with no alteration in therapeutic effect following IL-23 blockade. Notably, gene expression of IL1B, IL17A, or IL23 was not associated with clinical outcomes to doxorubicin treatment in human breast cancer patients, but perhaps this was not surprising given that the tumor biopsy samples were taken prior to doxorubicin treatment, and these cytokines were likely to be induced following treatment. In a parallel study, we have very recently shown that IL-17 from γδ T cells is critical for the efficacy of chemotherapy and preceded the activation and polarization of CD8 T cells for IFN-γ production (41). Also, γδ T cells that lacked IL-1R lost the capacity to amplify the action of chemotherapy, confirming the causal relationship with IL-1β production from adenomatous polyposis coli (APC) causing IL-17 production from γδ T cells and resulting in IFN-γ production from tumor-specific CD8 T cells. The current study provides important additional data in mammary tumor transplant and de novo fibrosarcoma models to support a role for IL-17A in doxorubicin mechanism of action. In doing so, we also show the relevance of γδ+ T cells but not of type I NKT cells.
Overall, our in vivo mouse data and human genetic analysis show the role of host CD8 T cells and IFN-γ in doxorubicin mechanism of action. The study also strongly supports our most recent work on the role of IL-17A in the mechanism of action of chemotherapy (41), particularly with respect to novel data concerning doxorubicin and mammary carcinoma. In addition, it extensively characterizes mechanism of action of doxorubicin in tumors established de novo by carcinogen, including comparative evaluation of the role of CD8+ T cells, IFN-γ, IL-1β, IL-17, and IL-23. These types of established tumors are arguably more relevant than any short-term transplanted tumors that have been used to previously establish the principle that chemotherapy may be immunogenic. The work corroborates recent studies showing that numbers of TILs in breast cancer are a predictor of response to neoadjuvant chemotherapy (49, 50). Similar outcomes in mouse sarcomas suggest more widespread applicability of these findings to other tumor cell and tissue types. Future work should address how to facilitate the potential development of sustained tumor-specific, adaptive immunity by anthracycline chemotherapy. This includes the generation of long-lived memory T cells and identification of the patients who are best candidates for such immunotherapeutic approaches, as enhancing tumor immunosurveillance by chemotherapy may allow extended immune-mediated control of the malignancy beyond the treatment period of the drug.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Qerime Mundrea and Ben Venville for maintenance of the mice, Nicole Haynes for generation of the AT3OVA cell line, Jennifer Towne (AMGEN, Inc.) for providing the anti-agp3, anti-IL-23, and anti-IL-17Rα mAbs, and Benjamin Haibe-Kains for bioinformatics assistance.
Grant Support
This work was supported by The Victorian Cancer Agency, The Victorian Breast Cancer Consortium, and the Susan G. Komen Breast Cancer Foundation. S.R. Mattarollo was supported by a Balzan Foundation Fellowship. S. Loi was supported by a National Health & Medical Research Council (NH&MRC) clinical fellowship. Y. Ma was supported by China Scholarship Council. L. Zitvogel was supported by LIGUE labellisee, INFLACARE FP7 EU grant, INCa, Fondation pour la Recherche Medicale, and Fondation de France. M.J. Smyth received support from an NH&MRC Australia Fellowship.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.