


Abstract
Histone deacetylase inhibitors (HDACi) are anticancer agents that induce hyperacetylation of histones, resulting in chromatin remodeling and transcriptional changes. In addition, nonhistone proteins, such as the chaperone protein Hsp90, are functionally regulated through hyperacetylation mediated by HDACis. Histone acetylation is thought to be primarily regulated by HDACs 1, 2, and 3, whereas the acetylation of Hsp90 has been proposed to be specifically regulated through HDAC6. We compared the molecular and biologic effects induced by an HDACi with broad HDAC specificity (vorinostat) with agents that predominantly inhibited selected class I HDACs (MRLB-223 and romidepsin). MRLB-223, a potent inhibitor of HDACs 1 and 2, killed tumor cells using the same apoptotic pathways as the HDAC 1, 2, 3, 6, and 8 inhibitor vorinostat. However, vorinostat induced histone hyperacetylation and killed tumor cells more rapidly than MRLB-223 and had greater therapeutic efficacy in vivo. FDCP-1 cells dependent on the Hsp90 client protein Bcr-Abl for survival, were killed by all HDACis tested, concomitant with caspase-dependent degradation of Bcr-Abl. These studies provide evidence that inhibition of HDAC6 and degradation of Bcr-Abl following hyperacetylation of Hsp90 is likely not a major mechanism of action of HDACis as had been previously posited. Mol Cancer Ther; 12(12); 2709–21. ©2013 AACR.
Introduction
Altered acetylation of the N-terminal regions of histone proteins through the reciprocal enzymatic activities of histone acetyltransferases (HAT) and histone deacetylases (HDAC) can facilitate the remodeling of chromatin and mediate changes in gene expression (1–3). In addition, HATs and HDACs can regulate the acetylation of numerous nonhistone proteins to affect their function, stability, or subcellular localization (4–8). Mammalian class I HDACs (1–3, 8) are predominantly expressed in the nucleus, and are the major mediators of histone deacetylation, whereas the Class IIa HDACs (4, 5, 7, 9) and Class IIb HDACs (6 and 10) either shuttle between the nucleus and the cytoplasm or are predominantly expressed in the cytoplasm and deacetylate nonhistone proteins (3, 9).
A large number HDAC inhibitors (HDACi) have been developed that can suppress HDAC enzymatic activity and mediate protein acetylation (1, 2). HDACis have pleiotropic anticancer activities by their inhibition of tumor cell proliferation and survival, through their effects on tumor angiogenesis, and their ability to modulate antitumor immunity (3, 10). Two HDACis, vorinostat and romidepsin, have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of hematologic malignancies (11).
Using genetic mouse models of cancer, we and others have demonstrated a direct link between HDACi-mediated apoptosis and therapeutic efficacy (12, 13), indicating that direct tumor cell killing by these agents plays a fundamental role in mediating antitumor responses in vivo. Importantly, the combined effects of HDACi on histone and nonhistone proteins may be important for the therapeutic activities of these compounds (3, 14). For example, HDAC6-mediated deacetylation of Hsp90 can regulate its protein chaperone activity. Accordingly, degradation of client oncoproteins such as ErbB2 and Bcr-Abl through HDACi-mediated hyperacetylation of Hsp90 has been proposed to be an important functional effector mechanism of HDACis (3).
Most HDACis have broad inhibitory activity against class I and II HDACs (15); however, isoform-selective HDACis have been developed (16). It is presently not clear if selectively targeting a single HDAC will provide a significant advantage over broad-acting HDACis in terms of increased efficacy and/or reduced toxicity. Indeed, treatment of tumor cells with the HDAC6-selective inhibitor tubacin did not affect tumor cell growth or survival (17). Moreover, gene deletion and knockdown studies indicate that inhibition of a single HDAC may not provide pronounced antitumor effects. For example, siRNA-mediated knockdown of HDAC1, 2, 3, or 6 failed to affect tumor cell growth or survival (18). In a study using Ras-mediated tumors derived from conditional Hdac1, 2, 3, and 8 knockout mice, combined knockout of Hdac1 and Hdac2, but not of any of the Hdacs1, 2, 3, or 8 alone, was sufficient to inhibit tumor growth in vitro and in vivo (19). However, although there have been some reports of single HDAC inhibition affecting tumor cell growth as transient knockdown of HDAC1 or HDAC3 suppressed HeLa cell proliferation (20), another group showed that transient knockdown of HDAC1, 2, or 3 decreased the rate of proliferation of colon carcinoma cell lines (21). Although these studies hint that in certain cellular contexts inactivation of a single HDAC may have an antitumor effect, most studies using genetic or pharmacologic approaches to inhibit HDAC function or delete HDAC expression indicate that targeting at least two HDACs may be necessary to mediate potent antitumor responses.
Hsp90 is an abundant cellular chaperone whose overexpression in tumor cells correlates with poor prognosis and drug resistance (22). As part of a multiprotein complex that includes Hsp70, Hsp90 binds a diverse array of client proteins including key oncogenic and antiapoptotic proteins, preventing their ubiquitination and proteosomal degradation (22, 23). Hsp90 is deacetylated by HDAC6 (24), and HDACis capable of inhibiting HDAC6 induce the hyperacetylation of Hsp90, resulting in proteosomal degradation of Hsp90 client proteins ErbB1, ErbB2/Her2, Akt, c-Raf, Bcr-Abl, and Flt-3 (25–28). Although it has been proposed that hyperacetylation of Hsp90 and subsequent degradation of oncogenic client proteins may be a major mediator of the antitumor effects of HDACis (3), there is counterevidence to refute this. In addition to the lack of antitumor effects of the HDAC6 inhibitor tubacin, siRNA-mediated knockdown of HDAC6 resulted in hyperacetylation and loss of chaperone function of Hsp90, but had little or no effect on tumor cell growth and survival (29) Moreover, a significant number of HDACis that are potent mediators of tumor cell death are very weak inhibitors of HDAC6 (3, 15).
