

Abstract
The rise in blood pressure during an acute aversive stress has been suggested to involve activation of angiotensin type 1A receptors (AT1ARs) at various sites within the brain, including the rostral ventrolateral medulla. In this study we examine the involvement of AT1ARs associated with a subclass of sympathetic premotor neurons of the rostral ventrolateral medulla, the C1 neurons. The distribution of putative AT1AR-expressing cells was mapped throughout the brains of three transgenic mice with a bacterial artificial chromosome-expressing green fluorescent protein under the control of the AT1AR promoter. The overall distribution correlated with that of the AT1ARs mapped by other methods and demonstrated that the majority of C1 neurons express the AT1AR. Cre-recombinase expression in C1 neurons of AT1AR-floxed mice enabled demonstration that the pressor response to microinjection of angiotensin II into the rostral ventrolateral medulla is dependent upon expression of the AT1AR in these neurons. Lentiviral-induced expression of wild-type AT1ARs in C1 neurons of global AT1AR knock-out mice, implanted with radiotelemeter devices for recording blood pressure, modulated the pressor response to aversive stress. During prolonged cage-switch stress, expression of AT1ARs in C1 neurons induced a greater sustained pressor response when compared to the control viral-injected group (22 ± 4 mmHg for AT1AR vs 10 ± 1 mmHg for GFP; p < 0.001), which was restored toward that of the wild-type group (28 ± 2 mmHg). This study demonstrates that AT1AR expression by C1 neurons is essential for the pressor response to angiotensin II and that this pathway plays an important role in the pressor response to aversive stress.
Introduction
The response to stress involves coordinated activation of the autonomic nervous system and the hypothalamic–pituitary–adrenal axis (Ulrich-Lai and Herman, 2009). While stress responses are essential physiology, there is a growing recognition that aberrant, or sustained, stress exposure is detrimental and can lead to the development of hypertension (Esler, 2009).
Both systemic and brain angiotensin systems play important roles in the responses to stress (Saavedra and Benicky, 2007). Treatment of hypertensive patients with an angiotensin converting enzyme inhibitor lowers blood pressure (BP) and reduces the pressor response to mental stress (Kahan and Eliasson, 1999). Experimental animal studies have demonstrated a role for brain angiotensin II (AngII), acting through its type 1A receptor (AT1AR), in the neuroendocrine and autonomic response to stress. This is particularly clear for corticotrophin-releasing hormone release induced by AT1AR stimulation in the hypothalamus (Aguilera et al., 1995; Armando et al., 2001). Studies in mice also implicate the AT1AR in the pressor response to aversive stress, as global genetic deletion of the AT1AR leads to an attenuated pressor response to aversive stress, but not to appetitive stimuli (Chen et al., 2009).
The rostral ventrolateral medulla (RVLM) is a critical site for regulation of sympathetic vasomotor activity (Dampney, 1994) and is proposed to be involved in the cardiovascular response to aversive stress, although this involvement remains controversial (Vianna and Carrive, 2010). Studies conducted in conscious rabbits showed that the stress-induced pressor response consists of at least two components acting in the RVLM—an initial response, mediated by glutamate, followed by a sustained response (Mayorov and Head, 2002, 2003). Local microinjections of AT1R antagonists in the RVLM attenuate the sustained pressor response (Mayorov and Head, 2003). These observations indicate that the RVLM may be an important node connecting the effects of AngII on stimulation of the autonomic nervous system in aversive stress.
The RVLM contains moderate to high densities of AT1Rs in most species, including humans (Allen et al., 1998). The RVLM contains at least two major subtypes of presympathetic neurons based on their expression of catecholamine synthetic enzymes. One group, the C1 neurons, expresses all of the enzymes required to synthesize adrenaline (Phillips et al., 2001), whereas the other group expresses none of these enzymes and is referred to as non-C1 neurons (Lipski et al., 1995; Schreihofer and Guyenet, 1997). The relative involvement of these different cell groups in the pressor response to aversive stress remains unclear. Recently, it was shown that C1 neurons of the RVLM have the capacity to induce a sympathetically mediated increase in BP (Abbott et al., 2009) and that expression of the AT1AR in C1 neurons of AT1AR knock-out (AT1AR−/−) mice reinstates the sympathoexcitation induced by AngII in the RVLM (Chen et al., 2010). Therefore, the aim of this current study is to examine the effect of transgenic expression of the AT1AR in the C1 neurons of the RVLM on the cardiovascular responses to acute aversive stress in conscious AT1AR−/− mice.
Materials and Methods
Experiments followed the Australian National Health and Medical Research Council Code of Practice for the Care and Use of Animals for Scientific Purposes and were approved by the University of Melbourne and the Alfred Medical Research Education Precinct Animal Ethics Committees. AT1AR−/− mice and their own control strain (AT1AR+/+) were bred in the animal facilities at the Baker IDI Heart and Diabetes Research Institute (Melbourne, VIC, Australia). Mice were originally obtained from Prof. T. Walther (Charité, Berlin, Germany), who produced F2 generation crosses of (129 × C57BL/6J) F1 AT1AR−/+ parents maintained on a C57Bl/6 background (Gembardt et al., 2008). Mice with a conditional Agtr1a allele (AT1ARfl/fl) were generated using standard gene targeting methods (Gurley et al., 2011) and obtained from Dr. S. Gurley and Prof. T. Coffman (Duke University, Durham, NC). They were maintained as a homozygote line on a C57BL/6 background at the Florey Neurosciences Institute (University of Melbourne, Melbourne, VIC, Australia). Cre-recombinase (Cre) reporter mice (ROSA26-eYFP, where eYFP is enhanced yellow fluorescent protein) were obtained from Prof. F. Costantini, Columbia University, New York, NY (Srinivas et al., 2001) and maintained at the Florey Neurosciences Institute, University of Melbourne. All mice were genotyped using standard PCR protocols. All mice were maintained under 12 h light/dark cycle (lights on from 6 A.M. to 6 P.M.) with ad libitum access to standard chow and water. Brains from three male adult mice, in which enhanced green fluorescent protein (GFP) and a polyadenylation signal were inserted before the ATG start codon of the Agtr1a gene (pAT1AR-GFP), were obtained from the GenSat BAC transgenic project (GENSAT). These had been deeply anesthetized, perfused with 4% paraformaldehyde, and stored in 20% sucrose solution for shipping from the United States to Australia. The brains were frozen in isopentane on dry ice and stored at −80°C before histological processing of serial sections taken every 300 μm throughout the entire neuraxis.
