
SPON1 Is Associated with Amyloid-β and APOE ε4-Related Cognitive Decline in Cognitively Normal Adults
Abstract
Abstract.
Background:
Genetic variation in Spondin-1, specifically rs11023139, has been associated with reduced rates of cognitive decline in individuals with Alzheimer’s disease.
Objective:
The aim of this study was to assess whether the association was present in cognitively normal older adults.
Methods:
Longitudinal cognitive decline was investigated using linear mixed modelling in a cohort of 590 cognitively normal older adults enrolled in the Australian Imaging, Biomarkers and Lifestyle Study.
Results:
No independent effect of Spondin-1 rs11023139 on cognitive decline was observed. However, significant associations were observed for the interaction between Apolipoprotein E (APOE) ɛ4 and rs11023139 in individuals with high amyloid-β burden. APOE ɛ4/rs11023139-A carriers declined significantly faster than APOE ɛ4/rs11023139-G_G carriers in measures of global cognition (p = 0.011) and verbal episodic memory (p = 0.020).
Conclusion:
These results suggest that carriage of the Spondin-1 rs11023139-A allele significantly contributes to a worsening of cognitive performance in APOE ɛ4 cognitively normal older adults with a high neocortical amyloid-β burden.
INTRODUCTION
Alzheimer’s disease (AD) is characterized by significant impairment in memory and cognition, which occurs gradually as the disease progresses [1]. Deterioration in cognition can also be observed in cognitively normal (CN) older adults, particularly those with evidence of pathological changes characteristic of AD, such as an elevated neocortical amyloid-β (Aβ) burden [2, 3], a feature included in the National Institute on Aging and Alzheimer’s Association (NIA/AA) recommended criteria for preclinical AD [4]. Although the presence of abnormally high levels of Aβ and tau, which define the preclinical AD stage, are associated with subtle decline in cognition, there remains substantial between person variability in the rate of this decline, suggesting other biological factors may act to influence this. Understanding these rates of change in cognition in preclinical AD and how they are influenced will not only assist with the development of disease intervention strategies but will also significantly contribute to furthering our understanding of the genesis of the disease itself.
In preclinical AD, rates of cognitive change have previously been shown to be influenced by genetic factors. Particularly, allelic variations within the genes encoding apolipoprotein E (ApoE; APOE) [5–7], brain derived neurotropic factor (BDNF) [8, 9], and Kidney and Brian expressed protein (KIBRA) [10] have all been associated with differences in cog-nitive performance over time at this stage of the disease. Cognitive domains known to deteriorate early in the disease process, i.e., episodic memory, have been observed to be influenced significantly by genetic variation.
In a genome-wide association study (GWAS) for the Alzheimer’s Disease Assessment Scale–cognitive subscale (ADAS-Cog) performance in the Alzhei-mer’s Disease Neuroimaging Initiative (ADNI), an association of rs11023139, within the Spondin-1 (F-Spondin; SPON1) gene located on chromosome 11 (11p15.5), in those clinically diagnosed with AD (though in the absence of reported Aβ status) was identified [11]. The study found associations of the minor allele (A) with reduced rates of decline on the ADAS-Cog and on subsequent analysis with reduced rates of decline on the Rey Auditory Verbal Learning Test (RAVLT) and Mini-Mental State Examination (MMSE) [11]. Spondin-1 is an extracellular matrix protein involved in the growth of axons during embryonic development [12, 13], differentiation of neural cells [14], and the promotion of axonal regeneration post-injury [15]. Overexpression of Spondin-1 in the brains of wild type and AD-transgenic mice has been associated with improvements in spatial learning and memory, and the reduction of Aβ plaque load [16]. Spondin-1 has also been implicated in pathways involved in AD pathology. Specifically, it was identified in a screen of proteins interacting with amyloid-β protein precursor (AβPP) in vivo [17]. In particular, Spondin-1 has been shown to bind to the extracellular domain of AβPP inhibiting its cleavage by beta-secretase [18].
As the previous study linking SPON1 rs11023139 with cognitive performance was undertaken at the advanced clinical stages of AD [11], the aim of this current study was to determine whether this association with cognitive decline was also present in the preclinical stages of AD. In this context the relationship between SPON1 rs11023139 and preclinical Aβ-related cognitive decline can be assessed in the absence of several potential confounds, including the influence of: consequent disease processes (e.g., inflammation, vascular deterioration), substantial generalized diminished intellectual ability that occurs in dementia that may reduce the reliability of cognitive assessment, or potential floor effects of cognitive assessments that may reduce sensitivity to any acceleration of disease related change in cognition. The hypothesis of the study was that SPON1 rs11023139 would exert a modifying effect on Aβ and APOE ɛ4 mediated cognitive decline. To address these research questions longitudinal cognitive data, collected as part of the Australian Imaging, Biomarkers and Lifestyle Study of Aging (AIBL) was utilized. This study focused on cognitively normal older adults with known neocortical Aβ burden.
MATERIALS AND METHODS
Participants
Participants for the study described here included 590 CN older adults recruited as part of the larger AIBL Study, for which information regarding enrolment, neuropsychological assessment and diagnostic criteria has been previously published [19]. Follow-up assessment occurred every 18-months, with 7.5 years of longitudinal data being utilized in this study. All assessments (neuropsychological, imaging, and laboratory) for each time-point were acquired within 3-months of each other. Ethics approval had been granted for the AIBL Study by all member institutions; Austin Health, St Vincent’s Health, Hollywood Private Hospital, and Edith Cowan University, and informed written consent was provided by all participants.
