MicroRNA-Related Strategies to Improve Cardiac Function in Heart Failure
 Huatao Zhou
Huatao Zhou Weijie Tang1
Weijie Tang1  Jinfu Yang
Jinfu Yang Jun Peng
Jun Peng Chengming Fan
Chengming Fan- 1Department of Cardiovascular Surgery, The Second Xiangya Hospital, Central South University, Changsha, China
- 2Department of Pharmacology, Hunan Provincial Key Laboratory of Cardiovascular Research, Xiangya School of Pharmaceutical Sciences, Central South University, Changsha, China
- 3Hunan Fangsheng Pharmaceutical Co., Ltd. Changsha, China
Heart failure (HF) describes a group of manifestations caused by the failure of heart function as a pump that supports blood flow through the body. MicroRNAs (miRNAs), as one type of non-coding RNA molecule, have crucial roles in the etiology of HF. Accordingly, miRNAs related to HF may represent potential novel therapeutic targets. In this review, we first discuss the different roles of miRNAs in the development and diseases of the heart. We then outline commonly used miRNA chemical modifications and delivery systems. Further, we summarize the opportunities and challenges for HF-related miRNA therapeutics targets, and discuss the first clinical trial of an antisense drug (CDR132L) in patients with HF. Finally, we outline current and future challenges and potential new directions for miRNA-based therapeutics for HF.
Introduction
Heart failure (HF) is a complex clinical syndrome with varied pathophysiology that occurs because cardiac output is insufficient to meet the metabolic needs of organs and tissues in the body (1). HF is very common, with an estimated prevalence of 1–3% in the adult population worldwide, and its incidence rises with advancing age (2). Despite substantial therapeutic advances over the past decades, HF remains a major cause of morbidity and mortality worldwide (1). HF survival rates are no better than they were a decade ago, with an ~53% 5-year survival rate from diagnosis (3). Hence, HF is a global public health problem that requires urgent attention to open a window of opportunity for early diagnosis/prognosis, prevention, and treatment (4).
MicroRNAs (miRNAs) are natural, endogenous non-coding single-stranded RNA molecules of around 22 nucleotides that regulate the expression levels of various genes through Watson-Crick base pairing with target messenger RNAs (mRNAs) at the posttranscriptional level, via binding to the 3′ untranslated regions (UTRs) of target mRNAs (5). The role of miRNAs was first described in Caenorhabditis elegans, where silencing of the lin-14 mRNA at various time points during growth resulted in the normal development of a worm from an embryo (6). During the last few decades, increasing numbers of investigations have indicated the regulatory functions of miRNAs in a diverse range of cellular biological processes (7). According to the latest release of miRBase, more than 2500 miRNAs have been reported to exist in the human genome to date (8). Furthermore, bioinformatic analyses have indicated that the expression levels of 30% of human protein-coding genes are regulated by miRNAs through a series of complex signaling pathways (9).
Encyclopedia of DNA Elements (ENCODE) is an international cooperative project that aims to establish a comprehensive database for human genome data research by integrating DNA, RNA, protein, epigenetic modification, and other levels of data (10–12). The ENCODE project is another tremendous achievement of the international scientific community in the field of genomics, following the Human Genome Project, and is an approach that facilitates improved understanding of miRNAs and their effects in disease pathogenesis (13, 14).
In this review, we give an overview of different types of chemical modifications and viral-vector-based delivery systems used for miRNA modulation in HF, as well as summarizing the opportunities and challenges for miRNAs as therapeutic targets for the treatment of HF. Furthermore, we discuss CDR132L, the first miRNA drug used to treat HF, and future prospects for such therapies.
MiRNA Biogenesis and Functions
The synthesis of miRNA begins with the transcription of miRNA genes by RNA polymerase II. This transcriptional process, which involves transcription factors in a similar way to protein-coding transcripts, leads to the generation of A-tail-capped long transcripts, referred to as primary microRNA or pri-microRNA (15, 16). In the nucleus, a microprocess complex containing two important constituents, the double-stranded RNase III enzyme, DROSHA, and the cofactor, DiGeorge syndrome critical region 8 (DGCR8), processes pri-miRNA to generate precursor miRNA or pre-miRNA (17, 18). In the cytoplasm, pre-microRNAs are further cleaved by the cytoplasmic nuclease, DICER, to produce double-stranded RNA molecules of 18–22 nucleotides, cut from the stem of the hairpin, similar to the double-stranded structure of small interfering RNA (siRNA) (19). The duplex comprises a guide strand (mature miRNA) and a passenger strand (miRNA*) (20). While loading on RISC, only the mature miRNA will become active in the silencing procedure, while the miRNA* is usually non-functional and is degraded (21). A multi-protein RNA-silencing complex composed of RISC and miRNA (22) recognizes target RNA molecules by Watson-Crick base pairing to partially complementary sites, mainly located in the 3′ UTR of target mRNAs, and can inhibit the function of corresponding genes in several ways, as follows: blocking initiation and elongation, forcing premature termination of translation, deadenylation of mRNAs to prevent their reuse, and (most importantly) mRNA degradation (23–27) (Figure 1).
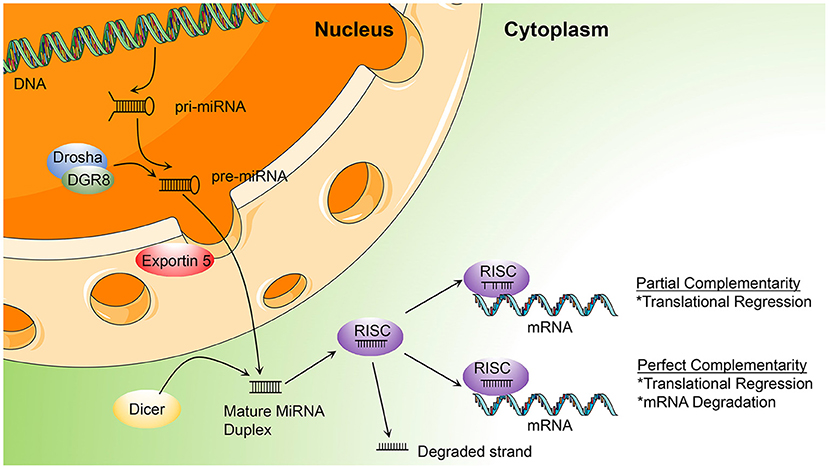
Figure 1. Illustration of the process of microRNA (miRNA) biogenesis and function, described in miRNA biogenesis and functions (DGCR8, DiGeorge syndrome critical region gene 8; RISC, RNA-induced silencing complex; pri-miRNA, primary miRNA; pre-miRNA, precursor microRNA).
MiRNA Functions in Cardiovascular System Development
The heart is the first organ to form and function during embryonic development. For normal morphogenesis and function, the development process must be uninterrupted (28). MiRNAs can manipulate cardiac gene expression at the posttranscriptional level (29). The requirement for miRNAs in cardiovascular development and function was initially proven by the deletion of the tissue-specific gene, Dicer1, in mice; Dicer1 encodes an enzyme essential for miRNA processing (30), and its deletion in vascular lineages and myocardial tissue results in embryonic lethality and defective heart morphogenesis (30, 31). Subsequently, numerous miRNAs that contribute to heart morphogenesis have been identified; for example, miR-1 and miR-133 contribute to cardiomyocyte proliferation, as well as the miR-15 family, thereby regulating congenital heart disease development or regeneration (32–35).
Several myomirs, including miR-1, miR-133a, miR-208a, miR-208b, and miR-499, play important roles in normal embryological development of the heart and precise regulation of these myomirs is crucial for normal cardiac development (36). The field of cellular reprogramming and trans-differentiation is rapidly evolving, and cardiac fibroblasts have been demonstrated to directly differentiate into induced cardiomyocytes (iCMs) under the combined influence of the transcription factors GATA4, MEF2C, and TBX5 (GMT) (37). Subsequently, Jayawardena et al. (38) demonstrated that the combination of miR-1, miR-133, miR-208, and miR-499 also function to directly convert fibroblasts to a cardiomyocyte-like phenotype in vitro. Among these miRNAs, miR-133 is considered to mediate the further maturation of transdifferentiated iCMs (39). The importance of miRNA-mediated post-transcriptional regulation in cardiovascular homeostasis, as well as its impact on the pathogenesis, diagnosis, and prognosis of heart disease, is established in the scientific community. Understanding the mechanisms underlying heart development may provide new perspectives relevant to cardiac reprogramming technology, which could, in turn, pave the way for development of miRNA-based approaches for heart disease therapy in the future.
Chemical Modifications of MiRNAs
MiRNAs are vulnerable to degradation by nucleases; therefore, chemical modifications to protect miRNAs from nucleases are major solutions that can enhance RNA stability and improve their efficacy (40). Optimal anti-miRNA oligonucleotides (AMOs, or anti-miRs) are designed to be completely complementary with specific mature miRNAs and are chemically modified. Lennox et al. (41) found that chemical modifications of anti-miRs could accelerate their invasion of RISC. At present, the main chemical modification methods used in preclinical studies are phosphorothioate (PS), locked nucleic acid (LNA), and ribose-2'-OH modification (Figure 2).
