Predictive Modeling of Estrogen Receptor Binding Agents Using Advanced Cheminformatics Tools and Massive Public Data
 Kathryn Ribay
Kathryn Ribay Marlene T. Kim1,2
Marlene T. Kim1,2  Wenyi Wang
Wenyi Wang Hao Zhu
Hao Zhu- 1Department of Chemistry, Rutgers University, Camden, NJ, USA
- 2The Rutgers Center for Computational and Integrative Biology, Camden, NJ, USA
Estrogen receptors (ERα) are a critical target for drug design as well as a potential source of toxicity when activated unintentionally. Thus, evaluating potential ERα binding agents is critical in both drug discovery and chemical toxicity areas. Using computational tools, e.g., Quantitative Structure-Activity Relationship (QSAR) models, can predict potential ERα binding agents before chemical synthesis. The purpose of this project was to develop enhanced predictive models of ERα binding agents by utilizing advanced cheminformatics tools that can integrate publicly available bioassay data. The initial ERα binding agent data set, consisting of 446 binders and 8307 non-binders, was obtained from the Tox21 Challenge project organized by the NIH Chemical Genomics Center (NCGC). After removing the duplicates and inorganic compounds, this data set was used to create a training set (259 binders and 259 non-binders). This training set was used to develop QSAR models using chemical descriptors. The resulting models were then used to predict the binding activity of 264 external compounds, which were available to us after the models were developed. The cross-validation results of training set [Correct Classification Rate (CCR) = 0.72] were much higher than the external predictivity of the unknown compounds (CCR = 0.59). To improve the conventional QSAR models, all compounds in the training set were used to search PubChem and generate a profile of their biological responses across thousands of bioassays. The most important bioassays were prioritized to generate a similarity index that was used to calculate the biosimilarity score between each two compounds. The nearest neighbors for each compound within the set were then identified and its ERα binding potential was predicted by its nearest neighbors in the training set. The hybrid model performance (CCR = 0.94 for cross validation; CCR = 0.68 for external prediction) showed significant improvement over the original QSAR models, particularly for the activity cliffs that induce prediction errors. The results of this study indicate that the response profile of chemicals from public data provides useful information for modeling and evaluation purposes. The public big data resources should be considered along with chemical structure information when predicting new compounds, such as unknown ERα binding agents.
Introduction
Estrogen receptors are cellular proteins that are activated when bound to estrogen molecules. When activated, estrogen receptors trigger the expression of gene products crucial to the endocrine system (Hall et al., 2001). These receptors can also be activated by certain endocrine disrupting chemicals (EDC), resulting in a disruption of normal estrogen signaling (Shanle and Xu, 2011). There are two unique estrogen receptors: ERα and ERβ. These two receptors are highly similar in the DNA binding domain, but differ more significantly in other regions. While there are many EDC that interact with both receptors, the difference between these two receptors allows some ligands specifically bind to only one receptor as well. Among all known binding agents, the ERα binders are much better characterized than ERβ binders (Hall et al., 2001; Shanle and Xu, 2011). Due to the nature of available data, this study focuses solely on ligands binding to ERα.
When estrogen receptors are activated by small molecules other than estrogens, the expression of the associated genes is deregulated leading to neurological, developmental, and reproductive toxicity (Mueller and Korach, 2001). There are many small molecules with different chemical structures which exhibit interaction with the ligand binding domain of the estrogen receptor (Blair et al., 2000; Schug et al., 2011). Considering the large number of compounds which needs to be evaluated for their estrogen receptor binding potentials, traditional experimental toxicology protocols can be costly and time-consuming. As a result, there is a strong need to effectively pre-screen and prioritize small molecules for potential endocrine disruption prior to more costly animal testing. In a 2007 publication, the U.S. National Research Council identified both high-throughput screening (HTS) and computational models as critical chemical toxicity evaluation tools in Twenty-First century toxicology (Committee on Toxicity Testing and Assessment of Environmental Agents N.R.C., 2007). HTS has been viewed as a potential alternative to animal models due to the ability to test many molecules at a rapid pace and lower cost. The large number of HTS studies has resulted in publically available bioassay databases which are a rich source of in vitro data (Zhu et al., 2014). Motivated by these available data, computational modeling, which costs even less than HTS, has been used as another important evaluation protocols for EDCs (Ding et al., 2010).
Quantitative structure-activity relationship (QSAR) modeling has been applied to develop estrogen receptor binding models in the past decade, as shown in Table 1 (Hong et al., 2002; Serafimova et al., 2007; Liu et al., 2008; Li and Gramatica, 2010; Taha et al., 2010; Vedani et al., 2012; Zang et al., 2013; Zhang et al., 2013, 2014; Deng et al., 2014; Ng et al., 2015). These studies have covered a wide range of modeling approaches and data set sizes, from a descriptor-based decision tree (Hong et al., 2002) to 3-D docking and multi-dimensional QSAR (Vedani et al., 2012). The number of compounds used for modeling purpose in these studies range from less than 100 to more than 8000. The QSAR modeling of estrogen receptor binding agents has also been reviewed (Lo Piparo and Worth, 2010).