Lentiviral injection in RVLM
Replication-deficient lentiviruses with transgene expression under the control of a synthetic phox2-selective promoter (PRSx8) were injected bilaterally into the RVLM of anesthetized mice (80 mg/kg ketamine, 10 mg/kg xylazine, i.p.) (Chen et al., 2010). Before surgery mice, were injected with a nonsteroidal analgesic (Carprofen, 5 mg/ml, 0.5 mg/100 g, i.p; Norbrook Laboratories) and anesthesia induced by inhalation of isoflurane (Rhodia Australia) in a sealed container. Three different viruses were used that induced expression of GFP (Lv-PRSx8-GFP; titer, 2 × 1011 viral particles per milliliter), wild-type hemagglutinin-tagged AT1AR (Lv-PRSx8-AT1AR; titer, 3.7 × 1011 viral particles per milliliter), or Cre (Lv-PRSx8-Cre; titer, 3.9 × 1010 viral particles per milliliter). A detailed description of the methods used to generate and characterize these viruses, including the demonstration that injection of Lv-PRSx8-AT1AR-induced expression of AT1ARs capable of altering neuronal activity upon stimulation, has been published previously (Chen et al., 2010). Anesthetized mice were placed in a stereotaxic apparatus (Benchmark stereotaxic instruments; MyNeurolab) in a prone position. The position of the head was adjusted so that the skull surface was horizontal. A midline incision was made over the occipital bone, and portions of the bone overlying the cerebellum were carefully removed with a dental drill while keeping the lambdoid suture intact. The lambdoid and midline sutures served as reference points for rostrocaudal and lateral coordinates, respectively. Microinjections (100 nl per site) were made to the RVLM, 1.2 mm lateral to the midline and 5.3 mm ventral to the dorsal surface of the brain at three rostrocaudal coordinates, 1.7, 1.9, and 2.1 mm caudal to lambda. Microinjections were made through pulled glass micropipettes (1B100F-4; World Precision Instruments), with tip diameters of 30–50 μm. Solutions were ejected using pressurized nitrogen gas delivered via a pneumatic pressure device (SYS-PV820; World Precision Instruments), and volumes were determined by observation of movement of the fluid meniscus using a monocular microscope fitted with an eye-piece graticule (Cole-Parmer). Upon completion of the injection, the incision was closed with surgical silk stitches. Mice received atipamezole hydrochloride (1 mg/kg, i.p.; Antisedan; VMS Supplies) to reverse the anesthesia. Each mouse was housed individually. ROSA26-eYFP mice were injected unilaterally with Lv-PRSx8-Cre. AT1ARfl/fl mice were injected bilaterally with either Lv-PRSx8-Cre or Lv-PRSx8-GFP. AT1AR−/− mice were injected bilaterally with either Lv-PRSx8-AT1AR or Lv-PRSx8-GFP and used in long-term studies to assess cardiovascular control and stress responses.
Verification of viral-induced Cre expression
Approximately 4 weeks after microinjection of Lv-PRSx8-Cre into the RVLM, the ROSA26-eYFP mice (n = 3) were deeply anesthetized using the anesthetic protocol described above and perfused with 4% paraformaldehyde, and the brains removed for histological processing.
RVLM microinjections in anesthetized mice
Approximately 4 weeks after bilateral microinjection of Lv-PRSx8-Cre (n = 6) or Lv-PRSx8-GFP (n = 6) into the RVLM, AT1ARfl/fl mice were anesthetized by inhalation of isoflurane (1.8–2%) to study the acute cardiovascular response to microinjection of glutamate and AngII into the RVLM. Standard protocols were used as described previously (Chen et al., 2010). Briefly, mice were maintained at a rectal temperature of 37°C using a servocontrolled heating blanket, tracheotomized, and artificially ventilated (UGO Basile mouse ventilator 28025) with oxygen. End-tidal CO2 was measured (CWE) and maintained between 2.5 and 3.5%. The left carotid artery was cannulated for measurement of BP (Neurolog System; Digitimer), and heart rate (HR) was derived from the digitized signal (Cambridge Electronic Design). Fluid balance was maintained by subcutaneous injection of Hartmann's solution (Baxter Healthcare). Mice were placed in a stereotaxic frame, as described for injections above, and either glutamate (10 nl of a 10 mm solution) or AngII (50 nl of a 1 mm solution; AusPep) were microinjected into the RVLM. Several injections of glutamate were made to identify the site that produced an immediate rise in arterial pressure. Injections were made independently into both sides of the brain, with a period of at least 30 min elapsing between AngII microinjections. The AngII solution contained a 1% concentration of rhodamine-labeled microspheres (Invitrogen) to enable histological identification of the injection site postmortem. At the completion of the experiment, mice were deeply anesthetized and perfused with 4% paraformaldehyde, and the brains removed for histological analysis of GFP or CD68 expression, to localize viral injection sites, and rhodamine-labeled microspheres.
Cardiovascular and stress responses in RVLM Lv-PRSx8-AT1A R-injected knock-out mice
Telemetry implantation.
Under isoflurane anesthesia (Abbott Laboratories), AT1AR−/− (n = 12) and AT1AR+/+ (n = 6) mice were implanted with PA-C10 telemeters (Data Sciences International) in the left carotid artery as described previously (Butz and Davisson, 2001). Both before surgery and on the first day following surgery, each mouse received pain relief (Carprofen, 0.5 mg/100 g, i.p.; Norbrook). Each mouse received 0.5 ml saline subcutaneously at the time of operation and then again 24 h after operation. Mice were kept warm on a heat pad and monitored until fully recovered from anesthesia. Each mouse was housed individually, and the mouse cages were half kept on a heated surface for 24–48 h to assist in their thermoregulation. Two weeks after telemetry implantation, baseline 24 h recordings of BP were made, and on the next day, bilateral injections of either Lv-PRSx8-AT1AR (n = 7) or Lv-PRSx8-GFP (n = 5) were made into the RVLM of AT1AR−/− mice.
Cardiovascular measurements.
