
Abstract
The objective of this study was to develop proliposomes and self-nanoemulsifying drug delivery system (SNEDDS) for a poorly bioavailable drug, valsartan, and to compare their in vivo pharmacokinetics. Proliposomes were prepared by thin-film hydration method using different lipids such as soy phosphatidylcholine (SPC), hydrogenated soy phosphatidylcholine (HSPC), distearyl phosphatidylcholine (DSPC), dimyristoylphosphatidylcholine (DMPC), and dimyristoyl phosphatidylglycerol sodium (DMPG) and cholesterol in various ratios. SNEDDS formulations were prepared using varying concentrations of capmul MCM, labrafil M 2125, and Tween 80. Both proliposomes and SNEDDS were evaluated for particle size, zeta potential, in vitro drug release, in vitro permeability, and in vivo pharmacokinetics. In vitro drug release was carried out in purified water and 0.1 N HCl using USP type II dissolution apparatus. In vitro drug permeation was studied using parallel artificial membrane permeation assay (PAMPA) and everted rat intestinal permeation techniques. Among the formulations, the proliposomes with drug/DMPG/cholesterol in the ratio of 1:1:0.5 and SNEDDS with capmul MCM (16.0% w/w), labrafil M 2125 (64.0% w/w), and Tween 80 (18.0% w/w) showed the desired particle size and zeta potential. Enhanced drug release was observed with proliposomes and SNEDDS as compared to pure valsartan. Valsartan permeability across PAMPA and everted rat intestinal permeation models was significantly higher with proliposomes and SNEDDS. Following single oral administration of proliposomes and SNEDDS, a relative bioavailability of 202.36 and 196.87%, respectively, was achieved compared to pure valsartan suspension. The study results indicated that both proliposomes and SNEDDS formulations are comparable in improving the oral bioavailability of valsartan.
Similar content being viewed by others

INTRODUCTION
Oral route is the most preferred route of administration because of its convenience. However, the delivery of drug by oral route may result in low bioavailability because of its poor solubility in gastrointestinal (GI) fluids, inadequate permeation across the GI membrane, and extensive first-pass affect. Proliposomes and SNEDDS are reported as drug delivery carriers for enhancing the oral bioavailability of drugs with poor bioavailability (1–7). Proliposomes are dry, free-flowing powders which can form liposomal suspension upon hydration with water. The stability of proliposomes is far superior to liposomes making them more suitable for oral delivery (8). SNEDDS is an isotropic mixture of oil, surfactants, and co-surfactants that form fine oil-in-water emulsion upon mild agitation in the presence of water (9). In the presence of GI fluids, these systems undergo rapid self-emulsification producing nanosized globules of high surface area resulting in enhanced rate and extent of absorption with consistent plasma concentration time profiles (10). Both proliposomes and SNEDDS are emerging platform technologies for improving the oral delivery of drugs with poor bioavailability.
Valsartan is an orally active non-peptide and highly selective angiotensin II antagonist acting on the AT1 receptor subtype (11). It relaxes blood vessels resulting in lowering blood pressure and improved blood flow. Valsartan exhibits poor solubility in water (0.18 mg/ml) and thus low oral bioavailability (~23%) (12,13). Peak plasma concentrations of valsartan have been reported between 2 and 4 h following oral administration, and approximately 94 to 97% of the drug was bound to plasma proteins, while the half-life of valsartan was reported in the range of 6–9 h (14). However, valsartan is soluble in neutral pH and shows low permeability. The low permeability of valsartan is reportedly due its two acidic centers, the carboxylic acid group (COOH), and the tetrazole ring with pKa values of 4.7 and 3.9 (15). Because of these two close pKa values, the drug exhibits poor solubility and low permeability in the gastrointestinal tract (GIT). Various formulation strategies have been reported to improve the oral bioavailability of valsartan, which includes the use of cyclodextrin complexes (16), nanoparticles (17), solid dispersions (18), micronization (19), self-microemulsifying drug delivery system (20), proniosomes (21), and mucoadhesive pellets (22). Most of these reports have been focused on improving the solubility of valsartan with limited success in improving the permeation and absorption across the GI leading to lower oral bioavailability. Therefore, our study was focused on developing an enhanced formulation for valsartan using proliposomes and SNEDDS that are capable of improving the drug permeation and thereby enhancing the absorption and oral bioavailability of valsartan. Although proliposomes and SNEDDS are well known for bioavailability enhancement, no study was reported till date comparing both the delivery technologies for improved oral delivery. SNEDDS is a well-established platform technology with successful products in the market; however, there is no approved proliposomal-based drug product for oral delivery in spite of its extensive utilization in the formulation research in last two decades. Therefore, in this study, we made an attempt to compare these drug delivery systems for improved in vitro and in vivo performance using poorly bioavailable drug, valsartan.
Lipids are known to promote the transport of drugs through lymphatic route (23). Many of the possible mechanisms of uptake include increasing transcellular absorption of drug by increasing the membrane fluidity, enhancing the paracellular transport by opening the tight junction, inhibiting the P-glycoprotein and cytochrome P450 to increase intracellular concentration, and formation of lipoprotein/chylomicron by phospholipid (24). In this study, the prepared formulations (proliposomes and SNEDDS) containing valsartan were measured for particle size, zeta potential, in vitro drug release, in vitro permeability, and in vivo pharmacokinetics.
MATERIALS AND METHODS
Materials
Valsartan was purchased from Tecoland Corporation, (Irvine, CA, USA). Soy phosphatidylcholine (SPC), distearoyl phosphatidylcholine (DSPC), hydrogenated soy phosphatidylcholine (HSPC), dimyristoylphosphatidylcholine (DMPC), and dimyristoyl phosphatidylglycerol sodium (DMPG) were purchased from Avanti Polar Lipids (Alabaster, AL, USA); cholesterol was purchased from Sigma-Aldrich, (St. Louis, MO, USA). Capmul MCM was purchased from Abitech Corporation (Janesville, WI, USA). Labrafil M 2125 was purchased from Alfa Chemicals (Binfield, Berkshire, UK). Tween 80 was purchased from EMD (Billerica, Massachussetts, USA). Avicel PH102 was purchased from FMC BioPolymers (Philadelphia, PA USA). Cell culture media were purchased from ATC Collection (Manassas, VA, USA); Transwell® plates (6-well) were purchased from Corning Life Sciences (Tewksbury, MA, USA). Cannulated Sprague–Dawley (SD) rats were purchased from Harlan Laboratories (Indianapolis, IN, USA); blank rat plasma was obtained from Biomedical (Winchester, VA, USA). Hard gelatin capsules (size 1) were purchased from Capsugel Inc., Morristown, NJ, USA, and all other reagents used are of analytical grade.
