
Abstract
This article considers the issue of personalized drug discovery for the orphan disease cystic fibrosis (CF) to deliver a candidate for therapeutic development. CF is a very complicated disease due to numerous anomalies of the gene leading to progressive severity and morbidity. Despite extensive research efforts, 20 years after the cloning of the CF gene, CF patients are still waiting for a curative treatment as prescribed medications still target the secondary manifestations of the disease rather than the gene or the CF transmembrane conductance regulator (CFTR) protein. New therapeutics aimed at improving mutant CFTR functions, also known as ‘protein repair therapy’ are nevertheless hoped and predicted to replace some of the currently used therapy, while improving the quality of life as well as life expectancy of CF patients. Although there is substantial variability in the cost of treating CF between countries, a protein repair therapy should also alleviate the financial burden of medical costs for CF patients and their families. Finding new drugs or rediscovering old ones for CF is critically dependent on the delivery of molecular and structural information on the CFTR protein, on its mutated version and on the network of CFTR-interacting proteins. The expertise needed to turn compounds into marketable drugs for CF will depend on our ability to provide biological information obtained from pertinent models of the disease and on our success in transferring safe molecules to clinical trials. Predicting a drug-induced response is also an attractive challenge that could be rapidly applied to patients.



Similar content being viewed by others
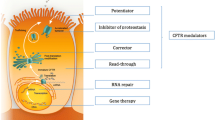
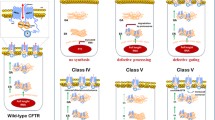

References
Bobadilla JL, Macek Jr M, Fine JP, et al. Cystic fibrosis: a worldwide analysis of CFTR mutations—correlation with incidence data and application to screening. Hum Mutat 2002; 19: 575–606
Rommens JM, Iannuzzi MC, Kerem BS, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989; 245: 1059–65
Kerem B, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science 1989; 245: 1073–80
Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245: 1066–73
Hanrahan JW, Gentzsch M, Riordan JR. The cystic fibrosis transmembrane conductance regulator (ABCC7). In: Holland IB, Cole S, Kuchler K, et al., editors. ABC proteins: from bacteria to man. New York: Academic Press, 2003: 589–618
Tabcharani JA, Chang XB, Riordan JR, et al. Phosphorylation-regulated Clchannel in CHO cells stably expressing the cystic fibrosis gene. Nature 1991; 352: 628–31
Gadsby DC, Nairn AC. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol Rev 1999; 79: 77–107
Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005; 352: 1992–2001
Boucher RC. Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends Mol Med 2007; 13: 231–40
Ribeiro CM. The role of intracellular calcium signals in inflammatory responses of polarised cystic fibrosis human airway epithelia. Drugs R D 2006; 7: 17–31
Jacquot J, Tabary O, Le Rouzic P, et al. Airway epithelial cell inflammatory signalling in cystic fibrosis. Int J Biochem Cell Biol 2008; 40: 1703–15
Simmonds NJ, Cullinan P, Hodson ME. Growing old with cystic fibrosis: the characteristics of long-term survivors of cystic fibrosis. Res Med 2009; 103: 629–35
Rosenblatt RL. Lung transplantation in cystic fibrosis. Resp Care 2009; 54: 777–87
Castellani C, Cuppens H, Macek Jr M, et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J Cyst Fibros 2008; 7: 179–96
De Araujo FG, Novaes FC, Dos Santos NPC, et al. Prevalence of DF508, G551D, G542X, and R553X mutations among cystic fibrosis patients in the north of Brazil. Braz J Med Biol Res 2005; 38: 11–5