Pulsatile arterial pressure was recorded continuously, sampled at 1 kHz using an analog-to-digital data acquisition card (PCI-8024E; National Instruments), and the beat-to-beat mean arterial pressure (MAP) and HR were detected on-line and analyzed using a program written in Labview (Head et al., 2001; Navakatikyan et al., 2002). An index of locomotor activity was obtained by monitoring changes in the received signal strength, which occurred upon movement of the animal. For detection of activity, the mouse had to move location. Therefore, with the transmitter implanted subcutaneously, slight movements during grooming or eating were not registered as activity. Systolic, mean, and diastolic BP, pulse interval, and locomotor activity were stored in text format on an IBM-compatible computer. Baseline cardiovascular parameters were observed for an additional 4 weeks with recordings over a consecutive 24 h period every week.
Autonomic activity was assessed by monitoring spontaneous changes in BP and HR. Beat-to-beat MAP and HR were analyzed between 10 A.M. and 1 P.M. (during the inactive period) on 2 consecutive days. The autopower and cross-power spectra were calculated for multiple overlapping (by 50%) segments of MAP and HR using fast Fourier transform (Head et al., 2001). The average value used as the estimate of the baroreflex sensitivity in this study is the mid-frequency band (0.3–0.5 Hz) and relates to autonomic function (Janssen and Smits, 2002). Other frequency bands analyzed were the low-frequency band (0.08–0.3 Hz) and the high-frequency band (0.5–3 Hz). Normalized power was obtained by dividing the cumulative power within each frequency band by the total power (Head et al., 2004; Baudrie et al., 2007).
Stress test protocols.
Blood pressure, HR, and locomotor activity were recorded continuously throughout the experiment. Restraint and feeding tests were performed on the same day but separated by at least 60 min of recovery period (or until BP returned to baseline levels) with a quiet rest period of at least 5 min before the commencement of each test. To test the reproducibility of stress responses, all tests were repeated, in random order, on the following day. As there was no difference in cardiovascular responses to stress on Experimental Days 1 and 2, the data were pooled and treated as repeated measures for subsequent analysis. The cage-switch test was conducted approximately 1 week later on the final day of experimental protocol. All mice were then perfused for immunohistochemistry.
Feeding test.
A piece of almond (∼0.5 g), a novel palatable stimulus, was placed in the home cage. This is accompanied by marked increases in BP and HR (Jackson et al., 2007; Chen et al., 2009). The mouse was observed from when it commenced eating until when it ceased eating. The average change in MAP and HR over the first 5 min was used to estimate the magnitude of cardiovascular response to feeding (De Matteo et al., 2006).
Restraint stress.
Mice were placed in a cylindrical Plexiglas container used for tail cuff BP measurements (length, 135 mm; diameter, 50 mm; Harvard Apparatus). The sliding plate was moved to confine the mouse without applying physical pressure to it. The restrained mouse was left in the restrainer in its home cage for 5 min before being released.
Cage-switch stress.
All mice were placed in a clean cage and maintained in that cage without further cleaning for at 7 d before cage-switch stress. This is a model of acute psychosocial stress (Lee et al., 2004). During the light period on the day of the study, 1 h of baseline recording was made with the mice undisturbed in their home cage. The mice were then placed into a cage occupied previously (for 7 d) by a different male mouse for a period of 1 h. The pressor response is reproducible, takes over 90 min after beginning cage-switch test to subside, and is accompanied by significant increases in HR and motor activity in mice (Lee et al., 2004). The cage-switch test was conducted approximately 1 week after the restraint and feed tests on the final day of the experimental protocol. All mice were then anesthetized and perfused for immunohistochemistry processing.
Data analysis.
Cardiovascular responses to microinjection of glutamate and AngII into the RVLM of viral-injected AT1ARfl/fl mice were measured as the 10 s average maximal response after microinjection of either glutamate or AngII and compared to a 60 s preinjection control period. For analysis of the responses to restraint and novel almond feeding stimuli, 30 s averages of MAP, HR, and activity signals were determined for the period 5 min before the stimulus, 5 min during the stimulus, and 5 min after the stimulus. For analysis of the responses to dirty-cage-switch stress, 10 min averages of MAP, HR, and activity signals were determined for the period 1 h before the cage switch and 1 h during the cage switch.
Statistical analysis.
All variables are presented as mean ± SEM. Changes in arterial pressure and heart rate in response to glutamate and AngII microinjection into the RVLM were compared by two-way repeated-measures ANOVA followed by a pairwise multiple comparison (Holm–Sidak method; SigmaPlot version 11; Systat Software). Cardiovascular data from the telemetered mice were analyzed by a split-plot repeated-measures ANOVA where the between-group sums of squares due to change over time or stress were used to compare the differences between Lv-PRSx8-GFP- and Lv-PRSx8-AT1AR-injected AT1AR−/− mice and control, uninjected AT1AR+/+ mice. For the 24 h recordings over 33 d before and after microinjection, individual between-group Bonferroni-adjusted contrasts were made to compare values at Days 0, 7, 13, 20, 27, and 33. The degrees of freedom of the ANOVA were adjusted by the Greenhouse–Geisser coefficient to reduce the interdependence of repeated variables. Values were considered significant at p < 0.05.
Histological processing.
At the completion of each experimental protocol, anesthesia was induced in all mice by inhalation of isoflurane and then injection of ketamine/xylazine as described above. They were perfused through the heart with saline (0.9% NaCl) followed by a solution of 4% formaldehyde in 0.1 mol/L sodium phosphate buffer. The brains were removed, postfixed in formaldehyde for 1–2 h, and then cryoprotected in 20% sucrose overnight at 4°C. Brainstem sections were cut on a cryostat (Microm International) and serial coronal 40 μm sections were collected in four series and stored in cryoprotectant [30% (w/v) sucrose, 30% (v/v) ethylene glycol, 1% (w/v) polyvinyl-pyrrolidone in 50 mm phosphate buffer, pH 7.2] until immunohistochemical staining as described previously (Llewellyn-Smith et al., 2003; Chen et al., 2010). Brains from pAT1AR-GFP, Lv-PRSx8-GFP-injected AT1AR−/− and AT1ARfl/fl and Lv-PRSx8-Cre-injected ROSA26-eYFP mice were processed for immunohistochemical double staining of tyrosine hydroxylase (TH) and GFP. The Lv-PRSx8-AT1AR-injected AT1AR−/− mice and Lv-PRSx8-Cre-injected AT1ARfl/fl mice were processed for TH and the macrophage marker CD68, which is a reliable marker for the injection site (Card et al., 2006). The primary antibodies were rat anti-mouse CD68 (1:1000; AbD Serotec), rabbit anti-TH (1:500; Millipore Bioscience Research Reagents), and chicken anti-GFP (1:1000; Millipore Bioscience Research Reagents). The secondary antibodies were Alexa Fluor 488-conjugated goat anti-chicken (1:200), Cy3-conjugated donkey anti-rabbit (1:200), Dylight 549 goat anti-rat (1:200; AbD Serotec), or Alexa Fluor 488 goat anti-rabbit (1:200; Vector Laboratories), as required. Immunofluorescence was imaged using an Axio Imager D.1 microscope connected to an AxioCam MRc5 digital camera (both from Zeiss) and mapping performed with reference to the mouse brain atlas (Paxinos and Franklin, 2004).
