
Abstract
Background
The National Institute on Aging and Alzheimer’s Association (NIA-AA) have proposed a new Research Framework: Towards a biological definition of Alzheimer’s disease, which uses a three-biomarker construct: Aß-amyloid, tau and neurodegeneration AT(N), to generate a biomarker based definition of Alzheimer’s disease.
Objectives
To stratify AIBL participants using the new NIA-AA Research Framework using cerebrospinal fluid (CSF) biomarkers. To evaluate the clinical and cognitive profiles of the different groups resultant from the AT(N) stratification. To compare the findings to those that result from stratification using two-biomarker construct criteria (AT and/or A(N)).
Design
Individuals were classified as being positive or negative for each of the A, T, and (N) categories and then assigned to the appropriate AT(N) combinatorial group: A−T−(N)−; A+T-(N)−; A+T+(N)−; A+T−(N)+; A+T+(N)+; A−T+(N)−; A−T−(N)+; A−T+(N)+. In line with the NIA-AA research framework, these eight AT(N) groups were then collapsed into four main groups of interest (normal AD biomarkers, AD pathologic change, AD and non-AD pathologic change) and the respective clinical and cognitive trajectories over 4.5 years for each group were assessed. In two sensitivity analyses the methods were replicated after assigning individuals to four groups based on being positive or negative for AT biomarkers as well as A(N) biomarkers.
Setting
Two study centers in Melbourne (Victoria) and Perth (Western Australia), Australia recruited MCI individuals and individuals with AD from primary care physicians or tertiary memory disorder clinics. Cognitively healthy, elderly NCs were recruited through advertisement or via spouses of participants in the study.
Participants
One-hundred and forty NC, 33 MCI participants, and 27 participants with AD from the AIBL study who had undergone CSF evaluation using Elecsys® assays.
Intervention (if any)
Not applicable.
Measurements
Three CSF biomarkers, namely amyloid β1–42, phosphorylated tau181, and total tau, were measured to provide the AT(N) classifications. Clinical and cognitive trajectories were evaluated using the AIBL Preclinical Alzheimer Cognitive Composite (AIBL-PACC), a verbal episodic memory composite, an executive function composite, California Verbal Learning Test–Second Edition; Long-Delay Free Recall, Mini-Mental State Examination, and Clinical Dementia Rating Sum of Boxes scores.
Results
Thirty-eight percent of the elderly NCs had no evidence of abnormal AD biomarkers, whereas 33% had biomarker levels consistent with AD or AD pathologic change, and 29% had evidence of non-AD biomarker change. Among NC participants, those with biomarker evidence of AD pathology tended to perform worse on cognitive outcome assessments than other biomarker groups. Approximately three in four participants with MCI or AD had biomarker levels consistent with the research framework’s definition of AD or AD pathologic change. For MCI participants, a decrease in AIBL-PACC scores was observed with increasing abnormal biomarkers; and increased abnormal biomarkers were also associated with increased rates of decline across some cognitive measures.
Conclusions
Increasing biomarker abnormality appears to be associated with worse cognitive trajectories. The implementation of biomarker classifications could help better characterize prognosis in clinical practice and identify those at-risk individuals more likely to clinically progress, for their inclusion in future therapeutic trials.
Similar content being viewed by others

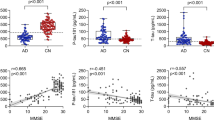
Alzheimer’s disease (AD) is a progressive, neurodegenerative disease characterized by neurodegeneration, synaptic loss, and the accumulation of extracellular-amyloid plaques and tau intracellular neurofibrillary tangles (1, 2). Several key imaging and cerebrospinal fluid (CSF) biomarkers have been identified in AD (3, 4). Deposition of beta-amyloid (Aβ-amyloid) plaques is one of the most important pathologic hallmarks of AD and is widely thought to be the initiating and primary driver of disease (amyloid hypothesis) (5, 6). Measures of Aβ-amyloid include amyloid imaging with positron emission tomography (PET) as well as CSF Aβ1–42, and studies have shown that these markers may be detectable over a decade before symptom onset (6, 7). Neurodegeneration and synaptic loss are also apparent prior to symptom onset, and may be visible on brain magnetic resonance imaging (MRI) as structural atrophy in regions consistent with AD (3). Other methods of assessing neurodegeneration include fluorodeoxyglucose [FDG]-PET, which measures brain metabolism as an indicator of synaptic activity (8, 9) and CSF total tau (t-tau), which is also indicative of synaptic loss and neurodegeneration (4, 10). Finally, tau pathology may be assessed using tau PET or CSF phosphorylated tau (p-tau), which has shown utility for predicting progression from mild cognitive impairment (MCI) to AD dementia as well as differentiating AD from other forms of dementia (3, 4, 11, 12).