Kabra SK, Kabra M, Lodha R, et al. Cystic fibrosis in India. Pediatr Pulmonol 2007; 42: 1087–94
Scotet V, Gollet D, Duguépéroux I, et al. Spatial and temporal distribution of the cystic fibrosis and of its mutations in Brittany, France: a retrospective study from 1960. Hum Genet 2002; 111: 247–54
Duguépéroux I, De Braekeleer M. Genotype-phenotype relationship for five CFTR mutations frequently identified in western France. J Cyst Fibros 2004; 4: 259–63
First Eastern European Conference on the diagnosis of cystic fibrosis [online]. Available from URL: http://www.klinickagenetika.org [Accessed 2009 May 25]
Yamashiro Y, Shimizu T, Oguchi S, et al. The estimated incidence of cystic fibrosis in Japan. J Pediatr Gastroenterol Nutr 1997; 24: 544–7
Morrow BM, Argent AC, Zar HJ, et al. Improvements in lung function of a pediatric cystic fibrosis population in a developing country. J Pediatr (Rio J) 2008; 84: 403–9
Ratbi I, Génin E, Legendre M, et al. Cystic fibrosis carrier frequency and estimated prevalence of the disease in Morocco. J Cyst Fibros 2008; 7: 440–3
Cystic fibrosis genetic analysis consortium [online]. Available from URL: http://www.genet.sickkid.on.ca/cftr [Accessed 2009 May 25]
Li N, Pei P, Bu D-F, et al. A novel CFTR mutation found in a Chinese patient with cystic fibrosis. Chin Med J 2006; 119: 103–9
Loumi O, Ferec C, Mercier B, et al. CFTR mutations in the Algerian population. J Cyst Fibros 2008; 7: 54–9
Messaoud T, Verlingue C, Denamur E, et al. Distribution of CFTR mutations in cystic fibrosis patients of Tunisian origin: identification of two novel mutations. Eur J Hum Genet 1996; 4: 20–4
Becq F. On the discovery and development of CFTR chloride channel activators. Curr Pharm Des 2006; 12: 471–84
Drumm ML, Wilkinson DJ, Smit LS, et al. Chloride conductance expressed by delta F508 and other mutant CFTRs in Xenopus oocytes. Science 1991; 254: 1797–9
Grubb B, Lazarowski E, Knowles M, et al. Isobutylmethylxanthine fails to stimulate chloride secretion in cystic fibrosis airway epithelia. Am J Respir Cell Mol Biol 1993; 8: 454–60
Verkman AS. Drug discovery in academia. Am J Physiol 2004; 286: 465–74
Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat Rev Drug Discov 2009; 8: 153–71
Becq F. Pharmacological interventions for the correction of ion transport defect in cystic fibrosis. Expert Opin Ther Patents 2004; 14: 1465–83
Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993; 73: 1251–4
Cheng SH, Gregory RJ, Marshall J, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990; 63: 827–34
Dalemans W, Barbry P, Champigny G, et al. Altered chloride ion channel kinetics associated with the DF508 cystic fibrosis mutation. Nature 1991; 354: 526–8
Cutting GR, Kasch LM, Rosenstein BJ, et al. A cluster of cystic fibrosis mutations in the first nucleotide binding fold domain of the cystic fibrosis conductance regulator protein. Nature 1990; 346: 366–9
Becq F, Jensen TJ, Chang X-B, et al. Phosphatase inhibitors activate normal and defective CFTR chloride channels. Proc Natl Acad Sci U S A 1994; 91: 9160–4
Sheppard DN, Rich DP, Ostedgaard LS, et al. Mutations in CFTR associated with mild-disease-form Cl− channels with altered pore properties. Nature 1993; 362: 160–4
Haardt M, Benharouga M, Lechardeur D, et al. C-terminal truncations destabilize the cystic fibrosis transmembrane conductance regulator without impairing its biogenesis. J Biol Chem 1999; 31: 21873–7
European Cystic Fibrosis Society [online]. Available from URL: http://www.ecfs.eu [Accessed 2009 May 25]