Results
Distribution of AT1AR-expressing neurons in the mouse brain
The distribution of neurons expressing GFP in the pAT1AR-GFP mouse was examined throughout the brain in serial sections, taken every 300 μm, in three mice. The distribution of GFP-expressing cells was consistent between all the mice examined. Green fluorescent protein labeling was found in the soma and processes of cells with neuronal morphology, and no labeling was obvious in cells that were morphologically similar to astrocytes or glia. The distribution of GFP immunoreactivity throughout the brain was consistent with previous studies examining AT1R distribution in the mouse brain (Jenkins et al., 1997) with no evidence of ectopic expression. Some regions, such as the area postrema, were expected to display GFP immunoreactivity but did not. The cerebellum and spinal cord were not examined in this study.
Forebrain
A dense population of GFP-labeled neurons occurred in the dorsal lateral septal nucleus (Fig. 1A), vascular organ of the lamina terminalis (Fig. 1B), median preoptic nucleus (Fig. 1B,C), and subfornical organ (Fig. 1D). Numerous GFP-expressing neurons were observed in the parvocellular subdivisions of the hypothalamic paraventricular nucleus (Fig. 1G), the periventricular hypothalamic nucleus (Fig. 1G), and the suprachiasmatic nucleus (Fig. 1H). Within the hypothalamic paraventricular nucleus, no colocalization was observed between GFP and TH, indicating that the AT1AR-expressing cells were not part of the A12 dopaminergic cell group (data not shown). Scattered GFP-labeled cells occurred in the anterior hypothalamic area (Fig. 1E), lateral hypothalamic area (Fig. 1F), dorsal medial hypothalamic area (data not shown), and arcuate nucleus. Scattered neurons in the caudate–putamen (data not shown) and central nucleus of the amygdala (Fig. 1I) also showed GFP labeling. No GFP labeling was observed in the hippocampus, thalamic nuclei, supraoptic nucleus, or magnocellular component of paraventricular hypothalamic nucleus.

A–K, Photomicrographs of coronal sections at different levels of the neuraxis showing GFP immunoreactivity in pAT1AR-GFP transgenic mice. The white cell profiles show GFP expression. Scale bars: 100 μm. ac, Anterior commissure; AHA, anterior hypothalamic area; cc, central canal; CeA, central nucleus of the amygdala; LH, lateral hypothalamus; LSD, dorsal subnucleus of the lateral septal nucleus; LV, lateral ventricle; MnPO, median preoptic nucleus; NTS, nucleus of the solitary tract; OVLT, organum vasculosum of the lamina terminalis; PVN, hypothalamic paraventricular nucleus; SCh, suprachiasmatic nucleus; SFO, subfornical organ; Sp5C, caudal part of the spinal trigeminal nucleus; 3V, third ventricle; 4V, fourth ventricle.
Midbrain and pons
A distinct cluster of GFP-positive cells occurred in the dorsomedial interpeduncular nucleus of the pons (Fig. 1B), and scattered GFP-positive cells were seen in the lateral superior olive (data not shown).
Medulla
Neurons expressing GFP were observed in the dorsal medulla in the nucleus of the solitary tract (Fig. 1J) and the dorsal motor nucleus of the vagus (data not shown). In addition, GFP-positive terminals were prominent within the nucleus of the solitary tract. Surprisingly, no GFP labeling was observed in area postrema. The nucleus ambiguus contained a compact group of GFP-expressing cells (data not shown). GFP-labeled neurons were also present throughout the caudal nucleus of the spinal trigeminal tract (Fig. 1K). Within the RVLM (Fig. 2A), caudal ventrolateral medulla (data not shown), and nucleus of the solitary tract (data not shown), the majority of GFP-positive cells also expressed TH immunoreactivity (Fig. 2B) and thus represent the C1, A1, and A2 cell groups, respectively. A small proportion (∼10%) of GFP-labeled cells that were non-TH-immunoreactive were observed in both the RVLM (Fig. 2A) and caudal ventrolateral medulla.

A–F, Photomicrographs of fluorescence immunohistochemistry images showing GFP (A, D), TH (B, E), and the merged images (C, F) at the level of the RVLM from a pAT1AR-GFP mouse (A–C) and a ROSA26-eYFP mouse injected with Lv-PRSx8-Cre virus (D–F). Within the RVLM of a pAT1AR-GFP mouse the majority of GFP-expressing neurons also expresses TH. The arrows point to the few neurons that are clearly GFP positive that do not express TH. Following microinjection of Lv-PRSx8-GFP into the RVLM of a ROSA26-eYFP mouse, the majority of YFP expression occurs in TH-expressing cells. G, H, Arterial pressure (AP; in millimeters mercury), MAP (in millimeters mercury), and HR (bpm) were recorded in anesthetized AT1ARfl/fl mice injected ∼4 weeks earlier with Lv-PRsx8-GFP (G) or Lv-PRSx8-Cre (H) into the RVLM. Microinjection of AngII into the RVLM (arrows) induced an increase in MAP and HR in the Lv-PRsx8-GFP-injected animals that was similar to that observed previously in wild-type mice. This response was markedly attenuated in Lv-PRSx8-Cre injected mice. I, The grouped data (n = 6 for each group) showing the response to microinjection of glutamate and AngII into the RVLM of these animals. Scale bars: 100. ns, No significant difference between groups. ***p < 0.001.