In this study, we used the recently developed HDACi MRLB-223, which preferentially inhibits recombinant HDAC1 and 2 at low nanomolar concentrations, to determine if selectively targeting certain class I HDACs would result in similar induction of apoptosis and therapeutic efficacy as seen using the broad-spectrum HDACi vorinostat. We show that although MRLB-223 can induce apoptosis in B-cell lymphomas derived from Eμ-myc transgenic mice and can provide a therapeutic benefit to mice bearing Eμ-myc lymphomas, the kinetics of histone hyperacetylation and tumor cell apoptosis induced by this compound is delayed, and its potency in vivo is inferior to that observed using vorinostat. In a different experimental setting using a panel of primary breast cancer cells, we found that there was incomplete overlap in sensitivity to vorinostat and MRLB-223, suggesting that molecular determinants of sensitivity to HDACis with different specificity exist within distinct tumor samples. Interestingly, MRLB-223 was capable of killing an interleukin 3 (IL-3)–dependent FDCP-1 mouse myeloid cell line that was engineered to grow independently of IL-3 through forced expression of the Hsp90 client protein Bcr-Abl. Moreover, both MRLB-223 and romidepsin, HDACis that potently target class I HDACs but not HDAC6, induced degradation of Bcr-Abl and hyperacetylation of Hsp90. Furthermore, the degradation of Bcr-Abl was caspase dependent and appeared to largely occur as a downstream consequence of tumor cell apoptosis. These data provide evidence that inhibition of HDAC6 and concomitant degradation of Bcr-Abl following hyperacetylation of Hsp90 is not necessary for HDACis to kill cells that are “addicted” to the expression of Hsp90 client oncoproteins. Our data provide important insights into the mechanisms of action of small molecules with diverse HDAC specificities. In addition, our studies bring into question the hypothesis that degradation of Hsp90 client proteins through hyperacetylation of Hsp90 following HDAC6 inhibition is a major effector mechanism of action of HDACi that target multiple HDACs.
Materials and Methods
Cell culture
Eμ-myc, Eμ-myc/Bcl-2, and Eμ-myc/p53−/− lymphomas were developed and grown in vitro as described previously (13). The IL-3–dependent mouse bone marrow-derived cell line FDCP-1 has been described previously (30). FDCP-1 cells were cultured in RPMI1640 and conditioned media from the WEHI-3B cell line as a source of IL-3, as previously outlined (30). The FDCP-1/Bcr-Abl and FDCP-1/Bcr-Abl(T315I) cells were engineered by retroviral transduction using MSCV-Bcr-Abl(p210) puro or MSCV-Bcr-Abl(T315I) puro (a gift from Scott Lowe, CSHL) and transduction methods that were previously described (13). Verification of cell lines used in this study was performed using a number of in-house biologic assays. For example, in vitro assays on FDCP-1 cells were performed to verify the requirement of IL-3 for the survival of this factor-dependent cell line. Western blot assays were performed to verify the expression or loss of appropriate genes in “compound mutant” Eμ-myc lymphomas. For further authentication of cells, transplantation and rejection studies of Eμ-myc and FDCP-1 cells were performed in genetically pure inbred strains of recipient mice to verify the genetic origin of the cells.
Gene knockdown using RNA interference
Targeting sequences for short hairpin RNA (shRNA)-miR30 constructs against murine HDAC6 (RefSeq NM_001130416) were identified using the previously described Designer of Small Interfering RNA (DSIR) algorithm (31). The top-ranked shRNAs were used to create 10 miR30 sequences (97mer, Sigma-Aldrich) and these were cloned into the pLMS plasmid (MSCV)LTR-miR30-SV40-GFP vector following the generation of approximately 110-bp shRNA-miR30s by amplification of 97mers using 5′mir30-XhoI (CAGAAGGCTCGAGAAGGTATATTGCTGTTGACAGTGAGCG) and 5′miR30-EcoRI (CTAAAGTAGCCCCTTGAATTCCGAGGCAGTAGGCA) primers. The miRNA oligomer sequences are in the format: mir-30 context-sense (underlined)-loop-antisense (underlined).
HDAC 6shRNAmiR30#1.
TGCTGTTGACAGTGAGCGCAAAGTTGGAGTTGCTACAAAATAGTGAAGCCACAGATGTATTTTGTAGCAACTCCAACTTTATGCCTACTGCCTCGGA
HDAC 6shRNAmiR30#2.
TGCTGTTGACAGTGAGCGCGAGGATGACCCTAGTGTATTATAGTGAAGCCACAGATGTATAATACACTAGGGTCATCCTCATGCCTACTGCCTCGGA
ScrambledshRNAmiR30.
TGCTGTTGACAGTGAGCGATCTCGCTTGGGCGAGAGTAAGTAGTGAAGCCACAGATGTACTTACTCTCGCCCAAGCGAGAGTGCCTACTGCCTCGGA
Clones were verified by sequencing using the Mouse Stem Cell Virus (MSCV) forward primer (CCCTTGAACCTCCTCGTTCGACC).
Reagents
Vorinostat and MRLB-223 (Merck) were dissolved in dimethyl sulfoxide (DMSO) for the preparation of 10 mmol/L stock solutions. Imatinib mesylate (Novartis) was dissolved in PBS for the preparation of a 100 mmol/L stock, and romidepsin (Celgene) was supplied as a 5-mg/mL solution and diluted in PBS or media before use. The structures of vorinostat, MRLB-223, romidepsin, and imatinib mesylate used in this study are shown in Fig. 1A and Supplementary Fig. S1. Tetramethylrhodamine ethyl (TMRE) ester was purchased from Molecular Probes, propidium iodide (PI) solution was purchased from Sigma, and Annexin V was purchased from BD PharMingen. [18F]fluoro-2-deoxy-D-glucose (FDG) was purchased from Cyclotek.

In vitro sensitivity of Eμ-myc lymphomas to isoform-selective HDACi. A, chemical structure of MRLB-223. B–E, Eμ-myc lymphomas were incubated in vitro for 24 and 48 hours with the indicated concentrations of either vorinostat or MRLB-223. Cell viability was assessed by PI staining (A and C) and TMRE (B and D) staining to assess mitochondrial outer membrane permeabilization (MOMP). Data are the mean ± SEM of at least three independent experiments.
In vitro sensitivity of Eμ-myc lymphomas to isoform-selective HDACi. A, chemical structure of MRLB-223. B–E, Eμ-myc lymphomas were incubated in vitro for 24 and 48 hours with the indicated concentrations of either vorinostat or MRLB-223. Cell viability was assessed by PI staining (A and C) and TMRE (B and D) staining to assess mitochondrial outer membrane permeabilization (MOMP). Data are the mean ± SEM of at least three independent experiments.
In vivo experiments
Six- to eight-week-old C57BL/6 mice were used for in vivo apoptosis assays (IVA), small-animal positron emission tomography (SA-PET), and therapy studies. The Peter MacCallum Cancer Centre Animal Ethics Committee approved all mouse protocols used in this study. Six- to eight-week-old C57BL/6 mice were injected intravenously with 5 × 105 Eμ-myc lymphomas. After the lymphomas became palpable, mice were used for IVAs (3 mice/group), SA-PET (3 mice per group), and therapy studies (10 mice/group). Therapy experiments were undertaken using MRLB-223 (15 mg/kg daily, intraperitoneal) and vorinostat (200 mg/kg i.p for 7 days, then 150 mg/kg i.p daily thereafter as previously optimized; ref. 13). Briefly, C57BL/6 mice were injected intravenously with 5 × 105 Eμ-myc lymphomas. Peripheral white blood cell counts (WBC) were monitored, and therapy commenced when counts exceeded 13 × 103 cells per microliter (Sysmex Hematology Analyzer K-1000; Sysmex). For IVAs, C57BL/6 mice were injected with 5 × 105 Eμ-myc lymphomas and, after 10 to 15 days during which lymph nodes became well palpable, either a 200-mg/kg dose of vorinostat or a 15-mg/kg dose of MRLB-223 was administered intraperitoneally. After the indicated time points, mice were sacrificed, and cells were harvested from brachial lymph nodes for fluorescence-activated cell sorting (FACS)–based assays to measure apoptotic signaling (13). For SA-PET analyses, mice were injected intraperitoneally with either a 200-mg/kg dose of vorinostat or a 15-mg/kg dose of MRLB-223, and then scanned on a small-animal PET scanner (Phillips) at 4, 24, and 48 hours following injection. For details, please refer Supplementary Materials. For all in vivo assays, the vehicle used to formulate vorinostat and MRLB-223 was DMSO. MRLB-223 was further diluted and administered with 50:50 (PEG400:H2O). Therefore, although the vehicle for vorinostat was DMSO, the vehicle for MRLB-223 was 50:50 (PEG400:H2O).