Preparation of Formulations
Proliposomes
Valsartan proliposomes were prepared using SPC, DSPC, DMPG, HSPC, DMPC, and cholesterol in various ratios. Formulation details and their respective compositions are described in Table I. Briefly, the specified amounts of valsartan, phospholipid, and cholesterol are weighed and dissolved in ethyl alcohol. The resultant solution was adsorbed onto microcrystalline cellulose (Avicel PH 102), and the excess solvent was removed using a rotavapor (Buchi R-210, Buchi Corporation, New Castle, DE, USA) to obtain dry proliposomes. The proliposomal formulations were passed through a sieve (50 mesh, 300 μm) to obtain free-flowing powders. The proliposomal formulations were filled into glass scintillation vials and stored at 4°C for further studies.
SNEDDS
Ternary phase diagrams were constructed using oils, surfactants, and co-surfactants; the selection of these excipients was based on the solubility of valsartan. A series of self-emulsifying systems were prepared with varying concentrations of oil (15–55% w/w), surfactant (35–65% w/w), and co-surfactant (5–25% w/w) at room temperature (25°C). For all the compositions, the total concentration of surfactant, co-surfactant, and oil added was always 100% (25). Several such compositions with varying concentrations were prepared to study the self-emulsifying regions. Ternary phase diagrams were constructed in the absence of valsartan using CHEMIX® ternary plot software.
Once the self-emulsifying region was identified, the formula compositions of SNEDDS were prepared with optimized concentrations of oil, surfactant, and co-surfactant (Table II). Briefly, valsartan was dissolved in required amount of capmul MCM in a glass vial with gentle heating at 40°C in a water bath with intermittent mixing using a vortex mixer to facilitate drug solubilization. Required amounts of labrafil M 2125 and Tween 80 are added to the glass vial and mixed well with gentle heating until the drug is completely dissolved. For all formulations, the level of valsartan was kept constant (i.e., 2%, w/w of the vehicle). The concentration of valsartan was maintained at this level taking into account of filling a therapeutic dose (5 mg) of drug into a size “1” hard gelatin capsule. The formulations were stored at room temperature for further studies.
Characterization of Formulations
Particle Size
The average particle size of the prepared proliposomes and SNEDDS formulations was measured by dynamic light scattering (DLS) technique using zetasizer (Nano ZS-90, Malvern Instruments, Malvern, Worcestershire, UK). All measurements (n = 3) were conducted at 25°C, using refractive index of 1.33.
Zeta Potential Measurement
The zeta potential of valsartan proliposomes and SNEDDS formulations were determined by photon correlation spectroscopy (PCS) using Malvern Zetasizer (Malvern Instruments, UK). The selected formulations were dispersed in water (diluted 100 times) and placed in an electrophoretic cell for measurement.
Analysis of Valsartan
Drug content, encapsulation efficiency, in vitro drug release, and in vitro permeation sample analysis was carried out by an HPLC analytical method using Waters-Alliance 2695 HPLC system (Waters Corporation, Milford, MA, USA). Inertsil ODS-3, C18, 250 × 4.6 mm, 5 μm stainless steel column (GL Sciences Inc., Torrance, CA, USA) was used for chromatographic elution of the samples. The mobile phase consisted of 0.02 M monobasic potassium phosphate and acetonitrile; pH adjusted to 3 using formic acid was pumped in a gradient fashion at 2 ml/min with detection at 265 nm. Sample injection volume was kept at 20 μl. The temperature of the column was 25°C, and sample run time was 15 min. A calibration curve was prepared in the concentration range of 0.25–50 μg/ml in acetonitrile. The method was validated for interday and intraday variations.
Formation of Liposomes
Cryogenic-transmission electron microscope (Cryo-TEM) was used to study the formation of vesicles upon hydration with purified water. Proliposomal powder was dispersed in purified water and hydrated by shaking gently for 5 min at room temperature to confirm the formation of liposomes. The hydrated sample was placed on to a sample grid (Lacey Formvar/carbon 300-mesh copper grids), and blotting was carried out using a Vitrobot Mark IV (FEI Company, Netherlands). Samples were observed under Cryo-TEM (JEOL-1230, JEOL, Tokyo, Japan) at a magnification of 15,000 at 100 kV.
Encapsulation Efficiency of Proliposomes
Samples of proliposomal formulations were reconstituted with purified water at room temperature. The hydrated proliposomal formulations of known concentration (0.5 mg/ml) were transferred to Centrifree® tubes (molecular weight cutoff of 30,000) (Merck Millipore Ltd., Cork, Ireland). The sample tubes were centrifuged at 2500 rpm for 20 min at room temperature. The encapsulated valsartan liposomes remain on the surface of the filter membrane, and the free drug was collected into the bottom tube of the filter (26). One milliliter of the separated aqueous phase was transferred to a 2 ml volumetric flask and diluted to volume with methanol and analyzed using the HPLC method. The amount of valsartan in the separated aqueous phase represented the amount of free drug. The experiment was performed in triplicate. The encapsulation efficiency was determined by calculating the difference between the initial amount taken and the amount recovered in the aqueous phase using the following equation (27)
Robustness of SNEDDS to Dilution
Robustness of valsartan SNEDDS to dilution was studied by diluting the formulations to 50 times with purified water, buffer pH 1.2, and buffer pH 6.8. The samples were measured for particle size following dilution. The diluted samples were stored for 6 h at room temperature and observed for any signs of phase separation or drug precipitation.