Lentiviral-induced Cre expression and conditional knock-out
Microinjection of Lv-PRSx8-Cre into the RVLM of ROSA26-eYFP mice
To verify the cell selectivity and level of Cre-recombinase expression when delivered as a lentiviral transgene, Lv-PRSx8-Cre was microinjected into the RVLM of ROSA26-eYFP mice. Approximately 4 weeks after injection, robust eYFP expression was observed within the RVLM, which predominantly (∼90%) colocalized with TH immunoreactivity (Fig. 2D–F).
Response to RVLM microinjection of angiotensin II following cell-selective AT1AR deletion
Four weeks after bilateral microinjection of Lv-PRSx8-Cre or Lv-PRSx8-GFP into the RVLM of AT1ARfl/fl mice, there was no difference in baseline BP or HR under isoflurane (1.8–2.0%) anesthesia [GFP vs Cre: MAP, 86 ± 8 mmHg and 76 ± 5 mmHg; HR, 538 ± 30 beats per minute (bpm) and 518 ± 16 bpm; not significant]. Microinjection of 10 mm glutamate (10 nl) resulted in a robust and rapid increase in BP (Fig. 2I) when localized within the RVLM. Comparison of maximal responses revealed no difference between Lv-PRSx8-Cre- and Lv-PRSx8-GFP-injected animals. Microinjection of 1 mm AngII (50 nl) similarly resulted in a robust increase in BP, although with a longer time to reach maximum than glutamate, in the Lv-PRSx8-GFP-injected animals (Fig. 2G). In contrast, the response to AngII was dramatically attenuated in Lv-PRSx8-Cre-injected AT1ARfl/fl mice (not significant compared to baseline) (Fig. 2H). Postmortem analysis revealed that all microinjections included in this study were within 50–400 μm of the caudal pole of the facial nucleus and correlated with the distribution of TH- immunoreactive neurons.
Behavioral experiments
Verification of lentiviral injection sites
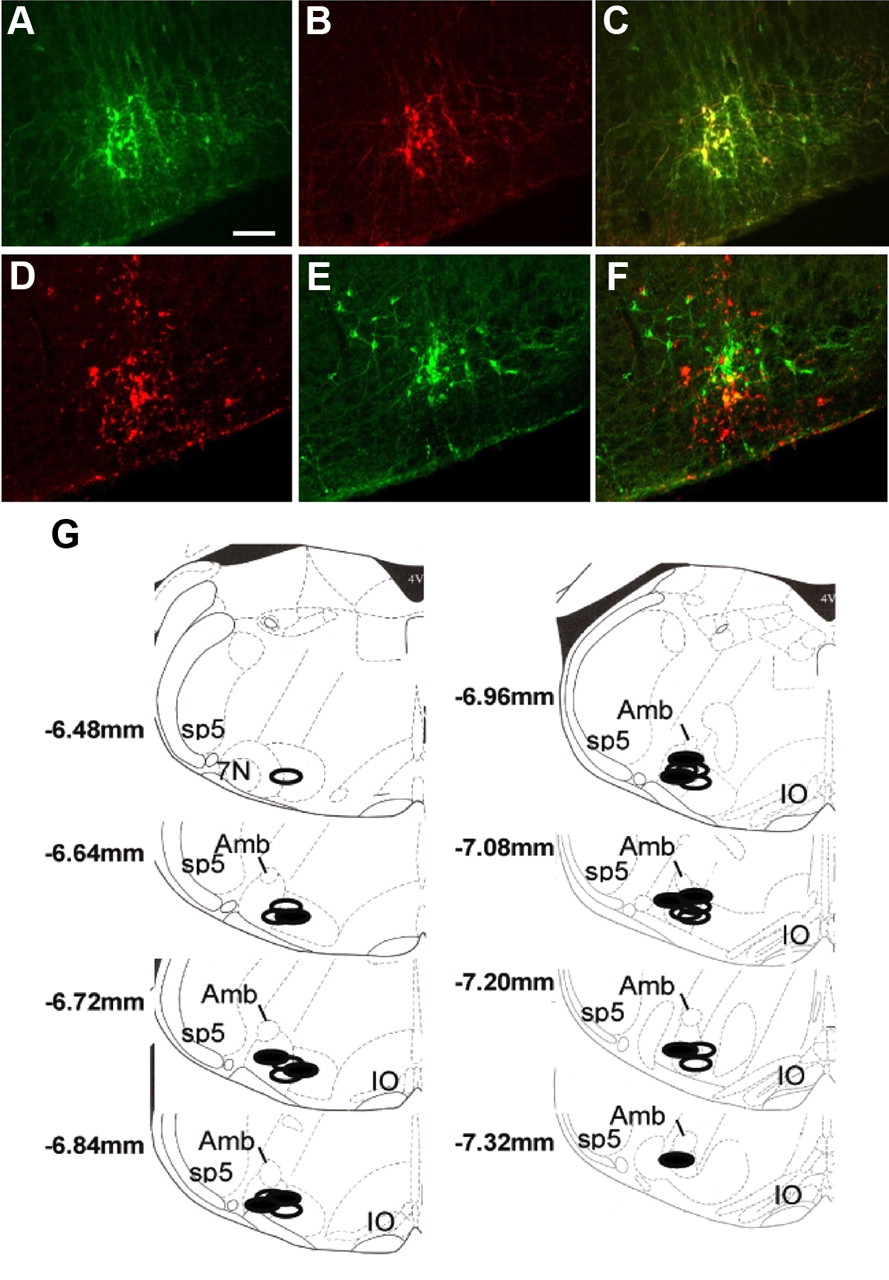
At the completion of the cage-switch stress tests, the mice were killed, and their brains were removed for histology. Verification of lentiviral-induced GFP expression was performed postmortem by immunohistochemistry with colocalization with TH (Fig. 3A–C). The site of Lv-PRSx8-AT1AR injections was visualized by examination of CD68 expression and colocalized with TH immunohistochemistry (Fig. 3D–F). For all mice included in this study, the microinjections were within the region of TH-expressing neurons and within 500 μm of the caudal pole of the facial motor nucleus. Schematic coronal sections of mouse medulla oblongata modified from the atlas of Paxinos and Franklin (2004), showing the range of all viral microinjections in mice from this study, are included (Fig. 3G). Previous studies using identical protocols have quantified expression of the AT1R binding site density in RVLM following lentiviral injections in AT1AR−/− mice (Chen et al., 2010). Those studies indicated that transgene expression resulted in an increase of 97% AT1AR expression above the level of endogenous receptor expression in the RVLM of a wild-type mouse. This remains less than the level of endogenous AT1R expression in other brain regions such as the nucleus of the solitary tract.