Pharmacokinetic analysis of vorinostat and MRLB-223
C57BL/6 mice were treated by intraperitoneal injection of either vorinostat (200 mg/kg) or MRLB-223 (15 mg/kg). Peripheral blood (∼200 μL) was obtained from individual mice (three per time point) at 2, 4, 8, 12, and 24 hours after treatment. Serum was then collected, and pharmacokinetic analysis was undertaken at the Merck Research Laboratories (Boston, MA). Briefly, plasma concentrations were determined using high-performance liquid chromatography/tandem mass spectrometry (HPLC/MS–MS). Then, 50-μl plasma samples were precipitated using 200 μL of acetonitrile containing an internal standard. The precipitated proteins were separated by filtration and the resulting supernatant was evaporated and then reconstituted in 100 μL acetonitrile:water (30:70 v/v). A 10-μL sample was injected on a C18 column (2.0 mm × 30 mm, 3 μm particle size) and eluted using a gradient LC method with water containing 0.1% formic acid as the aqueous mobile phase, and acetonitrile containing 0.1% formic acid as the organic phase. Electrospray ionization with multiple reaction monitoring was used for MS–MS detection. A standard curve ranging from 0.003 to 10 μmol/L was prepared and used to calculate the concentration of unknown samples.
Western blotting and immunoprecipitation
Western blot assays were performed as previously described (13) using the following antibodies: anti-acetylated tubulin (Sigma-Aldrich); anti-acetylated H3 and H4 (Millipore); anti-β-actin (Clone AC-74, Sigma-Aldrich); and anti-c-Abl (Calbiochem). For immunoprecipitation studies, the FDCP-1/Bcr-Abl cell lines were lysed in ice-cold immunoprecipitation lysis buffer (1% Triton X-100, 150 mmol/L NaCl, 2.0 mmol/L EDTA, 10 μg/mL leupeptin, 1 μg/mL Pepstatin-A, 2 μg/mL aprotinin, 25 mmol/L NaF, and 0.5 mmol/L sodium orthovanadate) for 30 minutes, and the nuclear and cellular debris was cleared by centrifugation. Immunoprecipitation was performed with sepharaose protein G beads (Zymed Laboratories) incubated with 1 mg of total protein plus either anti-HSP90 (Assay designs Stressgen) or normal mouse immunoglobulin G1 (IgG1) isotype control antibody (Santa Cruz Biotechnology) at 4°C for 16 hours. The immunoprecipitates were washed 3 times in lysis buffer, and proteins were eluted with the SDS sample-loading buffer. Proteins were separated by SDS-PAGE, and acetylated lysine proteins were detected using an anti-acetylated lysine antibody (Cell Signaling Technology). Secondary antibody incubations and chemiluminescence visualizations were performed as previously described (13).
In vitro cell death analysis
Cells (5–8 × 105 cells/mL) were incubated in the presence of the indicated compounds for 24 to 72 hours within 1 mL cell culture media in 24-well plates (Greiner Bio-One). Viability of cells as measured by PI uptake, Annexin V staining, cell-cycle analysis, or TMRE staining were performed as detailed previously (32, 33). FDCP1 cells (5–8 × 105 cells/mL) were incubated with or without 50 μmol/L Q-VD-OPh in the presence of the indicated HDACi for 24 to 48 hours in 5-mL culture flasks. Cell viability was measured by PI uptake, and cell suspensions were harvested for Western blot analysis.
Results
Molecular and biologic activities of broadly acting and HDAC1/2-specific HDACis
MRLB-223 (34) is an HDACi that selectively inhibits the enzymatic activity of HDAC1 and -2 (Supplementary Table S1). In comparison, vorinostat has broader HDAC inhibitory activities and is capable of inhibiting all four class I HDACs (1, 2, 3, and 8) as well as HDAC6 at concentrations lower than 500 nmol/L (Supplementary Table S1). We therefore, aimed to compare the ability of MRLB-223 and vorinostat to kill Eμ-myc cells. Vorinostat effectively killed Eμ-myc lymphomas in a concentration-dependent manner after a 24-hour incubation, as shown by the increase in the percentage of PI uptake (Fig. 1B) and an increase in the percentage mitochondrial outer membrane permeabilization (mitochondrial outer membrane permeabilization (MOMP); Fig. 1C). Following an additional 24-hour exposure with vorinostat, there was little change in the IC70 observed. In addition, MRLB-223 induced an increase in PI uptake (Fig. 1D) and an increase in MOMP (Fig. 1E) in the Eμ-myc cells after a 24-hour exposure. However, after a 48-hour exposure to the compound, the level of PI uptake mitochondrial membrane permeabilization significantly increased, reducing the IC70 concentration from 5 to 0.5 μmol/L. As a result, using MRLB-223 over a 48-hour period resulted in induction of cell death equivalent to that observed using an equivalent concentration of vorinostat for 24 hours. In experiments using both compounds, the change in MOMP correlated very well with the uptake of PI.
Our previous studies demonstrated that vorinostat induced tumor cell apoptosis via the intrinsic apoptosis pathway (13, 35). Vorinostat-mediated cell killing was not dependent on a functional p53 pathway, and was effectively blocked by overexpression of prosurvival proteins of the Bcl-2 family (13, 36). We, therefore, determined if MRLB-223 induced tumor cell apoptosis via a similar mechanism. Eμ-myc/p53−/− tumor cells were sensitive to vorinostat and MRLB-223 at similar concentrations and with the same kinetics as observed in Eμ-myc lymphomas (Fig. 2A and B). In contrast, overexpression of Bcl-2 suppressed tumor cell death mediated by both agents. These data indicate that, although vorinostat and MRLB-223 have a different repertoire of HDAC targets, they both induce apoptosis of Eμ-myc cells via the intrinsic apoptotic pathway and both function independently of p53.
