 Alan Harrison Anne Parle-McDermott*
Alan Harrison Anne Parle-McDermott*- Nutritional Genomics Group, School of Biotechnology, Dublin City University, Dublin, Ireland
DNA methylation is a biochemical process where a DNA base, usually cytosine, is enzymatically methylated at the 5-carbon position. An epigenetic modification associated with gene regulation, DNA methylation is of paramount importance to biological health and disease. Recently, the quest to unravel the Human Epigenome commenced, calling for a modernization of previous DNA methylation profiling techniques. Here, we describe the major developments in the methodologies used over the past three decades to examine the elusive epigenome (or methylome). The earliest techniques were based on the separation of methylated and unmethylated cytosines via chromatography. The following years would see molecular techniques being employed to indirectly examine DNA methylation levels at both a genome-wide and locus-specific context, notably immunoprecipitation via anti-5′methylcytosine and selective digestion with methylation-sensitive restriction endonucleases. With the advent of sodium bisulfite treatment of DNA, a deamination reaction that converts cytosine to uracil only when unmethylated, the epigenetic modification can now be identified in the same manner as a DNA base-pair change. More recently, these three techniques have been applied to more technically advanced systems such as DNA microarrays and next-generation sequencing platforms, bringing us closer to unveiling a complete human epigenetic profile.
Introduction
When the Human Genome Project was completed in 2003, 50 years after the discovery of the Double Helix, it was clear that the full picture had yet to be elucidated (Claverie, 2001; Kruglyak and Nickerson, 2001; Lander et al., 2001). The sequence of bases that make up the human genome alone was not enough to account for what makes the human populace so diverse. Five years later, giant leaps in technological advances paved the way for the announcement of the 1000 Genomes Project, aiming to sequence the genomes of 1000 anonymous individuals to visualize the genomic differences that make each person unique (Kaiser, 2008; Durbin et al., 2010). However, mounting evidence from the past few decades is pointing to a new set of variables that contribute to our individuality. The Human Genome Project has already unveiled the genetic hardware needed to create a person, but the search for the biochemical software is still underway. The next major milestone in defining life is The Epigenome: the sum of heritable chemical and chromosomal modifications to genetic material that influences the development of complex organisms. We focus on DNA methylation, the incorporation of a methyl group in mostly a CpG motif, which was first found to influence gene expression in 1975 (Holliday and Pugh, 1975; Riggs, 1975).
At the time, it had been accepted that bacteria were capable of methylating both adenine and cytosine residues while higher organisms possessed mainly methylated cytosines (Wyatt, 1950; Doskocil and Sorm, 1962; Meselson et al., 1972; Smith et al., 1973). The enzyme DNA adenine methylase (Dam) in E. coli specifically methylates GATC sequences, and DNA cytosine methylase (Dcm) methylates the duplex sequence CCWGG (W denotes A or T; Casadesús and Low, 2006). As a defence mechanism, bacteria use a plethora of very specific DNA digesting enzymes to ward off invading phages. These enzymes cleave DNA based on a target nucleotide sequence, usually a palindrome motif of several bases, so the enzymes have no way of differentiating between viral and bacterial DNA. A restriction/modification mechanism allows bacterial cells to protect their own DNA from restriction enzymes by introducing a DNA methylation signature into newly synthesized strands (reviewed in Bickle and Krüger, 1993). It was understood that these bacteria carried out methylation in a highly specific manner, but the significance of cytosine methylation in eukaryotes was not fully realized until later. Even though there was no direct evidence of a specific methylating enzyme, Holliday and Pugh (1975) based their early DNA methylation model in eukaryotes on the mechanisms of bacterial methylating enzymes, and the fact that methyl groups are distributed about the genome in a non-random manner. Amongst their concluding remarks, they suggest “it may be significant that the doublet CpG is the most highly methylated,” oblivious to how important this statement would be in context of the huge strides in the field to come in the following decades. Independently, a similar paper by Arthur Riggs presented the same hypothesis, this time focusing on the role of DNA methylation in X-inactivation and in mediating DNA binding proteins (Riggs, 1975). Both papers brought considerable attention to the phenomena of DNA methylation, whilst alluding to a new somatically heritable information system that lay within the genetic code.
