
Metals and Alzheimer’s Disease: How Far Have We Come in the Clinic?
Abstract
It is estimated that by the year 2050 there will be more than 1.5 billion people globally over the age of 65 years. Aging is associated with changes to a number of different cellular processes which are driven by a variety of factors that contribute to the characteristic decline in function that is seen across multiple physiological domains/tissues in the elderly (including the brain). Importantly, aging is also the primary risk factor for the development of neurodegenerative disorders such as Alzheimer’s disease. As such, there is an urgent need to provide a greater understanding of both the pathogenesis and treatment of these devastating neurodegenerative disorders. One of the key cellular processes that becomes dysregulated with age and participates both directly and indirectly in age-related dysfunction, is metal homeostasis and the neurochemistry of metalloproteins, the basic science of which has been extensively reviewed in the past. In this review, we will focus on the human clinical intervention trials that have been conducted over approximately the last four decades that have attempted to establish the efficacy of targeting metal ions in the treatment of AD.
INTRODUCTION
A little over a decade ago we published a review, titled “Metals and Alzheimer’s disease” [1], in which we reviewed the current literature around the mechanisms by which metal ions interacted with key Alzheimer’s disease (AD)-related proteins and pathways to foster an environment that can both precipitate and accelerate disease. This field has advanced over the last decade, providing new insights into the relevance of metals and metalloproteins not only to AD, but to other neurodegenerative diseases and disorders of the central nervous system (CNS). This literature has been reviewed extensively in recent years [2–7], with the majority of work focusing on cell-free, in vitro and in vivo animal model data, and human postmortem analyses. While compelling, ultimately this preclinical testing requires validation of the efficacy/relevance of such approaches to human disease though clinical investigation. In this review, we will summarize key outcomes from investigations into the role of metals in the pathogenesis of AD, focusing on the outcome of human clinical trials that have aimed to test many of the hypotheses that have arisen out of these basic science investigations. In this context, we will appraise that basic question of “how far have we come in the clinic”, and will also develop a perspective on the future of “metals and AD” based around the most recent emerging concepts that are headed to proof-of-principle clinical testing.
METALS: A TARGET IN ALZHEIMER’S DISEASE?
The metallobiology field is a broad one which is punctuated by diseases that have specific abnormalities in metals or metal transport proteins, such as acrodermatitis enteropathica (failure of zinc absorption across the intestine) [8] and Wilson’s disease (mutations in the copper transport gene, ATP7B) [9], through to those that have a failure in key enzymes that critically contain metal species such as occurs in amyotrophic lateral sclerosis (where Cu/Zn superoxide dismutase 1 plays a role) [10]. In the case of AD, there does not appear to be one metal species or metal transport protein that is of singular importance. Historically there have been a variety of different metals that have been proposed to be of relevance to AD [7], with each accompanied by varying amounts of preclinical and/or clinical evidence. This has included metals such as calcium [11], aluminum [12], magnesium [13], and selenium [14, 15]. While some of these data are compelling, the many reviews and original research publications in this area, and importantly the human clinical trials, have largely focused on the potential role of zinc, iron and copper in the pathogenesis of AD. As such, these metals will be the focus for this review.
METALS: TO SUPPLEMENT OR TO CHELATE?
As with any therapeutic approach the targeting of metals requires all the same considerations, such as the specificity of the compound used; the dose, timing, and administration route of the compound (and therefore the compound exposure); potential toxicity and off-target effects of the compound; and many other factors. One critical question to address, however, is what is the desired effect on the metal? Broadly speaking, is the approach designed to supplement metals or to bind/chelate metals.
In the case of supplementation, the implication is that there is a deficit in a key metal which can be rectified via its exogenous administration. The question here, however, becomes what form of metal is used (and indeed, which one), does it reach the area of deficit in the target tissue and will it be of sufficiently high concentration to support a clinical effect (e.g., zinc absorption is significantly impacted by the presence of dietary phytates), will this cause a non-specific and potentially toxic rise in metal levels in tissues that are not deficient, and so on.
The questions are no less complex with chelation, which implies that there is a toxic “pooling” of metals in a given cellular compartment and/or that normal homeostatic processes have become deranged and permit aberrant metal:protein interactions (both of which may also result in a tissue level deficiency in key metals). As metals are critical to so many cellular processes, the requirements for a metal chelator need to be carefully considered. High affinity chelators will effectively remove metals from “biological circulation” but may also strip metals from normal endogenous proteins/pathways. In contrast, moderate affinity chelators may have less of an impact on normal cellular processes, while still targeting the pathological metal pool or metal:protein interactions. In addition, careful design may result in compounds that can effectively re-distribute metals from areas of excess to areas of deficiency, thereby reducing “toxic” events and also ensuring that metal-dependent pathways remain functional to support ongoing neuronal health (a number of these such compounds, which have variously been referred to as “ionophores”, “modulators”, and “chaperones” will be discussed later). In AD, there is currently no definitive metal-targeted pathway (strict chelation, modulation, or supplementation of a specific metal) that has proven “optimal” or which should be pursued to the exclusion of all else. The trials, which will be discussed in the following sections, are summarized in Table 1. Furthermore, the breadth of both “normal” and “pathological” metal:protein and metal:metal interactions are such that altering metal ion homeostasis within the CNS is unlikely to be straightforward, entirely predictable in outcome or linear in its requirement over the course of age or disease, which may go someway to explain a number of the apparent clinical “failures” to date. As such, much of the path forward will rely on empirical evidence and the ongoing development and clinical translation of the most promising preclinical candidates/therapeutic strategies.