Based on these biomarkers of Aβ-amyloid (CSF Aβ1–42), neurodegeneration (t-tau) and tau pathology (p-tau), various constructs have been developed to accurately identify individuals in the earliest (pre-symptomatic) stages of disease who are likely to progress to MCI and AD. Initial diagnostic research criteria developed by the National Institute on Aging and Alzheimer’s Association (NIA-AA) classified individuals with evidence of Aβ-amyloid pathology (i.e., abnormal Aβ-amyloid PET and CSF Aβ-amyloid) into three stages of preclinical AD based on the presence or absence of markers of neuronal injury (i.e., FDG-PET, structural MRI, or measures of tau) and evidence of subtle cognitive change (13). The criteria were further expanded to include two additional categories for cognitively normal individuals, including those with no biomarkers of AD (i.e., normal Aβ-amyloid, neurodegeneration, and tau) and those without evidence of Aβ-amyloid pathology but who are positive for other markers of neuronal injury, also referred to as suspected non-AD pathophysiology (SNAP) (14). These classifications were able to characterize 97% of cognitively normal individuals from a population-based sample (14) and have been shown to correlate with the cognitive trajectories and disease progression of individuals over time (15, 16).
While previous iterations of the NIA-AA criteria were based on a two-marker construct using evidence of Aβ-amyloid pathology and neurodegeneration as a single category, it is thought that segregating measures of pathologic tau (i.e., tau PET, CSF p-tau) from other markers of neuronal injury may help to better distinguish AD-related pathology from other neurodegenerative conditions (3). The recent NIA-AA Research Framework: Towards a biological definition of Alzheimer’s disease (4) is therefore based on a three-marker construct. The recent framework uses normal (−) or abnormal (+) levels of Aβ-amyloid deposition (“A”), pathologic tau (“T”), and neurodegeneration (“(N)”) as constructs to create the AT(N) classification system. In this contribution, we interrogated the AT(N) classification system to improve understanding for its implementation and applicability in characterizing and understanding the pathogenesis of AD. Firstly, we apply the AT(N) classification system to CSF biomarkers from well-characterized participants in the longitudinal Australian Imaging, Biomarker & Lifestyle (AIBL) Flagship Study of Ageing. Secondly, we describe the long-term clinical and cognitive trajectories of AIBL elderly cognitively normal controls (NCs) as well as AIBL MCI individuals, using the three-marker construct.
Methods
The AIBL cohort
The AIBL cohort study of aging combines data from neuroimaging, biomarkers, lifestyle, clinical, and neuropsychological assessments. Two study centers in Melbourne (Victoria) and Perth (Western Australia), Australia recruited individuals with MCI and with AD from primary care physicians or tertiary memory disorders clinics. Cognitively healthy NC participants were recruited through advertisement or via spouses of participants in the study. Exclusion criteria included a history of non-AD dementia, Parkinson’s disease, schizophrenia, bipolar disorder, current depression, cancer in the past 2 years (with the exception of basal-cell skin carcinoma), symptomatic stroke, uncontrolled diabetes, or current regular alcohol use. Between November 3, 2006, and October 30, 2008, AIBL recruited 1112 eligible volunteers who were at least 60 years old and fluent in English. Full details on the study design and inclusion criteria have been reported elsewhere (17). An enrichment cohort of 86 participants with AD, 124 MCI participants, and 389 NC participants were recruited by AIBL between March 30, 2011, and June 29, 2015. At baseline, the AIBL study participants had an average age of 72 years, 58% were female, and 36% were Apolipoprotein E (APOE) ε4 carriers. APOE ε4 carriage was determined as previously described (18). Two hundred AIBL participants (140 NC, 33 MCI and 27 AD) with a mean age of 73 (50% Males) who had undergone lumbar puncture were included in the current study.