Cognitive measures
All 18-monthly AIBL study follow-ups involve neuropsychological, clinical and intelligence testing as previously described [19]. The AIBL neuropsychological test battery consists of the following; MMSE, Clock Drawing Test, California Verbal Learning Test-Second edition (CVLT-II), Logical Memory I and II (LMI; LMII; Story A only), D-KEFS verbal fluency, the 30-item version of the Boston Naming Test (BNT), Wechsler Test of Adult Reading (WTAR) for premorbid IQ, Digit Span and Digit Symbol-Coding subtests of the Wechsler Adult Intelligence Scale-Third edition (WAIS-III), the Stroop task (Victoria version), and the Rey Complex Figure Test (RCFT). In addition, data from the Clinical Dementia Rating (CDR), Geriatric Depression Scale (GDS), and WTAR-estimated premorbid IQ (WAIS-III Full Scale Intelligence Quotient (FSIQ)) were collected. Previously calculated cognitive composite scores, aiming to measure preclinical cognitive performance, were utilized in the current study [20, 21]. Burnham et al. developed a statistically derived composite measures of global cognition (CDRSB, MMSE, LMII, CVLTFP, and Clock) and verbal episodic memory (CDR sum of boxes (CDRSB), LMII, CVLT false positives (CVLTFP) and long delay free recall (CVLTLDFR)), both of which were corrected for age, gender, years of education, premorbid IQ and depressive symptoms [20]. Donohue et al. have previously published the Pre-Alzheimer’s cognitive composite (PACC (CVLT-II, LMII, MMSE, WAIS-III subscales)), which acted as an additional measure of global cognition specific to preclinical AD [21].
Amyloid-β imaging
As described previously [22–24], study participants underwent Aβ imaging with positron emission tomography (PET) using 11C-Pittsburgh Compound B (PiB), 18F-florbetapir or 18F-flutemetamol radiolabelled tracers. Generation of PET standardized uptake value (SUV) ratio (SUVR) data was through the use of CapAIBL, a web based freely availably MR-less methodology [25]. SUVs were summed and normalized to areas specific to the radiolabelled tracer used. That is, cerebellar cortex SUV for PiB, whole cerebellum SUV for florbetapir or pons SUV for flutemetamol, thus generating the target-region to reference-region SUVRs. The current study utilized a binary low (Aβlow) or high (Aβhigh) Aβ burden classification. This classification was determined by a participant’s SUVR being higher than the tracer-specific threshold at any time point for which they were imaged. The tracer-specific SUVR thresholds utilized in this study are; ≥ 1.5, ≥ 1.10 and ≥ 0.62 for PiB, florbetapir and flutemetamol, respectively, as previously described [26].
Genotyping
A 5 ml aliquot of whole blood taken as part of the AIBL protocol [19] and was utilized for DNA extraction and subsequent single nucleotide polymorphism (SNP) genotyping, as previously described [10, 27–29]. QIAamp DNA Blood Maxi Kits (Qiagen, Hilden, Germany) were used for DNA extraction and TaqMan® genotyping assays (Life Technologies, Carlsbad, CA) for APOE (rs7412, assay ID: C___904973_10; rs429358, assay ID: C___3084793_20) and SPON1 (rs11023139, assay ID: C___55174_30) genotyping. TaqMan® genotyping assays were performed using the QuantStudio 12K Flex™ Real-Time-PCR systems (Applied Biosystems, Foster City, CA) and TaqMan® GTXpress™ Master Mix (Life Technologies). Methodologies outlined in the manufacturer’s instructions were followed for kits and assays detailed above.
Data analyses
All analyses detailed in this study use a dicho-tomized APOE ɛ4 carrier status, being defined as the presence (1 or 2 copies; APOE ɛ4 + ve) or absence (0 copies; APOE ɛ4-ve) of the ɛ4 allele. Further, all analyses for SPON1 rs11023139 (henceforth referred to as SPON1) were performed based on the dominant model of the minor allele, A, in line with the findings of the initial GWAS [11]. That is, the absence of A (SPON1A–; G_G) compared with its presence (SPON1A+; A_A, G_A). The following statistical analyses were performed in RStudio (RStudio Team 2015) Version 0.98.1103 for Macintosh [30]. Analysis of baseline demographic data included means/counts and standard deviations/percentages for all variables. To determine whether significant differences between SPON1A– and SPON1A+ were present in demographic variables analysis of variance (ANOVA) and chi-squared tests were used.
To assess the influence of SPON1, Aβ, and APOE ɛ4 on longitudinal cognitive performance, linear mixed-effects (LME) models were performed using the R “nlme” package. Two planned interactions were conducted to investigate independent and interactional effects. In all analyses, cognitive composite scores are included in the models as dependent variables. In analyses assessing AIBL-PACC age is included as a covariate in addition to those specifically mentioned below. Initially, a SPON1×Time interaction was modelled in the complete cognitively normal sample, controlling for APOE ɛ4 and Aβ status. To determine whether there was a modifying effect of SPON1, on Aβ and APOE ɛ4 mediated decline, the sample was first stratified by Aβ status. Following this, an APOE×SPON1×Time interaction was modelled in each group. All analyses were corrected for the False Discovery Rate (FDR) using Q-Value (bootstrap method) [31].
RESULTS
SPON1 rs11023139 and cognition in cognitively normal older adults
Displayed in Table 1 is the demographic information for the cognitively normal sample utilized in the current study stratified by SPON1A carriage. No significant differences were observed on any demographic measures, including ɛ4 carriage (Table 1). The SPON1A+ group had a higher proportion of Aβhigh individuals, though this did not reach statistical significance (52% versus 42%; p = 0.1575).
Table 1
Demographic Information
| Overall n = 590 | SPON1A– n = 531 | SPON1A+ n = 59 | p | ||
| Age [y], mean (SD) | 70.87 (6.45) | 70.75 (6.42) | 71.71 (6.28) | 0.2743 | |
| Female, n (%) | 327 (55.42) | 288 (54.24) | 39 (66.10) | 0.1093 | |
| Years of Education, n (%) | 0–8 | 46 (7.82) | 41 (7.74) | 5 (8.62) | 0.8405 |
| 9–12 | 216 (36.73) | 194 (36.63) | 22 (37.93) | ||
| 13–15 | 124 (21.09) | 110 (20.75) | 14 (24.14) | ||
| 15+ | 202 (34.35) | 185 (34.91) | 17 (29.31) | ||
| Premorbid IQ [FSIQ], mean (SD) | 108.00 (7.24) | 107.84 (7.30) | 108.98 (6.85) | 0.2567 | |
| Depressive Symptoms [GDS], mean (SD) | 1.06 (1.27) | 1.06 (1.29) | 0.98 (1.13) | 0.6781 | |
| APOE ɛ4 carriage, n (%) | 167 (28.31) | 153 (28.81) | 14 (23.73) | 0.5027 | |
| High amyloid-β burden, n (%) | 254 (43.05) | 223 (42.00) | 31 (52.54) | 0.1575 |
Baseline demographic and clinical characteristics of all imaged cognitively normal adults in the AIBL study and based on SPON1 rs11023139 A carriage (SPON1A+; A_A and G_A) and non-carriage (SPON1A–; G_G). p values represent statistical significance when comparing SPON1A– and SPON1A+. SPON1, Spondin-1; APOE, Apolipoprotein E; FSIQ, Wechsler Adult Intelligence Scale 3rd Edition (WAIS-III) Full Scale Intelligence Quotient; GDS, Geriatric Depression Scale.