DNA methylation is now considered to be an important molecular mechanism in a number of biological processes including genomic imprinting, X-inactivation, tissue specific gene expression, and possibly trans-generational effects (Riggs, 1975; Razin and Ceder, 1991; Li et al., 1993). However, the methods to analyze genome-wide DNA methylation patterns is still evolving. We review the development of DNA methylation methodologies from the late 1970s to the present day (Figure 1).
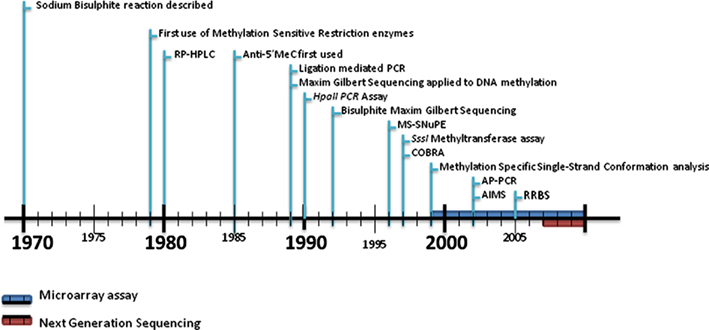
Figure 1. Timeline of DNA methylation analysis. The techniques for DNA methylation analysis have developed from the ability to simply measure the amount of 5-methylcytosine within a particular genome in the early 1980s to a variety of basic comparative methods involving methylation-sensitive restriction enzymes, immunoprecipitation or bisulfite sequencing usually in combination with PCR up to the late 1990s. The introduction of microarray technology and next-generation sequencing saw the adaption of these earlier methods to these newer platforms during the 2000s. More details on microarray/beadchip technologies and next-generation sequencing are described in Tables 1 and 2. RP-HPLC, reversed-phase high performance liquid chromatography; 5mC, 5-methylcytosine; MS-SnuPe, methylation-sensitive single nucleotide primer extension; COBRA, combined bisulfite restriction analysis; AP-PCR, arbitrarily primed PCR; AIMS, amplification of inter-methylated sites; RRBS, reduced representation bisulfite sequencing.
Early Non-Specific Methods
Early non-specific methods are summarized in Table 1 and described in more detail below.

Table 1. Summary of early non-specific methods.
HPLC and TLC Methods
Ambitious attempts to map the Epigenome started long before the era of the Human Genome. In the fallout of the Holliday and Riggs papers(Holliday and Pugh, 1975; Riggs, 1975), methods for measuring and profiling these epigenetic variations were put forward. The earliest breaches into the epigenetic landscape were based on the separation of methylated and unmethylated deoxynucleosides. The most significant technique at the time was the separation of purines and pyrimidines by Vischer and Chargaff (1948) through paper chromatography. In the context of DNA methylation, Kuo et al. (1980) established an analytical technique to measure 5-methylcytosine (5mC) quantitatively using reversed-phase high performance liquid chromatography (RP-HPLC). This method is based on the quantitative hydrolysis of DNA using DNase I and nuclease P1, followed by treatment with alkaline phosphatase. The individual bases can then be monitored based on their UV absorbances at 254 and 280 nm. The RP-HPLC method was further improved throughout the 1980s (Gomes and Chang, 1983; Patel and Gopinathan, 1987) with incorporation of mass spectrometry with standard HPLC by Annan et al. (1989). Of course, HPLC based methods require specialized machinery, so naturally, alternative separation techniques came into use. Bestor et al. (1984) used two restriction endonucleases, Msp1 and Taq1 to discriminate between methylated and unmethylated-CpG residues in their restriction sites, CCGG and TCGA respectively. Digested DNA was 5′ end-labeled with a 32P isotope and subsequently hydrolyzed to deoxyribonucleotide monophosphate followed by separation in two dimensions via thin-layer chromatography (TLC). Quantitative measurement of DNA methylation is based on the relative intensity between C and 5mC fractions after separation.
