
Abstract
Background and the purpose of the study
There has been increscent interest in the field of cancer chemotherapy by discovery and development of novel agents with high efficacy, low toxicity, and minimum side effects. In order to find new anticancer agents, we replaced the pyrazolone part of well-known cytotoxic agent SJ-172550 with 7-methoxychroman-4-one. Thus, a novel series of 3-benzylidene-4-chromanones were synthesized and tested in vitro against human cancer cell lines.
Methods
The title compounds were prepared by condensation of 7-methoxychroman-4-one with suitable aldehydes in appropriate alcohol in the presence of gaseous HCl. The antiproliferative activity of target compounds were evaluated against MDA-MB-231 (breast cancer), KB (nasopharyngeal epidermoid carcinoma) and SK-N-MC (human neuroblastoma) cell lines using MTT assay.
Results
Although the direct analog of SJ-172550 (compound 5d) did not show any cytotoxic activity against tested cell lines, but 2-(2-chloro-6-methoxyphenoxy)acetic acid methyl ester analog 5c showed some activity against MDA-MB-231 and SK-N-MC cells. Further modification of compound 5c resulted in the 3-chloro-4,5-dimethoxybenzylidene derivative 5b which demonstrated better cytotoxic profile against all tested cell lines (IC50 values = 7.56–25.04 μg/ml).
Conclusion
The results demonstrated that the cytotoxic activity of compound 5b against MDA-MB-231 and SK-N-MC cells is more than etoposide. Therefore, compound 5b prototype could be considered as novel cytotoxic agent for further developing new anticancer chemotherapeutics.
Similar content being viewed by others


Introduction
Cancer has been known as one of the most impressive clinical problems in both developing and developed countries. In spite of improved diagnostic techniques and advances in prevention and chemotherapeutic management of cancer, the disease still afflicts millions of peoples in the world [1]. Cancer cells are defined by uncontrolled replications associated with self-sufficiency in growth signals, hyposensitivity to anti-growth signals, ongoing angiogenesis, metastasis, and evasion of apoptosis [2]. Anti-cancer agents cannot recognize cancer cells from normal cells, as a matter of fact, these agents usually act on metabolically active or rapidly proliferating cells [3]. Thus, there has been increscent interest in the field of cancer chemotherapy by discovery and development of novel agents with high efficacy, low toxicity, and minimum side effects.
During recent years, several researchers developed different chalcone-like compounds with anticancer activity through the introduction of heterocyclic scaffolds [4, 5]. The chemical structure of chalcone is characterized by two aromatic rings connected by a three carbon, α,β-unsaturated carbonyl system (1,3-diphenyl-2-propen-1-one) [6–8]. The highly significant advantage of chalcone derivatives as cytotoxic agents is the low propensity to interact with DNA; which omits the risk of mutagenesity as the common side effect of current chemotherapeutic agents [9].
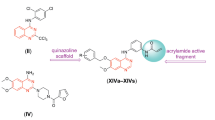
Previously, Perjési et al. have reported cytotoxicity of 3-benzylidene-4-chromanones as rigid analogs of chalcones (Figure 1) [10]. Recently, high-throughput screening of drug libraries results in the identification of SJ-172550 that exhibited p53-dependent cytotoxic activity against cancer cell lines [11]. Structurally, SJ-172550 is characterized by having α,β-unsaturated carbonyl system attached to the 2-(2-chloro-6-ethoxyphenoxy)acetic acid methyl ester. Accordingly, in continuation of our research program to find novel anti-cancer agents [12–16] and considering the diverse biological activities of rigid chalcones [17], we have synthesized a series of 3-benzylidene-4-chromanones bearing 2-(2-chloro-6-alkoxyphenoxy) acetic acid esters. The related analogs of 3-benzylidene-4-chromanones were also prepared for more studying of structure-activity relationships (Figure 1).

Structure of 3-benzylidene-4-chromanones as rigid analogs of chalcones exhibiting cytotoxic activity, structure of SJ-172550 as a lead compound that showed cytotoxic effects against tumour cell lines and designed compounds as cytotoxic agents.
Material and methods
Chemistry
All chemical reagents and solvents were provided from Merck AG (Darmstadt, Germany). The general procedures for the synthesis of 3-benzylidene-4-chromanones 5a–k, and aldehyde intermediates (compounds 7–9 and 11) are illustrated in Schemes 1 and 2, respectively. 7-Methoxychroman-4-one (4) was prepared as literature method [18, 19]. Melting points of compounds were determined using Kofler hot stage apparatus and are uncorrected. The IR spectra were recorded on a Shimadzu 470 spectrometer by using potassium bromide disks. The NMR spectra were obtained using a Bruker 400 MHz spectrometer (Bruker Bioscience, Billerica, MA, USA). Tetramethylsilane (TMS) was used as internal standard and chemical shifts (δ) are reported in ppm. Mass spectra were recorded on a Finnigan TSQ 70 spectrometer at 70 eV. Elemental analyses were carried out by using a HERAEUS CHN-O rapid elemental analyzer (Heraeus GmbH, Hanau, Germany) for C, H and N and the results are within ± 0.4% of the theoretical values.

General synthetic route to 3-benzylidene-4-chromanones 5a–k. Reagents and conditions: (a) 3-chloropropionic acid, CF3SO3H; (b) 2.0 M NaOH; (c) methyl iodide, K2CO3, DMF; (d) appropriate aldehyde, HCl (gas), ROH.

Synthesis of aldehyde intermediates 7–9 and 11. Reagents and conditions: (a) Cl2 , CH3COOH; (b) R2OCOCH2Br, K2CO3, CH3COCH2CH3; (c) methyl iodide, K2CO3, DMF.