Cystic Fibrosis Foundation [online]. Available from URL: http://www.cff.org [Accessed 2009 May 25]
Doring G, Elborn JS, Johannesson M, et al. Clinical trials in cystic fibrosis. J Cyst Fibros 2007; 6: 85–99
Goss CH, Mayer-Hamblett N, Williams J, et al. The Cystic Fibrosis Foundation Therapeutics Development Network: a national effort by the Cystic Fibrosis Foundation to build a clinical trials network. Child Health Care 2008; 37: 5–20
Bedwell DM, Kaenjak A, Benos DJ, et al. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med 1997; 3: 1280–4
Wilchanski M, Yahav Y, Yaacov Y, et al. Gentamicininduced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med 2003; 349: 1433–41
PTC Therapeutic [online]. Available from URL: http://www.ptcbio.com [Accessed 2009 May 25]
Aurino S, Nigro V. Readthrough strategies for stop codons in Duchenne muscular dystrophy. Acta Myol 2006; 25: 5–12
Kerem E, Hirawat S, Armoni S, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet 2008; 372: 719–27
Sermet-Gaudelus I, De Boeck K, Casimir G, et al. Children with nonsense-mutation-mediated cystic fibrosis respond to investigational treatment with PTC 124 [abstract]. Pediatr Pulmonol 2008; Suppl. 31: 313
Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 1995; 83: 122–7
Heda GD, Tanwani M, Marino CR. The delta F508 mutation shortens the biochemical half-life of plasma membrane CFTR in polarized epithelial cells. Am J Physiol Cell Physiol 2001; 280: 166–74
Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing DF508-CFTR. J Clin Invest 1997; 100: 2457–65
Rubenstein RC, Zeitlin PL. Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of DF508-CFTR. Am J Physiol 2000; 278: 259–67
Choo-Kang LR, Zeitlin PL. Induction of HSP70 promotes deltaF508 CFTR trafficking. Am J Physiol 2001; 281: L58–68
Egan ME, Glockner-Pagel J, Ambrose CA, et al. Calciumpump inhibitors induce functional surface expression of DF508-CFTR protein in cystic fibrosis epithelial cells. Nat Med 2002; 8: 485–92
Egan ME, Pearson M, Weiner SA, et al. Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science 2004; 304: 600–2
Norez C, Antigny F, Becq F, et al. Maintaining low Ca2+ level in the endoplasmic reticulum restores abnormal endogenous F508del-CFTR trafficking in airway epithelial cells. Traffic 2006; 7: 562–73
Dormer RL, Harris CM, Clark Z, et al. Sildenafil (Viagra) corrects DeltaF508-CFTR location in nasal epithelial cells from patients with cystic fibrosis. Thorax 2005; 60: 55–9
Lubamba B, Lecourt H, Lebacq J, et al. Preclinical evidence that sildenafil and vardenafil activate chloride transport in cystic fibrosis. Am J Respir Crit Care Med 2008; 177: 506–15
Poschet JF, Timmins GS, Taylor-Cousar JL, et al. Pharmacological modulation of cGMP levels by phosphodiesterase 5 inhibitors as a therapeutic strategy for treatment of respiratory pathology in cystic fibrosis. Am J Physiol Lung Cell Mol Physiol 2007; 293: 712–9
Robert R, Carlile GW, Pavel C, et al. Structural analogue of sildenafil identified as a novel corrector of the F508del-CFTR trafficking defect. Mol Pharm 2008; 73: 478–89
Becq F, Zegarra-Moran O. Specific pharmacological therapy in cystic fibrosis. Workpackage 5: novel therapies [online]. Available from URL: http://www.eurocarecf.eu/ [Accessed 2009 May 25]
Van Goor F, Straley KS, Cao D, et al. Rescue of DF508 CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol 2006; 290: 1117–30
Vertex Pharmaceuticals [online]. Available from URL: http://www.vpharm.com [Accessed 2009 May 25]
Pedemonte N, Lukacs GL, Du K, et al. Small-molecule correctors of defective DF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest 2005; 115: 2564–71