In Vitro Dissolution Studies
The in vitro release studies of the prepared formulations were conducted in USP Type II dissolution apparatus (VK7000, Varian Inc., Cary, NC, USA) connected to an autosampler (VK8000, Varian Inc., Cary, NC, USA). The paddle speed was set at 75 rpm, and temperature of the media was programmed to 37 ± 0.2°C. The dissolution medium used was 900 ml of purified water or SGF (pH 1.2 without enzymes). Pure valsartan, proliposomes, and SNEDDS formulations equivalent to 5 mg dose filled in to size 1 hard gelatin capsules were subjected to dissolution testing. Samples were collected at predetermined time intervals up to 1 h, filtered using a 0.45 μm filter, and analyzed using HPLC method described earlier. All experiments were carried out in triplicate. The percentage drug released from the formulations was estimated, and the average of six samples was used for analyzing the data.
Parallel Artificial Membrane Permeability Assay Studies
A 96-well filter plate (Millipore, Billerica, MA, USA) was used for parallel artificial membrane permeability assay (PAMPA) study. The filter plate consists of two parts: a receiver plate and a donor plate. Dimethyl sulfoxide (5% v/v) solution in water was used as receptor vehicle, and purified water was used as the donor vehicle in this study. Five microliters of 1% w/v solution of lecithin in dodecane was added into each donor plate well to enable formation of artificial membrane. The selected proliposomal and SNEDDS formulations were added to purified water to obtain a known concentration of the drug. Pure drug suspension of strength equivalent to the formulation was prepared using 0.5% methyl cellulose solution in water. One hundred fifty microliters of the prepared formulations (1 mg/ml) was added into donor plate wells, and 300 μl of DMSO solution (5% v/v) was added into each receptor plate well. The donor plate containing the sample was placed on the receptor cell plate carefully and incubated for 16 h at room temperature. Samples from the donor and the receptor plates were collected, filtered through 0.45 μm filter, and analyzed using a HPLC method. The permeation of drug across the membrane was estimated using Eq. 2 (2).
where P e is permeability in cm/s, A is effective filter area (0.24 cm2), V D is the well volume (0.3 ml) of the donor plate, V A is the well volume (0.2 ml) of the receptor plate, t is incubation time(s), CA (t) is the amount of drug in receptor plate well at time t, CD (t) is the amount of drug in donor plate well at time t, and Cequilibrium was estimated using Eq. 3.
Everted Rat Intestinal Permeation Studies
Everted rat intestinal permeation studies (ex vivo) were conducted according to the method reported by Ravis and Feldman (28). The anesthetized, non-treated SD rats were taken, and insertions were made in the midline of the stomach to remove the intestine. The first 15 cm of the intestine portion was discarded, and the isolated intestine was cleaned using saline solution (cold) to remove its contents. The cleaned isolated intestine was then everted using a glass rod. The everted intestine was cut into small portions of 6 cm long. Each portion of intestine was mounted in the perfusion apparatus housed with two ports. One port was used for addition and removal of serosal fluid (29), and the second port was used to maintain continuous circulation of carbon dioxide (5%) and oxygen (95%) during the study. Samples were withdrawn at regular time intervals for 1 h. The collected samples were centrifuged at 1500 rpm for 10 min and analyzed for valsartan using HPLC method.
Permeability Analysis
The absorption rate constant (K a) of valsartan from pure drug suspension, proliposomes, and SNEDDS was estimated by plotting the slope of concentration of drug remaining versus time. The effective permeability coefficient (P eff) of valsartan across the membrane was estimated using Eq. 4.
where Q in is the rate of intestinal fluid (0.2 ml/min) and A is the intestinal surface area which is considered to be the area of a cylinder (2πrl) with the length (l) and radius (r) of 0.18 cm. C in and C out are the concentrations at the inlet and corrected outlet, respectively. The C out (corrected) was measured using Eq. 5 (30).
where Q out is the estimated flow of the intestinal fluid (ml/min) at specific time point. The enhancement ratio (ER) was estimated using Eq. 6 (31).
Estimation of Valsartan in Plasma using LC/MS/MS
The stock solution of 1 mg/ml of valsartan was prepared by dissolving the pure valsartan in methanol, and working solutions in the concentration range of 10 to 500 ng/ml were prepared using methanol. The plasma samples for construction of the standard curves were spiked with these working standard solutions. Hydrochlorothiazide was used as the internal standard (IS). A protein precipitation method was employed for the extraction of valsartan and IS. Briefly, the working standard and IS solutions were added to 100 μl of plasma and mixed well. To the mixture, 500 μl of methanol was added and mixed vigorously by vortex mixing, followed by centrifugation at 10,000 rpm for 10 min at 4°C. The organic supernatant was transferred and evaporated to dryness by flushing nitrogen at room temperature. The final residue was reconstituted using 100 μl of methanol, and 5 μl of the sample was injected into LC/MS/MS system for analysis.
The chromatographic separation was carried out using a Zorbax SB-C18 column (Agilent, Santa Clara, CA, USA, particle size 5 μm; 2.1 × 150 mm) protected with a guard column (12.5 × 2.1 mm) at room temperature. The mobile phase consisted of acetonitrile and 0.1% formic acid in water (80:20). The flow rate used was 0.3 ml/min. LC/MS/MS system (API 3200, Framingham, MA, USA) was used for sample analysis. The mass ion spectrophotometer was operated in the negative-ion mode to monitor the transition of 434.033–178.800 for valsartan and 295.851–268.800 for IS. The ratios of corresponding peak heights of valsartan and IS were calculated. Precision and accuracy within-run and between-run was carried out using three different series at concentrations of 10, 50, and 100 ng/ml of mixture of valsartan and hydrochlorothiazide.
