Dysregulation of Neuronal Calcium Signaling via Store-Operated Channels in Huntington's Disease
 Magdalena Czeredys
Magdalena Czeredys- Laboratory of Neurodegeneration, International Institute of Molecular and Cell Biology in Warsaw, Warsaw, Poland
Huntington's disease (HD) is a progressive neurodegenerative disorder that is characterized by motor, cognitive, and psychiatric problems. It is caused by a polyglutamine expansion in the huntingtin protein that leads to striatal degeneration via the transcriptional dysregulation of several genes, including genes that are involved in the calcium (Ca2+) signalosome. Recent research has shown that one of the major Ca2+ signaling pathways, store-operated Ca2+ entry (SOCE), is significantly elevated in HD. SOCE refers to Ca2+ flow into cells in response to the depletion of endoplasmic reticulum Ca2+ stores. The dysregulation of Ca2+ homeostasis is postulated to be a cause of HD progression because the SOCE pathway is indirectly and abnormally activated by mutant huntingtin (HTT) in γ-aminobutyric acid (GABA)ergic medium spiny neurons (MSNs) from the striatum in HD models before the first symptoms of the disease appear. The present review summarizes recent studies that revealed a relationship between HD pathology and elevations of SOCE in different models of HD, including YAC128 mice (a transgenic model of HD), cellular HD models, and induced pluripotent stem cell (iPSC)-based GABAergic medium spiny neurons (MSNs) that are obtained from adult HD patient fibroblasts. SOCE in MSNs was shown to be mediated by currents through at least two different channel groups, Ca2+ release-activated Ca2+ current (ICRAC) and store-operated Ca2+ current (ISOC), which are composed of stromal interaction molecule (STIM) proteins and Orai or transient receptor potential channel (TRPC) channels. Their role under physiological and pathological conditions in HD are discussed. The role of Huntingtin-associated protein 1 isoform A in elevations of SOCE in HD MSNs and potential compounds that may stabilize elevations of SOCE in HD are also summarized. Evidence is presented that shows that the dysregulation of molecular components of SOCE or pathways upstream of SOCE in HD MSN neurons is a hallmark of HD, and these changes could lead to HD pathology, making them potential therapeutic targets.
Introduction
Calcium (Ca2+) ions are important universal second messengers that regulate numerous cellular processes. Ca2+ concentrations are strictly regulated by multiple Ca2+ channels, pumps, exchangers, and protein buffers. Under resting conditions, intracellular Ca2+ levels oscillate within the range of 100–300 nM. Upon cell activation, an increase in cytosolic Ca2+ concentrations in the range of 50–100 μM can occur in the form of microdomains by both the influx of Ca2+ ions through the plasma membrane (PM) or Ca2+ release from intracellular stores, such as the endoplasmic reticulum (ER) (Targos et al., 2005; Kiselyov et al., 2006; McCarron et al., 2006; Majewski and Kuznicki, 2015). Store-operated Ca2+ entry (SOCE) is the main Ca2+ entry pathway in non-excitable cells (Parekh and Putney, 2005; Prakriya and Lewis, 2015; Putney et al., 2017), which was characterized in detail in immune cells (Parekh and Penner, 1997; Feske et al., 2001, 2005; Feske, 2011). Initially, the SOCE process was known as capacitative Ca2+ entry (Putney, 1986). SOCE is described as Ca2+ flow into cells through PM Ca2+ channels in response to the depletion of ER Ca2+ stores. Ca2+ is released from the ER upon the activation of inositol-1,4,5-triphosphate receptors (IP3Rs) (Berridge, 2002, 2009; Bezprozvanny, 2005; Mikoshiba, 2007) and ryanodine receptors (RyRs) (Rossi and Sorrentino, 2002; Amador et al., 2013) and through passive leakage (van Coppenolle et al., 2004; Tu et al., 2006; Supnet and Bezprozvanny, 2011). The Ca2+ response via IP3Rs is known as IP3-induced Ca2+ release (IICR) (Miyazaki et al., 1993; Mikoshiba, 2007), whereas RyRs act according to Ca2+-induced Ca2+ release (CICR) (Berridge, 1998, 2002; Verkhratsky, 2005). Ca2+ release by IICR is often amplified by CICR (Berridge, 2002; Chen et al., 2011). RyRs are stimulated to transport Ca2+ into the cytoplasm by recognizing Ca2+ on its cytoplasmatic side, thus creating a positive feedback mechanism (Santulli and Marks, 2015). Ca2+ concentrations in the ER are detected by two stromal interaction molecule (STIM) isoforms, STIM1, and STIM2 (Liou et al., 2005; Roos et al., 2005; Zhang et al., 2005). They are localized in ER membranes and function as sensors of Ca2+ concentrations. Upon a decrease in Ca2+ in the ER, STIM proteins oligomerize, and migrate to ER-PM junctions where they interact with highly selective Ca2+ channels, Orai1-3, at the PM (Feske et al., 2006; Peinelt et al., 2006; Prakriya et al., 2006; Vig et al., 2006) and form large complexes that are visible as puncta under a microscope (Potier and Trebak, 2008; Klejman et al., 2009; Gruszczynska-Biegala and Kuznicki, 2013; Shim et al., 2015). This interaction causes the SOCE process, specifically Ca2+ flow from the extracellular space into the cytoplasm (Liou et al., 2005; Cahalan, 2009). To refill ER stores, the sarco-endoplasmic reticulum Ca2+ adenosine triphosphatase (SERCA) pump transports Ca2+ ions to the ER (Berridge, 2002; Periasamy and Kalyanasundaram, 2007; Brini et al., 2013a; Majewski and Kuznicki, 2015). SOCE is mediated by two different currents. One of them is Ca2+ release-activated Ca2+ current (ICRAC), which is highly selective for Ca2+, non-voltage activated, and inwardly rectifying (Hoth and Penner, 1993; Lewis and Cahalan, 1995). The second is store-operated Ca2+ current (ISOC), which is characterized by non-selective outward current with distinct biophysical features, including greater conductance than ICRAC (Golovina et al., 2001; Trepakova et al., 2001; Strübing et al., 2003; Ma et al., 2015; Lopez et al., 2016, 2020). STIM-regulated Orai channels contribute to ICRAC-mediated SOCE, whereas STIM operated transient receptor potential channel 1 (TRPC1) activity mediates ISOC (Majewski and Kuznicki, 2015; Moccia et al., 2015; Secondo et al., 2018).
Ca2+ signaling regulates multiple cellular processes (Brini et al., 2013b). Its dysregulation is the postulated underlying mechanism of many disorders, including neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis (Pchitskaya et al., 2018; Secondo et al., 2018), and Huntington's disease (HD) (Raymond, 2017; Pchitskaya et al., 2018).
Huntington's disease is a progressive neurodegenerative disorder with autosomal-dominant heritability that is caused by CAG trinucleotide repeat expansion in the huntingtin gene (HTT) (MacDonald et al., 1993). The HTT gene encodes huntingtin protein (HTT), which is around 350 kDa in size and ubiquitously expressed in the cytoplasm of all cell types (MacDonald et al., 1993). Mutant HTT (mHTT) contains an expansion of polyglutamine residues (polyQ) in its amino-terminal part (Ross, 2002). Expansion longer than 35 repeats in mHTT results in a polyglutamine tract that leads to mHTT aggregation and earlier HD onset (DiFiglia et al., 1997; Krobitsch and Lindquist, 2000; Langbehn et al., 2004; Gusella and MacDonald, 2006). CAG repeats in mHTT between 40 and 60 cause the onset of HD at 30–50 years of age. The onset of HD before the age of 21 and CAG repeats over 60 are characteristic of the juvenile form of HD (Quigley, 2017), which resembles a neurodevelopmental disorder (Switońska et al., 2018; Wiatr et al., 2018). The most affected cells in HD are γ-aminobutyric acid (GABA)ergic medium spiny neurons (MSNs) in the striatum (Vonsattel and DiFiglia, 1998; Zoghbi and Orr, 2000). The clinical manifestations of HD include chorea, dementia, and mood and cognitive impairments (Zoghbi and Orr, 2000; Bates et al., 2015). Juvenile HD patients often present with rigidity, dystonia, seizures, cognitive alterations, and psychiatric symptoms (Quigley, 2017). No effective treatments have been developed for HD. The available medications only delay progression of the disease or alleviate its symptoms. Therefore, identification of the molecular mechanisms of HD and potential treatment targets are needed. In HD, the cascade of neurodegenerative processes was suggested to be caused by disturbances in Ca2+ signaling (Pchitskaya et al., 2018) that appear to be related to HTT function. Although its function remains unclear, the highest levels of wildtype HTT are found in the brain. Mutant HTT forms aggregates in neuronal nuclei. mHTT inhibits the function of various proteins, such as key transcription factors and Ca2+ signaling components, thereby affecting Ca2+ homeostasis (Giacomello et al., 2013). Disturbances in the Ca2+ signalosome were found in HD models and post-mortem samples from HD patients (Hodges et al., 2006; Wu et al., 2011, 2016; Czeredys et al., 2013). Abnormal Ca2+ signaling is considered an early event in HD pathology (Pchitskaya et al., 2018), particularly the SOCE pathway that is elevated in HD (Wu et al., 2011, 2016, 2018; Czeredys et al., 2013).
The present review provides an overview of Ca2+ signaling via store-operated Ca2+ channels under physiological conditions in neurons and under pathological conditions, namely HD. The distribution of STIM, Orai, and TRPC proteins in neurons and their functions in both the maintenance of ER Ca2+ concentrations and the activity of SOCE are discussed. The dysregulation of neuronal SOCE channels (nSOCs) has been implicated in HD pathology, especially affecting dendritic spines. The role of SOCE and SOCE components in the formation and maturation of dendritic spines and their contribution to synaptic plasticity under physiological conditions are also discussed. Finally, recent findings are presented that support the role of molecular components of neuronal SOCE (nSOCE) and upstream pathways that regulate these processes in dendritic spine pathology in HD. Potential drug candidates are proposed that may restore normal SOCE in HD. An argument is made that nSOCE may be a novel therapeutic target for HD.
Neuronal Ca2+ Signaling VIA Store-Operated Ca2+ Channels Under Physiological Conditions
The role of SOCE as the main Ca2+ entry pathway in non-excitable cells is well-established. An important role for SOCE in excitable cells in the central nervous system (CNS), such as cortical pyramidal neurons (Klejman et al., 2009; Gruszczynska-Biegala et al., 2011), dorsal root ganglion neurons (Gemes et al., 2011), striatal MSNs (Wu et al., 2011, 2016; Czeredys et al., 2017), hippocampal pyramidal neurons (Emptage et al., 2001; Baba et al., 2003; Samtleben et al., 2015), and cerebellar Purkinje neurons (Hartmann et al., 2014), has also been recently demonstrated. Moreover, growing evidence suggests the existence of synaptic nSOCE (Sun et al., 2014; Wu et al., 2016, 2018; Ryskamp D. et al., 2019). The presence of molecular components of SOCE in both the neuronal cell body (Klejman et al., 2009; Gruszczynska-Biegala and Kuznicki, 2013) and dendritic spines (Garcia-Alvarez et al., 2015; Korkotian et al., 2017) has been reported. Additionally, the ER was shown to extend over the entire neuron, including numerous spines that encrust the dendrites (Berridge, 2002). Both STIM isoforms, STIM1 and STIM2, are broadly but differentially expressed in the CNS. STIM1 is the predominant isoform in the cerebellum (Klejman et al., 2009; Skibinska-Kijek et al., 2009; Hartmann et al., 2014). STIM2 is most prominent in the hippocampus (Berna-Erro et al., 2009; Skibinska-Kijek et al., 2009) and cortex (Skibinska-Kijek et al., 2009; Kraft, 2015). In immune cells, STIM2.1 and STIM2.2 isoforms were identified, which have different properties in the regulation of SOCE. STIM2.1 is a negative regulator of SOCE, and STIM2.2 is the counterpart of STIM2 (Miederer et al., 2015). Both isoforms are equally expressed in MSNs (Czeredys et al., 2018), but their functions in the regulation of SOCE in neurons are unknown.
STIM1 has high affinity for Ca2+ and requires the higher depletion of Ca2+ from the ER during SOCE. STIM2 is characterized by slower aggregation kinetics, and the weaker depletion of ER Ca2+ stores is sufficient to activate it (Brandman et al., 2007; Parvez et al., 2008; Stathopulos et al., 2009; Gruszczynska-Biegala et al., 2011; Gruszczynska-Biegala and Kuznicki, 2013). STIM2 was found to stabilize basal Ca2+ levels in rat neuronal cortical cultures and cell lines of peripheral origin (Brandman et al., 2007; Gruszczynska-Biegala et al., 2011; Gruszczynska-Biegala and Kuznicki, 2013). The interaction between STIM2 and Orai1 is weak and causes poor channel activation, whereas STIM2 was found to trigger remodeling of the STIM1 C-terminus and facilitate STIM1/Orai1 coupling and the enhancement of Orai1 function when alterations of Ca2+ levels in the ER are not sufficiently low to initiate STIM1 responses (Subedi et al., 2018).
Three members of the Orai channel family have been identified: Orai1, Orai2, and Orai3. They all interact with STIM1 but have different inactivation and permeability properties (DeHaven et al., 2007; Lis et al., 2007). Orais are known as Ca2+ release-activated Ca2+ channels (CRACs) (Prakriya et al., 2006). Among them, Orai1 is a crucial pore subunit of the CRAC channel (Prakriya et al., 2006) that mediates higher currents compared with the other isoforms (Putney et al., 2017). The Orai1 channel functions as a hexamer (Hou et al., 2012; Cai et al., 2016; Yen et al., 2016). Orai1 is the dominant form in immune cells (Vaeth et al., 2017), but the highest expression levels of the Orai2 isoform were detected in the brain (Chen-Engerer et al., 2019). Orai3 is highly expressed in cancer cells (Vashisht et al., 2018). Apart from STIM-regulated Orai channels that contribute to Ca2+ release-activated Ca2+ current (ICRAC)-mediated SOCE, recent studies indicated that STIM-operated TRPC1 activity mediates ISOC (Liu et al., 2003; Ambudkar et al., 2017). Among six known TRPC proteins, TRPC1, TRPC3, and TRPC4 are SOCE partners that are activated by the depletion of Ca2+ from stores, whereas the operation of TRPC5, TRPC6, and TRPC7 is store-independent (Ambudkar et al., 2007; Liu et al., 2007; Venkatachalam and Montell, 2007; Trebak et al., 2009; Putney and Tomita, 2012). Phospholipase C (PLC)-mediated phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) hydrolysis upon the (S)-3,5-dihydroxyphenylglycine (DHPG)-induced activation of metabotropic glutamate receptor 1/5 (mGluR1/5) may activate TRPCs in a store-independent manner with diacylglycerol (DAG) (Itsuki et al., 2014). TRPC1 is recruited to the PM by Rab4-dependent recycling, which is critical for TRPC1-STIM1 clustering within ER-PM junctions (de Souza et al., 2015). TRPC1 function depends on Orai1-mediated Ca2+ entry, which enables TRPC1 recruitment to the PM where it is subsequently activated by STIM1 (Ambudkar et al., 2017). TRPC proteins may form different heteromeric structures that are involved in SOCE (Goel et al., 2002; Hofmann et al., 2002; Strübing et al., 2003; Wu et al., 2004; Liu et al., 2005; Zagranichnaya et al., 2005; Sundivakkam et al., 2012). The most prevalent TRPC channels in the mammalian brain are TRPC1, TRPC4, and TRPC5, which are mainly expressed in the hippocampus, prefrontal cortex, and lateral septum (Fowler et al., 2007, 2012), whereas TRPC3 is highly expressed in cerebellar Purkinje cells (Hartmann et al., 2014).