Table 1
Completed human clinical trials that target metals, via either supplementation, chelation or
| Compound | Dose | Duration | Cohort* | Outcome | Reference |
| Zinc bis-DL-hydrogenaspartate | 3×50 mg/day (n = 7) or 3×50 mg/day + 30 mg IV every 2 days (n = 3) | Three months to one year | Presenile (n = 5) and senile (n = 5) AD | Improved memory, understanding, communication and social interaction in 8/10 patients | 10 |
| Zinc methionine | 2×15 mg/day | One year | AD (n = 4) | MMSE and ADAS-Cog improved after three months only | 13 |
| reaZin | Daily (equivalent to 150 mg Zn) | Six months | AD (n = 42) | A post-hoc subset analysis in the oldest three quartiles revealed significant improvements in ADAS-Cog and CRD sum of boxes | – |
| Cu-(II)-orotate-dihydrate | Daily (equivalent to 8 mg Cu) | One year | Mild AD (n = 57) | No significant benefit on MMSE or ADAS-Cog | 16 |
| Desferrioxamine mesylate (chelation study focussed on aluminum, with reference to iron) | I.M injection (500 mg) every 12 hours for three days, one day of rest, two days of twice daily injections, a day of rest (for 16 days), then twice daily I.M injections (125 mg) for remaining two years | Two years | AD (n = 48) | Rate of decline, based on in-home measures of daily living activities, in the no-treatment group was double that of the DFO group | 21 |
| D-pencillamine (chelation study targeting copper) | 600 mg/day | 24 weeks | Probable AD (n = 18) | No significant benefit on most tests, including MMSE, verbal fluency, immediate visual memory, Rey’s immediate recall and Rey’s delayed recall. However, “copy drawing with landmark” did not show the same worsening in the treatment group as shown in placebos | |
| Clioquinol (divalent metal “chaperone”) | 125 mg×2 (weeks 0 to 12), 250 mg×2 (weeks 13 to 24) and 375 mg×2 (weeks 25 to 36) | Thirty six weeks | Probable AD (n = 36) | When stratified by the level of impairment at baseline, significant improvements in MMSE | 25 |
| PBT2 (divalent metal “chaperone”) | 50 mg or 250 mg/day | Twelve weeks | Early AD (n = 78) | Significantly improved executive function in high dose group; Likelihood of improvement on the NTB composite, Composite z-score and executive factor z-score significantly greater in the high dose PBT2 group | 26–28 |
| PBT2 (divalent metal “chaperone”) | 250 mg/day | One year | Mild AD (n = 40) | No significant benefit on secondary outcomes of MMSE or NTB | – |
| Combination | Daily (containing 400 mg zinc sulphate and 2 mg Sodium selenite in addition to 6 g primrose oil) | Twenty weeks | AD (n = 15) | Significant improvements in anomalous sentences repetition test, coloured progressive matrices, graded naming test and digit copying test | 17 |
| Zinc oxide + cupric oxide (AREDS study) | 80 mg zinc + 2 mg copper/day | Median of 6.9 years | Older individuals (n = 2166) | No significant benefit on neuropsychiatric battery, which included modified MMSE, animal category, letter fluency, logical memory, Wechsler memory scale and word recall. | 15 |
| Zinc gluconate (Zenith study) | 30 mg/day | Six months | Healthy aged (n = 387) | A significant treatment×time interaction for aspects of CANTAB, although long-term benefit unclear | 14 |
*Cohort sizes include numbers for placebo controls where relevant.
STUDIES WITH METAL SUPPLEMENTATION
Initial studies of zinc supplementation (ZnSO4) in AD patients, which ultimately were not completed due to adverse incidents, were originally undertaken by Constantinidis [16, 17] and also later suggested by Burnet [18], who hypothesized that zinc supplementation could prevent or delay the onset of dementia. In subsequent years a number of human trials were undertaken using zinc as a therapeutic approach to enhance performance across different functional endpoints in a variety of neurodegenerative and other disorders of the CNS [19, 20].
Later, Constantinidis reported on follow-on studies from his original work in 1980, but this time using zinc aspartate (zinc bis-DL-hydrogenaspartate; reviewed in [21]) in a very limited cohort of patients. These studies trialed either oral (3×50 mg pills/day; n = 10 total), or a combination of oral (3×50 mg/day, n = 3 as a subset of the n = 10 just described) and IV (30 mg once every 2 days) administration of zinc from 3 months to one year. This small group of patients had a diverse age range (56–82 years) and clinical diagnoses (five presenting as presenile, and five as senile AD). Assessments were made in the form of neuropsychiatric tests and caregiver interviews, and revealed that two of the patients given only oral dosing showed no improvements, whereas the remaining 8 patients demonstrated improved memory, understanding, communication, and social interaction. The discontinuation of treatment resulted in a decrease and disappearance of any apparent benefit. These studies, and others around the same time [19], led to the hypothesis that zinc supplementation might be beneficial to both the plaque and tangle pathology of AD, which in turn might improve functional outcomes [22, 23].
A later study by Potocnik and colleagues [24] assessed the impact of oral dosing with zinc-methionine (2×15 mg/day) in just four AD patients (63–72 years) over the course of a year. Neuropsychiatric tests [Mini-Mental State Examinatio (MMSE) and Alzheimer’s Disease Assessment Scale-Cog (ADAS-cog)] were performed every quarter, and all patients showed a spike with improved performance on the cognitive tests relative to their baseline after three months of dosing. Patients then followed the expected trajectory of decline in function on these tasks over the remainder of the year, although the decline was less than expected had they not received zinc supplementation (this was based on historical data, no concurrent placebo controls were included in the trial).
These investigator-driven studies no doubt contributed to the development of Adeona Pharmaceuticals’ compound reaZIN (originally coined Zinthionein). This compound was a proprietary formulation containing zinc (150 mg) and cysteine (100 mg) that was designed to be a gastroretentive sustained release tablet that was taken once daily, but which would achieve both high oral zinc bioavailability and superior tolerability (as noted, some of the early zinc supplementation studies were limited by gastrointestinal side effects). There were a total of 42 patients (aged 52 to 86 years) that completed the prospective, randomized, double-blind, placebo-controlled 6 month trial, which included the secondary neuropsychiatric endpoints of ADAS-Cog, MMSE, and Clinical Dementia Rating (CDR) assessments. The outcomes, presented at the 63rd Annual Meeting of the American Academy of Neurology (2011, Honolulu, Hawaii), demonstrated no significant benefit of the treatment on any of the cognitive tests. The study authors, however, reported that there were minor, but not significant, trends to improvement in the ADAS-Cog, the CDR sum of boxes, and the MMSE. None of these differences were close to statistical significance, nor were the differences clinically meaningful. A subsequent subset analysis in the ∼oldest three quartiles of patients (aged >70 years), however, revealed significant effects of treatment in the ADAS-Cog (p = 0.037) and CDR sum of boxes (p = 0.032), with the MMSE not reaching significance (p = 0.067). These analyses prompted the announcement of a subsequent larger clinical trial at the time, although this now appears to have been abandoned.