Pharmacokinetic Study
Jugular vein-cannulated male SD rats (230–250 g body weight) were used for the pharmacokinetic study. The male SD rats were divided into three subgroups of five animals each. All animals were fasted overnight prior to the day of experiment with free access to water. Using an oral gavage, group 1 animals were administered with 0.5 ml of pure valsartan suspension at a dose of 10 mg/kg and group 2 animals were administered with proliposomal formulation. Group 3 animals were administered with SNEDDS formulation of equal dose. Following oral administration, 0.3 ml of blood samples were collected from the cannula at 0.5, 1, 2, 4, 6, and 24 h and transferred into a heparinized eppendorf tubes. A “zero” time sample was collected prior to administration of the dose. The collected blood samples were centrifuged at 13,000 rpm for 8 min at 4°C, and supernatant plasma was collected and stored at −80°C until analysis. Food was provided to the rats after 4 h following oral dosing. The animal study protocol was approved by the IACUC, Western University of Health Sciences, Pomona, California, USA. Animals were sacrificed after completion of the study according to the procedure defined in the IACUC protocol.
Statistical Analysis
Statistical analysis of the in vitro drug release was conducted by two-way analysis of variance (ANOVA) and permeation studies using PAMPA, everted intestinal sac, and in vivo pharmacokinetics was performed by one-way ANOVA. Tukey’s multiple comparison tests was used as the post hoc test. A P value less than 0.05 was considered statistically significant.
Formulation Stability
Valsartan proliposomes and SNEDDS formulation stability was evaluated by analyzing the physical appearance, particle size, and drug content upon storage at room temperature (25°C) and refrigerated (5 ± 3°C) condition for 3 months. At predetermined time intervals, the samples of proliposomes and SNEDDS formulations were analyzed for particle size distribution and drug content.
RESULTS AND DISCUSSION
Valsartan is a poorly water-soluble drug with two acid dissociation constants (pKa): The first pKa of 4.7 is due to the presence acid group of the tetrazole derivative, and a second pKa of 3.9 is due to the presence of carboxylic group (12,13). These two close pKas allows the drug to exist in un-dissociated form at pH 1.2 and completely ionized to dianion form at pH > 6.7 (32). The poor bioavailability of valsartan may be attributed to its poor solubility and permeability due to its presence in ionized form across the GI.
Preparation of Proliposomes
The compositions selected for developing the proliposomal formulations are summarized in Table I. The formulated dry proliposomes were hydrated with distilled water at 37°C under sonication for 10 min using a bath sonicator (Branson Ultrasonics, Danbury, CT, USA) for performing the characterization studies.
Preparation of SNEDDS
The formula compositions of SNEDDS were selected based on the self-emulsifying region identified using the ternary phase diagram. The phase behavior of all the compositions was observed at room temperature, and the shadow region of ternary phase diagram in Fig. 1 represents the self-emulsifying region. The compositions selected based on the ternary phase diagram for formula optimization are summarized in Table II.

Ternary phase diagram of SNEDDS
Characterization
Particle Size
The particle size of proliposomes and SNEDDS formulations is shown in Tables I and II, respectively. The mean particle size of the liposomal formulations was in the range of 364.1 ± 14.9 to 879.2 ± 63.4 nm. The liposomal formulation F-III containing drug/DMPG/cholesterol in the ratio of 1:1:0.5 showed the smaller particle size (364.1 ± 14.9), and larger particle size was observed with formulation F-I (879.2 ± 63.4) containing drug/DSPC/cholesterol in the ratio of 1:1:0.5. These results indicated that the size of vesicles formed differs based on the type of lipid used in the formulation. Packing of lipid in the liposome membranes has a major role on the physical properties such as membrane permeability, elasticity, surface charge, and binding properties of proteins (33). The mean particle size of the SNEDDS formulations was in the range of 133.7 ± 42.8 to 348.4 ± 71.4 nm. The smallest being observed with formulation F-IX and the largest was with formulation F-VI. The smaller particle size of formulation F-IX may be attributed to the increased surfactant and co-surfactant concentrations in the composition. Polydispersity index (PDI) gives a measure of the width of particle size distribution (34). A PDI of greater than 0.1 is considered to indicate a heterogeneous system. From Tables I and II, it is evident that PDI of the particle size produced from the proliposomal formulation ranged from 0.523 ± 0.021 to 0.770 ± 0.014, and for SNEDDS, the observed PDI was 0.321 ± 0.021 to 0.547 ± 0.022, respectively, indicating a polydisperse system.
Zeta Potential
Formulations having high zeta potential will oppose aggregation and remain stable due to the net charge on the dispersed particles (35). Zeta potential of the selected formulations (proliposomes and SNEDDS) is summarized in Tables I and II, respectively. The results indicate that formulation F-III containing valsartan/DMPG/cholesterol has higher zeta potential (27.8 ± 2.8) as compared to other formulations. Generally, liposomes with cationic lipids are more prone to binding cells than liposomes with anionic lipids due to electrostatic interaction with negatively charged cell membrane (sialic acids and phospholipid head groups) (36). Because of this reason, cationic liposomes were often preferred to improve the in vivo efficacy of drugs with poor bioavailability (37,38).
Among SNEDDS formulations, F-IX has shown a zeta potential of 24.48 ± 4.8. The high positive charge of the prepared nanoemulsion may be attributed to the presence of free fatty acids. In general, a zeta potential value more than ±30 mV is reported to provide good formulation stability (39).
Formation of Liposomes
The Cryo-TEM images of hydrated proliposomes (Fig. 2a, b) showed the presence of multilamellar and oligolamellar vesicles in polynomial distribution correlating with our results from particle size analysis (Table II).