In the CNS, Ca2+ influx is mainly controlled by voltage-gated Ca2+ channels (VGCCs) and ionotropic glutamate receptors, such as glutamate-sensitive N-methyl-D-aspartate receptors (NMDARs) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) (Catterall, 2011; Paoletti et al., 2013; Henley and Wilkinson, 2016), whereas the SOCE pathway is activated under resting conditions to refill ER stores and governs spontaneous neurotransmitter release (Emptage et al., 2001; Baba et al., 2003; Gruszczynska-Biegala and Kuznicki, 2013; Moccia et al., 2015; Wegierski and Kuznicki, 2018). In Purkinje neurons, STIM1-mediated SOCE replenishes intracellular Ca2+ levels when VGCC activity is low (Hartmann et al., 2014). Recent data suggest that SOCE may play a role in synaptic plasticity in the CNS and participate in regulating spine morphogenesis, neuronal excitability, and gene expression (Moccia et al., 2015). The role of STIM2 in maintaining post-synaptic mushroom spines in hippocampal neurons was also reported (Sun et al., 2014; Kraft, 2015). Ca2+ influx via STIM2-mediated SOCE regulates Ca2+/calmodulin-dependent protein kinase II (CaMKII) and stabilizes mushroom spines that play a role in memory storage (Sun et al., 2014). STIM2 also regulates AMPAR trafficking and plasticity at hippocampal synapses (Yap et al., 2017). Garcia-Alvarez et al. showed that STIM2 localizes to dendritic spines, is enriched in the post-synaptic density, and is required for regular synaptic activity. STIM2 mediates cyclic adenosine monophosphate (cAMP)-dependent phosphorylation and trafficking of the GluA1 subunit of AMPARs to PM-ER junctions independently from SOCE (Garcia-Alvarez et al., 2015). The role of STIM2 in synaptic plasticity has been established (Moccia et al., 2015), but the roles of STIM1 and Orai1 in spine architecture are just emerging. Korkotian et al. recently reported the role of Orai1 in the formation, maturation, and plasticity of dendritic spines in developing hippocampal neurons (Korkotian et al., 2017). These authors found that upon store depletion, STIM2 co-localized with Orai1 in spines, and STIM1 was less mobile in moving to spines than STIM2. Furthermore, the presence of clusters of Orai1 correlated with the emergence of nascent spines on dendrites following the transient elevation of extracellular Ca2+ concentrations (Korkotian et al., 2017). The role of STIM1 in regulating the structural plasticity of L-type VGCC-dependent dendritic spines was reported (Dittmer et al., 2017). The NMDAR activation of L-type VGCCs was proposed to release Ca2+ from the ER, which consequently causes STIM1 aggregation, inhibits L-type VGCCs, enhances ER spine content, and stabilizes mushroom spines (Dittmer et al., 2017). These findings were consistent with previous studies by two independent research groups that found a direct interaction between STIM1 protein and L-type VGCCs and the role of STIM1 in inhibiting the depolarization-mediated opening of L-type VGCC depolarization (Park et al., 2010; Wang et al., 2010). The role of STIM proteins in synaptic plasticity is further supported by findings by our group that STIMs can interact with and induce Ca2+ influx through AMPARs (Gruszczynska-Biegala et al., 2016). Additionally, the role of STIM proteins as potential negative regulators of NMDA-stimulated Ca2+ signaling was shown in cortical neurons (Gruszczynska-Biegala et al., 2020). The overexpression of STIM1 in brain neurons in transgenic mice improved contextual learning and impaired long-term depression (Majewski et al., 2017). Furthermore, STIM1 protein is responsible for mGluR1-dependent synaptic transmission in cerebellar Purkinje neurons (Hartmann et al., 2014). Accumulating evidence indicates a role for STIM1 in neurogenesis (Somasundaram et al., 2014) and the proliferation and early differentiation of neural progenitor cells (NPCs) (Somasundaram et al., 2014; Gopurappilly et al., 2018). The knockdown of both STIM isoforms reduced SOCE and inhibited the entry of mouse embryonic stem cells into a neural lineage (Hao et al., 2014). In human embryonic stem cells (hESCs), SOCE but not VGCC-mediated Ca2+ entry was observed, thus confirming that SOCE could play an essential role in Ca2+ signaling that is important for the self-renewal and differentiation of hESCs (Huang et al., 2017) and supporting the role of molecular components of SOCE in brain development.
Neuronal Ca2+ signaling is a complex process that involves Ca2+ inflow from the extracellular space and Ca2+ discharge from ER. Apart from the main molecular components of SOCE and PM receptors, several other molecules regulate Ca2+ signaling in neurons (Figure 1). The list of Ca2+ signalosomes directly or indirectly regulating Ca2+ signaling in neurons and their main physiological function were listed in Table 1. One of them is HTT binding partner, Huntingtin-associated protein-1 (HAP1), which facilitates functional effects of HTT on IP3R1 in planar lipid bilayers (Tang et al., 2004). In neurons, both HTT and HAP1 are involved in cytoskeleton regulation (Ma et al., 2011) and intracellular trafficking (Caviston and Holzbaur, 2009; Wu and Zhou, 2009). Another Ca2+ signalosome that does not directly regulate Ca2+ signaling is calcyclin binding protein and Siah-1 interacting protein (CacyBP/SIP), which interacts with Ca2+ binding protein calcyclin (Filipek et al., 2002), and its role was found, among others, in the cytoskeleton regulation (Jurewicz et al., 2013). There are several important signaling molecules in the ER that include presenilin 1 (PS1), which is a Ca2+ leak channel in the ER (Tu et al., 2006), the sigma-1 receptor (S1R), which is an ER-resident transmembrane protein regulating ER Ca2+ homeostasis (Su et al., 2010), and binding immunoglobulin protein (BiP), a chaperone protein, which maintains high Ca2+ levels in the ER (Hendershot, 2004). S1R interacts with BiP and prolong Ca2+ signaling from ER into mitochondria by stabilizing IP3R3s at the mitochondria-associated membranes (MAMs) (Hayashi and Su, 2007). It also blocks the inhibitory actions of ankyrin on IP3R3 (Wu and Bowen, 2008). In MSNs, where IP3R1 is a predominant neuronal isoform (Czeredys et al., 2018) S1R resisting in MAMs and stabilizes IP3R3 (Ryskamp D. A. et al., 2019). The S1R directly binds to IP3R1 and leads to the stimulation of protein kinase C (PKC) activity and suppression of IP3 synthesis in hepatocytes (Abou-Lovergne et al., 2011).
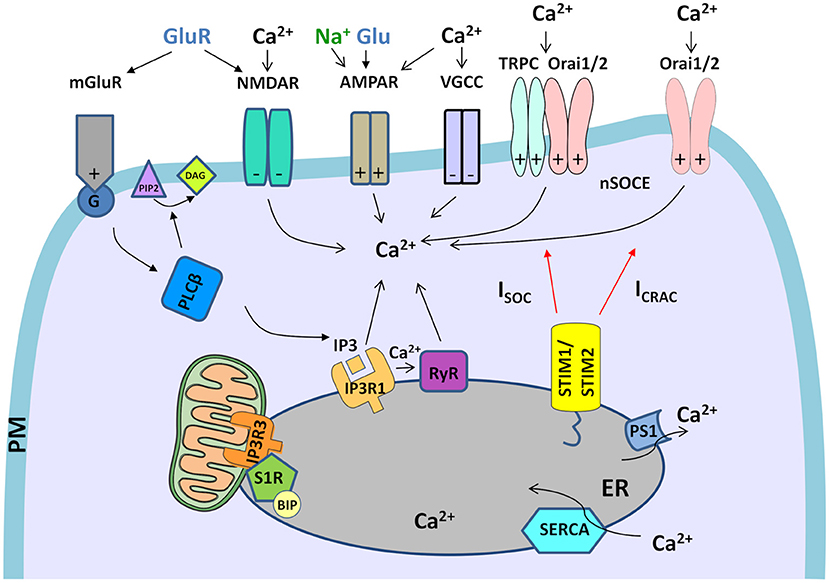
Figure 1. Neuronal Ca2+ signalosomes in physiology. Neuronal Ca2+ signaling is a complex process that involves Ca2+ influx from the extracellular space and Ca2+ release from ER, which contains the largest endogenous Ca2+ pool. Glutamate (Glu) is released from presynaptic neuronal terminals, which leads to stimulation of glutamate receptors (NMDARs, AMPARs, and mGluR1s), which are present in post-synaptic terminals of the excitatory synapses. Activation of Ca2+ efflux by NMDAR and VGCCs requires the excitatory post-synaptic potential that arises as a result of AMPAR activity that gates Na+ entry. VGCCs are activated only by depolarized membrane potential. In NMDAR activation excitatory post-synaptic potential removes the Mg2+ block from the channel what allows its opening in response to Glu stimulation and mediates Ca2+ influx. mGluR is activated by Glu and coupled to trimeric G-protein, which activates a membrane-associated enzyme, PLCβ, that mediates the hydrolysis of PI(4,5)P2 phospholipid to diacylglycerol (DAG) and signaling molecule, IP3. Then, IP3 interacts with IP3R receptors in the ER what causes the release of Ca2+ from ER stores. Further, Ca2+ release from the ER is exaggerated by RyR receptors in the ER, which are activated by Ca2+ release via IP3Rs through a mechanism called Ca2+-induced Ca2+ release (CICR). The activation of RyRs via NMDARs or VGCCs in the plasma membrane (PM) could also be involved in CICR (not shown in the figure). Upon depletion of ER Ca2+ stores, Ca2+ influx from extracellular space is caused by neuronal store-operated Ca2+ entry (nSOCE), which is mediated by the interaction between the ER Ca2+-sensors, Stim1 and Stim2, and the Ca2+-permeable channels, consisting of Orai1 and Orai2 and/or Orai and TRPC channels. These interactions mediate two types of currents, ICRAC and ISOC. To refill ER Ca2+ stores SERCA pumps Ca2+ from the cytosol to the ER, while presenilin 1 (PS1) causes Ca2+ leak from the ER. Crosstalk between STIM proteins and receptors/channels in PM that was observed in neurons is marked on each receptor/channel as “+”–positive regulator/effector of STIM or “–“–negative effector of STIM. The only mGluR is the regulator of STIM. Within mitochondria-associated membranes (MAMs), the S1R receptor interacts with binding immunoglobulin protein (BIP), thereby regulates lipid dynamics and chaperones IP3R3 to the MAMs and facilitates Ca2+ flux from the ER to mitochondria. Filled black and red arrows represent interaction mechanisms. Open black arrows represent Ca2+/Na+ flux. Parts of the Figures 1–3 were drawn by using pictures from Servier Medical Art (http://smart.servier.com/), licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
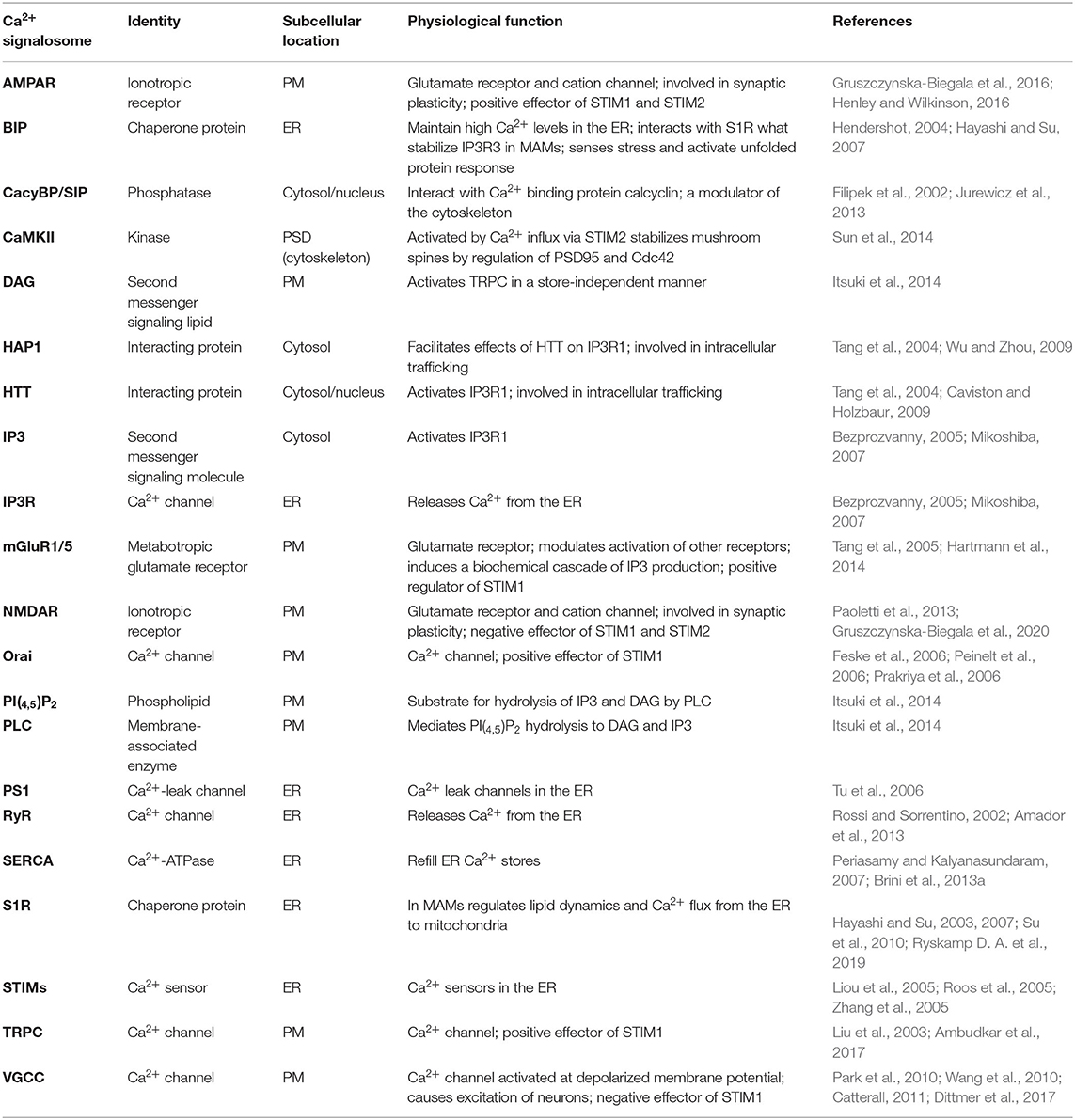
Table 1. Ca2+ signalosome directly or indirectly regulating Ca2+ signaling in neurons.
Besides the molecules that regulate SOCE, these processes might be regulated by rearrangement in cytoskeleton proteins. The role of the cytoskeleton in SOCE regulation through the modulation of the interaction between their main molecular components was demonstrated by several groups (Smyth et al., 2007; Grigoriev et al., 2008; Vaca, 2010; Galán et al., 2011; Giurisato et al., 2014). Septins are one of the cytoskeletal components that have been shown to facilitate interactions between STIM1 and Orai1 proteins (Sharma et al., 2013). In Drosophila neurons it was shown that dSEPT7 prevents dOrai-mediated spontaneous Ca2+ entry. Lower dSEPT7 levels lead to Ca2+ store-independent constitutive opening of dOrai channels and higher cytosolic Ca2+ in resting neurons, while dSEPT7 overexpression resulted in lower SOCE (Deb et al., 2016). Furthermore, it was shown in Drosophila neurons that microtubule stabilization by expression of a dominant-negative variant of the microtubule-severing protein spastin reduces SOCE, and decreases ER Ca2+ content. These effects were restored upon the application of a drug that stabilized microtubules (Vajente et al., 2019).
Dysregulation of SOCE Causes Synaptic Loss in HD Models
Elevations of SOCE have been reported in several models of HD, including YAC128 MSNs (Wu et al., 2011, 2016; Czeredys et al., 2017). In YAC128 MSNs, an increase in STIM2 expression was observed in both YAC128 MSN cultures and 12-month-old YAC128 mice compared with controls. Increases in STIM2 expression elevated synaptic nSOCE, which likely was involved in synaptic loss in MSNs (Wu et al., 2016), whereas STIM1 expression was unchanged in HD MSNs. Indeed the CRISPR-Cas9-mediated knockdown of STIM2 caused the stabilization of dendritic spines in YAC128 MSNs. A key role for TRPC1 channels in supporting the SOC pathway was also shown in an HD model. The RNA interference (RNAi)-mediated knockdown of TRPC1 in YAC128 mouse MSNs exerted a significant protective effect against glutamate-induced apoptosis (Wu et al., 2011). Patch-clamp experiments in SK-N-SH cells transfected with full-length mutant HTT with 138Q (HTT-138Q) revealed that the current-voltage relationship of ISOC currents was consistent with the involvement of TRP channels, and TRPC1 knockdown significantly reduced the magnitude of ISOC currents in these cells (Wu et al., 2011). Additionally, crossing YAC128 mice with TRPC1 knockout mice improved motor performance and rescued MSN spines in vitro and in vivo (Wu et al., 2018). The RNAi-mediated knockdown of channels that are involved in SOCE other than TRPC1, such as TRPC6, Orai1, and Orai2, and knockout of the ER Ca2+ sensor STIM2 (Wu et al., 2016, 2018) resulted in the stabilization of spines and suppressed abnormal nSOCE in YAC128 MSNs. These results indicate that the molecular composition of the SOCE pathway in HD is complex and suggest the involvement of other players beyond STIM2 and TRPC1.
Apart from YAC128 transgenic mice, elevations of SOCE have also been described in a cellular model of HD (Wu et al., 2011; Vigont et al., 2014, 2015). In human neuroblastoma cells, expression of the N-terminal fragment of mHTT (exon-1 HTT with 138Q) increased STIM1-mediated SOCE (Vigont et al., 2014). Similar findings were reported in mouse neuroblastoma cells and primary cultures of mouse MSNs upon the lentiviral expression of N-terminal mHTT. Elevations of SOCE through actions of the sensor STIM1 and both TRPC1 and Orai1 subunits that comprise a heteromeric channel were found in these in vitro cultures (Vigont et al., 2015). Recent studies found that iPSC-based GABAergic MSNs from HD patient fibroblasts, which recapitulate some aspects of HD pathology, were characterized by elevations of SOC currents in vitro (Nekrasov et al., 2016). The SOCE of iPSC-based GABAergic HD MSNs was shown to be mediated by currents through at least two different channel groups, ICRAC and ISOC. These currents were upregulated compared with wildtype iPSC-based GABAergic MSNs. Thapsigargin-induced intracellular Ca2+ store depletion in iPSC-based GABAergic MSNs resulted in the simultaneous activation of both ICRAC and ISOC (Vigont et al., 2018).