That zinc might be of benefit to cognition is something that has also been explored in the absence of dementia [25, 26]. The Age-Related Eye Disease Study (AREDS) was a large multicenter randomized trial primarily designed to provide greater understanding around age-related macular degeneration [26]. Part of this study involved an assessment of cognitive function in 2,166 individuals (from 3,640 that were originally enrolled; ages 61 to 87) that had received daily oral dosing of one of several formulations (antioxidant (500 mg vitamin C + 400 IU vitamin E + 15 mg beta carotene); zinc/copper mix (80 mg zinc oxide + 2 mg cupric oxide); antioxidants plus zinc/copper; placebo) for a median period of 6.9 years. The neuropsychiatric battery included the modified MMSE, animal category, letter fluency, logical memory part I and part II, Wechsler memory scale revised, immediate recall and word list mean, Buschke selective reminding test, and digits backwards. There were no significant differences between the four treatment groups across all the cognitive tests assessed. Furthermore, there were 97 individuals that met the criteria for cognitive impairment, and while there was a trend to a slight benefit of zinc it was not significant, and similarly the likelihood of being impaired was not influenced by any of the treatment groups.
In contrast, the ZENITH study was a randomized double-blind placebo-controlled study conducted in 387 healthy older adults (aged 55–87 years) in which an oral dose of 0, 15, or 30 mg/day zinc gluconate was given to individuals for a period of 6 months [25]. Cognitive testing was conducted at baseline, 3 and 6 months, and consisted of the Cambridge Automated Neuropsychological Test Battery (CANTAB) (visual memory was tested by pattern recognition memory; working memory by spatial span and spatial working memory and attention by reaction time and matching to sample visual search). The data analysis revealed that there was an effect in both the treatment groups, with a significant treatment×time interaction for spatial working memory errors (requiring frontal and temporal lobe activation), although it was not clear that either treatment produced any significant prolonged benefit above that seen in the placebo group. There was also an effect in the matching to sample visual search (attention, requiring activation of several brain regions) test, but this was a detrimental effect in the 15 mg/day group, which did not show the decreased latency over the study that was observed in the placebo and the 30 mg/day groups. Thus, there were few significant cognitive benefits observed in this selective study in healthy control subjects, and it was postulated by the authors that a greater existing zinc deficiency may have been required to observe a more profound impact of the supplementation on cognitive performance.
A more recent trial in a disease population examined the effect of copper intake alone on cognition and other parameters in patients with mild AD [27]. This prospective, randomized, double-blind and placebo-controlled trial assessed the impact of once daily dosing with Cu-(II)-orotate-dihydrate (51.62 mg, which equated to 8 mg Cu) for 12 months in a total of 29 patients (average age of 69 years), with a further 28 patients (average age of 69 years) on placebo (cohort sizes reflect the individuals that completed the study). Clinical testing was conducted at baseline and then quarterly thereafter, and consisted of MMSE and ADAS-cog evaluations. Over the course of the trial there was no impact of the elevated copper intake on cognitive endpoints, and there were no significant differences between the two groups when comparing back to baseline scores on the neuropsychiatric scores at any of the quarterly visits. Further studies, examining different doses, duration or combinations (e.g., with zinc) of treatment, might of course yield different results, but in this snapshot study there appeared to be no benefit of copper supplementation. Conversely, attempts have also been made to chelate copper with D-penicillamine [28]. This study, whist demonstrating efficacy in increasing urinary copper excretion and also reducing peroxide levels in those able to tolerate the treatment (n = 34 were initially recruited, but as a result of patient dropout the study had to be stopped before finishing recruitment for ethical reasons. Only nine patients from each group completed the trial), also had no widespread benefit on cognitive outcomes over the 24 week study period (primary outcome measures were assessed using the Mental Deterioration Battery scale, with secondary outcomes measures of MMSE, NeuroPsychiatric Inventory, Geriatric Depression Scale, and the Gottfries Brane Steen scale; the sub-test of “Copy Drawing with Landmark” showed a worsening over time in the placebo group which was not present in the D-penicillamine group).
Other studies utilizing combination therapies that included zinc also fueled speculation that zinc supplementation might be of benefit in AD. A study by Van Rhijn and colleagues [29] reported that individuals (n = 15) taking a combination (12 capsules each) of primrose oil (500 mg), zinc sulphate (400 mg), and sodium selenite (2 mg) for 20 weeks had significant improvements in a number of neuropsychiatric measures (anomalous sentences repetition test, colored progressive matrices, graded naming test and digit copying test), but not other tests (fluid object memory evaluation or Cambridge Cognitive Examination). It should also be noted that the authors observed some benefit in the primrose oil alone control group, but that the majority of benefit was observed in the combination therapy that included zinc (although it also included sodium selenite, which among other effects, has been reported to impact the secretase processing of amyloid-β (Aβ) [30] and to also prevent cognitive decline in a streptozotocin model of memory impairment [31]). Thus, while intriguing, the trial did not specifically address the impact of individual metals alone and so it is difficult to draw firm conclusions from this work.
All the aforementioned studies utilized metal or formulations that included metal to assess the impact on cognitive outcomes across both normal and pathological aging. There have been further related studies conducted which will not be discussed here, in part because of the complexity of some of the compounds assessed (e.g., where metals were a minor component of a complex formulation) or where the impact of the metal itself was not an endpoint that was examined. Several of these additional reports, together with other correlational studies have been previously reviewed [32]. Another strategy that has been utilized is the administration of compounds that alter endogenous brain metal levels, or their localization, to correct a disease-related abnormality.