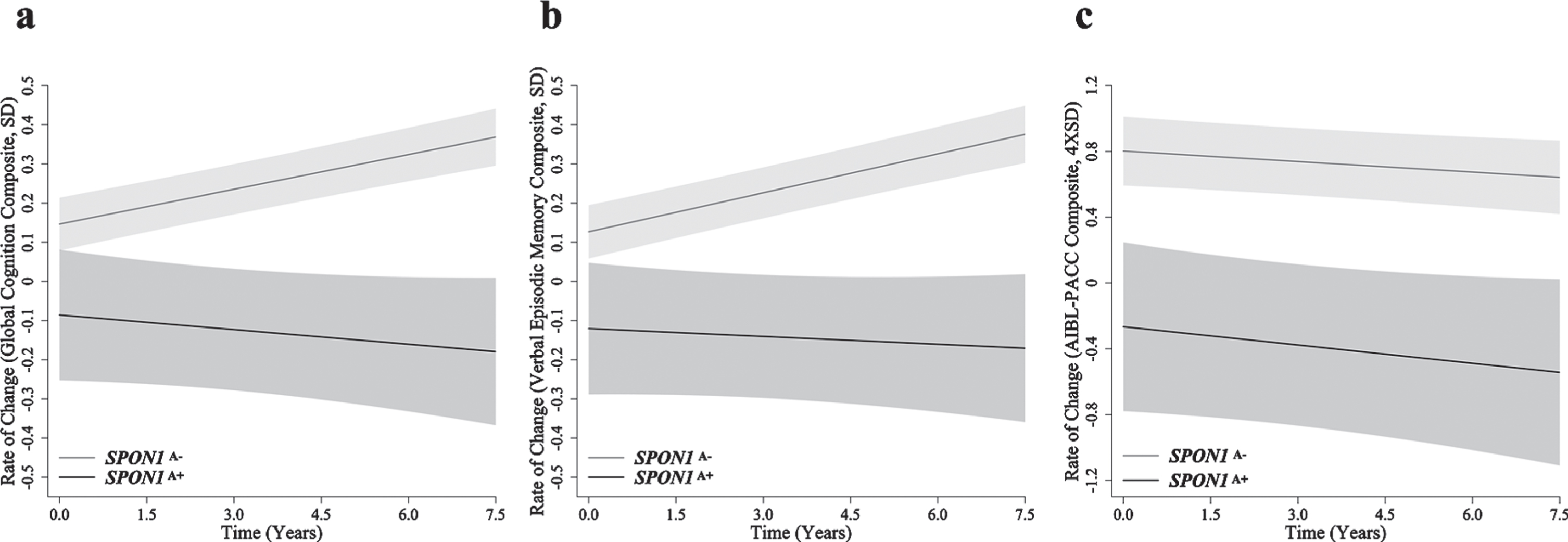
When investigating the independent effect of SPON1, differences in longitudinal cognitive performance between SPON1A+ and SPON1A– were observed, however these differences did not reach statistical significance (Table 2, Fig. 1). In the measure of global cognition (p = 0.075, q = 0.105) and verbal episodic memory (p = 0.084; q = 0.114), decline was observed in the SPON1A+ group, but not the SPON1A– group, this difference trended towards significant (Table 2, Fig. 1). No significant differences are reported in the AIBL-PACC based on carriage of SPON1A (p = 0.797; Table 2, Fig. 1).
Table 2
Slopes for cognitive composites in cognitively normal older adults
| SPON1A– | SPON1A+ | ||
| n = 531 | n = 59 | ||
| β | β | p | |
| Global Cognition | 0.030 | –0.012 | 0.075 |
| Verbal Episodic Memory | 0.033 | –0.007 | 0.084 |
| AIBL-PACC | –0.021 | –0.037 | 0.797 |
Mean slopes for global cognition (SD/year), verbal episodic memory (SD/year) and AIBL-PACC (4×SD/year) in all imaged cognitively normal participants (n = 590), controlling for APOE ɛ4 carrier, amyloid-β status and additionally age in AIBL-PACC. *Represents a nominally statistically significant difference in slope of the SPON1A– (G_G) group when compared to the SPON1A+ (A_A and G_A) group. §q < 0.05 for those reporting nominal significance at p < 0.05. SPON1, Spondin-1; AIBL-PACC, Australian Imaging, Biomarkers and Lifestyle Study Pre-Alzheimer’s Cognitive Composite.
Fig. 1
Rates of change in cognitively normal older adults. Rates of change are presented for (a) global cognition (SD/year), (b) verbal episodic memory (SD/year) and (c) AIBL-PACC (4×SD/year) in cognitively normal older adults (n = 590). SPON1A– (grey) and SPON1A+ (black). Controlling for APOE ɛ4 and amyloid-β status, AIBL-PACC analysis also controlled for age. Error bars represent time dependent standard error, *p < 0.05.