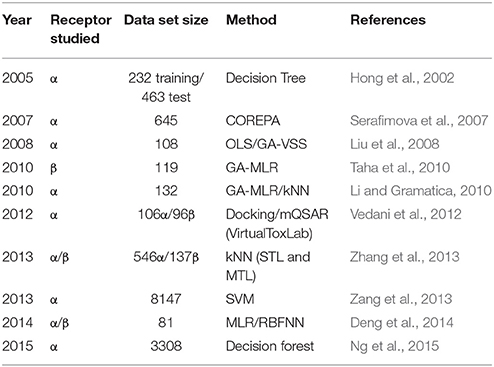
Table 1. A sampling of QSAR studies on estrogen receptor interaction.
Although, there have been many promising models developed to predict ER binding data, these QSAR models are all based on data derived from chemical structure alone. As a result, there is increasing evidence that the applicability of these models is limited to certain compounds (Johnson, 2008; Scior et al., 2009). In certain cases, compounds with similar structures may show significantly different activities, leading to prediction errors in QSAR models. These pairs of molecules are known as “activity cliffs” in QSAR studies (Maggiora, 2006). QSAR models predict the activity of compounds only based on their chemical structure information, but the presence of activity cliffs can lead to unavoidable prediction errors if there is no other information than chemical structures (Cruz-Monteagudo et al., 2014).
Inspired by the biosimilarity study reported by Low and her coworkers (Low et al., 2013), in this study, we developed enhanced computational models for estrogen receptor binding agents using both QSAR approaches and a biosimilarity search, which is based on publically available bioassay data. The initial QSAR models developed using the combination of various chemical descriptors and modeling approaches, were integrated with the biosimilarity information to generate hybrid predictions. Using the resulting hybrid models, the new compounds can be directly predicted for their estrogen receptor binding potential. The incorporation of a biosimilarity search based on additional bioassay data can solve the activity cliffs issue of QSAR modeling and improve the prediction accuracy of new compounds.
Materials and Methods
Data Curation
The original dataset used in this study was obtained in two parts separately from the National Center for Advancing the Translational Science (NCATS) via the Tox21 Challenge project. The dataset (PubChem assay AID 743077) consisted of the results of the quantitative High Throughput Screening (qHTS) to identify agonists of the ERα signaling pathway by measuring the expression of a beta lactamase reporter gene controlled by an ERα ligand binding domain (ER-LBD) fusion protein (National Center for Biotechnology Information, 2015). This dataset was used as the training set in the Tox21 Challenge. The original dataset consisted of 8753 compounds, of which 446 were categorized as active (ERα binders) and 8307 were categorized as inactive (non-binders). The compounds were processed by the CaseUltra® (www.multicase.com) structure checker tool to remove duplicates and inorganic compounds, resulting in 5647 unique organic compounds (259 actives and 5388 inactives). All the active compounds were selected for the training set and combined with a randomly selected 259 inactive compounds to produce a balanced training set of 518 compounds. An additional but much smaller set of compounds not included in the original qHTS data was provided by the Tox21 Challenge project as an external test set to validate the resulting models (see Figure 1 for modeling workflow). This external test set of 297 compounds (25 actives and 272 inactives) was also processed by the CaseUltra® structure checker to remove duplicates and inorganics, resulting in 264 unique compounds (24 actives and 240 inactives).
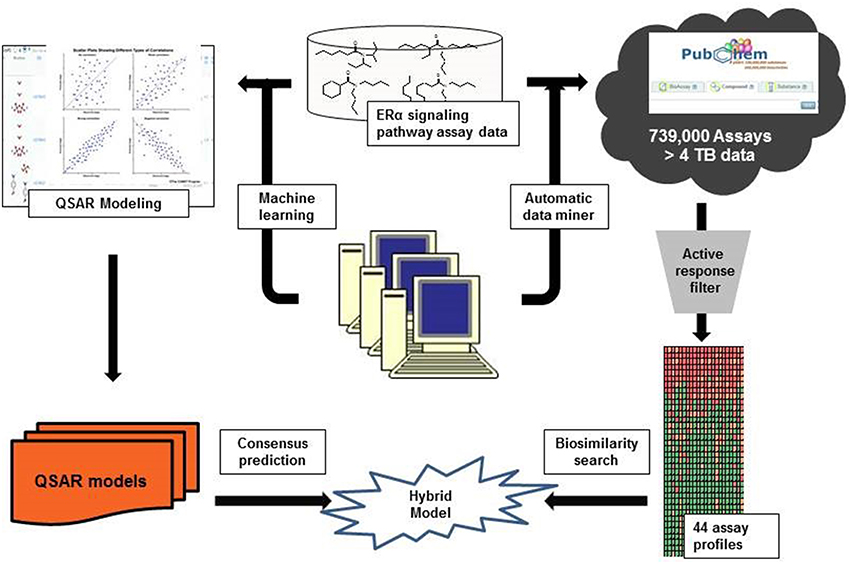
Figure 1. The hybrid modeling workflow.
Chemical Descriptors
Once the datasets were curated, chemical descriptors were calculated using two commercial descriptor generators. A total of 192 2-D Molecular Operating Environment® (MOE) (www.chemcomp.com) descriptors were generated using MOE version 2013, which include physical properties, atom and bond counts, connectivity and shape indices, adjacency and distance matrix descriptors, etc. Dragon® (www.talete.mi.it/) version 6 was used to generate 1259 descriptors including constitutional indices, drug-like indices, connectivity indices, functional group counts, etc. All descriptors were normalized to (0,1) and any redundant descriptors were removed by deleting those with low variance (standard deviation < 0.01 for the whole training set) and randomly keeping one of any pairs of descriptors that had high correlation (R2 > 0.95 between two descriptor values for the training set compounds), leaving 132 unique MOE descriptors and 594 unique Dragon descriptors for both data sets. In order to calculate the chemical similarity among compounds, MOE 2013 was used to calculate 166 MACCS fingerprints of each compound. These fingerprints were used as descriptors to calculate the Tanimoto coefficient of each compound pair to determine their chemical similarity (Willett, 2006).