TREATMENTS DESIGNED TO MODULATE ENDOGENOUS METALS
A well-documented early report on the use of a metal modulating compound (as opposed to just supplementation with a metal itself) in AD was conducted by Crapper McLachlan and colleagues [33]. This was a small study consisting of a total of 48 patients at baseline (n = 23, no treatment, 63±6.2 years of age; n = 25, treatment, 63.2±6.4 years). The compound under study in this single-blind trial was desferrioxamine mesylate (DFO), a trivalent metal chelator, that was being used to assess the role of brain aluminum in the progression of AD. A key point here is that while DFO has been reported to bind a variety of metals [34–36], it is currently the preferred treatment for iron overload disorders and has six orders of magnitude greater affinity for iron(III) than aluminum(III). It is likely, then, that any effect seen in this trial was being driven by an impact on iron, which will be discussed later in the review. The treatment arms in this study were either no treatment, oral placebo (500 mg lecithin, twice daily) or DFO. The DFO dosing followed the following schedule for the first 16 days; 500 mg intramuscular injection every 12 hours for three days, one day of rest, two days of twice daily DFO injections, a day of rest. On day 17, the DFO dose was dropped to 125 mg, given twice a day (12 hourly) for the remaining period of the two-year study. There were a number of safety/tolerability criteria in the study, but the clinical outcome measurements included the Wechsler Adult Intelligence Scale-revised, the Wechsler Memory Scale form 1, the Western Aphasia Battery (all completed at baseline, 12 and 24 months) and a recorded behavioral assessment conducted in the patient’s home (completed at baseline, 6, 12, 18, and 24 months). These in-home assessments were designed to assess various activities of daily living, semantic representations of the body, left/right orientation, and the recognition of, and capacity to count, money. Due to insufficient patient numbers, the neuropsychiatric evaluations were only conducted at baseline, and revealed no differences between groups. The behavioral assessments, therefore, formed the primary clinical outcome for this trial. The principal finding from this work was that rate of decline present in the no-treatment group (which comprised both the no-treatment and the lecithin placebo control group, which were found to be no different from one another) was significantly greater (double) compared to that present in the DFO-treated patients, consistent with a slowing of clinical deterioration/increased patient activities of daily living in the treatment group. While the authors acknowledged the potential for iron-mediated mechanisms underlying the apparent clinical benefit, their conclusion was that aluminum chelators should be further developed for AD. While the use of chelation therapy for AD and other neurodegenerative diseases has proven a popular area of research, another approach that relies on metal delivery by “chaperone”-like compounds or “ionophores” has also shown great utility in vitro and in in vivo models and has subsequently been translated into human clinical trials.
Much has been written about the use of compounds that were intended to intervene in abnormal metal-protein interactions in the AD brain, which would then decrease the pathological accumulation of proteins like amyloid and tau and ultimately facilitate the cellular redistribution of metals such that the function of critical metal-dependent signaling pathways was improved/normalized and would effect an improvement in brain health and function [2]. The first well reported clinical trial designed to test this hypothesis was conducted by Prana Biotechnology [37], who assessed the effect of clioquinol in a small cohort of patients that had a diagnosis of probable AD (n = 18 on treatment and n = 18 placebo). This was a double-blind, placebo-controlled, parallel-group randomized study design that lasted for 36 weeks. The daily dose of clioquinol given was escalated over the trial, 125 mg×2 (weeks 0 to 12), 250 mg×2 (weeks 13 to 24) and 375 mg×2 (weeks 25 to 36). The primary cognitive output was ADAS-cog, although MMSE was also captured, measured at baseline, 4, 12, 24, and 36 weeks. While there was no statistically significant effect of clioquinol treatment on cognitive outcomes at any of the timepoints assessed, there were trends to improvement in the clioquinol group at weeks 4 and 24. When the groups were further stratified by their level of impairment at baseline (based on their ADAS-cog score), these results did achieve statistical significance, with the trend to improvement in the clioquinol group continuing to the end of the trial (although not reaching statistical significance at week 36). These data, together with a significant impact of the treatment on plasma Aβ levels (which was confined to the less severely affected patients, as shown by stratification of the data based upon their ADAS-cog score at baseline), prompted further clinical development of follow-on compounds by Prana Biotechnology. This led to a superior compound, PBT2, which has subsequently been tested in two Phase II human clinical trials for AD and also one for Huntington’s disease.
The original PBT2 trial [38–40] was a double-blind, placebo-controlled trial conducted in a cohort of early AD patients (based upon MMSE and ADAS-cog scores; aged ∼72 years) that were randomly assigned to one of three treatment arms (placebo (n = 29), 50 mg PBT2 (n = 20), or 250 mg PBT2 (n = 29)). The treatments were oral, once per day dosing for a total period of 12 weeks, with a follow-up visit on week 14. Consistent with the earlier clioquinol trial, there were significant biomarker readouts, which included reductions in cerebrospinal fluid (CSF) Aβ levels in the high dose PBT2 group. The initial publication [39] reported that there was no significant difference in the Neuropsychiatric Test Battery (NTB) between placebo and PBT2 treatment arms, but that an analysis of the different components of the NTB revealed that category fluency and Trail Making Test Part B, both in the executive function domain of the test, were significantly improved in the high dose PBT2 group compared to placebo at the completion of the trial (it is of note that a subsequent clinical trial using PBT2 in a Huntington’s disease cohort also revealed a significant effect of 250 mg PBT2 on trails B [41]). The effect of PBT2 on the trails B score was also dose-dependent (p < 0.05). An erratum published the following year [40] also demonstrated that there was a significant benefit of 250 mg PBT2 on the executive factor Z score at the conclusion of the trial, as compared to placebo. Subsequent analyses [38] examined the NTB Composite and Executive Factor z-scores and demonstrated that of those patients showing improvement, a significantly greater proportion of them were in the 250 mg PBT2 treatment group. Similarly, Receiver-operator curve analysis demonstrated that the likelihood of an individual improving (on the Composite z-score and Executive Factor z-score) was significantly greater in the 250 mg PBT2 group (and approached significance for the ADAS-cog—which also showed a trend to improvement in the high dose PBT2 group, as compared to placebo, in the original report). These data helped progress a clinical trial in Huntington’s disease [41] and also a subsequent brain imaging trial in AD.