Synthesis of 3-chloro-4-hydroxy-5-methoxybenzaldehyde (7a)
To a solution of vanillin (6a, 2.5 g, 16.4 mmol) in glacial acetic acid (15 ml) was slowly introduced a stream of chlorine gas over 30 min. White solid product was filtrated, washed with n-hexane (50 ml) to give compound 7a (1.9 g). The acetic acid filtrate was again treated with chlorine gas flow as above for 30 min to give 0.7 g of compound 7a[20]. A total of 2.6 g (85% yield) of white solid 7a was obtained and used in next step without purification. 1H NMR (CDCl3, 400 MHz) δ: 9.7 (s, 1H, CHO), 7.5 (d, J = 1.6 Hz, 1H, aromatic), 7.3 (d, J = 1.6 Hz, 1H, aromatic), 3.9 (s, 3H, OCH3).
Synthesis of 3-chloro-5-ethoxy-4-hydroxybenzaldehyde (7b)
3-Ethoxy-4-hydroxybenzaldehyde (6b, 4 g, 5 mmol) was dissolved in a solution of glacial acetic acid (30 ml) and chloroform (10 ml) at 0°C and a stream of chlorine gas was slowly introduced over 15 min. Then, the solvents was evaporated and the residue was purified by silica gel column, eluting with a mixture of ethyl acetate/ petroleum ether (40:60) to give compound 7b in 65% yield. 1H NMR (CDCl3, 400 MHz) δ: 10.3 (s, 1H, CHO), 7.5 (br s, 1H, aromatic), 6.9 (br s, 1H, aromatic), 5.7 (s, 1H, OH), 4.2 (q, J = 7.2 Hz, 2H, CH2), 1.5 (t, J = 7.2 Hz, 3H, CH3).
Synthesis of methyl 2-(2-chloro-4-formyl-6-methoxyphenoxy)acetate (8a)
A mixture of 5-chlorovanillin (7a, 2 g, 10.7 mmol) and K2CO3 (1.5 g, 10.8 mmol) in ethyl methyl ketone (40 ml) was stirred under reflux. After 10 min, methyl bromoacetate (1.64 g, 10.8 mmol) was added, and the mixture was allowed to stir under reflux for another 4 h. After the reaction was completed, ethyl methyl ketone was removed, and the residue was extracted with EtOAc (3 × 10 ml). The organic layer was dried over Na2SO4 and evaporated to give compound 8a in 88.8% yield. 1H NMR (CDCl3, 400 MHz) δ: 9.8 (s, 1H, CHO), 7.4 (d, J = 1.6 Hz, 1H, aromatic), 7.3 (d, J = 1.6 Hz, 1H, aromatic), 4.8 (s, 2H, CH2), 3.9 (s, 3H, OCH3), 3.8 (s, 3H, OCH3).
Synthesis of methyl 2-(2-chloro-6-ethoxy-4-formylphenoxy)acetate (8b)
To a solution of compound 7b (200 mg, 1 mmol) in ethyl methyl ketone (4 ml), was added potassium carbonate (147 mg, 1 mmol) and methyl bromoacetate (153 mg, 1 mmol) successively. The mixture was stirred under reflux for 2 h, cooled to room temperature and the solvent was removed under reduced pressure. The residue was added to 5 ml of water and extracted three times with ethyl acetate (5 ml). The combined extracts was dried (Na2SO4), and concentrated to give 8b as a white solid in 91% yield. 1H NMR (CDCl3, 400 MHz) δ: 10.3 (s, 1H, CHO), 7.3 (s, 1H, aromatic), 6.8 (s, 1H, aromatic), 4.7 (s, 2H, CH2), 4.2 (q, J = 7.2 Hz, 2H, CH2), 3.8 (s, 3H, OCH3), 1.5 (t, J = 7.2 Hz, 3H, CH3).
Synthesis of ethyl 2-(2-chloro-6-ethoxy-4-formylphenoxy)acetate (8c)
To a solution of compound 7b (500 mg, 2.5 mmol) in ethyl methyl ketone (10 ml), was added potassium carbonate (370 mg, 2.5 mmol) and ethyl bromoacetate (2.5 mmol) successively. The mixture was stirred under reflux for 3 h and then the solvent was removed under reduced pressure. The residue was added to 10 ml of water and extracted three times with ethyl acetate (10 ml). The combined extracts was dried (Na2SO4) and concentrated to give 8c as a white solid in 88% yield. 1H NMR (CDCl3, 400 MHz) δ: 10.2 (s, 1H, CHO), 7.3 (s, 1H, aromatic), 6.9 (s, 1H, aromatic), 4.7 (s, 2H, CH2), 4.25 (q, J = 7.2 Hz, 2H, CH2), 1.5 (t, J = 7.2 Hz, 3H, CH3), 1.3 (t, J = 7.2 Hz, 3H, CH3).
Synthesis of 3-chloro-4,5-dimethoxybenzaldehyde (9a)
To a solution of 5-chlorovanillin (7a, 5 g, 26.8 mmol) in DMF (40 ml) was added potassium carbonate (3.7 g, 26.8 mmol) and iodomethane (4.56 g, 32.16 mmol) successively. The mixture was stirred at 80°C for 3 h, cooled to room temperature and poured to water (100 ml). The precipitated white solid was filtrated and washed with water to give 5.1 g of compound 9a in 94.8 yield. 1H NMR (CDCl3, 400 MHz) δ: 9.85 (s, 1H, CHO), 7.5 (s, 1H, aromatic), 7.35 (s, 1H, aromatic), 3.97 (s, 3H, OCH3), 3.94 (s, 3H, OCH3).
General procedure for the preparation of compounds 11a-c
A mixture of hydroxybenzaldehyde 10a-c (5 g, 40.95 mmol) and K2CO3 (6 g, 40.95 mmol) in ethyl methyl ketone (100 ml) was stirred under reflux. After 1 h, methyl bromoacetate was added, and the mixture was allowed to stir under reflux for another 3 h. After the reaction was completed, ethyl methyl ketone was removed, and the residue was extracted with EtOAc (3 × 20 ml). The organic layer was dried (Na2SO4) and evaporated to give methyl (formylphenoxy)acetate 11a-c[21].