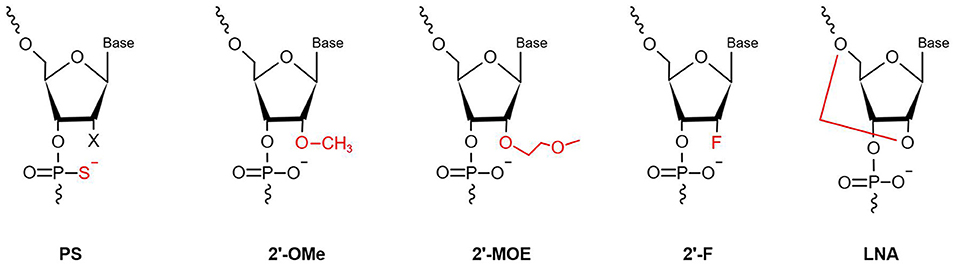
Figure 2. Chemical modifications applied in the AMOs design (PS, phosphorothioate; 2′-OMe, 2′-O-methyl; 2′-F, 2′-fluoro-RNA; 2′-MOE, 2′-O-methoxyethyl; LNA, Locked Nucleic Acid. In red is highlighted the site and the type of modification).
PS modification, also referred to as oligonucleotide backbone modification, involves replacement of a non-bridging oxygen atom in the phosphodiester bond with a sulfur atom (42). PS can reduce exonuclease- and endonuclease-mediated miRNA degradation; therefore, PS bonds usually are designed near the 5′ and 3′ ends of anti-miRs, to enhance their stability (43). As well as resistance to nucleases, PS-modification can reduce plasma clearance of anti-miRs, increase their stability in serum, and lengthen their half-life, to improve their pharmacokinetic properties, by giving them high affinity with serum albumin (44). Every PS-antimiR substitution reduces the melting temperature of the heteroduplex by 0.5°C (45); however, although the thermal stability of PS-antimiRs is reduced, their increased hybridization specificity and greater nuclease resistance can compensate for this shortcoming. Partial PS oligodeoxynucleotides (ODNs) targeted against the AT1 receptor mRNA are more effective than full PS ODNs in decreasing blood pressure, as well as having lower cost and improved specificity (46). Compared with other modification strategies, PS modification of anti-miRs is more effective in vitro. Two PS modifications of 2'-O-methyl RNA/LNA (2'OMe/LNA) mixtures were reported to be the most efficient of all chemical compounds tested in vitro (43).
LNA, as a modified nucleotide analog, is also termed inaccessible RNA, and uses one of its methylene bridges to fix the ribose ring, by linking an oxygen atom at the 2′ position to a carbon atom at the 4′ position. The methylene bridge is usually in the C3′-endo conformation (47). LNA modifications reduce nuclease degradation by increasing RNA stability and avoid off-target effects through 5′ end modification, which prevents molecules from merging into RISC (48, 49). The high affinity of LNA allows shorter anti-miR sequences to be designed for applications, with similar efficiency (50). Obad et al. (50) first demonstrated that it is feasible for tiny LNAs (including those of only 8 nucleotides) to silence miRNAs with negligible off-target effects. Subsequently, Bernardo et al. (51) showed that the seed-targeting 8-mer LNA-modified antimiR-34 (LNA-antimiR-34) could silence the miR-34 family to prevent pathological cardiac remodeling and improve cardiac function. Another study found that silencing of miR-34a using LNA-antimiR-34a in mice with moderate cardiac pathology reduced atrial dilatation and prevented failure of cardiac function (52). A recent study also found that LNA-antimiR-34 doubled the cardiac progenitor growth rate by inhibiting miR-34 expression (53). The first identified miRNA, miR-1, has a critical role in cardiac development and disease (54); it can significantly inhibit the expression of PKCe and HSP60 to promote cardiac injury. Pan et al. (55) verified that LNA modified antimiR-1 (LNA-antimiR-1), at a dose of 1 mg/kg, down-regulated miR-1 expression by 83% and efficiently reduced cardiac ischemia/reperfusion injury. Wang et al. (56) found that LNA-antimiR-1 remarkably inhibits the production of reactive oxygen species in neonatal Wistar rat cardiomyocytes.
Ribose-2′-OH modification describes the substitution of alternative chemical groups in the 2′ position, primarily 2′-O-methyl (2′-O-Me), 2′-meth-oxyethyl (2′-O-MOE), and 2′-fluoro (2′-F) (57). Ribose-2′-OH modification can enhance nuclease resistance and reduce immunoreactivity (58); however, in most cases, modification is conducted together with LNA or PS modification, or occasionally even both together. Intravenous administration of 2′-O-Me-anti-miRs targeting miR-16, miR-122, miR-192, and miR-194 could decrease the corresponding miRNA levels in mice (59). Similarly, inhibition of miR-133 using an anti-miR with 2′-O-Me modification leads to hypertrophy in vivo (60).
All of the chemical modifications described above have the common advantage of improving nuclease resistance, while PS modification alone also reduces thermal stability and affinity. LNA has become the most commonly used anti-miR chemical modification; however, use of LNA modification alone may decrease RNase H activity. Most anti-miRs tend to be designed with these three chemical modifications in the first and last five nucleotides, referred to as gapmers. These molecules have a good affinity for their target RNAs, effectively improve RNase H activity, and are widely used in many miRNA therapeutics (61, 62).
Overall, AMOs are an effective means of functionalizing miRNAs, both in vivo and in vitro. The differential effects of chemical modifications of AMOs are summarized in Table 1.
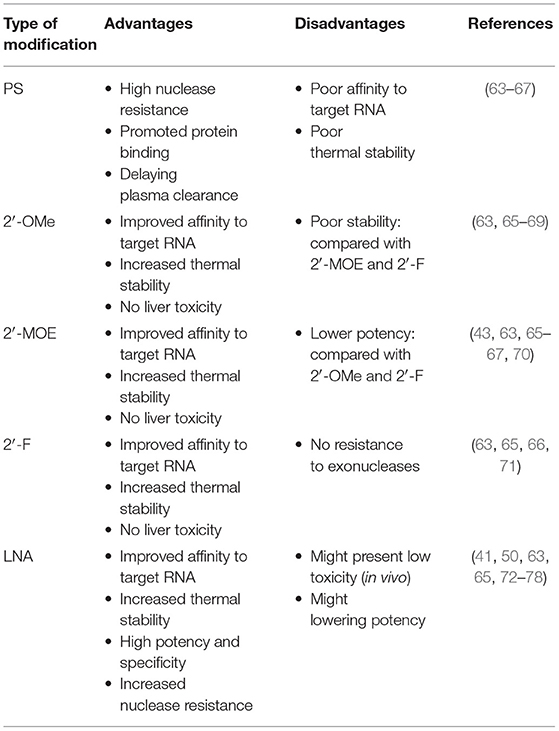
Table 1. Advantages and disadvantages of chemical modifications applied to the AMOs.
Design of MiRNA Delivery Vehicles
When miRNA therapies are applied for heart disease, it is necessary to consider the non-polar and hydrophobic properties of the myocardial cytomembrane, which represent an enormous challenge to miRNA delivery, as miRNAs have a negative charge and are hydrophilic. Therefore, crossing the myocardial cytomembrane is a vital step in transferring miRNAs to their targets successfully and generating effective miRNA therapies. Effective miRNA delivery requires the molecules to exhibit hypocytotoxicity, high transfection efficiency, and specificity. MiRNA delivery vehicles are mainly divided into viral and non-viral vector-based approaches. In this review, we discuss viral-vector-based delivery systems; see refs (79–82) for detailed information on non-viral miRNA vectors. Viral vector delivery aims to reach the target cell using the genome of the virus itself, and has been used extensively for delivery of miRNA therapies. The main viruses used for this purpose are adenoviruses, adeno-associated viruses (AAVs), and lentiviruses.
Adenoviruses, are double-stranded DNA viruses commonly used as viral vectors. As early as 1992, Stratford-Perricaudet et al. (83) found that injecting adenovirus LacZ vectors into neonatal mice resulted in extensive gene transfer in cardiomyocytes. Subsequent studies have shown that adenovirus-mediated miRNA-24 upregulation can enhance myocardial angiogenesis and blood perfusion in myocardial infarction (MI) tissue. While this approach can induce cardiomyocyte and fibroblast apoptosis, overall, it produced a good MI treatment effect (84). Another study using miRNA therapy for heart failure demonstrated that adenovirus vector contributed to up-regulation of AMPKα2, the direct target of miR-195a-3p, helping to relieve cardiac hypertrophy and avoid heart failure (85). Nevertheless, serious flaws have emerged on wide application of adenovirus delivery in clinical studies; for example, use of adenovirus vectors for treatment of malignant intracranial tumors resulted in serious side effects, including headache, change of mental status, and relapsing seizures (86) and such side effects limit the use of adenoviruses for delivery of miRNA therapies.
AAVs are single-stranded, non-enveloped DNA viruses belonging to the “Parvoviridae” family, and are among the most promising vector delivery systems. Unlike other viruses, AAVs are not pathogenic to humans, have minimal immunogenicity, and are low molecular weight (83). AAV2, AAV6, and AAV9 are the most cardiogenic AAVs among AAV1 to AAV9, with AAV6 vector exhibiting the most efficient transduction (87). Bian et al. (88) found that AAV6-mediated over-expression of miR-199a promoted the proliferation of human induced-pluripotent stem cells (hiPSC-CMs), which could improve cardiac function and fibrosis. Further, a recent study demonstrated that myocardial tissue in aorta-constricted mice transfected with miR-124 via AAV9 inhibited the influence of shikonin on sympathetic remodeling, revealing the mechanism underlying the use of shikonin for treatment of chronic heart failure (89). AAV6 and AAV9 have become the main AAVs serotypes used for heart disease therapy. In addition, Gao et al. found that delivery of miR-19a/19b using AAV vectors may reduce the cardiac trauma induced by myocardial infarction and protect heart function (90).