Actelion Pharmaceuticals [online]. Available from URL: http://www.actelion.com [Accessed 2009 May 25]
McCormack PL, Goa KL. Miglustat. Drugs 2003; 63: 2427–34
Dwek RA, Butters TD, Platt FM, et al. Targeting glycosylation as a therapeutic approach. Nat Rev Drug Discov 2002; 1: 65–75
Ficicioglu C. Review of miglustat for clinical management in Gaucher disease type I. Ther Clin Risk Manag 2008; 4: 425–31
Norez C, Noel S, Wilke M, et al. Rescue of functional delF508-CFTR channels in cystic fibrosis epithelial cells by the α-glucosidase inhibitor miglustat. FEBS Lett 2006; 580: 2081–6
Noel S, Wilke M, Bot A, et al. Parallel improvement of sodium and chloride transport defects by miglustat in cystic fibrosis epithelial cells. J Pharmacol Exp Ther 2008; 325: 1016–23
Norez C, Antigny F, Noel S, et al. A cystic fibrosis respiratory epithelial cell chronically treated by miglustat acquires a non-cystic fibrosis-like phenotype. Am J Respir Cell Mol Biol 2009; 41: 217–25
Lubamba B, Lebacq J, Lebecque P, et al. Airway delivery of low-dose miglustat normalizes nasal potential difference in F508del cystic fibrosis mice. Am J Respir Crit Care Med 2009; 179(11): 1022–8
Antigny F, Norez C, Becq F, et al. Calcium homeostasis is abnormal in cystic fibrosis airway epithelial cells but is normalized after rescue of F508del-CFTR. Cell Calcium 2008; 43: 175–83
Dechecchi MC, Nicolis E, Norez C, et al. Anti-inflammatory effect of miglustat in bronchial epithelial cells. J Cyst Fibros 2008; 7: 555–65
Pedemonte N, Sondo E, Caputo A, et al. Dual activity of aminoarylthiazoles on trafficking and gating defects caused by CF mutations [abstract]. Pediatr Pulmonol 2008; Suppl. 31: 311
Norez C, Bilan F, Kitzis A, et al. Proteasome-dependent pharmacological rescue of cystic fibrosis transmembrane conductance regulator revealed by mutation of glycine 622. J Pharmacol Exp Ther 2008; 325: 89–99
Becq F, Mettey Y, Gray MA, et al. Development of substituted benzo[c]quinolizinium compounds as novel activators of the cystic fibrosis chloride channel. J Biol Chem 1999; 274: 27415–25
Marivingt-Mounir C, Norez C, Derand R, et al. Synthesis, SAR, crystal structure, and biological evaluation of benzoquinoliziniums as activators of wild-type and mutant cystic fibrosis transmembrane conductance regulator channels. J Med Chem 2004; 47: 962–72
Dormer RL, Dérand R, McNeilly C, et al. Correction of delF508-CFTR activity with benzoquinolizinium compounds through facilitation of its processing in cystic fibrosis airway cells. J Cell Sci 2001; 114: 4073–81
Dérand R, Bulteau-Pignoux L, Mettey Y, et al. Activation of G551D CFTR channel with MPB-91: regulation by ATPase activity and phosphorylation. Am J Physiol Cell Physiol 2001; 281: 1657–66
Stratford FLL, Pereira MMC, Becq F, et al. Benzo(c)quinolizinium drugs inhibit degradation of DF508-CFTR cytoplasmic domain. Biochem Bioph Res Com 2003; 300: 524–30
Young A, Gentzsch M, Abban CY, et al. Dynasore inhibits removal of wild-type and DeltaF508 CFTR from the plasma membrane. Biochem J 2009 Jul 15; 421(3): 377–85
Loo MA, Jensen TJ, Cui L, et al. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J 1998; 17: 6879–87
Vij N, Fang S, Zeitlin PL. Selective inhibition of endoplasmic reticulum-associated degradation rescues DeltaF508-cystic fibrosis transmembrane regulator and suppresses interleukin-8 levels: therapeutic implications. J Biol Chem 2006; 281: 17369–78
Bortezomib information [online]. Available from URL: http://www.bortezomib.org [Accessed 2009 May 25]
Jiang C, Fang SL, Xiao YF, et al. Partial restoration of cAMP-stimulated CFTR chloride channel activity in DeltaF508 cells by deoxyspergualin. Am J Physiol 1998; 275: C171–8