Ca2+ Signaling Pathways That May Affect Soce and Cause Synaptic Loss in HD
Previous studies demonstrated that mHTT affects Ca2+ signaling in MSNs by increasing the sensitivity of IP3R1 to IP3 (Tang et al., 2003), facilitating Ca2+ release from internal Ca2+ stores (Ca2+ leakage) through RyRs (Suzuki et al., 2012), stimulating the activity of NR1/NR2B NMDARs (Chen et al., 1999; Sun et al., 2001; Zeron et al., 2002, 2004; Tang et al., 2005; Fan et al., 2007; Milnerwood and Raymond, 2007; Zhang et al., 2008), and disturbing mitochondrial Ca2+ handling (Panov et al., 2002; Choo et al., 2004; Oliveira et al., 2007). The increase in SOC pathway activity in HD may be caused directly by an increase in STIM2 expression in the presence of mHTT (Wu et al., 2016) or may result from a compensatory response of cells to the destabilization of Ca2+ signaling, such as an increase in the activity of IP3R1 by mHTT in YAC128 mice (Tang et al., 2003, 2004; Wu et al., 2016) or the modulation of SOC channels by receptors in the ER, such as S1Rs (Su et al., 2010).
A physiological mechanism that is responsible for activating SOCE results from the stimulation of G-protein-coupled receptors that are associated with the IP3 and PLC cascade, resulting in the release of Ca2+ from the ER via the IP3Rs (Streb et al., 1983). IP3R1 signaling is one of the pathways that act upstream of SOCE. Upon the release of Ca2+ from the ER via IP3R1, STIMs activate channels in the PM and cause Ca2+ influx to the cytosol. IP3R1 is abnormally activated in HD models that dysregulate ER Ca2+ dynamics. IP3R1 is the main IP3R isoform that is expressed in the striatum (Czeredys et al., 2018), and its activity is elevated in YAC128 MSNs (Wu et al., 2016). In MSNs, the overexpression of HTT-138Q activates IP3R1 more strongly compared with HTT that contains only 82Q repeats (Tang et al., 2003). Wu et al. reported that mHTT caused supranormal IP3R1 activity, which reduced ER Ca2+ levels (Wu et al., 2016). The depletion of ER Ca2+ leads to an increase in the activation of nSOCE in YAC128 MSN spines. Wu et al. showed that the inhibition of IP3R1 expression attenuated abnormal nSOCE and rescued spine loss in YAC128 MSNs (Wu et al., 2016), supporting the role of IP3R1 in neurite development (Fiedler and Nathanson, 2011). To inhibit IP3R1 activity, antisense oligonucleotides were applied, which resulted in the normalization of nSOCE and rescued spine loss in YAC128 MSNs (Wu et al., 2016). In summary, the role of IP3R1 that acts upstream of SOCE was established in synaptic loss in HD, in addition to known components of ISOC, including STIM2 and TRPC1 (Wu et al., 2016).
In our previous studies, we observed several abnormalities of Ca2+ signaling in YAC128 mice, such as the upregulation of some members of Ca2+ signalosomes in the striatum, including HAP1 and CacyBP/SIP (Czeredys et al., 2013). Their role has been recently established in neurodegenerative or neurodevelopmental diseases (Wasik et al., 2013; Xiang et al., 2017; Wang et al., 2019; Bohush and Filipek, 2020; Liu et al., 2020). Our group also observed the elevation of SOCE and an increase in IP3R1 activity in YAC128 MSNs (Czeredys et al., 2017). Our recent research focused on regulatory mechanisms of elevations of SOCE in MSNs from YAC128 mice (Czeredys et al., 2018). We found that the HAP1A isoform was responsible for the increase in SOC channel activity when it was overexpressed in MSNs from YAC128 mice. The HAP1B isoform differs from HAP1A in its C-terminal part and cannot interact with IP3R1 (Tang et al., 2003). HAP1B did not affect SOCE (Czeredys et al., 2018). We also observed a decrease in SOC channel activity when HAP1 protein was silenced. Upon HAP1A overexpression, an increase in IP3R1 activity and a decrease in the ionomycin-sensitive ER Ca2+ pool were observed. When the IP3R1 inhibitor IP3 sponge (Uchiyama et al., 2002) was applied via lentiviruses in YAC128 MSN cultures that overexpressed HAP1A, the increase in the release of Ca2+ from the ER and consequently the elevation of SOCE were both restored to wildtype levels. These experiments revealed that HAP1A elevates SOCE in the HD model through its interaction with mHTT and facilitation of the effects of mHTT on IP3R1 (Tang et al., 2003, 2004). To unravel the role of HAP1A in SOCE dysregulation in HD, we also used a cellular model of HD, SK-N-SH cells that express HTT138Q. We found that HAP1A overexpression constitutively activated SOC channels and upregulated STIM2 protein (Czeredys et al., 2018). Therefore, STIM2 may also be involved in the activation of DHPG-induced SOCE in YAC128 MSN cultures that overexpress HAP1A. The upregulation of STIM2 and hyperactivity of IP3R1 may underlie the constitutive activity of SOC channels in SK-N-SH-HTT-138Q-overexpressing HAP1A. Overall, our data indicate that HAP1A causes the aberrant activation of SOC channels in HD models. Thus, HAP1A appears to be an important player in HD pathology. Published data indicate that HAP1A dysregulates IP3R1 and upregulates STIM2. Therefore, we predict that elevations of SOCE in HD models may also underlie synaptic loss and neurodegeneration in HD.
In addition to IP3R1 signaling, which contributes to elevations of SOCE in HD models, another mechanism that involves S1Rs was recently reported (Ryskamp et al., 2017). The striatal upregulation of S1R was detected in aged YAC128 transgenic mice and HD patients, which caused ER Ca2+ dysregulation by IP3R1 modulation, an increase in SOCE, and abnormal dendritic spine morphology (Ryskamp et al., 2017). It was previously reported that upon ER Ca2+ depletion or ER stress or at high concentrations of S1R agonists, S1R is released from BIP, dislocates beyond the MAM domain (Su et al., 2010), and modulates additional targets, including IP3R1, STIM1, and ion channels and receptors on the PM (Kourrich et al., 2012; Ryskamp D. A. et al., 2019). The S1R was also reported to have functions in neuromodulation (Maurice et al., 2006) and neuroplasticity (Takebayashi et al., 2004; Tsai et al., 2009; Kourrich et al., 2012). The CRISPR/Cas9-mediated deletion of S1Rs resulted in MSN spine loss in wildtype corticostriatal co-cultures, thus indicating an essential role for S1Rs in the development or maintenance of synaptic connections between cortical and striatal neurons (Ryskamp et al., 2017).
Concentrations of Ca2+ in the ER are a determinant of the magnitude of Ca2+ signaling through IP3R and RyR channels. In HD models, the dysregulation of Ca2+ release from the ER by RyR signaling was also observed. Abnormal Ca2+ leakage by RyRs was detected in striatal and cortical neurons from an R6/2 mouse model of HD that overexpresses exon-1 mHTT (Suzuki et al., 2012). The involvement of RyRs in mHTT-induced neuronal death was also shown in R6/2 HD mice (Suzuki et al., 2012) and YAC128 mice (Chen et al., 2011). The association between SOCE and IP3R-gated Ca2+ stores has been thoroughly examined, but the role of RyR-gated stores in SOCE is less well-known. RyRs might play a role in synaptic plasticity (Baker et al., 2013; Johenning et al., 2015). A study that used an Orai1 dominant-negative mutant revealed the role of Orai1 in dendritic spine formation following the chemical induction of long-term potentiation (LTP) (Tshuva et al., 2017). This finding was attributable to the release of Ca2+ from RyR-associated ER stores. Spine formation in control neurons was reduced by the RyR antagonist dantrolene (Zucchi and Ronca-Testoni, 1997). Tshuva et al. postulated that Ca2+ stores are important for the formation of new dendritic spines. Under conditions of Orai1 deficiency, there is less Ca2+ in the stores, and Ca2+ release decreases, leading to decreases in LTP and spine formation (Tshuva et al., 2017).
The activation of SOCE by RyRs has been intensively studied in different cell types. RyR2-gated Ca2+ stores were shown to contribute to SOCE in pulmonary artery smooth muscle cells. However, depletion of RyR-sensitive Ca2+ store with caffeine was insufficient to activate Ca2+ entry but required a particular RyR conformation that was modify by ryanodine binding (Lin et al., 2016). Additionally, in primary human T cells, the RyR is related to CRAC machinery such that SOCE triggers RyR activation via a CICR mechanism following the attenuation of Ca2+ concentrations within the ER lumen in the proximity of STIM1, thereby facilitating SOCE by diminishing store-dependent CRAC inhibition (Thakur et al., 2012). Moreover, the RyR agonist 4-chloro-3-ethylphenol blocked Orai store-operated channels in rat L6 myoblasts and HEK293 cells (Zeng et al., 2014).
Apart from the effect of an increase in SOCE or its upstream pathways on the dysregulation of spines in HD, other pathways that involve disturbances in Ca2+ signaling have been proposed to be involved in HD pathology (Tang et al., 2005). One of them is the dysregulation of neurotransmitter release in synapses of HD neurons. A few recent studies found that mHTT can modulate N-type VGCCs (Cav2.2), which are essential for presynaptic neurotransmitter release. In young BACHD mice that expressed full-length mHTT, an increase in striatal glutamate release was found, which was reduced to wildtype levels by Cav2.2 inhibition. Cav2.2 Ca2+ current density and PM expression also increased in these mice, which could be responsible for the elevation of glutamate release (Silva et al., 2017). The increases in synaptic vesicle release and elevations of Ca2+ influx at presynaptic terminals in primary cortical neurons were also found in a knock-in mouse model of HD (zQ175) (Chen et al., 2018). Moreover, under experimental conditions, the application of glutamate in vitro resulted in the apoptosis of YAC128 MSNs but not wildtype MSNs (Tang et al., 2005). Tang et al. found that abnormal glutamate release from corticostriatal projection neurons stimulated NR1/NR2B NMDARs and mGluR1/5 in YAC128 MSNs (Tang et al., 2005). Activation of NR1/NR2B NMDAR leads to Ca2+ influx and activation of mGluR5 receptors, leading to the production of IP3, which caused the subsequent activation of Ca2+ release via IP3R1. The stimulation of glutamate receptors results in supranormal Ca2+ responses in HD MSNs, which contributes to cytosolic Ca2+ overload and consequently exceeds mitochondrial Ca2+ storage capacity. This, in turn, guides the opening of mitochondrial permeability transition pores (MPTPs), release of cytochrome c into the cytosol, and activation of caspases 9 and 3 and apoptosis (Zeron et al., 2004; Tang et al., 2005). Although, the effect of the abnormal glutamate stimulation of NMDARs on dendritic spines of YAC128 MSNs have not been examined (Tang et al., 2005), these receptors may contribute to dendritic spine pathology in HD. Accumulating evidence suggests crosstalk between NMDARs and STIMs (Dittmer et al., 2017; Gruszczynska-Biegala et al., 2020). STIM1 was shown to regulate the structural plasticity of L-type VGCC-dependent dendritic spines. The NMDAR activation of L-type Ca2+ channels triggers Ca2+ release from the ER, which then regulates STIM1 aggregation, downregulates L-type channels, and enhances ER spine content and the stabilization of mushroom spines (Dittmer et al., 2017). The activation of NMDARs in pyramidal neurons causes the recruitment of IP3 through interactions with IP3R, leading to Ca2+ release from ER stores and SOCE stimulation. SOCE inhibitors reduced NMDA-dependent Ca2+ influx and synaptic plasticity in the hippocampus (Baba et al., 2003).
A few recent studies reported the role of AMPARs in HD pathology. In the striatum in HTTQ111/+ HD knock-in mice, synaptic dysfunction and disturbances in AMPAR-mediated signaling were observed. In the striatum in these mice, a decrease in synapse density, an increase in post-synaptic density thickness, and an increase in synaptic cleft width were observed. Acute slice electrophysiology showed alterations of spontaneous AMPAR-mediated post-synaptic currents, an increase in evoked NMDAR-mediated excitatory post-synaptic currents, and elevations of extrasynaptic NMDAR currents (Kovalenko et al., 2018). Cognitive impairments in rodent models of HD are linked to the improper diffusion of AMPARs in dendritic spines of hippocampal neurons through the dysregulation of brain-derived neurotrophic factor (BDNF)-TrkB-CaMKII signaling (Zhang et al., 2018). Moreover, in YAC128 mice, the AMPAR-dependent formation of new synapses through BDNF signaling is also disturbed (Smith-Dijak et al., 2019). Ca2+ influx via STIM2-mediated SOCE regulates CaMKII and stabilizes mushroom spines (Sun et al., 2014), but we cannot exclude the possible effects of other players of SOCE on abnormal AMPAR diffusion in spines in HD that were shown to act through BDNF-TrkB-CaMKII signaling (Zhang et al., 2018). Recent studies in rat cortical pyramidal neurons suggested that Ca2+ influx through AMPARs is induced by STIMs, which might confirm the relationship between SOCE and AMPARs (Gruszczynska-Biegala et al., 2016). These authors found that AMPAR antagonists inhibited SOCE, SOCE inhibitors decreased AMPA-induced Ca2+ influx, and both STIM1 and STIM2 proteins cooperated with GluA1 and GluA2 subunits of AMPARs (Gruszczynska-Biegala et al., 2016). However, other studies showed that STIM2 protein can interact with AMPARs in a SOCE-independent manner (Garcia-Alvarez et al., 2015). It was found that STIM2 regulated the phosphorylation of GluA1 at both Ser845 and Ser831, which promoted the cAMP/protein kinase A (PKA)-dependent surface delivery of GluA1 via exocytosis and endocytosis (Garcia-Alvarez et al., 2015).
Potential Therapeutic Strategies to Stabilize SOCE in HD
In the striatum in HD transgenic mice, elevations of synaptic nSOCE were suggested to underlie post-synaptic dendritic spines loss in MSNs in aged corticostriatal co-cultures that were established from YAC128 mice (Wu et al., 2016). Several studies postulated that early neuropathological features of HD include perturbances of corticostriatal synaptic function and connectivity (Milnerwood and Raymond, 2007, 2010; Miller and Bezprozvanny, 2010; Orth et al., 2010; Murmu et al., 2013) and might lead to the accelerated neurodegeneration of MSNs in the striatum (Myers et al., 1988; Vonsattel and DiFiglia, 1998). Disturbances in the stability of synaptic spines have been suggested to underlie the development of HD symptoms (Bezprozvanny and Hiesinger, 2013; Murmu et al., 2013). The pharmacological inhibition of nSOCE that is abnormally elevated in HD could be a potential way to block neurodegeneration. A few SOCE inhibitors have been tested in HD models to date. One of these inhibitors is 6-amino-4-(4-phenoxyphenethyl-amino)quinazoline (EVP4593), which is a specific inhibitor of nSOC entry (Wu et al., 2011). A screen of a library of quinazoline-derived compounds was performed in HD flies that developed a progressive motor phenotype upon the induction of HTT-128Q transgene expression in the nervous system. In these flies, the HD phenotype was quantified using an automated climbing assay that automatically monitors motor functions. The application of EVP4593 normalized motor behavior in this fly model of HD and exerted neuroprotective effects in a glutamate toxicity assay in YAC128 MSN cultures, whereas its inactive analog EVP14808 failed to inhibit the SOC pathway in HD models (Wu et al., 2011). Further research revealed that EVP4593 reduces synaptic nSOCE and recovers abnormal spines in YAC128 MSNs. The intraventricular administration of EVP4593 in YAC128 mice in vivo rescued age-dependent striatal spine loss (Wu et al., 2016). Additionally, EVP4593 reduced SOCE to normal levels in MSNs that expressed exon-1 HTT with 138Q (Vigont et al., 2015). This compound also potently stabilized SOC entry in HD iPSC-based GABAergic MSNs (Nekrasov et al., 2016), thus confirming its specificity in neurons that were reprogrammed from fibroblasts from HD patients. The molecular target of EVP4593 is still unknown, but recent data showed that EVP4593 equally affected different SOC channels in HD iPSC-based GABAergic MSNs, suggesting that this compound could target SOCE regulatory proteins that are involved in both ICRAC and ISOC (e.g., STIM proteins) (Vigont et al., 2018). Huntington's disease iPSC-based GABAergic MSNs that were characterized by both excessive SOCE and progressive HD pathology (Nekrasov et al., 2016) were recently proposed to serve as a platform for personal drug screening in HD (Bezprozvanny and Kiselev, 2017; Vigont et al., 2018).
The stabilization of SOCE by tetrahydrocarbazoles was reported by our group in MSNs from YAC128 mice (Czeredys et al., 2017). We previously showed that these compounds decreased carbachol-induced Ca2+ efflux from the ER in HEK293-PS1-M146L cells, a cellular model of familial Alzheimer's disease (Honarnejad et al., 2014). Among seven selected tetrahydrocarbazoles, the most active compound in the HD model was 6-bromo-N-(2-phenylethyl)-2,3,4,9-tetrahydro-1H-carbazol-1-aminehydrochloride (C20H22BrClN2). This compound was able to restore disturbances in Ca2+ homeostasis and stabilize both DHPG- and CPA-induced SOCE in YAC128 MSN cultures. C20H22BrClN2 was added just 5 min before the measurements, and we concluded that it acts through the post-translational modification of proteins that are involved in SOCE, stabilization of the cytoskeleton, or the inhibition of SOCE or ER Ca2+ release channels. However, the effect of this tetrahydrocarbazole on the increase in Ca2+ release from the ER was not statistically significant, in contrast to its effect on SOCE. It specifically attenuated SOCE in YAC128 MSNs and did not affect SOCE in wildtype MSNs. We also found beneficial effects of this tetrahydrocarbazole on the function of mitochondria from YAC128 MSNs (Czeredys et al., 2017), which was consistent with its protective effects in the Alzheimer's disease model (Honarnejad et al., 2014). Our recent studies showed that the SOCE stabilizer C20H22BrClN2 reversed the elevation of SOCE in YAC128 MSN cultures that overexpressed HAP1A (Czeredys et al., 2018). In conclusion, this tetrahydrocarbazole stabilized SOCE in MSNs from YAC128 mice and might be a promising compound for the treatment of HD.