Methyl (2-formylphenoxy)acetate (11a)
This compound was obtained using general procedure as a pale yellow oil without further purification in 85% yield. IR (KBr, cm-1): 1767 (C = O, ester), 1690 (C = O, aldehyde); 1H NMR (CDCl3, 400 MHz) δ: 10.56 (s, 1H, CHO), 7.87 (dd, J = 5.6 and 2 Hz, 1H, H6-phenyl), 7.54 (t, J = 7.6 Hz, 1H, H4-phenyl), 7.09 (t, J = 7.6 Hz, 1H, H5-phenyl), 6.86 (t, J = 8.8 Hz, 1H, H3-phenyl), 4.7 (s, 2H, CH2), 3.8 (s, 3H, OCH3).
Methyl (3-formylphenoxy)acetate (11b)
This compound was obtained using general procedure as a pale yellow oil without further purification in 63% yield. IR (KBr, cm-1): 1761 (C = O, ester), 1682 (C = O, aldehyde); 1H NMR (CDCl3, 400 MHz) δ: 10 (s, 1H, CHO), 7.51 (m, 2H, H5, H6-phenyl), 7.36 (s, 1H, H2-phenyl), 7.25 (br s, 1H, H4-phenyl), 4.71 (s, 2H, CH2), 3.92 (s, 3H, OCH3).
Methyl (4-formylphenoxy)acetate (11c)
This compound was obtained using general procedure as a pale yellow oil without further purification in 93% yield. IR (KBr, cm-1): 1742 (C = O, ester), 1695 (C = O, aldehyde); 1H NMR (CDCl3, 400 MHz) δ: 9.9 (s, 1H, CHO), 7.9 (d, J = 8 Hz, 2H, aromatic), 7.0 (d, J = 8 Hz, 2H, aromatic), 4.73 (s, 2H, CH2), 3.82 (s, 3H, CH3O).
Synthesis of (E)-3-(3-chloro-4-hydroxy-5-methoxybenzylidene)-7-methoxychroman-4-one (5a)
A solution of 7-methoxychroman-4-one (4, 100 mg, 0.56 mmol), 5-chlorovanillin (7a, 105 mg, 0.56 mmol) in EtOH (2 ml) was stirred at room temperature for 5 min, while a stream of HCl gas was introduced. After 24 h at room temperature, the precipitation was filtrated, crystallized from EtOH to give 5a as red solid in 52% yield. m.p. 171–173°C; IR (KBr, cm-1): 3428 (OH), 1655 (C = O); 1H NMR (DMSO-d6, 400 MHz) δ: 7.8 (d, J = 8.8 Hz, 1H, H5-chromanone), 7.6 (br s, 1H, CH-vinylic), 7.07 (d, J = 1.5 Hz, 1H, H2-phenyl), 7.03 (d, J = 1.6 Hz, 1H, H6-phenyl), 6.7 (dd, J = 6.4 and 2.4 Hz, 1H, H6-chromanone), 6.5 (d, J = 2.3 Hz, 1H, H8-chromanone), 5.4 (d, J = 1.6 Hz, 2H, H2-chromanone), 3.89 (s, 3H, OCH3), 3.83 (s, 3H, OCH3); Anal. Calcd for C18H15ClO5: C, 62.35; H, 4.36. Found: C, 62.04; H, 4.40.
Synthesis of (E)-3-(3-chloro-4,5-dimethoxybenzylidene)-7-methoxychroman-4-one (5b)
A solution of 7-methoxychroman-4-one (4, 500 mg, 2.8 mmol), 3-chloro-4,5-dimethoxybenzaldehyde (9a, 562 mg, 2.8 mmol) in EtOH (10 ml) was stirred at room temperature for 25 min, while a stream of HCl gas was introduced. After 24 h at room temperature, the precipitated solid was filtrated, crystallized from EtOH to afford compound 5b as orange solid in 85% yield. m.p. 125–127°C; IR (KBr, cm-1): 1670 (C = O); 1H NMR (CDCl3, 400 MHz) δ: 7.96 (d, J = 8.8 Hz, 1H, H5-chromanone), 7.7 (br s, 1H, CH-vinylic), 6.8 (br s, 1H, H2-phenyl), 6.7 (br s, 1H, H6-phenyl), 6.6 (d, J = 8 Hz, 1H, H6-chromanone), 6.4 (br s, 1H, H8-chromanone), 5.3 (s, 2H, H2-chromanone), 3.92 (s, 3H, OCH3), 3.9 (s, 3H, OCH3), 3.85 (s, 3H, OCH3); Anal. Calcd for C19H17ClO5: C, 63.25; H, 4.75. Found: C, 62.88; H, 4.41.
Synthesis of (E)-methyl 2-(2-chloro-6-methoxy-4-((7-methoxy-4-oxo-2H-chromen-3(4H)-ylidene)methyl)phenoxy)acetate (5c)
A solution of 7-methoxychroman-4-one (4, 300 mg, 1.68 mmol), methyl 2-(2-chloro-4-formyl-6-methoxyphenoxy)acetate (8a, 435 mg, 1.68 mmol) in MeOH (6 ml) was stirred at room temperature for 20 min, while a stream of HCl gas was introduced. After 24 h at room temperature, the precipitation was filtrated, crystallized from MeOH to give compound 5c as pink solid in 64% yield. m.p. 118–121°C; IR (KBr, cm-1): 1767 (C = O), 1750 (C = O); 1H NMR (CDCl3, 400 MHz) δ: 7.9 (d, J = 9.2 Hz, 1H, H5-chromanone), 7.7 (br s, 1H, CH-vinylic), 6.9 (s, 1H, H3-phenyl), 6.7 (s, 1H, H5-phenyl), 6.6 (d, J = 8.8 Hz, 1H, H6-chromanone), 6.4 (br s, 1H, H8-chromanone), 5.3 (s, 2H, OCH2CO), 4.7 (s, 2H, H2-chromanone), 3.87 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 3.83 (s, 3H, OCH3); Anal. Calcd for C21H19ClO7: C, 60.22; H, 4.57. Found: C, 60.36; H, 4.71.