Assessment of CSF biomarkers
Lumbar puncture was used to collect CSF from 200 AIBL participants in the morning after overnight fasting, with a protocol aligned to the Alzheimer’s Biomarkers Standardization Initiative (ABSI). Lumbar puncture was performed in the sitting position using a strictly aseptic technique and gravity drip collection. CSF was collected into a polypropylene tube and placed on ice prior to centrifugation (2000 ×g at 4°C for 10 minutes), and the supernatant was transferred to a second polypropylene tube and gently inverted. Samples were aliquoted (500 µL) into Nunc cryobank polypropylene tubes (NUN374088) and stored in liquid nitrogen vapor tanks within 1 hour (kept on dry ice prior to storage) and only thawed once, immediately before analysis. CSF levels of Aβ1-42, t-tau, and p-tau were measured by electrochemiluminescence Elecsys® immunoassay (Roche Diagnostics, Penzberg, Germany) that uses a quantitative sandwich principle. Levels were measured using the Roche cobas® e601 analyzer (Roche Diagnostics) with a total assay duration of 18 minutes.
Application of the NIA-AA Research Framework
The NIA-AA Research Framework (4), details grouping of individuals based on AT(N) criteria, where: ‘A’ represents Aβ-amyloid or associated pathologic state—here ‘A’ is defined using CSF Aβ1-42; ‘T’ represents aggregated tau (neurofibrillary tangles) or associated pathologic state—in this current study ‘T’ is defined using CSF p-tau; ‘(N)’ represents neurodegeneration or neuronal injury—here ‘(N)’ is defined using CSF t-tau. Individuals were classified as being positive or negative for each of the A, T, and (N) criteria. A+ was defined as having a CSF Aβ1–42 level ≤1054.00pg/mL and A− as having a CSF Aβ1–42 level >1054.00 pg/mL. T+ was defined as having a CSF p-tau level ≥21.34 pg/mL and T− as having a CSF p-tau level <21.34 pg/mL. (N)+ was defined as having a CSF t-tau level ≥212.60 pg/mL and T− as having a CSF p-tau level <212.60 pg/mL. Individuals were then classified as belonging to one of the eight AT(N) combinatorial groups: A−T−(N)−; A+T−(N)−; A+T+(N)−; A+T−(N)+; A+T+(N)+; A−T+(N)−; A−T−(N)+; A−T+(N)+. In line with the NIA-AA Research Framework (4), the eight AT(N) groups were collapsed into four main groups of interest: those with normal AD biomarkers (A−T−(N)−), those with non-AD pathologic change (A−T+(N)−; A−T+(N)+; A−T+(N)−), those with AD pathologic change (A+T−(N)−; A+T−(N)+), and those with AD (A+T+(N)−; A+T+(N)+).
Cognitive markers
All participants underwent extensive neuropsychological testing, as previously described (17). Briefly, the tests comprising the AIBL clinical and neuropsychological battery were selected to cover the main domains of cognition affected by AD and other dementias, and are all internationally recognized as having good reliability and validity. The full battery comprised: the Clinical Dementia Rating (CDR) Scale, Mini-Mental State Examination (MMSE) (19), Clock-Drawing Test, California Verbal Learning Test — Second Edition (CVLT-II) (20), Logical Memory (LM) I and II (Wechsler Memory Scale [WMS]-III; Story A only) (21–23), Delis–Kaplan Executive Function System (D-KEFS) verbal fluency (24), 30-item Boston Naming Test (BNT) (25), the Stroop Test (Victoria version) (22), the Rey Complex Figure Test (RCFT) (26), Digit Span and Digit Symbol-Coding subtests of the Wechsler Adult Intelligence Scale — Third Edition (WAIS–III) (27), the Wechsler Test of Adult Reading (WTAR) (28), the Hospital Anxiety and Depression Scale (HADS), and the Geriatric Depression Scale (GDS).