SPON1 rs11023139 and cognition in cognitively normal older adults stratified by Aβ
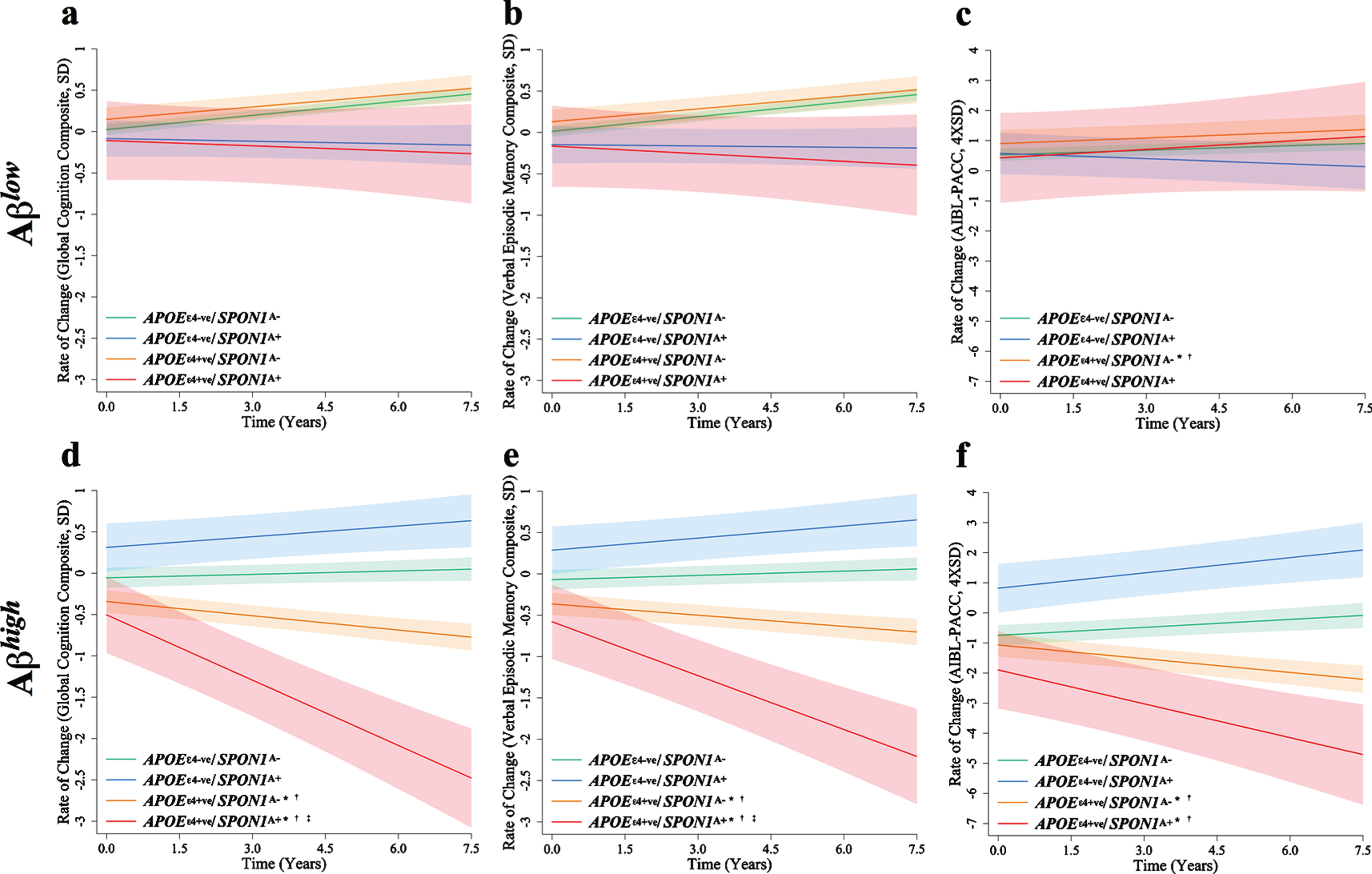
In Aβlow CN older adults, the APOE ɛ4-ve/SPON1A+ group declined significantly faster when compared to APOE ɛ4-ve/SPON1A– in measures of global cognition (p = 0.037, q = 0.062) and verbal episodic memory (p = 0.047, q = 0.071; Table 3, Fig. 2). However, these significances did not survive correction for the false discovery rate. When investigating Aβhigh CN older adults, the APOE ɛ4 + ve/SPON1A+ group showed a significantly greater rate of decline on the global composite (p = 0.0005, q = 0.004; p = 0.0004 q = 0.004), verbal episodic memory (p = 0.001, q = 0.006; p = 0.001, q = 0.005), and the AIBL-PACC (p = 0.030, q = 0.059; p = 0.019 q = 0.048), over seven and a half years relative to the APOE ɛ4-ve/SPON1A– and APOE ɛ4-ve/SPON1A+ groups (Table 3, Fig. 2). Similarly, assessment of the APOE ɛ4 + ve/SPON1A– group showed a significantly greater rate of decline on all composites; global (p = 0.007, q = 0.02; p = 0.021, q = 0.048), verbal episodic memory (p = 015, q = 0.042; p = 0.027, q = 0.056), and AIBL-PACC (p = 0.0008, q = 0.005; p = 0.006, q = 0.022), relative to the APOE ɛ4-ve/SPON1A– and APOE ɛ4-ve/SPON1A+ groups (Table 3, Fig. 2). When compared to the APOE ɛ4 + ve/SPON1A– group, the APOE ɛ4 + ve/SPON1A+ group showed a significantly greater rate of decline on the global composite (p = 0.011, q = 0.033), and verbal episodic memory (p = 0.020, q = 0.048; Table 3, Fig. 2).