QSAR Model Development and Model Validation
Three machine learning algorithms were used to develop QSAR models: support vector machines (SVM), random forest (RF), and k nearest neighbor (kNN; Mitchell, 2014). In this study, the RF (Breiman, 2001) and SVM (Vapnik, 2000) algorithms available in R® 3.0.2 using the packages “e1071” and “randomForest” (Dalgaard, 2008) were implemented. The available SVM algorithm was tuned to identify the optimal inputs for model performance. The kNN models (Zheng and Tropsha, 2000) were built using in-house modeling tools, also available at Chembench (http://chembench.mml.unc.edu; Walker et al., 2010). This model uses a genetic algorithm selection procedure to predict the activity of a target compound by identifying the k most similar compounds within the chemical descriptor space and using their activity to predict that of the target compound. The best model of each run is kept, while inferior models are discarded. In our modeling process, a random selection of 50 chemical descriptors was used in each iteration of the algorithm. Each method was performed with both MOE and Dragon descriptors, as shown in the modeling workflow in Figure 1. The six resulting models (SVM-Dragon; SVM-MOE; RF-Dragon; RF-MOE; kNN-Dragon; and kNN-MOE) were averaged to give a consensus prediction, as described in previous publications (Solimeo et al., 2012; Kim et al., 2014). All models were validated using a five-fold cross validation. In this procedure, the training set was randomly split into five equal selected subsets. Four subsets (80%) were used as a training set and the compounds in the fifth subset (20%) were used as a test set. The training set was used to develop QSAR models and the resulting models were used to predict the test set. This procedure was repeated five times until all compounds were used in the test set once (Golbraikh et al., 2003; Tropsha and Golbraikh, 2007).
Biosimilarity Calculation
An in-house profiling tool (Zhang et al., 2014) was used to extract relevant bioassay data from PubChem for each compound in both the training and test sets. The PubChem assays were ranked by the numbers of active responses for the compounds in our training set. The resulting PubChem bioassay profile consisted of 44 bioassays, which contain the largest number of active responses in the training set, and was then used to calculate the biosimilarity between pairs of two compounds using the following formula:
where p is the number of assays in which both compounds show active results, n is the number of assays in which both compounds show inactive results, and d is the number of assays in which the two compounds show opposite results. Inconclusive data were not considered in the calculation. The negative response data (inactives) are weighted less than positive responses (actives) in the biosimilarity calculation. In this study, the weight parameter ω was given the value of 0.06. The resulting WEBS values range from 0 to 1 and were used to determine the nearest neighbors in the training set for each test set compound. Any compound with WEBS similarity score over 0.6 was considered as a potential nearest neighbor for the target compound. The ERα binding activities of up to the top five nearest neighbors were used to calculate the predicted activity of the relevant test set compound. When fewer than five nearest neighbors existed within the training set, all nearest neighbors were used.
In order to form a hybrid model, the biosimilarity prediction was averaged with the QSAR prediction for each compound. For compounds which were not able to be predicted by the biosimilarity tool due to missing data, the QSAR consensus prediction was used as the predicted value. Compounds with opposite results from QSAR consensus models and biosimilarity search were considered as inconclusive and removed. This method returned a prediction for 192 of the 264 test set compounds.
Results
QSAR Results
The modeling set was used to develop six individual QSAR models and their predictions were averaged as a consensus prediction. The model performance was indicated by five-fold cross validation of the modeling set itself and external prediction of a set of 264 unknown compounds. The performance was evaluated by calculating the sensitivity, specificity, and CCR for all models, as shown in Figure 2.
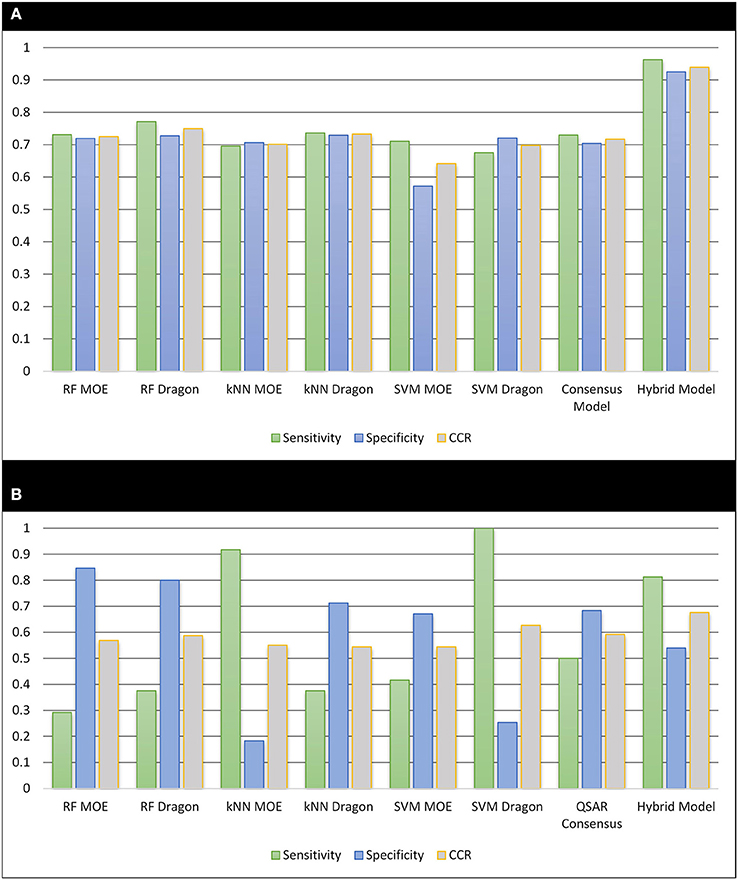
Figure 2. The performance of all resulting models. (A) Cross-validation of the 518 training set compounds; (B) external validation of 264 unknown compounds.