Synthesis of (E)-methyl 2-(2-chloro-6-ethoxy-4-((7-methoxy-4-oxo-2H-chromen-3(4H)-ylidene)methyl)phenoxy)acetate (5d)
A solution of 7-methoxychroman-4-one (4, 200 mg, 1.12 mmol), methyl 2-(2-chloro-6-ethoxy-4-formylphenoxy)acetate (8b, 288 mg, 1.12 mmol) in MeOH (4 ml) was stirred at room temperature for 25 min, while a stream of HCl gas was introduced. After 24 h at room temperature, the precipitated solid was filtrated, crystallized from MeOH to give pure compound 5d as a pink solid in 52% yield. m.p. 151–153°C; IR (KBr, cm-1): 1759 (C = O), 1737 (C = O); 1H NMR (CDCl3, 400 MHz) δ: 7.9 (d, J = 8.8 Hz, 1H, H5-chromanone), 7.8 (br s, 1H, CH-vinylic), 6.9 (s, 1H, H3-phenyl), 6.6 (br s, 2H, H6-chromanone and H5-phenyl), 6.4 (br s, 1H, H8-chromanone), 5.1 (s, 2H, CH2), 4.7 (s, 2H, H2-chromanone), 4.1 (m, 2H, CH2), 3.85 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 1.5 (t, J = 6.8 Hz, 3H, CH3); Anal. Calcd for C22H21ClO7: C, 61.05; H, 4.89. Found: C, 60.83; H, 5.03.
Synthesis of (E)-ethyl 2-(2-chloro-6-ethoxy-4-((7-methoxy-4-oxo-2H-chromen-3(4H)-ylidene)methyl)phenoxy)acetate (5e)
A solution of 7-methoxychroman-4-one (4, 50 mg, 0.28 mmol), ethyl 2-(2-chloro-6-ethoxy-4-formylphenoxy)acetate (8c, 76 mg, 0.28 mmol) in EtOH (2 ml) was stirred at room temperature for 15 min, while a stream of HCl gas was introduced. After 24 h at room temperature, the precipitation was filtrated, crystallized from EtOH to give compound 5e as a white solid in 43% yield. m.p. 122–123°C; IR (KBr, cm-1): 1748 (C = O); 1H NMR (CDCl3, 400 MHz) δ: 7.9 (d, J = 8.0, 1H, H5-chromanone), 7.8 (br s, 1H, CH-vinylic), 6.9 (s, 1H, H3-phenyl), 6.6 (br s, 2H, H6-chromanone and H5-phenyl), 6.4 (s, 1H, H8-chromanone), 5.1 (s, 2H, H2-chromanone), 4.6 (s, 2H, OCH2CO), 4.2 (m, 2H, CH2), 4.1 (m, 2H, CH2), 3.85 (s, 3H, OCH3), 1.4 (m, 3H, CH3), 1.3 (s, 3H, CH3); Anal. Calcd for C23H23ClO7: C, 61.82; H, 5.19. Found: C, 62.01; H, 4.89.
Synthesis of (E)-methyl 2-(4-((7-methoxy-4-oxo-2H-chromen-3(4H)-ylidene)methyl)phenoxy)acetate (5f)
A solution of 7-methoxychroman-4-one (4, 100 mg, 0.56 mmol), methyl 2-(4-formylphenoxy)acetate (11c, 109 mg, 0.56 mmol) in anhydrous MeOH (2 ml) was stirred at room temperature for 25 min, while a stream of HCl gas was introduced. After 24 h at room temperature, the precipitation was filtrated, crystallized from methanol to afford pure compound 5f as a red solid in 71% yield. m.p. 131–133°C; IR (KBr, cm-1): 1760 (C = O), 1667 (C = O); 1H NMR (CDCl3, 400 MHz) δ: 7.8 (d, J = 8.4 Hz, 1H, H5-chromanone), 7.6 (s, 1H, CH-vinylic), 7.41 (d, J = 8.8 Hz, 2H, H2- and H6-phenyl), 7.05 (d, J = 8.4 Hz, 2H, H3- and H5-phenyl), 6.7 (d, J = 8.2 Hz, 1H, H6-chromanone), 6.5 (s, 1H, H8-chromanone), 5.4 (s, 2H, OCH2CO), 4.8 (s, 2H, H2-chromanone), 3.8 (s, 3H, OCH3), 3.7 (s, 3H, OCH3); MS, m/z: 354, 295, 281, 265, 253, 151, 131, 115, 77, 63, 45. Anal. Calcd for C20H18O6: C, 67.79; H, 5.12; Found: C, 68.00; H, 4.98.