Norez C, Pasetto M, Dechecchi MC, et al. Chemical conjugation of DeltaF508-CFTR corrector deoxyspergualin to transporter human serum albumin enhances its ability to rescue Cl− channel functions. Am J Physiol Lung Cell Mol Physiol 2008; 295: L336–47
Pedemonte N, Sonawane ND, Taddei A, et al. Phenylglycine and sulfonamide correctors of defective DF508 and G551D cystic fibrosis transmembrane conductance regulator chloride-channel gating. Mol Pharmacol 2005; 67: 1797–807
Sammelson RE, Ma T, Galietta LJV, et al. 3-(2-benzyphenyl) isoxazoles and isoxazolines: synthesis and evaluation as CFTR activators. Bio Med Chem Lett 2003; 13: 2509–12
Ma T, Vetrivel L, Yang H, et al. High-affinity activators of cystic fibrosis transmembrane conductance regulator (CFTR) chloride conductance identified by high-throughput screening. J Biol Chem 2002; 277: 37235–41
Accurso FJ, Rowe SM, Durie PR, et al. Interim results of Phase 2A study of VX770 to evaluate safety, pharmacokinetics, and biomarkers of CFTR activity in cystic fibrosis subjects with G551D [abstract]. Pediatr Pulmonol 2008; Suppl. 31: 295
Noël S, Faveau C, Norez C, et al. Discovery of pyrrolo[2,3-b]pyrazines derivatives as submicromolar affinity activators of wild-type, G551D and F508del CFTR chloride channels. J Pharmacol Exp Ther 2006; 319: 349–59
Hofmann T, Senier I, Bittner P, et al. Aerosolized amiloride: dose effect on nasal bioelectric properties, pharmacokinetics, and effect on sputum expectoration in patients with cystic fibrosis. J Aerosol Med 1997; 10: 147–58
Hofmann T, Stutts MJ, Ziersch A, et al. Effects of topically delivered benzamil and amiloride on nasal potential difference in cystic fibrosis. Am J Respir Crit Care Med 1998; 157: 1844–9
Graham A, Hasani A, Alton EW, et al. No added benefit from nebulized amiloride in patients with cystic fibrosis. Eur Respir J 1993; 6: 1243–8
Pons G, Marchand MC, D’Athis P, et al. French multicenter randomised double-blind placebo-controlled trial on nebulized amiloride in cystic fibrosis patients. Pediatr Pulmonol 2000; 30: 25–31
Rodgers HC, Knox AJ. The effect of topical benzamil and amiloride on nasal potential difference in cystic fibrosis. Eur J Respir 1999; 14: 693–6
Parion Sciences [online]. Available from URL: http://www.parion.com [Accessed 2009 May 25]
Hirsh AJ, Zhang J, Zamurs A, et al. Pharmacological properties of N-(3,5-diamino-6-chloropyrazine-2-carbonyl)-N′-4-[4-(2,3-dihydroxypropoxy) phenyl]butyl-guanidine methanesulfonate (552-02), a novel epithelial sodium channel blocker with potential clinical efficacy for cystic fibrosis lung disease. J Pharmacol Exp Ther 2008; 325: 77–88
Planès C, Caughey GH. Regulation of the epithelial Na+ channel by peptidases. Curr Top Dev Biol 2007; 78: 23–46
Bridges RJ, Newton BB, Pilewski JM, et al. Na+ transport in normal and CF human bronchial epithelial cells is inhibited by BAY 39–9437. Am J Physiol 2001; 281: 16–23
Clinicaltrials.gov [online]. Available from URL: http://www.clinicaltrials.gov [Accessed 2009 May 25]
Rowe SM, Reeves G, Young H, et al. Correction of sodium transport with nasal administration of the prostasin inhibitor QUA145 in CF subjects [abstract]. Pediatr Pulmonol 2008; Suppl. 31: 295
Rubenstein RC, Zeitlin PL. A pilot clinical trial of oral sodium 4-phenylbutyrate (biphenyl) in DF508-homozygous cystic fibrosis patients. Am J Respir Crit Care Med 1998; 157: 484–90
Hwang TH, Schwiebert EM, Guggino WB. Apical and basolateral ATP stimulates tracheal epithelial chloride secretion via multiple purinergic receptors. Am J Physiol 1996; 270: C1611–23
Kellerman D, Mospan AR, Engens J, et al. Denofosol: a review of studies with inhaled P2Y2 agonists that led to phase 3. Pulm Pharm Ther 2008; 21: 600–7