Clinical and cognitive trajectories were evaluated using the AIBL-Preclinical Alzheimer Cognitive Composite (AIBL-PACC) (29), a verbal episodic memory composite, an executive function composite (30), CVLT-II Long-Delay Free Recall (CVLT-II LDFR), MMSE, and CDR Sum of Boxes (CDR SoB) measures. The AIBL-PACC was constructed by summing Z-score measures of CVLT-II LDFR, LM-II, MMSE, and Digit Symbol-Coding. The verbal episodic memory composite was created from Z-scores of CVLT-II LDFR, CVLT-II recognition false positives, and LM-II, and the executive function composite was generated from Z-scores of D-KEFS letter fluency and category switching totals as well as the colors/dots interference measure from the Stroop Test (Victoria version).
Analysis
Demographic information was assessed across clinical classifications for 200 AIBL participants who had undergone CSF evaluation. Participants were classified into one of eight categories based on the three-construct model of AT(N) in the NIA-AA Research Framework. The prevalence of the AT(N) groups was assessed across the clinical classification groups. The eight AT(N) groups were then collapsed into four main groups of interest: those with normal AD biomarkers, those with non-AD pathologic change, those with AD pathologic change, and those with AD. Baseline cognitive performance was assessed across these four groups within the NC and MCI clinical classification groups using boxplots and one-way t-tests. Longitudinal change in cognitive performance over time, separately for the NC and MCI, was assessed using boxplots and one-way t-tests of the random slopes obtained from linear mixed-effect models. In the linear mixed-effect models, the cognitive measure represented the dependent variable; age, sex, and APOE ε4 status were included as interacting independent factors and time since CSF evaluation was included as a random factor. The dependent variable was evaluated every 18 months for a mean follow-up of 4.5 years. The number of participants progressing towards more advanced disease (i.e., NC to MCI/AD and MCI to AD) within each of these four groups was also evaluated using descriptive statistics, due to the small number of conversions more sophisticated analyses such as Cox proportional hazards analyses could not be undertaken.
Sensitivity Analysis I
Participants were assigned to one of four groups (A−T−; A+T−; A−T+; A+T+) based on their CSF Aα1–42 and p-tau levels as described above. Baseline cognitive performance was assessed across these four AT groups within each clinical classification group using boxplots and one-way t-tests. Longitudinal change in cognitive performance over time was assessed using boxplots and one-way t-tests of the random slopes obtained from linear mixed-effect models. In the linear mixed-effect models, the cognitive measure represented the dependent variable; age, sex, and APOE ε4 status were included as interacting independent factors and time since CSF evaluation was included as a random factor.
Sensitivity Analysis II
Participants were assigned to one of four groups (A−N−; A+N−; A−N+; A+N+) based on their CSF Aα1–42 and t-tau levels as described above. Baseline cognitive performance was assessed across these four A(N) groups within each clinical classification group using boxplots and one-way t-tests. Longitudinal change in cognitive performance over time was assessed using boxplots and one-way t-tests of the random slopes obtained from linear mixed-effect models. In the linear mixed-effect models, the cognitive measure represented the dependent variable; age, sex, and APOE ε4 status were included as interacting independent factors and time since CSF evaluation was included as a random factor.
Results
Demographics
The majority of participants (140/200) were cognitively healthy (NC) and the remaining comprised MCI or AD (n=33 and n=27, respectively) (Table 1). There was a higher prevalence of males in the MCI and AD samples compared to the NC sample. Reported ages at baseline did not differ across the three samples (averaging around 73 years). The NC participants had a higher level of education and had fewer APOE ε4 carriers. The mean duration of follow-up for all participants was 4.54 years.