a Cryo-TEM image showing the multilamellar vesicles of valsartan proliposomal formulation (F-V) (bar = 50 nm). b Cryo-TEM image showing the oligolamellar vesicles of valsartan proliposomal formulation (F-V) (bar = 100 nm)
Encapsulation Efficiency of Proliposomes
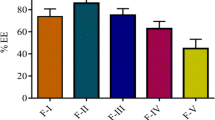
The encapsulation efficiency of the selected formulations is shown in Fig. 3. The results envisage that encapsulation efficiency of the proliposomal formulations was ranged between 81.2 ± 3.9 and 95.6 ± 2.9%. The encapsulation efficiency depends on physicochemical properties of the drug, lipid composition, and amount of cholesterol used in the formulation. Osmotic stability of the proliposomes in the GI environment is one of the important factors to be considered in the design of liposomal formulation. The effect of osmotic shock may result in structural rearrangement of vesicles in the GI. It has been reported that liposomes prepared using only phospholipids are usually not rigid enough, mainly due to low phase transition temperature and/or unsaturation of the fatty alkyl chains, which causes imperfections in the bilayer packing. Such liposomes tend to leak the encapsulated drug in the GI due to osmotic shock. Addition of bilayer additives (cholesterol or α-tocopherol) can result in tight packing of the bilayers and increased rigidity of the membrane thus preventing the drug leakage in the GI environment (40). The results indicated that liposomal formulation F-III containing drug/DMPG/cholesterol in the ratio of 1:1:0.5 showed the highest encapsulation efficiency (95.6 ± 2.9%) as compared to other proliposomal formulations. This difference in entrapment values might be due to the bonding forces between valsartan and phospholipid (head group) with varying carbon chain length. The increase in fatty acid chain length improves stability of the vesicle membrane and prevents the expulsion of drug from vesicles leading to greater efficiency (33). Formulation F-I containing drug/SPC/cholesterol in the ratio of 1:1:0.5 showed lowest encapsulation efficiency (81.2 ± 3.9%), and this could be due to weak bilayer structure and decreased packaging space for accommodating the drug molecule which might have resulted in drug leaking (31).

Encapsulation efficiency of proliposomal formulations
Robustness of SNEDDS to Dilution
The tendency of SNEDDS to precipitate at higher dilution may significantly affect the in vivo absorption; therefore, study of the formation of uniform emulsion from SNEDDS after dilution is very important (41). The prepared formulations were exposed to different media to mimic the in vivo conditions. Each formulation was subjected to 50 times dilution with purified water and pH 1.2 and pH 6.8 buffers. The mean particle size of valsartan SNEDDS following 50 times dilution with various media is shown in Fig. 4. The time required for the formation of nanoemulsions after dilution with various media was 3 min. The acceptable size range for SNEDDS is 20–200 nm (20,42,43). The resulting emulsions were found to be in the range of 133.7 ± 42.8 to 348.4 ± 71.4. Among the SNEDDS formulations, F-IX indicated the lowest particle size distribution following dilution with purified water, pH 1.2 buffer, and pH 6.8 buffer. After 6 h, all the formulations showed no signs of precipitation or separation (44).

Dilution study data of valsartan SNEDDS
Formula Optimization
The optimized formulations of proliposomes and SNEDDS were selected based on the particle size, zeta potential, encapsulation efficiency, and robustness to dilution studies. Among the formulations, proliposomal formulation F-III and SNEDDS formulation F-IX showed the desired particle size, zeta potential encapsulation efficiency, and robustness to dilution studies. Therefore, these optimized formulations were selected for further studies.
In Vitro Drug Release Study
The in vitro drug release profiles of pure valsartan, proliposomes, and SNEDDS in purified water and pH 1.2 buffer are depicted in Figs. 5 and 6 respectively. After 60 min, the amount of drug released in purified water from the capsules containing proliposomes and SNEDDS was significantly higher as compared to the pure drug. The release of drug from proliposomes and SNEDDS formulations was 2.5 and 2.6 fold higher, respectively, as compared to pure valsartan (Fig. 5). Similar study carried out in SGF (pH 1.2) showed that the proliposomal and SNEDDS formulations were 14.6 and 16.0-fold higher, respectively, as compared to pure valsartan (Fig. 6). The enhanced dissolution with proliposomes may be attributed to the presence of DMPG in the composition which is reported to rapidly undergo phase transition and thus demonstrating an increased dissolution of valsartan. Mirza and co-workers have reported DMPG as a promising carrier for enhancing the bioavailability of poorly soluble drugs (45). The improved dissolution of SNEDDS in comparison to pure valsartan is due to the presence of capmul MCM, which improves the water penetration and self-dispersibility of lipid formulation, augment the interfacial fluidity of surfactant (46). The similarity factor (F2) calculated on the dissolution data of proliposomes and SNEDDS versus pure valsartan in 0.1 N HCl was 9.7 and 6.5, respectively. In purified water, the F2 was 12.4 and 10.5 in comparison to pure valsartan indicating dissimilarity between the formulations.

Comparative dissolution profile of pure valsartan, proliposomes, and SNEDDS formulations in purified water (n = 3, mean ± SD)

Comparative dissolution profile of pure valsartan, proliposomes, and SNEDDS formulations in 0.1 N HCl (n = 3, mean ± SD)
Due to the poor solubility of valsartan in both SGF (pH 1.2) and water, dissolution studies were performed in these media to discriminate the dissolution of valsartan from different proliposomal formulations. Earlier, Gurrapu et al. have reported studies on proniosome-based delivery system to enhance the dissolution of valsartan in gastric pH. However, the results showed about 30–-35% drug release after 2 h (21). Dixit and co-workers reported self-microemulsifying drug delivery system (SMEDDS) to improve dissolution and bioavailability of valsartan. This report also shows less than 20% drug release in SGF (pH 1.2) after 12 h (20). In another study by Cao et al., an attempt was made to improve the dissolution of valsartan in gastric pH by incorporating poloxamer 188 and sodium hydroxide in mucoadhesive pellet formulation. However, the dissolution data was reported only in simulated intestinal fluid (pH 6.8) (22). In our study, using proliposomal and SNEDDS formulations, we have demonstrated that the dissolution of valsartan can be significantly improved in both simulated gastric fluid and purified water within 1 h (P < 0.05).
