The Role of Ferroptosis in Cardiovascular Disease and Its Therapeutic Significance
 Zhenzhen Chen
Zhenzhen Chen  Youyou Yan
Youyou Yan Jia Liu
Jia Liu Junnan Wang
Junnan Wang- Department of Cardiology, Second Hospital of Jilin University, Changchun, China
Cardiovascular diseases (CVDs) are the leading cause of deaths worldwide with regulated cell death playing an important role in cardiac pathophysiology. However, the classical mode of cell death cannot fully explain the occurrence and development of heart disease. In recent years, much research has been performed on ferroptosis, a new type of cell death that causes cell damage and contributes to the development of atherosclerosis, myocardial infarction, heart failure, and other diseases. In this review, we discuss the role of different organelles in ferroptosis and also focus on the relationship between autophagy and ferroptosis. Additionally, we describe the specific mechanism by which ferroptosis contributes to the development of CVD. Finally, we summarize the current research on ferroptosis-related pathway inhibitors and the applications of clinically beneficial cardiovascular drugs.
Introduction
The term “cardiovascular disease” (CVD) refers to a group of diseases that include heart disease, vessel disease, heart attack, stroke, heart failure, arrhythmia, and heart valve disorders. The prevalence of CVD-associated morbidity and mortality are increasing each year with the associated healthcare burden being a leading problem worldwide. Therefore, the prevention, treatment, and prognosis of CVDs have emerged as key areas of medical research in recent years (1–3). Previous studies have shown that various forms of regulated cell death (RCD), including apoptosis, pyrolysis, and necrosis, can destroy the structure of the heart and blood vessels and disrupt their physiological functions, thereby promoting the development of CVDs (4). Recent studies have reported that emricasan (an inhibitor of apoptosis), necrostatin-1 (an inhibitor of necrosis), or 3 -methyladenine (3-MA; an inhibitor of autophagy) fail to improve the survival of mice exposed to doxorubicin (DOX)-induced cardiotoxicity, whereas ferrostatin-1 (a ferroptosis inhibitor) is able to reduce mortality of these mice (5). Further studies found that lipid peroxidation occurred in the mitochondrial membrane. Although this occurs in multiple regulated death modalities, the use of a Fer-1 was found to ameliorate the cellular damage (5). This suggests that ferroptosis, which is a novel type of cell death, may play an important role in the development of CVD.
Ferroptosis was first proposed by Dixon et al. (6). Inhibition of cystine uptake by erastin induces oxidative death and results in the accumulation of lipid reactive oxygen species (ROS), smaller mitochondria, and increased membrane density. This occurs without typical apoptotic features, such as cell shrinkage, nuclear fragmentation, and apoptotic body formation, without necrotic morphological features, such as cytoplasmic and organelle swelling, and without typical features of autophagy, such as membrane-encapsulated vesicle formation (6). Deferoxamine (DFO) can inhibit this type of cell death (6). Another compound, RSL3, has also been shown to induce a similar cell death process (7). Researchers have found that ferroptosis is widely involved in the pathophysiology of a variety of conditions, including nervous system diseases, ischemia/reperfusion injury, kidney injury, and blood disorders (8). Changes in ROS levels are significantly associated with ferroptosis; this association is of concern as increased ROS levels are an important pathophysiological basis for the development of various CVDs. Therefore, an insight into the mechanism underlying ferroptosis-induced cell damage is crucial for a detailed understanding of CVD pathogenesis.
Basic Characteristics of Ferroptosis
The biochemical aspects of ferroptosis manifest as the accumulation of lipid peroxides and increased Fe2+ levels. Mitochondrial morphology relative to ferroptosis is characterized by smaller mitochondria, increased membrane density, reduced or disappearance of mitochondrial cristae, and ruptured outer membranes (8). However, while iron can promote lipid peroxidation and ferroptosis by generating free radicals through the Fenton reaction, it is not essential for ferroptosis. For instance, it was recently determined that metallic copper ions can induce cell death by reducing glutathione (GSH) levels via the inhibition of glutamate-cysteine ligase (GCL) activity (9). That noted, the effect of copper ions on ferroptosis requires further study. Accumulated lipid peroxides generally exert their toxic effects as a key factor in ferroptosis induction through two mechanisms, by destroying membrane integrity and by altering the fluidity and permeability of the cell membrane. As highly reactive molecules, lipid peroxides interact with Fe2+ to further generate ROS. The lipid degradation products 4-hydroxynonenal (4-HNE), malondialdehyde (MDA), and acrolein form covalent adducts with proteins, DNA, and phospholipids, thereby altering the structure and function of the proteins and nucleic acids and inducing the development of a range of diseases (10, 11).
Role of Organelles in the Regulation of Ferroptosis
Mitochondria, lysosomes, and endoplasmic reticulum play significant roles in the regulation of ferroptosis. Mitochondria provide energy for cells through oxidative phosphorylation and play a key role in cell metabolism and signal transduction regulation (12), with most free radicals in the body being produced in mitochondria. The morphological changes associated with ferroptosis mainly manifest as small mitochondrial and the reduction or disappearance of cristae (8, 13). The activation of mitochondrial voltage-dependent anion channels (VDACs) and siderofexin-1 (SFXN1) can significantly promote ferroptosis (14, 15), suggesting mitochondria are an important part of ferroptosis. However, there have been some recent studies that challenge this concept. For instance, Gao et al. found that inhibition of the mitochondrial tricarboxylic acid cycle (TCA) or electron transport chain (ETC) can reduce cysteine deficiency-induced ferroptosis whereas ferroptosis induced by the inhibition of glutathione peroxidase 4 (GPX4) with Ras-selective lethal small molecule 3 (RLS3) has no clear mitochondrial involvement (16). This may be explained by the fact that lipid ROS can be rapidly amplified through the Fenton reaction and subsequently induce ferroptosis. In contrast, another study found that the mitochondria-targeted ROS scavenger mitoquinone (MitoQ) attenuates RLS3-induced lipid peroxidation, mitochondrial ROS production, and loss of mitochondrial membrane potential in neuronal HT22 cells and mouse embryonic fibroblasts (17). Thus, the role of mitochondria in ferroptosis may be cell-type dependent and change under different conditions. As mitochondria participate in various aspects of redox reactions, understanding the role mitochondria play in ferroptosis may provide important cues for the direction of future research.
Ferroptosis is considered an autophagy-dependent mode of cell death (18, 19). Lysosomes are the main organelles for autophagic degradation and thus play a key role in ferroptosis. Torii et al. found that the lysosomal inhibitor Baf-A1 reduces ferroptosis of tumor cells (20). It has been previously suggested that the autophagy-lysosome pathway inhibits CVD progression (21, 22). However, most of the current studies on the relationship between the autophagy-lysosome pathway and ferroptosis have focused on tumor cells with the findings suggesting that autophagy promotes ferroptosis. Accordingly, the autophagy-lysosome pathway need to be further studied in the backdrop of CVDs.
In addition to mitochondria and lysosomes, we found that the endoplasmic reticulum may also be involved in ferroptosis. Cells make adaptive changes when exposed to hypoxia, oxidative stress, and toxic substances that cause protein misfolding, unfolded protein aggregation, and dysregulation of calcium homeostasis in the lumen of the endoplasmic reticulum. The process is known as endoplasmic reticulum stress and is mediated by three main signaling pathways, the IRE1, ATF6, and PERK/eIF2α pathways (23, 24). PERK/eIF2α can upregulate the expression of activating transcription factor 4 (ATF4), which is an important molecule involved in ferroptosis. ATF4 can promote the expression of heat shock protein 5 (HSPA5) and further upregulate GPX4 to protect glioma cells from ferroptosis (25). Meanwhile, ATF4 can also promote the expression of cation transport regulator–like protein 1 (CHAC1) and degrade GSH in Burkitt lymphoma cells to induce ferroptosis (26). The dual effect of the PERK/eIF2α/ATF4 signaling on ferroptosis may be due to different pathological conditions; however, this regulatory pathway has not been fully studied in the context of CVDs.
Disease progression is often accompanied by the occurrence of multiple cell death modes. As organelles are structures that perform important biological functions in cells, determining their role in RCD is of interest. Clarifying the relationship between different cell death pathways and evaluating the incidence of specific death modes in the course of disease will help gain critical insights on the theoretical basis for the use of targeted therapies and the use of different combinations of drugs in preventing and treating CVDs.
Connecting the Links Between Ferroptosis and Autophagy
The main function of autophagy is to remove the damaged organelles and proteins. Moderate levels of autophagy protect cells by digesting their senescent organelles and metabolic wastes; however, under excessive damage, autophagy can lead to apoptosis and necrosis, which may subsequently induce the development of various clinical diseases (27). The ferroptosis inducer erastin promotes the conversion of autophagy-related protein LC3 from LC3-I to LC3-II. In turn, autophagy regulates ferroptosis by affecting cellular iron homeostasis and ROS production (28). We summarize the interaction of regulatory molecules of autophagy and ferroptosis (Figure 1).