Synaptic nSOCE is suggested to underlie spines loss in YAC128 MSNs. Therefore, compounds that can inhibit elevations of SOCE could be beneficial for synapse maintenance in HD and possibly attenuating HD pathology. The effects of EVP4593 have already been established in the restoration of spines in HD models (Wu et al., 2016), but the effects of tetrahydrocarbazoles require further studies. Potential drugs that may inhibit elevations of SOCE and consequently attenuate spine abnormalities in HD neurons are summarized in Table 2.

Table 2. Potential strategies to stabilize SOCE in HD models.
Potential Therapeutic Strategies to Stabilize Ca2+ Signaling in HD
There are several points of mHTT interference with MSN Ca2+ signaling that are upstream of the SOCE pathway (Figure 2), and their dysregulation may cause elevations of SOCE in HD models, such as IP3R1 (Wu et al., 2016; Czeredys et al., 2018) and S1R (Ryskamp et al., 2017) signaling. Therefore, inhibitors of these pathways may also be promising drug candidates in HD.
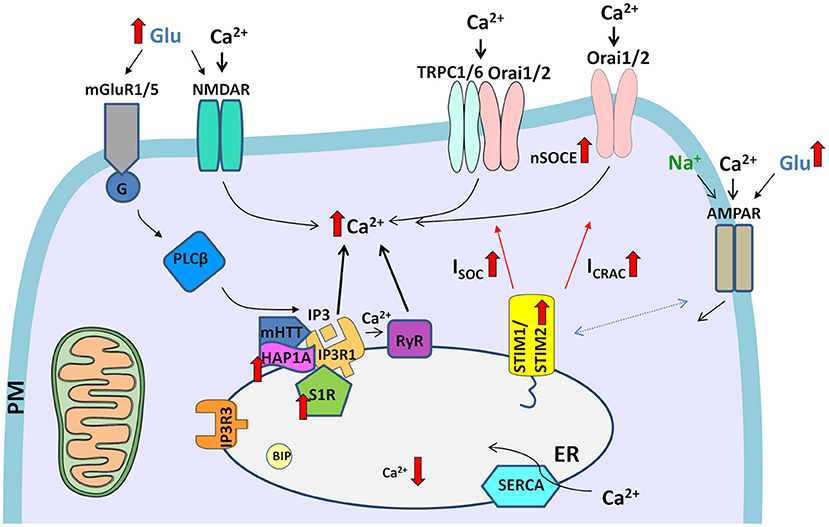
Figure 2. Dysregulation of Ca2+ signaling pathways in HD neurons. The elevation of glutamate (Glu) release from presynaptic neuronal terminals leads to the excessive stimulation of glutamate receptors (e.g., NMDARs and mGluR1/5) in post-synaptic terminals. The overactivation of NMDARs results in excessive Ca2+ influx. In turn, mGluRs couple to G-protein, which activates PLCβ and induces the formation of IP3, which interacts with IP3Rs and causes the release of Ca2+ from ER stores. HAP1A interacts with mHTT and exaggerates the effect of mHTT on IP3R1, which leads to the greater sensitivity of IP3R1 to IP3 and an increase in the release of Ca2+ from the ER, resulting in the attenuation of ER Ca2+ levels. Additionally, Ca2+ release from the ER is exaggerated by RyRs, which are activated by abnormal Ca2+ release via IP3Rs. Activated S1Rs via several processes, such as ER stress, Ca2+ release from the ER, or ligands, dissociate from binding immunoglobulin protein (BIP), thereby disconnecting from IP3R3 in mitochondria-associated membranes. Subsequently, upregulated S1R interacts with IP3R1. Upon the abnormal depletion of ER Ca2+ stores, upregulated STIM2 senses ER Ca2+ content and activates SOC channels in the plasma membrane (PM), which mediate two currents, ISOC and ICRAC. Elevations of neuronal SOCE (nSOCE) contribute to Ca2+ overload. The surface distribution of AMPARs is disturbed in HD neurons. Crosstalk that was observed between AMPARs and STIMs in wildtype neurons might contribute to abnormal Ca2+ influx through AMPARs in HD pathology in the presence of upregulated STIM2. Filled black and red arrows represent interaction mechanisms. Open black arrows represent Ca2+/Na+ flux. Thick red arrows represent an increase or decrease in expression/concentration. Dashed arrows represent suspected interactions.
One such drug is pridopidine, which was postulated to be a “dopamine stabilizer.” It has been shown to improve motor symptoms in clinical trials of HD (Lundin et al., 2010; de Yebenes et al., 2011; Esmaeilzadeh et al., 2011; Kieburtz et al., 2013). In previous studies, pridopidine improved motor functions and prolonged the survival in R6/2 HD mice, and exerted neuroprotective effects in a mouse striatal knock-in (STHdh111/111) model of HD (Squitieri et al., 2015). Further research has shown that the pharmacological activation of S1Rs with pridopidine and the chemically similar S1R agonist R(+)-3-(3-hydroxyphenyl)-N-propylpiperidine (3-PPP) prevents MSN spine loss in aging YAC128 co-cultures through the normalization of IP3R1 hyperactivity, suppression of abnormal ER Ca2+ release, restoration of ER Ca2+ levels, and reduction of excessive nSOCE in spines (Ryskamp et al., 2017). Pridopidine normalized ER Ca2+ levels, but its actions were prevented by S1R deletion. To evaluate the long-term effects of pridopidine, the expression profiles of Ca2+ signaling genes were analyzed. Pridopidine elevated the striatal expression of proteins that regulate Ca2+, such as calbindin and homer1a, whereas their striatal expression decreased in aged Q175KI and YAC128 HD mouse models compared with wildtype mice. Both pridopidine and 3-PPP restored Ca2+ dysregulation and synaptic loss in corticostriatal co-cultures from YAC128 mice (Ryskamp et al., 2017). Moreover, another S1R agonist, PRE-084, exerted neuroprotective effects in PC6.3 cells that expressed N-terminal mHTT (Hyrskyluoto et al., 2013).
An RyR antagonist and the clinically relevant intracellular Ca2+ stabilizer dantrolene was shown to protect cultured YAC128 MSNs against glutamate-induced apoptosis. Feeding dantrolene to YAC128 mice significantly attenuated age-dependent motor deficiency and decreased both the death of NeuN-positive striatal neurons and nuclear aggregation of mHTT (Chen et al., 2011). These results indicate that inhibiting RyR-mediated CICR may be a possible therapeutic approach for the treatment of HD, and dantrolene could be a potent compound that can be applied as an HD medication. An increase in Ca2+ leakage was observed in striatal and cortical neurons from R6/2 HD mice that overexpressed exon-1 mHTT. The application of RyR inhibitors, such as dantrolene, ryanodine, 1,1′-diheptyl-4,4′-bipyridinium dibromide (DHBP), and ruthenium red, suppressed cell death in cortical neurons that overexpressed the N-terminal fragment of mHTT, whereas the addition of the potent RyR activator 4-chloro-m-cresol (CMC) increased mHTT toxicity. In contrast, 2-aminoethoxydiphenyl borate (2-APB), an inhibitor of IP3Rs, failed to protect these neurons (Suzuki et al., 2012). These results suggest that the inhibition of Ca2+ release from RyRs but not IP3Rs alleviates neuronal death that is induced by exon-1 mHTT. Additionally, intracellular Ca2+ imaging revealed that exon-1 mHTT caused excessive basal Ca2+ release (Ca2+ leakage) through RyRs, leading to the depletion of internal Ca2+ stores. Moreover, the expression of FK506-binding protein 12 (FKBP12), which interacts and stabilizes RyR1 by decreasing channel open permeability (Brillantes et al., 1994), suppressed both Ca2+ leakage and cell death. These results provide evidence of the role of RyRs in neuronal cell death and suggest that the stabilization of RyRs might be beneficial for the treatment of HD (Suzuki et al., 2012).
Apart from small-molecule compounds that have been proposed for the treatment of HD, antisense oligonucleotides (ASOs) may also target mHTT in HD patients (Skotte et al., 2014; van Roon-Mom et al., 2018; Barker et al., 2020). Antisense oligonucleotides are short, synthetic, single-stranded oligodeoxynucleotides that can alter mRNA expression through various mechanisms and modify protein expression (Rinaldi and Wood, 2018). In addition to the ability of ASOs to decrease mHTT levels, other strategies to normalize the dysregulation of Ca2+ signaling have been proposed. The knockdown of IP3R1 by ASOs (536178-2) stabilized the supranormal steady-state activity of IP3R1 in YAC128 MSNs in corticostriatal co-cultures (Wu et al., 2016). Abnormal IP3R1 activation is a major cause of elevations of ER Ca2+ leakage that consequently leads to abnormal nSOCE in HD MSNs. Treatment with ASOs against IP3R1 rescued YAC128 MSN spines, increasing their numbers to the same levels as in wildtype cultures (Wu et al., 2016). Therefore, the ASO-mediated suppression of IP3R1 expression was sufficient to prevent spine loss in YAC128 MSNs, presumably through the stabilization of Ca2+ levels in the ER and nSOCE in neuronal spines. Wu et al. proposed that ASOs may be a promising treatment for HD because they can modify IP3R1 levels (Wu et al., 2016).
Moreover, IP3R1 sponge that was delivered using a lentivirus system in vitro into YAC128 MSNs that overexpressed HAP1A restored abnormal SOCE. IP3R1 sponge, through its ability to inhibit IP3R1 activity, attenuated the effects of mHTT and HAP1A on this receptor, which led to the normalization of SOCE in an HD model (Czeredys et al., 2018). Mutant HTT was previously shown to specifically bind to the C-terminal cytosolic region of IP3R1 (a 122-amino acid-long IC10 fragment) (Tang et al., 2003). These findings led Tang et al. to propose a novel therapy for HD, who introduced IC10 peptide into HD MSNs in trans to disrupt the pathogenic association between IP3R1 and mHTT to normalize neuronal Ca2+ signaling and prevent the cell death of HD MSNs. Indeed, infection with lenti-GFP-IC10 virus stabilized Ca2+ signaling in YAC128 MSNs in vitro and protected these neurons from glutamate-induced apoptosis. Intrastriatal injections of AAV1-GFP-IC10 attenuated motor deficits and diminished MSN loss and shrinkage in YAC128 mice in vivo (Tang et al., 2009). These results demonstrated the importance of IP3R1 and mHTT in the pathogenesis of HD and suggested that IC10 peptide may be useful for HD treatment. Additionally, IC10 peptide was shown to reduce mHTT aggregation and nuclear accumulation, which may suggest a novel protective mechanism of IC10 that works jointly with its stabilizing effects on Ca2+ signaling (Tang et al., 2009). Additionally, the IP3R blocker 2-APB (Maruyama et al., 1997) protected YAC128 MSNs from glutamate-induced apoptosis (Tang et al., 2005). Furthermore, low-molecular-weight heparin sulfate Enoxaparin inhibited IP3R1 activity and protected YAC128 MSNs from glutamate-induced apoptosis (Tang et al., 2005).
In summary, potential strategies to stabilize elevations of Ca2+ signaling pathways in the ER might be beneficial for HD patients because they may prevent abnormal SOCE and dendritic spine abnormalities. Possible treatment options that target Ca2+ signaling receptors in the ER, which may restore spine pathology in HD, are summarized in Table 3.
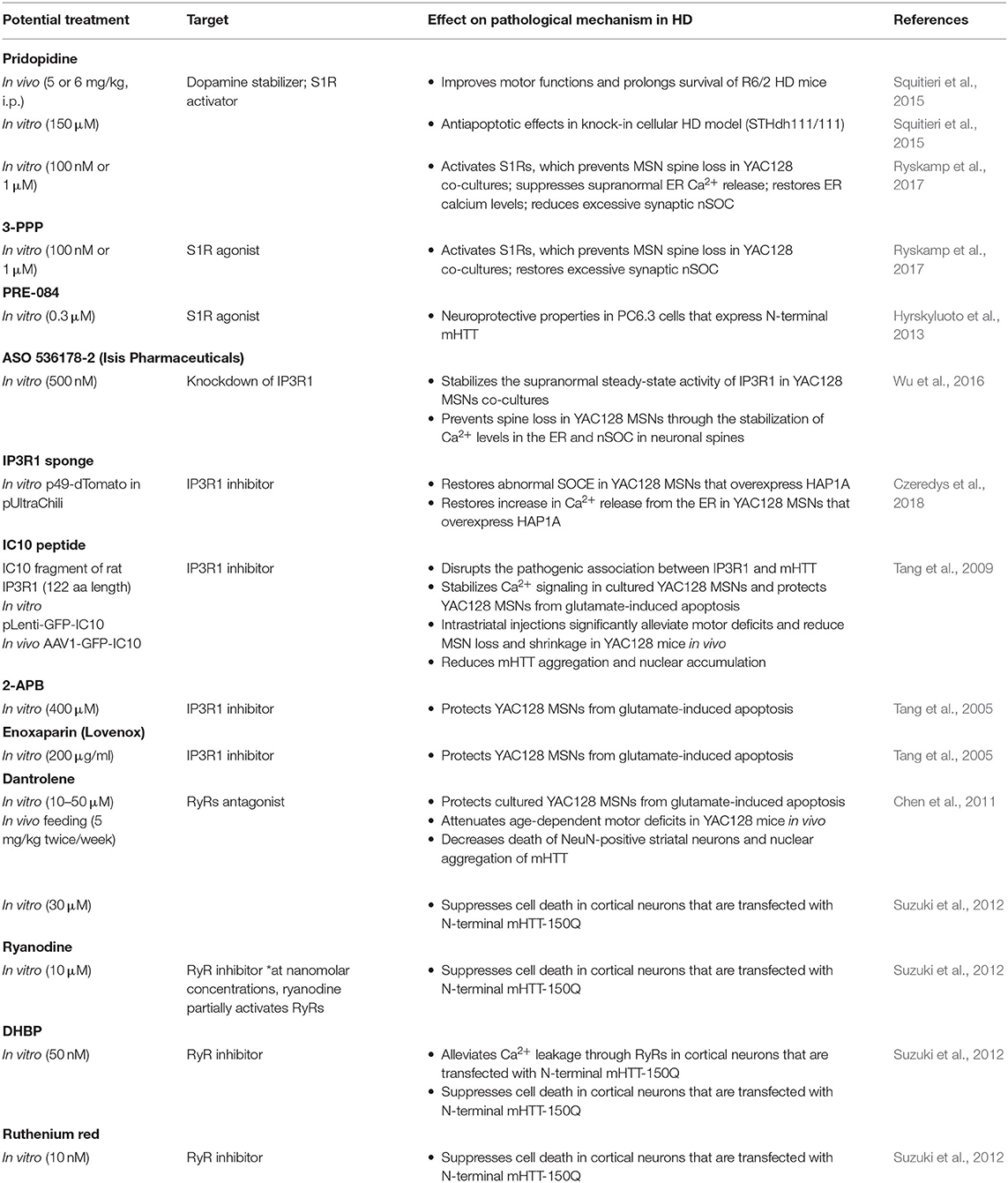
Table 3. Potential strategies to stabilize Ca2+ signaling pathways in the ER that may prevent abnormal SOCE in HD models.
One of the causes of MSN degeneration in HD is dysregulation of the glutamate/Ca2+ signaling pathway. Antagonists of glutamate signaling pathways might have beneficial effects for the treatment of HD, especially because crosstalk between glutamate receptors and neuronal SOCE was recently reported (Serwach and Gruszczynska-Biegala, 2019). The non-competitive NMDAR antagonist memantine (Lipton, 2006) protected YAC128 MSNs from glutamate-induced cell death. Furthermore, another antagonist of glutamate signaling pathways, riluzole, which acts on the inhibition of glutamate release, was also protective against glutamate-induced cell death, although it was less potent than memantine (Wu et al., 2006). Memantine was also examined in a 2-year-long human clinical study of HD. Memantine retarded the progression of HD in patients in motor, functional, and behavioral tests (Beister et al., 2004). Neuroprotective effects of the NMDAR antagonist (+)MK801 and NR2B-specific NMDAR antagonist ifenprodil were also found in a glutamate-induced cell death assay in YAC128 MSNs (Tang et al., 2005). Tang et al. found that mGluR1/5 inhibition by a combination of MPEP and CPCCOEt reduced the glutamate-induced apoptosis of YAC128 MSNs to wildtype MSN levels, and a mixture of mGluR1/5 and NMDAR blockers [MPEP, CPCCOEt, and (+)MK801] abolished glutamate-dependent cell death in both wildtype and YAC128 MSNs (Tang et al., 2005).
Although crosstalk between AMPARs and SOCE has been shown (Serwach and Gruszczynska-Biegala, 2019), an inhibitor of AMPARs, the antidepressant tianeptine ([3-chloro-6-methyl-5,5-dioxo-6,11-dihydro-(c,f)-dibenzo-(1,2-thiazepine)-11-yl)amino]-7 heptanoic acid) restored synaptic deficits in HD models independently from the SOCE process. Tianeptine improved hippocampal synaptic and memory deficits and anxiety/depression-like behavior in HD mice, possibly through the modulation of BDNF signaling and AMPAR surface diffusion (Zhang et al., 2018). The AMPAR-dependent formation of new synapses in cortical neurons from HD mice through BDNF signaling was restored by pridopidine, a drug that enhances BDNF signaling through the stimulation of S1Rs, and the S1R agonist 3-PPP (Smith-Dijak et al., 2019). Potential strategies to stabilize glutamate receptors in the PM that might affect SOCE in HD models are presented in Table 4.