The most recent clinical trial of PBT2 (the IMAGINE trial) in an AD population was primarily designed to assess the impact of PBT2 on brain amyloid burden (by PiB PET imaging), with secondary outcomes on endpoints that included cognition (NTB) (http://pranabio.com/research-and-development/imagine-trial/#.WXVgc2XnBsY). This was a 12-month, randomized, double-blind, placebo controlled study examining a single dose of PBT2 (250 mg/d orally, n = 25) as compared to placebo (n = 15). In this trial PBT2 did not significantly reduce brain amyloid burden in the prodromal/mild AD patients, although there was a reduction in the overall levels of the PiB PET signal in patients treated with PBT2 which was confounded by an atypical reduction of levels of the PiB PET signal in the placebo group (http://pranabio.com/news/prana-biotechnology-announces-top-line-results-phase-2-imagine-trial-pbt2-alzheimers-disease/#.Wbc44BijUUE). The cognitive assessments similarly did not show any improvement in the PBT2-treated group, although the company did note a trend towards preserving hippocampal brain volume (i.e., less atrophy) in the PBT2 group (2.6% compared to 4.0% in the placebo group) which is potentially relevant for maintaining hippocampal-dependent functions, such as learning and memory. This lack of difference on the cognitive outcomes may have been impacted both by the individual variances present and the small number of patients involved in the study. Furthermore, the baseline characteristics of the patients recruited, based on the PiB SUVR, would suggest that they had a much greater amyloid burden as compared to patients enrolled in previous studies with PBT2 and also other recent AD clinical trials (Master et al., submitted). These data, therefore, are difficult to interpret and appear to have been largely confounded by being underpowered for the analysis of what is, by its very nature, a very heterogeneous disease. At completion, the IMAGINE trial participants were allowed to enroll in an open-label extension trial for an additional 12-month period. There were 33 out of the 40 eligible participants that elected to continue dosing, with 27 patients completing the trial. The compound was well tolerated during the full two years of high dose treatment with PBT2. Similarly, PBT2 has completed four Phase I and four Phase II trials, and each was reviewed by an independent Data Safety Monitoring Board, which did not identify any safety concerns. Despite this, it should be noted that PBT2 is currently on a partial clinical hold by the US FDA (February 2015), which placed a cap on the maximum dose that can be used in humans, due to non-clinical neurotoxicology findings in dog studies. Whether this compound will be tested at higher doses in future trials remains uncertain. Despite these setbacks, the concept of safely targeting brain metal retains its promise, and one being pursued with other compounds/targets.
FUTURE TREATMENTS DESIGNED TO MODULATE ENDOGENOUS METAL
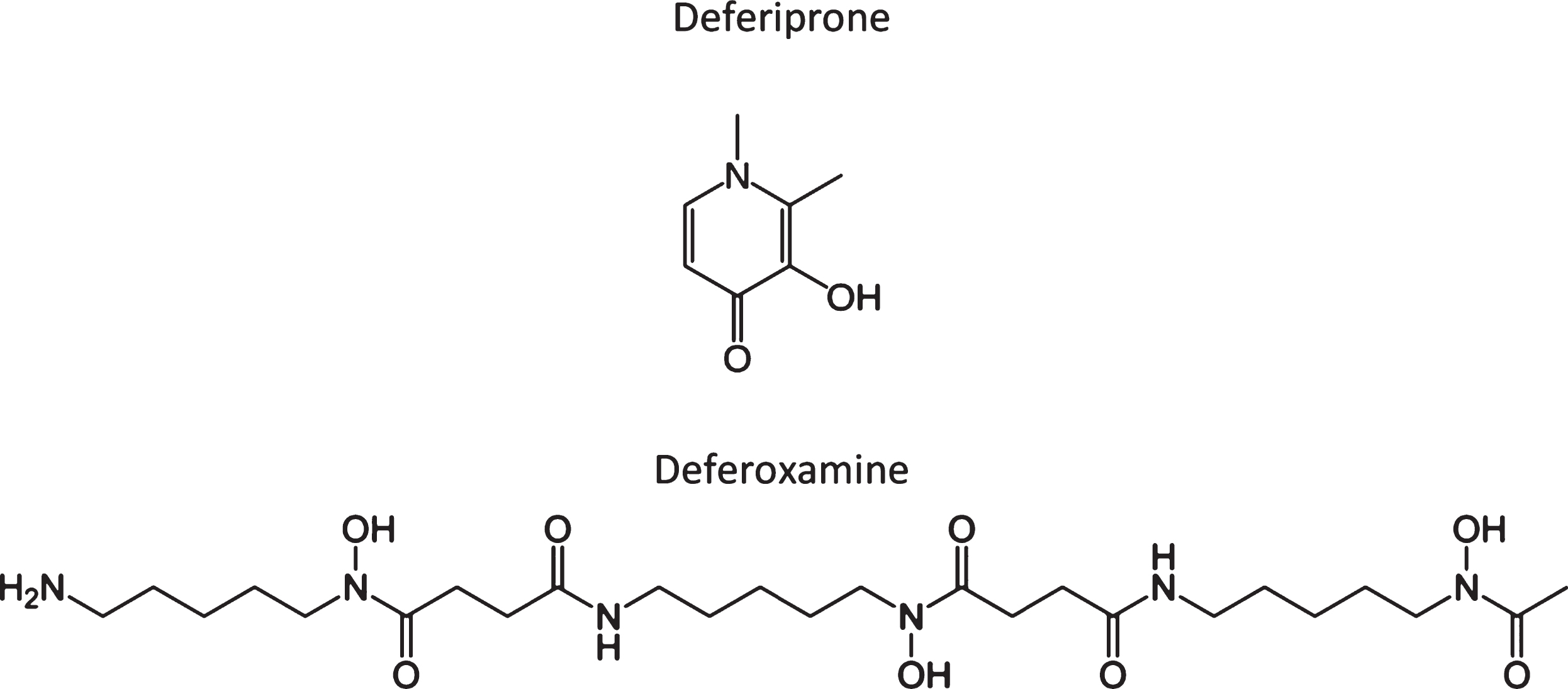
The “3D Study” (Deferiprone Delays Dementia, Ashley Bush and colleagues) is predicated on the hypothesized toxic role of iron in AD, in which iron is shown to accumulate in affected regions of the postmortem brain [42–46], as well as in vivo by MRI [47–51]. Brain iron levels also increase with aging and are associated with cognitive decline prior to disease [52, 53]. In neurodegenerative diseases, this may contribute to the associated proteinopathies (e.g., involving Aβ and tau), but the sub-region specific accumulation of iron is a neurotoxic event [43, 46, 54–56] which does contribute to longitudinal AD outcomes [57]. Furthermore, elevated CSF ferritin, which is a reporter of brain iron [58, 59], independently predicted progression to AD. CSF ferritin was also associated with poorer cognition (e.g., ADAS-Cog, a composite performance score) and brain atrophy across a 6-7-year period in a cohort of patients, including cognitively normal individuals (n = 91) and patients with either mild cognitive impairment (n = 144) or AD (n = 67). These data, together with the early suggestion from McLachlan and colleagues, has helped progress this clinical trial forward. The trial itself is a randomized, double-blind, placebo-controlled, multicenter, Phase II proof-of-concept clinical trial for patients with AD, which will be focused on testing deferiprone (DFP), which is currently in clinical use for treating iron overload in patients with thalassemia. DFP is a moderate iron-binding chelator (unlike desferoxamine; see Fig. 1 for a comparison of the chemical structures) that readily crosses the blod-brain barrier and which can penetrate cellular membranes to form a neutral and lipophilic complex with iron that readily exits cells, and which can redistribute iron to transferrin [44, 45]. There will be 170 males and females with positive amyloid burden (defined by CSF or PET criteria), objective memory impairment indicated by cognitive assessment, and a diagnosis of prodromal AD (mild cognitive impairment) or mild AD (NIAAA criteria) that will be recruited (and which also satisfy other inclusion/exclusion criteria) and then randomly assigned to one of the two treatment arms: DFP [15 mg/kg, twice daily] versus placebo, randomized 2:1 DFP:placebo; and treated for 12 months. The primary readout will be the NTB, which is a composite of six tests assessing episodic memory, executive function and attention, with other assessments (e.g., biomarker studies) also made. This trial is expected to get underway in 2017/2018 and will assess the role of iron in AD, with a perspective on the impact of its chelation on a number of key clinical outcomes. Given our most recent findings [60], in which we demonstrate that brain iron may accelerate cognitive decline via an interaction with Aβ (assessed in 117 patients from the Australian Imaging, Biomarkers and Lifestyle study), together with our earlier work in this space, then iron-targeted therapeutics may become an increased focus in the field of the metallobiology of AD. There are also other clinical trials with metal-targeted therapeutics for neurodegenerative disease (AD, Huntington’s disease, Parkinson’s disease, etc.) that are both in the pipeline and recruiting for clinical trial that will hopefully provide validation for continued research in this area.