The RP-HPLC and TLC methods described above were only capable of measuring the relative ratio of methylated cytosine residues against unmethylated cytosines. Although this has been useful for many applications, such as comparing the DNA methylation amongst different animal or plant species (Wagner and Capesius, 1981; Gama-Sosa et al., 1983), fully charting the epigenome was far out of reach using these methods. More specific and informative methods are now in practice to detect 5-methylcytosine, but today, HPLC and TLC based methods are now best suited to detecting hydroxymethylcytosine, an epigenetic modification once believed to be only found in bacteriophages, but recently discovered to be abundant in humans and animals (Kriaucionis and Heintz, 2009; Tahiliani et al., 2009).
Radiolabeling
Instead of trying to separate and observe individually methylated bases at a high resolution, more indirect approaches have been devised. It is possible to enzymatically incorporate tritium labeled methyl groups from S-adenosylmethionine to unmethylated cytosines. Assays have been developed using bacterial SssI methyltransferase to incorporate radiolabeled methyl groups into CpG sites. The level of radioactivity measured is inversely proportional to the level of DNA methylation of a sample (Wu et al., 1993; Duthie et al., 2000).
Anti-Methylcytosine
Other alternative methods include the wide range of immunological DNA methylation assays that suddenly appeared after it was first found that methylcytosine was accessible to specific antibodies in 1985 (Adouard et al., 1985). This vital development paved the way for the possibility to chart the DNA methylation landscape on a cell to cell basis. The Adouard paper introduces the quantification of radiolabeled DNA retained by leporine polyclonal antibodies, visualized under electron microscopy. Later, confocal fluorescence microscopy was used to detect global changes in methylation patterns. Using anti-5mC monoclonal antibodies and secondary antibodies labeled with fluorescent isothiocyanate, Oakeley et al. (1997) devised an efficient method to study global changes in DNA methylation during tobacco pollen maturation. The use of anti-5mC has been widely applied since its introduction but most notably in the investigation of DNA methylation changes during embryonic development. The mammalian genome undergoes a mass loss of DNA methylation, followed by remethylation during early embryonic development. Although this was established using methylation-sensitive restriction enzymes (Monk et al., 1987) a more precise profile was obtained with anti-5mC antibody in conjunction with confocal imaging (Santos et al., 2002). They found that the paternal genome undergoes selective demethylation immediately after sperm decondensation, and is complete after 90–120 min. The de novo methylases DNMT3a and DNMT3b restore DNA methylation later in development, which is maintained by DNMT1 throughout life (Bestor, 2000).
Out of the methods discussed so far, the immunological approach has seen the most significant improvements and novel applications over the past decade alone, due to advances in microarray technology. These will be discussed in more detail later.
Early Differential Gene Methylation Analysis
Early differential gene methylation methods are summarized in Table 2 and described in more detail below.

Table 2. Summary of early differential gene methylation analysis.
Methylation-Sensitive Restriction Enzymes
Restriction enzymes cleave DNA through recognition of specific nucleotide motifs. Amongst the variety of different types of restriction enzymes that exist, only some are sensitive to DNA methylation. Given the considerable amount of redundancy amongst the many different palindrome motifs targeted by restriction enzymes, many pairs of restriction enzymes exist that both cut at the same nucleotide sequence but with differing sensitivity to DNA methylation signatures. Isoschizomer pairs like this can be used to discriminate between methylated and unmethylated regions of the genome in a laboratory setting (Bird and Southern, 1978) and initially exemplified in 1979 with HpaII and MspI (Cedar et al., 1979). Both recognize and cut at the same sequence, CCGG, but methylation of the second C in this motif prevents digestion by HpaII. Detection of digested DNA fragments was initially by radiolabeling and two dimensional TLC. Later Southern blotting was employed (Southern, 1975) for visualization followed by the introduction of methylation-sensitive PCR-based methods in 1990 (Singer-Sam et al., 1990). However, efficiency of the restriction enzymes was a likely issue for these techniques.