Synthesis of (E)-ethyl 2-(4-((7-methoxy-4-oxo-2H-chromen-3(4H)-ylidene)methyl)phenoxy)acetate (5g)
A solution of 7-methoxychroman-4-one (4, 100 mg, 0.56 mmol), methyl 2-(4-formylphenoxy)acetate (11c, 109 mg, 0.56 mmol) in anhydrous EtOH (2 ml) was stirred at room temperature for 20 min, while a stream of HCl gas was introduced. After 24 h at room temperature, the precipitation was filtrated, crystallized from ethanol to give compound 5 g as a red solid in 71% yield. m.p. 133–135°C; IR (KBr, cm-1): 1760 (C = O), 1659 (C = O); 1H NMR (DMSO-d6, 400 MHz) δ: 7.8 (d, J = 8.4 Hz, 1H, H5-chromanone), 7.6 (s, 1H, CH-vinylic), 7.41 (d, J = 8.8 Hz, 2H, H2- and H6-phenyl), 7.05 (d, J = 8.4 Hz, 2H, H3- and H5-phenyl), 6.7 (d, J = 8.2 Hz, 1H, H6-chromanone), 6.5 (s, 1H, H8-chromanone), 5.4 (s, 2H, OCH2CO), 4.8 (s, 2H, H2-chromanone), 3.8 (s, 3H, OCH3), 3.7 (s, 3H, OCH3); MS, m/z: 368, 281, 253, 164, 151, 131, 115, 77. Anal. Calcd for C21H20O6: C, 68.47; H, 5.47. Found: C, 68.27; H, 5.51.
Synthesis of (E)-propyl 2-(4-((7-methoxy-4-oxo-2H-chromen-3(4H)-ylidene)methyl)phenoxy)acetate (5h)
A solution of 7-methoxychroman-4-one (4, 300 mg, 1.68 mmol), methyl 2-(4-formylphenoxy)acetate (11c, 327 mg, 1.68 mmol) in n-PrOH (6 ml) was stirred at room temperature for 15 min, while a stream of HCl gas was introduced. After 24 h at room temperature, the precipitation was filtrated, crystallized from PrOH to give compound 5 h as a pink solid in 47% yield. m.p. 102–105°C; IR (KBr, cm-1): 1758 (C = O), 1660 (C = O); 1H NMR (DMSO-d6, 400 MHz) δ: 7.8 (d, J = 8.4 Hz, 1H, H5-chromanone), 7.6 (s, 1H, CH-vinylic), 7.4 (d, J = 8.0 Hz, 2H, H2- and H6-phenyl), 7.0 (d, J = 8.4 Hz, 2H, H3- and H5-phenyl), 6.7 (d, J = 8.0 Hz, 1H, H6-chromanone), 6.5 (s, 1H, H8-chromanone), 5.4 (s, 2H, OCH2CO), 4.89 (s, 2H, H2-chromanone), 4.09 (t, J = 6.4 Hz, 2H, OCH2), 3.8 (s, 3H,OCH3), 1.6 (m, 2H, CH2), 0.87 (t, J = 7.2 Hz, 3H, CH3); MS, m/z: 382, 354, 295, 281, 265, 151, 131, 115, 77, 69, 57, 43. Anal. Calcd for C22H22O6: C, 69.10; H, 5.80. Found: C, 68.90; H, 6.07.
Synthesis of (E)-butyl 2-(4-((7-methoxy-4-oxo-2H-chromen-3(4H)-ylidene)methyl)phenoxy)acetate (5i)
A solution of 7-methoxychroman-4-one (4, 100 mg, 0.56 mmol), methyl 2-(4-formylphenoxy)acetate (11c, 109 mg, 0.56 mmol) in n-BuOH (2 ml) was stirred at room temperature for 10 min, while a stream of HCl gas was introduced. After 24 h at room temperature, the precipitation was filtrated, crystallized from n-BuOH to afford compound 5i as a pink solid in 36% yield. m.p. 85–87°C; IR (KBr, cm-1): 1766 (C = O), 1664 (C = O); 1H NMR (DMSO-d6, 400 MHz) δ: 7.8 (d, J = 8.8 Hz, 1H, H5-chromanone), 7.6 (br s, 1H, CH-vinylic), 7.4 (d, J = 8.0 Hz, 2H, H2- and H6-phenyl), 7.0 (d, J = 8.4 Hz, 2H, H3- and H5-phenyl), 6.7 (d, J = 7.6 Hz, 1H, H6-chromanone), 6.5 (br s, 1H, H8-chromanone), 5.4 (s, 2H, OCH2CO), 4.8 (s, 2H, H2-chromanone), 4.1 (t, J = 7.6 Hz, 2H, OCH2), 3.8 (s, 3H, OCH3), 1.5 (m, 2H, CH2), 1.3 (m, 2H, CH2), 0.8 (t, J = 7.6 Hz, 3H, CH3); MS, m/z: 396, 281, 265, 253, 167, 149, 131, 115, 107, 81, 57, 41. Anal. Calcd for C23H24O6: C, 69.68; H, 6.10. Found: C, 69.80; H, 6.37.
Synthesis of (E)-methyl 2-(2-((7-methoxy-4-oxo-2H-chromen-3(4H)-ylidene)methyl)phenoxy)acetate (5j)
A solution of 7-methoxychroman-4-one (4, 100 mg, 0.56 mmol), methyl 2-(2-formylphenoxy)acetate (11a, 109 mg, 0.56 mmol) in anhydrous MeOH (2 ml) was stirred at room temperature for 35 min, while a stream of HCl gas was introduced. After 24 h at room temperature, the precipitation was filtrated, crystallized from methanol to give compound 5j as yellow viscous oil in 32% yield. IR (KBr, cm-1): 1755 (C = O), 1664 (C = O); 1H NMR (CDCl3, 400 MHz) δ: 7.98 (br s, 1H, H5-chromanone), 7.96 (s, 1H, CH-vinylic), 7.35 (d, J = 7.6 Hz, 1H, H3-phenyl), 7.07 (m, 2H, H4- and H5-phenyl), 6.81 (d, J = 8 Hz, 1H, H6-phenyl), 6.62 (d, J = 7.6 Hz, 1H, H6-chromanone), 6.4 (s, 1H, H8-chromanone), 5.22 (s, 2H, OCH2CO), 4.7 (s, 2H, H2-chromanone), 3.8 (s, 3H, OCH3), 3.79 (s, 3H, OCH3); MS, m/z: 354, 295, 281, 265, 151, 131, 77, 67, 57, 43. Anal. Calcd for C20H18O6: C, 67.79; H, 5.12. Found: C, 68.02; H, 4.98.