A number of studies have reported the use of AAV vectors for miRNA therapy in heart disease. The latest research showed that varying levels of cardiac miRNA-122 have a crucial effect on the treatment efficiency of AAV vectors regulated by miRNAs (91); however, another report noted severe cardiotoxicity in AAV6 vector-mediated shRNA cardiac gene transfer (92). Notably, AAV antibodies are present in most people, which may influence the effectiveness of vector entry and transgenic expression.
Lentiviruses are a subgroup of retroviruses, and lentivirus vectors are often used for miRNA delivery into myocardial cells. Yang et al. (93) demonstrated that downregulation of miR-322 via lentiviral transduction could further prevent cardiomyocyte apoptosis induced by hypoxia. Similarly, Wang et al. (94) showed that reduction of miR-137 levels, mediated by lentivirus vectors, decreased cardiomyocyte apoptosis. Although lentivirus-mediated miRNA therapy has been applied for the treatment of heart disease, the disadvantages of insertional mutagenesis may limit its use (95, 96).
MiRNA-related therapy based on viral vectors has certain advantages. For example, it is easy to generate vectors and they exhibit high transduction efficiency. Moreover, the long-term and stable gene expression mediated by these vectors makes them potentially ideal in this context; however, safety issues represent a huge obstacle that requires further research.
The Role of MiRNAs in Heart Failure
Cardiac remodeling, a basic mechanism underlying HF, is the process of generating changes in the size, shape, and function of the heart, and responds to internal and external cardiovascular injury or risk factors (97). Pathological ventricular remodeling has three main characteristics: extensive fibrosis, pathological cardiomyocyte hypertrophy, and myocardial cell apoptosis (98). In this review, we summarize the regulatory effects of newly-discovered miRNAs in cardiac remodeling, particularly cardiac hypertrophy (Figure 3), fibrosis (Figure 4), and apoptosis (Figure 5).
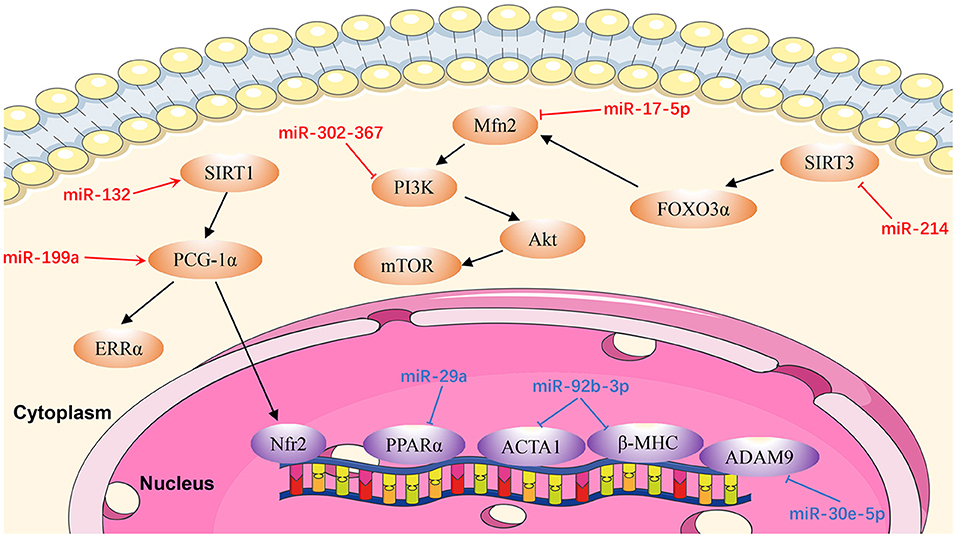
Figure 3. Reviewed miRNAs in cardiac hypertrophic pathways (Red color miRNA, pro-hypertrophic function; Blue color miRNA, anti-hypertrophic function).
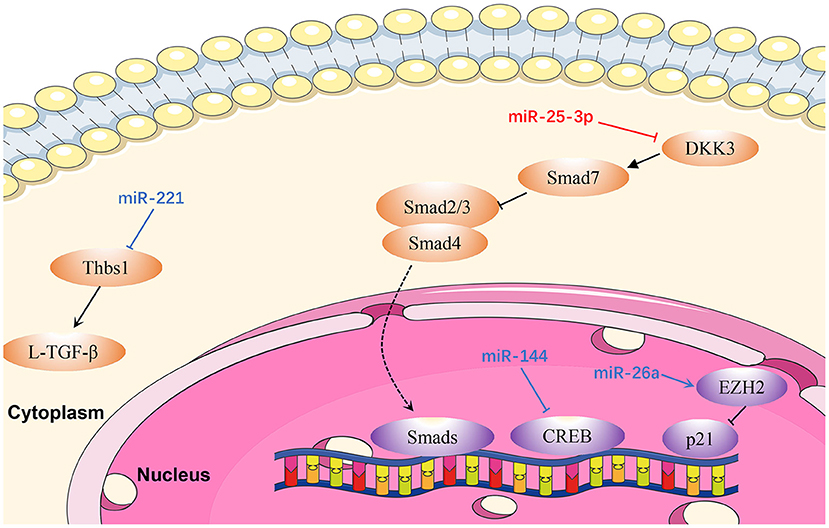
Figure 4. Reviewed miRNAs in cardiac fibrosis pathways (Red color miRNA, pro- fibrosis function; Blue color miRNA, anti- fibrosis function; Dash lines indicate translocation of molecules from cytoplasm to nucleus).
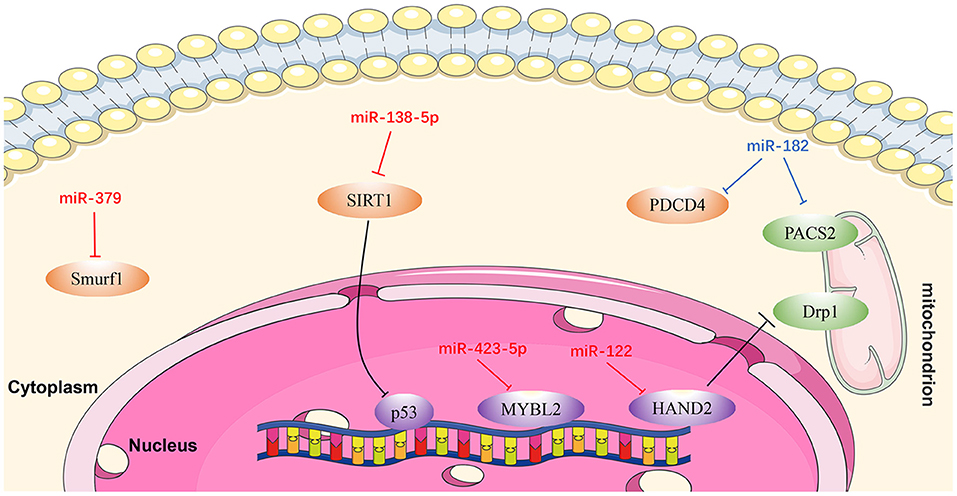
Figure 5. Reviewed miRNAs in cardiomyocyte apoptosis pathways (Red color miRNA, pro-apoptosis function; Blue color miRNA, anti-apoptosis function).
MiRNAs in Cardiac Hypertrophy
MiR-17-5p
Xu et al. (99) identified miR-17-5p as a critical miR that regulates the expression and function of the mitochondrial fusion protein, mitofusin 2 (MFN2). They found that miR-17-5p expression in cardiomyocytes was upregulated in rat hearts at 4 weeks after transverse aortic constriction and in an angiotensin II (Ang II)-induced cell hypertrophy model. Mechanistic in vivo and in vitro studies demonstrated that miR-17-5p suppresses autophagy to promote cardiac hypertrophy by inhibiting Mfn2 expression, and activating the phosphatidylinositol-3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway. Furthermore, Ang II-induced cell hypertrophy in neonatal rat left ventricle myocytes was significantly reversed by an miR-17-5p inhibitor. These findings suggest an essential role for miR-17-5p in cardiovascular disease and present potential new therapeutic targets in patients with pathological cardiac hypertrophy (99).
MiR-29a
MiR-29a is significantly associated with cardiac hypertrophy and a potent therapeutic target for the treatment of cardiac hypertrophy (100). Upregulation of miR-29a in vivo can attenuate the cardiac hypertrophy response induced by isoproterenol hydrochloride via targeting the nuclear receptor peroxisome proliferator-activated receptor δ (PPARD) and downregulating atrial natriuretic factor, suggesting that miR-29a can protect the myocardium in cardiac hypertrophy (100).
MiR-30e-5p
Upregulation of miR-30e-5p has anti-hypertrophic effects in hypertrophic cardiomyocytes induced by Ang II, via targeting a disintegrin and metallopeptidase domain 9 (meltrin gamma) (101). Overexpression of miR-30e-5p also attenuates hypoxia-induced apoptosis in an hiPSC-CM injury model by targeting the 3'-UTR of BCL2L11, which is an apoptosis activator and autophagy suppressor (102).
MiR-92b-3p
According to Yu et al. (103), miR-92b-3p is up-regulated in mouse cardiomyocytes under Ang II-induced conditions. MiR-92b-3p, is downstream of heart and neural crest derivatives-expressed 2 (HAND2), and is involved in cardiac hypertrophy through targeting ACTA1 and MYH7, which are both genes involved in hypertrophy. In Ang II-induced cardiomyocyte hypertrophy, therapeutic studies using miR-92b-3p mimics reduced cardiomyocyte cell size and inhibited the expression levels of ACTA1 and MYH7.