Differential Genome-Wide Scanning
Restriction landmark genomic scanning (RLGS) is a genomic scanning method that takes advantage of the specificity of restriction endonucleases and allows a low resolution comparison of genome-wide differences between individuals (Hatada et al., 1991). Radiolabeled DNA is digested with two restriction enzymes and separated in two dimensions. This produces an autoradiograph profile of thousands of spots spread through the gel, each spot representing a restriction site. This method was adapted for DNA methylation analysis (RLGS-M) by employing methylation-sensitive restriction enzymes (Hayashizaki et al., 1993; Kawai et al., 1993) to differentiate methylation differences between individuals. Later, simpler and less expensive genome-wide screening strategies came into use. Using a single primer and two low-stringency annealing steps, Liang et al. (2002) found that methylation profiles could be obtained by digesting DNA with methylation-specific endonucleases followed by a PCR reaction with random primers. This process is known as arbitrarily primed PCR (AP-PCR), and is based on a method developed by Welsh and McClelland (1990) initially used to identify bacterial species. AP-PCR was adapted in order to scour tumor genomes for new differential methylation sites (Gonzalgo et al., 1997; Liang et al., 2002). Amplification of inter-methylated sites (AIMS) is a similar but more effective PCR-based approach. Methylation-sensitive isoschizomers are employed that cleave DNA leaving a blunt end or an overhang. These properties are exploited by the addition of linkers that only ligate to the methylated sites with subsequent PCR amplification. Fingerprints composed of multiple anonymous bands represent methylated regions of the genome are generated and can be excised out and characterized individually (Frigola et al., 2002).
It should be noted that the methods discussed so far are limited in the context of other genetic techniques at the time. This is because in vitro amplification of methylated DNA strands via PCR causes the target strand to lose its methylation status. The methods so far have aimed to detect 5-methylcytosine as it manifests naturally. On a genome-wide or gene-specific scale, these approaches are limited. In order to advance to the stage of possibly sequencing the epigenome, a new approach was needed.
The Sodium Bisulfite Era
In 1970, a chemical interaction between sodium bisulfite and pyrimidines was described that would have a colossal impact on how DNA methylation is studied (Hayatsu et al., 1970). It was found that uracil, thymidine, and deoxycytidine were subjected to sulfonation at position six of their pyrimidine rings. Ten years later, this model was extended to 5-methylcytosine although the reaction takes place at a slower rate than cytosine (Wang et al., 1980). Frommer et al. (1992) described in a classic paper that the differing reaction rates of 5mC to C could be exploited to analyze DNA methylation patterns in genomic DNA. Treating DNA with sodium bisulfite, they proposed, will deaminate cytosine residues into uracil at a much faster rate than 5mC. This phenomenon made it possible to change a chemical modification of DNA to an easily detected genetic element. At the time, Maxim and Gilbert sequencing was used to pinpoint the changes, but the methods put forward by Frommer and colleagues would be revised and refined as technological advances in the subsequent years would pave the way for large scale, next-generation sequencing.
The Frommer et al. (1992) paper marked somewhat of a revolution in the field. Now the elusive biochemical software could be converted to more tangible genetic hardware. Although it was initially described how bisulfite modification could be used to augment sequencing-based methods, the concept itself would be used to formulate entirely new methods to probe the genome for DNA methylation in the following years. These methods are based on the treatment of DNA with bisulfite such that unmethylated cytosines are converted to uracil and methylated cytosines remain as cytosines. The approaches to detect these conversions are various and are summarized in Table 3 and described in more detail below.

Table 3. Summary of methods using sodium bisulfite treatment.