Synthesis of (E)-methyl 2-(3-((7-methoxy-4-oxo-2H-chromen-3(4H)-ylidene)methyl)phenoxy)acetate (5k)
A solution of 7-methoxychroman-4-one (4, 100 mg, 0.56 mmol), methyl 2-(3-formylphenoxy)acetate (11b, 109 mg, 0.56 mmol) in anhydrous MeOH (2 ml) was stirred at room temperature for 30 min, while a stream of HCl gas was introduced. After 24 h at room temperature, the precipitation was filtrated, crystallized from methanol to give compound 5 k as a pink solid in 79% yield. m.p. 111–114°C; IR (KBr, cm-1): 1758 (C = O), 1670 (C = O); 1H NMR (DMSO-d6, 400 MHz) δ: 7.8 (d, J = 8.8 Hz, 1H, H5-chromanone), 7.7 (s, 1H, CH-vinylic), 7.4 (t, J = 6.8 Hz, 1H, H6-phenyl), 7.0 (m, 3H, H2 and H4- and H5-phenyl), 6.7 (dd, J = 6.8 and 2.0 Hz, 1H, H6-chromanone), 6.5 (d, J = 2.0 Hz, 1H, H8-chromanone), 5.4 (s, 2H, OCH2CO), 4.9 (s, 2H, H2-chromanone), 3.8 (s, 3H, OCH3), 3.7 (s, 3H, OCH3); MS, m/z: 354, 281, 178, 167, 150, 122, 107, 79, 69, 57, 43. Anal. Calcd for C20H18O6: C, 67.79; H, 5.12. Found: C, 67.65; H, 5.33.
Cytotoxicity assay
The in-vitro cytotoxic activity of each synthesized compounds 5a-k was assessed using MTT colorimetric assay according to the literature method [22]. Each set of experiments was independently performed three times. For each compound, the concentration causing 50% cell growth inhibition (IC50) compared with the control was calculated from concentration-response curves by regression analysis.
Results and discussion
Chemistry
Reaction sequence employed for the synthesis of (E)-3-benzylidene-7-methoxychroman-4-one derivatives 5a-k is shown in Scheme 1. The reaction of resorcinol 1 with 3-chloropropionic acid in the presence of trifluoromethane sulfonic acid furnished 2′,4′-dihydroxy-3-chloropropiophenone (2) which was cyclized using 2 M NaOH to give 7-hydroxy-4-chromanone (3). Compound 4 was obtained by reacting intermediate 3 with iodomethane in the presence of potassium carbonate in DMF. Condensation of 7-methoxychroman-4-one (4) with suitable aldehydes 7–9 and 11 in appropriate alcohol in the presence of gaseous HCl gave the target compounds 5a-k.
The corresponding aldehydes 7–9 and 11 were prepared as shown in Scheme 2. Chlorination of 3-alkoxy-4-hydroxybenzaldehyde 6a,b using acetic acid as a solvent gave 3-chloro-4-hydroxy-5-alkoxybenzaldehyde 7a,b which was reacted with suitable alkyl bromoacetate, in the presence of potassium carbonate to give compounds 8a-c. On the other hand, O-methylation of compound 7a afforded dimethoxybenzaldehyde derivative 9a. Methyl (formylphenoxy)acetate 11a-c was synthesized by heating compounds 10a-c with methyl bromoacetate and potassium carbonate in ethyl methyl ketone.
In vitro cytotoxic activity
The cytotoxic activity of synthesized compounds 5a-k was evaluated against three cell lines namely MDA-MB-231 (breast cancer), KB (nasopharyngeal epidermoid carcinoma) and SK-N-MC (human neuroblastoma) cells. The results of cytotoxic assay were mentioned as IC50 (μg/ml) of compounds in comparison with reference drug etoposide in Table 1.
In the case of MDA-MB-231 cell line, the IC50 values of all compounds were ≤20 μg/ml with the exception of compounds 5d and 5e. Furthermore, compounds 5b and 5i exhibited the highest cytotoxic activity against this cell line (IC50 < 10 μg/ml). Compound 5b was also the most potent derivative against KB cell line with IC50 value of 25.04 μg/ml. Beside compound 5b, compound 5a exhibited good activity against KB cells, but remaining compounds 5c-k showed no activity against this cell line (IC50 >100 μg/ml). Against SK-N-MC cells, compound 5b followed by compounds 5a and 5c showed significant inhibitory activity with IC50 values of 9.64, 12.6 and 58.04 μg/ml, respectively.
Overall, it is clear that among the test compounds described in this study, the 3-chloro-4,5-dimethoxybenzylidene derivative 5b demonstrated better cytotoxic profile against all tested cell lines (IC50 values = 7.56–25.04 μg/ml). Generally, the comparison of IC50 values of compound 5b with those of etoposide demonstrated that the cytotoxic activity of compound 5b against MDA-MB-231 and SK-N-MC cells is more than etoposide.
In this work, as part of an ongoing program to find new cytotoxic agents, we have focused our attention on modification of the 3-benzylidene-4-chromanones and introducing new functionality on the benzylidene moiety. Thus, we designed novel 3-benzylidene-4-chromanones that possessed a 2-(2-chloro-6-alkoxyphenoxy)acetic acid ester. These modifications were made on the basis of SJ-172550, a new cytotoxic agent possessing 2-(2-chloro-6-ethoxyphenoxy)acetic acid methyl ester attached to the pyrazolone ring. Surprisingly, compound 5d, the chromanone analog of SJ-172550 showed no activity against tested cell lines. Also, the ethyl ester counterpart of 5d (compound 5e) was inactive against tumor cell lines. However, the 2-(2-chloro-6-methoxyphenoxy)acetic acid methyl ester analog 5c was active against MDA-MB-231 and SK-N-MC cells. We have briefly investigated the SAR of compounds by simplification of the functionality on the benzylidene part of the basic molecule.