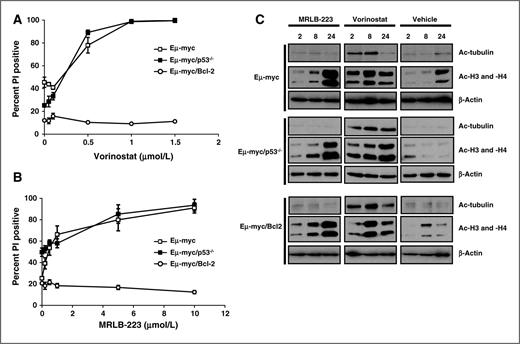
Apoptosis induced by MRLB-223 and vorinostat is blocked by Bcl2 but is independent of a functional p53 pathway. Eμ-myc, Eμ-myc/p53−/−, and Eμ-myc/Bcl-2 lymphomas were incubated in vitro for 24 hours with the indicated concentrations of (A) vorinostat or (B) MRLB-223. Cell viability was assessed by PI staining. Data are the mean ± SEM of at least three independent experiments. C, Eμ-myc, Eμ-myc/p53−/−, and Eμ-myc/Bcl-2 lymphomas were incubated in vitro with 0.5 μmol/L vorinostat or 3 μmol/L MRLB-223 for 2, 8, and 24 hours. Cells were harvested, and Western blot analysis was performed with antibodies to detect acetylated tubulin and acetylated histones H3 and H4. Anti-β-actin was used as a loading control.
Apoptosis induced by MRLB-223 and vorinostat is blocked by Bcl2 but is independent of a functional p53 pathway. Eμ-myc, Eμ-myc/p53−/−, and Eμ-myc/Bcl-2 lymphomas were incubated in vitro for 24 hours with the indicated concentrations of (A) vorinostat or (B) MRLB-223. Cell viability was assessed by PI staining. Data are the mean ± SEM of at least three independent experiments. C, Eμ-myc, Eμ-myc/p53−/−, and Eμ-myc/Bcl-2 lymphomas were incubated in vitro with 0.5 μmol/L vorinostat or 3 μmol/L MRLB-223 for 2, 8, and 24 hours. Cells were harvested, and Western blot analysis was performed with antibodies to detect acetylated tubulin and acetylated histones H3 and H4. Anti-β-actin was used as a loading control.
To demonstrate the differences in HDAC specificity between vorinostat and MRLB-223 in a cellular setting and to formally show that loss of expression of p53 or overexpression of Bcl-2 did not affect the HDAC inhibitory functions of the compounds, Western blot assays were performed to assess the acetylation status of histones and α-tubulin. Deacetylation of α-tubulin is mediated by HDAC6 and, therefore, the acetylation state of α-tubulin serves as a surrogate readout for HDAC6 activity in cell-based systems (37, 38). Both vorinostat and MRLB-223 induced hyperacetylation of histones H3 and H4 although the kinetics of response were different (Fig. 2C). Hyperacetylation of histone H3 and H4 occurred after a 2-hour exposure to vorinostat in Eμ-myc and Eμ-myc/Bcl-2 lymphomas and reached a maximum level following 8 hours of exposure whereas, in Eμ-myc/p53−/− lymphomas, the maximum level of histone H3 and H4 acetylation was seen at the 24-hour time point. In contrast, MRLB-223 induced only minimal histone hyperacetylation after 2-hour exposure, and this gradually increased over time. Vorinostat induced hyperacetylation of α-tubulin with maximal acetylation that was observed following 8-hour exposure to the compound. Consistent with the HDAC1/2 binding specificity of MRLB-223 shown in Supplementary Table S1, there was no increase in acetylation of α-tubulin following exposure of Eμ-myc, Eμ-myc/Bcl-2, or Eμ-myc/p53−/− lymphomas to the compound. Taken together, these data demonstrate that both vorinostat and MRLB-223 inhibit HDACs that deacetylate histones although the kinetics of the MRLB-223 response were slower than that observed with vorinostat. In addition, vorinostat, but not MRLB-223, inhibited HDAC6 as measured by the hyperacetylation of α-tubulin.
In vivo effects of vorinostat and MRLB-223
We, next, wanted to compare the antitumor effects of vorinostat and MRLB-223 in vivo. Pharmacokinetic studies using vorinostat and MRLB-223 at their maximum-tolerated doses (MTD) revealed that the maximum serum concentration of MRLB-223 was significantly greater than vorinostat (Supplementary Table S2). To determine the therapeutic effect of vorinostat and MRLB-223, Eμ-myc lymphomas were transplanted into C57BL/6 mice and treatment with the HDACi or vehicle commenced when WBC counts reached 13 × 106/mL or greater (day 7 post lymphoma injection). The WBC counts in tumor-bearing mice treated with vehicle increased over time to an average of 39.2 × 103/μL on day 21 that increased to 54.3 by day 28 (Fig. 3A). Treatment of mice with MRLB-223 decreased the blood tumor load and, by day 21, the average WBC count was 23.9 × 103/μL and increased to 33.8 × 103/μL by day 35. In contrast, treatment of mice bearing Eμ-myc lymphomas with vorinostat reduced the WBC counts to normal levels, and the reduction in blood tumor load was maintained throughout the vorinostat treatment period. After treatment with vorinostat ceased, the WBC counts increased over a 2-week period to a maximum level of 31.5 × 103/μL by day 55.

In vivo sensitivity of Eμ-myc lymphomas to isoform-selective HDACi. Eμ-myc lymphomas were transplanted into C57BL/6 mice, and treatment with vehicle, vorinostat, or MRLB-223 commenced when WBC counts reached 13 × 106/mL or higher. A, WBC counts from mice transplanted with Eμ-myc lymphomas from the commencement of therapy and over time with vehicle, vorinostat, or MRLB-223. Data are the mean ± SEM of at least three independent experiments. B, Kaplan–Meier survival curves of vehicle-treated mice (green line), MRLB-223–treated mice (blue line), and vorinostat-treated mice (brown line) are shown. Median survival was: 33 days for vehicle; 42 days for MRLB-223, P = 0.0015; and 63 days for vorinostat, P = 0.0026. There was a statistically significant difference in median survival in vorinostat- versus MRLB-223–treated mice (P = 0.0050). Statistical analyses were performed using MedCalc statistical software. C, C57Bl/6 mice bearing Eμ-myc lymphomas were injected with one dose of either vorinostat (200 mg/kg) or MRLB-223 (15 mg/kg). Lymphoma cells were harvested at various time points (hours) indicated following HDACi treatment and Western blot analysis was performed with antibodies to detect acetylated H3 and acetylated H4. Anti-β-actin was used as a loading control. D, FDG-based PET uptake was measured in C57Bl/6 mice bearing Eμ-myc lymphomas at baseline as well as 4 and 48 hours after a 200-mg/kg dose of vorinostat (top) or a 15-mg/kg dose of MRLB-223 (bottom). All time points in each panel represent the same mouse, and each panel is a single representative from three mice/group.