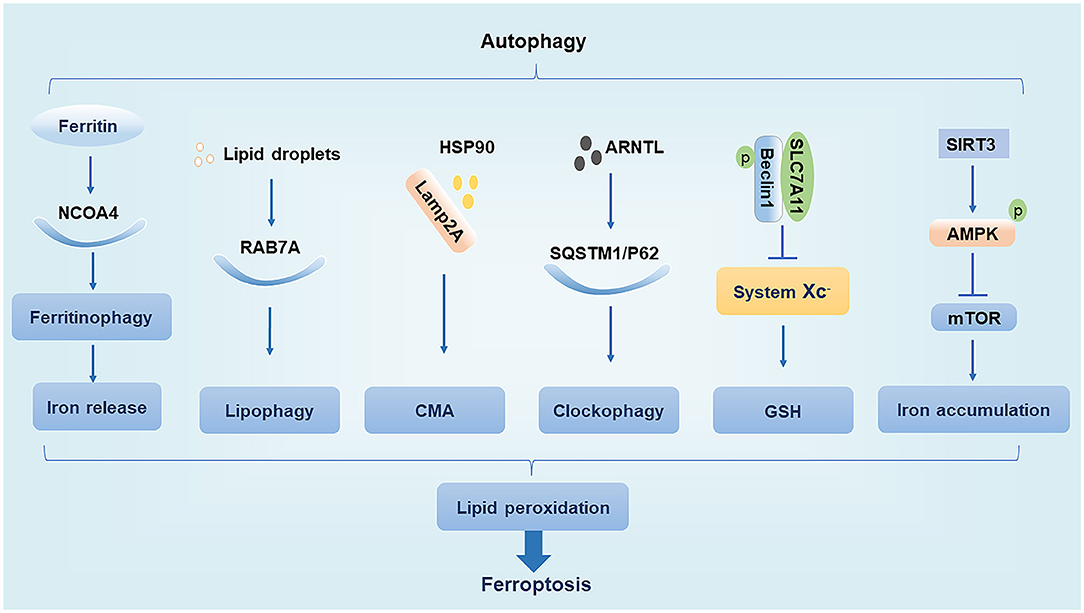
Figure 1. Relationship between autophagy and ferroptosis. NCOA4, nuclear receptor coactivator 4; Lamp2A, lysosome associated membrane protein 2A; HSP90, heat shock protein 90; AMPK, AMP-activated protein kinase.
Nuclear Receptor Coactivator 4
NCOA4 is a cargo receptor for autophagic ferritin degradation. It participates in ferritinophagy, thereby promoting the release of Fe3+ and increasing the intracellular concentration of free iron, which promotes ferroptosis. When cellular iron levels are high, NCOA4 is degraded by the ubiquitin E3 ligase HERC2, leading to reduced iron release. In contrast, when intracellular iron levels are low, the degradation of NCOA4 decreases and ferritinophagy increases, leading to increased iron release (19). Moderate levels of ferritinophagy can maintain the balance of intracellular iron, but excessive ferritinophagy can cause ferroptosis by increasing ROS accumulation (14, 28).
RAB7A
Ras-related protein RAB7A is a member of the RAS oncogene family and is a cargo receptor required for lipophagy. Lipophagy can selectively recognize and degrade lipid droplets, increase free fatty acid production, promote lipid peroxidation, and induce ferroptosis. Studies have found that tumor protein D52 (TPD52) inhibits RSL3-induced ferroptosis by promoting lipid droplet storage, whereas knockdown of autophagy-related genes ATG5 and RAB7A restricts RSL3-induced ferroptosis by inhibiting lipid droplet degradation (29). This suggests that the balance between lipid synthesis, storage, and degradation can regulate ferroptosis.
Chaperone-Mediated Autophagy
CMA is a selective autophagy pathway for soluble cytosolic proteins that is mediated through molecular chaperones. Cytosolic heat shock cognate protein 70 (HSC70), also known as HSPA8, recognizes the KFERQ amino acid sequence to form a complex and bind to lysosome associated membrane protein 2A (Lamp2A), which is subsequently transported to the lysosomal membrane under HSC70 guidance where the target substrate is unfolded and translocated to the lysosome for degradation (30). Heat shock protein 90 (HSP90) binds to Lamp2A on the lysosomal membrane and activates CMA. It was found that GPX4 is degraded in vitro by HSP90-induced CMA to induce ferroptosis in cells (31). Interestingly, another study found that HSPA5 can in turn upregulate GPX4 to inhibit ferroptosis (25). Thus, various types of heat shock proteins appear to have different effects on ferroptosis.
Ubiquitin-Binding Protein p62
The p62 protein, which is also known as sequestosome 1 (SQSTM1), is a selective autophagy connector protein that serves as a bridge between autophagy-related LC3 proteins and polyubiquitinated proteins. It then transports damaged proteins, mitochondria, and invading bacteria to autophagosomes for degradation (32). It has been determined that p62 can be used as a cargo receptor required for circadian clock autophagy and that it participates in autophagic degradation of ubiquitinated substrates. The term clockophagy is used to refer to the selective autophagy degradation of the core clock protein Aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL). Studies have found that ARNTL promotes the expression of Egl nine homolog 2 (EGLN2) via p62-mediated selective autophagic degradation and destroys the stability of hypoxia-inducible factor 1-alpha (HIF1A), which promotes lipid peroxidation and ferroptosis to exert antitumor effects (33). EGLN2, also known as prolyl hydroxylase 1 (PHD1), is an oxygen sensor that degrades HIF1A, which in turn inhibits lipid storage to promote ferroptosis (33, 34).
Beclin1
Beclin1, the mammalian homolog of yeast autophagy-related gene 6 (ATG6), is a key protein necessary for the formation of autophagosomes (35). Studies have shown that phosphorylated Beclin1 interacts with SLC7A11, inhibits system Xc– activity, promotes GSH depletion, induces tumor cell lipid peroxidation, promotes ferroptosis, and increases antitumor efficacy (36, 37). Phosphorylated Beclin1 can also bind to phosphoinositol-3-kinase catalytic subunit type 3 (PIK3C3) to promote autophagy (38, 39). However, it is unclear whether phosphorylated Beclin1 preferentially interacts with SLC7A11 over that of PIK3C3 to promote ferroptosis.
AMP-Activated Protein Kinase
AMPK is a serine/threonine-protein kinase whose activity is regulated by the AMP/ATP ratio, hypoxia, oxidative stress, and other factors. The mechanistic target of rapamycin (mTOR) is a negative regulator of autophagy, and AMPK enhances autophagy by inhibiting mTOR activity (40). In trophoblast cells treated with high glucose, SIRT3 has been shown to inhibit the activity of mTOR by enhancing the phosphorylation of AMPK to promote autophagy, thereby resulting in iron accumulation, lipid peroxidation, and induction of ferroptosis (41). However, the effect of AMPK activation on ferroptosis may have a dual effect as treatment of cells with glucose-free medium revealed that AMPK activation under conditions of energetic stress promotes the phosphorylation of acetyl coenzyme A carboxylase (ACC) to inhibit unsaturated fatty acid synthesis and ultimately inhibit ferroptosis (42). Thus, the regulation of ferroptosis by AMPK may depend on different stimulatory conditions, which needs to be further clarified in future studies.
In summary, several studies have investigated the connecting links between autophagy and ferroptosis, particularly in tumor cells. Ferroptosis promoted by autophagy can inhibit tumor cell growth, which is of great significance in the context of tumorigenesis and the development of resistance. However, it must be noted that in other diseases, autophagy is usually considered to be a protective phenomenon that can delay the development of diseases by inhibiting pyrolysis and apoptosis (21, 22). The role of autophagy-mediated ferroptosis in these diseases remains to be further studied. Owing to the contrary biological outcomes, there is a need to determine the relationship between autophagy and ferroptosis in non-tumor cells and the regulation of ferroptosis under different autophagic conditions.
Mechanism of Ferroptosis
The global prevalence of CVD-related mortality has been increasing each year. This makes it important to understand the basic molecular mechanisms underlying CVDs and to develop effective therapeutic drugs. Injury caused by oxidative stress is a key trigger for the development of CVDs (43). An imbalance between free radical production and scavenging is also encountered in ferroptosis, a phenomenon that is currently thought to be related to lipid, amino acid, and iron metabolism. For instance, when unsaturated fatty acids are oxidized into harmful lipid peroxides, they may damage the endothelium. In addition, excessive consumption of GSH will lead to increased accumulation of lipid peroxides, a phenomenon that affects cellular function. Furthermore, ROS generated by Fenton reaction can induce cell damage (44). In this review, we introduce the main mechanisms of abnormal lipid, glutamate acid, and iron metabolisms involved in inducing ferroptosis (Figure 2).