Table 3
Slopes for cognitive composites in cognitively normal older adults stratified by amyloid-β, APOE ɛ4 interaction
| APOE ɛ4-ve / | APOE ɛ4-ve / | APOE ɛ4 + ve / | APOE ɛ4 + ve / | |
| SPON1A– | SPON1A+ | SPON1A– | SPON1A+ | |
| β | β | β | β | |
| Aβlow | n = 255 | n = 23 | n = 53 | n = 5 |
| Global Cognition | 0.057 | –0.011* | 0.050 | –0.021 |
| Verbal Episodic Memory | 0.059 | –0.005* | 0.051 | –0.031 |
| AIBL-PACC | 0.051 | –0.059 | 0.062 | 0.095 |
| Aβhigh | n = 123 | n = 22 | n = 100 | n = 9 |
| Global Cognition | 0.014 | 0.043 | –0.058*†§ | –0.263*†‡§ |
| Verbal Episodic Memory | 0.017 | 0.049 | –0.045*†§ | –0.217*†‡§ |
| AIBL-PACC | 0.088 | 0.169 | –0.152*†§ | –0.375*†§ |
Mean slopes for global cognition (SD/year), verbal episodic memory (SD/year) and AIBL-PACC (4× SD/year) in imaged cognitively normal adults with low (n = 336) or high (n = 254) amyloid-β. Controlling for age in AIBL-PACC. *p < 0.05 when comparing to the APOE ɛ4-ve/SPON1A–group, †p < 0.05 when comparing to the APOE ɛ4-ve/SPON1A+group, ‡p < 0.05 when comparing to the APOE ɛ4 + ve/SPON1A–. §q < 0.05 for those reporting nominal significance at p < 0.05. SPON1, Spondin-1; APOE, Apolipoprotein E; Aβ, Amyloid-β; AIBL-PACC, Australian Imaging, Biomarkers and Lifestyle Study Pre-Alzheimer’s Cognitive Composite.
Fig. 2
Rates of change in cognitively normal adults stratified by amyloid-β status, APOE ɛ4 interaction. Rates of change are presented for (a, d) global cognition (SD/year), (b, e) verbal episodic memory (SD/year) and (c, f) AIBL-PACC (4×SD/year) in cognitively normal older adults with low (a, b, c; n = 336) or high (d, e, f; n = 254) amyloid-β. APOE ɛ4-ve/SPON1A– (green), APOE ɛ4-ve/SPON1A+ (blue), APOE ɛ4 + ve/SPON1A– (orange), APOE ɛ4 + ve/SPON1A+ (red). Controlling for age in AIBL-PACC. Error bars represent time dependent standard error. *p < 0.05 when comparing to the APOE ɛ4-ve/SPON1A– carrier group, †p < 0.05 when comparing to the APOE ɛ4-ve/SPON1A+ group, ‡p < 0.05 when comparing to the APOE ɛ4 + ve/SPON1A– carrier.
DISCUSSION
While this study did not observe an independent association between SPON1 and cognitive decline in a preclinical AD sample, as previously reported [11], the hypothesis that SPON1 modifies Aβ and APOE ɛ4 driven cognitive decline in the preclinical stages of AD was supported. However, whilst Sherva and colleagues [11] reported that carriage of the minor allele of rs11023139 (SPON1A+) was associated with slower decline in advanced stages of AD, the opposite was observed, though in preclinical AD, in the current study. Specifically, whilst no significant associations were observed in Aβlow individuals for the assessment of the combinatory effect of Aβ, APOE ɛ4, and SPON1 on cognitive decline, in the Aβhigh CN older adults both APOE ɛ4 + ve groups had significantly increased rates of decline when compared to the APOE ɛ4-ve groups in all measures of cognition. Further, within Aβhigh CN older adults, global cognition and verbal episodic memory declined significantly faster in the APOE ɛ4 + ve/SPON1A+ group when compared to the APOE ɛ4 + ve/SPON1A– group. These results suggest that at the preclinical stages of AD, carriage of SPON1A+ increases rates of decline, in these measures of cognition, beyond what is expected by the carriage of APOE ɛ4 alone.
Several studies suggest that Spondin-1 may play a role in AD pathogenesis. In vivo screening identified Spondin-1 as a protein that interacts with AβPP [17]. It has been reported to interact with AβPP, thereby regulating beta-secretase of AβPP [18]. Hoe et al. published an interaction between Spondin-1 and, AβPP and ApoE receptor 2 [32]. This interaction was reported to result in the increased cleavage of AβPP and ApoE receptor-2, which was speculated by the authors to result in reduced Aβ production [32]. The overexpression of Spondin-1 in mice has been associated with improvements in cognition [16, 33], particularly spatial learning and memory [16]. Spondin-1 overexpression has also been associated with reduction in Aβ plaque deposition in in vivo mouse models [16] and reduced Aβ in in vitro murine neural cell models [33]. However, despite these findings, another study has reported increased levels of Spondin-1 in the cerebrospinal fluid (CSF) of AD patients when compared to healthy controls [34]. The biological effects exhibited by Spondin-1 support the hypothesis that genetic variation may moderate the AD pathogenesis, the impact of genetic variation in SPON1 on gene expression and protein function are largely unknown, though a variant (rs2618516) has been associated with patterns of brain connectivity in young adults [35] and subsequent impact on severity of AD [35] and cognitive performance in APOE ɛ4 carriers [36]. However, this variant is not in complete linkage disequilibrium as has very low correlation (D’ = 0.45, r2 = 0.006) with the variant investigated by Sherva and colleagues [11] and this study. As such, studies aimed at increasing the understanding of the specific functional effects of genetic variants would be an important next step.
There are several factors that may have contributed to the contradictory allelic associations reported in preclinical AD, in the current study, and in more advanced stages, by Sherva and colleagues [11]. The differences in the stages of the disease studied may be one factor contributing to the contrasting associations reported here. Cognitive assessments at the preclinical stages of AD are less likely to be significantly confounded by consequent disease processes and other factors that may reduce sensitivity or reliability of cognitive assessments, such as diminished intellectual ability or floor effects of cognitive assessments that may occur when assessing advanced stages of AD. A further factor may be the inclusion of brain Aβ status in analyses. The current study found associations with cognitive decline in Aβhigh CN older adults who were APOE ɛ4 + ve, whilst Aβ status was not included in the original analyses and therefore the AD diagnosis was not confirmed by imaging findings. Finally, the duration over which cognitive decline was assessed, in addition to the cognitive assessment themselves, were significantly different. The current study used a combination of assessments over a 90-month period, with 18-months between visits, to define composite measures of function compared to the ADAS-Cog over only 24 (AD participants) to 48-months (mildly cognitively impaired participants), with 6 to12-months between visits. These factors, when combined with rs11023139’s minor allele frequency of approximately 5% may explain the disparate associations. Overall, the findings presented in this study provide further evidence to support the role of genetics in contributing to variable rates of cognitive decline seen at the preclinical stages of AD. While the clinical relevance of this SPON1 variant in isolation may be limited due to its minor allele frequency, we have previously reported its predictive role in combination with other genetic variants, particularly when weighted by their impact on cognitive decline [27, 29].