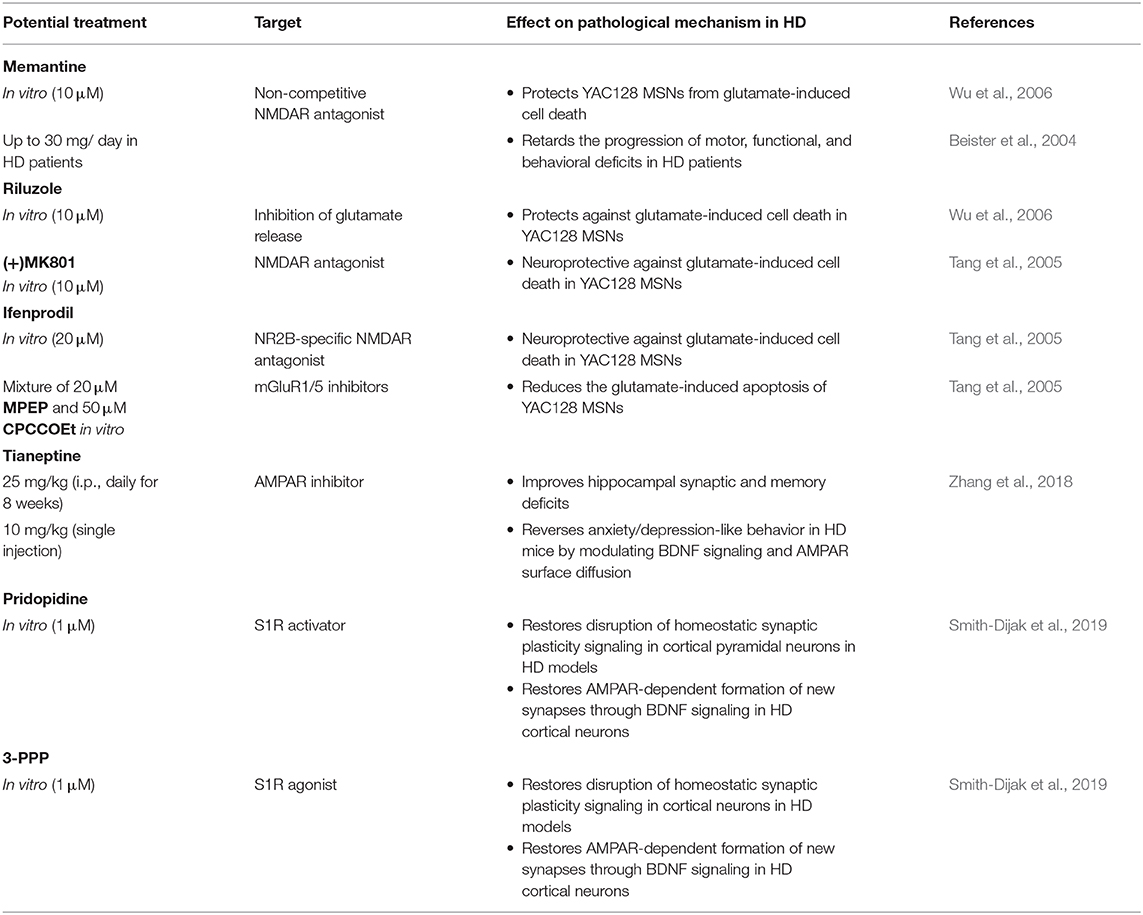
Table 4. Potential strategies to stabilize receptors in the plasma membrane that may affect nSOCE in HD models.
The nSOC pathway plays an important role in supporting the stability of MSN spines under physiological conditions, and this process is disturbed in HD pathology. Both SOCE players and other Ca2+ signaling pathways that regulate nSOCE may contribute to the synaptic dysregulation of MSNs in HD (Figure 3). Substantial evidence suggests that the pharmacological inhibition of nSOCE, its upstream pathways, and other receptors that may affect the SOCE process may be beneficial for synapse maintenance in HD models. Although SOCE inhibition in the striatum might be protective in HD patients, the same compound might be detrimental in other brain regions that contain cortical and hippocampal pyramidal neurons where SOCE is unchanged (Secondo et al., 2018).
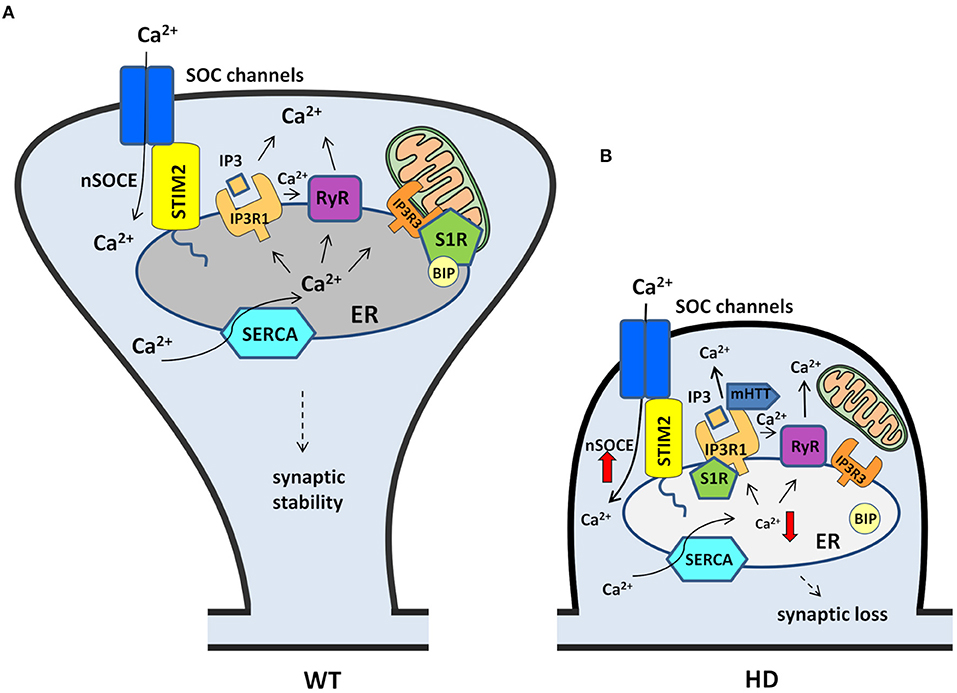
Figure 3. Contribution of nSOCE components and Ca2+ signaling pathways to the dysregulation of MSN spines in HD. (A) In wildtypes, MSN synaptic spine stability is maintained by the nSOCE pathway. nSOCE is gated by STIM2, which regulates ER Ca2+ levels by activating SOC channels in the plasma membrane, and SERCA pumps Ca2+ from the cytosol to the ER to refill ER Ca2+ stores. Ca2+ is released from the ER to the cytosol by IP3R1 and RyRs, and S1R facilitates Ca2+ flux from the ER to mitochondria via IP3R3. (B) In HD MSNs, supranormal synaptic nSOCE causes spine loss. mHTT sensitizes IP3R1 to IP3, causing excessive Ca2+ leakage from the ER. Ca2+ release from the ER is exaggerated by RyRs that are activated by abnormal Ca2+ release via IP3R1. Upon activation, S1Rs redistribute to the entire ER network where they interact with additional targets, including IP3R1, to affect their function. STIM2 upregulation compensates for the abnormal depletion of Ca2+ from the ER and causes the aberrant activation of nSOCE. Finally, supranormal nSOCE activates pathways and effectors that destabilize spines and lead to their loss in HD MSNs. Black arrows represent Ca2+ flux. The thick red arrows represent an increase or decrease in Ca2+ concentration.
Concluding Remarks
The dysregulation of Ca2+ signaling via store-operated Ca2+ channels is involved in the pathogenesis of HD. Abnormal nSOCE leads to synaptic dysregulation in HD MSNs. Several SOCE components have been identified in dendritic spine pathology. Among these, STIM2 and TRPC1 deserve special attention as potential HD treatment targets. Elevations of SOCE in HD models also result from abnormal Ca2+ signaling responses at the ER. The activity of two receptors in the ER, IP3R1, and S1R, is significantly elevated in HD models, which contributes to elevations of nSOCE and consequently MSN dendritic spine pathology. Additionally, the role of HAP1A protein should be emphasized because it can exaggerate the effect of mHTT on IP3R1, which leads to an increase in the release of Ca2+ from the ER and consequently elevations of SOCE in HD MSNs. One function of SOCE in neurons is to refill ER Ca2+ that is released during cell activation to initiate specific signaling pathways. However, SOCE also triggers various other signaling pathways. Evidence suggests crosstalk in neurons between glutamate receptors and SOCE components. Elevations of SOCE in HD might also be affected by glutamate receptors that are abnormally activated, which consequently causes MSN cell death. Under physiological conditions, SOCE components appear to be crucial in several aspects of neuronal development, including the proliferation and early differentiation of NPCs. Additionally, SOCE components play a role in synaptic formation and maturation, and they contribute to synaptic plasticity. Therefore, the dysregulation of SOCE appears to underlie dendritic spine abnormalities that have been identified in HD models. Recent findings that were reviewed herein support the role of molecular components of nSOCE and upstream pathways that regulate these processes in dendritic spine pathology in HD, and these components may be potential targets for HD treatment. Several drug candidates may restore abnormal SOCE (e.g., EVP4593 and tetrahydrocarbazoles) or other disturbances in Ca2+ signaling pathways, including IP3R1, RyR, and S1R. A few known compounds may prevent dendritic spine loss in HD by stabilizing abnormal nSOCE (e.g., EVP4593) and S1R activity (e.g., pridopidine and 3-PPP) or by attenuating supranormal IP3R1 activity (e.g., ASOs). The arguments that nSOC pathways could be novel therapeutic targets for HD treatment are convincing because elevations of SOCE were observed not only in transgenic and cellular HD models but also in iPSC-based GABAergic MSNs that were obtained from adult HD patient fibroblasts. However, such proposed drug candidates or other therapeutic strategies that may act on SOCE components in HD deserve further scrutiny to provide clear evidence that they can successfully and specifically target these proteins to improve or reverse the dysregulation of Ca2+ homeostasis and signaling without causing major side effects.
Author Contributions
MC wrote the review and discussed literature data in this paper.
Funding
This study was supported by the National Science Center in Poland (Grant No. 2019/33/B/NZ3/02889 and 2014/15/D/NZ3/05181 to MC).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author thanks Prof. Jacek Kuznicki and Dr. Tomasz Wegierski for critically reading the manuscript.
Abbreviations
AMPARs, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors; 2-APB, 2-aminoethoxydiphenyl borate; ASOs, antisense oligonucleotides; BDNF, brain-derived neurotrophic factor; BIP, binding immunoglobulin protein; CacyBP/SIP, calcyclin binding protein and Siah-1 interacting protein; CaMKII, Ca2+/calmodulin-dependent protein kinase II; cAMP, cyclic adenosine monophosphate; Cav2.2, N-type voltage-gated calcium channel; Cdc42, cell division cycle 42; C20H22BrClN2, 6-bromo-N-(2-phenylethyl)-2,3,4,9-tetrahydro-1H-carbazol-1-aminehydrochloride; CICR, Ca2+-induced Ca2+ release; CMC, 4-chloro-m-cresol; CNS, central nervous system; CPA, cyclopiazonic acid; CRACs, Ca2+ release-activated Ca2+ channels; CRISPR-Cas9, clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9; DAG, diacylglycerol; DHBP, 1,1′-diheptyl-4,4′-bipyridinium dibromide; DHPG, (S)-3,5-dihydroxyphenylglycine; ER, endoplasmic reticulum; EVP4593, 6-amino-4-(4-phenoxyphenethyl-amino)quinazoline; FKBP12, FK506-binding protein 12; HAP1, huntingtin-associated protein 1; HD, Huntington's disease; hESCs, human embryonic stem cells; HTT, huntingtin; mHTT, mutant huntingtin; ICRAC, Ca2+ release-activated Ca2+ current; IICR, IP3-induced Ca2+ release; IP3, inositol trisphosphate; IP3Rs, inositol-1,4,5-triphosphate receptors; iPSCs, induced pluripotent stem cells; ISOC, store-operated Ca2+ current; LTP, long-term potentiation; MAM, mitochondria-associated membrane; mGluR1/5, metabotropic glutamate receptors1/5; MPTPs, mitochondrial permeability transition pores; MSNs, γ-aminobutyric acid (GABA)ergic medium spiny neurons; NMDARs, N-methyl-D-aspartate receptors; NPCs, neural progenitor cells; nSOCE, neuronal store-operated calcium entry; nSOCs, neuronal store-operated calcium channels; PKA, protein kinase A; PKC, protein kinase C; PI(4,5)P(2), phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; PM, plasma membrane; polyQ, polyglutamine residues; 3-PPP, R(+)-3-(3-hydroxyphenyl)-N-propylpiperidine; PS1, presenilin 1; PSD95, PDZ-domain scaffold protein post-synaptic density 95; Rab4, Ras-related protein Rab4; RyRs, ryanodine receptors; SERCA, sarco-endoplasmic reticulum calcium-adenosine triphosphatase; SOCE, store-operated calcium entry; S1Rs, sigma-1 receptors; STIM, stromal interaction molecule; TrkB, tropomyosin receptor kinase B; TRPCs, transient receptor potential channels; VGCCs, voltage-gated calcium channels.