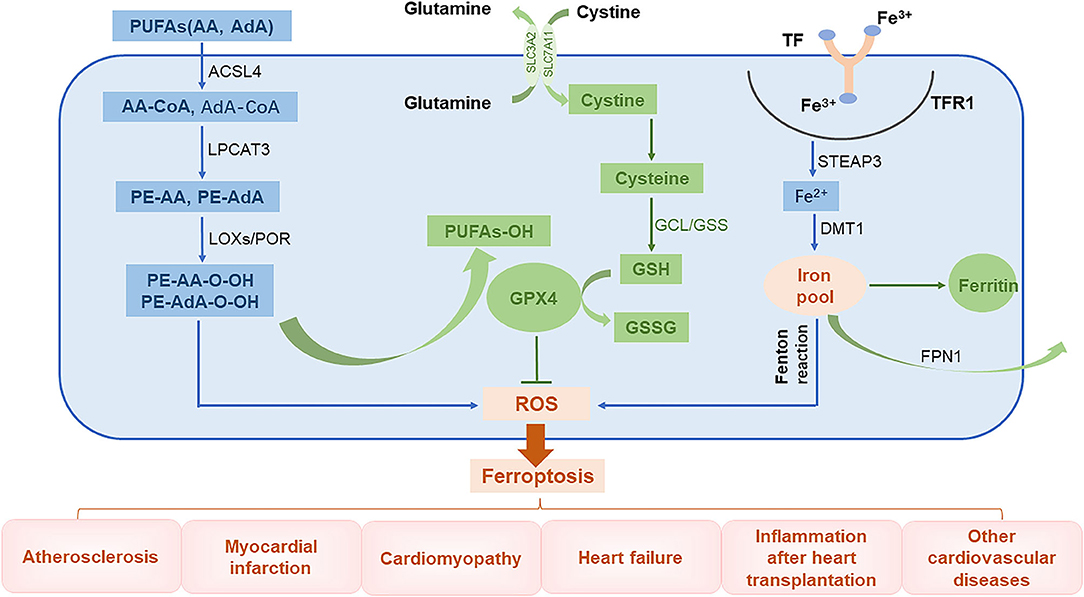
Figure 2. The role of ferroptosis in the pathophysiology of CVDs. PUFAs, polyunsaturated fatty acids; AA, arachidonic acid; AdA, adrenal acid; ACSL4, long-chain fatty acyl-CoA synthase 4; LPCAT3, lysolecithin acyltransferase 3; LOXs, lipoxygenases; POR, cytochrome P450 oxidoreductase; GCL, glutamate-cysteine ligase; GPX4, glutathione peroxidase 4; TF, transferrin; TFR1, transferrin receptor 1; Steap3, six-transmembrane epithelial antigen of prostate 3; DMT1, divalent metal transporter 1; FPN1, ferroportin 1.
Abnormal Lipid Metabolism
The lipid bilayer of cell membranes is essential for maintaining the integrity of membrane function. Cell membranes are mainly composed of lipids, proteins, and carbohydrates with polyunsaturated fatty acids (PUFAs) being an important component (45). Lipid peroxidation caused by interactions between unsaturated fatty acids and free radicals is the major cause of ferroptosis. Arachidonic acid (AA) and adrenal acid (AdA) are catalyzed by long-chain fatty acyl-CoA synthase 4 (ACSL4) to synthesize corresponding fatty acyl-coenzymes AA-CoA and AdA-CoA, respectively, which are involved in the synthesis of membrane phospholipids (46). These products are catalyzed by lysolecithin acyltransferase 3 (LPCAT3) through esterification reactions to form phosphatidylethanolamine PE-AA and PE-AdA, which are then oxidized by lipoxygenases (LOXs) or cytochrome P450 oxidoreductase (POR) into harmful lipid peroxidation products PE-AA-OOH and PE-AdA-OOH, respectively. Normally, these harmful substances are reduced by GPX4 to form non-toxic lipid alcohols; however, when the levels of GPX4 are low, these peroxide products excessively accumulate, leading to ferroptosis (47–49).
Abnormal Glutamate Metabolism
Glutathione is an important antioxidant and free radical scavenger that is widely distributed in biological tissues and is catalytically produced from glutamate, cysteine, and glycine by glutamate-cysteine ligase (GCL) and glutathione synthase (GSS). Glutathione is an essential cofactor of glutathione peroxidase 4 (GPX4) and is required for its antioxidant activity (50). Glutathione has two forms, a reduced form (GSH) and an oxidized disulfide form (GSSG). Under the stimulation of free radicals and heavy metals, GPX4 converts GSH into GSSG and reduces lipid peroxides (L-OOHs) to its corresponding lipid alcohols (L-OHs). This limits the spread of lipid peroxidation in the membrane (8, 51). When GSH levels are reduced, ROS and lipid peroxides can accumulate, inducing ferroptosis (41, 50).
Abnormal Iron Metabolism
Intracellular iron homeostasis depends on the dynamic balance between iron absorption, excretion, utilization, and storage. The distribution and content of iron in the body affects multiple physiological processes (52). Normally, the body's daily need for iron to maintain the balance of iron metabolism comes from the lysis of aging red blood cells and from ingested food. Gastric acids reduce Fe3+ in food to Fe2+, which is then absorbed in the duodenum and jejunum. The Fe2+ entering the body is then oxidized through the action of ceruloplasmin to Fe3+, which combines with transferrin (TF) on the cell membrane to form TF-Fe3+. This subsequently forms a complex with transferrin receptor 1 (TFR1). After being endocytosed into the cells, the iron is converted into Fe2+ by six-transmembrane epithelial antigen of prostate 3 (Steap3). Unstable Fe2+ is released into the cytoplasm by divalent metal transporter 1 (DMT1), also known as SLC11A2, and it then either enters the mitochondria or is directly utilized in the cytoplasm. The iron can also be stored in ferritin or secreted by ferroportin 1 (FPN1) (8, 53). Ferritin is composed of two subunits, a ferritin heavy chain (FTH1) and ferritin light chain (FTL) and is the most important iron storage protein in the cell. Iron that is not used in the cytoplasm or discharged from the cell is stored in ferritin (54). Iron levels are regulated by key proteins, such as TFR1, DMT1, and FTH1, to maintain intracellular iron homeostasis. Because iron has unpaired electrons, it can participate in redox reactions. In the Fenton reaction, Fe2+ and hydrogen peroxide (H2O2) react to generate hydroxyl radicals with strong oxidative capacity and Fe3+, which is then reduced by peroxide (O2−) back to Fe2+ to re-participate in the Fenton reaction. This cycle is called the Haber–Weiss reaction, where a large number of free hydroxyl radicals (a type of ROS) are produced (55). These free hydroxyl radicals can cause a series of clinical diseases (13, 56, 57).
Overall, ferroptosis is mainly caused by an imbalance between free radicals and the antioxidant systems, resulting in RCD. Most of the existing studies describe the mechanism of ferroptosis occurring in terms of lipid peroxide sources and degradation. The regulation of the above-mentioned metabolic molecules can clearly affect ferroptosis; however, there are many questions that need to be addressed by conducting further investigations. The first is whether other metal ions participate in ferroptosis, as noted before (9). The second question is that lipid peroxidation also occurs in other types of RCD, and whether more specific diagnostic methods could be developed for identifying ferroptosis.
Ferroptosis is Involved in CVDs
As mentioned previously, ferroptosis plays various roles in the pathophysiology of CVDs. We elaborate on this in this section of the review.
Atherosclerosis
AS is a pathological process characterized by lipid metabolism disorders. Endothelial injury, oxidative stress, inflammation, and immune dysfunction can each contribute to the development and progression of AS (58, 59). Clinically, coronary heart disease (CHD) is the most common type of organ lesion caused by atherosclerosis. Studies have shown that the iron content in tissues of individuals with AS is significantly increased compared to that of healthy arteries, and such iron overload promotes oxidative stress and inflammatory responses (60, 61). This suggests that ferroptosis may be involved in the development of AS. Researchers have found that in vivo, the ferroptosis inhibitor Fer-1 alleviates atherosclerotic lesions and lipid peroxidation induced by a high-fat diet in ApoE−/− mice. Similarly, in vitro studies have demonstrated that Fer-1 can improve ferroptosis and endothelial dysfunction induced by ox-LDL and can delay the progression of AS (62). Therefore, inhibition of ferroptosis may be a new strategy for the treatment of CHD.
Myocardial Infarction
MI is a common cause of death worldwide. In mice with MI, the levels of FTH1 in the infarct area decreased and free iron levels increased (63). Other studies have also found that the amount of GPX4 protein in MI tissue decreased compared with that of normal tissue and that the use of the GPX4 inhibitor RSL3 induces ferroptosis in H9c2 cells (64). These findings suggest that increased iron or decreased GPX4 can promote ferroptosis of cardiomyocytes and lead to MI. Currently, the most preferred treatment method for MI is recanalization being performed as soon as possible (65). Although the timely restoration of blood flow can partially save the damaged myocardium, prolongation of ischemia time can lead to damage, even when blood flow is restored, and this ischemia/reperfusion injury can increase mortality rates of patients with MI (66). In addition to calcium overload, oxidative stress, inflammation, and energy metabolism disorders, ferroptosis can also cause ischemia/reperfusion injury (67). Tang et al. found that ferroptosis mainly occurs in the reperfusion phase in a rat myocardial ischemia/reperfusion injury model, and further studies revealed that the use of DFO can reduce myocardial reperfusion injury (68). Surprisingly, the levels of the ferroptosis-associated proteins ACSL4 and GPX4 do not significantly change during the ischemic phase. The reason for this difference between the ischemia and reperfusion phases may be that large numbers of free radicals are produced during reperfusion to induce ferroptosis. In addition, it has been determined that the ferroptosis inhibitors liproxstatin 1 (Lip-1) and Fer-1 reduce ischemia/reperfusion injury by reducing ROS production (69, 70). Autophagy has also been found to promote ferroptosis during myocardial ischemia/reperfusion injury, leading to myocardial damage (71, 72). Therefore, actively preventing the occurrence of ferroptosis may become an important consideration after recanalization of patients suffering from MI.