In vivo sensitivity of Eμ-myc lymphomas to isoform-selective HDACi. Eμ-myc lymphomas were transplanted into C57BL/6 mice, and treatment with vehicle, vorinostat, or MRLB-223 commenced when WBC counts reached 13 × 106/mL or higher. A, WBC counts from mice transplanted with Eμ-myc lymphomas from the commencement of therapy and over time with vehicle, vorinostat, or MRLB-223. Data are the mean ± SEM of at least three independent experiments. B, Kaplan–Meier survival curves of vehicle-treated mice (green line), MRLB-223–treated mice (blue line), and vorinostat-treated mice (brown line) are shown. Median survival was: 33 days for vehicle; 42 days for MRLB-223, P = 0.0015; and 63 days for vorinostat, P = 0.0026. There was a statistically significant difference in median survival in vorinostat- versus MRLB-223–treated mice (P = 0.0050). Statistical analyses were performed using MedCalc statistical software. C, C57Bl/6 mice bearing Eμ-myc lymphomas were injected with one dose of either vorinostat (200 mg/kg) or MRLB-223 (15 mg/kg). Lymphoma cells were harvested at various time points (hours) indicated following HDACi treatment and Western blot analysis was performed with antibodies to detect acetylated H3 and acetylated H4. Anti-β-actin was used as a loading control. D, FDG-based PET uptake was measured in C57Bl/6 mice bearing Eμ-myc lymphomas at baseline as well as 4 and 48 hours after a 200-mg/kg dose of vorinostat (top) or a 15-mg/kg dose of MRLB-223 (bottom). All time points in each panel represent the same mouse, and each panel is a single representative from three mice/group.
Vorinostat- and MRLB-223–treated mice demonstrated a significant increase in survival compared with vehicle-treated mice (Fig. 3B; median survival vehicle = 33 days, median survival MRLB-223 = 42 days, median survival vorinostat = 63 days). Clearly, MRLB-223 provided a therapeutic benefit to the mice; however, the effect was not nearly as robust as that provided by vorinostat. Consistent with our in vitro data (Fig. 2C), the kinetics of histone hyperacetylation mediated by MRLB-223 in vivo was delayed compared with the response to vorinostat (Fig. 3C).
In addition, the more rapid and robust response to vorinostat in vivo was demonstrated by molecular imaging using FDG-PET. The uptake of FDG at baseline and following 4- and 48-hour exposure to vorinostat and MRLB-223 is shown in Fig. 3D. At baseline, there was specific uptake of FDG in the lymph nodes and spleen of tumor-bearing mice as well as nonspecific uptake in the heart and bladder. The FDG signal in the lymphoid organs diminished significantly following 4-hour treatment with vorinostat (Supplementary Table S3) and remained low at 48 hours. In contrast, treatment with MRLB-223 had little or no effect on FDG uptake at the 4-hour time point (Supplementary Table S3); however, uptake in the lymph nodes at 48 hours was decreased.
HDAC6 inhibition is not required to kill cells dependent on the Hsp90 client protein Bcr-Abl
To test the hypothesis that inhibition of HDAC6 activity and concomitant degradation of Hsp90 client oncoproteins following hyperacetylation of Hsp90 plays an important mechanistic role in HDACi-induced cell death (25, 26, 29), we expressed wild-type Bcr-Abl and a mutant form of the fusion protein (T315I) that does not respond to imatinib (39) in the IL-3–dependent cell line FDCP-1. FDCP-1/Bcr-Abl and FDCP-1/Bcr-Abl(T315I) cells grew in the absence of IL-3 whereas parental FDCP-1 cells died (Supplementary Fig. S2A). We confirmed the expression of Bcr-Abl and Bcr-Abl(T315I) in FDCP-1 cells by Western blot analysis (Supplementary Fig. S1B). In addition, we showed the functional activity of Bcr-Abl by treating FDCP-1, FDCP-1/Bcr-Abl, and FDCP-1/Bcr-Abl(T315I) cells with imatinib (Supplementary Fig. S2C). Only cells expressing wild-type Bcr-Abl were sensitive to imatinib whereas the T315I substitution rendered Bcr-Abl insensitive to imatinib, but maintained its prosurvival function.
As with the Eμ-myc cells, the kinetics of MRLB-223-induced death of FDCP-1 cells were slower than that observed using vorinostat (Fig. 4A and B). Vorinostat induced hyperacetylation of histones H3 and H4 and α-tubulin following a 2-hour incubation, whereas MRLB-223 induced only acetylation of histone H3 and H4 with delayed kinetics compared with vorinostat and no acetylation of α-tubulin was observed (Fig. 4C). In addition, we tested the sensitivity of FDCP-1 cells to romidepsin, an HDACi that potently inhibits HDACs 1, 2, and 3 (see Supplementary Table S1; ref. 15). Romidepsin effectively killed FDCP-1 cells (Fig. 4D) and induced acetylation of histone H3 following 24-hour incubation, but did not affect acetylation of α-tubulin (Fig. 4E).

In vitro sensitivity of FDCP1 cells to HDACi. FDCP-1 cells were incubated with the indicated concentrations of: A, vorinostat; B, MRLB-223; or D, romidepsin; and time points were taken at 24, 48, and/or 72 hours. Cell viability was assessed by PI staining, and data are the mean ± SEM of at least three independent experiments. In addition, FDCP1 cells were incubated in vitro with (C) 3 μmol/L vorinostat and 5 μmol/L MRLB-223 for 2, 8, and 24 hours; (C) or (E) 3 μmol/L vorinostat and 3 nmol/L romidepsin for 0 and 24 hours. The cells were harvested and Western blot analysis was performed with antibodies that detect acetylated tubulin and acetylated histones H3 and H4. Anti-β-actin was used as a loading control.
In vitro sensitivity of FDCP1 cells to HDACi. FDCP-1 cells were incubated with the indicated concentrations of: A, vorinostat; B, MRLB-223; or D, romidepsin; and time points were taken at 24, 48, and/or 72 hours. Cell viability was assessed by PI staining, and data are the mean ± SEM of at least three independent experiments. In addition, FDCP1 cells were incubated in vitro with (C) 3 μmol/L vorinostat and 5 μmol/L MRLB-223 for 2, 8, and 24 hours; (C) or (E) 3 μmol/L vorinostat and 3 nmol/L romidepsin for 0 and 24 hours. The cells were harvested and Western blot analysis was performed with antibodies that detect acetylated tubulin and acetylated histones H3 and H4. Anti-β-actin was used as a loading control.