References
Abou-Lovergne, A., Collado-Hilly, M., Monnet, F. P., Koukoui, O., Prigent, S., Coquil, J. F., et al. (2011). Investigation of the role of sigma1-receptors in inositol 1,4,5-trisphosphate dependent calcium signaling in hepatocytes. Cell Calcium 50, 62–72. doi: 10.1016/j.ceca.2011.05.008
Amador, F. J., Stathopulos, P. B., Enomoto, M., and Ikura, M. (2013). Ryanodine receptor calcium release channels: lessons from structure-function studies. FEBS J. 280, 5456–5470. doi: 10.1111/febs.12194
Ambudkar, I. S., de Souza, L. B., and Ong, H. L. (2017). TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium 63, 33–39. doi: 10.1016/j.ceca.2016.12.009
Ambudkar, I. S., Ong, H. L., Liu, X., Bandyopadhyay, B. C., Bandyopadhyay, B., and Cheng, K. T. (2007). TRPC1: the link between functionally distinct store-operated calcium channels. Cell Calcium 42, 213–223. doi: 10.1016/j.ceca.2007.01.013
Baba, A., Yasui, T., Fujisawa, S., Yamada, R. X., Yamada, M. K., Nishiyama, N., et al. (2003). Activity-evoked capacitative Ca2+ entry: implications in synaptic plasticity. J. Neurosci. 23, 7737–7741. doi: 10.1523/JNEUROSCI.23-21-07737.2003
Baker, K. D., Edwards, T. M., and Rickard, N. S. (2013). The role of intracellular calcium stores in synaptic plasticity and memory consolidation. Neurosci. Biobehav. Rev. 37, 1211–1239. doi: 10.1016/j.neubiorev.2013.04.011
Barker, R. A., Fujimaki, M., Rogers, P., and Rubinsztein, D. C. (2020). Huntingtin-lowering strategies for Huntington's disease. Expert Opin. Investig. Drugs 29, 1125–1132. doi: 10.1080/13543784.2020.1804552
Bates, G. P., Dorsey, R., Gusella, J. F., Hayden, M. R., Kay, C., Leavitt, B. R., et al. (2015). Huntington disease. Nat. Rev. Dis. Primers 1:15005. doi: 10.1038/nrdp.2015.5
Beister, A., Kraus, P., Kuhn, W., Dose, M., Weindl, A., and Gerlach, M. (2004). The N-methyl-D-aspartate antagonist memantine retards progression of Huntington's disease. J. Neural Transm. Suppl. 117–22. doi: 10.1007/978-3-7091-0579-5_14
Berna-Erro, A., Braun, A., Kraft, R., Kleinschnitz, C., Schuhmann, M. K., Stegner, D., et al. (2009). STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal 2:ra67. doi: 10.1126/scisignal.2000522
Berridge, M. J. (1998). Neuronal calcium signaling. Neuron. 21, 13–26. doi: 10.1016/S0896-6273(00)80510-3
Berridge, M. J. (2002). The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32, 235–249. doi: 10.1016/S0143416002001823
Berridge, M. J. (2009). Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta. 1793, 933–940. doi: 10.1016/j.bbamcr.2008.10.005
Bezprozvanny, I. (2005). The inositol 1,4,5-trisphosphate receptors. Cell Calcium 38, 261–272. doi: 10.1016/j.ceca.2005.06.030
Bezprozvanny, I., and Hiesinger, P. R. (2013). The synaptic maintenance problem: membrane recycling, Ca2+ homeostasis and late onset degeneration. Mol. Neurodegener. 8:23. doi: 10.1186/1750-1326-8-23
Bezprozvanny, I., and Kiselev, S. L. (2017). Neurons from skin mimic brain holes. Oncotarget 8, 8997–8998. doi: 10.18632/oncotarget.13709
Bohush, A., and Filipek, A. (2020). HSP90 co-chaperone, CacyBP/SIP, protects α-synuclein from aggregation. Cells 9:2254. doi: 10.3390/cells9102254
Brandman, O., Liou, J., Park, W. S., and Meyer, T. (2007). STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 131, 1327–1339. doi: 10.1016/j.cell.2007.11.039
Brillantes, A. B., Ondrias, K., Scott, A., Kobrinsky, E., Ondriasov,á, E., Moschella, M. C., et al. (1994). Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell 77, 513–523. doi: 10.1016/0092-8674(94)90214-3
Brini, M., Calì, T., Ottolini, D., and Carafoli, E. (2013a). The plasma membrane calcium pump in health and disease. FEBS J. 280, 5385–5397. doi: 10.1111/febs.12193
Brini, M., Calì, T., Ottolini, D., and Carafoli, E. (2013b). Intracellular calcium homeostasis and signaling. Met. Ions Life Sci. 12, 119–168. doi: 10.1007/978-94-007-5561-1_5
Cahalan, M. D. (2009). STIMulating store-operated Ca(2+) entry. Nat. Cell Biol. 11, 669–677. doi: 10.1038/ncb0609-669
Cai, X., Zhou, Y., Nwokonko, R. M., Loktionova, N. A., Wang, X., Xin, P., et al. (2016). The orai1 store-operated calcium channel functions as a Hexamer. J. Biol. Chem. 291, 25764–25775. doi: 10.1074/jbc.M116.758813
Catterall, W. A. (2011). Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 3:a003947. doi: 10.1101/cshperspect.a003947
Caviston, J. P., and Holzbaur, E. L. (2009). Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 19, 147–155. doi: 10.1016/j.tcb.2009.01.005
Chen, N., Luo, T., Wellington, C., Metzler, M., McCutcheon, K., Hayden, M. R., et al. (1999). Subtype-specific enhancement of NMDA receptor currents by mutant huntingtin. J. Neurochem. 72, 1890–1898. doi: 10.1046/j.1471-4159.1999.0721890.x
Chen, S., Yu, C., Rong, L., Li, C. H., Qin, X., Ryu, H., et al. (2018). Altered synaptic vesicle release and Ca2+ influx at single presynaptic terminals of cortical neurons in a knock-in mouse model of Huntington's disease. Front. Mol. Neurosci. 11:478. doi: 10.3389/fnmol.2018.00478
Chen, X., Wu, J., Lvovskaya, S., Herndon, E., Supnet, C., and Bezprozvanny, I. (2011). Dantrolene is neuroprotective in Huntington's disease transgenic mouse model. Mol. Neurodegener. 6:81. doi: 10.1186/1750-1326-6-81
Chen-Engerer, H. J., Hartmann, J., Karl, R. M., Yang, J., Feske, S., and Konnerth, A. (2019). Two types of functionally distinct Ca. Nat. Commun. 10:3223. doi: 10.1038/s41467-019-11207-8
Choo, Y. S., Johnson, G. V., MacDonald, M., Detloff, P. J., and Lesort, M. (2004). Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 13, 1407–1420. doi: 10.1093/hmg/ddh162
Czeredys, M., Gruszczynska-Biegala, J., Schacht, T., Methner, A., and Kuznicki, J. (2013). Expression of genes encoding the calcium signalosome in cellular and transgenic models of Huntington's disease. Front. Mol. Neurosci. 6:42. doi: 10.3389/fnmol.2013.00042
Czeredys, M., Maciag, F., Methner, A., and Kuznicki, J. (2017). Tetrahydrocarbazoles decrease elevated SOCE in medium spiny neurons from transgenic YAC128 mice, a model of Huntington's disease. Biochem. Biophys. Res. Commun. 483, 1194–1205. doi: 10.1016/j.bbrc.2016.08.106
Czeredys, M., Vigont, V. A., Boeva, V. A., Mikoshiba, K., Kaznacheyeva, E. V., and Kuznicki, J. (2018). Huntingtin-associated protein 1A regulates store-operated calcium entry in medium spiny neurons from transgenic YAC128 mice, a model of Huntington's disease. Front. Cell. Neurosci. 12:381. doi: 10.3389/fncel.2018.00381
de Souza, L. B., Ong, H. L., Liu, X., and Ambudkar, I. S. (2015). Fast endocytic recycling determines TRPC1-STIM1 clustering in ER-PM junctions and plasma membrane function of the channel. Biochim. Biophys. Acta 1853, 2709–2721. doi: 10.1016/j.bbamcr.2015.07.019
de Yebenes, J. G., Landwehrmeyer, B., Squitieri, F., Reilmann, R., Rosser, A., Barker, R. A., et al. (2011). Pridopidine for the treatment of motor function in patients with Huntington's disease (MermaiHD): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 10, 1049–1057. doi: 10.1016/S1474-4422(11)70233-2
Deb, B. K., Pathak, T., and Hasan, G. (2016). Store-independent modulation of Ca(2+) entry through Orai by Septin 7. Nat. Commun. 7:11751. doi: 10.1038/ncomms11751
DeHaven, W. I., Smyth, J. T., Boyles, R. R., and Putney, J. W. (2007). Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J. Biol. Chem. 282, 17548–17556. doi: 10.1074/jbc.M611374200
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi: 10.1126/science.277.5334.1990
Dittmer, P. J., Wild, A. R., Dell'Acqua, M. L., and Sather, W. A. (2017). STIM1 Ca2+ sensor control of L-type Ca2+ channel-dependent dendritic spine structural plasticity and nuclear signaling. Cell Rep. 19, 321–334. doi: 10.1016/j.celrep.2017.03.056
Emptage, N. J., Reid, C. A., and Fine, A. (2001). Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron 29, 197–208. doi: 10.1016/S0896-6273(01)00190-8
Esmaeilzadeh, M., Kullingsjö, J., Ullman, H., Varrone, A., and Tedroff, J. (2011). Regional cerebral glucose metabolism after pridopidine (ACR16) treatment in patients with Huntington disease. Clin. Neuropharmacol. 34, 95–100. doi: 10.1097/WNF.0b013e31821c31d8
Fan, M. M., Fernandes, H. B., Zhang, L. Y., Hayden, M. R., and Raymond, L. A. (2007). Altered NMDA receptor trafficking in a yeast artificial chromosome transgenic mouse model of Huntington's disease. J. Neurosci. 27, 3768–3779. doi: 10.1523/JNEUROSCI.4356-06.2007
Feske, S. (2011). Immunodeficiency due to defects in store-operated calcium entry. Ann. N. Y. Acad. Sci. 1238, 74–90. doi: 10.1111/j.1749-6632.2011.06240.x
Feske, S., Giltnane, J., Dolmetsch, R., Staudt, L. M., and Rao, A. (2001). Gene regulation mediated by calcium signals in T lymphocytes. Nat. Immunol. 2, 316–324. doi: 10.1038/86318
Feske, S., Gwack, Y., Prakriya, M., Srikanth, S., Puppel, S. H., Tanasa, B., et al. (2006). A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185. doi: 10.1038/nature04702
Feske, S., Prakriya, M., Rao, A., and Lewis, R. S. (2005). A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J. Exp. Med. 202, 651–662. doi: 10.1084/jem.20050687
Fiedler, M. J., and Nathanson, M. H. (2011). The type I inositol 1,4,5-trisphosphate receptor interacts with protein 4.1N to mediate neurite formation through intracellular Ca waves. Neurosignals 19, 75–85. doi: 10.1159/000324507
Filipek, A., Jastrzebska, B., Nowotny, M., and Kuznicki, J. (2002). CacyBP/SIP, a calcyclin and Siah-1-interacting protein, binds EF-hand proteins of the S100 family. J. Biol. Chem. 277, 28848–28852. doi: 10.1074/jbc.M203602200
Fowler, M. A., Sidiropoulou, K., Ozkan, E. D., Phillips, C. W., and Cooper, D. C. (2007). Corticolimbic expression of TRPC4 and TRPC5 channels in the rodent brain. PLoS ONE 2:e573. doi: 10.1371/journal.pone.0000573
Fowler, M. A., Varnell, A. L., Dietrich, A., Birnbaumer, L., and Cooper, D. C. (2012). Deletion of the trpc1 gene and the effects on locomotor and conditioned place-preference responses to cocaine. Nat. Prec. doi: 10.1038/npre.2012.7153
Galán, C., Dionisio, N., Smani, T., Salido, G. M., and Rosado, J. A. (2011). The cytoskeleton plays a modulatory role in the association between STIM1 and the Ca2+ channel subunits Orai1 and TRPC1. Biochem. Pharmacol. 82, 400–410. doi: 10.1016/j.bcp.2011.05.017
Garcia-Alvarez, G., Lu, B., Yap, K. A., Wong, L. C., Thevathasan, J. V., Lim, L., et al. (2015). STIM2 regulates PKA-dependent phosphorylation and trafficking of AMPARs. Mol. Biol. Cell 26, 1141–1159. doi: 10.1091/mbc.E14-07-1222
Gemes, G., Bangaru, M. L., Wu, H. E., Tang, Q., Weihrauch, D., Koopmeiners, A. S., et al. (2011). Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. J. Neurosci. 31, 3536–3549. doi: 10.1523/JNEUROSCI.5053-10.2011
Giacomello, M., Oliveros, J. C., Naranjo, J. R., and Carafoli, E. (2013). Neuronal Ca(2+) dyshomeostasis in Huntington disease. Prion 7, 76–84. doi: 10.4161/pri.23581
Giurisato, E., Gamberucci, A., Ulivieri, C., Marruganti, S., Rossi, E., Giacomello, E., et al. (2014). The KSR2-calcineurin complex regulates STIM1-ORAI1 dynamics and store-operated calcium entry (SOCE). Mol. Biol. Cell. 25, 1769–1781. doi: 10.1091/mbc.e13-05-0292
Goel, M., Sinkins, W. G., and Schilling, W. P. (2002). Selective association of TRPC channel subunits in rat brain synaptosomes. J. Biol. Chem. 277, 48303–48310. doi: 10.1074/jbc.M207882200
Golovina, V. A., Platoshyn, O., Bailey, C. L., Wang, J., Limsuwan, A., Sweeney, M., et al. (2001). Upregulated TRP and enhanced capacitative Ca(2+) entry in human pulmonary artery myocytes during proliferation. Am. J. Physiol. Heart Circ. Physiol. 280, H746–H755. doi: 10.1152/ajpheart.2001.280.2.H746
Gopurappilly, R., Deb, B. K., Chakraborty, P., and Hasan, G. (2018). Stable STIM1 knockdown in self-renewing human neural precursors promotes premature neural differentiation. Front Mol Neurosci. 11:178. doi: 10.3389/fnmol.2018.00178
Grigoriev, I., Gouveia, S. M., van der Vaart, B., Demmers, J., Smyth, J. T., Honnappa, S., et al. (2008). STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr. Biol. 18, 177–182. doi: 10.1016/j.cub.2007.12.050
Gruszczynska-Biegala, J., and Kuznicki, J. (2013). Native STIM2 and ORAI1 proteins form a calcium-sensitive and thapsigargin-insensitive complex in cortical neurons. J. Neurochem. 126, 727–738. doi: 10.1111/jnc.12320
Gruszczynska-Biegala, J., Pomorski, P., Wisniewska, M. B., and Kuznicki, J. (2011). Differential roles for STIM1 and STIM2 in store-operated calcium entry in rat neurons. PLoS ONE 6:e19285. doi: 10.1371/journal.pone.0019285
Gruszczynska-Biegala, J., Sladowska, M., and Kuznicki, J. (2016). AMPA receptors are involved in store-operated calcium entry and interact with STIM proteins in rat primary cortical neurons. Front. Cell. Neurosci. 10:251. doi: 10.3389/fncel.2016.00251
Gruszczynska-Biegala, J., Strucinska, K., Maciag, F., Majewski, L., Sladowska, M., and Kuznicki, J. (2020). STIM protein-NMDA2 receptor interaction decreases NMDA-dependent calcium levels in cortical neurons. Cells 9:160. doi: 10.3390/cells9010160
Gusella, J. F., and MacDonald, M. E. (2006). Huntington's disease: seeing the pathogenic process through a genetic lens. Trends Biochem. Sci. 31, 533–540. doi: 10.1016/j.tibs.2006.06.009
Hao, B., Lu, Y., Wang, Q., Guo, W., Cheung, K. H., and Yue, J. (2014). Role of STIM1 in survival and neural differentiation of mouse embryonic stem cells independent of Orai1-mediated Ca2+ entry. Stem Cell Res. 12, 452–466. doi: 10.1016/j.scr.2013.12.005
Hartmann, J., Karl, R. M., Alexander, R. P., Adelsberger, H., Brill, M. S., Rühlmann, C., et al. (2014). STIM1 controls neuronal Ca2+ signaling, mGluR1-dependent synaptic transmission, and cerebellar motor behavior. Neuron 82, 635–644. doi: 10.1016/j.neuron.2014.03.027
Hayashi, T., and Su, T. P. (2003). Sigma-1 receptors (sigma(1) binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum: roles in endoplasmic reticulum lipid compartmentalization and export. J. Pharmacol. Exp. Ther. 306, 718–725. doi: 10.1124/jpet.103.051284
Hayashi, T., and Su, T. P. (2007). Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610. doi: 10.1016/j.cell.2007.08.036
Hendershot, L. M. (2004). The ER function BiP is a master regulator of ER function. Mt. Sinai J. Med. 71, 289–297.