Photomicrographs of images obtained from fluorescence immunohistochemistry. A–C, The top row shows immunohistochemistry for GFP produced following microinjection of Lv-PRSx8-GFP in the RVLM (A) with colocalization of TH (B) on the same section and the merged image (C). D–F, The next row shows immunohistochemistry for the macrophage marker CD68 (D) from an animal injected with Lv-PRSx8-AT1AR in the RVLM, with colocalization of TH on the same section (E) and the merged image (F). Correlation of the two markers indicates correct microinjection placement. Scale bar: A (for A–F), 100 μm. G, Schematic diagrams of coronal sections of the mouse medulla oblongata [modified from the atlas of Paxinos and Franklin (2004)] showing the range of all viral microinjections in the AT1AR−/− mice included in this study. The open areas are RVLM Lv-PRSx8-GFP-injected AT1AR−/− mice, and the blocked areas are RVLM Lv-PRSx8-AT1AR-injected AT1AR−/− mice. Some injections from different mice were in identical regions and are represented only by a single symbol. The numbers represent the distance from bregma. Amb, Nucleus ambiguus; 7N, facial motor nucleus; Sp5, spinal trigeminal tract; IO, inferior olivary nucleus.
Average body weights of Lv-PRSx8-GFP- and Lv-PRSx8-AT1AR-injected AT1AR−/− mice were not different throughout the experiment (Week 0, 23.2 ± 1.0 g vs 23.3 ± 0.6 g; Week 5, 24 ± 1.2 g vs 24.0 ± 0.7 g, respectively). The AT1AR+/+ mice weighed 30 ± 0.5 g.
Basal cardiovascular measurements
The 24 h average basal MAP and HR were similar between both groups of AT1AR−/− mice at Week 0, before RVLM microinjection of Lv-PRSx8-GFP or Lv-PRSx8-AT1AR (Fig. 4). Seven days after viral microinjections, Lv-PRSx8-GFP mice had a transient increase in MAP and HR compared to Lv-PRSx8-AT1AR mice (100 ± 1 mmHg vs 83 ± 1 mmHg, p < 0.001; 585 ± 6 bpm vs 489 ± 5 bpm, p < 0.001). At all other times there was no difference in 24 h average MAP, HR, or locomotor activity between groups (Fig. 4).

A, B, Average hourly values for MAP, HR, and activity (arbitrary units) in AT1AR−/− mice at Day 0 (open circles) and at Day 33 (closed circles) after RVLM microinjection of either (A) Lv-PRSx8-AT1AR (n = 7) or (B) Lv-PRSx8-GFP (n = 5). The closed bar on the x-axis represents the dark period (active period), and the open bar represents the light period (inactive period). Error bars indicate SEM.
A distinct circadian pattern of MAP, HR, locomotor activity, and time spent active was observed in both groups, characterized by higher values of these parameters during the night (Fig. 5). Analysis of the circadian variation of BP showed that at Weeks 3–5 the Lv-PRSx8-AT1AR-injected mice had a small decrease in MAP during the inactive, light period (−6.6 ± 3.3 mmHg; p < 0.05), with no change in the dark period (−2.8 ± 3.4 mmHg) (Fig. 5B). There was no change in any of the other measured parameters. At the time of the stress responses, the AT1AR+/+ mice had a significantly higher resting blood pressure and heart rate compared to the Lv injected AT1AR−/− mice (MAP, AT1AR+/+, 108 ± 0.2 mmHg; Lv-PRSx8-GFP, 77 ± 1 mmHg; Lv-PRSx8-AT1AR, 74 ± 1 mmHg; HR: AT1AR+/+, 508 ± 7 bpm; Lv-PRSx8-GFP, 435 ± 10 bpm; Lv-PRSx8-AT1AR, 411 ± 11 bpm).

A, Grouped data for MAP during the day (6 A.M. to 6 P.M.) and night (6 P.M. to 6 A.M.) periods at baseline (control) and 3, 4, and 5 weeks following RVLM microinjection of either Lv-PRSx8-GFP (n = 5) or Lv-PRSx8-AT1AR (n = 7). B, The bar graph shows the change in MAP observed when the values for Weeks 3, 4, and 5 are averaged and compared to baseline. Values are mean ± SEM. *p < 0.05.
Autonomic activity was assessed indirectly by monitoring spontaneous changes in BP and HR using power spectral analysis. The average power of MAP and HR variability in all frequency spectral bands was similar in all mice before RVLM microinjections of Lv-PRSx8-AT1AR or Lv-PRSx8-GFP during the light period (inactive period) (Table 1). The average power of HR variability in the low-, mid-, and high-frequency bands during the inactive period was similar between Lv-PRSx8-AT1AR and Lv-PRSx8-GFP RVLM-microinjected mice (Table 1). However, the total power of HR variability (the average across low, middle, and high frequencies) was smaller in the Lv-PRSx8-AT1AR- compared to Lv-PRSx8-GFP-microinjected mice at Day 33 (926 ± 126 bpm2 vs 1538 ± 158bpm2; p < 0.05) (Table 1). The spontaneous baroreflex sensitivity (midfrequency of the HR variability spectra) was similar before and after viral microinjections, as well as between Lv-PRSx8-AT1AR and Lv-PRSx8-GFP at Day 33 (Table 1).
Average spectral power in low-, mid-, and high-frequency bands for MAP, HR, coherence, and gain in Lv-PRSx8-AT1A and Lv-PRSx8-GFP RVLM-microinjected mice assessed during the inactive period
Behavioral tests
Cage-switch stress
Placing a male mouse in a cage occupied previously for 1 week by another male mouse is a strong aversive stressor that induces an increase in BP that is maintained for at least 90 min. All mice showed similar initial pressor, tachycardic, and activity responses to the cage-switch stress (Fig. 6). Differences between cohorts were observed in the sustained phase of the response, which we measured as the average value over the entire 60 min cage-switch period. Lv-PRSx8-GFP mice showed a significant reduction in the sustained component of these responses, indicating that their BP and HR responses returned to baseline levels more quickly than the AT1AR+/+ mice (Fig. 6). Reexpression of the AT1AR in C1 neurons induced a significantly greater sustained pressor response to the 1 h period of dirty-cage-switch stress compared to Lv-PRSx8-GFP mice (average change in MAP, 22 ± 4 mmHg vs 10 ± 1 mmHg, respectively; p < 0.001) (Fig. 6). While significantly increased relative to Lv-PRSx8-GFP, the sustained pressor response in Lv-PRSx8-AT1AR mice remained reduced compared to the AT1AR+/+ (28 ± 2 mmHg; p < 0.05). The cage-switch-stress-induced HR (155 ± 12 bpm vs 163 ± 11 bpm, respectively) and locomotor responses were similar between Lv-PRSx8-AT1AR and Lv-PRSx8-GFP groups and significantly reduced compared to AT1AR+/+ mice.