MiR-132
Another anti-hypertrophic miR, miR-132, controls cardiac hypertrophy in a novel porcine model of pressure-overload-induced heart failure via PPARGC1A/NFE2 signaling by targeting SIRT1 (104). Furthermore, Li et al. (105) proved that NFE2 protects the murine heart against pathological cardiac hypertrophy and heart failure. Furthermore, NFE2 is increased in myocardium of anti-miR-132-treated porcine hearts with percutaneous transverse aortic constriction compared with the control group at the 8-week time point, indicating that this could be an effective therapeutic strategy for patients with heart failure (104). Therefore, antimiR-132 may be pivotal in mediation of pathologic heart hypertrophy.
MiR-199a
Yan et al. (106) demonstrated that miR-199a is increased in pressure-induced cardiac hypertrophy, while inhibition of miR-199a mitigates cardiac hypertrophy in vitro. Furthermore, anti-miR-199a attenuates cardiac hypertrophy and restores cardiac function in vivo through the PPARGC1A (PGC-1α)/ESRRA (ERRα) axis. The mechanism underlying restoration of mitochondrial structure and function in anti-miR-199a-treated mice involves the downstream pathways of mitochondrial fatty acid oxidation and oxidative phosphorylation (107).
MiR-214
MiR-214 levels are dramatically raised in Ang II-infused mice (108). Hypertrophic stimuli cause increased expression of miR-214 in cardiomyocytes, resulting in miR-214-mediated hypertrophic growth. In contrast, inhibition of miR-214 confers protection from Ang II-mediated hypertrophy in vivo. These effects are attributable to increased expression of SIRT3, which participates in cardiomyopathy pathogenesis by inducing mitochondrial injury and energy metabolism disorder. Using a dual-luciferase reporter assay Ding et al. (108) demonstrated that SIRT3 was a direct target of miR-214. These results provide information about the biological functions of miR-214 and indicate that it may be a promising target for cardiac hypertrophy therapy.
MiR-302/367
An initial study of the role of miR-302/367 focused on its function in H9c2 cells treated with Ang II. The expression of miR-302/367 is up-regulated and leads to autophagy in an in vitro hypertrophic model (109). In addition, loss- and gain-of-function assays demonstrated that miR302/367 aggravates cardiac hypertrophy by inhibiting autophagy. Jin et al. (109) demonstrated that upregulation of miR302/367 expression acts as an endogenous inhibitor of autophagy in hypertrophic H9c2 cells through PTEN/PI3K/AKT/mTORC1 signaling. Anti-hypertrophic effects were observed in Ang II-induced hypertrophic H9c2 cells via the inhibition of miR302/367. These findings reveal the critical role of miR302/367 in mediating hypertrophy of the heart through PTEN/PI3K/ AKT/mTORC1 signaling (109).
MiRNAs in Cardiac Fibrosis
MiR-25-3p
Zeng et al. (110) proved that miR-25-3p expression is mediated by NF-κB signaling in cardiac fibrosis, and the regulation of fibrosis-related gene expression by miR-25-3p is observed both in vitro and in vivo. Mechanistically, DKK3 expression is reduced in cardiac fibrosis under modulation by miR-25-3p via inhibition of SMAD7 and promotion of SMAD3 and fibrosis-related gene expression (110). Hence, miR-25-3p represents a potentially promising drug target in cardiac fibrosis.
MiR-26a
MiR-26a is downregulated in the plasma and myocardium of spontaneously hypertensive rats (111). Zhang et al. (111) demonstrated that miR-26a-deficient mice exhibit increased myocardial fibrosis, whereas overexpression of miR-26a significantly inhibited myocardial fibrosis in vivo and Ang II-induced fibrogenesis in cardiac fibroblasts by directly targeting connective tissue growth factor and SMAD4 (111). In addition, cardiac fibroblast proliferation is inhibited by miR-26a via the EZH2/P21 pathway. These results reveal a novel role for miR-26a in hypertensive myocardial fibrosis and provide a possible treatment strategy for this condition.
MiR-144
Li et al. (112) demonstrated that miR-144 was dramatically down-regulated in response to pathological stimuli. Upregulation of miR-144 significantly decreased the proliferation and migration ability of cardiac fibroblasts, and also reduced the transformation from fibroblasts to myofibroblasts, whereas downregulation of miR-144 could reverse these effects (113). In that study, bioinformatics analysis and luciferase reporter assays demonstrated that miR-144 directly targets and downregulates CREB expression in cardiac fibroblasts treated with Ang II (113).
MiR-221
Zhou et al. (114) proved that miR-221 inhibits the activation of L-TGF-β1 by directly targeting THBS1, thus mitigating cardiac fibrosis and improving cardiac function. Similarly, miR-221 mimics transfected into rat cardiac fibroblasts induced by kidney failure resulted in reduction of cardiac fibrosis. Therefore, miR-221 mimics are a promising therapeutic target in cardiac fibrosis (114).
MiRNAs in Cardiomyocyte Apoptosis
MiR-122
MiR-122 is one of several miRNAs elevated in patients with HF, and plays a pivotal role in cardiac insufficiency by inducing cardiomyocyte apoptosis (115). Shi et al. (116) found that DRP1 levels were increased in response to upregulation of miR-122, indicating that apoptosis (and cardiac dysfunction) increase through DRP1-mediated up-regulation of mitochondrial fission. Furthermore, miR-122 interacts directly with a transcription factor regulating its own expression, and the effect of miR-122 on DRP1 is mediated by HAND2 (116). These findings demonstrate that miR-122 exerts a regulatory role in cardiomyocyte apoptosis via suppressing HAND2, which results in increased expression of DRP1, and ultimately leads to apoptosis (116).
MiR-138-5p
Recent research showed that SIRT1 is a potential candidate target gene of miR-138-5p and that miR-138-5p levels were negatively correlated with those of SIRT1 in cardiomyocytes (117). SIRT1 alleviates HF by enhancing P53 deacetylation, thereby inhibiting cardiomyocyte apoptosis. MiR-138-5p can decrease SIRT1 expression in H2O2-induced AC-16 and HCM cells by activating P53 signaling. By contrast, in vitro knockdown of miR-138-5p has a clear protective effect on cardiomyocytes in HF models (118). In summary, miR-138-5p inhibits SIRT1 enzyme activity by activating P53 signaling, resulting in deterioration of HF (118).
MiR-182
Rapid ventricular pacing downregulates miR-182 levels and induces cardiomyocyte apoptosis and HF in rats (119). An in vitro study illustrated that PDCD4 can promote tumor cell apoptosis by affecting the translation of eukaryotic initiation factor-4A (eIF4A) and eIF4G (120). PACS2 is an initiator of apoptosis, which accelerates displacement of mitochondrial death pathway agonists (121, 122). Expression levels of both PDCD4 and PACS2 were inhibited following upregulation of miRNA-182, while the apoptotic rate of cardiomyocytes in HF rats decreased. These results suggest that miR-182 inhibits cardiomyocyte apoptosis induced by non-ischemic HF via downregulating PDCD4 and PACS2 (119).
MiR-379
Chen et al. (123) discovered that the KLOTHO gene is a suppressor of aging whose deficiency can damage heart function and lead to heart failure. In addition, an antimir for miR-379 can prevent H9c2 cell apoptosis induced by KLOTHO deficiency, while miR-379 mimics can induce apoptosis of H9c2 cells (123). These authors also proved that the inhibition of SMURF1 may be related to H9c2 cell apoptosis induced by miR-379. In summary, miR-379 promotes cardiomyocyte apoptosis via targeting SMURF1, which is essential for mir-379-induced apoptosis. Anti-mir-379 represents a potential therapeutic target for cardiomyocyte apoptosis (123).
Mir-423-5p
A recent study confirmed that downregulation of miR-423-5p reduced hypoxia/reoxygenation-mediated cardiomyocyte injury by targeting MYBL2 in cardiomyocytes through the WNT/β-catenin signaling pathway, and that this process can be reversed by treatment with an miR-423-5p inhibitor (124).
Overall, various miRNAs modulate different mechanisms and signaling pathways that promote or protect against heart failure and those miRNAs are potential therapeutic targets in patients with heart failure.
However, a number of obstacles limit the clinical applicability of anti-miRNA agents as cardiovascular disease therapeutics. In particular, their potential off-target effects, which will result in unwanted toxicity, remain major challenges to be overcome (125). For example, Hinkel et al. (126) demonstrated that the administration of antimiR-21 in a pig model of HF caused considerable downregulation of miR-21 in the lung and kidney, which may cause unwanted side effects in these organs. This finding had also been reported in other investigations and was not unexpected (57). For the treatment of HF, it is necessary to develop a non-invasive and efficient tissue-specific drug delivery method. Moreover, it should be noted that many microRNA's systems effect is complicated by the fact that some microRNAs are protective in cancer but detrimental in cardiovascular setting. For example miR-34 (127, 128). Cardiotoxicity of a microRNA therapeutic is definitely something that limited the full potential of a specific microRNA therapeutics. The underlying mechanisms of a microRNA's effect or just with its seed sequence still need to be elucidated.
Identification of the specific target of an miRNA is one approach for determining the role of an miRNA in biological or pathological processes (129); however, a single miRNA can have thousands of targets. Further, experimentally determined miRNA genes and targets are far removed from clinical and translational demands. At present, machine learning-based computational targets and gene predictions have become an intense focus in this field (130), and have the potential to initiate a new era in miRNA research in various diseases (131). A summary of all miRNAs known to be involved in human cardiovascular diseases and their predicted target genes is presented in Table 2.