Inspire Pharmaceuticals [online]. Available from URL: http://www.inspirepharm.com [Accessed 2009 May 25]
ISM Therapeutics [online]. Available from URL: http://www.ismtherapeutics.com [Accessed 2009 May 25]
Rudolf MT, Dinkel C, Traynor-Kaplan AE, et al. Antagonists of myo-Inositol 3,4,5,6-tetrakisphophate allow repeated epithelial chloride secretion. Bioorg Med Chem 2003; 11: 3315–29
Moody M, Pennington C, Schultz C, et al. Inositol polyphosphate derivatives inhibits Na+ transport and improves fluid dynamics in cystic fibrosis airway epithelia. Am J Physiol 2005; 289: C512–20
Traynor-Kaplan AE, Moody M, Nur M, et al. INO-4995 therapeutic efficacy is enhanced with repeat dosing in cystic fibrosis knockout mice and human epithelia. Am J Respir Cell Mol Biol 2010; 42(1): 105–12
Grasemann H, Stehling F, Brunar H, et al. Inhalation of Moli1901 in patients with cystic fibrosis. Chest 2007; 131: 1461–6
Cloutier MM, Guernsey L, Sha’afi RI. Duramycin increases intracellular calcium in airway epithelium. Membr Biochem 1993; 10: 107–18
Zeitlin PL, Boyle MP, Guggino WB, et al. A phase I trial of intranasal Moli1901 for cystic fibrosis. Chest 2004; 125: 143–9
AOP Orphan Pharmaceuticals AG [online]. Available from URL: http://www.aoporphan.com/ [Accessed 2009 May 25]
Norez C, Vandebrouck C, Antigny F, et al. Guanabenz, an alpha2-selective adrenergic agonist, activates Ca2+-dependent chloride currents in cystic fibrosis human airway epithelial cells. Eur J Pharmacol 2008; 592: 33–40
Holmes B, Brogden RN, Hell RC, et al. Guanabenz: a review of its pharmacodynamic properties and therapeutic efficacy in hypertension. Drugs 1983; 26: 212–29
Perez A, Issler AC, Cotton CU, et al. CFTR inhibition mimics the cystic fibrosis inflammatory profile. Am J Physiol Lung Cell Mol Physiol 2007; 292: 383–95
Dechecchi MC, Nicolis E, Bezzerri V, et al. MPB-07 reduces the inflammatory response to Pseudomonas aeruginosa in cystic fibrosis bronchial cells. Am J Respir Cell Mol Biol 2007; 36: 615–24
Talebian L, Coutermarsh B, Channon JY, et al. Corr4a and VRT325 do not reduce the inflammatory response to P. Aeruginosa in human cystic fibrosis airway epithelial cells. Cell Physiol Biochem 2009; 23: 199–204
Eidelman O, Guay-Broder C, Van Galen PJM, et al. A1 adenosine-receptor antagonists activate chloride efflux from cystic fibrosis cells. Proc Natl Acad Sci U S A 1992; 89: 5562–6
Srivastava M, Eidelman O, Zhang J, et al. Digitoxin mimics gene therapy with CFTR and suppresses hypersecretion of IL-8 from cystic fibrosis lung epithelial cells. Proc Natl Acad Sci U S A 2004; 101: 7693–8
Bernard C. Introduction à l’étude de la médecine exprimentale. Paris: JB Baillière et Fils, 1865
Schultz SG. A century of (epithelial) transport physiology: from vitalism to molecular cloning. Am J Physiol 1998; 274: C12–23
Acknowledgements
The author wishes to thank past and present students and researchers of his laboratory who have contributed to the ongoing drug discovery project and findings. The author’s laboratory work is currently supported by Vaincre La Mucoviscidose, Mucovie66, ABCF2 and Région Poitou-Charentes. The author is a member of EuroCareCF and a workpackage 5 co-leader. CNRS and the University of Poitiers own a patent on the use of miglustat in cystic fibrosis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Becq, F. Cystic Fibrosis Transmembrane Conductance Regulator Modulators for Personalized Drug Treatment of Cystic Fibrosis. Drugs 70, 241–259 (2010). https://doi.org/10.2165/11316160-000000000-00000
Published:
Issue Date:
DOI: https://doi.org/10.2165/11316160-000000000-00000