Prevalence of AT(N) groups
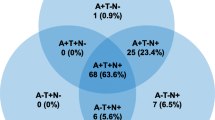
The prevalence of each of the eight AT(N) classifications within the AIBL NC, MCI, and AD samples are given in Figure 1. The highest proportion of NC participants (38%) had normal AD biomarkers; 13% had AD pathologic change, 20% have AD, and 29% had non-AD pathologic change. In the MCI and AD samples, 75% and 70% of participants had AD pathologic change, respectively.

Prevalence of the AT(N) groups across clinical classifications
Cross-sectional cognitive performance in NC
In general, NC participants with biomarkers consistent with AD performed the worst on the cognitive composite markers and MMSE (Figure 2A–C and E). Differences were not observed for CDR SoB with all NCs scoring 0 on this test (Figure 2D). The NC participants with normal AD biomarkers had the lowest scores on the CVLT-II LDFR (Figure 2F). In general, within the NC sample those classified as having non-AD pathologic change had similar scores to those with normal AD biomarkers. Regarding the sensitivity analyses, The A+T+ group had significantly (p=0.03) lower baseline scores for AIBL-PACC in comparison to the A−T− group and the A+T+ group had significantly lower baseline scores for the Verbal Episodic Memory composite than the A−T+ group. Also, the A+N+ group had significantly lower baseline scores for the Verbal Episodic Memory composite than the A−N+ group. No other differences were observed in the sensitivity analyses of differences in the NC at baseline.

Cross-sectional performance on the six cognitive measures (A: AIBL-PACC; B: Verbal Episodic Memory; C: Executive Function; D: CDR Sum of Boxes; E: MMSE; F: CVLT-II LDFR) for the four contracted AT(N) groups in NC
Cross-sectional cognitive performance in MCI
For MCI participants there was a decrease in performance from those with normal AD biomarkers, to those with AD pathologic change and then AD for the AIBL-PACC (Figure 3A). This trend was not observed in the other five clinical and cognitive markers considered (Figure 3B–F). No baseline differences were obsevered for the MCI in the sensitivity analyses.

Cross-sectional performance on the six cognitive measures (A: AIBL-PACC; B: Verbal Episodic Memory; C: Executive Function; D: CDR Sum of Boxes; E: MMSE; F: CVLT-II LDFR) for the four contracted At(n) groups in MCI
Longitudinal change in cognitive performance
For both the NC and MCI participants, systematic differences were not observed in the rates of decline for the four groups considered (Supplementary Figures 1 and 2). No differences were observed in the sensitivity analyses.
Progression to disease
Over the period of follow-up (mean=4.54 years), of the 53 NC individuals with normal AD biomarkers, one progressed to MCI due to AD and one progressed to MCI not due to AD. Of the 18 NC individuals with AD pathologic change, two progressed to MCI due to AD. Of the 28 NC individuals with AD biomarkers, one participant died and there were no other transitions. Of the 41 individuals with non-AD pathologic change, one participant died, one progressed to MCI, and one progressed to vascular dementia. Of the nine MCI individuals with AD pathologic change, one progressed to AD. Of the 13 MCI individuals with AD biomarkers, two participants died and two progressed to AD. There were not enough events of progression to ascertain any statistically significant differences in progression between the groups.
Discussion
This analysis evaluated the AT(N) classification system in a well-characterized population from the AIBL cohort, including cognitively healthy NC participants as well as those with MCI and AD. Approximately two in five of the elderly NC had no evidence of abnormal AD biomarkers, whereas one in three had biomarker levels consistent with AD or AD pathological change and almost one in three had evidence of non-AD pathological change. Twenty-three percent of the NC participants had biomarker levels aligned with the SNAP category (A−(N+)), which aligns with other reports in the literature (3, 16).
Among NC participants, those with biomarker evidence of AD pathology tended to perform worse on composite cognitive outcome assessments and the MMSE compared with other biomarker groups. Participants with abnormal non-AD-specific biomarkers performed similarly to those with or without normal AD biomarkers across endpoints. No differences were observed across the four biomarker groups with respect to rate of decline on any outcome assessment.