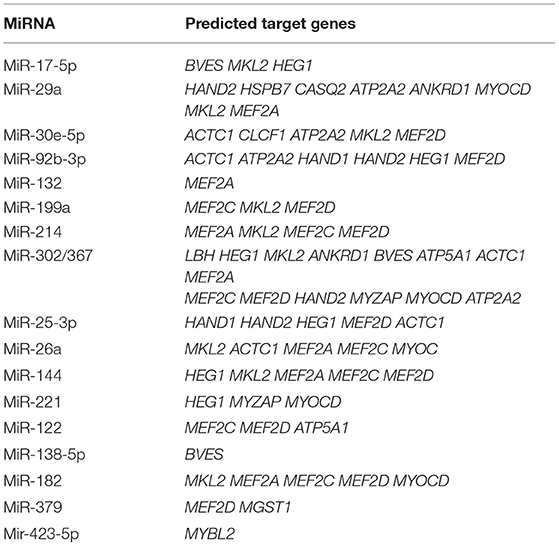
Table 2. All miRNAs summarized in this review and their predicted target genes in human cardiovascular diseases (TargetScanHuman Release 7.2).
A Milestone Breakthrough in the Treatment of Heart Failure: First-in-human Evidence (CDR132L)
Progress over the past three decades has significantly improved the development of nucleic acid therapeutics. The first-in-human study of an miRNA-based therapy was MRX34, a liposome-based miR-34a mimic, for the treatment of advanced solid tumors in April 2013 (132, 133). This provided valuable insights into the potential for application of new oligonucleotide-based drugs in oncology (134). To date, several RNA-targeted drugs have been approved for commercial use, while others are in the final phases of clinical trials (135–137).
MiRNA-132-3p (miR-132) is a non-coding RNA whose cardiac expression is up-regulated in patients under cardiomyocyte stress. High expression of miR-132 in heart tissue leads to progressive cardiac remodeling, and thereby HF events. In addition, preclinical animal experiments show that miR-132 can affect signaling pathways related to cardiomyocyte growth, autophagy, calcium handling and contraction and, more significantly, can down-regulate FOXO3 levels and inhibit the expression of genes related to intracellular calcium handling and contraction, which can lead to cardiac remodeling (138). Hence, miR-132 has attracted the attention of clinicians as a promising molecular target for HF treatment.
CDR132L, a synthetic lead-optimized oligonucleotide targeting miR-132, was the first miRNA-132 inhibitor. In related preclinical studies, the application of CDR132L significantly improved cardiac function, and can attenuate, or even reverse, HF (139).
Based on these findings, Täubel et al. published the first in-human clinical trial of CDR132L in patients with HF. The study was a randomized, double-blind, placebo-controlled, dose-escalation clinical trial. This Phase 1b clinical trial confirmed for the first time that CDR132L is safe and well-tolerated in humans, with no obvious toxicity. Simultaneously, CDR132L can significantly reduce the level of NT-proBNP, a biomarker of heart failure and narrow QRS complex, and lead to positive trends in myocardial fibrosis markers (140). This represents a huge transformation in treatment for patients with heart disease, from symptomatic and supportive therapies, to etiological treatments (141). As clinical researchers and medical workers, while maintaining a cautious attitude toward data generated from small sample sizes, we should also actively recognize the “encouraging” pharmacodynamic performance of CDR132L. Larger-scale clinical studies are needed in the future to further confirm the positive role of CDR132L in the treatment of heart failure. More broadly, this research can inspire more studies investigating RNA-based therapeutics for cardiovascular disease.
Conclusion
As miRNAs can target a wide range of genes (mRNAs), and an individual gene can be regulated by multiple miRNAs, the complex communication networks between these two molecule types indicate that miRNAs can regulate numerous different biological phenomena, ranging from hypertrophy and fibrosis to angiogenesis, among other processes. Therefore, miRNAs have significant potential for preventing, alleviating, and even restoring cardiac dysfunction, such as adverse ventricular remodeling. Further, therapeutic cocktails including miRNA inhibitors and treatment strategies targeting multiple genes involved in disease development may produce remarkable results; however, the broad clinical application of miRNA-related therapy must first overcome several obstacles, including targeted delivery, off-target effects, and hepatic/renal toxicity. Thus, further attempts to promote clinical application of miRNA-related therapeutics are urgently required.
Author Contributions
HZ drafted the manuscript. CF designed the study. WT, JY, JP, and JG revised the manuscript. HZ and WT were responsible for the collection of data or analysis. All authors read and approved the final manuscript.
Funding
This work was supported by the Key Project of Science and Technology of Hunan Province (No. 2020SK53420 to JY). Hunan Province Outstanding Postdoctoral Innovative Talent Project (2021RC2106 to CF).
Conflict of Interest
JG and CF was employed by the company, Hunan Fangsheng Pharmaceutical Co. Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AAVs, adeno-associated viruses; AMOs, anti-miRNA oligonucleotides; Ang II, angiotensin II; HF, Heart failure; hiPSC-CMs, human induced-pluripotent stem cells; iCMs, induced cardiomyocytes; LNA, locked nucleic acid; MI, myocardial infarction; miRNAs, microRNAs; mRNAs, messenger RNAs; mTOR, mammalian target of rapamycin; ODNs, oligodeoxynucleotides; PS, phosphorothioate; PI3K, phosphatidylinositol-3-kinase; UTR, untranslated region.
References
1. Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, et al. Heart disease and stroke statistics-2021 update: a report from the American Heart Association. Circulation. (2021) 143:e254–743.
2. Dunlay SM, Roger VL, Redfield MM. Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. (2017) 14:591–602. doi: 10.1038/nrcardio.2017.65
3. Gerber Y, Weston SA, Berardi C, McNallan SM, Jiang R, Redfield MM, et al. Contemporary trends in heart failure with reduced and preserved ejection fraction after myocardial infarction: a community study. Am J Epidemiol. (2013) 178:1272–80. doi: 10.1093/aje/kwt109
4. Chen YT, Wong LL, Liew OW, Richards AM. Heart Failure with Reduced Ejection Fraction (HFrEF) and Preserved Ejection Fraction (HFpEF): the diagnostic value of circulating MicroRNAs. Cells. (2019) 8:1651. doi: 10.3390/cells8121651
5. Small EM, Olson EN Pervasive roles of microRNAs in cardiovascular biology. Nature. (2011) 469:336–42. doi: 10.1038/nature09783
6. Lee RC, Feinbaum RL, Ambros V The Celegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. (1993) 75:843–54. doi: 10.1016/0092-8674(93)90529-Y
7. Espinoza-Lewis RA, Wang DZ. Functional evolution of cardiac MicroRNAs in heart development and functions. Heart Dev. (2012) 100:279–317. doi: 10.1016/B978-0-12-387786-4.00009-9
8. Kozomara AS. Griffiths-Jones, miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. (2014) 42:D68–73. doi: 10.1093/nar/gkt1181
9. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. (2005) 120:15–20. doi: 10.1016/j.cell.2004.12.035
10. Rosenbloom KR, Dreszer TR, Long JC, Malladi VS, Sloan CA, Raney BJ, et al. ENCODE whole-genome data in the UCSC Genome Browser: update 2012. Nucleic Acids Res. (2012) 40:D912–7. doi: 10.1093/nar/gkr1012
12. Gerstein MB, Bruce C, Rozowsky JS, Zheng D, Du J, Korbel JO, et al. What is a gene, post-ENCODE? History and updated definition. Genome Res. (2007) 17:669–81. doi: 10.1101/gr.6339607
13. Adhikari S, Nice EC, Deutsch EW, Lane L, Omenn GS, Pennington SR, et al. A high-stringency blueprint of the human proteome. Nat Commun. (2020) 11:5301. doi: 10.1038/s41467-020-19045-9
14. Diehl AG, Boyle AP. Deciphering ENCODE. Trends Genet. (2016) 32:238–49. doi: 10.1016/j.tig.2016.02.002
15. Lee Y, Kim M, Han JJ, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. Embo J. (2004) 23:4051–60. doi: 10.1038/sj.emboj.7600385
16. Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Structural Mol Biol. (2006) 13:1097–101. doi: 10.1038/nsmb1167
17. Almeida MI, Reis RM, Calin GA. MicroRNA history: discovery, recent applications, next frontiers. Mutation Res Fundamental Mol Mechanisms Mutagenesis. (2011) 717:1–8. doi: 10.1016/j.mrfmmm.2011.03.009
18. Knight SW, Bass BL A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science. (2001) 293:2269–71. doi: 10.1126/science.1062039
19. Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. (2015) 15:321–33. doi: 10.1038/nrc3932
20. Xie S, Yu H, Li E, Wang Y, Liu J, Jiang H. Identification of miRNAs Involved in Bacillus velezensis FZB42-Activated Induced Systemic Resistance in Maize. Int J Mol Sci. (2019) 20:5057. doi: 10.3390/ijms20205057
21. Zhao C, Zhang Y, Popel AS. Mechanistic computational models of MicroRNA-mediated signaling networks in human diseases. Int J Mol Sci. (2019) 20:421. doi: 10.3390/ijms20020421
22. Holley CL, Topkara VK. An introduction to small non-coding RNAs: miRNA and snoRNA. Cardiovascular Drugs Therapy. (2011) 25:151–9. doi: 10.1007/s10557-011-6290-z