Parallel Artificial Membrane Permeability Assay Studies
PAMPA is an in vitro drug permeability model (non-cell based) that gives information on permeability of drug through passive transcellular mechanism. The PAMPA model lacks transporters and pore-mediated permeability. A logP (lipophilicity) value of >3 is a prerequisite for transportation of drug across the phospholipid membranes by passive diffusion. The effect of formulations (proliposomes and SNEDDS) on valsartan permeability in PAMPA model is depicted in Fig. 7. The results show enhanced permeability of proliposomes and SNEDDS indicating improved passive permeation of drug across the simulated membrane barrier. The effective permeability (P e) values for the pure drug, proliposomal, and SNEDDS formulations was found to be 1.0 × 10−5, 1.7 × 10−5, and 1.8 × 10−5, respectively, which indicates that liposomes and SNEDDS have higher permeability compared to pure valsartan (Fig. 7). Statistical analysis of permeability coefficient of liposomal, SNEDDS formulation versus pure drug showed a significant increase in the transport of valsartan when formulated into proliposomes (P < 0.05). The increased permeability may be due to change in the ratio of ionized versus unionized form of drug in the studied condition. In addition, the increased lipophilicity of the encapsulated drug may also have resulted in improved passive diffusion (47).

PAMPA membrane transport profile of valsartan from pure drug suspension, proliposomes, and SNEDDS formulations
Everted Rat Intestinal Permeation Studies
In order to confirm the data obtained from the in vitro permeation studies, an ex vivo rat intestinal permeation model was performed. The studies indicated a 4.8-fold increase in the drug transport with proliposomal formulation against the pure drug and 5.1 fold greater in the transport with SNEDDS formulation against the pure valsartan (Fig. 8). The intestinal permeation studies showed that the amount of drug perfused across the serosal membrane was found increased exponentially over the time as compared to pure valsartan. The increased concentration across serosal membrane clearly indicates that proliposomal and SNEDDS formulations demonstrated enhanced drug permeation compared to pure valsartan. The increased permeation of drug with the proliposomes and SNEDDS may be attributed to the increased lipophilicity of the drug to permeate across the GI membrane (Fig. 8).

Everted rat intestinal permeation profile of valsartan from pure drug suspension, proliposomes, and SNEDDS formulations (n = 3, mean ± SD)

Mean plasma concentrations of valsartan following oral administration of pure valsartan suspension, proliposomes, and SNEDDS formulations to male SD rats (n = 5, mean ± SD)
The P eff, K a, and ER of pure drug, proliposomes, and SNEDDS were estimated, and the results were summarized in Table III. The estimated P eff values for pure drug, proliposomes, and SNEDDS were 3.20 ± 0.25 × 10−6, 5.15 ± 0.43 × 10−6, and 5.81 ± 0.38 × 10−6 cm/s, respectively. The higher P eff for valsartan from proliposomes and SNEDDS in comparison to pure drug demonstrated the potential for overcoming the membrane barriers of the GIT and facilitating the absorption of valsartan. The rate of absorption, K a, for proliposomes and SNEDDS was 4.2 and 4.6-fold higher, respectively, as compared to the pure valsartan. The ER value greater than 1 generally indicates an improved permeation (31), and in our investigation, proliposomal and SNEDDS formulations have an ER value higher than 1 which indicates the role of proliposomes and SNEDDS as carriers for improving the oral bioavailability of poorly soluble drugs.
PAMPA provides transcellular diffusion of drug across lipid bilayers (48) and is considered to be passive absorption model since it is an artificially prepared membrane and does not contain cellular tight junctions and transporters. Whereas, the rat intestinal permeation model comprises of various influx and efflux transporters or paracellular pores for effective transportation of drug across the membranes. The in vitro transport studies in PAMPA and everted rat intestinal permeation models revealed that liposomal and SNEDDS formulations showed increased permeability of valsartan. It has been reported that small polar molecules are permeated across the intestinal barriers predominantly through the paracellular pathway (49). Valsartan’s poor aqueous solubility and molecular weight (435.5) may favor the transport through transcellular route. The transport of drugs from paracellular route is controlled by the tight cell junctions; therefore, widening of these tight junctions may provide a path to improve paracellular transport (50). Bile salts and cholesterol are reported to facilitate the uptake of drugs through transcellular route (51).
Pharmacokinetic Study
Formulation of valsartan into proliposomes and SNEDDS showed improved in vitro dissolution and in vitro permeation. To confirm these findings, in vivo studies were carried out in male SD rats following oral administration. The mean plasma concentration versus time profiles of valsartan after oral administration of proliposomes, SNEDDS, and pure drug suspension at a dose of 10 mg/kg are depicted in Fig. 9. The pharmacokinetic parameters obtained from the study are summarized in Table IV. The results indicate that T max of valsartan from pure valsartan formulation was 1.02 ± 0.61 h, whereas, for proliposomes and SNEDDS, it was 1.38 ± 0.75 and 0.75 ± 0.31 h, respectively. The C max of proliposomes and SNEDDS formulations was 5.1 and 5.4 fold higher, respectively, as compared to pure valsartan. The systemic exposure (AUC0–24) of proliposomes, SNEDDS, and pure valsartan suspension was 673.51 ± 21.78, 655.24 ± 29.15, and 332.82 ± 12.42, respectively, showing nearly a 2-fold increase for proliposomes and SNEDDS in comparison to pure valsartan suspension. This increase in the overall exposure of the proliposomal and SNEDDS formulations corresponds to an increase in its relative bioavailability which was 202.36 and 196.87%, respectively.
The increased C max observed with proliposomal and SNEDDS formulations further substantiated the in vitro and ex vivo findings. The delay in T max with the proliposomal formulation (1.38 h) when compared to pure valsartan (1 h) may be attributed to the time taken by the drug to release from the multilamellar or oligolamellar liposomes. The increased AUC0–24 by proliposomes and SNEDDS indicate the improved absorption of valsartan from the GI. The in vivo pharmacokinetic results further confirms our in vitro findings that by formulating proliposomes and SNEDDS, we can deliver the drug in a well-dispersed form in the upper GI, to enable rapid absorption of drug from the site. The in vivo pharmacokinetic results reveal that there is a potential for improving the absorption of valsartan by formulating as proliposomes and SNEDDS.