Fig.1
Chemical structure of two iron chelators, deferiprone and deferoxamine (also known as desferrioxamine), highlighting the large structural differences in many of these compounds that chelate or modulate metals.
CONCLUSION
There is an abundance of preclinical in vitro and in vivo evidence to support the role of transition metals, such as copper, zinc, and iron, in both the onset and progression of neurodegenerative disorders such as AD. Furthermore, there is strong evidence that these metals are also critically involved in cellular processes that mediate neuronal/brain health, with specific interactions on key pathways responsible for functional outcomes such as learning and memory (in both healthy and pathological states). As such, the natural extension of these preclinical studies has involved translation into human clinical trials. Unfortunately for the field, as the area of “metals and Alzheimer’s disease” remains a niche area of interest from both a wet bench, but also bedside, perspective—then there has continued to be only limited support available to a small number of groups/companies to pursue these concepts. As such, many of the interventional trials that have been conducted to date have been underpowered or otherwise compromised in some way such that we still have yet to see a sufficiently rigorous, in-depth, and controlled study completed that would definitively answer the question of whether targeting metal dyshomeostasis in the AD brain is an efficacious therapeutic approach worth pursuing. Confounding the field of course, is the issue that the clinical trials that have been conducted have taken very different therapeutic approaches, either supplementing, chelating, or modulating a variety of metals (be it copper, zinc, iron, or some other metal or combination thereof). Given the complex interplay between different metals and the multitude of metal:protein interactions that subserve critical cellular pathways, not to mention the heterogeneous nature of AD and the likely non-linear change in metals that will occur across both age and the course of the disease (and which may be different depending on whether the disease is progressing or slowed/halted), then arguably there may be too many variables to ever allow a single “definitive” trial to be conducted.
In an environment of limited funding, largely characterized by small “snapshot” style clinical trials, we can only hope that incremental investigator driven trials can provide sufficiently compelling data to generate excitement in this space that will prompt a significant investment from the biotech sector to facilitate the large scale human trials that are required to properly interrogate the potential therapeutic efficacy of metal targeting compounds in AD.
ACKNOWLEDGMENTS
PAA and AIB are variously supported by funds from the National Health and Medical Research Council of Australia and The Australian Research Council. In addition, the Florey Institute of Neuroscience and Mental Health acknowledge the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0662r1).
REFERENCES
[1] | Adlard PA , Bush AI ((2006) ) Metals and Alzheimer’s disease. J Alzheimers Dis 10: , 145–163. |
[2] | Adlard PA , Bush AI ((2012) ) Metal chaperones: A holistic approach to the treatment of Alzheimer’s disease. Front Psychiatry 3: , 15. |
[3] | Ayton S , Lei P , Bush AI ((2013) ) Metallostasis in Alzheimer’s disease. Free Radic Biol Med 62: , 76–89. |
[4] | Ayton S , Lei P , Bush AI ((2015) ) Biometals and their therapeutic implications in Alzheimer’s disease. Neurotherapeutics 12: , 109–120. |
[5] | Cicero CE , Mostile G , Vasta R , Rapisarda V , Signorelli SS , Ferrante M , Zappia M , Nicoletti A ((2017) ) Metals and neurodegenerative diseases. A systematic review. Environ Res 159: , 82–94. |
[6] | Cristovao JS , Santos R , Gomes CM ((2016) ) Metals and neuronal metal binding proteins implicated in Alzheimer’s disease. Oxid Med Cell Longev 2016: , 9812178. |
[7] | Wang P , Wang ZY ((2017) ) Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res Rev 35: , 265–290. |
[8] | Kasana S , Din J , Maret W ((2015) ) Genetic causes and gene-nutrient interactions in mammalian zinc deficiencies: Acrodermatitis enteropathica and transient neonatal zinc deficiency as examples. J Trace Elem Med Biol 29: , 47–62. |
[9] | Bandmann O , Weiss KH , Kaler SG ((2015) ) Wilson’s disease and other neurological copper disorders. Lancet Neurol 14: , 103–113. |
[10] | Lovejoy DB , Guillemin GJ ((2014) ) The potential for transition metal-mediated neurodegeneration in amyotrophic lateral sclerosis. Front Aging Neurosci 6: , 173. |
[11] | Popugaeva E , Pchitskaya E , Bezprozvanny I ((2017) ) Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease – A therapeutic opportunity? Biochem Biophys Res Commun 483: , 998–1004. |
[12] | Walton JR ((2013) ) Aluminum involvement in the progression of Alzheimer’s disease. J Alzheimers Dis 35: , 7–43. |
[13] | Veronese N , Zurlo A , Solmi M , Luchini C , Trevisan C , Bano G , Manzato E , Sergi G , Rylander R ((2016) ) Magnesium status in Alzheimer’s disease: A systematic review. Am J Alzheimers Dis Other Demen 31: , 208–213. |
[14] | Cardoso BR , Roberts BR , Bush AI , Hare DJ ((2015) ) Selenium, selenoproteins and neurodegenerative diseases. Metallomics 7: , 1213–1228. |