Whilst this study uses a highly characterized longi-tudinal cohort, the following limitations are acknowledged. The AIBL cohort, like ADNI, represent a predominantly Caucasian population, thus the findings from these studies should be replicated in additional ethnically diverse cohorts to gain a broader understanding of the SPON1 rs11023139 associations. Additionally, the voluntary nature of the AIBL Study has resulted in a cohort that is highly educated; therefore, the observations made here may only be observable in similar cohorts. Due to the unavailability of sufficient tau imaging or CSF data for participants in this study, the definition of preclinical AD was based on NIA-AA recommended criteria [4] rather than more recent criteria that includes amyloidosis, tau and neurodegeneration. This study focused particularly on cognitive changes that occur early in the disease process through the use of cognitive composite scores. Different findings may result from the use of different cognitive assessments in the calculation of composite scores or when using single cognitive assessments and when assessing different cognitive domains. Finally, as alluded to earlier, rs11023139 has a minor allele frequency of approximately 0.05, resulting in small sample sizes for the groups, most notably after interactions with Aβ and APOE ɛ4, possibly influencing the results. The results reported here should be replicated in other preclinical AD cohorts with Aβ imaging and longitudinal cognitive data comparative to what is utilized here.
CONCLUSIONS
The results presented in this study are to our knowledge the first to attempt replicate the findings in the ADNI GWAS implicating SPON1A+ with cognitive function. This is the first study to observe an effect of the interaction of Aβ, APOE ɛ4, and SPON1 on cognitive performance in a cohort of CN older adults. Reported here are results implicating SPON1 rs11023139 as factor in preclinical cognitive decline. Investigation into the possible functional mechanisms of SPON1 rs11023139 is required to properly understand its influence over Aβ and APOE ɛ4 mediated cognitive decline. However, this study does provide additional strong evidence for the study of genetic variation with respect to cognitive rates of change at the earliest stages of AD.
CONFLICT OF INTEREST
CLM is an advisor to Prana Biotechnology Ltd and a consultant to Eli Lilly. PM is a full-time employee of Cogstate Ltd. DA has served on scientific advisory boards for Novartis, Eli Lilly, Janssen, and Pfizer Inc. RNM is a consultant to Alzhyme. HRS has received/is receiving remuneration from activities with Pfizer and Takeda pharmaceuticals. CCR has served on scientific advisory boards for Bayer Pharma, Elan Corporation, GE Healthcare and AstraZeneca; has received speaker honoraria from Bayer Pharma and GE Healthcare; and has received research support from Bayer Pharma, GE Healthcare, Piramal Lifesciences and Avid Radiopharmaceuticals. VLV served as a consultant for Bayer Pharma; and received research support from a NEDO grant from Japan. SML serves on the scientific advisory board of Cytox Ltd. All other authors have no conflicts of interest to report.
AVAILABILITY OF DATA AND MATERIALS
All data and samples used in this study were collected as part of the Australian Imaging, Biomarkers and Lifestyle (AIBL) Study of Ageing. AIBL data can be accessed at https://aibl.csiro.au/awd through an Expression of Interest procedure, for more information please see https://aibl.csiro.au/research/support/.
ACKNOWLEDGMENTS
We thank all those who took part as subjects in the study for their commitment and dedication to helping advance research into the early detection and causation of AD. We kindly thank all AIBL Research Group members (http://aibl.csiro.au/about/aibl-research-team/).
Funding for the AIBL study was provided in part by the study partners [Commonwealth Scientific Industrial and research Organization (CSIRO), Edith Cowan University (ECU), Mental Health Research institute (MHRI), National Ageing Research Institute (NARI), Austin Health, CogState Ltd.]. The AIBL study has also received support from the National Health and Medical Research Council (NHMRC) and the Dementia Collaborative Research Centres program (DCRC), as well as funding from the Science and Industry Endowment Fund (SIEF) and the Cooperative Research Centre (CRC) for Mental Health –funded through the CRC Program (Grant ID:20100104), an Australian Government Initiative. This specific study was also supported by NHMRC Project Grant funding (APP1161706) awarded to SML.