Approximately three in four participants with MCI or AD had biomarker levels consistent with AD or AD pathologic change. For MCI participants, a decrease in AIBL-PACC scores was observed with increasing abnormal biomarkers; increased abnormal biomarkers were also associated with increased rates of decline across some cognitive measures. There were not enough events of disease progression (i.e., NC to MCI/AD or MCI to AD) to draw any conclusions about the risk of disease progression based on the biomarker constructs.
Despite the lack of statistically significant trends, which is likely to be related to the small numbers of participants included, observations from the current study are qualitatively consistent with previous work showing that biomarkers of AD evident before clinical symptoms appear to predict cognitive deficit. In a natural history study classifying NC participants (N=166) with a two-marker construct, using Aβ-amyloid (assessed using amyloid PET imaging) and markers of neurodegeneration (hippocampus volume seen on MRI, FDG-PET), those with normal AD biomarkers showed improvement over time on a composite cognitive measure derived from eight neuropsychological tests, likely due to practice effects (15). Conversely, participants who either had evidence of Aβ-amyloid pathology or were considered SNAP participants had reduced practice effects, and those positive for both Aβ-amyloid pathology and markers of neurodegeneration showed cognitive decline (15). An analysis of a larger group of NC individuals from the AIBL cohort (N=573) also applied the two-marker construct, using amyloid PET as a marker of Aβ-amyloid pathology and hippocampal volume on MRI to assess neurodegeneration, and showed that amyloid-PET positivity conferred significant risk for cognitive decline, with structural evidence of neurodegeneration further compounding this risk (16). Applying this two-marker construct here in a sensitivity analysis, highlighted some baseline differences: individuals with abnormal CSF levels for Aβ-amyloid and one of the tau markers performed worse than participants with less biomarker abnormality on two of the cognition measures. No longitudinal differences were observed in the sensitivity analysis.
The composite AT(N) system for classifying AD used in the present analysis separates markers of tau pathology from other neurodegenerative markers which is thought to improve specificity in terms of differentiating patients with AD vs. non-AD pathology. However, our inconclusive findings suggest that further study of the AT(N) classification system and its comparison to the two-biomarker constructs in larger groups of participants across the disease spectrum is needed.
Our construct employed CSF-based immunoassay measures for determining A, T, and (N) status, in comparison to the imaging metrics employed in the previous studies discussed (15,16). The availability of immunoassay methodology for evaluating AD and neurodegeneration biomarkers could have important implications for clinical practice as this type of testing may be more widely accessible and cheaper than imaging-based methodologies. In turn, this potential for great accessibility vs. imaging methodologies may facilitate wider application of AT(N) classification in clinical trial methodology to screen more potential participants and further enrich study populations with AD biomarker-positive individuals who are most likely to show AD-related disease progression within the duration of the study. A much wider application would be achievable once blood biomarkers become available.
There are a number of limitations to this study, including the small sample size, which may preclude any statistically significant differences being observed. Further, only a small number of disease progression events occurred precluding any evaluations to be made regarding the power of the AT(N) criteria to predict progression to disease. The participants were volunteers who were not randomly selected from the community, and were generally well educated; thus, these findings might only be valid in similar cohorts and this limitation precludes the generalization of the findings. In view of the stringent selection criteria in AIBL, which excluded individuals with cerebrovascular disease or other dementias, the effect of other comorbidities on the trajectories might be underestimated. Longitudinal cognitive performance was based on three composite measures as well as two clinical scores and one standard measure, which were corrected using within-study norms; however, other cognitive tests, or combinations thereof, might yield different results. Further, biomarker levels were obtained from a CSF immunoassay and different techniques may yield different results. The cutoffs used for dichotomous stratification were somewhat arbitrary and continuous variables might provide better predictors of progression. Another potential limitation is the non-specificity of t-tau for the (N) classification and other markers, such as neurofilament light, either in CSF of plasma, may provide a more robust assessment of (N).