For the five-fold cross-validation procedures, the predictivity was similar across all the models (CCR = 0.642−0.749). However, the external predictions of the 264 unknown compounds showed a significant decrease in accuracy (CCR = 0.544−0.627), as observed in previous QSAR studies (Zhu et al., 2008a; Solimeo et al., 2012; Ng et al., 2015). Compared to individual models, the consensus model gave similar performance to the best individual models for both five-fold cross validation (sensitivity = 0.730, specificity = 0.704, and CCR = 0.717) and external predictions (sensitivity = 0.500, specificity = 0.683, and CCR = 0.592). Applying an applicability domain (AD), as described in previous studies (Zhu et al., 2008a, 2009), to both validation procedures did not show an improvement in predictive ability, so all predictions (100%) were retained when analyzing the QSAR models.
Bio-Assay Profile and Predictions
Our previous studies have shown improvements of QSAR models by incorporating biological data as extra descriptors into the modeling procedure (Sedykh et al., 2011; Kim et al., 2014). Relevant bioassay activity has been shown to be useful for the bioactivity predictions (Zhu et al., 2008b; Wang et al., 2015; Kim et al., 2016). In this study, the in-house profiling tool was used to automatically extract and optimize a biological profile containing 44 PubChem assays for 518 modeling set compounds. Using the WEBS score to calculate the biological similarity of each two compounds, those most similar compounds with WEBS scores over the nearest neighbor cut-off were identified for each test set compound and then used to predict the ERα binding potential. When combining the biosimilarity search with the QSAR consensus model as a hybrid model, the cross validation demonstrated a significant improvement of the accuracy over traditional QSAR modeling only based on chemical descriptors. Compared to the QSAR consensus model, the sensitivity, specificity and CCR of the hybrid model increased from 0.730 to 0.963, from 0.704 to 0.925, and from 0.717 to 0.939, respectively.
The external test set was also predicted by including up to five of the most biosimilar compounds in the training set. These hybrid predictions showed a noticeable improvement over the QSAR based solely on chemical descriptors. The external test set predictions returned a sensitivity = 0.813, specificity = 0.540, and CCR = 0.676 with a coverage of 73% (192 out of 264). The increase of sensitivity in both cross validation and external predictions brings considerable benefit when prioritizing potential EDCs for experimental testing.
Discussion
The estrogen receptor has been the target of many modeling studies due to the effects of endocrine disruption that occur when a compound present in the environment or in a consumer product activates the receptor. While recent modeling studies (Ng et al., 2015) have demonstrated impressive relative balanced accuracy and specificity based on only chemical structures, these models are still challenged by the high prevalence of false negative results when testing an external set, leading to a low sensitivity. There is a need for methods that can quickly and effectively screen a wide range of chemicals to correctly identify potential EDCs before a product is brought to market. This is a particular challenge when screening new compound sets, such as that used as an external test set in this study, where only a small fraction of the new compounds may be active binders. The attempt to use QSAR models based on only chemical descriptors to fill this need has been hindered by the structural diversity of the estrogen receptor binders and has reached a bottleneck due to the existence of activity cliffs. In this study, the noticeable improvement of the sensitivity of the model when predicting an external test set using the hybrid model suggests that the use of biological response data may be of particular importance in lowering the rate of false negative predictions from a model. Although this study focuses on activation of ERα only, there is a wide variety of chemical structures that are able to activate this receptor due to its large ligand binding domain (Shanle and Xu, 2011). The lack of experimental data, especially for active compounds (ERα binders), has resulted in activity cliffs in QSAR models based solely on chemical structures and limited the applicability of traditional QSAR modeling methods.
The QSAR models all showed acceptable predictivity when considering the cross validation of the training set. However, the external prediction of 264 unknown compounds had significantly decreased prediction accuracy, especially for individual models. Although the consensus model shows relatively stable performance, the sensitivity of its external test set prediction is much lower than the cross validation results due to the high proportion of false negatives. Table 2 displays examples of compounds that were consistently predicted incorrectly by the original QSAR models along with both their chemical nearest neighbor and biological nearest neighbor in the training set. The first active compound, A-315456 (PubChem CID 6603710), an α-1D-adrenoceptor antagonist, is an ERα binder that was incorrectly predicted as inactive by all QSAR models. This compound's chemical nearest neighbor in the training set is the inactive compound sulfamethoxazole (PubChem CID 5329). Dimethoxynaphtoquinone (PubChem CID 3136) is also an active ERα binder that was incorrectly predicted by the QSAR consensus model. Its chemical nearest neighbor dichlofop-methyl (PubChem CID 39985) is an inactive compound in this assay. Similarly, the compound N-methyl-2,3-diphenyl-1,2,4-thiadiazol-5-imine (PubChem CID 682802) is an inactive compound. However, its chemical nearest neighbor, in the training set, dichlorodiphenyltrichloroethane (DDT) (PubChem CID 3036), is an ERα binder. These prediction errors cannot be avoided if only chemical structure information is used for modeling.