Cardiomyopathy
Cardiomyopathy is a group of heterogeneous myocardial diseases that causes mechanical and/or electrical dysfunction and occurs mostly due to hereditary causes. Such conditions often manifest from ventricular hypertrophy or dilation that may be confined to the heart itself or may be the result of systemic diseases. The outcome of cardiomyopathy may lead to cardiac arrest or progressive heart failure (73). The accumulation of iron in the myocardium can cause iron-overload cardiomyopathy, which usually presents in the early stages as diastolic dysfunction. As the disease progresses, the heart may enlarge and contraction may decrease (74). Ferroptosis is associated with many types of cardiomyopathies. Although there are differences in the pathogenesis of the different types, it is ultimately due to an imbalance in the production and scavenging of free radicals that causes an increase in lipid peroxides.
Dox-Induced Cardiomyopathy
Studies have found that the chemotherapeutic drug DOX induces DNA double-strand breaks in cardiomyocytes and increases Fe2+ and Ca2+ levels in mitochondria. The resulting increase in ROS and iron may be the main cause of DOX-induced ferroptosis, leading to cardiomyopathy (75–80). As mitochondria are the main organelles of DOX-induced myocardial damage, the mitochondrial antioxidant MitoTEMPO can be used to eliminate DOX-induced lipid peroxidation and ferroptosis (5). Iron chelators targeting Fe2+ may also be an ideal method to treat myocardial damage caused by DOX (81).
Hypertrophic Cardiomyopathy
Studies have also shown that high-iron feeding mice (FthMCK/MCK and FthMyh6/Myh6) that knock out the gene encoding the ferritin heavy chain (Ferritin H, Fth) of cardiomyocytes are prone to induce hypertrophic cardiomyopathy (82). Further studies suggest that the deficiency of GSH due to the downregulation of SLC7A11 is the main mechanism that induces ferroptosis in cardiomyocytes (82).
Heart Failure
HF is the terminal stage of CVD, but the timely prevention of cardiomyocyte hypertrophy can maintain heart function and delay progression. Ferroptosis has been observed in mice with heart failure (83). Toll-like receptor 4 (TLR4) binds to NADPH oxidase 4 (NOX4), which promotes the production of superoxide anion and H2O2 and induces ferroptosis in cardiomyocytes. Therefore, blocking the TLR4-NOX4 signaling pathway may serve as a potential therapeutic strategy for treating patients with HF by inhibiting ferroptosis (84). It is strongly believed that iron is a cofactor involved in the energy metabolism of cardiomyocytes (85). Iron supplementation is recommended as it can improve the cardiac function in some patients with chronic HF who have iron deficiency (85). Therefore, determining the mechanism of ferroptosis will also be beneficial for the treatment of patients with HF.
Inflammation After Heart Transplantation
Heart transplantation is the most effective method for treating patients with end-stage heart disease. However, the restoration of coronary blood flow after transplantation may result in aseptic inflammation and reduce the success rate of transplantation (86). After mice with heart transplantation, damage-associated molecular patterns (DAMPs) released from injured tissue promote neutrophil recruitment and infiltration via the TLR4/Trif signaling pathway. Fer-1 decreases the number of neutrophils adhering to blood vessels and inhibits inflammation. These findings demonstrate that inhibition of ferroptosis may be a viable therapeutic strategy for treating heart transplant recipients by reducing both inflammation and release of DAMPs (86).
Other CVDs
Myocardial damage caused by sepsis may also be related to ferroptosis. In the myocardium of mice with sepsis-induced myocardial damage, the expression of GPX4 and GSH are reduced. Meanwhile, iron concentration and transferrin receptor levels increase and ferritin levels decrease during sepsis. According to Wang et al., these changes may be related to heme degradation caused by heme oxygenase-1 (HO-1) (87). It had been previously suggested that HO-1 has anti-cancer, anti-inflammatory, anti-apoptotic, and anti-oxidation effects (88). In the study by Wang et al., dexmedetomidine (DEX) was found to exert its protective effects possibly by reducing the expression of HO-1 and decreasing iron overload (87). HO-1 may exhibit dual roles in the regulation of ferroptosis under different pathological conditions. However, the role of HO-1 requires further study.
Regulation of Ferroptosis for the Treatment of CVDs
As ferroptosis plays a detrimental role in CVD, intervening in this process may help prevent the development and progression of CVDs. Existing ferroptosis inhibitors, including clinically approved drugs and small-molecule drugs currently in the research phase of development, mainly eliminate free radicals, reduce free iron levels, or inhibit lipid peroxidation. This section of the review will focus on the above noted aspects of inhibiting ferroptosis (Table 1).
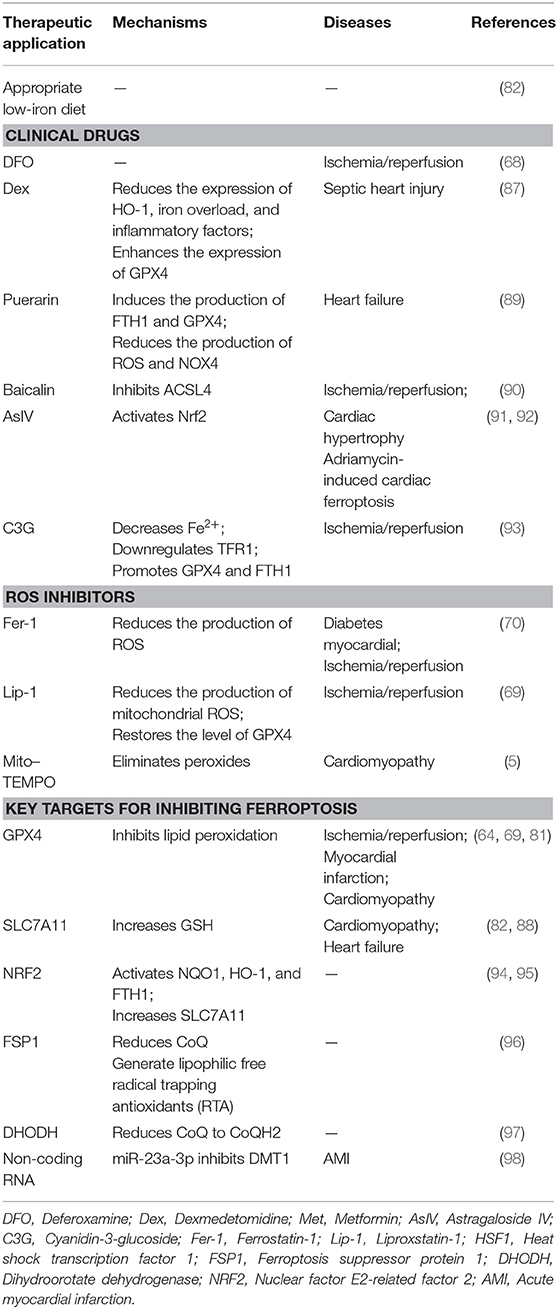
Table 1. Strategies to target ferroptosis in cardiovascular diseases.
Appropriate Low-Iron Diet
The normal iron content in the body is 3–5 g and an insufficient supply of iron may impair the synthesis of ferritin at levels required for normal cell physiology, leading to a series of adverse consequences (99). In contrast, iron overload may result in the production of large amounts of ROS and induce lipid peroxidation and ferroptosis (82). Therefore, appropriate low-iron diets with consideration of minimum requirements may help prevent CVDs caused by ferroptosis.
Clinical Drugs
DFO is a high-affinity iron chelator that reduces intracellular iron and has been approved by the US Food and Drug Administration for the treatment of iron overload. In rats with ischemia/reperfusion injury, DFO can reduce lipid peroxides in cardiac tissue and attenuated myocardial injury (68).
DEX is an α2 adrenergic receptor agonist. Studies have shown that pretreatment with DEX can reduce myocardial ischemia/reperfusion injury by inhibiting the release of inflammatory factors IL-1β, IL-6, and TNF-α through the TLR4-MyD88-NF-κB signaling pathway (100). DEX can inhibit apoptosis and oxidative stress by activating the phosphatidylinositide 3-kinase/protein kinase B (PI3K/Akt) signaling pathway, thereby protecting diabetic rats from ischemia/reperfusion injury (101). It also has anti-inflammatory, antioxidant, and protective effects on ischemia/reperfusion organs. Recently, DEX was found to reduce the expression of HO-1, reduce iron overload and inflammatory factors, and enhance the expression of GPX4, thereby inhibiting ferroptosis and alleviating myocardial cell damage induced by sepsis (87).