REFERENCES
[1] | Forstl H , Kurz A ((1999) ) Clinical features of Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci 249: , 288–290. |
[2] | Lim YY , Maruff P , Pietrzak RH , Ames D , Ellis KA , Harrington K , Lautenschlager NT , Szoeke C , Martins RN , Masters CL , Villemagne VL , Rowe CC , Group AR ((2014) ) Effect of amyloid on memory and non-memory decline from preclinical to clinical Alzheimer’s disease. Brain 137: , 221–231. |
[3] | Lim YY , Laws SM , Villemagne VL , Pietrzak RH , Porter T , Ames D , Fowler C , Rainey-Smith S , Snyder PJ , Martins RN , Salvado O , Bourgeat P , Rowe CC , Masters CL , Maruff P ((2016) ) Aβ-related memory decline in APOE epsilon4 noncarriers: Implications for Alzheimer disease. Neurology 86: , 1635–1642. |
[4] | Sperling RA , Aisen PS , Beckett LA , Bennett DA , Craft S , Fagan AM , Iwatsubo T , Jack CR Jr. , Kaye J , Montine TJ , Park DC , Reiman EM , Rowe CC , Siemers E , Stern Y , Yaffe K , Carrillo MC , Thies B , Morrison-Bogorad M , Wagster MV , Phelps CH ((2011) ) Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 280–292. |
[5] | Lim YY , Ellis KA , Ames D , Darby D , Harrington K , Martins RN , Masters CL , Rowe C , Savage G , Szoeke C , Villemagne VL , Maruff P , AIBL Research Group ((2013) ) Abeta amyloid, cognition, and APOE genotype in healthy older adults. Alzheimers Dement 9: , 538–545. |
[6] | Lim YY , Villemagne VL , Pietrzak RH , Ames D , Ellis KA , Harrington K , Snyder PJ , Martins RN , Masters CL , Rowe CC , Maruff P , Australian Imaging, Biomarkers and Lifestyle (AIBL) Research Group ((2015) ) APOE epsilon4 moderates amyloid-related memory decline in preclinical Alzheimer’s disease. Neurobiol Aging 36: , 1239–1244. |
[7] | Lim YY , Williamson R , Laws SM , Villemagne VL , Bourgeat P , Fowler C , Rainey-Smith S , Salvado O , Martins RN , Rowe CC , Masters CL , Maruff P , AIBL Research Group ((2017) ) Effect of APOE genotype on amyloid deposition, brain volume, and memory in cognitively normal older individuals. J Alzheimers Dis 58: , 1293–1302. |
[8] | Lim YY , Villemagne VL , Laws SM , Ames D , Pietrzak RH , Ellis KA , Harrington KD , Bourgeat P , Salvado O , Darby D , Snyder PJ , Bush AI , Martins RN , Masters CL , Rowe CC , Nathan PJ , Maruff P , Australian Imaging, Biomarkers and Lifestyle (AIBL) Research Group ((2013) ) BDNF Val66Met, Abeta amyloid, and cognitive decline in preclinical Alzheimer’s disease. Neurobiol Aging 34: , 2457–2464. |
[9] | Lim YY , Villemagne VL , Laws SM , Pietrzak RH , Snyder PJ , Ames D , Ellis KA , Harrington K , Rembach A , Martins RN , Rowe CC , Masters CL , Maruff P ((2015) ) APOE and BDNF polymorphisms moderate amyloid beta-related cognitive decline in preclinical Alzheimer’s disease. Mol Psychiatry 20: , 1322–1328. |
[10] | Porter T , Burnham SC , Dore V , Savage G , Bourgeat P , Begemann K , Milicic L , Ames D , Bush AI , Maruff P , Masters CL , Rowe CC , Rainey-Smith S , Martins RN , Groth D , Verdile G , Villemagne VL , Laws SM ((2018) ) KIBRA is associated with accelerated cognitive decline and hippocampal atrophy in APOE epsilon4-positive cognitively normal adults with high Abeta-amyloid burden. Sci Rep 8: , 2034. |
[11] | Sherva R , Tripodis Y , Bennett DA , Chibnik LB , Crane PK , de Jager PL , Farrer LA , Saykin AJ , Shulman JM , Naj A , Green RC , GENAROAD Consortium; Alzheimer’s Disease Neuroimaging Initiative; Alzheimer’s Disease Genetics Consortium ((2014) ) Genome-wide association study of the rate of cognitive decline in Alzheimer’s disease. Alzheimers Dement 10: , 45–52. |
[12] | Klar A , Baldassare M , Jessell TM ((1992) ) F-spondin: A gene expressed at high levels in the floor plate encodes a secreted protein that promotes neural cell adhesion and neurite extension. Cell 69: , 95–110. |
[13] | Feinstein Y , Borrell V , Garcia C , Burstyn-Cohen T , Tzarfaty V , Frumkin A , Nose A , Okamoto H , Higashijima S , Soriano E , Klar A ((1999) ) F-spondin and mindin: Two structurally and functionally related genes expressed in the hippocampus that promote outgrowth of embryonic hippocampal neurons. Development 126: , 3637–3648. |
[14] | Schubert D , Klar A , Park M , Dargusch R , Fischer WH ((2006) ) F-spondin promotes nerve precursor differentiation. J Neurochem 96: , 444–453. |
[15] | Burstyn-Cohen T , Frumkin A , Xu YT , Scherer SS , Klar A ((1998) ) Accumulation of F-spondin in injured peripheral nerve promotes the outgrowth of sensory axons. J Neurosci 18: , 8875–8885. |
[16] | Hafez DM , Huang JY , Richardson JC , Masliah E , Peterson DA , Marr RA ((2012) ) F-spondin gene transfer improves memory performance and reduces amyloid-beta levels in mice. Neuroscience 223: , 465–472. |
[17] | Bai Y , Markham K , Chen F , Weerasekera R , Watts J , Horne P , Wakutani Y , Bagshaw R , Mathews PM , Fraser PE , Westaway D , St George-Hyslop P , Schmitt-Ulms G ((2008) ) The in vivo brain interactome of the amyloid precursor protein. Mol Cell Proteomics 7: , 15–34. |
[18] | Ho A , Sudhof TC ((2004) ) Binding of F-spondin to amyloid-beta precursor protein: A candidate amyloid-beta precursor protein ligand that modulates amyloid-beta precursor protein cleavage. Proc Natl Acad Sci U S A 101: , 2548–2553. |
[19] | Ellis KA , Bush AI , Darby D , De Fazio D , Foster J , Hudson P , Lautenschlager NT , Lenzo N , Martins RN , Maruff P , Masters C , Milner A , Pike K , Rowe C , Savage G , Szoeke C , Taddei K , Villemagne V , Woodward M , Ames D , AIBL Research Group ((2009) ) The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: Methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer’s disease. Int Psychogeriatr 21: , 672–687. |