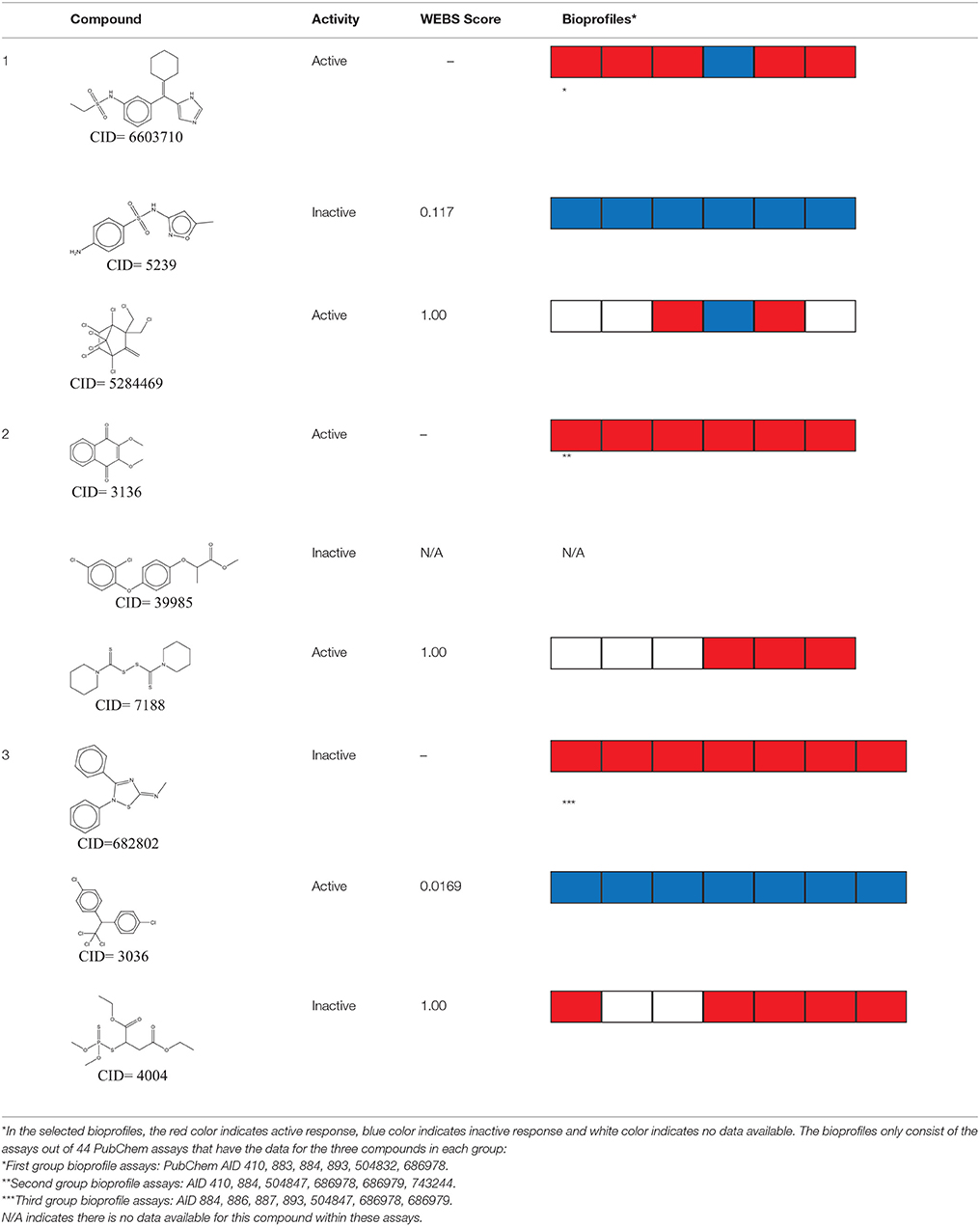
Table 2. Three test set compounds (the first compound in each group) with their chemical nearest neighbor (the second compound) and biological nearest neighbor (the third compound).
The prediction of the test set compounds improved when biosimilarity results were combined with the QSAR consensus model to form a hybrid model. Of particular note, the sensitivity of the external test set prediction increased from 0.500 for the QSAR consensus model alone to 0.813 for the hybrid model. In these examples, the biological nearest neighbors, as determined by WEBS score, provide more useful information for the predictions of external compounds. For example, the biological nearest neighbor in the training set of A-315456 (PubChem CID 6603710), an ERα binder, is toxaphene (PubChem CID 5284469), also an active compound (Table 2). For the other external test set compounds in Table 2, their biological nearest neighbors show the same ERα binding activities as the relevant target compounds. Furthermore, the WEBS scores for these test set compounds show dissimilarity to their chemical nearest neighbors. For example, the inactive compound N-methyl-2,3-diphenyl-1,2,4-thiadiazol-5-imine (PubChem CID 682802) has a biological nearest neighbor, malathion (PubChem CID 4004), a widely used insecticide that also showed inactive response in the ERα binding assay. Its chemical nearest neighbor, DDT (PubChem CID 3036), a now-banned insecticide, has a very low biosimilarity (WEBS = 0.0169) to N-methyl-2,3-diphenyl-1,2,4-thiadiazol-5-imine. Seven PubChem assays with testing data for both compounds show opposite results between these two compounds. The above analysis indicates that the activity cliffs are chemically similar compounds but have different biological effects (i.e., ERα binding). The hybrid model, using biosimilarity search as additional information in the modeling process, was able to differentiate them.