Puerarin is an isoflavone compound extracted from plants that has been used in traditional Chinese medicine. Puerarin has been shown to prevent ischemia/reperfusion injury by inhibiting pyrolysis (102), reducing the area of MI, improving diastolic cardiac function (103), and preventing myocardial fibrosis (104). Puerarin has been approved by the China Food and Drug Administration as a clinical treatment drug for CVDs (105). Recent studies by Liu et al. have found that puerarin can also improve cardiac function in rats with HF by reducing iron content and lipid peroxidation, thereby inhibiting ferroptosis (89). Liu et al. found that treatment of H9c2 cells with erastin or isoproterenol can cause ferroptosis. Puerarin was shown to inhibit ferroptosis in cardiomyocytes by inducing the production of FTH1 and GPX4 and reducing the production of ROS and NOX4, thus improving the cardiac function of rats with HF (68).
Baicalin is a natural flavonoid compound isolated from the root of the plant Scutellaria baicalensis Georgi and has anti-inflammatory, anti-viral, anti-cancer, antioxidant, and other pharmacological effects (106). Studies have shown that baicalin has a protective effect on myocardial ischemia/reperfusion, reducing myocardial apoptosis and inflammation by activating the PI3K/AKT pathway (107). Furthermore, baicalin protects the microvascular endothelial cells in the heart of ischemia/reperfusion rats by activating the PI3K-AKT-eNOS pathway (108). Fan et al. found that baicalin can also play a cardioprotective role by inhibiting ferroptosis induced by ACSL4 (90). ACSL4 is a key enzyme in fatty acid metabolism and synthesizes AA and AdA into AA-CoA and AdA-CoA, respectively, which are involved in membrane phospholipid synthesis and promote ferroptosis (109).
Astragaloside IV (AsIV) is extracted from Astragalus spp. and is used in traditional Chinese medicine. It protects the myocardium by inhibiting myocardial cell hypertrophy, apoptosis, and fibrosis. Studies have confirmed that Nrf2 may be a key molecule targeted by AsIV for it to exert its protective effect. Upregulation of Nrf2 can inhibit cardiac hypertrophy in an abdominal aortic constriction model of chronic HF (91). Furthermore, the activation of Nrf2 signaling and the expression of GPX4 can inhibit ferroptosis, thus further preventing adriamycin-induced cardiac fibrosis (92).
Cyanidin-3-glucoside (C3G) is a member of the anthocyanin family, is found in a wide variety of vegetables and fruits, and has anti-inflammatory and antioxidant effects. C3G can reduce DOX-induced cardiotoxicity in mice and prevent cardiac hypertrophy and diastolic cardiac dysfunction (110, 111). Experiments have shown that C3G alleviates oxidative stress in rats with ischemia/reperfusion injury and in H9C2 cells with oxygen-glucose deprivation/reoxygenation (OGD/R). Treatment with C3G leads to decreased Fe2+ content, downregulation of TFR1 expression, and promotion of GPX4 and FTH1 expressions. Therefore, C3G is a potential drug that may be used to protect the myocardium from ischemia/reperfusion injury (93).
ROS Inhibitor
Fer-1 is a specific inhibitor of ferroptosis that was identified through high-throughput screening of small-molecule libraries (112). Fer-1 can inhibit the accumulation of lipid ROS and reduce cell ferroptosis induced by erastin or RSL3. It has also been has found that Fer-1 can inhibit ferroptosis and improve DOX-induced myocardial damage in CVDs and alleviate myocardial ischemia/reperfusion injury in diabetic rats (5, 70).
Lip-1 has a mechanism of action similar to that of Fer-1. Without affecting the Ca2+-induced mitochondrial permeability transition pore opening, Lip-1 reduces the production of mitochondrial ROS, restores GPX4 levels by downregulating the level of VDAC1 (without affecting VDAC2/3) and oligomerization to protect myocardial mitochondria from ischemia/reperfusion injury (69). VDACs are located in the outer membrane of mitochondria, regulate mitochondrial metabolism and cell-signal transduction, and are transmembrane channels used for the transport of ions and metabolites (113). After erastin acts on VDACs, permeability of the outer mitochondria membrane increases, membrane ion channels open, and intracellular homeostasis becomes disturbed, leading to mitochondrial metabolism and oxidation dysfunction, increased ROS production, enhanced lipid peroxidation, and the subsequent induction of cell ferroptosis (15).
Mito-TEMPO is a cell-permeable mitochondria-targeted antioxidant that reduces the accumulation of superoxides (114). Mito-TEMPO can also reduce cell apoptosis by reducing Ca2+ release (115). In recent years, Mito-TEMPO has been shown to effectively inhibit DOX-induced ferroptosis of cardiomyocytes by eliminating lipid peroxidation in mitochondria, which is a cardioprotective effect similar to that of Fer-1 (5).
Key Targets for Inhibiting Ferroptosis
GPX4 can reduce lipid peroxides (L-OOHs) to lipid alcohols (L-OH), which inhibit the spread of lipid hydroperoxides and protect cells from damage. When levels of GPX4 is reduced, ferroptosis is promoted and ischemia/reperfusion injury (69), MI (64), and cardiomyopathy (81) can be induced.
SLC7A11, also known as xCT, is a multichannel transmembrane protein that serves as a component of the cystine/glutamate reverse transporter, system Xc–. System Xc– is an antiporter, consisting of SLC7A11 as the light chain subunit and SLC3A2 as the heavy chain subunit. System Xc– transfers intracellular glutamate to the outside of cells and extracellular cystine into the cells. The cystine entering the cell is converted to cysteine by a reduction reaction and then used in the synthesis of GSH, which protects the cells from oxidative stress damage. The transport direction of system Xc– is driven by substrate concentrations. When the concentration of extracellular glutamate is high, the uptake of cystine by system Xc– is inhibited, which leads to a reduction in the synthesis of intracellular GSH and decreases the antioxidant capacity of the cells, eventually leading to the accumulation of lipid ROS and the induction of ferroptosis (50, 82).
Nuclear factor E2-related factor 2 (NRF2) is a transcriptional activator that plays an important role in antioxidant responses and regulates ferroptosis through the p62-KEAP1-NRF2 signaling pathway (116). Under normal physiological conditions, NRF2 and KEAP1 bind one another and are anchored in the cytoplasm in an inactive state. When the cell is under oxidative stress, the conformation of KEAP1 changes, KEAP1 dissociates from NRF2, and NRF2 enters the nucleus where it recognizes components of antioxidant reactions and regulates the expression of genes related to oxidative damage, thereby playing an antioxidant role. The structure of p62 includes an STGE sequence that can bind to the Kelch domain of KEAP1, which reduces the stability of the KEAP1/NRF2 complex causing NRF2 to dissociate from the complex and become activated (117). This further activates the transcription of NQO1, HO-1, and FTH1, or increases the expression of SLC7A11 to inhibit ferroptosis (94, 95). While these results demonstrate that HO-1 has a protective effect, a previous study found that HO-1 can degrade heme and promote ferroptosis (87). Therefore, HO-1 may have a dual role in ferroptosis. The mechanisms of these different models require further investigation.
At least three defense mechanisms have been found to inhibit ferroptosis. These include (1) GPX4 using reduced GSH to eliminate lipid peroxidation and inhibiting ferroptosis; (2) Ferroptosis suppressor protein 1 (FSP1), also known as AIFM2, acting as an oxidoreductase that primarily reduces CoQ to a lipophilic radical, trapping antioxidant (RTA) at the plasma membrane and preventing the spread of lipid peroxidation (96); and (3) Dihydroorotate dehydrogenase (DHODH), an enzyme located on the outer surface of mitochondrial intima, reducing CoQ to CoQH2 while oxidizing dihydroorotate (DHO) to orotate (OA) and cooperating with GPX4 to inhibit lipid peroxidation and ferroptosis in mitochondria (97). The first mechanism has been verified in CVDs; it remains to be determined whether the latter two defense mechanisms also play protective roles in CVDs.
Non-coding RNA
ncRNAs are classified as long non-coding RNA (lncRNA), microRNA (miRNA), and small interfering RNA (siRNA) according to their length. Recent studies have shown that ncRNAs are differentially expressed in CVDs and play an important role in CVD development (118). A previous study found that exosomes derived from human umbilical cord mesenchymal stem cells (HUCMSCs) can suppress DMT1 expression through the delivery of miR-23a-3p, thereby inhibiting ferroptosis and attenuating myocardial injury (98). DMT1 promotes iron metabolism by transporting Fe2+, and the knockout of DMT1 significantly inhibits ferroptosis (98). This suggests ncRNAs may be an important factor in the intervention of ferroptosis and that exosomes may serve as transport carriers to deliver the ncRNAs (119).
In the cardiovascular field, existing research has shown that it is possible to suppress the occurrence of ferroptosis and prevent the development of the disease through various mechanisms. However, most of the experiments remain at the animal or cell level, so the application of these findings to clinical treatment requires additional detailed analysis and appropriate clinical trials.