As can be deduced from the cytotoxic data of compounds 5f-k which characterized by the lack of 2-chloro-6-alkoxy functionality, the cytotoxic activity against MDA-MB-231 can be served by the simple phenoxyacetic acid ester derivatives. However, the lack of 2-chloro-6-alkoxy functionality results in the lack of activity against KB and SK-N-MC cells. Among the compounds 5f-k, butyl ester derivative 5i showed the highest activity against MDA-MB-231 being 3-fold more potent than standard drug etoposide.
To determine the effect of acetic acid ester substitution in compound 5c, we prepared both the 4-hydroxy derivative 5a and 4-methoxy analog 5b. When compared to 5c, both compounds had similar or better in vitro activities against tested cell lines.
The cytotoxic activities of regio-isomeric compounds 5f, 5j and 5 k against MDA-MB-231cells revealed that changing the position of oxyesteric group has led to non-significant changes in activities. As seen from data, in poly-substituted compounds changing of methoxy group on phenyl ring to ethoxy group (for example 5d versus 5c) dramatically decreased the cytotoxic potency in MDA-MB-231 and SK-N-MC cells.
In summary, in the pursuit for finding new cytotoxic agents, we replaced the pyrazolone part of well-known cytotoxic agent SJ-172550 with 7-methoxychroman-4-one. Although the direct analog of SJ-172550 (compound 5d) did not show any cytotoxic activity against tested cell lines, but 2-(2-chloro-6-methoxyphenoxy)acetic acid methyl ester analog 5c showed some activity against MDA-MB-231 and SK-N-MC cells. Further modification of compound 5c resulted in the 3-chloro-4,5-dimethoxybenzylidene derivative 5b which demonstrated better cytotoxic profile against all tested cell lines (IC50 values = 7.56–25.04 μg/ml).
It is worthwhile to mention that, since we have originally designed the target compounds based on p53-dependent cytotoxic agent SJ-172550, it was better using the latter compound as standard drug in our cytotoxic assay. However, our primary cytotoxic experiments on the closest compound to SJ-172550 (compound 5d) in a side-by-side comparison manner with etoposide revealed that compound 5d had no activity against cancer cell lines. On the other hand, simplified compounds 5a and 5b with more dissimilarity respect to the SJ-172550 showed better profile of cytotoxicity. Based on these results, it seems that a different mechanism is responsible for potential cytotoxic activity of compound 5b prototype.
We employed MTT cell viability assay as a standard and well-documented in vitro method for evaluation of the cytotoxic potential of designed compounds. Although these types of in vitro models are beneficial and promising as early screening tools for finding new lead compounds, but these models are associated with some limitations [23]. Thus, for efficacy and safety evaluation of lead compounds, conducting a method based on animal model is necessary in the next steps of study.
In conclusion, the results demonstrated that the cytotoxic activity of 3-(3-chloro-4,5-dimethoxybenzylidene)-7-methoxychroman-4-one (5b) against MDA-MB-231 and SK-N-MC cells is more than standard drug etoposide. Therefore, compound 5b prototype bearing 3-chloro-4,5-dimethoxybenzylidene moiety could be considered as novel lead compound for further developing new anticancer chemotherapeutics. Although, compound 5b showed promising activity in vitro, but to identify a promising anticancer drug candidate that has good pharmacokinetic and toxicological profiles, the in vivo ADME-Tox studies of compound 5b prototype should be conducted.
References
Chen YL, Lin SZ, Chang JY, Cheng YL, Tsai NM, Chen SP, Chang WL, Harn HJ: In vitro and in vivo studies of a novel potential anticancer agent of isochaihulactone on human lung cancer A549 cells. Biochem Pharmacol. 2006, 72: 308-319. 10.1016/j.bcp.2006.04.031.
Hanahan D, Weinberg RA: The hallmarks of cancer. Cell. 2000, 100: 57-70. 10.1016/S0092-8674(00)81683-9.
Ananda Kumar CS, Nanjunda Swamy S, Thimmegowda NR, Benaka Prasad SB, Yip GW, Rangappa KS: Synthesis and evaluation of 1-benzhydryl-sulfonyl-piperazine derivatives as inhibitors of MDA-MB-231 human breast cancer cell proliferation. Med Chem Res. 2007, 16: 179-187. 10.1007/s00044-007-9022-y.
Reddy MV, Su C-R, Chiou W-F, Liu Y-N, Chen RY-H, Bastow KF, Lee K-H, Wu T-S: Design, synthesis, and biological evaluation of Mannich bases of heterocyclic chalcone analogs as cytotoxic agents. Bioorg Med Chem. 2008, 16: 7358-7370. 10.1016/j.bmc.2008.06.018.
Firoozpour L, Edraki N, Nakhjiri M, Emami S, Safavi M, Ardestani SK, Khoshneviszadeh M, Shafiee A, Foroumadi A: Cytotoxic activity evaluation and QSAR study of chromene-based chalcones. Arch Pharm Res. 2012, 35: 2117-2125. 10.1007/s12272-012-1208-2.
Patil CB, Mahajan SK, Katti SA: Chalcone: A versatile molecule. J Pharm Sci Res. 2009, 1: 11-22.
Nowakowska Z: A review of anti-infective and anti-inflammatory chalcones. Eur J Med Chem. 2007, 42: 125-137. 10.1016/j.ejmech.2006.09.019.
Akihisa T, Tokuda H, Hasegawa D, Ukiya M, Kimura Y, Enjo F, Suzuki T, Nishino H: Chalcones and other compounds from the exudates of and their cancer chemopreventive effects. J Nat Prod. 2006, 69: 38-42. 10.1021/np058080d.