The bioassay response profile of the compounds shows promising potential to improve traditional QSAR models. Furthermore, when examining the PubChem assays used in the profile of this study, many targets of the assays regulate or are regulated by ERα. This provides additional useful information as to the types of bioassays which may be most useful in developing hybrid prediction models for ERα. The highest ranked assay, which consists of the highest number of active responses for our training set compounds, was used to screen potential inhibitors of histone lysine methyltransferase G9a (PubChem AID 504332). This assay acts as a co-regulator in the estradiol-induced activation or repression of gene transcription by ERα (Métivier et al., 2003; Purcell et al., 2011). Several other assays used in this profile specifically target enzymes in the cytochrome P450 (CYP450) family. These assays include screening inhibitors for CYP1A2 (PubChem AID 410) and CYP3A4 (PubChem AID 884), and a composite screening results for various CYP450 inhibitors (PubChem AID 1851). These proteins modulate ERα signaling by helping to maintain the androgen/estrogen balance (Tsuchiya et al., 2005). By analyzing the bioassays within the response profile, it indicates the future direction of gathering useful data for evaluating potential ERα binders.
The biosimilarity methodology used in this project shows a promising way to improve the predictivity of traditional QSAR modeling, particularly for increasing the sensitivity of the prediction results. However, since many compounds may not have been tested and have no data available in public resources, the usefulness of biosimilarity is limited by its coverage. A potential strategy to address the limitation of missing data is by using “read-across” methods (Patlewicz et al., 2014) to fill gaps in bioassay data for unknown compounds. Another pitfall of using the public data is the presence of experimental errors and the redundancy between various assay results. Currently, we are developing multiple novel data mining approaches to address this issue and will report them in future studies.
Conclusion
In this study, we first developed QSAR models for the qHTS assay data, which identify agonists for the ERα signaling pathway, provided in the Tox21 challenge. The external test set prediction of all QSAR models, including the consensus model, is lower than the cross validation results of the training set. However, by combining the biosimilarity search, developed using the bioassay response profile automatically extracted from PubChem, with the QSAR consensus predictions, a hybrid model was created. The resulting hybrid model showed a noticeable improvement in both cross-validation and external prediction results compared to QSAR models based only on chemical descriptors. This result demonstrated that integrating extra biological data in the modeling process can improve traditional QSAR models when predicting ERα binding potentials for unknown compounds. This strategy can be used to develop enhanced models to evaluate other types of toxicity for compounds of interest.
Author Contributions
Substantial contributions to the conception or design of the work: HZ. Acquisition, analysis, or interpretation of data for the work: KR, MK, WW, and DP. Drafting the work: KR and HZ. Final approval of the version to be published: HZ. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: HZ.
Funding
This research was supported in part by National Institutes of Health grants P30ES005022 and R15ES023148, and the Colgate-Palmolive Grant for Alternative Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors want to thank Dr. Ruili Huang and Dr. Menghang Xia at NCATS to help with the data curation and result discussion.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fenvs.2016.00012
References
Blair, R. M., Fang, H., Branham, W. S., Hass, B. S., Dial, S. L., Moland, C. L., et al. (2000). The estrogen receptor relative binding affinities of 188 natural and xenochemicals: structural diversity of ligands. Toxicol. Sci. 54, 138–153. doi: 10.1093/toxsci/54.1.138
Committee on Toxicity Testing Assessment of Environmental Agents N.R.C. (2007). Toxicity Testing in the 21st Century: A Vision and a Strategy. Washington, DC: The National Academies Press.
Cruz-Monteagudo, M., Medina-Franco, J., Pérez-Castillo, Y., Nicolotti, O., Cordeiro, M. N., and Borges, F. (2014). Activity cliffs in drug discovery: Dr Jekyll or Mr Hyde? Drug Discov. Today 19, 1069–1080. doi: 10.1016/j.drudis.2014.02.003
Dalgaard, P. (2008). Introductory Statistics with R. New York, NY: Springer Science & Business Media.