[15] | Du X , Wang C , Liu Q ((2016) ) Potential roles of selenium and selenoproteins in the prevention of Alzheimer’s disease. Curr Top Med Chem 16: , 835–848. |
[16] | Constantinidis J ((1980) ) Neurochimie des demences. Evol Psychiatr (Paris) 45: , 785–803. |
[17] | Constantinidis J ((1980) ) Maladie de Pick, demences a plaques seniles et lesions neurofibrillaires d’Alzheimer, metabolisme du zinc et acides amines neurotransmetteurs. In Etats Deficitaires Cerebraux Lies a l’Age, Tissot R, ed. Georg, Geneve, pp. 201–228. |
[18] | Burnet FM ((1981) ) A possible role of zinc in the pathology of dementia. Lancet 1: , 186–188. |
[19] | Fuenfgeld EW ((1992) ) The trace elemtn zinc enhancing brain metabolism. Neurobiol Aging 13: , 98. |
[20] | Nachdev CK , Bonchev P , Kirov GK , Kissiova K ((1990) ) Zinc deficiency and Alzheimer’s disease: A new approach. In Psychiatry, A world perspective, Stefanis C, ed. Elsevier Science, New York, pp.496–504. |
[21] | Constantinidis J ((1992) ) Treatment of Alzheimer’s disease by zinc compounds. Drug Dev Res 27: , 1–14. |
[22] | Constantinidis J ((1991) ) The hypothesis of zinc deficiency in the pathogenesis of neurofibrillary tangles. Med Hypotheses 35: , 319–323. |
[23] | Constantinidis J ((1991) ) Hypothesis regarding amyloid and zinc in the pathogenesis of Alzheimer disease: Potential for preventive intervention. Alzheimer Dis Assoc Disord 5: , 31–35. |
[24] | Potocnik FC , van Rensburg SJ , Park C , Taljaard JJ , Emsley RA ((1997) ) Zinc and platelet membrane microviscosity in Alzheimer’s disease. The in vivo effect of zinc on platelet membranes and cognition. S Afr Med J 87: , 1116–1119. |
[25] | Maylor EA , Simpson EE , Secker DL , Meunier N , Andriollo-Sanchez M , Polito A , Stewart-Knox B , McConville C , O’Connor JM , Coudray C ((2006) ) Effects of zinc supplementation on cognitive function in healthy middle-aged and older adults: The ZENITH study. Br J Nutr 96: , 752–760. |
[26] | Yaffe K , Clemons TE , McBee WL , Lindblad AS , Age-Related Eye Disease Study Research G ((2004) ) Impact of antioxidants, zinc, and copper on cognition in the elderly: A randomized, controlled trial. Neurology 63: , 1705–1707. |
[27] | Kessler H , Bayer TA , Bach D , Schneider-Axmann T , Supprian T , Herrmann W , Haber M , Multhaup G , Falkai P , Pajonk FG ((2008) ) Intake of copper has no effect on cognition in patients with mild Alzheimer’s disease: A pilot phase 2 clinical trial. J Neural Transm (Vienna) 115: , 1181–1187. |
[28] | Squitti R , Rossini PM , Cassetta E , Moffa F , Pasqualetti P , Cortesi M , Colloca A , Rossi L , Finazzi-Agro A ((2002) ) d-penicillamine reduces serum oxidative stress in Alzheimer’s disease patients. Eur J Clin Invest 32: , 51–59. |
[29] | van Rhijn AG , Prior CA , Corrigan FM ((1990) ) Dietary supplementation with zinc sulphate, sodium selenite and fatty acids in early dementia of Alzheimer’s type. J Nutr Med 1: , 259–266. |
[30] | Tung YT , Hsu WM , Wang BJ , Wu SY , Yen CT , Hu MK , Liao YF ((2008) ) Sodium selenite inhibits gamma-secretase activity through activation of ERK. Neurosci Lett 440: , 38–43. |
[31] | Ishrat T , Parveen K , Khan MM , Khuwaja G , Khan MB , Yousuf S , Ahmad A , Shrivastav P , Islam F ((2009) ) Selenium prevents cognitive decline and oxidative damage in rat model of streptozotocin-induced experimental dementia of Alzheimer’s type. Brain Res 1281: , 117–127. |
[32] | Loef M , von Stillfried N , Walach H ((2012) ) Zinc diet and Alzheimer’s disease: A systematic review. Nutr Neurosci 15: , 2–12. |
[33] | Crapper McLachlan DR , Dalton AJ , Kruck TP , Bell MY , Smith WL , Kalow W , Andrews DF ((1991) ) Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337: , 1304–1308. |
[34] | Hernlem BJ , Vane LM , Sayles GD ((1996) ) Stability constants for complexes of the siderophore desferrioxamine B with selected heavy metal cations. Inorganica Chim Acta 244: , 179–184. |
[35] | Kiss T , Farkas E ((1998) ) Metal-binding ability of desferrioxamine B. J Incl Phenom Mol Recognit Chem 32: , 385–403. |
[36] | Liu ZD , Hider RC ((2002) ) Design of clinically useful iron(III)-selective chelators. Med Res Rev 22: , 26–64. |
[37] | Ritchie CW , Bush AI , Mackinnon A , Macfarlane S , Mastwyk M , MacGregor L , Kiers L , Cherny R , Li QX , Tammer A , Carrington D , Mavros C , Volitakis I , Xilinas M , Ames D , Davis S , Beyreuther K , Tanzi RE , Masters CL ((2003) ) Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: A pilot phase 2 clinical trial. Arch Neurol 60: , 1685–1691. |
[38] | Faux NG , Ritchie CW , Gunn A , Rembach A , Tsatsanis A , Bedo J , Harrison J , Lannfelt L , Blennow K , Zetterberg H , Ingelsson M , Masters CL , Tanzi RE , Cummings JL , Herd CM , Bush AI ((2010) ) PBT2 rapidly improves cognition in Alzheimer’s Disease: Additional phase II analyses. J Alzheimers Dis 20: , 509–516. |
[39] | Lannfelt L , Blennow K , Zetterberg H , Batsman S , Ames D , Harrison J , Masters CL , Targum S , Bush AI , Murdoch R , Wilson J , Ritchie CW , PBT2-201-EURO study group ((2008) ) Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol 7: , 779–786. |