We next determined the effect of vorinostat, MRLB-223, and romidepsin on FDCP-1/Bcr-Abl and FDCP-1/Bcr-Abl(T315I) cells grown in the absence of IL-3 (Fig. 5A–C and Supplementary Fig. S3). FDCP-1 cells expressing either wild-type or mutant Bcr-Abl were equivalently sensitive to the three HDACis. Tumor cell death mediated by MRLB-223 required longer exposure times compared with vorinostat and romidepsin. The effect of different HDACi on Bcr-Abl expression was assessed by Western blot analysis. Treatment with 5 μmol/L vorinostat for 24 hours induced degradation of Bcr-Abl concomitant with substantial apoptosis (Fig. 5D). In addition, treatment with MRLB-223 and romidepsin induced cell death and, interestingly, Bcr-Abl expression was lost concomitant with induction of apoptosis (Fig. 5D). Moreover, inhibition of HDACi-induced apoptosis using the caspase inhibitor Q-VD-OPh resulted in maintenance of Bcr-Abl expression following HDACi treatment (Fig. 5E).

HDACis with different isoform specificities induce degradation of Bcr-Abl–expressing cells after death. FDCP-1, FDCP-1/Bcr-Abl, and FDCP-1/Bcr-Abl(T315I) cells were incubated with the indicated concentrations of (A) vorinostat (24 hours), (B) MRLB-223 (72 hours), or (C) romidepsin (48 hours). Cell viability was assessed by PI staining, and data are the mean ± SEM of at least three independent experiments. D, FDCP-1/Bcr-abl cells were incubated with 5 μmol/L vorinostat for 24 hours (left), or 5 and 10 μmol/L of MRLB-223 and 3 nmol/L romidepsin for 48 hours (right). The cells were harvested, cell viability was assessed by PI staining (shown as percentage of PI-positive cells at the bottom of the Western blots), and degradation of Bcr-Abl was analyzed via Western blot. Expression of β-actin was used as a loading control. The results are representative of at least three independent experiments. E, FDCP-1/Bcr-Abl cells were incubated for 24 hours with 5 μmol/L vorinostat with or without 50 μmol/L Q-VD-OPh (left), or 48 hours with either 5 or 10 μmol/L MRLB-223, and 3 nmol/L romidepsin, with or without 50 μmol/L Q-VD-OPh (right). The cells were harvested, cell viability was assessed by PI staining, and the degradation of Bcr-Abl was analyzed via Western blot. Expression of β-actin was used as a loading control.
HDACis with different isoform specificities induce degradation of Bcr-Abl–expressing cells after death. FDCP-1, FDCP-1/Bcr-Abl, and FDCP-1/Bcr-Abl(T315I) cells were incubated with the indicated concentrations of (A) vorinostat (24 hours), (B) MRLB-223 (72 hours), or (C) romidepsin (48 hours). Cell viability was assessed by PI staining, and data are the mean ± SEM of at least three independent experiments. D, FDCP-1/Bcr-abl cells were incubated with 5 μmol/L vorinostat for 24 hours (left), or 5 and 10 μmol/L of MRLB-223 and 3 nmol/L romidepsin for 48 hours (right). The cells were harvested, cell viability was assessed by PI staining (shown as percentage of PI-positive cells at the bottom of the Western blots), and degradation of Bcr-Abl was analyzed via Western blot. Expression of β-actin was used as a loading control. The results are representative of at least three independent experiments. E, FDCP-1/Bcr-Abl cells were incubated for 24 hours with 5 μmol/L vorinostat with or without 50 μmol/L Q-VD-OPh (left), or 48 hours with either 5 or 10 μmol/L MRLB-223, and 3 nmol/L romidepsin, with or without 50 μmol/L Q-VD-OPh (right). The cells were harvested, cell viability was assessed by PI staining, and the degradation of Bcr-Abl was analyzed via Western blot. Expression of β-actin was used as a loading control.
HDAC6 inhibition is not required for HDACi to induce the hyperacetylation of Hsp90 in FDCP-1/Bcr-Abl cells
Our data indicated that inhibition of HDAC6 and subsequent degradation of Hsp90 client oncoproteins such as Bcr-Abl was not a major apoptosis effector mechanism of HDACi action in our experimental system. The current literature suggests that HDAC6 is a direct deacetylase for HSP90 and that HDAC6 inhibition leads to the hyperacetylation of Hsp90 (24, 40, 41). To investigate this in our system, we performed immunoprecipitation/Western blotting assays to assess the acetylation state of Hsp90 in FDCP-1/Bcr-Abl cells after treatment with HDACis of different isoform specificities (Fig. 6A). Vorinostat and romidepsin induced hyperacetylation of Hsp90 following an 8-hour incubation compared with the DMSO-treated controls. Importantly, although MRLB-223 required a longer incubation time of 24 hours, it was also able to induce hyperacetylation of Hsp90. Our results indicate that direct inhibition of HDAC6 by HDACis is not necessary for these agents to induce Hsp90 hyperacetylation and subsequent degradation of client proteins.

Regulated acetylation of Hsp90 and expression of Bcr-Abl by HDACis and knockdown of HDAC6. A, whole-cell lysates from FDCP-1/Bcr-Abl cells treated with 10 μmol/L vorinostat for 8 hours, 10 μmol/L MRLB-223 for 24 hours, or 3 nmol/L romidepsin for 8 hours were immunoprecipitated with an anti-Hsp90 antibody and Western blot assays for acetylated proteins was performed. The blot was stripped and re-probed for expression of Hsp90. Results are representative of at least three independent experiments. B, Western blot analyses were performed using lysates from an FDCP-1/Bcr-Abl (lane 1), FDCP-1/Bcr-Abl/scrambledshRNA-miR30 (lane 2), FDCP-1/Bcr-Abl/HDAC6shRNA-miR30#1 (lane 3), and FDCP1/Bcr-Abl/HDAC6shRNA-miR30#2 (lane 4) with antibodies that detect HDAC6 (left) and acetylated tubulin (right). The expression of β-actin was used as a loading control. C, whole-cell lysates from FDCP-1/Bcr-Abl/scrambledshRNA-miR30 (lane 1) and FDCP-1/Bcr-Abl/HDAC6shRNA-miR30#1 (lane 2) cells were immunoprecipitated with an anti-HSP90 antibody and Western blot assays for acetylated proteins was performed. The blot was stripped and re-probed for expression of Hsp90. Results are representative of at least three independent experiments. D, Western blot analyses were performed using lysates from an FDCP-1/Bcr-Abl (lane 1), FDCP-1/Bcr-Abl/scrambledshRNA-miR30 (lane 2), FDCP-1/Bcr-Abl/HDAC6shRNA-miR30#1 (lane 3), and FDCP-1/Bcr-Abl/HDAC6shRNA-miR30#2 (lane 4) with antibodies that detect Bcr-Abl. The expression of β-actin was used as a loading control. E and F, FDCP-1/Bcr-Abl, FDCP-1/Bcr-Abl/scrambledshRNA-miR30, and FDCP-1/Bcr-Abl/HDAC6shRNA-miR30#1 cells were incubated with the indicated concentrations of vorinostat or MRLB-223 for 48 and 72 hours, respectively. Cell viability was assessed by PI staining. Data are the mean ± SEM of at least three independent experiments.