Deng, C. L., Chen, X. X., Lu, H. Y., Yang, X., Luan, F., and Cordeiro, M. (2014). Prediction of the Estrogen Receptor Binding Affinity for both hER(alpha) and hER(beta) by QSAR Approaches. Lett. Drug Des. Disc. 11, 265–278. doi: 10.2174/15701808113109990067
Ding, D., Xu, L., Fang, H., Hong, H., Perkins, R., Harris, S., et al. (2010). The EDKB: an established knowledge base for endocrine disrupting chemicals. BMC Bioinformatics 11(Suppl 6):S5. doi: 10.1186/1471-2105-11-S6-S5
Golbraikh, A., Shen, M., Xiao, Z., Xiao, Y. D., Lee, K. H., and Tropsha, A. (2003). Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des. 17, 241–253. doi: 10.1023/A:1025386326946
Hall, J. M., Couse, J. F., and Korach, K. S. (2001). The multifaceted mechanisms of estradiol and estrogen receptor signaling. J. Biol. Chem. 276, 36869–36872. doi: 10.1074/jbc.r100029200
Hong, H., Tong, W., Fang, H., Shi, L., Xie, Q., Wu, J., et al. (2002). Prediction of estrogen receptor binding for 58,000 chemicals using an integrated system of a tree-based model with structural alerts. Environ. Health Perspect. 110, 29–36. doi: 10.1289/ehp.0211029
Johnson, S. R. (2008). The trouble with QSAR (or how I learned to stop worrying and embrace fallacy). J. Chem. Inf. Model. 48, 25–26. doi: 10.1021/ci700332k
Kim, M., Huang, R., Sedykh, A., Zhang, J., Xia, M., and Zhu, H. (2016). Mechanism profiling of hepatotoxicity caused by oxidative stress using the antioxidant response element reporter gene assay models and big data. Environ. Health Perspect. doi: 10.1289/ehp.1509763. [Epub ahead of print].
Kim, M. T., Sedykh, A., Chakravarti, S. K., Saiakhov, R. D., and Zhu, H. (2014). Critical evaluation of human oral bioavailability for pharmaceutical drugs by using various cheminformatics approaches. Pharm. Res. 31, 1002–1014. doi: 10.1007/s11095-013-1222-1
Li, J., and Gramatica, P. (2010). The importance of molecular structures, endpoints' values, and predictivity parameters in QSAR research: QSAR analysis of a series of estrogen receptor binders. Mol. Divers. 14, 687–696. doi: 10.1007/s11030-009-9212-2
Liu, H., Papa, E., and Gramatica, P. (2008). Evaluation and QSAR modeling on multiple endpoints of estrogen activity based on different bioassays. Chemosphere 70, 1889–1897. doi: 10.1016/j.chemosphere.2007.07.071
Lo Piparo, E., and Worth, A. (2010). Review of QSAR Models and Software Tools for Predicting Developmental and Reproductive Toxicity. Luxemborg: Publications Office of the European Union. doi: 10.2788/9628
Low, Y., Sedykh, A., Fourches, D., Golbraikh, A., Whelan, M., Rusyn, I., et al. (2013). Integrative chemical-biological read-across approach for chemical hazard classification. Chem. Res. Toxicol. 26, 1199–1208. doi: 10.1021/tx400110f
Maggiora, G. M. (2006). On outliers and activity cliffs–why QSAR often disappoints. J. Chem. Inf. Model. 46, 1535–1535. doi: 10.1021/ci060117s
Métivier, R., Penot, G., Hübner, M. R., Reid, G., Brand, H., Kos, M., et al. (2003). Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115, 751–763. doi: 10.1016/S0092-8674(03)00934-6
Mitchell, J. B. O. (2014). Machine learning methods in chemoinformatics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 4, 468–481. doi: 10.1002/wcms.1183
Mueller, S. O., and Korach, K. S. (2001). Estrogen receptors and endocrine diseases: lessons from estrogen receptor knockout mice. Curr. Opin. Pharmacol. 1, 613–619. doi: 10.1016/S1471-4892(01)00105-9
National Center for Biotechnology Information (2015). PubChem BioAssay Database; AID=743077. (Accessed September 15, 2015).
Ng, H. W., Luo, H., Ye, H., Ge, W., Tong, W., Hong, H., et al. (2015). Development and validation of decision forest model for estrogen receptor binding prediction of chemicals using large data sets. Chem. Res. Toxicol. 28, 2343–2351. doi: 10.1021/acs.chemrestox.5b00358
Patlewicz, G., Ball, N., Becker, R. A., Booth, E. D., Cronin, M. T. D., Kroese, D., et al. (2014). Read-across approaches - Misconceptions, promises and challenges ahead. Arch. Med. Vet. 46, 387–396. doi: 10.14573/altex.1410071
Purcell, D. J., Jeong, K. W., Bittencourt, D., Gerke, D. S., and Stallcup, M. R. (2011). A distinct mechanism for coactivator versus corepressor function by histone methyltransferase G9a in transcriptional regulation. J. Biol. Chem. 286, 41963–41971. doi: 10.1074/jbc.m111.298463
Schug, T. T., Janesick, A., Blumberg, B., and Heindel, J. J. (2011). Endocrine disrupting chemicals and disease susceptibility. J. Steroid Biochem. Mol. Biol. 127, 204–215. doi: 10.1016/j.jsbmb.2011.08.007
Scior, T., Medina-Franco, J., Do, Q. T., Martínez-Mayorga, K., Rojas, J., and Bernard, P. (2009). How to recognize and workaround pitfalls in QSAR studies: a critical review. Curr. Med. Chem. 16, 4297–4313. doi: 10.2174/092986709789578213
Sedykh, A., Zhu, H., Tang, H., Zhang, L., Richard, A., Rusyn, I., et al. (2011). Use of in vitro HTS-derived concentration-response data as biological descriptors improves the accuracy of QSAR models of in vivo toxicity. Environ. Health Perspect. 119, 364–370. doi: 10.1289/ehp.1002476
Serafimova, R., Todorov, M., Nedelcheva, D., Pavlov, T., Mekenyan, O., Akahori, Y., et al. (2007). QSAR and mechanistic interpretation of estrogen receptor binding. SAR QSAR Environ. Res. 18, 389–421. doi: 10.1080/10629360601053992
Shanle, E. K., and Xu, W. (2011). Endocrine disrupting chemicals targeting estrogen receptor signaling: identification and mechanisms of action. Chem. Res. Toxicol. 24, 6–19. doi: 10.1021/tx100231n
Solimeo, R., Kim, M., Zhu, H., Zhang, J., and Sedykh, A. (2012). Predicting chemical ocular toxicity using a combinatorial QSAR approach. Chem. Res. Toxicol. 25, 2763–2769. doi: 10.1021/tx300393v
Taha, M. O., Tarairah, M., Zalloum, H., and Abu-Sheikha, G. (2010). Pharmacophore and QSAR modeling of estrogen receptor β ligands and subsequent validation and in silico search for new hits. J. Mol. Graph. Model. 28, 383–400. doi: 10.1016/j.jmgm.2009.09.005
Tropsha, A., and Golbraikh, A. (2007). Predictive QSAR modeling workflow, model applicability domains, and virtual screening. Curr. Pharm. Des. 13, 3494–3504. doi: 10.2174/138161207782794257
Tsuchiya, Y., Nakajima, M., and Yokoi, T. (2005). Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 227, 115–124. doi: 10.1016/j.canlet.2004.10.007
Vapnik, V. (2000). The Nature of Statistical Learning theory. New York, NY: Springer Science & Business Media.