[40] | Lannfelt L , Blennow K , Zetterberg H , Batsman S , Ames D , Harrison J , Masters CL , Targum S , Bush AI , Murdoch R , Wilson J , Ritchie CW , PBT2-201-EURO study group ((2009) ) erratum: Safety, Efficacy, and biomarker findings of PBT2 in targeting Abeta modifying therapy for Alzheimer’s disease: A controlled Phase 2a, double-blind, randomized, placebo-controlled trial. Lancet Neurol 8: , 981. |
[41] | Huntington Study Group Reach2HD Investigators ((2015) ) Safety, tolerability, and efficacy of PBT2 in Huntington’s disease: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol 14: , 39–47. |
[42] | Connor JR , Snyder BS , Beard JL , Fine RE , Mufson EJ ((1992) ) Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer’s disease. J Neurosci Res 31: , 327–335. |
[43] | Duce JA , Tsatsanis A , Cater MA , James SA , Robb E , Wikhe K , Leong SL , Perez K , Johanssen T , Greenough MA , Cho HH , Galatis D , Moir RD , Masters CL , McLean C , Tanzi RE , Cappai R , Barnham KJ , Ciccotosto GD , Rogers JT , Bush AI ((2010) ) Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 142: , 857–867. |
[44] | Good PF , Perl DP , Bierer LM , Schmeidler J ((1992) ) Selective accumulation of aluminum and iron in the neurofibrillary tangles of Alzheimer’s disease: A laser microprobe (LAMMA) study. Ann Neurol 31: , 286–292. |
[45] | Jellinger K , Paulus W , Grundke-Iqbal I , Riederer P , Youdim MB ((1990) ) Brain iron and ferritin in Parkinson’s and Alzheimer’s diseases. J Neural Transm Park Dis Dement Sect 2: , 327–340. |
[46] | Smith MA , Harris PL , Sayre LM , Perry G ((1997) ) Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A 94: , 9866–9868. |
[47] | Bartzokis G , Sultzer D , Cummings J , Holt LE , Hance DB , Henderson VW , Mintz J ((2000) ) In vivo evaluation of brain iron in Alzheimer disease using magnetic resonance imaging. Arch Gen Psychiatry 57: , 47–53. |
[48] | Luo Z , Zhuang X , Kumar D , Wu X , Yue C , Han C , Lv J ((2013) ) The correlation of hippocampal T2-mapping with neuropsychology test in patients with Alzheimer’s disease. PLoS One 8: , e76203. |
[49] | Moon Y , Han SH , Moon WJ ((2016) ) Patterns of brain iron accumulation in vascular dementia and Alzheimer’s dementia using quantitative susceptibility mapping imaging. J Alzheimers Dis 51: , 737–745. |
[50] | Raven EP , Lu PH , Tishler TA , Heydari P , Bartzokis G ((2013) ) Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer’s disease detected in vivo with magnetic resonance imaging. J Alzheimers Dis 37: , 127–136. |
[51] | van Rooden S , Doan NT , Versluis MJ , Goos JD , Webb AG , Oleksik AM , van der Flier WM , Scheltens P , Barkhof F , Weverling-Rynsburger AW , Blauw GJ , Reiber JH , van Buchem MA , Milles J , van der Grond J ((2015) ) 7T T(2)*-weighted magnetic resonance imaging reveals cortical phase differences between early- and late-onset Alzheimer’s disease. Neurobiol Aging 36: , 20–26. |
[52] | Acosta-Cabronero J , Betts MJ , Cardenas-Blanco A , Yang S , Nestor PJ ((2016) ) In vivo MRI mapping of brain iron deposition across the adult lifespan. J Neurosci 36: , 364–374. |
[53] | Ghadery C , Pirpamer L , Hofer E , Langkammer C , Petrovic K , Loitfelder M , Schwingenschuh P , Seiler S , Duering M , Jouvent E , Schmidt H , Fazekas F , Mangin JF , Chabriat H , Dichgans M , Ropele S , Schmidt R ((2015) ) R2* mapping for brain iron: Associations with cognition in normal aging. Neurobiol Aging 36: , 925–932. |
[54] | Ayton S , Lei P , Duce JA , Wong BX , Sedjahtera A , Adlard PA , Bush AI , Finkelstein DI ((2013) ) Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease. Ann Neurol 73: , 554–559. |
[55] | Ayton S , Lei P , Hare DJ , Duce JA , George JL , Adlard PA , McLean C , Rogers JT , Cherny RA , Finkelstein DI , Bush AI ((2015) ) Parkinson’s disease iron deposition caused by nitric oxide-induced loss of beta-amyloid precursor protein. J Neurosci 35: , 3591–3597. |
[56] | Lei P , Ayton S , Finkelstein DI , Spoerri L , Ciccotosto GD , Wright DK , Wong BX , Adlard PA , Cherny RA , Lam LQ , Roberts BR , Volitakis I , Egan GF , McLean CA , Cappai R , Duce JA , Bush AI ((2012) ) Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med 18: , 291–295. |
[57] | Ayton S , Faux NG , Bush AI , Alzheimer’s Disease Neuroimaging I ((2015) ) Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat Commun 6: , 6760. |
[58] | Devos D , Moreau C , Devedjian JC , Kluza J , Petrault M , Laloux C , Jonneaux A , Ryckewaert G , Garcon G , Rouaix N , Duhamel A , Jissendi P , Dujardin K , Auger F , Ravasi L , Hopes L , Grolez G , Firdaus W , Sablonniere B , Strubi-Vuillaume I , Zahr N , Destee A , Corvol JC , Poltl D , Leist M , Rose C , Defebvre L , Marchetti P , Cabantchik ZI , Bordet R ((2014) ) Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid Redox Signal 21: , 195–210. |
[59] | Earley CJ , Connor JR , Beard JL , Malecki EA , Epstein DK , Allen RP ((2000) ) Abnormalities in CSF concentrations of ferritin and transferrin in restless legs syndrome. Neurology 54: , 1698–1700. |
[60] | Ayton S , Fazlollahi A , Bourgeat P , Raniga P , Ng A , Lim Y , Diouf I , Farquharson S , Fripp J , Ames D , Doecke J , Desmond P , Ordidge R , Masters CL , Rowe CC , Maruff P , Villemagne VL , Australian Imaging Biomarkers Lifestyle (AIBL) Research Group, Salvado O , Bush AI ((2017) ) Cerebral quantitative susceptibility mapping predicts amyloid-β-related cognitive decline. Brain 140: , 2112–2119. |