Regulated acetylation of Hsp90 and expression of Bcr-Abl by HDACis and knockdown of HDAC6. A, whole-cell lysates from FDCP-1/Bcr-Abl cells treated with 10 μmol/L vorinostat for 8 hours, 10 μmol/L MRLB-223 for 24 hours, or 3 nmol/L romidepsin for 8 hours were immunoprecipitated with an anti-Hsp90 antibody and Western blot assays for acetylated proteins was performed. The blot was stripped and re-probed for expression of Hsp90. Results are representative of at least three independent experiments. B, Western blot analyses were performed using lysates from an FDCP-1/Bcr-Abl (lane 1), FDCP-1/Bcr-Abl/scrambledshRNA-miR30 (lane 2), FDCP-1/Bcr-Abl/HDAC6shRNA-miR30#1 (lane 3), and FDCP1/Bcr-Abl/HDAC6shRNA-miR30#2 (lane 4) with antibodies that detect HDAC6 (left) and acetylated tubulin (right). The expression of β-actin was used as a loading control. C, whole-cell lysates from FDCP-1/Bcr-Abl/scrambledshRNA-miR30 (lane 1) and FDCP-1/Bcr-Abl/HDAC6shRNA-miR30#1 (lane 2) cells were immunoprecipitated with an anti-HSP90 antibody and Western blot assays for acetylated proteins was performed. The blot was stripped and re-probed for expression of Hsp90. Results are representative of at least three independent experiments. D, Western blot analyses were performed using lysates from an FDCP-1/Bcr-Abl (lane 1), FDCP-1/Bcr-Abl/scrambledshRNA-miR30 (lane 2), FDCP-1/Bcr-Abl/HDAC6shRNA-miR30#1 (lane 3), and FDCP-1/Bcr-Abl/HDAC6shRNA-miR30#2 (lane 4) with antibodies that detect Bcr-Abl. The expression of β-actin was used as a loading control. E and F, FDCP-1/Bcr-Abl, FDCP-1/Bcr-Abl/scrambledshRNA-miR30, and FDCP-1/Bcr-Abl/HDAC6shRNA-miR30#1 cells were incubated with the indicated concentrations of vorinostat or MRLB-223 for 48 and 72 hours, respectively. Cell viability was assessed by PI staining. Data are the mean ± SEM of at least three independent experiments.
We complemented the studies detailed in Fig. 6A using a gene knockdown approach. FDCP-1 cells were transduced with retroviruses expressing shRNAs embedded within the miR30 miRNA that targeted HDAC6 (HDAC6shRNAmiR#1 and HDAC6shRNAmiR#2) or with a control scrambled shRNA (scrambledshRNAmiR). FDCP-1/Bcr-Abl/HDAC6shRNAmiR#1, FDCP-1/Bcr-Abl/HDAC6shRNAmiR#2 and FDCP-1/Bcr-Abl/scrambledshRNAmiR cells were viable and analyzed for expression of HDAC6 and tubulin acetylation. Both FDCP-1 cell lines expressing HDAC6shRNAmiRs had knockdown of HDAC6 to below detectable levels and concomitant hyperacetylation of α-tubulin (Fig. 6B). Consistent with previous data using a specific HDAC6 inhibitor (17), knockdown of HDAC6 did not induce histone acetylation (Supplementary Fig. S4A). However, knockdown of HDAC6 resulted in hyperacetylation of Hsp90 (Fig. 6C). Interestingly, and in contrast to the expected result, the expression of Bcr-Abl was increased in FDCP-1/Bcr-Abl/HDAC6shRNAmiR#1 and FDCP-1/Bcr-Abl/HDAC6shRNAmiR#2 (Fig. 6D and Supplementary Fig. S4B). Our data, therefore, indicate that inhibition of HDAC6 activity or loss of HDAC6 expression is sufficient to induce hyperacetylation of Hsp90 but not necessary nor sufficient to cause a decrease in expression of Bcr-Abl.
To formally determine if inhibition of HDAC6 was important for mediating the apoptotic effects of a multi-HDAC inhibitor such as vorinostat or if loss of HDAC6 could functionally interact with a more selective HDACi such as MRLB-223, we tested FDCP-1/Bcr-Abl/HDAC6shRNAmiR#1, FDCP-1/Bcr-Abl/HDAC6shRNAmiR#2, and FDCP-1/Bcr-Abl/scrambledshRNAmiR cells for sensitivity to vorinostat and MRLB-223. As shown in Fig. 6E and F, decreased expression of HDAC6 to below detectable levels neither suppressed the apoptotic effects of vorinostat nor enhanced the apoptotic effects of MRLB-223.
Discussion
HDACis are a new class of anticancer agents that are gaining widespread use in the clinic for the treatment of hematologic malignancies (42). There are two HDACi for cutaneous T-cell lymphoma (CTCL) that have been approved by the FDA—the Merck compound vorinostat (SAHA, Zolinza) and the Celgene agent romidepsin (depsipeptide, Istodax). At least 16 other companies have HDACis in clinical trials, mostly as oncology agents, and the majority of these compounds are somewhat nonselective, inhibiting more than 1 of the 11 class I, II, and IV HDACs (3). For example, vorinostat equivalently inhibits HDACs 1, 2, 3, and 6, whereas romidepsin has very high affinity for HDACs 1, 2, and 3 and more weakly inhibits HDAC4 (15). In addition to histones, the function, localization, or stability of a number of nonhistone proteins can be regulated by acetylation (5) and, thus, the combined effects of HDACi on histone and nonhistone proteins may be important for the therapeutic activities of these compounds (14). For example, the protein chaperone activity of Hsp90 can be regulated through HDAC6-mediated deacetylation of Hsp90. Accordingly, degradation of client oncoproteins such as ErbB2 and Bcr-Abl through HDACi-mediated hyperacetylation of Hsp90 has also been proposed to be a major functional effector mechanism of HDACi action (3).
It is not yet known if single HDAC isoform-selective inhibitors would provide better exposure and higher dose levels in vivo, which might lead to better efficacy in the clinic. In order to answer this question and determine if inhibition of HDAC6 and subsequent hyperacetylation of Hsp90 and degradation of client proteins such as Bcr-Abl was a major mechanism of action of HDACi, we compared and contrasted the molecular, biologic, and therapeutic activities of two compounds, vorinostat and MRLB-223, that possess different HDAC inhibitory profiles. Specifically, we aimed to determine if inhibition of HDACs 1 and 2 by MRLB-223 had the same apoptotic and therapeutic effects as vorinostat, which can inhibit HDACs 1, 2, 3, and 6 (Supplementary Table S1; ref. 15). We showed that both agents induced tumor cell death in a p53-independent manner that was inhibited by overexpression of Bcl-2. This is consistent with our previous studies and those of others showing that HDACis induce apoptosis via the intrinsic apoptotic pathway (13, 15, 32, 35, 43–50).