Vedani, A., Dobler, M., and Smieško, M. (2012). VirtualToxLab — A platform for estimating the toxic potential of drugs, chemicals and natural products. Toxicol. Appl. Pharmacol. 261, 142–153. doi: 10.1016/j.taap.2012.03.018
Walker, T., Grulke, C. M., Tropsha, A., and Pozefsky, D. (2010). Chembench: a cheminformatics workbench. Bioinformatics 26, 3000–3001. doi: 10.1093/bioinformatics/btq556
Wang, W., Kim, M., Sedykh, A., and Zhu, H. (2015). Developing enhanced blood-brain barrier permeability models: integrating external bio-assay data in QSAR modeling. Pharm. Res. 32, 3055–3065. doi: 10.1007/s11095-015-1687-1
Willett, P. (2006). Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 11, 1046–1053. doi: 10.1016/j.drudis.2006.10.005
Zang, Q., Rotroff, D. M., and Judson, R. S. (2013). Binary classification of a large collection of environmental chemicals from estrogen receptor assays by quantitative structure-activity relationship and machine learning methods. J. Chem. Inf. Model. 53, 3244–3261. doi: 10.1021/ci400527b
Zhang, J., Zhu, H., and Hsieh, J. H. (2014). Profiling animal toxicants by automatically mining public bioassay data: a big data approach for computational toxicology. PLoS ONE 9:e99863. doi: 10.1371/journal.pone.0099863
Zhang, L., Sedykh, A., Tripathi, A., Zhu, H., Afantitis, A., Mouchlis, V. D., et al. (2013). Identification of putative estrogen receptor-mediated endocrine disrupting chemicals using QSAR- and structure-based virtual screening approaches. Toxicol. Appl. Pharmacol. 272, 67–76. doi: 10.1016/j.taap.2013.04.032
Zheng, W., and Tropsha, A. (2000). Novel variable selection quantitative structure-property relationship approach based on the k-nearest-neighbor principle. J. Chem. Inf. Comput. Sci. 40, 185–194. doi: 10.1021/ci980033m
Zhu, H., Martin, T. M., Ye, L., Sedykh, A., Young, D. M., and Tropsha, A. (2009). Quantitative structure-activity relationship modeling of rat acute toxicity by oral exposure. Chem. Res. Toxicol. 22, 1913–1921. doi: 10.1021/tx900189p
Zhu, H., Rusyn, I., Richard, A., and Tropsha, A. (2008b). Use of cell viability assay data improves the prediction accuracy of conventional quantitative structure-activity relationship models of animal carcinogenicity. Environ. Health Perspect. 116, 506–513. doi: 10.1289/ehp.10573
Zhu, H., Tropsha, A., Fourches, D., Varnek, A., Papa, E., Gramatical, P., et al. (2008a). Combinatorial QSAR modeling of chemical toxicants tested against Tetrahymena pyriformis. J. Chem. Inf. Model. 48, 766–784. doi: 10.1021/ci700443v
Keywords: QSAR modeling, estrogen receptor α, bioassay profiling, endocrine disrupting chemicals, biosimilarity
Citation: Ribay K, Kim MT, Wang W, Pinolini D and Zhu H (2016) Predictive Modeling of Estrogen Receptor Binding Agents Using Advanced Cheminformatics Tools and Massive Public Data. Front. Environ. Sci. 4:12. doi: 10.3389/fenvs.2016.00012
Received: 30 September 2015; Accepted: 19 February 2016;
Published: 08 March 2016.
Edited by:
Juergen Pilz, Alpen-Adria Universitaet Klagenfurt, AustriaReviewed by:
Ijaz Hussain, Quaid-i-Azam University, PakistanVenkata Krishna Jandhyala, Washington State University, USA
Copyright © 2016 Ribay, Kim, Wang, Pinolini and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Zhu, hao.zhu99@rutgers.edu