23. Eulalio A, Huntzinger E, Izaurralde E. Getting to the root of miRNA-Mediated gene silencing. Cell. (2008) 132:9–14. doi: 10.1016/j.cell.2007.12.024
24. Filipowicz W, Bhattacharyya SN, Sonenberg N Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. (2008) 9:102–14. doi: 10.1038/nrg2290
25. Murchison EP, Hannon GJ MiRNAs on the move: miRNA biogenesis and the RNAi machinery. Curr Opinion Cell Biol. (2004) 16:223–9. doi: 10.1016/j.ceb.2004.04.003
26. Okamura K, Chung WJ, Lai EC. The long and short of inverted repeat genes in animals: microRNAs, mirtrons and hairpin RNAs. Cell Cycle. (2008) 7:2840–5. doi: 10.4161/cc.7.18.6734
27. Kim VN, Han J, Siomi MC Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. (2009) 10:126–39. doi: 10.1038/nrm2632
28. Olson EN, Schneider MD. Sizing up the heart: development redux in disease. Genes Dev. (2003) 17:1937–56. doi: 10.1101/gad.1110103
29. Callis TE, Wang DZ Taking microRNAs to heart. Trends Mol Med. (2008) 14:254–60. doi: 10.1016/j.molmed.2008.03.006
30. Chen JF, Murchison EP, Tang R, Callis TE, Tatsuguchi M, Deng Z, et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc Natl Acad Sci USA. (2008) 105:2111–6. doi: 10.1073/pnas.0710228105
31. Albinsson S, Suarez Y, Skoura A, Offermanns S, Miano JM, Sessa WC. MicroRNAs are necessary for vascular smooth muscle growth, differentiation, and function. Arterioscler Thrombosis Vascular Biol. (2010) 30:1118–80. doi: 10.1161/ATVBAHA.109.200873
32. Zhao Y, Ransom JF, Li AV, Vedantham von Drehle M, Muth AN, et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. (2007) 129:303–17. doi: 10.1016/j.cell.2007.03.030
33. Torrini C, Cubero RJ, Dirkx E, Braga L, Ali H, Prosdocimo G, et al. Common regulatory pathways mediate activity of MicroRNAs inducing cardiomyocyte proliferation. Cell Rep. (2019) 27:2759–71.e2755. doi: 10.1016/j.celrep.2019.05.005
34. Porrello ER, Johnson BA, Aurora AB, Simpson E, Nam YJ, Matkovich SJ, et al. miR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circulation Res. (2011) 109:670–U208. doi: 10.1161/CIRCRESAHA.111.248880
35. Hoelscher SC, Doppler SA, Dressen M, Lahm H, Lange R, Krane M. MicroRNAs: pleiotropic players in congenital heart disease and regeneration. J Thoracic Dis. (2017) 9:S64–81. doi: 10.21037/jtd.2017.03.149
36. Ohtani K, Dimmeler S. Control of cardiovascular differentiation by microRNAs. Basic Res Cardiol. (2011) 106:5–11. doi: 10.1007/s00395-010-0139-7
37. Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. (2010) 142:375–86. doi: 10.1016/j.cell.2010.07.002
38. Jayawardena TM, Egemnazarov B, Finch EA, Zhang LN, Payne JA, Pandya K, et al. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circul Res. (2012) 110:1465. doi: 10.1161/CIRCRESAHA.112.269035
39. Muraoka N, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Isomi M, et al. MiR-133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures. Embo J. (2014) 33:1565–81. doi: 10.15252/embj.201387605
40. Lima JF, Cerqueira L, Figueiredo C, Oliveira C, Azevedo NF. Anti-miRNA oligonucleotides: a comprehensive guide for design. RNA Biol. (2018) 15:338–52. doi: 10.1080/15476286.2018.1445959
41. Lennox KA, Behlke MA. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. (2011) 18:1111–20. doi: 10.1038/gt.2011.100
42. Stein CA, Cheng YC. Antisense oligonucleotides as therapeutic agents–is the bullet really magical? Science. (1993) 261:1004–12. doi: 10.1126/science.8351515
43. Lennox KA, Behlke MA. A direct comparison of anti-microRNA oligonucleotide potency. Pharm Res. (2010) 27:1788–99. doi: 10.1007/s11095-010-0156-0
44. van Rooij E, Kauppinen S. Development of microRNA therapeutics is coming of age. EMBO Mol Med. (2014) 6:851–64. doi: 10.15252/emmm.201100899
45. Piao X, Wang H, Binzel DW, Guo P. Assessment and comparison of thermal stability of phosphorothioate-DNA, DNA, RNA, 2'-F RNA, and LNA in the context of Phi29 pRNA 3WJ. RNA. (2018) 24:67–76. doi: 10.1261/rna.063057.117
46. Piegari E, Galderisi U, Berrino L, Di Bernardo G, Cipollarog M, Esposito F, et al. In vivo effects of partial phosphorothioated AT1 receptor antisense oligonucleotides in spontaneously hypertensive and normotensive rats. Life Sci. (2000) 66:2091–9. doi: 10.1016/S0024-3205(00)00535-X
47. Kaur H, Arora A, Wengel J, Maiti S. Thermodynamic, counterion, and hydration effects for the incorporation of locked nucleic acid nucleotides into DNA duplexes. Biochemistry. (2006) 45:7347–55. doi: 10.1021/bi060307w
48. Mook OR, Baas F, de Wissel MB, Fluiter K. Evaluation of locked nucleic acid-modified small interfering RNA in vitro and in vivo. Mol Cancer Ther. (2007) 6:833–43. doi: 10.1158/1535-7163.MCT-06-0195
49. Elmen J, Thonberg H, Ljungberg K, Frieden M, Westergaard M, Xu Y, et al. Locked nucleic acid (LNA) mediated improvements in siRNA stability and functionality. Nucleic Acids Res. (2005) 33:439–47. doi: 10.1093/nar/gki193
50. Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet. (2011) 43:371–8. doi: 10.1038/ng.786
51. Bernardo BC, Gao XM, Winbanks CE, Boey EJ, Tham YK, Kiriazis H, et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc Natl Acad Sci USA. (2012) 109:17615–20. doi: 10.1073/pnas.1206432109
52. Bernardo BC, Gao XM, Tham YK, Kiriazis H, Winbanks CE, Ooi JY, et al. Silencing of miR-34a attenuates cardiac dysfunction in a setting of moderate, but not severe, hypertrophic cardiomyopathy. PLoS ONE. (2014) 9:e90337. doi: 10.1371/journal.pone.0090337
53. Iannolo G, Sciuto MR, Raffa GM, Pilato M, Conaldi PG MiR34 inhibition induces human heart progenitor proliferation. Cell Death Dis. (2018) 9:368. doi: 10.1038/s41419-018-0400-9
54. Tang Y, Zheng J, Sun Y, Wu Z, Liu Z, Huang G MicroRNA-1 regulates cardiomyocyte apoptosis by targeting Bcl-2. Int Heart J. (2009) 50:377–87. doi: 10.1536/ihj.50.377
55. Pan Z, Sun X, Ren J, Li X, Gao X, Lu C, et al. miR-1 exacerbates cardiac ischemia-reperfusion injury in mouse models. PLoS ONE. (2012) 7:e50515. doi: 10.1371/journal.pone.0050515
56. Wang L, Yuan Y, Li J, Ren H, Cai Q, Chen X, et al. MicroRNA-1 aggravates cardiac oxidative stress by post-transcriptional modification of the antioxidant network. Cell Stress Chaperones. (2015) 20:411–20. doi: 10.1007/s12192-014-0565-9
57. Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S. Inhibition of microRNA function by antimiR oligonucleotides. Silence. (2012) 3:1. doi: 10.1186/1758-907X-3-1
58. Judge AD, Bola G, Lee AC, MacLachlan I. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol Ther. (2006) 13:494–505. doi: 10.1016/j.ymthe.2005.11.002
59. Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, et al. Silencing of microRNAs in vivo with 'antagomirs'. Nature. (2005) 438:685–9. doi: 10.1038/nature04303
60. Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. (2007) 13:613–8. doi: 10.1038/nm1582
61. Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. (2012) 11:125–40. doi: 10.1038/nrd3625
62. Adachi H, Hengesbach M, Yu YT, Morais P. From antisense RNA to RNA modification: therapeutic potential of RNA-based technologies. Biomedicines. (2021) 9:550. doi: 10.3390/biomedicines9050550
63. Duygu B, Juni R, Ottaviani L, Bitsch N, Wit JBM, de Windt LJ, et al. Comparison of different chemically modified inhibitors of miR-199b in vivo. Biochem Pharmacol. (2019) 159:106–15. doi: 10.1016/j.bcp.2018.11.013
64. Chae DK, Ban E, Yoo YS, Baik JH, Song EJ. Evaluation of inhibition of miRNA expression induced by anti-miRNA oligonucleotides. Anal Bioanal Chem. (2016) 408:4829–33. doi: 10.1007/s00216-016-9611-z
65. Montgomery RL, Hullinger TG, Semus HM, Dickinson BA, Seto AG, Lynch JM, et al. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation. (2011) 124:1537–U1125. doi: 10.1161/CIRCULATIONAHA.111.030932
66. Esau CC. Inhibition of microRNA with antisense oligonucleotides. Methods. (2008) 44:55–60. doi: 10.1016/j.ymeth.2007.11.001
67. Davis S, Lollo B, Freier S, Esau C. Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res. (2006) 34:2294–304. doi: 10.1093/nar/gkl183
68. Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. (2004) 10:544–50. doi: 10.1261/rna.5235104
69. Hutvagner G, Simard MJ, Mello CC, Zamore PD. Sequence-specific inhibition of small RNA function. PLoS Biol. (2004) 2:E98. doi: 10.1371/journal.pbio.0020098
70. Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metabolism. (2006) 3:87–98. doi: 10.1016/j.cmet.2006.01.005
71. Davis S, Propp S, Freier SM, Jones LE, Serra MJ, Kinberger G, et al. Potent inhibition of microRNA in vivo without degradation. Nucleic Acids Res. (2009) 37:70–7. doi: 10.1093/nar/gkn904