Consistent with the HDAC-specific inhibitory properties of the two agents, both vorinostat and MRLB-223 induced histone hyperacetylation; however, only vorinostat induced acetylation of α-tubulin, a function known to be regulated by HDAC6 (37). Interestingly, the kinetics of histone hyperacetylation and subsequent tumor cell apoptosis induced by MRLB-223 was much slower than vorinostat. Histone acetylation can be regulated by multiple class I HDACs, and the ability of vorinostat to inhibit HDAC3 may confer a more robust and rapid histone hyperacetylation effect than that observed with MRLB-223. Consistent with this hypothesis, HDAC3 is a known regulator of histone acetylation (51). Moreover, romidepsin specifically inhibits HDACs 1, 2, and 3 (15), and we have previously demonstrated that vorinostat and romidepsin induce histone hyperacetylation and tumor cell death with very similar kinetics (32). Taken together, these data demonstrated that although the multi-HDACi vorinostat induced histone acetylation and killed tumor cells with faster kinetics than the HDAC 1/2-selective inhibitor MRLB-223, both agents appeared to mediate their biologic effects through the same molecular pathway—in this case, the intrinsic apoptotic pathway. Whether the delayed kinetics of MRLB-223 is the result of its unique HDACi specificity or due to another characteristic, such as slower cell penetration, remains to be determined.
The differences in antitumor potency and kinetics of histone hyperacetylation observed between vorinostat and MRLB-223 in vitro were also apparent when the agents were tested in vivo in mice bearing Eμ-myc lymphomas. Although MRLB-223 reached higher serum concentrations and was more persistent at the MTD than vorinostat, the therapeutic effects of the agent were inferior to those observed using vorinostat. These data imply that selective inhibition of HDACs 1 and 2 may not be the optimal strategy for the clinical development of more potent and tolerable HDACi.
To determine the importance of HDAC6 inhibition in mediating the antitumor effects of HDACi, we established an “oncogene-addiction” model by expressing Bcr-Abl in IL-3–dependent FDCP-1 cells and growing the cells in the absence of IL-3. We showed that vorinostat, MRLB-223, and romidepsin were all capable of killing FDCP-1 cells that overexpressed wild-type or an imatinib-insensitive form of Bcr-Abl (Bcr-Abl/T315I), and that MRLB-223 showed a similar delay in inducing histone hyperacetylation and tumor cell apoptosis as observed using Eμ-myc lymphomas. Interestingly, we showed that all three HDACi could induce degradation of Bcr-Abl, but this effect was suppressed by cotreatment with a caspase inhibitor. These data indicate that degradation of the Hsp90 client protein Bcr-Abl occurs as a result of induction of apoptosis pathways and is not a primary mediator of HDACi-induced tumor cell death as has been previously proposed. Moreover, we showed that Hsp90 was hyperacetylated following treatment with romidepsin and MRLB-223 although these agents did not induce acetylation of α-tubulin. These data are consistent with another study showing that MS-275, an HDACi with specificity for HDACs 1, 2, and 3 (3), can also induce Hsp90 acetylation and degradation of the Flt3 client protein (52) and are inconsistent with a proposed mode of action of HDACi which posited that a specific inhibition of HDAC6 leads to acetylation of HSP90 and disruption of its chaperone function, resulting in depletion of client proteins such as Bcr-Abl and Flt-3 (25–28).
In summary, we have used MRLB-223 (inhibits HDACs 1 and 2) as a novel tool compound to demonstrate that the inhibition of multiple HDACs by FDA-approved agents such as romidepsin (inhibits HDACs 1, 2, and 3) and vorinostat (inhibits HDACs 1, 2, 3, 6, and 8) provided a superior antitumor response. Moreover, our studies allowed us to investigate the functional importance of inhibition of HDAC6 in mediating the antitumor effects of HDACis. It has been proposed that inhibition of HDAC6 resulting in hyperacetylation of Hsp90 and release and degradation of client oncoproteins such as Bcr-Abl is important for the antitumor activities of HDACis (3, 10). However, our studies, using sophisticated “oncogene-addiction” models provide clear evidence that inhibition of HDAC6 is not necessary for HDACis to kill tumor cells that rely on the Hsp90 client protein Bcr-Abl for survival. Moreover, we demonstrate that although depletion of HDAC6 was sufficient to induce hyperacetylation of HDAC6, this did not result in degradation of Bcr-Abl nor loss of viability of tumor cells “addicted” to Bcr-Abl. This information is very important to the field as it not only helps define the molecular events that are necessary for HDACi-mediated antitumor responses, but provides a cautionary note regarding any proposed use of acetylated Hsp90 as a biomarker for response to HDACis.
Disclosure of Potential Conflicts of Interest
A. M. Kral, N. D. Ozerova, T. A. Miller, J. L. Methot, V. M. Richon, and J. P. Secrist are current or former employees of Merck. No potential conflicts of interest were disclosed by the other authors.
Authors' Contributions
Conception and design: R.W. Johnstone, A. Newbold, G.M. Matthews, S. Chiocca, T.A. Miller, J.L. Methot, V.M. Richon, P. Secrist, S. Minucci
Development of methodology: A. Newbold, G.M. Matthews, L.A. Cluse, C.J.P. Clarke, C. Cullinane, J.E. Bolden, R.A. Dickins, S. Chiocca
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): A. Newbold, G.M. Matthews, L.A. Cluse, K-M. Banks, C. Cullinane, A.J. Christiansen, A.M. Kral, N.D. Ozerova, S. Minucci
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): R.W. Johnstone, A. Newbold, M. Bots, C.J.P. Clarke, J.E. Bolden, A.J. Christiansen, A.M. Kral, V.M. Richon, P. Secrist, S. Minucci
Writing, review, and/or revision of the manuscript: A. Newbold, G.M. Matthews, C.J.P. Clarke, T.A. Miller, S. Minucci
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): A. Newbold, G.M. Matthews, C. Miccolo
Study supervision: R.W. Johnstone, A. Newbold, S. Minucci
Acknowledgments
The authors thank Donna Dorow, Ekaterina Bogatyreva, Susan Jackson, Laura Kirby, Agnese Collino, and Alfonso Passafaro for technical help, and Salvatore Pece and Pier Paolo Di Fiore of the Molecular Medicine Program at the European Institute of Oncology for technical help and discussions.
Grant Support
R.W. Johnstone is a principal research fellow of the National Health and Medical Research Council of Australia (NHMRC) and supported by the NHMRC Program and Project Grants, Cancer Council Victoria, The Leukemia Foundation of Australia, Victorian Breast Cancer Research Consortium, and Victorian Cancer Agency. R.A. Dickins is supported by the NHMRC, Victorian Endowment for Science, Knowledge and Innovation (VESKI) (Australia), and The Sylvia and Charles Viertel Foundation. Work in the laboratory of S. Minucci has been supported by the Association for International Cancer Research (AIRC), Ministry of Health.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.