Effect of expression of AT1ARs in RVLM C1 neurons on the response to dirty-cage-switch stress. MAP and HR and activity (arbitrary units) were measured before and for 60 min after placing each male mouse in a cage occupied previously by another male mouse. Each point represents the mean ± SEM of the average data for 10 min. Gray circles are AT1AR+/+ mice (n = 6), open circles are AT1AR−/− mice injected with Lv-PRSx8-GFP (n = 5), and filled circles are AT1AR−/− mice injected with Lv-PRSx8-AT1AR (n = 7). The same color scheme applies to the bar graphs, which show average changes in each parameter over with the initial 5 min period or the entire 60 min period compared to the control period. Plus signs denote comparison to the AT1AR+/+ mice, while asterisks denote comparison with Lv-PRSx8-GFP-injected mice. One symbol represents p < 0.05, two symbols represent p < 0.01, and three symbols represent p < 0.001.
Restraint stress
Restraint stress was induced by placing mice in a tail cuff BP restrainer. No difference in the MAP or HR responses to this stress was observed between AT1AR+/+ mice and the Lv-PRS-GFP group, but the Lv-PRSx8-AT1AR group showed a significantly diminished pressor response and increased tachycardia (p < 0.001) (Fig. 7).

A, B, Effect of expression of AT1ARs in RVLM C1 neurons on the short-term MAP and HR responses to restraint stress (A) and novel food stimulus (B) in AT1AR+/+ (gray symbols) and AT1AR−/− mice microinjected in the RVLM with either Lv-PRSx8-AT1AR (n = 7; closed circles) or Lv-PRSx8-GFP (n = 5; open circles). Each symbol represents an average (mean ± SEM) for each 30 s period. The bar graphs represent the average change in the measured parameters in each group. These average responses were calculated from a 5 min control period and 5 min of each stressor (the period between the dotted lines). Plus signs denotes comparison to the AT1AR+/+ mice, while asterisks denotes comparison with Lv-PRSx8-GFP-injected mice. One symbol represents p < 0.05, two symbols represent p < 0.01, and three symbols represet p < 0.001.
Novel feeding stimulus
The pressor response to presentation and eating of a novel, palatable food (almond) was increased in Lv-PRSx8-GFP compared to both AT1AR+/+ and Lv-PRSx8-AT1AR mice (MAP, 27 ± 1 mmHg vs 25 ± 3 mmHg; HR, 198 ± 15 bpm vs 171 ± 11 bpm) (Fig. 7). The HR response was significantly elevated in the Lv-PRSx8-AT1AR and Lv-PRSx8-GFP compared to AT1AR+/+ mice (Fig. 7).
Discussion
Using several transgenic mouse models, we demonstrated that the AT1AR is endogenously expressed in RVLM C1 neurons and required for the pressor response to microinjection of AngII into this nucleus. Lentiviral expression of this AT1AR in the C1 neurons of adult AT1AR−/− mice did not alter basal BP or HR but significantly increased the sustained pressor response to cage-switch stress toward that of AT1AR+/+ mice. Lentiviral expression of the AT1AR in C1 neurons did not alter the HR or activity response to cage-switch stress. The effect of AT1AR expression in C1 neurons on the immediate pressor response to cage-switch or restraint stressors or the appetitive stimulus was more variable. These observations argue that endogenous AngII acts on C1 neurons to modulate the BP response to aversive stress. In addition, our results clearly link activation of the RVLM C1 neurons to the sustained elevation of BP elicited by cage-switch stress.
Two lines of evidence support the conclusion that RVLM C1 neurons express AT1AR endogenously. First, in a transgenic mouse with a bacterial artificial chromosome, in which the AT1AR promoter drives expression of GFP, transgene expression occurs in rostral RVLM C1 neurons. These are presumptive sympathetic premotor neurons. Autoradiographic and in situ hybridization histochemical studies have demonstrated expression of AT1R in the RVLM of several species, including humans (Allen et al., 1988; Lenkei et al., 1997). These approaches did not provide the resolution or compatibility that enabled neurochemical characterization of the AT1R-expressing cells. Immunohistochemical approaches have been difficult due to the low-level expression of the receptor and general issues surrounding specificity of antibodies directed to the AT1AR (Smith et al., 1998). The transgenic mouse used in this study provides an alternative approach. Our observations concur with a previous immunohistochemical study (Wang et al., 2008) and support electrophysiological observations in slices of RVLM that demonstrate that AngII directly excites C1 neurons (Li and Guyenet, 1996). While transgenic mice represent a novel approach to examining the distribution of proteins for which standard approaches are problematic, there are methodological limitations due to potential ectopic expression. In this study, we mapped the distribution of GFP-expressing cells throughout the mouse CNS and compared it to the known distribution observed using in vitro autoradiography (Jenkins et al., 1997). The maps are very similar, although there were a few sites where, on the basis of autoradiography, we expected to see GFP but didn't. These tend to be regions of lower level expression, such as the area postrema and parabrachial nucleus. This may be due to reduced sensitivity or that the AT1BR may be responsible for the majority of AT1R binding observed in some nuclei.
Second, we used lentiviruses with the PRSx8 promoter to drive Cre expression in the RVLM of mice containing a conditional AT1AR allele. These mice, which showed no change in their response to RVLM microinjection of glutamate, did not show a pressor response to microinjection of AngII. The response to AngII was not affected by injection of Lv-PRSx8-GFP. Together, these data strongly support the conclusion that AngII acts in the RVLM to increase BP via the AT1AR which is endogenously expressed on C1 neurons.