[20] | Burnham SC , Raghavan N , Wilson W , Baker D , Ropacki MT , Novak G , Ames D , Ellis K , Martins RN , Maruff P , Masters CL , Romano G , Rowe CC , Savage G , Macaulay SL , Narayan VA , Alzheimer’s Disease Neuroimaging Initiative; AIBL Research Group ((2015) ) Novel statistically-derived composite measures for assessing the efficacy of disease-modifying therapies in prodromal Alzheimer’s disease trials: An AIBL study. J Alzheimers Dis 46: , 1079–1089. |
[21] | Donohue MC , Sperling RA , Salmon DP , Rentz DM , Raman R , Thomas RG , Weiner M , Aisen PS , Australian Imaging, Biomarkers, and Lifestyle Flagship Study of Ageing; Alzheimer’s Disease Neuroimaging Initiative; Alzheimer’s Disease Cooperative Study ((2014) ) The preclinical Alzheimer cognitive composite: Measuring amyloid-related decline. JAMA Neurol 71: , 961–970. |
[22] | Rowe CC , Ellis KA , Rimajova M , Bourgeat P , Pike KE , Jones G , Fripp J , Tochon-Danguy H , Morandeau L , O’Keefe G , Price R , Raniga P , Robins P , Acosta O , Lenzo N , Szoeke C , Salvado O , Head R , Martins R , Masters CL , Ames D , Villemagne VL ((2010) ) Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 31: , 1275–1283. |
[23] | Clark CM , Schneider JA , Bedell BJ , Beach TG , Bilker WB , Mintun MA , Pontecorvo MJ , Hefti F , Carpenter AP , Flitter ML , Krautkramer MJ , Kung HF , Coleman RE , Doraiswamy PM , Fleisher AS , Sabbagh MN , Sadowsky CH , Reiman EP , Zehntner SP , Skovronsky DM , AV45-A07 Study Group ((2011) ) Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA 305: , 275–283. |
[24] | Vandenberghe R , Van Laere K , Ivanoiu A , Salmon E , Bastin C , Triau E , Hasselbalch S , Law I , Andersen A , Korner A , Minthon L , Garraux G , Nelissen N , Bormans G , Buckley C , Owenius R , Thurfjell L , Farrar G , Brooks DJ ((2010) ) 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: A phase 2 trial. Ann Neurol 68: , 319–329. |
[25] | Bourgeat P , Villemagne VL , Dore V , Brown B , Macaulay SL , Martins R , Masters CL , Ames D , Ellis K , Rowe CC , Salvado O , Fripp J , AIBL Research Group ((2015) ) Comparison of MR-less PiB SUVR quantification methods. Neurobiol Aging 36: (Suppl 1), S159–166. |
[26] | Rowe CC , Bourgeat P , Ellis KA , Brown B , Lim YY , Mulligan R , Jones G , Maruff P , Woodward M , Price R , Robins P , Tochon-Danguy H , O’Keefe G , Pike KE , Yates P , Szoeke C , Salvado O , Macaulay SL , O’Meara T , Head R , Cobiac L , Savage G , Martins R , Masters CL , Ames D , Villemagne VL ((2013) ) Predicting Alzheimer disease with beta-amyloid imaging: Results from the Australian imaging, biomarkers, and lifestyle study of ageing. Ann Neurol 74: , 905–913. |
[27] | Porter T , Villemagne VL , Savage G , Milicic L , Ying Lim Y , Maruff P , Masters CL , Ames D , Bush AI , Martins RN , Rainey-Smith S , Rowe CC , Taddei K , Groth D , Verdile G , Burnham SC , Laws SM ((2018) ) Cognitive gene risk profile for the prediction of cognitive decline in presymptomatic Alzheimer’s disease. Pers Med Psychiatry 7: , 14–20. |
[28] | Porter T , Burnham SC , Milicic L , Savage G , Maruff P , Lim YY , Li QX , Ames D , Masters CL , Rainey-Smith S , Rowe CC , Salvado O , Groth D , Verdile G , Villemagne VL , Laws SM , AIBL Research Group ((2018) ) Utility of an Alzheimer’s disease risk-weighted polygenic risk score for predicting rates of cognitive decline in preclinical Alzheimer’s disease: A prospective longitudinal study. J Alzheimers Dis 66: , 1193–1211. |
[29] | Porter T , Burnham SC , Savage G , Lim YY , Maruff P , Milicic L , Peretti M , Ames D , Masters CL , Martins RN , Rainey-Smith S , Rowe CC , Salvado O , Taddei K , Groth D , Verdile G , Villemagne VL , Laws SM ((2018) ) A polygenic risk score derived from episodic memory weighted genetic variants is associated with cognitive decline in preclinical Alzheimer’s disease. Front Aging Neurosci 10: , 423. |
[30] | RStudio: Integrated Development for R. RStudio I, Boston, MA. |
[31] | Storey JD ((2002) ) A direct approach to false discovery rates. J R Stat Soc Series B Stat Methodol 64: , 479–498. |
[32] | Hoe HS , Wessner D , Beffert U , Becker AG , Matsuoka Y , Rebeck GW ((2005) ) F-spondin interaction with the apolipoprotein E receptor ApoEr2 affects processing of amyloid precursor protein. Mol Cell Biol 25: , 9259–9268. |
[33] | Park SY , Kang JY , Lee T , Nam D , Jeon CJ , Kim JB ((2020) ) SPON1 can reduce amyloid beta and reverse cognitive impairment and memory dysfunction in Alzheimer’s disease mouse model. Cells 9: , 1275. |
[34] | Zhang J , Goodlett DR , Quinn JF , Peskind E , Kaye JA , Zhou Y , Pan C , Yi E , Eng J , Wang Q , Aebersold RH , Montine TJ ((2005) ) Quantitative proteomics of cerebrospinal fluid from patients with Alzheimer disease.125-133; discussion. J Alzheimers Dis 7: , 173–180. |
[35] | Jahanshad N , Rajagopalan P , Hua X , Hibar DP , Nir TM , Toga AW , Jack CR Jr. , Saykin AJ , Green RC , Weiner MW , Medland SE , Montgomery GW , Hansell NK , McMahon KL , de Zubicaray GI , Martin NG , Wright MJ , Thompson PM , Alzheimer’s Disease Neuroimaging Initiative ((2013) ) Genome-wide scan of healthy human connectome discovers SPON1 gene variant influencing dementia severity. Proc Natl Acad Sci U S A 110: , 4768–4773. |
[36] | Liu Z , Dai X , Tao W , Liu H , Li H , Yang C , Zhang J , Li X , Chen Y , Ma C , Pei J , Mao H , Chen K , Zhang Z ((2018) ) APOE influences working memory in non-demented elderly through an interaction with SPON1 rs2618516. Hum Brain Mapp 39: , 2859–2867. |

