
Abstract
The lysosomal storage disorder (LSD) mucopolysaccharidosis type I (MPS I, McKusick 25280, Hurler syndrome, Hurler-Scheie syndrome, Scheie syndrome) is caused by a deficiency in the lysosomal enzyme, α-L-iduronidase (EC 3.2.1.76). MPS I patients can present within a diverse clinical spectrum, ranging from classical Hurler syndrome to attenuated Scheie syndrome. Laronidase (Aldurazyme®) enzyme replacement therapy has been developed as a treatment strategy for MPS I patients and has been approved for clinical practice. Here we review the pre-clinical studies and clinical trials that have been used to demonstrate that intravenous laronidase therapy is well tolerated and effective for treating MPS I patients who do not have neuronal pathology. Current challenges for a viable treatment strategy for all MPS I patients include development of an early screening protocol that identifies patients before the onset of irreversible pathology, methods to predict disease severity, appropriate treatment for neuropathology, and an effective patient monitoring regimen.

Similar content being viewed by others
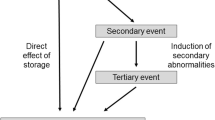
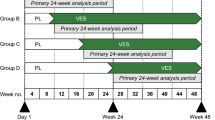

Notes
International Union of Biochemistry and Molecular Biology [IUBMB] nomenclature; (see URL: http://www.chem.qmul.ac.uk/iubmb/enzyme/)
The use of trade names is for product identification purposes only and does not imply endorsement
References
McKusick VA, Neufeld EF. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, et al., editors. The metabolic and molecular basis of inherited diseases. 8th ed. New York: McGraw-Hill, 2001
Berkhan O. Zwei falle von skaphokephalie. Arch Anthrop 1907; 34: 8
Hurler G. Ueber einen Typ multiper Abartungen, vor-weigend am skelettsystem. Z Kinderheilkd 1919; 24: 220–34
Brante G. Gargoylism: a mucopolysaccharidosis. Scand J Clin Lab Invest 1952; 4: 43–6
Hopwood JJ, Morris CP. The mucopolysaccharidoses: diagnosis, molecular genetics and treatment. Mol Biol Med 1990; 7: 381–404
DeDuve C. From cytases to lysosomes. Fed Proc 1964; 23: 1045–9
Hers HG. Inborn lysosomal diseases. Prog Gastroent 1965; 48: 625–33
Frantantoni JC, Hall CW, Neufeld EF. Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science 1968; 162: 570–2
Kornfeld S. Trafficking of lysosomal enzymes. FASEB J 1987; 1: 462–8
BioMarin Pharmaceutical Inc. Laronidase™ Clin Pharmacol Ther 2001; 70: 101
Durand P, Lehn P, Callebaut I, et al. Active-site motifs of lysosomal acid hydrolases: invariant features of clan GH-A glycosyl hydrolases deduced from hydrophobic cluster analysis. Glycobiology 1997; 7: 277–84
Durand P, Fabrega S, Henrissat B, et al. Structural features of normal and mutant human lysosomal glycoside hydrolases deduced from bioinformatics analysis. Hum Mol Genet 2000; 9: 967–77
McCarter JD, Withers SG. Mechanisms of enzymatic glycoside hydrolysis. Curr Opin Struct Biol 1994; 4: 885–92
Davies G, Henrissat B. Structures and mechanisms of glycosyl hydrolases. Structure 1995; 3: 853–9
Taylor JA, Gibson GJ, Brooks DA, et al. α-L-iduronidase in normal and mucopolysaccharidosis-type-I human skin fibroblasts. Biochem J 1991; 274: 263–8
Kakkis ED, Matynia A, Jonas AJ, et al. Over-expression of the human lysosomal enzyme α-L-iduronidase in Chinese hamster ovary cells. Protein Expr Purif 1994; 5: 225–32
Unger EG, Durrant J, Anson DS, et al. Recombinant α-L-iduronidase: characterisation of the purified enzyme and correction of mucopolysaccharidosis type I fibroblasts. Biochem J 1994; 304: 43–9
Bach G, Friedman R, Weissmann B, et al. The defect in the Hurler and Scheie syndromes: deficiency of α-L-iduronidase. Proc Natl Acad Sci U S A 1972; 69: 2048–51