The pressor response induced by aversive stressors has two phases—an initial rapid phase reliant upon glutamatergic transmission in the RVLM and a sustained phase, which partially involves AngII action in the RVLM (Mayorov and Head, 2002, 2003). Blockade of RVLM AT1R attenuates the sustained cardiovascular response to aversive air-jet stress in conscious rabbits, without affecting the initial response (Mayorov and Head, 2003). Similarly, the sustained pressor response to aversive stress is diminished in AT1AR−/− mice, with an associated decrease in Fos expression in RVLM neurons (Davern et al., 2009). In the current study, cage-switch stress in AT1AR−/− mice expressing AT1AR in C1 neurons induced a sustained elevation in BP compared to Lv-PRSx8-GFP-injected AT1AR−/− mice. This mimics the BP response to cage-switch stress in wild-type mice (Davern et al., 2009). In contrast, the pressor response in the Lv-PRSx8-GFP-injected AT1AR−/− mice starts to return to baseline by 20 min and has reached baseline by the end of the cage-switch test. This observation is consistent with that seen in the AT1AR−/− mice (Davern et al., 2009). We conclude that the sustained pressor response to cage-switch stress is dependent upon AngII acting on the AT1AR in C1 neurons. The outstanding question relates to the source of the AngII that provides this activation. Unfortunately our understanding of the biochemistry of the brain renin–angiotensin system and the localization of its components remains incomplete such that this question cannot be definitively answered.
The response of the lentiviral-injected AT1AR−/− mice to short-term stress is more variable, both within this study and in comparison to results from previous studies (Chen et al., 2009). In this study, the initial pressor response to aversive stress is either not altered (cage switch) or significantly reduced (restraint) in the Lv-PRSx8-AT1AR-expressing AT1AR−/− mice, while the pressor response to an appetitive stimulus is slightly reduced relative to the GFP-expressing mice and not different from the AT1AR+/+ mice. In published observations (Chen et al., 2009), the AT1AR−/− mice showed a diminished response to the appetitive and restraint stimuli compared to AT1AR+/+ mice, which is not replicated here by the Lv-PRSx8-GFP-injected mice. It is possible that GFP expression has altered the response to restraint stress, but this is unlikely as none of the other observations related to the Lv-PRSx8-GFP mice show any substantial difference compared to AT1AR−/− mice from previous studies. The reason for this discrepancy is not clear and suggests caution in relation to drawing strong conclusions about the responses to short-term stressors.
The link between acute aversive stress and cardiovascular events such as myocardial infarction is clearly established (Willich et al., 1993; Mittleman et al., 1995; Leor et al., 1996). There is also a growing recognition that chronic inescapable or unpredictable stress contributes to the development of essential hypertension in some people (Esler, 2009). The mechanisms underpinning this involvement in hypertension remain unclear. We propose that the sustained increase in BP induced by AngII in RVLM has significance for understanding how exposure to repeated aversive stressors could lead to hypertension. If BP remains elevated from one aversive stress exposure to the next, it is possible that a tetanic response could occur. This, combined with potential trophic effects of the elevated SNA (Hardebo, 1992; Mancia et al., 1999), could lead to sustained elevations of BP and consequent cardiovascular disease.
Cage-switch stress also induced tachycardia and increases in activity. Injection of Lv-PRSx8-AT1AR in RVLM did not alter either of these parameters. This suggests that the tachycardia induced by cage-switch stress is produced by an alternative pathway activating cardiac sympathetic activity, or by withdrawal of cardiac vagal activity. Cage-switch stress in mice activates neurons in raphe pallidus (Davern et al., 2009), which is involved in the sympathetic HR response to some stressors (Zaretsky et al., 2003). While our studies do not provide further information about the pathways involved in the cardiac response to aversive stress, the lack of effect on this pathway adds considerable strength to the specificity of our observations on the role of C1 neurons.
Appetitive stimuli induce a distinct rise in BP and HR in both experimental animals and humans (Del Bo et al., 1985; Brosschot and Thayer, 2003; Braesicke et al., 2005). Feeding-associated arousal is initiated by the activation of descending inputs from the CNS rather than the viscerosympathetic reflex responses (Diamond and LeBlanc, 1987; Matsukawa and Ninomiya, 1987). In our study, presentation of a novel palatable food elicited a pressor and tachycardic response, with only minor differences between groups. In addition, the cardiovascular responses to the appetitive stimulus are comparable to those seen in the naive AT1AR−/− and AT1AR+/+ mice (Chen et al., 2009). The lack of BP difference in the appetitive stimulus suggests that either the RVLM C1 neurons or AngII is not essential to this response.
We had hypothesized that expression of AT1AR in C1 neurons might increase resting BP in the AT1AR−/− mice. However, the observed lack of effect, and potentially a small decrease in daytime resting BP, is supported by previous observations. Little or no BP change occurs in response to microinjection of AT1R antagonists into the RVLM of normotensive animals under basal conditions (Allen et al., 2009). Transgenic mice with neuronal overexpression of the AT1AR have normal resting BP, despite having exaggerated pressor responses to intracerebroventricular infusion of AngII (Lazartigues et al., 2002). Thus, it seems that under basal conditions AngII has little effect on RVLM neurons to regulate BP. In contrast, under conditions where the AngII input to RVLM is activated, either by physiological stimuli or in pathological states such as hypertension, AT1R antagonists produce significant decreases in BP through inhibition of sympathetic efferent activity (Allen et al., 2009). This supports the observation that activation of the receptor, and not just increased expression, is required for an altered phenotype.
In this study, we demonstrate that the AT1AR is expressed in C1 neurons and required for the pressor response to AngII in this nucleus. Furthermore, transgenic expression of the AT1AR in C1 neurons in adult AT1AR−/− mice modulates the pressor response to cage-switch stress, implicating both these cells and this receptor system in the stress pathway. Of importance is the demonstration that expression of the AT1AR in C1 neurons returns the BP response to cage-switch stress toward that of the AT1AR+/+ mouse. This implies that AngII is released within the RVLM in response to cage-switch stress and serves to sustain the pressor response. We propose that this sustained activation of autonomic activity could underpin the development of hypertension induced by repeated aversive stressors.
Footnotes
↵*G.A.H. and A.M.A. are joint senior authors for this publication
This work was supported by National Heart Foundation of Australia Grant G07M3186, Australian Research Council Grant DP1094301, and National Health and Medical Research Council of Australia Grants 566563 and 1007451.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Andrew M. Allen, Department of Physiology, University of Melbourne, Melbourne, VIC 3010, Australia. a.allen{at}unimelb.edu.au





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}