From the present investigation and the earlier reported studies (13,52), it is evident that the poor bioavailability of valsartan is primarily due to poor permeation of ionized drug in the gastrointestinal pH and not due to increased hepatic first-pass metabolism as reported (20,53). Relatively small particle size, modified lipophilicity, and similarity between the lipids used and biomembranes might have favored in improving the absorption of valsartan from the liposomes and SNEDDS. The study results indicate that by designing an optimal formulation, the oral bioavailability of valsartan can be improved which may further translate into reduced dose and dose-related toxicity.
Formulation Stability
The stability results of the selected proliposomal and SNEDDS formulations stored at room temperature (25°C) and refrigerated (5 ± 3°C) conditions are summarized in Table V. There were no significant changes observed in the particle size distribution and drug content values indicating the stability of the developed formulations on storage at room temperature and refrigerated conditions.
CONCLUSIONS
The present investigation reports the development of proliposomal and SNEDDS formulations for a poorly bioavailable drug, valsartan. The developed formulations showed enhanced drug release as compared to the pure valsartan under the same study conditions. The in vitro transport studies in PAMPA and everted rat intestine permeation models revealed significant enhancement in drug absorption from the developed formulations. The in vivo pharmacokinetic results of proliposomes and SNEDDS showed comparable drug plasma concentration time profiles indicating the effectiveness of these formulations in improving the in vivo performance of the drug. SNEDDS is an established platform technology with considerable success commercially, whereas proliposomes is yet to hit the market. This investigational study results indicate that both proliposomes and SNEDDS formulations are comparable and equally promising in improving the oral bioavailability of valsartan.
References
Bobbala SKR, Veerareddy PR. Formulation, evaluation, and pharmacokinetics of isradipine proliposomes for oral delivery. J Liposome Res. 2012;22(4):285–94.
Hiremath PS, Soppimath KS, Betageri GV. Proliposomes of exemestane for improved oral delivery: formulation and in vitro evaluation using PAMPA, Caco-2 and rat intestine. Int J Pharm. 2009;380(1):96–104.
Katare O, Vyas S, Dixit V. Effervescent granule based proliposomes of ibuprofen. J Microencapsul. 1990;7(4):455–62.
Katare O, Vyas S, Dixit V. Proliposomes of indomethacin for oral administration. J Microencapsul. 1991;8(1):1–7.
Potluri P, Betageri GV. Mixed-micellar proliposomal systems for enhanced oral delivery of progesterone. Drug Deliv. 2006;13(3):227–32.
Basalious EB, Shawky N, Badr-Eldin SM. SNEDDS containing bioenhancers for improvement of dissolution and oral absorption of lacidipine. I: development and optimization. Int J Pharm. 2010;391(1):203–11.
Parmar N, Singla N, Amin S, Kohli K. Study of cosurfactant effect on nanoemulsifying area and development of lercanidipine loaded (SNEDDS) self nanoemulsifying drug delivery system. Colloids Surf B: Biointerfaces. 2011;86(2):327–38.
Nekkanti V, Venkatesan N, Betageri GV. Proliposomes for oral delivery: progress and challenges. Curr Pharm Biotechnol. 2014;16(4):303–12.
Wang L, Dong J, Chen J, Eastoe J, Li X. Design and optimization of a new self-nanoemulsifying drug delivery system. J Colloid Interface Sci. 2009;330(2):443–8.
Porter CJ, Pouton CW, Cuine JF, Charman WN. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv Drug Deliv Rev. 2008;60(6):673–91.
Smith DG, Cerulli A, Frech FH. Use of valsartan for the treatment of heart-failure patients not receiving ACE inhibitors: a budget impact analysis. Clin Ther. 2005;27(6):951–9.
Criscione L, Bradley WA, Bühlmayer P, Whitebread S, Glazer R, Lloyd P, et al. Valsartan: preclinical and clinical profile of an antihypertensive angiotensin‐II antagonist. Cardiovasc Drug Rev. 1995;13(3):230–50.
Flesch G, Müller P, Lloyd P. Absolute bioavailability and pharmacokinetics of valsartan, an angiotensin II receptor antagonist, in man. Eur J Clin Pharmacol. 1997;52(2):115–20.
Brookman LJ, Rolan PE, Benjamin IS, Palmer KR, Wyld PJ, Lloyd P, et al. Pharmacokinetics of valsartan in patients with liver disease. Clin Pharmacol Ther. 1997;62(3):272–8.
Wu C-Y, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22(1):11–23.
Cappello B, Di Maio C, Iervolino M, Miro A. Improvement of solubility and stability of valsartan by hydroxypropyl-\boldbeta-cyclodextrin. J Incl Phenom Macrocycl Chem. 2006;54(3–4):289–94.
Parmar B, Mandal S, Petkar K, Patel L, Sawant K. Valsartan loaded solid lipid nanoparticles: development, characterization, and in vitro and ex vivo evaluation. Int J Pharm Sci Nanotechnol. 2011;4:1483–90.
Yan Y-D, Sung JH, Kim KK, Kim DW, Kim JO, Lee B-J, et al. Novel valsartan-loaded solid dispersion with enhanced bioavailability and no crystalline changes. Int J Pharm. 2012;422(1):202–10.
Rane BS, Rao N, Sathe DG, Sawant KD, Tarur VR, Thoovara SKM. Process for the preparation of micronized valsartan. Google Patents; 2007.
Date AA, Desai N, Dixit R, Nagarsenker M. Self-nanoemulsifying drug delivery systems: formulation insights, applications and advances. Nanomedicine. 2010;5(10):1595–616.
Gurrapu A, Jukanti R, Bobbala SR, Kanuganti S, Jeevana JB. Improved oral delivery of valsartan from maltodextrin based proniosome powders. Adv Powder Technol. 2012;23(5):583–90.
Cao Q-R, Liu Y, Xu W-J, Lee B-J, Yang M, Cui J-H. Enhanced oral bioavailability of novel mucoadhesive pellets containing valsartan prepared by a dry powder-coating technique. Int J Pharm. 2012;434(1):325–33.
Porter CJ, Charman WN. Intestinal lymphatic drug transport: an update. Adv Drug Deliv Rev. 2001;50(1):61–80.