Shull RM, Kakkis ED, McEntee MF, et al. Enzyme replacement in a canine model of Hurler syndrome. Proc Natl Acad Sci U S A 1994; 91: 12937–41
Kakkis ED, McEntee MF, Schmidtchen A, et al. Long-term and high-dose enzyme replacement therapy in the canine model of mucopolysaccharidosis I. Biochem Mol Med 1996; 58: 156–67
Kakkis ED, Schuchman E, He X, et al. Enzyme replacement therapy in feline mucopolysaccharidosis I. Mol Genet Metab 2001; 72: 199–208
Turner CT, Hopwood JJ, Brooks DA. Enzyme replacement therapy in mucopolysaccharidosis I: altered distribution and targeting of α-L-iduronidase in immunized rats. Mol Genet Metab 2000; 69: 277–85
Kakkis ED, Muenzer J, Tiller GE, et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med 2001; 344: 182–8
Wraith JE. Enzyme replacement therapy in mucopolysaccharidosis type I: progress and emerging difficulties. J Inherit Metab Dis 2001; 24: 245–50
Yogalingam G, Guo XH, Muller VJ, et al. Identification and molecular characterisation of a-L-iduronidase mutations present in mucopolysaccharidosis I patients undergoing enzyme replacement therapy. Hum Mutat 2004; 24: 199–207
Wraith JE, Clarke LA, Beck M, et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human α-L-iduronidase (Laronidase™). J Pediatr 2004; 144: 581–8
Clarke LA, Wraith JE, Beck M, et al. Aldurazyme® (Laronidase™) enzyme replacement therapy for MPS I: 72-week extension data [abstract]. Mol Genet Metab 2004; 81: 169
Beck M, Guffon N, Wraith JE, et al. Aldurazyme® (Laronidase™) enzyme replacement therapy in mucopolysaccharidosis I: preliminary data in children less than 5 years of age [abstract P42]. 8th International Symposium on Mucopolysaccharide and Related Diseases; 2004 Jun 10–13; Mainz: 29
Wright J, Motwani J, Gray G, et al. Hurler syndrome (MPSIH): enzyme replacement therapy pre-bone marrow transplantation [abstract]. Mol Genet Metab 2004; 81: 168
Kakavanos R, Turner CT, Hopwood JJ, et al. Immune tolerance in mucopolysaccharidosis I patients after long term enzyme replacement therapy. Lancet 2003; 361: 1608–13
Kakkis ED, Lester T, Yang R, et al. Successful induction of immune tolerance to enzyme replacement therapy in canine mucopolysaccharidosis I. Proc Natl Acad Sci U S A 2004; 101: 829–34
Chamoles NA, Blanco M, Gaggioli D, et al. Hurler-like phenotype: enzymatic diagnosis in dried blood spots on filter paper. Clin Chem 2001; 47: 2098–102
Meikle PJ, Hopwood JJ, Clague AE, et al. Prevalence of lysosomal storage disorders. JAMA 1999; 281: 249–54
Meikle PJ, Brooks DA, Ravenscroft EM, et al. Diagnosis of lysosomal storage disorders: evaluation of lysosome-associated membrane protein LAMP-1 as a diagnostic marker. Clin Chem 1997; 43: 1325–35
Meikle PJ, Yan M, Ravenscroft EM, et al. Altered trafficking and turnover of LAMP-1 in Pompe disease-affected cells. Mol Genet Metab 1999; 66: 179–88
Ranieri E, Gerace RL, Ravenscroft EM, et al. Pilot neonatal screening program for lysosomal storage disorders, using LAMP-1. Southeast Asian J Trop Med Public Health 1999; 30(Suppl. 2): 111–3
Chang MH, Bindloss CA, Grabowski GA, et al. Saposins A, B, C, and D in plasma of patients with lysosomal storage disorders. Clin Chem 2000; 46: 167–74
Umapathysivam K, Whittle AM, Ranieri E, et al. Determination of acid aglucosidase protein: evaluation as a screening marker for Pompe disease and other lysosomal storage disorders. Clin Chem 2000; 46: 1318–25
Gerber SA, Scott CR, Turecek F, et al. Direct profiling of multiple enzyme activities in human cell lysates by affinity chromatography/electrospray ionization mass spectrometry: application to clinical enzymology. Anal Chem 2001; 73: 1651–7
Scott HS, Bunge S, Gal A, et al. Molecular genetics of mucopolysaccharidosis type I: diagnostic, clinical, and biological implications. Hum Mutat 1995; 6: 288–302