Dimmock JR, Elias DW, Beazely MA, Kandepu NM: Bioactivities of chalcones. Curr Med Chem. 1999, 6: 1125-1149.
Perjési P, Das U, De Clercq E, Balzarini J, Kawase M, Sakagami H, Stables JP, Lorand T, Rozmer Z, Dimmock JR: Design, synthesis and antiproliferative activity of some 3-benzylidene-2,3-dihydro-1-benzopyran-4-ones which display selective toxicity for malignant cells. Eur J Med Chem. 2008, 43: 839-845. 10.1016/j.ejmech.2007.06.017.
Reed D, Shen Y, Shelat AA, Arnold LA, Ferreira AM, Zhu F: Identification and characterization of the first small molecule inhibitor of MDMX. J Biol Chem. 2010, 285: 10786-10796. 10.1074/jbc.M109.056747.
Nakhjiri M, Safavi M, Alipour E, Emami S, Atash AF, Jafari-Zavareh M, Ardestani SK, Khoshneviszadeh M, Foroumadi A, Shafiee A: Asymmetrical 2,6-bis(benzylidene)cyclohexanones: Synthesis, cytotoxic activity and QSAR study. Eur J Med Chem. 2012, 50: 113-123.
Mahmoodi M, Aliabadi A, Emami S, Safavi M, Rajabalian S, Mohagheghi MA, Khoshzaban A, Samzadeh-Kermani A, Lamei N, Shafiee A, Foroumadi A: Synthesis and in-vitro cytotoxicity of poly-functionalized 4-(2-arylthiazol-4-yl)-4H-chromenes. Arch Pharm. 2010, 7: 411-416.
Fallah-Tafti A, Tiwari R, Shirazi AN, Akbarzadeh T, Mandal D, Shafiee A, Parang K, Foroumadi A: 4-Aryl-4H-chromene-3-carbonitrile derivatives: evaluation of Src kinase inhibitory and anticancer activities. Med Chem. 2011, 7: 466-472. 10.2174/157340611796799258.
Bazl R, Ganjali M, Saboury A, Foroumadi A, Nourozi P, Amanlou M: A new strategy based on pharmacophore-based virtual screening in adenosine deaminase inhibitors detection and in-vitro study. DARU J Pharmaceut Sci. 2012, 20: 64-10.1186/2008-2231-20-64.
Rafinejad A, Fallah-Tafti A, Tiwari R, Nasrolahi Shirazi A, Mandal D, Shafiee A, Parang K, Foroumadi A, Akbarzadeh T: 4-Aryl-4H-naphthopyrans derivatives: One-pot synthesis, evaluation of Src kinase inhibitory and anti-proliferative activities. DARU J Pharmaceut Sci. 2012, 20: 100-10.1186/2008-2231-20-100.
Nadri H, Pirali-Hamedani M, Moradi A, Sakhteman A, Vahidi A, Sheibani V, Asadipour A, Hosseinzadeh N, Abdollahi M, Shafiee A, Foroumadi A: 5,6-Dimethoxybenzofuran-3-one derivatives: a novel series of dual Acetylcholinesterase/Butyrylcholinesterase inhibitors bearing benzyl pyridinium moiety. DARU J Pharmaceut Sci. 2013, 21: 15-10.1186/2008-2231-21-15.
Foroumadi A, Samzadeh-kermani A, Emami S, Dehghan G, Sorkhi M, Arabsorkhi F, Heidari MR, Abdollahi M, Shafiee A: Synthesis and antioxidant properties of substituted 3-benzylidene-7-alkoxychroman-4-ones. Bioorg Med Chem Lett. 2007, 17: 6764-6769. 10.1016/j.bmcl.2007.10.034.
Ayati A, Falahati M, Irannejad H, Emami S: Synthesis, in vitro antifungal evaluation and in silico study of 3-azolyl-4-chromanone phenylhydrazones. DARU J Pharmaceut Sci. 2012, 20: 46-10.1186/2008-2231-20-46.
Hua DH, Huang X, Chen Y, Battina SK, Tamura M, Noh SK, Koo SI, Namatame I, Tomoda H, Perchellet EM, Perchellet JP: Total syntheses of (+)-chloropuupehenone and (+)-chloropuupehenol and their analogues and evaluation of their bioactivities. J Org Chem. 2004, 69: 6065-6078. 10.1021/jo0491399.
Cheng M-F, Fang J-M: Liquid-phase combinatorial synthesis of 1,4-benzodiazepine-2,5-diones as the candidates of endothelin receptor antagonism. J Comb Chem. 2004, 6: 99-104. 10.1021/cc030034d.
Mosmann T: Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983, 65: 55-63. 10.1016/0022-1759(83)90303-4.
Shetab-Boushehri SV, Abdollahi M: Current concerns on the validity of in vitro models that use transformed neoplastic cells in pharmacology and toxicology. Int J Pharmacol. 2012, 8: 594-595. 10.3923/ijp.2012.594.595.
Acknowledgements
This work was financially supported by grants from Tehran University of Medical Sciences and Iran National Science Foundation (INSF).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SN: Synthesis of target compounds. EA: Supervision of the synthetic part. SE: Collaboration in design and identifying of the structures of target compounds, manuscript preparation. MS: Performed the cytotoxic tests. SKA: Supervision of the cytotoxic tests. ARG: Collaboration in identifying the structures of target compounds. AS: Collaboration in identifying the structures of target compounds. AF: Design of target compounds and supervision of the synthetic and pharmacological parts. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Noushini, S., Alipour, E., Emami, S. et al. Synthesis and cytotoxic properties of novel (E)-3-benzylidene-7-methoxychroman-4-one derivatives. DARU J Pharm Sci 21, 31 (2013). https://doi.org/10.1186/2008-2231-21-31
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2008-2231-21-31