72. Zhang Y, Roccaro AM, Rombaoa C, Flores L, Obad S, Fernandes SM, et al. LNA-mediated anti-miR-155 silencing in low-grade B-cell lymphomas. Blood. (2012) 120:1678–86. doi: 10.1182/blood-2012-02-410647
73. Thum T, Chau N, Bhat B, Gupta SK, Linsley PS, Bauersachs J, et al. Comparison of different miR-21 inhibitor chemistries in a cardiac disease model. J Clin Invest. (2011) 121, 461–2; author reply 462–463. doi: 10.1172/JCI45938
74. Yamamichi N, Shimomura R, Inada K, Sakurai K, Haraguchi T, Ozaki Y, et al. Locked nucleic acid in situ hybridization analysis of miR-21 expression during colorectal cancer development. Clin Cancer Res. (2009) 15:4009–16. doi: 10.1158/1078-0432.CCR-08-3257
75. Fabani MM, Gait MJ. miR-122 targeting with LNA/2'-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), PNA-peptide conjugates. RNA. (2008) 14:336–46. doi: 10.1261/rna.844108
76. Elmen J, Lindow M, Silahtaroglu A, Bak M, Christensen M, Lind-Thomsen A, et al. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. (2008) 36:1153–62. doi: 10.1093/nar/gkm1113
77. Orom UA, Kauppinen S, Lund AH. LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene. (2006) 372:137–41. doi: 10.1016/j.gene.2005.12.031
78. Elmen J, Lindow M, Schutz S, Lawrence M, Petri A, Obad S, et al. LNA-mediated microRNA silencing in non-human primates. Nature. (2008) 452:896–U810. doi: 10.1038/nature06783
79. Liu Y, Wu W, Wang Y, Han S, Yuan Y, Huang J, et al. Recent development of gene therapy for pancreatic cancer using non-viral nanovectors. Biomater Sci. (2021) 9:6673–90. doi: 10.1039/D1BM00748C
80. Forterre A, Komuro H, Aminova S, Harada M. A comprehensive review of cancer MicroRNA therapeutic delivery strategies. Cancers. (2020) 12:1852. doi: 10.3390/cancers12071852
81. Bai Z, Wei J, Yu C, Han X, Qin X, Zhang C, et al. Non-viral nanocarriers for intracellular delivery of microRNA therapeutics. J Mater Chem B. (2019) 7:1209–25. doi: 10.1039/C8TB02946F
82. Labatut AE, Mattheolabakis G. Non-viral based miR delivery and recent developments. Eur J Pharm Biopharm. (2018) 128:82–90. doi: 10.1016/j.ejpb.2018.04.018
83. Stratford-Perricaudet LD, Makeh I, Perricaudet M, Briand P. Widespread long-term gene transfer to mouse skeletal muscles and heart. J Clin Invest. (1992) 90:626–30. doi: 10.1172/JCI115902
84. Meloni M, Marchetti M, Garner K, Littlejohns B, Sala-Newby G, Xenophontos N, et al. Local inhibition of microRNA-24 improves reparative angiogenesis and left ventricle remodeling and function in mice with myocardial infarction. Mol Ther. (2013) 21:1390–402. doi: 10.1038/mt.2013.89
85. Wang S, Wu J, You J, Shi H, Xue X, Huang J, et al. HSF1 deficiency accelerates the transition from pressure overload-induced cardiac hypertrophy to heart failure through endothelial miR-195a-3p-mediated impairment of cardiac angiogenesis. J Mol Cell Cardiol. (2018) 118:193–207. doi: 10.1016/j.yjmcc.2018.03.017
86. Vorburger SA, Hunt KK. Adenoviral gene therapy. Oncologist. (2002) 7:46–59. doi: 10.1634/theoncologist.7-1-46
87. Zincarelli C, Soltys S, Rengo G, Koch WJ, Rabinowitz JE. Comparative cardiac gene delivery of adeno-associated virus serotypes 1-9 reveals that AAV6 mediates the most efficient transduction in mouse heart. Clin Transl Sci. (2010) 3:81–9. doi: 10.1111/j.1752-8062.2010.00190.x
88. Bian W, Chen W, Nguyen T, Zhou Y, Zhang J. miR-199a overexpression enhances the potency of human induced-pluripotent stem-cell-derived cardiomyocytes for myocardial repair. Front Pharmacol. (2021) 12:673621. doi: 10.3389/fphar.2021.673621
89. Liu WL, Liu Q. Shikonin attenuates sympathetic remodeling in chronic heart failure mice via regulating miR-124. Biochem Biophys Res Commun. (2019) 520:359–65. doi: 10.1016/j.bbrc.2019.10.038
90. Gao F, Kataoka M, Liu N, Liang T, Huang ZP, Gu F, et al. Therapeutic role of miR-19a/19b in cardiac regeneration and protection from myocardial infarction. Nat Commun. (2019) 10:1802. doi: 10.1038/s41467-019-09530-1
91. Kraszewska I, Tomczyk M, Andrysiak K, Biniecka M, Geisler A, Fechner H, et al. Variability in cardiac miRNA-122 level determines therapeutic potential of miRNA-regulated AAV vectors. Mol Ther Methods Clin Dev. (2020) 17:1190–201. doi: 10.1016/j.omtm.2020.05.006
92. Bish LT, Sleeper MM, Reynolds C, Gazzara J, Withnall E, Singletary GE, et al. Cardiac gene transfer of short hairpin RNA directed against phospholamban effectively knocks down gene expression but causes cellular toxicity in canines. Hum Gene Ther. (2011) 22:969–77. doi: 10.1089/hum.2011.035
93. Yang L, Song S, Lv H. MicroRNA-322 protects hypoxia-induced apoptosis in cardiomyocytes via BDNF gene. Am J Transl Res. (2016) 8:2812–9.
94. Wang J, Xu R, Wu J, Li Z. MicroRNA-137 negatively regulates H2O2-induced cardiomyocyte apoptosis through CDC42. Med Sci Monit. (2015) 21:3498–504. doi: 10.12659/MSM.894648
95. Pauwels K, Gijsbers R, Toelen J, Schambach A, Willard-Gallo K, Verheust C, et al. State-of-the-art lentiviral vectors for research use: risk assessment and biosafety recommendations. Curr Gene Ther. (2009) 9:459–74. doi: 10.2174/156652309790031120
96. Baum C, Kustikova O, Modlich U, Li Z, Fehse B. Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum Gene Ther. (2006) 17:253–63. doi: 10.1089/hum.2006.17.253
97. Deng J, Zhong Q. Advanced research on the microRNA mechanism in heart failure. Int J Cardiol. (2016) 220:61–4. doi: 10.1016/j.ijcard.2016.06.185
98. Wang J, Liew OW, Richards AM, Chen YT. Overview of MicroRNAs in cardiac hypertrophy, fibrosis, and apoptosis. Int J Mol Sci. (2016) 17:749. doi: 10.3390/ijms17050749
99. Xu X, Su YL, Shi JY, Lu Q, Chen C. MicroRNA-17-5p promotes cardiac hypertrophy by targeting Mfn2 to inhibit autophagy. Cardiovasc Toxicol. (2021) 21:759–71. doi: 10.1007/s12012-021-09667-w
100. Zhang S, Yin Z, Dai FF, Wang H, Zhou MJ, Yang MH, et al. miR-29a attenuates cardiac hypertrophy through inhibition of PPARδ expression. J Cell Physiol. (2019) 234:13252–62. doi: 10.1002/jcp.27997
101. Wang WW, Wu CW, Ren LN, Bao YD, Han YC, Li C, et al. MiR-30e-5p is sponged by Kcnq1ot1 and represses Angiotensin II-induced hypertrophic phenotypes in cardiomyocytes by targeting ADAM9. Exp Cell Res. (2020) 394:112140. doi: 10.1016/j.yexcr.2020.112140
102. Mo B, Wu X, Wang X, Xie J, Ye Z, Li L. miR-30e-5p mitigates hypoxia-induced apoptosis in human stem cell-derived cardiomyocytes by suppressing Bim. Int J Biol Sci. (2019) 15:1042–51. doi: 10.7150/ijbs.31099
103. Yu XJ, Huang YQ, Shan ZX, Zhu JN, Hu ZQ, Huang L, et al. MicroRNA-92b-3p suppresses angiotensin II-induced cardiomyocyte hypertrophy via targeting HAND2. Life Sci. (2019) 232:116635. doi: 10.1016/j.lfs.2019.116635
104. Hinkel R, Batkai S, Bahr A, Bozoglu T, Straub S, Borchert T, et al. AntimiR-132 attenuates myocardial hypertrophy in an animal model of percutaneous aortic constriction. J Am College Cardiol. (2021) 77:2923–35. doi: 10.1016/j.jacc.2021.04.028
105. Li J, Ichikawa T, Villacorta L, Janicki JS, Brower GL, Yamamoto M, et al. Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. Arterioscler Thromb Vasc Biol. (2009) 29:1843–50. doi: 10.1161/ATVBAHA.109.189480
106. Yan HL, Li YF, Wang C, Zhang Y, Liu C, Zhou KY, et al. Contrary microRNA expression pattern between fetal and adult cardiac remodeling: therapeutic value for heart failure. Cardiovascular Toxicol. (2017) 17:267–76. doi: 10.1007/s12012-016-9381-z
107. Yan HL, Wang H, Zhu XX, Huang JB, Li YF, Zhou KY, et al. Adeno-associated virus-mediated delivery of anti-miR-199a tough decoys attenuates cardiac hypertrophy by targeting PGC-1alpha. Mol Therapy-Nucleic Acids. (2021) 23:406–17. doi: 10.1016/j.omtn.2020.11.007
108. Ding YQ, Zhang YH, Lu J, Li B, Yu WJ, Yue ZB, et al. MicroRNA-214 contributes to Ang II-induced cardiac hypertrophy by targeting SIRT3 to provoke mitochondrial malfunction. Acta Pharmacol Sin. (2021) 42:1422–36. doi: 10.1038/s41401-020-00563-7
109. Jin LH, Zhou Y, Han LZ, Piao JH. MicroRNA302-367-PI3K-PTEN-AKT-mTORC1 pathway promotes the development of cardiac hypertrophy through controlling autophagy. In Vitro Cell Dev Biol Animal. (2020) 56:112–9. doi: 10.1007/s11626-019-00417-5
110. Zeng N, Wen YH, Pan R, Yang J, Yan YM, Zhao AZ, et al. Dickkopf 3: a novel target gene of miR-25-3p in promoting fibrosis-related gene expression in myocardial fibrosis. J Cardiovasc Transl Res. (2021). doi: 10.1007/s12265-021-10116-w. [Epub ahead of print].