Xu H, He L, Nie S, Guan J, Zhang X, Yang X, et al. Optimized preparation of vinpocetine proliposomes by a novel method and in vivo evaluation of its pharmacokinetics in New Zealand rabbits. J Control Release. 2009;140(1):61–8.
Balakumar K, Raghavan CV, Abdu S. Self nanoemulsifying drug delivery system (SNEDDS) of rosuvastatin calcium: design, formulation, bioavailability and pharmacokinetic evaluation. Colloids Surf B: Biointerfaces. 2013;112:337–43.
Chu C, Tong S-S, Xu Y, Wang L, Fu M, Ge Y-R, et al. Proliposomes for oral delivery of dehydrosilymarin: preparation and evaluation in vitro and in vivo. Acta Pharmacol Sin. 2011;32(7):973–80.
Janga KY, Jukanti R, Velpula A, Sunkavalli S, Bandari S, Kandadi P, et al. Bioavailability enhancement of zaleplon via proliposomes: role of surface charge. Eur J Pharm Biopharm. 2012;80(2):347–57.
Ravis WR, Feldman S. Effect of enzyme‐inducing and enzyme‐inhibiting agents on drug absorption I: 3‐O‐methylglucose transport in rats. J Pharm Sci. 1978;67(2):245–8.
Barthe L, Woodley J, Houin G. Gastrointestinal absorption of drugs: methods and studies. Fundam Clin Pharmacol. 1999;13(2):154–68.
Issa C, Gupta P, Bansal AK. Implications of density correction in gravimetric method for water flux determination using rat single-pass intestinal perfusion technique: a technical note. AAPS Pharmscitech. 2003;4(2):44–9.
Jukanti R, Mateti A, Bandari S, Veerareddy PR. Transdermal delivery of acyclovir sodium via carbopol gels: role of chemical permeation enhancers. Lett Drug Des Discov. 2011;8(4):381–9.
Tosco P, Rolando B, Fruttero R, Henchoz Y, Martel S, Carrupt PA, et al. Physicochemical profiling of sartans: a detailed study of ionization constants and distribution coefficients. Helvetica Chimica Acta. 2008;91(3):468–82.
Betageri G. Liposomal encapsulation and stability of dideoxyinosine triphosphate. Drug Dev Ind Pharm. 1993;19(5):531–9.
Elorza M, Garcia de la Cruz F, San Juan R, Marcilla A, Rico H, Mormeneo S, et al. Linkages between macromolecules in Candida albicans cell wall. Arch Med Res. 1992;24(3):305–10.
Patel J, Patel A, Raval M, Sheth N. Formulation and development of a self-nanoemulsifying drug delivery system of irbesartan. J Adv Pharm Technol Res. 2011;2(1):9.
Nekkanti V, Kalepu S. Recent advances in liposomal drug delivery: a review. Pharm Nanotechnol. 2015;3(1):35–55.
Dragicevic-Curic N, Gräfe S, Gitter B, Winter S, Fahr A. Surface charged temoporfin-loaded flexible vesicles: in vitro skin penetration studies and stability. Int J Pharm. 2010;384(1):100–8.
Wu J, Lee A, Lu Y, Lee RJ. Vascular targeting of doxorubicin using cationic liposomes. Int J Pharm. 2007;337(1):329–35.
Müller R, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy: rationale for development and what we can expect for the future. Adv Drug Deliv Rev. 2001;47(1):3–19.
Sackmann E. Membrane bending energy concept of vesicle-and cell-shapes and shape-transitions. FEBS Lett. 1994;346(1):3–16.
Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12(11):1561–72.
Date AA, Nagarsenker M. Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil. Int J Pharm. 2007;329(1):166–72.
Dabhi MR, Limbani MD, Sheth NR. Preparation and in vivo evaluation of self-nanoemulsifying drug delivery system (SNEDDS) containing ezetimibe. Curr Nanosci. 2011;7(4):616–27.
Gupta S, Chavhan S, Sawant KK. Self-nanoemulsifying drug delivery system for adefovir dipivoxil: design, characterization, in vitro and ex vivo evaluation. Colloids Surf A Physicochem Eng Asp. 2011;392(1):145–55.
Mirza S, Miroshnyk I, Habib MJ, Brausch JF, Hussain MD. Enhanced dissolution and oral bioavailability of piroxicam formulations: modulating effect of phospholipids. Pharmaceutics. 2010;2(4):339–50.
Pouton C, editor. Properties and uses of common formulation lipids, surfactants and cosolvents. AAPS, Workshop; 2007.
Sandhya KV, Devi GS, Mathew ST. Liposomal formulations of serratiopeptidase: in vitro studies using PAMPA and Caco-2 models. Mol Pharm. 2007;5(1):92–7.
Kansy M, Senner F, Gubernator K. Physicochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes. J Med Chem. 1998;41(7):1007–10.
Artursson P, Palm K, Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv Drug Deliv Rev. 2001;46(1):27–43.
Hochman J, Artursson P. Mechanisms of absorption enhancement and tight junction regulation. J Control Release. 1994;29(3):253–67.
Werner U, Kissel T, Reers M. Effects of permeation enhancers on the transport of a peptidomimetic thrombin inhibitor (CRC 220) in a human intestinal cell line (Caco-2). Pharm Res. 1996;13(8):1219–27.
Waldmeier F, Flesch G, Mu Ller P, Winkler T, Kriemler H-P, Buhlmayer P, et al. Pharmacokinetics, disposition and biotransformation of [14C]-radiolabelled valsartan in healthy male volunteers after a single oral dose. Xenobiotica. 1997;27(1):59–71.
Bissessor N, White H. Valsartan in the treatment of heart failure or left ventricular dysfunction after myocardial infarction. Vasc Health Risk Manag. 2007;3(4):425.
Conflict of Interest
The authors report no declarations of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Nekkanti, V., Wang, Z. & Betageri, G.V. Pharmacokinetic Evaluation of Improved Oral Bioavailability of Valsartan: Proliposomes Versus Self-Nanoemulsifying Drug Delivery System. AAPS PharmSciTech 17, 851–862 (2016). https://doi.org/10.1208/s12249-015-0388-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-015-0388-8