Henley, J. M., and Wilkinson, K. A. (2016). Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 17, 337–350. doi: 10.1038/nrn.2016.37
Hodges, A., Strand, A. D., Aragaki, A. K., Kuhn, A., Sengstag, T., Hughes, G., et al. (2006). Regional and cellular gene expression changes in human Huntington's disease brain. Hum. Mol. Genet. 15, 965–977. doi: 10.1093/hmg/ddl013
Hofmann, T., Schaefer, M., Schultz, G., and Gudermann, T. (2002). Subunit composition of mammalian transient receptor potential channels in living cells. Proc. Natl. Acad. Sci. U.S.A. 99, 7461–7466. doi: 10.1073/pnas.102596199
Honarnejad, K., Daschner, A., Gehring, A. P., Szybinska, A., Giese, A., Kuznicki, J., et al. (2014). Identification of tetrahydrocarbazoles as novel multifactorial drug candidates for treatment of Alzheimer's disease. Transl. Psychiatry 4:e489. doi: 10.1038/tp.2014.132
Hoth, M., and Penner, R. (1993). Calcium release-activated calcium current in rat mast cells. J. Physiol. 465, 359–386. doi: 10.1113/jphysiol.1993.sp019681
Hou, X., Pedi, L., Diver, M. M., and Long, S. B. (2012). Crystal structure of the calcium release-activated calcium channel Orai. Science 338, 1308–1313. doi: 10.1126/science.1228757
Huang, J. J., Wang, Y. J., Zhang, M., Zhang, P., Liang, H., Bai, H. J., et al. (2017). Functional expression of the Ca2+ signaling machinery in human embryonic stem cells. Acta Pharmacol. Sin. 38, 1663–1672. doi: 10.1038/aps.2017.29
Hyrskyluoto, A., Pulli, I., Törnqvist, K., Ho, T. H., Korhonen, L., and Lindholm, D. (2013). Sigma-1 receptor agonist PRE084 is protective against mutant huntingtin-induced cell degeneration: involvement of calpastatin and the NF-κB pathway. Cell Death Dis. 4:e646. doi: 10.1038/cddis.2013.170
Itsuki, K., Imai, Y., Hase, H., Okamura, Y., Inoue, R., and Mori, M. X. (2014). PLC-mediated PI(4,5)P2 hydrolysis regulates activation and inactivation of TRPC6/7 channels. J. Gen. Physiol. 143, 183–201. doi: 10.1085/jgp.201311033
Johenning, F. W., Theis, A. K., Pannasch, U., Rückl, M., Rüdiger, S., and Schmitz, D. (2015). Ryanodine receptor activation induces long-term plasticity of spine calcium dynamics. PLoS Biol. 13:e1002181. doi: 10.1371/journal.pbio.1002181
Jurewicz, E., Ostrowska, Z., Jozwiak, J., Redowicz, M. J., Lesniak, W., Moraczewska, J., et al. (2013). CacyBP/SIP as a novel modulator of the thin filament. Biochim. Biophys. Acta 1833, 761–766. doi: 10.1016/j.bbamcr.2012.12.010
Kieburtz, K., McGarry, A., McDermott, M., Kayson, E., Harrison, M., Marder, K., et al. (2013). A randomized, double-blind, placebo-controlled trial of pridopidine in Huntington's disease. Mov. Disord. 28, 1407–1415. doi: 10.1002/mds.25362
Kiselyov, K., Wang, X., Shin, D. M., Zang, W., and Muallem, S. (2006). Calcium signaling complexes in microdomains of polarized secretory cells. Cell Calcium 40, 451–459. doi: 10.1016/j.ceca.2006.08.009
Klejman, M. E., Gruszczynska-Biegala, J., Skibinska-Kijek, A., Wisniewska, M. B., Misztal, K., Blazejczyk, M., et al. (2009). Expression of STIM1 in brain and puncta-like co-localization of STIM1 and ORAI1 upon depletion of Ca(2+) store in neurons. Neurochem. Int. 54, 49–55. doi: 10.1016/j.neuint.2008.10.005
Korkotian, E., Oni-Biton, E., and Segal, M. (2017). The role of the store-operated calcium entry channel Orai1 in cultured rat hippocampal synapse formation and plasticity. J. Physiol. 595, 125–140. doi: 10.1113/JP272645
Kourrich, S., Su, T. P., Fujimoto, M., and Bonci, A. (2012). The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci. 35, 762–771. doi: 10.1016/j.tins.2012.09.007
Kovalenko, M., Milnerwood, A., Giordano, J., St. Claire, J., Guide, J. R., Stromberg, M., et al. (2018). HttQ111/+ Huntington's disease knock-in mice exhibit brain region-specific morphological changes and synaptic dysfunction. J. Huntingtons. Dis. 7, 17–33. doi: 10.3233/JHD-170282
Kraft, R. (2015). STIM and ORAI proteins in the nervous system. Channels 9, 245–252. doi: 10.1080/19336950.2015.1071747
Krobitsch, S., and Lindquist, S. (2000). Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 1589–1594. doi: 10.1073/pnas.97.4.1589
Langbehn, D. R., Brinkman, R. R., Falush, D., Paulsen, J. S., Hayden, M. R., Group, I. H., et al. (2004). A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin. Genet. 65, 267–277. doi: 10.1111/j.1399-0004.2004.00241.x
Lewis, R. S., and Cahalan, M. D. (1995). Potassium and calcium channels in lymphocytes. Annu. Rev. Immunol. 13, 623–653. doi: 10.1146/annurev.iy.13.040195.003203
Lin, A. H., Sun, H., Paudel, O., Lin, M. J., and Sham, J. S. (2016). Conformation of ryanodine receptor-2 gates store-operated calcium entry in rat pulmonary arterial myocytes. Cardiovasc. Res. 111, 94–104. doi: 10.1093/cvr/cvw067
Liou, J., Kim, M. L., Heo, W. D., Jones, J. T., Myers, J. W., Ferrell, J. E., et al. (2005). STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241. doi: 10.1016/j.cub.2005.05.055
Lipton, S. A. (2006). Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat. Rev. Drug Discov. 5, 160–170. doi: 10.1038/nrd1958
Lis, A., Peinelt, C., Beck, A., Parvez, S., Monteilh-Zoller, M., Fleig, A., et al. (2007). CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr. Biol. 17, 794–800. doi: 10.1016/j.cub.2007.03.065
Liu, D. Y., Thilo, F., Scholze, A., Wittstock, A., Zhao, Z. G., Harteneck, C., et al. (2007). Increased store-operated and 1-oleoyl-2-acetyl-sn-glycerol-induced calcium influx in monocytes is mediated by transient receptor potential canonical channels in human essential hypertension. J. Hypertens. 25, 799–808. doi: 10.1097/HJH.0b013e32803cae2b
Liu, Q., Cheng, S., Yang, H., Zhu, L., Pan, Y., Jing, L., et al. (2020). Loss of Hap1 selectively promotes striatal degeneration in Huntington disease mice. Proc. Natl. Acad. Sci. U.S.A. 117, 20265–20273. doi: 10.1073/pnas.2002283117
Liu, X., Bandyopadhyay, B. C., Singh, B. B., Groschner, K., and Ambudkar, I. S. (2005). Molecular analysis of a store-operated and 2-acetyl-sn-glycerol-sensitive non-selective cation channel. Heteromeric assembly of TRPC1-TRPC3. J. Biol. Chem. 280, 21600–21606. doi: 10.1074/jbc.C400492200
Liu, X., Singh, B. B., and Ambudkar, I. S. (2003). TRPC1 is required for functional store-operated Ca2+ channels. Role of acidic amino acid residues in the S5-S6 region. J. Biol. Chem. 278, 11337–11343. doi: 10.1074/jbc.M213271200
Lopez, J. J., Albarran, L., Gómez, L. J., Smani, T., Salido, G. M., and Rosado, J. A. (2016). Molecular modulators of store-operated calcium entry. Biochim. Biophys. Acta 1863, 2037–2043. doi: 10.1016/j.bbamcr.2016.04.024
Lopez, J. J., Jardin, I., Sanchez-Collado, J., Salido, G. M., Smani, T., and Rosado, J. A. (2020). TRPC channels in the SOCE scenario. Cells 9:126. doi: 10.3390/cells9010126
Lundin, A., Dietrichs, E., Haghighi, S., Göller, M. L., Heiberg, A., Loutfi, G., et al. (2010). Efficacy and safety of the dopaminergic stabilizer Pridopidine (ACR16) in patients with Huntington's disease. Clin. Neuropharmacol. 33, 260–264. doi: 10.1097/WNF.0b013e3181ebb285
Ma, B., Savas, J. N., Yu, M. S., Culver, B. P., Chao, M. V., and Tanese, N. (2011). Huntingtin mediates dendritic transport of β-actin mRNA in rat neurons. Sci. Rep. 1:140. doi: 10.1038/srep00140
Ma, G., Wei, M., He, L., Liu, C., Wu, B., Zhang, S. L., et al. (2015). Inside-out Ca(2+) signalling prompted by STIM1 conformational switch. Nat. Commun. 6:7826. doi: 10.1038/ncomms8826
MacDonald, M. E., Ambrose, C. M., Duyao, M. P, Myers, R. H., Lin, C., Srinidhi, L., et al. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 72, 971–983. doi: 10.1016/0092-8674(93)90585-E
Majewski, L., and Kuznicki, J. (2015). SOCE in neurons: signaling or just refilling? Biochim. Biophys. Acta 1853, 1940–1952. doi: 10.1016/j.bbamcr.2015.01.019
Majewski, Ł., Maciag, F., Boguszewski, P. M., Wasilewska, I., Wiera, G., Wójtowicz, T., et al. (2017). Overexpression of STIM1 in neurons in mouse brain improves contextual learning and impairs long-term depression. Biochim Biophys Acta Mol Cell Res. 1864, 1071–1087. doi: 10.1016/j.bbamcr.2016.11.025
Maruyama, T., Kanaji, T., Nakade, S., Kanno, T., and Mikoshiba, K. (1997). 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J. Biochem. 122, 498–505. doi: 10.1093/oxfordjournals.jbchem.a021780
Maurice, T., Grégoire, C., and Espallergues, J. (2006). Neuro(active)steroids actions at the neuromodulatory sigma1 (sigma1) receptor: biochemical and physiological evidences, consequences in neuroprotection. Pharmacol. Biochem. Behav. 84, 581–597. doi: 10.1016/j.pbb.2006.07.009
McCarron, J. G., Chalmers, S., Bradley, K. N., MacMillan, D., and Muir, T. C. (2006). Ca2+ microdomains in smooth muscle. Cell Calcium 40, 461–493. doi: 10.1016/j.ceca.2006.08.010
Miederer, A. M., Alansary, D., Schwär, G., Lee, P. H., Jung, M., Helms, V., et al. (2015). A STIM2 splice variant negatively regulates store-operated calcium entry. Nat. Commun. 6:6899. doi: 10.1038/ncomms7899
Mikoshiba, K. (2007). IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J. Neurochem. 102, 1426–1446. doi: 10.1111/j.1471-4159.2007.04825.x
Miller, B. R., and Bezprozvanny, I. (2010). Corticostriatal circuit dysfunction in Huntington's disease: intersection of glutamate, dopamine and calcium. Future Neurol. 5, 735–756. doi: 10.2217/fnl.10.41
Milnerwood, A. J., and Raymond, L. A. (2007). Corticostriatal synaptic function in mouse models of Huntington's disease: early effects of huntingtin repeat length and protein load. J. Physiol. 585, 817–831. doi: 10.1113/jphysiol.2007.142448
Milnerwood, A. J., and Raymond, L. A. (2010). Early synaptic pathophysiology in neurodegeneration: insights from Huntington's disease. Trends Neurosci. 33, 513–523. doi: 10.1016/j.tins.2010.08.002
Miyazaki, S., Shirakawa, H., Nakada, K., and Honda, Y. (1993). Essential role of the inositol 1,4,5-trisphosphate receptor/Ca2+ release channel in Ca2+ waves and Ca2+ oscillations at fertilization of mammalian eggs. Dev. Biol. 158, 62–78. doi: 10.1006/dbio.1993.1168
Moccia, F., Zuccolo, E., Soda, T., Tanzi, F., Guerra, G., Mapelli, L., et al. (2015). Stim and Orai proteins in neuronal Ca(2+) signaling and excitability. Front. Cell. Neurosci. 9:153. doi: 10.3389/fncel.2015.00153
Murmu, R. P., Li, W., Holtmaat, A., and Li, J. Y. (2013). Dendritic spine instability leads to progressive neocortical spine loss in a mouse model of Huntington's disease. J. Neurosci. 33, 12997–13009. doi: 10.1523/JNEUROSCI.5284-12.2013
Myers, R. H., Vonsattel, J. P., Stevens, T. J., Cupples, L. A., Richardson, E. P., Martin, J. B., et al. (1988). Clinical and neuropathologic assessment of severity in Huntington's disease. Neurology 38, 341–347. doi: 10.1212/WNL.38.3.341
Nekrasov, E. D., Vigont, V. A., Klyushnikov, S. A., Lebedeva, O. S., Vassina, E. M., Bogomazova, A. N., et al. (2016). Manifestation of Huntington's disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 11:27. doi: 10.1186/s13024-016-0092-5
Oliveira, J. M., Jekabsons, M. B., Chen, S., Lin, A., Rego, A. C., Gonçalves, J., et al. (2007). Mitochondrial dysfunction in Huntington's disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. J. Neurochem. 101, 241–249. doi: 10.1111/j.1471-4159.2006.04361.x
Orth, M., Schippling, S., Schneider, S. A., Bhatia, K. P., Talelli, P., Tabrizi, S. J., et al. (2010). Abnormal motor cortex plasticity in premanifest and very early manifest Huntington disease. J. Neurol. Neurosurg. Psychiatr. 81, 267–270. doi: 10.1136/jnnp.2009.171926
Panov, A. V., Gutekunst, C. A., Leavitt, B. R., Hayden, M. R., Burke, J. R., Strittmatter, W. J., et al. (2002). Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat. Neurosci. 5, 731–736. doi: 10.1038/nn884
Paoletti, P., Bellone, C., and Zhou, Q. (2013). NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400. doi: 10.1038/nrn3504
Parekh, A. B., and Penner, R. (1997). Store depletion and calcium influx. Physiol. Rev. 77, 901–930. doi: 10.1152/physrev.1997.77.4.901
Parekh, A. B., and Putney, J. W. (2005). Store-operated calcium channels. Physiol. Rev. 85, 757–810. doi: 10.1152/physrev.00057.2003
Park, C. Y., Shcheglovitov, A., and Dolmetsch, R. (2010). The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 330, 101–105. doi: 10.1126/science.1191027
Parvez, S., Beck, A., Peinelt, C., Soboloff, J., Lis, A., Monteilh-Zoller, M., et al. (2008). STIM2 protein mediates distinct store-dependent and store-independent modes of CRAC channel activation. FASEB J. 22, 752–761. doi: 10.1096/fj.07-9449com
Pchitskaya, E., Popugaeva, E., and Bezprozvanny, I. (2018). Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 70, 87–94. doi: 10.1016/j.ceca.2017.06.008
Peinelt, C., Vig, M., Koomoa, D. L., Beck, A., Nadler, M. J., Koblan-Huberson, M., et al. (2006). Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat. Cell Biol. 8, 771–773. doi: 10.1038/ncb1435
Periasamy, M., and Kalyanasundaram, A. (2007). SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve 35, 430–442. doi: 10.1002/mus.20745
Potier, M., and Trebak, M. (2008). New developments in the signaling mechanisms of the store-operated calcium entry pathway. Pflugers Arch. 457, 405–415. doi: 10.1007/s00424-008-0533-2
Prakriya, M., Feske, S., Gwack, Y., Srikanth, S., Rao, A., and Hogan, P. G. (2006). Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233. doi: 10.1038/nature05122
Prakriya, M., and Lewis, R. S. (2015). Store-operated calcium channels. Physiol. Rev. 95, 1383–1436. doi: 10.1152/physrev.00020.2014
Putney, J. W. (1986). A model for receptor-regulated calcium entry. Cell Calcium 7, 1–12. doi: 10.1016/0143-4160(86)90026-6
Putney, J. W., Steinckwich-Besançon, N., Numaga-Tomita, T., Davis, F. M., Desai, P. N., D'Agostin, D. M., et al. (2017). The functions of store-operated calcium channels. Biochim. Biophys. Acta Mol. Cell Res. 1864, 900–906. doi: 10.1016/j.bbamcr.2016.11.028
Putney, J. W., and Tomita, T. (2012). Phospholipase C signaling and calcium influx. Adv. Biol. Regul. 52, 152–164. doi: 10.1016/j.advenzreg.2011.09.005
Quigley, J. (2017). Juvenile Huntington's disease: diagnostic and treatment considerations for the psychiatrist. Curr. Psychiatry Rep. 19:9. doi: 10.1007/s11920-017-0759-9
Raymond, L. A. (2017). Striatal synaptic dysfunction and altered calcium regulation in Huntington disease. Biochem. Biophys. Res. Commun. 483, 1051–1062. doi: 10.1016/j.bbrc.2016.07.058
Rinaldi, C., and Wood, M. J. A. (2018). Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 14, 9–21. doi: 10.1038/nrneurol.2017.148
Roos, J., DiGregorio, P. J., Yeromin, A. V., Ohlsen, K., Lioudyno, M., Zhang, S., et al. (2005). STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 169, 435–445. doi: 10.1083/jcb.200502019
Ross, C. A. (2002). Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron 35, 819–822. doi: 10.1016/S0896-6273(02)00872-3
Rossi, D., and Sorrentino, V. (2002). Molecular genetics of ryanodine receptors Ca2+-release channels. Cell Calcium 32, 307–319. doi: 10.1016/S0143416002001987
Ryskamp, D., Wu, J., Geva, M., Kusko, R., Grossman, I., Hayden, M., et al. (2017). The sigma-1 receptor mediates the beneficial effects of pridopidine in a mouse model of Huntington disease. Neurobiol. Dis. 97, 46–59. doi: 10.1016/j.nbd.2016.10.006
Ryskamp, D., Wu, L., Wu, J., Kim, D., Rammes, G., Geva, M., et al. (2019). Pridopidine stabilizes mushroom spines in mouse models of Alzheimer's disease by acting on the sigma-1 receptor. Neurobiol. Dis. 124, 489–504. doi: 10.1016/j.nbd.2018.12.022
Ryskamp, D. A., Korban, S., Zhemkov, V., Kraskovskaya, N., and Bezprozvanny, I. (2019). Neuronal Sigma-1 receptors: signaling functions and protective roles in neurodegenerative diseases. Front. Neurosci. 13:862. doi: 10.3389/fnins.2019.00862
Samtleben, S., Wachter, B., and Blum, R. (2015). Store-operated calcium entry compensates fast ER calcium loss in resting hippocampal neurons. Cell Calcium 58, 147–159. doi: 10.1016/j.ceca.2015.04.002
Santulli, G., and Marks, A. R. (2015). Essential roles of intracellular calcium release channels in muscle, brain, metabolism, and aging. Curr. Mol. Pharmacol. 8, 206–222. doi: 10.2174/1874467208666150507105105
Secondo, A., Bagetta, G., and Amantea, D. (2018). On the role of store-operated calcium entry in acute and chronic neurodegenerative diseases. Front. Mol. Neurosci. 11:87. doi: 10.3389/fnmol.2018.00087
Serwach, K., and Gruszczynska-Biegala, J. (2019). STIM proteins and glutamate receptors in neurons: role in neuronal physiology and neurodegenerative diseases. Int. J. Mol. Sci. 20:2289. doi: 10.3390/ijms20092289
Sharma, S., Quintana, A., Findlay, G. M., Mettlen, M., Baust, B., Jain, M., et al. (2013). An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature 499, 238–242. doi: 10.1038/nature12229
Shim, A. H., Tirado-Lee, L., and Prakriya, M. (2015). Structural and functional mechanisms of CRAC channel regulation. J. Mol. Biol. 427, 77–93. doi: 10.1016/j.jmb.2014.09.021
Silva, F. R., Miranda, A. S., Santos, R. P. M., Olmo, I. G., Zamponi, G. W., Dobransky, T., et al. (2017). N-type Ca2+ channels are affected by full-length mutant huntingtin expression in a mouse model of Huntington's disease. Neurobiol. Aging 55, 1–10. doi: 10.1016/j.neurobiolaging.2017.03.015
Skibinska-Kijek, A., Wisniewska, M. B., Gruszczynska-Biegala, J., Methner, A., and Kuznicki, J. (2009). Immunolocalization of STIM1 in the mouse brain. Acta Neurobiol. Exp. 69, 413–428.