111. Zhang WQ, Wang QZ, Feng YJ, Chen XG, Yang LJ, Xu M, et al. MicroRNA-26a protects the heart against hypertension-induced myocardial fibrosis. J Am Heart Assoc. (2020) 9:e017970. doi: 10.1161/JAHA.120.017970
112. Li J, Cai S, He Q, Zhang H, Friedberg D, Wang FF, et al. Intravenous miR-144 reduces left ventricular remodeling after myocardial infarction. Basic Res Cardiol. (2018) 113:36. doi: 10.1007/s00395-018-0694-x
113. Liu ZY, Lu MJ, Liu J, Wang ZN, Wang WW, Li Y, et al. MicroRNA-144 regulates angiotensin II-induced cardiac fibroblast activation by targeting CREB. Exp Therapeutic Med. (2020) 20:2113–21. doi: 10.3892/etm.2020.8901
114. Zhou Y, Ng DYE, Richards AM, Wang P. microRNA-221 inhibits latent TGF-β1 activation through targeting thrombospondin-1 to attenuate kidney failure-induced cardiac fibrosis. Mol Ther Nucleic Acids. (2020) 22:803–14. doi: 10.1016/j.omtn.2020.09.041
115. Liu Y, Song JW, Lin JY, Miao R, Zhong JC. Roles of MicroRNA-122 in cardiovascular fibrosis and related diseases. Cardiovascular Toxicol. (2020) 20:463–73. doi: 10.1007/s12012-020-09603-4
116. Shi YJ, Zhang Z, Yin QQ, Fu C, Barszczyk A, Zhang XF, et al. Cardiac-specific overexpression of miR-122 induces mitochondria-dependent cardiomyocyte apoptosis and promotes heart failure by inhibiting Hand2. J Cell Mol Med. (2021) 25:5326–34. doi: 10.1111/jcmm.16544
117. Ma J, Zhang Y, Ji H, Chen L, Chen T, Guo C, et al. Overexpression of miR-138-5p suppresses MnCl(2) -induced autophagy by targeting SIRT1 in SH-SY5Y cells. Environ Toxicol. (2019) 34:539–47. doi: 10.1002/tox.22708
118. Sun S, Wang C, Weng J. MicroRNA-138-5p drives the progression of heart failure via inhibiting sirtuin 1 signaling. Mol Med Rep. (2021) 23:276. doi: 10.3892/mmr.2021.11915
119. Zhou F, Fu WD, Chen L. MiRNA-182 regulates the cardiomyocyte apoptosis in heart failure. Eur Rev Med Pharmacol Sci. (2019) 23:4917–23.
120. Zakowicz H, Yang HS, Stark C, Wlodawer A, Laronde-Leblanc N, Colburn NH. Mutational analysis of the DEAD-box RNA helicase eIF4AII characterizes its interaction with transformation suppressor Pdcd4 and eIF4GI. RNA. (2005) 11:261–74. doi: 10.1261/rna.7191905
121. Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, et al. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. Embo J. (2005) 24:717–29. doi: 10.1038/sj.emboj.7600559
122. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. (1998) 94:491–501. doi: 10.1016/S0092-8674(00)81590-1
123. Chen K, Zhang B, Sun ZJ MicroRNA 379 regulates klotho deficiency-induced cardiomyocyte apoptosis via repression of Smurf1. Hypertension. (2021) 78:342–52. doi: 10.1161/HYPERTENSIONAHA.120.16888
124. Zhu X, Lu X. MiR-423-5p inhibition alleviates cardiomyocyte apoptosis and mitochondrial dysfunction caused by hypoxia/reoxygenation through activation of the wnt/β-catenin signaling pathway via targeting MYBL2. J Cell Physiol. (2019) 234:22034–43. doi: 10.1002/jcp.28766
125. Seok H, Lee H, Jang ES, Chi SW. Evaluation and control of miRNA-like off-target repression for RNA interference. Cell Mol Life Sci. (2018) 75:797–814. doi: 10.1007/s00018-017-2656-0
126. Hinkel R, Ramanujam D, Kaczmarek V, Howe A, Klett K, Beck C, et al. AntimiR-21 prevents myocardial dysfunction in a pig model of ischemia/reperfusion injury. J Am College Cardiol. (2020) 75:1788–800. doi: 10.1016/j.jacc.2020.02.041
127. Piegari E, Cozzolino A, Ciuffreda LP, Cappetta D, De Angelis A, Urbanek K, et al. Cardioprotective effects of miR-34a silencing in a rat model of doxorubicin toxicity. Sci Rep. (2020) 10:12250. doi: 10.1038/s41598-020-69038-3
128. Zhu JN, Fu YH, Hu ZQ, Li WY, Tang CM, Fei HW, et al. Activation of miR-34a-5p/Sirt1/p66shc pathway contributes to doxorubicin-induced cardiotoxicity. Sci Rep. (2017) 7:11879. doi: 10.1038/s41598-017-12192-y
129. Stegmayer G, Di Persia LE, Rubiolo M, Gerard M, Pividori M, Yones C, et al. Predicting novel microRNA: a comprehensive comparison of machine learning approaches. Brief Bioinform. (2019) 20:1607–20. doi: 10.1093/bib/bby037
130. Chen L, Heikkinen L, Wang C, Yang Y, Sun H, Wong G. Trends in the development of miRNA bioinformatics tools. Brief Bioinform. (2019) 20:1836–52. doi: 10.1093/bib/bby054
131. Seyhan AA, Carini C. Are innovation and new technologies in precision medicine paving a new era in patients centric care? J Transl Med. (2019) 17:114. doi: 10.1186/s12967-019-1864-9
132. Hong DS, Kang YK, Borad M, Sachdev J, Ejadi S, Lim HY, et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br J Cancer. (2020) 122:1630–7. doi: 10.1038/s41416-020-0802-1
133. Beg MS, Brenner AJ, Sachdev J, Borad M, Kang YK, Stoudemire J, et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest New Drugs. (2017) 35:180–8. doi: 10.1007/s10637-016-0407-y
134. Zhang L, Liao Y, Tang L. MicroRNA-34 family: a potential tumor suppressor and therapeutic candidate in cancer. J Exp Clin Cancer Res. (2019) 38:53. doi: 10.1186/s13046-019-1059-5
135. Stephenson ML, Zamecnik PC. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci USA. (1978) 75:285–8. doi: 10.1073/pnas.75.1.285
136. Crooke ST, Witztum JL, Bennett CF, Baker BF, RNA-Targeted therapeutics. Cell Metab. (2018) 27:714–39. doi: 10.1016/j.cmet.2018.03.004
137. Setten RL, Rossi JJ, Han SP. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov. (2019) 18:421–46. doi: 10.1038/s41573-019-0017-4
138. Wang G, Wang R, Ruan Z, Liu L, Li Y, Zhu L. MicroRNA-132 attenuated cardiac fibrosis in myocardial infarction-induced heart failure rats. Biosci Rep. (2020) 40:BSR20201696. doi: 10.1042/BSR20201696
139. Batkai S, Genschel C, Viereck J, Rump S, Bär C, Borchert T, et al. CDR132L improves systolic and diastolic function in a large animal model of chronic heart failure. Eur Heart J. (2021) 42:192–201. doi: 10.1093/eurheartj/ehaa791
140. Täubel J, Hauke W, Rump S, Viereck J, Batkai S, Poetzsch J, et al. Novel antisense therapy targeting microRNA-132 in patients with heart failure: results of a first-in-human Phase 1b randomized, double-blind, placebo-controlled study. Eur Heart J. (2021) 42:178–88. doi: 10.1093/eurheartj/ehaa898
Keywords: heart failure, non-coding RNA, microRNA, modification, therapy
Citation: Zhou H, Tang W, Yang J, Peng J, Guo J and Fan C (2021) MicroRNA-Related Strategies to Improve Cardiac Function in Heart Failure. Front. Cardiovasc. Med. 8:773083. doi: 10.3389/fcvm.2021.773083
Received: 09 September 2021; Accepted: 25 October 2021;
Published: 19 November 2021.
Edited by:
Katherine Athayde Teixeira De Carvalho, Pelé Pequeno Príncipe Research Institute, BrazilReviewed by:
Murugavel Ponnusamy, Qingdao University, ChinaRuby C. Y. Lin, Westmead Institute for Medical Research, Australia
Copyright © 2021 Zhou, Tang, Yang, Peng, Guo and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chengming Fan, fanchengming@csu.edu.cn