Conclusions and Prospects
Since the discovery of ferroptosis as a new type of RCD, in-depth biological research has provided new ways to explore its occurrence and role in a variety of diseases. A growing number of studies have shown that lipid peroxidation-induced ferroptosis is an important pathogenetic mechanism of CVD development. Although, this discovery poses a challenge to the treatment of CVDs, it also provides a new therapeutic target. Current research continues to reveal details regarding the mechanism of action of ferroptosis in a variety of diseases and also raises many questions that are worthy of researchers' consideration. First, further research is needed on the interaction between ferroptosis and other modes of RCD. The key ferroptosis proteins P53, GPX4, and SLC7A11 are also involved in other modes of death. When these proteins are changed, we need to be able to distinguish their interactions among the different types of RCD through experimental means. Second, further insight into the manifestation of ferroptosis is needed. For instance, lipid peroxidation is also involved in other forms of RCD and may be only an intermediate link in ferroptosis. Third, defining the threshold peroxidation reaction is essential. Oxygen-free radicals in humans and other species are necessary for metabolism and can also induce ferroptosis. The threshold of oxygen free radicals required for inducing ferroptosis needs to be further studied. Fourth, ways need to be determined to regulate ferroptosis. As there are different regulatory pathways for ferroptosis, it is necessary to clarify whether there are intersecting points between each pathway and the final executive molecule. Finally, we need to be able to ultimately control the “double-edged sword” of ferroptosis. Their effects must be carefully balanced while developing or administering drugs and other therapeutic interventions.
The significance of the basic research regarding ferroptosis and CVD lies in its ultimate clinical relevance. Experimental research needs to continuously improve on available strategies before it can provide hope for new treatment options for patients suffering from CVDs. We have a reason to believe that with the advancement of science, research on ferroptosis will help provide a solid theoretical basis on the occurrence and development of CVDs. Furthermore, we believe that the specific targeting of ferroptosis will emerge as an important means of treating CVD.
Author Contributions
ZC and YY researched the article and wrote the manuscript. CQ, JL, LL, and JW reviewed and edited the manuscript before submission. All authors provided substantial contribution to the discussion of content.
Funding
This work was supported by Development and Reform Commission of Jilin Province (2020C036-3) and the Science and Technology Development of Jilin Province (20200301003RQ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: update from the GBD 2019 Study. J Am College Cardiol. (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
2. GBD 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. (2020) 396:1204–22. doi: 10.1016/S0140-6736(20)30925-9
3. Li N, Jiang W, Wang W, Xiong R, Wu X, Geng Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharmacol Res. (2021) 166:105466. doi: 10.1016/j.phrs.2021.105466
4. Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. (2019) 99:1765–817. doi: 10.1152/physrev.00022.2018
5. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci USA. (2019) 116:2672–80. doi: 10.1073/pnas.1821022116
6. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
7. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. (2008) 15:234–45. doi: 10.1016/j.chembiol.2008.02.010
8. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. (2020) 11:88. doi: 10.1038/s41419-020-2298-2
9. Maher P. Potentiation of glutathione loss and nerve cell death by the transition metals iron and copper: Implications for age-related neurodegenerative diseases. Free Rad Biol Med. (2018) 115:92–104. doi: 10.1016/j.freeradbiomed.2017.11.015
10. Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. (2017) 482:419–25. doi: 10.1016/j.bbrc.2016.10.086
11. Gegotek A, Skrzydlewska E. Biological effect of protein modifications by lipid peroxidation products. Chem Phys Lipids. (2019) 221:46–52. doi: 10.1016/j.chemphyslip.2019.03.011
12. Wang H, Liu C, Zhao Y, Gao G. Mitochondria regulation in ferroptosis. Euro J Cell Biol. (2020) 99:151058. doi: 10.1016/j.ejcb.2019.151058
13. Zhou RP, Chen Y, Wei X, Yu B, Xiong ZG, Lu C, et al. Novel insights into ferroptosis: Implications for age-related diseases. Theranostics. (2020) 10:11976–97. doi: 10.7150/thno.50663
14. Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Rad Biol Med. (2020) 160:303–18. doi: 10.1016/j.freeradbiomed.2020.08.009
15. Yang Y, Luo M, Zhang K, Zhang J, Gao T, Connell DO, et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat Commun. (2020) 11:433. doi: 10.1038/s41467-020-14324-x
16. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. (2019) 73:354–63.e3. doi: 10.1016/j.molcel.2018.10.042
17. Jelinek A, Heyder L, Daude M, Plessner M, Krippner S, Grosse R, et al. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Rad Biol Med. (2018) 117:45–57. doi: 10.1016/j.freeradbiomed.2018.01.019
18. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. (2020) 66:89–100. doi: 10.1016/j.semcancer.2019.03.002
19. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. (2016) 12:1425–8. doi: 10.1080/15548627.2016.1187366
20. Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J. (2016) 473:769–77. doi: 10.1042/BJ20150658
21. Meng Q, Li Y, Ji T, Chao Y, Li J, Fu Y, et al. Estrogen prevent atherosclerosis by attenuating endothelial cell pyroptosis via activation of estrogen receptor α-mediated autophagy. J Adv Res. (2021) 28:149–64. doi: 10.1016/j.jare.2020.08.010
22. Li CF, Pan YK, Gao Y, Shi F, Wang YC, Sun XQ. Autophagy protects HUVECs against ER stress-mediated apoptosis under simulated microgravity. Apoptosis. (2019) 24:812–25. doi: 10.1007/s10495-019-01560-w
23. Omidkhoda N, Wallace Hayes A, Reiter RJ, Karimi G. The role of MicroRNAs on endoplasmic reticulum stress in myocardial ischemia and cardiac hypertrophy. Pharmacol Res. (2019) 150:104516. doi: 10.1016/j.phrs.2019.104516
24. Donnelly N, Gorman AM, Gupta S, Samali A. The eIF2α kinases: their structures and functions. Cell Mol Life Sci. (2013) 70:3493–511. doi: 10.1007/s00018-012-1252-6
25. Chen Y, Mi Y, Zhang X, Ma Q, Song Y, Zhang L, et al. Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J Experi Clin Cancer Res. (2019) 38:402. doi: 10.1186/s13046-019-1413-7
26. Wang N, Zeng GZ, Yin JL, Bian ZX. Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects ferroptosis in Burkitt's Lymphoma. Biochem Biophys Res Commun. (2019) 519:533–9. doi: 10.1016/j.bbrc.2019.09.023
27. Yun HR, Jo YH, Kim J, Shin Y, Kim SS, Choi TG. Roles of Autophagy in Oxidative Stress. Int J Mol Sci. (2020) 21:93289. doi: 10.3390/ijms21093289
28. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. (2016) 26:1021–32. doi: 10.1038/cr.2016.95
29. Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao H, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. (2019) 508:997–1003. doi: 10.1016/j.bbrc.2018.12.039
30. Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. (2018) 19:365–81. doi: 10.1038/s41580-018-0001-6
31. Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci USA. (2019) 116:2996–3005. doi: 10.1073/pnas.1819728116
32. Berkamp S, Mostafavi S, Sachse C. Structure and function of p62/SQSTM1 in the emerging framework of phase separation. FEBS J. (2020). doi: 10.1111/febs.15672 [Epub ahead of print].
33. Yang M, Chen P, Liu J, Zhu S, Kroemer G, Klionsky DJ, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. (2019) 5:eaaw2238. doi: 10.1126/sciadv.aaw2238
34. Chiba N, Sunamura M, Nakagawa M, Koganezawa I, Yokozuka K, Kobayashi T, et al. Overexpression of hydroxyproline via EGLN/HIF1A is associated with distant metastasis in pancreatic cancer. Am J Cancer Res. (2020) 10:2570–81.