In conclusion, increasing CSF biomarker abnormality appears to be associated with worse cognitive trajectories. The implementation of the AT(N) classification could help better characterize prognosis in clinical practice and identify those at-risk individuals more likely to progress, for inclusion in future therapeutic trials. However, our inconclusive findings suggest that further study of the AT(N) classification system in larger groups of participants is warranted.
References
Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet 2006;368:387–403.
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 2011;1:a006189.
Jack CR, Bennett DA, Blennow K, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016;87:539–547.
Jack CR, Bennett DA, Blennow K, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018;14:535–562.
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002;297:353–356.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 2016;8:595–608.
Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol 2013;12:357–367.
Apostolova LG, Hwang KS, Andrawis JP, Green AE, Babakchanian S, Morra JH, et al. 3D PIB and CSF biomarker associations with hippocampal atrophy in ADNI subjects. Neurobiol Aging 2010;31:1284–1303.
Johnson KA, Fox NC, Sperling RA, Klunk WE. Brain imaging in Alzheimer disease. Cold Spring Harb Perspect Med 2012;2:a006213.
Blennow K. CSF biomarkers in Alzheimer’s disease — use in clinical diagnosis and to monitor treatment effects. Eur Neuropsychopharmacol 2010;20:S159–S.
Hampel H, Buerger K, Zinkowski R, et al. Measurement of phosphorylated tau epitopes in the differential diagnosis of Alzheimer disease: A comparative cerebrospinal fluid study. Arch Gen Psychiatry 2004;61:95–102.
Blom ES, Giedraitis V, Zetterberg H, et al. Rapid progression from mild cognitive impairment to Alzheimer’s disease in subjects with elevated levels of tau in cerebrospinal fluid and the APOE ε4/ε4 genotype. Dement Geriatr Cogn Disord 2009;27:458–464.
Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7:280–292.
Jack CR Jr, Knopman DS, Weigand SD, et al. An operational approach to National Institute on Aging-Alzheimer’s Association criteria for preclinical Alzheimer disease. Ann Neurol 2012;71:765–775.
Mormino EC, Betensky RA, Hedden T, et al. Synergistic effect of β-amyloid and neurodegeneration on cognitive decline in clinically normal individuals. JAMA Neurol 2014;71:1379–1385.
Burnham SC, Bourgeat P, Doré V, et al. Clinical and cognitive trajectories in cognitively healthy elderly individuals with suspected non-Alzheimer’s disease pathophysiology (SNAP) or Alzheimer’s disease pathology: a longitudinal study. Lancet Neurol 2016;15:1044–1053.
Ellis KA, Bush AI, Darby D, De Fazio D, Foster J, Hudson P, et al. The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer’s disease. Int Psychogeriatr 2009;21:672–687.
Porter T, Villemagne VL, Savage G, et al. Cognitive gene risk profile for the prediction of cognitive decline in presymptomatic Alzheimer’s disease. Pers Med Psychiatry 2018;7–8:14–20.
Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J psychiatr Res 1975;12:189–198.
Delis D, Kramer J, Kaplan E, Ober B. California Verbal Learning Test–Second Edition. 2000. The Psychological Corporation, San Antonio, Tx.
Wechsler D. A standardized memory scale for clinical use. J Psychol 1945;19:87–95.
Strauss E, Sherman, Spreen O. A Compendium of Neuropsychological Tests: Administration, Norms, and Commentary (3rd edn). 2006. Oxford University Press, New York.
Wechsler D. Wechsler Memory Scale (3rd edn). 1997. The Psychological Corporation, San Antonio, Tx.
Delis D, Kaplan E, Kramer J. Delis-Kaplan Executive Function System (D-KEFS). 2001. The Psychological Corporation, San Antonio, Tx.
Saxton J, Ratcliff G, Munro CA, et al. Normative data on the Boston Naming Test and two equivalent 30-item short forms. Clin Neuropsychol 2000;14:526–534.
Meyers J, Meyers K. Rey Complex FigureTest and Recognition Trial. Professional Manual. 1995. Psychological Assessment Resource.
Wechsler D. Wechsler Adult Intelligence Scale, 3rd Edition (WAIS III). 1997. The Psychological Corporation, San Antonio, Tx.