Skotte, N. H., Southwell, A. L., Østergaard, M. E., Carroll, J. B., Warby, S. C., Doty, C. N., et al. (2014). Allele-specific suppression of mutant huntingtin using antisense oligonucleotides: providing a therapeutic option for all Huntington disease patients. PLoS ONE 9:e107434. doi: 10.1371/journal.pone.0107434
Smith-Dijak, A. I., Nassrallah, W. B., Zhang, L. Y. J., Geva, M., Hayden, M. R., and Raymond, L. A. (2019). Impairment and restoration of homeostatic plasticity in cultured cortical neurons from a mouse model of Huntington disease. Front. Cell. Neurosci. 13, 209. doi: 10.3389/fncel.2019.00209
Smyth, J. T., DeHaven, W. I., Bird, G. S., and Putney, J. W. (2007). Role of the microtubule cytoskeleton in the function of the store-operated Ca2+ channel activator STIM1. J. Cell Sci. 120, 3762–3771. doi: 10.1242/jcs.015735
Somasundaram, A., Shum, A. K., McBride, H. J., Kessler, J. A., Feske, S., Miller, R. J., et al. (2014). Store-operated CRAC channels regulate gene expression and proliferation in neural progenitor cells. J Neurosci. 34, 9107–9123. doi: 10.1523/JNEUROSCI.0263-14.2014
Squitieri, F., Di Pardo, A., Favellato, M., Amico, E., Maglione, V., and Frati, L. (2015). Pridopidine, a dopamine stabilizer, improves motor performance and shows neuroprotective effects in Huntington disease R6/2 mouse model. J. Cell. Mol. Med. 19, 2540–2548. doi: 10.1111/jcmm.12604
Stathopulos, P. B., Zheng, L., and Ikura, M. (2009). Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J. Biol. Chem. 284, 728–732. doi: 10.1074/jbc.C800178200
Streb, H., Irvine, R. F., Berridge, M. J., and Schulz, I. (1983). Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature. 306, 67–69. doi: 10.1038/306067a0
Strübing, C., Krapivinsky, G., Krapivinsky, L., and Clapham, D. E. (2003). Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J. Biol. Chem. 278, 39014–39019. doi: 10.1074/jbc.M306705200
Su, T. P., Hayashi, T., Maurice, T., Buch, S., and Ruoho, A. E. (2010). The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol. Sci. 31, 557–566. doi: 10.1016/j.tips.2010.08.007
Subedi, K. P., Ong, H. L., Son, G. Y., Liu, X., and Ambudkar, I. S. (2018). STIM2 induces activated conformation of STIM1 to control orai1 function in ER-PM junctions. Cell Rep. 23, 522–534. doi: 10.1016/j.celrep.2018.03.065
Sun, S., Zhang, H., Liu, J., Popugaeva, E., Xu, N. J., Feske, S., et al. (2014). Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 82, 79–93. doi: 10.1016/j.neuron.2014.02.019
Sun, Y., Savanenin, A., Reddy, P. H., and Liu, Y. F. (2001). Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-D-aspartate receptors via post-synaptic density 95. J. Biol. Chem. 276, 24713–24718. doi: 10.1074/jbc.M103501200
Sundivakkam, P. C., Freichel, M., Singh, V., Yuan, J. P., Vogel, S. M., Flockerzi, V., et al. (2012). The Ca(2+) sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca(2+) entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol. Pharmacol. 81, 510–526. doi: 10.1124/mol.111.074658
Supnet, C., and Bezprozvanny, I. (2011). Presenilins function in ER calcium leak and Alzheimer's disease pathogenesis. Cell Calcium 50, 303–309. doi: 10.1016/j.ceca.2011.05.013
Suzuki, M., Nagai, Y., Wada, K., and Koike, T. (2012). Calcium leak through ryanodine receptor is involved in neuronal death induced by mutant huntingtin. Biochem. Biophys. Res. Commun. 429, 18–23. doi: 10.1016/j.bbrc.2012.10.107
Switońska, K., Szlachcic, W. J., Handschuh, L., Wojciechowski, P., Marczak, Ł., Stelmaszczuk, M., et al. (2018). Identification of altered developmental pathways in human juvenile HD iPSC with 71Q and 109Q using transcriptome profiling. Front. Cell. Neurosci. 12:528. doi: 10.3389/fncel.2018.00528
Takebayashi, M., Hayashi, T., and Su, T. P. (2004). A perspective on the new mechanism of antidepressants: neuritogenesis through sigma-1 receptors. Pharmacopsychiatry 37 (Suppl. 3), S208–S213. doi: 10.1055/s-2004-832679
Tang, T. S., Guo, C., Wang, H., Chen, X., and Bezprozvanny, I. (2009). Neuroprotective effects of inositol 1,4,5-trisphosphate receptor C-terminal fragment in a Huntington's disease mouse model. J. Neurosci. 29, 1257–1266. doi: 10.1523/JNEUROSCI.4411-08.2009
Tang, T. S., Slow, E., Lupu, V., Stavrovskaya, I. G., Sugimori, M., Llinás, R., et al. (2005). Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proc. Natl. Acad. Sci. U.S.A. 102, 2602–2607. doi: 10.1073/pnas.0409402102
Tang, T. S., Tu, H., Chan, E. Y., Maximov, A., Wang, Z., Wellington, C. L., et al. (2003). Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39, 227–239. doi: 10.1016/S0896-6273(03)00366-0
Tang, T. S., Tu, H., Orban, P. C., Chan, E. Y., Hayden, M. R., and Bezprozvanny, I. (2004). HAP1 facilitates effects of mutant huntingtin on inositol 1,4,5-trisphosphate-induced Ca release in primary culture of striatal medium spiny neurons. Eur. J. Neurosci. 20, 1779–1787. doi: 10.1111/j.1460-9568.2004.03633.x
Targos, B., Barańska, J., and Pomorski, P. (2005). Store-operated calcium entry in physiology and pathology of mammalian cells. Acta Biochim. Pol. 52, 397–409. doi: 10.18388/abp.2005_3452
Thakur, P., Dadsetan, S., and Fomina, A. F. (2012). Bidirectional coupling between ryanodine receptors and Ca2+ release-activated Ca2+ (CRAC) channel machinery sustains store-operated Ca2+ entry in human T lymphocytes. J. Biol. Chem. 287, 37233–37244. doi: 10.1074/jbc.M112.398974
Trebak, M., Lemonnier, L., DeHaven, W. I., Wedel, B. J., Bird, G. S., and Putney, J. W. (2009). Complex functions of phosphatidylinositol 4,5-bisphosphate in regulation of TRPC5 cation channels. Pflugers Arch. 457, 757–769. doi: 10.1007/s00424-008-0550-1
Trepakova, E. S., Gericke, M., Hirakawa, Y., Weisbrod, R. M., Cohen, R. A., and Bolotina, V. M. (2001). Properties of a native cation channel activated by Ca2+ store depletion in vascular smooth muscle cells. J. Biol. Chem. 276, 7782–7790. doi: 10.1074/jbc.M010104200
Tsai, S. Y., Hayashi, T., Harvey, B. K., Wang, Y., Wu, W. W., Shen, R. F., et al. (2009). Sigma-1 receptors regulate hippocampal dendritic spine formation via a free radical-sensitive mechanism involving Rac1xGTP pathway. Proc. Natl. Acad. Sci. U.S.A. 106, 22468–22473. doi: 10.1073/pnas.0909089106
Tshuva, R. Y., Korkotian, E., and Segal, M. (2017). ORAI1-dependent synaptic plasticity in rat hippocampal neurons. Neurobiol. Learn. Mem. 140, 1–10. doi: 10.1016/j.nlm.2016.12.024
Tu, H., Nelson, O., Bezprozvanny, A., Wang, Z., Lee, S. F., Hao, Y. H., et al. (2006). Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell 126, 981–993. doi: 10.1016/j.cell.2006.06.059
Uchiyama, T., Yoshikawa, F., Hishida, A., Furuichi, T., and Mikoshiba, K. (2002). A novel recombinant hyperaffinity inositol 1,4,5-trisphosphate (IP(3)) absorbent traps IP(3), resulting in specific inhibition of IP(3)-mediated calcium signaling. J. Biol. Chem. 277, 8106–8113. doi: 10.1074/jbc.M108337200
Vaca, L. (2010). SOCIC: the store-operated calcium influx complex. Cell Calcium 47, 199–209. doi: 10.1016/j.ceca.2010.01.002
Vaeth, M., Yang, J., Yamashita, M., Zee, I., Eckstein, M., Knosp, C., et al. (2017). ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat. Commun. 8:14714. doi: 10.1038/ncomms14714
Vajente, N., Norante, R., Redolfi, N., Daga, A., Pizzo, P., and Pendin, D. (2019). Microtubules stabilization by mutant spastin affects ER morphology and Ca. Front. Physiol. 10:1544. doi: 10.3389/fphys.2019.01544
van Coppenolle, F., vanden Abeele, F., Slomianny, C., Flourakis, M., Hesketh, J., Dewailly, E., et al. (2004). Ribosome-translocon complex mediates calcium leakage from endoplasmic reticulum stores. J. Cell Sci. 117, 4135–4142. doi: 10.1242/jcs.01274
van Roon-Mom, W. M. C., Roos, R. A. C., and de Bot, S. T. (2018). Dose-dependent lowering of mutant huntingtin using antisense oligonucleotides in huntington disease patients. Nucleic Acid Ther. 28, 59–62. doi: 10.1089/nat.2018.0720
Vashisht, A., Tanwar, J., and Motiani, R. K. (2018). Regulation of proto-oncogene Orai3 by miR18a/b and miR34a. Cell Calcium 75, 101–111. doi: 10.1016/j.ceca.2018.08.006
Venkatachalam, K., and Montell, C. (2007). TRP channels. Annu. Rev. Biochem. 76, 387–417. doi: 10.1146/annurev.biochem.75.103004.142819
Verkhratsky, A. (2005). Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol. Rev. 85, 201–279. doi: 10.1152/physrev.00004.2004
Vig, M., Peinelt, C., Beck, A., Koomoa, D. L., Rabah, D., Koblan-Huberson, M., et al. (2006). CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 312, 1220–1223. doi: 10.1126/science.1127883
Vigont, V., Kolobkova, Y., Skopin, A., Zimina, O., Zenin, V., Glushankova, L., et al. (2015). Both orai1 and TRPC1 are involved in excessive store-operated calcium entry in striatal neurons expressing mutant huntingtin exon 1. Front. Physiol. 6:337. doi: 10.3389/fphys.2015.00337
Vigont, V., Nekrasov, E., Shalygin, A., Gusev, K., Klushnikov, S., Illarioshkin, S., et al. (2018). Patient-specific iPSC-based models of Huntington's disease as a tool to study store-operated calcium entry drug targeting. Front. Pharmacol. 9:696. doi: 10.3389/fphar.2018.00696
Vigont, V. A., Zimina, O. A., Glushankova, L. N., Kolobkova, J. A., Ryazantseva, M. A., Mozhayeva, G. N., et al. (2014). STIM1 protein activates store-operated calcium channels in cellular model of Huntington's disease. Acta Nat. 6, 40–47. doi: 10.32607/20758251-2014-6-4-40-47
Vonsattel, J. P., and DiFiglia, M. (1998). Huntington disease. J. Neuropathol. Exp. Neurol. 57, 369–384. doi: 10.1097/00005072-199805000-00001
Wang, T., Wang, J., Mao, L., Tang, B., Vanderklish, P. W., Liao, X., et al. (2019). HAP1 is an in vivo UBE3A target that augments autophagy in a mouse model of Angelman syndrome. Neurobiol. Dis. 132, 104585. doi: 10.1016/j.nbd.2019.104585
Wang, Y., Deng, X., Mancarella, S., Hendron, E., Eguchi, S., Soboloff, J., et al. (2010). The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 330, 105–109. doi: 10.1126/science.1191086
Wasik, U., Schneider, G., Mietelska-Porowska, A., Mazurkiewicz, M., Fabczak, H., Weis, S., et al. (2013). Calcyclin binding protein and Siah-1 interacting protein in Alzheimer's disease pathology: neuronal localization and possible function. Neurobiol. Aging. 34, 1380–1388. doi: 10.1016/j.neurobiolaging.2012.11.007
Wegierski, T., and Kuznicki, J. (2018). Neuronal calcium signaling via store-operated channels in health and disease. Cell Calcium 74, 102–111. doi: 10.1016/j.ceca.2018.07.001
Wiatr, K., Szlachcic, W. J., Trzeciak, M., Figlerowicz, M., and Figiel, M. (2018). Huntington disease as a neurodevelopmental disorder and early signs of the disease in stem cells. Mol. Neurobiol. 55, 3351–3371. doi: 10.1007/s12035-017-0477-7
Wu, J., Ryskamp, D., Birnbaumer, L., and Bezprozvanny, I. (2018). Inhibition of TRPC1-dependent store-operated calcium entry improves synaptic stability and motor performance in a mouse model of Huntington's disease. J. Huntingtons. Dis. 7, 35–50. doi: 10.3233/JHD-170266
Wu, J., Ryskamp, D. A., Liang, X., Egorova, P., Zakharova, O., Hung, G., et al. (2016). Enhanced store-operated calcium entry leads to striatal synaptic loss in a Huntington's disease mouse model. J. Neurosci. 36, 125–141. doi: 10.1523/JNEUROSCI.1038-15.2016
Wu, J., Shih, H. P., Vigont, V., Hrdlicka, L., Diggins, L., Singh, C., et al. (2011). Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington's disease treatment. Chem. Biol. 18, 777–793. doi: 10.1016/j.chembiol.2011.04.012
Wu, J., Tang, T., and Bezprozvanny, I. (2006). Evaluation of clinically relevant glutamate pathway inhibitors in in vitro model of Huntington's disease. Neurosci. Lett. 407, 219–223. doi: 10.1016/j.neulet.2006.08.036
Wu, L. L., and Zhou, X. F. (2009). Huntingtin associated protein 1 and its functions. Cell Adh. Migr. 3, 71–76. doi: 10.4161/cam.3.1.7511
Wu, X., Zagranichnaya, T. K., Gurda, G. T., Eves, E. M., and Villereal, M. L. (2004). A TRPC1/TRPC3-mediated increase in store-operated calcium entry is required for differentiation of H19-7 hippocampal neuronal cells. J. Biol. Chem. 279, 43392–43402. doi: 10.1074/jbc.M408959200
Wu, Z., and Bowen, W. D. (2008). Role of sigma-1 receptor C-terminal segment in inositol 1,4,5-trisphosphate receptor activation: constitutive enhancement of calcium signaling in MCF-7 tumor cells. J. Biol. Chem. 283, 28198–28215. doi: 10.1074/jbc.M802099200
Xiang, J., Yang, S., Xin, N., Gaertig, M. A., Reeves, R. H., Li, S., et al. (2017). DYRK1A regulates Hap1-Dcaf7/WDR68 binding with implication for delayed growth in Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 114, E1224–E1233. doi: 10.1073/pnas.1614893114
Yap, K. A., Shetty, M. S., Garcia-Alvarez, G., Lu, B., Alagappan, D., Oh-Hora, M., et al. (2017). STIM2 regulates AMPA receptor trafficking and plasticity at hippocampal synapses. Neurobiol. Learn. Mem. 138, 54–61. doi: 10.1016/j.nlm.2016.08.007
Yen, M., Lokteva, L. A., and Lewis, R. S. (2016). Functional analysis of orai1 concatemers supports a hexameric stoichiometry for the CRAC channel. Biophys. J. 111, 1897–1907. doi: 10.1016/j.bpj.2016.09.020
Zagranichnaya, T. K., Wu, X., and Villereal, M. L. (2005). Endogenous TRPC1, TRPC3, and TRPC7 proteins combine to form native store-operated channels in HEK-293 cells. J. Biol. Chem. 280, 29559–29569. doi: 10.1074/jbc.M505842200
Zeng, B., Chen, G. L., Daskoulidou, N., and Xu, S. Z. (2014). The ryanodine receptor agonist 4-chloro-3-ethylphenol blocks ORAI store-operated channels. Br. J. Pharmacol. 171, 1250–1259. doi: 10.1111/bph.12528
Zeron, M. M., Fernandes, H. B., Krebs, C., Shehadeh, J., Wellington, C. L., Leavitt, B. R., et al. (2004). Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington's disease. Mol. Cell. Neurosci. 25, 469–479. doi: 10.1016/j.mcn.2003.11.014
Zeron, M. M., Hansson, O., Chen, N., Wellington, C. L., Leavitt, B. R., Brundin, P., et al. (2002). Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron 33, 849–860. doi: 10.1016/S0896-6273(02)00615-3
Zhang, H., Li, Q., Graham, R. K., Slow, E., Hayden, M. R., and Bezprozvanny, I. (2008). Full length mutant huntingtin is required for altered Ca2+ signaling and apoptosis of striatal neurons in the YAC mouse model of Huntington's disease. Neurobiol. Dis. 31, 80–88. doi: 10.1016/j.nbd.2008.03.010
Zhang, H., Zhang, C., Vincent, J., Zala, D., Benstaali, C., Sainlos, M., et al. (2018). Modulation of AMPA receptor surface diffusion restores hippocampal plasticity and memory in Huntington's disease models. Nat. Commun. 9:4272. doi: 10.1038/s41467-018-06675-3
Zhang, S. L., Yu, Y., Roos, J., Kozak, J. A., Deerinck, T. J., Ellisman, M. H., et al. (2005). STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905. doi: 10.1038/nature04147
Zoghbi, H. Y., and Orr, H. T. (2000). Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 23, 217–247. doi: 10.1146/annurev.neuro.23.1.217
Keywords: Huntington disease, huntingtin, Ca2+ signaling, neuronal store-operated Ca2+ channels, neuronal store-operated Ca2+ entry, medium spiny neurons, spines, huntingtin-associated protein 1 isoform A
Citation: Czeredys M (2020) Dysregulation of Neuronal Calcium Signaling via Store-Operated Channels in Huntington's Disease. Front. Cell Dev. Biol. 8:611735. doi: 10.3389/fcell.2020.611735
Received: 29 September 2020; Accepted: 01 December 2020;
Published: 23 December 2020.
Edited by:
Agnese Secondo, University of Naples Federico II, ItalyReviewed by:
Emanuele Giurisato, University of Siena, ItalyRosely Oliveira Godinho, Federal University of São Paulo, Brazil
Copyright © 2020 Czeredys. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Magdalena Czeredys, mczeredys@iimcb.gov.pl