35. Shi B, Ma M, Zheng Y, Pan Y, Lin X. mTOR and Beclin1: Two key autophagy-related molecules and their roles in myocardial ischemia/reperfusion injury. J Cell Physiol. (2019) 234:12562–8. doi: 10.1002/jcp.28125
36. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system X(c)(-) Activity. Curr Biol. (2018) 28:2388–99.e5. doi: 10.1016/j.cub.2018.05.094
37. Liu R, Li X, Zhao G. Beclin1-mediated ferroptosis activation is associated with isoflurane-induced toxicity in SH-SY5Y neuroblastoma cells. Acta Biochim Biophys Sinica. (2019) 51:1134–41. doi: 10.1093/abbs/gmz104
38. Zhang D, Wang W, Sun X, Xu D, Wang C, Zhang Q, et al. AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy. (2016) 12:1447–59. doi: 10.1080/15548627.2016.1185576
39. Kang R, Zhu S, Zeh HJ, Klionsky DJ, Tang D. BECN1 is a new driver of ferroptosis. Autophagy. (2018) 14:2173–5. doi: 10.1080/15548627.2018.1513758
40. Sukumaran A, Choi K, Dasgupta B. Insight on transcriptional regulation of the energy sensing AMPK and biosynthetic mTOR pathway genes. Front Cell Dev Biol. (2020) 8:671. doi: 10.3389/fcell.2020.00671
41. Han D, Jiang L, Gu X, Huang S, Pang J, Wu Y, et al. SIRT3 deficiency is resistant to autophagy-dependent ferroptosis by inhibiting the AMPK/mTOR pathway and promoting GPX4 levels. J Cell Physiol. (2020) 235:8839–51. doi: 10.1002/jcp.29727
42. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. (2020) 22:225–34. doi: 10.1038/s41556-020-0461-8
43. Senoner T, Dichtl W. Oxidative stress in cardiovascular diseases: still a therapeutic target? Nutrients. (2019) 11:2090. doi: 10.3390/nu11092090
44. Wu X, Li Y, Zhang S, Zhou X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics. (2021) 11:3052–9. doi: 10.7150/thno.54113
45. Casares D, Escribá PV, Rosselló CA. Membrane lipid composition: effect on membrane and organelle structure, function and compartmentalization and therapeutic avenues. Int J Mol Sci. (2019) 20:92167. doi: 10.3390/ijms20092167
46. Cheng J, Fan YQ, Liu BH, Zhou H, Wang JM, Chen QX. ACSL4 suppresses glioma cells proliferation via activating ferroptosis. Oncol Rep. (2020) 43:147–58. doi: 10.3892/or.2019.7419
47. Latunde-Dada GO. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gene Subj. (2017) 1861:1893–900. doi: 10.1016/j.bbagen.2017.05.019
48. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. (2017) 13:81–90. doi: 10.1038/nchembio.2238
49. Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, et al. membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mo Cell. (2021) 81:355–69.e10. doi: 10.1016/j.molcel.2020.11.024
50. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. (2020) 12:599–620. doi: 10.1007/s13238-020-00789-5
51. Bajic VP, Van Neste C, Obradovic M, Zafirovic S, Radak D, Bajic VB, et al. Glutathione “redox homeostasis” and its relation to cardiovascular disease. Oxidative Med Cell Longevity. (2019) 2019:5028181. doi: 10.1155/2019/5028181
52. Ward DM, Cloonan SM. Mitochondrial Iron in Human Health and Disease. Annual review of physiology. (2019) 81:453–82. doi: 10.1146/annurev-physiol-020518-114742
53. Han C, Liu Y, Dai R, Ismail N, Su W, Li B. Ferroptosis and its potential role in human diseases. Front Pharmacol. (2020) 11:239. doi: 10.3389/fphar.2020.00239
54. Li X, Si W, Li Z, Tian Y, Liu X, Ye S, et al. miR-335 promotes ferroptosis by targeting ferritin heavy chain 1 in in vivo and in vitro models of Parkinson's disease. Int J Mol Med. (2021) 47:4894. doi: 10.3892/ijmm.2021.4894
55. Torti SV, Manz DH, Paul BT, Blanchette-Farra N, Torti FM. Iron and cancer. Ann Rev Nutr. (2018) 38:97–125. doi: 10.1146/annurev-nutr-082117-051732
56. Tian Y, Lu J, Hao X, Li H, Zhang G, Liu X, et al. FTH1 inhibits ferroptosis through ferritinophagy in the 6-OHDA model of Parkinson's disease. Neurotherapeutics. (2020) 17:1796–812. doi: 10.1007/s13311-020-00929-z
57. Capelletti MM, Manceau H, Puy H, Peoc'h K. Ferroptosis in liver diseases: an overview. Int J Mol Sci. (2020) 21:4908. doi: 10.3390/ijms21144908
58. Wolf D, Ley K. Immunity and Inflammation in Atherosclerosis. Circul Res. (2019) 124:315–27. doi: 10.1161/CIRCRESAHA.118.313591
59. Marchio P, Guerra-Ojeda S, Vila JM, Aldasoro M, Victor VM, Mauricio MD. Targeting early atherosclerosis: a focus on oxidative stress and inflammation. Oxidat Med Cell Longevity. (2019) 2019:8563845. doi: 10.1155/2019/8563845
60. Xu S. Iron and atherosclerosis: the link revisited. Trends Mol Med. (2019) 25:659–61. doi: 10.1016/j.molmed.2019.05.012
61. Hu X, Cai X, Ma R, Fu W, Zhang C, Du X. Iron-load exacerbates the severity of atherosclerosis via inducing inflammation and enhancing the glycolysis in macrophages. J Cell Physiol. (2019) 234:18792–800. doi: 10.1002/jcp.28518
62. Bai T, Li M, Liu Y, Qiao Z, Wang Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Rad Biol Med. (2020) 160:92–102. doi: 10.1016/j.freeradbiomed.2020.07.026
63. Omiya S, Hikoso S, Imanishi Y, Saito A, Yamaguchi O, Takeda T, et al. Downregulation of ferritin heavy chain increases labile iron pool, oxidative stress and cell death in cardiomyocytes. J Mol Cell Cardiol. (2009) 46:59–66. doi: 10.1016/j.yjmcc.2008.09.714
64. Park TJ, Park JH, Lee GS, Lee JY, Shin JH, Kim MW, et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. (2019) 10:835. doi: 10.1038/s41419-019-2061-8
65. Lu L, Liu M, Sun R, Zheng Y, Zhang P. Myocardial infarction: symptoms and treatments. Cell Biochem Biophys. (2015) 72:865–7. doi: 10.1007/s12013-015-0553-4
66. Chen M, Li X, Yang H, Tang J, Zhou S. Hype or hope: Vagus nerve stimulation against acute myocardial ischemia-reperfusion injury. Trends Cardiovasc Med. (2020) 30:481–8. doi: 10.1016/j.tcm.2019.10.011
67. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. (2015) 59:298–308. doi: 10.1016/j.molcel.2015.06.011
68. Tang LJ, Luo XJ, Tu H, Chen H, Xiong XM, Li NS, et al. Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn-Schmiedeberg's Arch Pharmacol. (2021) 394:401–10. doi: 10.1007/s00210-020-01932-z
69. Feng Y, Madungwe NB, Imam Aliagan AD, Tombo N, Bopassa JC. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem Biophys Res Commun. (2019) 520:606–11. doi: 10.1016/j.bbrc.2019.10.006
70. Li W, Li W, Leng Y, Xiong Y, Xia Z. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol. (2020) 39:210–25. doi: 10.1089/dna.2019.5097
71. Baba Y, Higa JK, Shimada BK, Horiuchi KM, Suhara T, Kobayashi M, et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circulatory Physiol. (2018) 314:H659–68. doi: 10.1152/ajpheart.00452.2017
72. Chen HY, Xiao ZZ, Ling X, Xu RN, Zhu P, Zheng SY. ELAVL1 is transcriptionally activated by FOXC1 and promotes ferroptosis in myocardial ischemia/reperfusion injury by regulating autophagy. Mol Med. (2021) 27:14. doi: 10.1186/s10020-021-00271-w
73. Seferović PM, Polovina M, Bauersachs J, Arad M, Gal TB, Lund LH, et al. Heart failure in cardiomyopathies: a position paper from the Heart Failure Association of the European Society of Cardiology. Euro J Heart Fail. (2019) 21:553–76. doi: 10.1002/ejhf.1611
74. Gujja P, Rosing DR, Tripodi DJ, Shizukuda Y. Iron overload cardiomyopathy: better understanding of an increasing disorder. J Am College Cardiol. (2010) 56:1001–12. doi: 10.1016/j.jacc.2010.03.083
75. Vejpongsa P, Yeh ET. Topoisomerase 2β: a promising molecular target for primary prevention of anthracycline-induced cardiotoxicity. Clin Pharmacol Therap. (2014) 95:45–52. doi: 10.1038/clpt.2013.201
76. Mu H, Liu H, Zhang J, Huang J, Zhu C, Lu Y, et al. Ursolic acid prevents doxorubicin-induced cardiac toxicity in mice through eNOS activation and inhibition of eNOS uncoupling. J Cell Mol Med. (2019) 23:2174–83. doi: 10.1111/jcmm.14130
77. Wu YZ, Zhang L, Wu ZX, Shan TT, Xiong C. Berberine ameliorates doxorubicin-induced cardiotoxicity via a SIRT1/p66Shc-mediated pathway. Oxidative Med Cell Longevity. (2019) 2019:2150394. doi: 10.1155/2019/2150394
78. Guo Q, Guo J, Yang R, Peng H, Zhao J, Li L, et al. Cyclovirobuxine D attenuates doxorubicin-induced cardiomyopathy by suppression of oxidative damage and mitochondrial biogenesis impairment. Oxidative Med Cell Longevity. (2015) 2015:151972. doi: 10.1155/2015/151972
79. Kalyanaraman B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: Have we been barking up the wrong tree? Redox Biol. (2020) 29:101394. doi: 10.1016/j.redox.2019.101394
80. Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. (2012) 52:1213–25. doi: 10.1016/j.yjmcc.2012.03.006
81. Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. (2020) 5:132747. doi: 10.1172/jci.insight.132747
82. Fang X, Cai Z, Wang H, Han D, Cheng Q, Zhang P, et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circul Res. (2020) 127:486–501. doi: 10.1161/CIRCRESAHA.120.316509
83. Wang J, Deng B, Liu Q, Huang Y, Chen W, Li J, et al. Pyroptosis and ferroptosis induced by mixed lineage kinase 3 (MLK3) signaling in cardiomyocytes are essential for myocardial fibrosis in response to pressure overload. Cell Death Dis. (2020) 11:574. doi: 10.1038/s41419-020-02777-3
84. Chen X, Xu S, Zhao C, Liu B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem Biophys Res Commun. (2019) 516:37–43. doi: 10.1016/j.bbrc.2019.06.015
85. McDonagh T, Damy T, Doehner W, Lam CSP, Sindone A, van der Meer P, et al. Screening, diagnosis and treatment of iron deficiency in chronic heart failure: putting the 2016 European Society of Cardiology heart failure guidelines into clinical practice. Euro J Heart Fail. (2018) 20:1664–72. doi: 10.1002/ejhf.1305
86. Li W, Feng G, Gauthier JM, Lokshina I, Higashikubo R, Evans S, et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Investig. (2019) 129:2293–304. doi: 10.1172/JCI126428
87. Wang C, Yuan W, Hu A, Lin J, Xia Z, Yang CF, et al. Dexmedetomidine alleviated sepsis-induced myocardial ferroptosis and septic heart injury. Mol Med Rep. (2020) 22:175–84. doi: 10.3892/mmr.2020.11114
88. Chiang SK, Chen SE, Chang LC. A dual role of heme oxygenase-1 in cancer cells. Int J Mol Sci. (2018) 20:39. doi: 10.3390/ijms20010039
89. Liu B, Zhao C, Li H, Chen X, Ding Y, Xu S. Puerarin protects against heart failure induced by pressure overload through mitigation of ferroptosis. Biochem Biophys Res Commun. (2018) 497:233–40. doi: 10.1016/j.bbrc.2018.02.061
90. Fan Z, Cai L, Wang S, Wang J, Chen B. Baicalin prevents myocardial ischemia/reperfusion injury through inhibiting ACSL4 mediated ferroptosis. Front Pharmacol. (2021) 12:628988. doi: 10.3389/fphar.2021.628988
91. Nie P, Meng F, Zhang J, Wei X, Shen C. Astragaloside IV exerts a myocardial protective effect against cardiac hypertrophy in rats, partially via activating the Nrf2/HO-1 signaling pathway. Oxidat Med Cell Longevity. (2019) 2019:4625912. doi: 10.1155/2019/4625912
92. Luo LF, Guan P, Qin LY, Wang JX, Wang N, Ji ES. Astragaloside IV inhibits adriamycin-induced cardiac ferroptosis by enhancing Nrf2 signaling. Mol Cell Biochem. (2021) 476:2603–11. doi: 10.1007/s11010-021-04112-6
93. Shan X, Lv ZY, Yin MJ, Chen J, Wang J, Wu QN. The protective effect of cyanidin-3-glucoside on myocardial ischemia-reperfusion injury through ferroptosis. Oxidative Med Cell Longevity. (2021) 2021:8880141. doi: 10.1155/2021/8880141
94. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. (2016) 63:173–84. doi: 10.1002/hep.28251
95. Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. (2017) 6:e371. doi: 10.1038/oncsis.2017.65
96. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
97. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. (2021) 593:586–90. doi: 10.1038/s41586-021-03539-7
98. Song Y, Wang B, Zhu X, Hu J, Sun J, Xuan J, et al. Human umbilical cord blood-derived MSCs exosome attenuate myocardial injury by inhibiting ferroptosis in acute myocardial infarction mice. Cell Biol Toxicol. (2021) 37:51–64. doi: 10.1007/s10565-020-09530-8
99. Anderson GJ, Frazer DM. Current understanding of iron homeostasis. Am J Clin Nutrit. (2017) 106:1559s−66. doi: 10.3945/ajcn.117.155804
100. Gao JM, Meng XW, Zhang J, Chen WR, Xia F, Peng K, et al. Dexmedetomidine protects cardiomyocytes against hypoxia/reoxygenation injury by suppressing TLR4-MyD88-NF-κB signaling. BioMed Res Int. (2017) 2017:1674613. doi: 10.1155/2017/1674613
101. Cheng XY, Gu XY, Gao Q, Zong QF, Li XH, Zhang Y. Effects of dexmedetomidine postconditioning on myocardial ischemia and the role of the PI3K/Akt-dependent signaling pathway in reperfusion injury. Mol Med Rep. (2016) 14:797–803. doi: 10.3892/mmr.2016.5345
102. Wang ZK, Chen RR, Li JH, Chen JY, Li W, Niu XL, et al. Puerarin protects against myocardial ischemia/reperfusion injury by inhibiting inflammation and the NLRP3 inflammasome: The role of the SIRT1/NF-κB pathway. Int Immunopharmacol. (2020) 89:107086. doi: 10.1016/j.intimp.2020.107086
103. Li X, Yuan T, Chen D, Chen Y, Sun S, Wang D, et al. Cardioprotective effects of puerarin-V on isoproterenol-induced myocardial infarction mice is associated with regulation of PPAR-γ/NF-κB pathway. Molecules. (2018) 23:322. doi: 10.3390/molecules23123322
104. Cai SA, Hou N, Zhao GJ, Liu XW, He YY, Liu HL, et al. Nrf2 is a key regulator on puerarin preventing cardiac fibrosis and upregulating metabolic enzymes UGT1A1 in rats. Front Pharmacol. (2018) 9:540. doi: 10.3389/fphar.2018.00540
105. Zhou YX, Zhang H, Peng C. Puerarin: a review of pharmacological effects. Phytother Res. (2014) 28:961–75. doi: 10.1002/ptr.5083
106. Wu J, Chen H, Qin J, Chen N, Lu S, Jin J, et al. Baicalin improves cardiac outcome and survival by suppressing Drp1-mediated mitochondrial fission after cardiac arrest-induced myocardial damage. Oxidative Med Cell Longevity. (2021) 2021:8865762. doi: 10.1155/2021/8865762
107. Luan Y, Sun C, Wang J, Jiang W, Xin Q, Zhang Z, et al. Baicalin attenuates myocardial ischemia-reperfusion injury through Akt/NF-κB pathway. J Cell Biochem. (2019) 120:3212–9. doi: 10.1002/jcb.27587
108. Bai J, Wang Q, Qi J, Yu H, Wang C, Wang X, et al. Promoting effect of baicalin on nitric oxide production in CMECs via activating the PI3K-AKT-eNOS pathway attenuates myocardial ischemia-reperfusion injury. Phytomedicine. (2019) 63:153035. doi: 10.1016/j.phymed.2019.153035
109. Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian D, et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Different. (2019) 26:2284–99. doi: 10.1038/s41418-019-0299-4
110. Petroni K, Trinei M, Fornari M, Calvenzani V, Marinelli A, Micheli LA, et al. Dietary cyanidin 3-glucoside from purple corn ameliorates doxorubicin-induced cardiotoxicity in mice. Nutr Meta Cardiovasc Dis. (2017) 27:462–9. doi: 10.1016/j.numecd.2017.02.002
111. Aloud BM, Raj P, McCallum J, Kirby C, Louis XL, Jahan F, et al. Cyanidin 3-O-glucoside prevents the development of maladaptive cardiac hypertrophy and diastolic heart dysfunction in 20-week-old spontaneously hypertensive rats. Food Funct. (2018) 9:3466–80. doi: 10.1039/C8FO00730F
112. Miotto G, Rossetto M, Di Paolo ML, Orian L, Venerando R, Roveri A, et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. (2020) 28:101328. doi: 10.1016/j.redox.2019.101328
113. Fang D, Maldonado EN. VDAC regulation: a mitochondrial target to stop cell proliferation. Adv Cancer Res. (2018) 138:41–69. doi: 10.1016/bs.acr.2018.02.002
114. Rocha VC, França LS, de Araújo CF, Ng AM, de Andrade CM, Andrade AC, et al. Protective effects of mito-TEMPO against doxorubicin cardiotoxicity in mice. Cancer Chemother Pharmacol. (2016) 77:659–62. doi: 10.1007/s00280-015-2949-7
115. Olgar Y, Billur D, Tuncay E, Turan B. MitoTEMPO provides an antiarrhythmic effect in aged-rats through attenuation of mitochondrial reactive oxygen species. Experi Gerontol. (2020) 136:110961. doi: 10.1016/j.exger.2020.110961
116. Sun Y, He L, Wang T, Hua W, Qin H, Wang J, et al. Activation of p62-Keap1-Nrf2 pathway protects 6-hydroxydopamine-induced ferroptosis in dopaminergic cells. Mol Neurobiol. (2020) 57:4628–41. doi: 10.1007/s12035-020-02049-3
117. Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. (2013) 51:618–31. doi: 10.1016/j.molcel.2013.08.003
118. Poller W, Dimmeler S, Heymans S, Zeller T, Haas J, Karakas M, et al. Non-coding RNAs in cardiovascular diseases: diagnostic and therapeutic perspectives. Eur Heart J. (2018) 39:2704–16. doi: 10.1093/eurheartj/ehx165
Keywords: ferroptosis, reactive oxygen species, lipid peroxidation, autophagy, cardiovascular diseases
Citation: Chen Z, Yan Y, Qi C, Liu J, Li L and Wang J (2021) The Role of Ferroptosis in Cardiovascular Disease and Its Therapeutic Significance. Front. Cardiovasc. Med. 8:733229. doi: 10.3389/fcvm.2021.733229
Received: 02 July 2021; Accepted: 17 September 2021;
Published: 26 October 2021.
Edited by:
Paul Poirier, Laval University, CanadaReviewed by:
Pamela Maher, Salk Institute for Biological Studies, United StatesSeitaro Nomura, The University of Tokyo, Japan
Copyright © 2021 Chen, Yan, Qi, Liu, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junnan Wang, jdeywjn@163.com