Wechsler D. Wechsler Test of Adult Reading:Examiner’s Manual. 2001. The Psychological Corporation, San Antonio, Tx.
Donohue MC, Sperling RA, Salmon DP, et al. The preclinical Alzheimer cognitive composite: measuring amyloid-related decline. JAMA Neurol 2014;71:961–970.
Burnham SC, Raghavan N, Wilson W, et al. Novel statistically-derived composite measures for assessing the efficacy of disease-modifying therapies in prodromal Alzheimer’s disease trials: an AIBL study. J Alzheimers Dis 2015;46:1079–1089.
Acknowledgments
We thank the participants who took part in the AIBL study and their families. Editorial assistance was provided by Liz LaFlamme, PhD, and Rachel Johnson, PhD, of Health Interactions, funded by F. Hoffmann-La Roche Ltd, Basel, Switzerland.
Funding
Funding: Core funding for the AIBL study was provided by the CSIRO Flagship Collaboration Fund and the Science and Industry Endowment Fund (SIEF) in partnership with the CRC for Mental Health, Edith Cowan University (ECU), Mental Health Research Institute (MHRI), Alzheimer’s Australia (AA), National Ageing Research Institute (NARI), Austin Health, Macquarie University, CogState Ltd, Hollywood Private Hospital, and Sir Charles Gairdner Hospital. The study also received funding from the National Health and Medical Research Council (NHMRC), Dementia Collaborative Research Centre (DCRC) program, and McCusker Alzheimer’s Research Foundation, and operational infrastructure support from the Government of Victoria. This specific study was funded in part by F. Hoffmann-La Roche Ltd, Basel, Switzerland. The sponsors had no role in the design and conduct of the study; in the collection, analysis, and interpretation of data; in the preparation of the manuscript; or in the review or approval of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest: Samantha C. Burnham: reports speaker honoraria from Novartis outside the scope of the submitted work and research funding paid to her employers from F. Hoffmann-La Roche Ltd. Preciosa M. Coloma: is a full-time employee of, and own shares in, F. Hoffmann-La Roche Ltd. Simon Laws: received personal fees from Alzhyme outside the scope of the submitted work. James Doecke: reports research funding paid to his employers from F. Hoffmann-La Roche Ltd. David Ames: reports receipt of financial assistance to his employer to assist with an international drug trial of an anti-Alzheimer’s agent, owned by Eli Lilly. Christopher C. Rowe: reports speaker honoraria from GE Healthcare and Avid Radiopharmaceuticals, consulting fees from Avid Radiopharmaceuticals, AstraZeneca, and Piramal Imaging, and research grants from Avid Radiopharmaceuticals, GE Healthcare, and Piramal Imaging all outside the scope of the submitted work. Colin L. Masters: reports personal fees from Prana Biotechnology, Eli Lilly, and Actinogen outside the scope of the submitted work. Victor L. Villemagne: reports speaker honoraria from GE Healthcare, Piramal Imaging, and Avid Radiopharmaceuticals, and consulting fees from Lundbeck, AbbVie, Shanghai Green Valley Pharmaceutical Co. outside the scope of the submitted work and consulting fees from F. Hoffmann-La Roche Ltd. All other authors declare no conflicts of interest
Ethical standards: This work was conducted in accordance with the principles set forth by the Declaration of Helsinki. The institutional ethics committees of Austin Health, St Vincent’s Health, Hollywood Private Hospital, and Edith Cowan University in Australia approved the AIBL study, and all volunteers gave written informed consent before participating.
Electronic supplementary material
Rights and permissions
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
About this article

Cite this article
Burnham, S.C., Coloma, P.M., Li, QX. et al. Application of the NIA-AA Research Framework: Towards a Biological Definition of Alzheimer’s Disease using Cerebrospinal Fluid Biomarkers in the AIBL Study. J Prev Alzheimers Dis 6, 248–255 (2019). https://doi.org/10.14283/jpad.2019.25
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.14283/jpad.2019.25