Cardiff University. The human gene mutation database [online]. Available from URL: http://archive.uwcm.ac.uk/uwcm/mg/hgmdO.html [Accessed 2004 Dec 14]
Beesley CE, Meaney CA, Greenland G, et al. Mutational analysis of 85 mucopolysaccharidosis type I families: frequency of known mutations, identification of 17 novel mutations and in vitro expression of missense mutations. Hum Genet 2001; 109: 503–11
Hopwood JJ, Harrison JR. High-resolution electrophoresis of urinary glycosaminoglycans: an improved screening test for the mucopolysaccharidoses. Anal Biochem 1982; 119: 120–7
Hopwood JJ, Muller V. Diagnostic enzymology of α-L-iduronidase with special reference to a sulphated disaccharide derived from heparin. Clin Sci (Lond) 1982; 62: 193–201
Fuller M, Meikle PJ, Hopwood JJ. Glycosaminoglycan degradation fragments in mucopolysaccharidosis I. Glycobiology 2004; 14: 443–50
Watts RW, Spellacy E, Adams JH. Neuropathological and clinical correlations in Hurler disease. J Inherit Metab Dis 1986; 9: 261–72
Mendez-Otero R, Santiago MF. Functional role of a specific ganglioside in neuronal migration and neurite outgrowth. Braz J Med Biol Res 2003; 36: 1003–13
Hopwood JJ, Vellodi A, Scott HS, et al. Long-term clinical progress in bone marrow transplanted mucopolysaccharidosis type I patients with a defined genotype. J Inherit Metab Dis 1993; 16: 1024–33
Peters C, Balthazor M, Shapiro EG, et al. Outcome of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood 1996; 87: 4894–902
Peters C, Shapiro EG, Anderson J, et al. Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children: the Storage Disease Collaborative Study Group. Blood 1998; 91: 2601–8
Staba SL, Escolar ML, Poe M, et al. Cord-blood transplants from unrelated donors in patients with Hurler’s syndrome. N Engl J Med 2004; 350: 1960–9
Fairbairn LJ, Lashford LS, Spooncer E, et al. Towards gene therapy of Hurler syndrome. Proc Natl Acad Sci U S A 1996; 93: 2025–30
Huang MM, Wong A, Yu X, et al. Retrovirus-mediated transfer of the human α-L-iduronidase cDNA into human hematopoietic progenitor cells leads to correction in trans of Hurler fibroblasts. Gene Ther 1997; 4: 1150–9
Kolter T, Sandhoff K. Glycosphingolipid degradation and animal models of GM2-gangliosidoses. J Inherit Metab Dis 1998; 21: 548–63
Matsuda J, Suzuki O, Oshima A, et al. Chemical chaperone therapy for brain pathology in G(M1)-gangliosidosis. Proc Natl Acad Sci U S A 2003; 100: 15912–7
Keeling KM, Brooks DA, Hopwood JJ, et al. Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of α-L-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation. Hum Mol Genet 2001; 10: 291–9
Hein LK, Bawden M, Muller VJ, et al. α-L-iduronidase premature stop codons and potential read-through in mucopolysaccharidosis type I patients. J Mol Biol 2004; 338: 453–62
Kakkis ED, Peinovich M, Lester T, et al. Successful treatment of the brain and meninges with immune tolerance in canine MPS I [abstract V32]. 8th International Symposium on Mucopolysaccharide and Related Diseases; 2004 Jun 10–13; Mainz: 14
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia, by Fellowship and Program grant funding. D.A. Brooks and J.J. Hopwood have developed intellectual property relating to α-L-iduronidase and mucopolysaccharidosis I. J.J. Hopwood is a consultant for Transkaryotic Therapies. J.J. Hopwood and P.J. Meikle currently receive research funds from Genzyme Corporation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wraith, E.J., Hopwood, J.J., Fuller, M. et al. Laronidase Treatment of Mucopolysaccharidosis I. BioDrugs 19, 1–7 (2005). https://doi.org/10.2165/00063030-200519010-00001
Published:
Issue Date:
DOI: https://doi.org/10.2165/00063030-200519010-00001