Gene-Specific Approaches
Methylation-specific PCR (MS-PCR) was one of the first innovative methods to incorporate bisulfite conversion outside the context of sequencing (Herman et al., 1996). Primers were designed to discriminate between methylated and unmethylated regions of DNA after bisulfite treatment, so primer sites that were originally methylated would undergo amplification only. The nature of this rapid assay eliminated the frequent false positives associated with previous PCR-based endonuclease methods; however PCR bias was an issue. Technical advances in genomics and molecular biology in more recent years have allowed MS-PCR take on a new form.
Many of the new techniques introduced during the Sodium Bisulfite Era followed a similar strategy; using well established genetic techniques to detect DNA methylation since the elusive epigenetic modification could be converted into the more tangible nucleotide variant. Methylation-sensitive single nucleotide primer extension (MS-SNuPE) is based on a conventional genotyping technique, single nucleotide primer extension (Kuppuswamy et al., 1991). MS-SNuPE (Gonzalgo and Jones, 1997) uses a PCR step after bisulfite conversion to amplify a desired fragment. Once the product is isolated, primers specific for the amplified fragments are used in another PCR stage, this time incorporating 32P dNTPs which can be used to quantify the nucleotides that have been converted during bisulfite treatment, therefore quantifying the level of DNA methylation in the initial genomic DNA.
Based on another well-known PCR method for resolving single-base restriction fragment length polymorphisms (Poduslo et al., 1991), methylation-sensitive single-strand conformation analysis (MS-SSCA) is a method to screen and analyze DNA methylation in a gene-specific manner (Bianco et al., 1999). Genomic DNA is bisulfite treated and the gene of interest is amplified with PCR, and then cut with frequently cutting restriction enzymes. The digestion patterns of samples are compared to a methylation standard and variations in pattern imply changes in DNA methylation. Methylation differences are characterized using a gel stabbing technique and sequencing (Wilton et al., 1997). This method has been expanded by Suzuki et al. (2000) to include high performance capillary electrophoresis (HPCE).
High resolution melting (HRM) was originally used to genotype Single Nucleotide Polymorphisms (Wittwer et al., 2003) but was adopted to detect DNA methylation changes in bisulfite treated DNA (Wojdacz and Dobrovic, 2007). Single base differences can be detected by their distinct melting profiles utilizing specific fluorescent dyes. The difference between 5-methylcytosine and cytosine, manifests as a single base change after DNA is treated with sodium bisulfite. With careful primer design to eliminate PCR bias, it is possible to estimate the methylation levels of a test sample by comparison of its melting curve with that of a series of controls of known methylated and unmethylated percentages (Wojdacz et al., 2008).
The “bisulfite revolution” was not limited to the importation of early genetic techniques to the field of DNA methylation; methylation analysis mentioned earlier in this review would also receive a renewal. The endonuclease-based protocols used up until the mid 1990s were limited to the detection of a negative result: the absence of a band indicates a methylated site. This was first improved by Sadri and Hornsby (1996), where DNA was first treated with bisulfite according to a revised version of the 1992 bisulfite reaction (Feil et al., 1994), then exposed to two rounds of endonuclease digestion including a newly created restriction site following bisulfite treatment. This innovation was expanded by Xiong and Liard (1997) to determine the methylation status of individual loci. Combined bisulfite restriction analysis (COBRA), is based on the creation of new methylation dependant restriction sites, or the retention of pre-existing ones, by bisulfite conversion followed by PCR. With phosphorimaging, the relative ratio of digested products can be determined. Although it is a powerful technique, COBRA is limited to the restriction sites of the enzymes used. Laird et al. (2004) devised a technique known as “hairpin-bisulfite PCR” to investigate DNA methylation symmetry at a specific locus. With bisulfite treatment, the required denaturation steps make it difficult to analyze the methylation pattern of two complementary DNA strands from one molecule. By ligating a hairpin linker to restriction-enzyme cleaved DNA, the team were able to establish a covalent bond between complementary strands of the DNA molecule, which would allow a PCR product to span the linker and cover both strands.
Regional Methylation Levels
Another method already mentioned here that has received a sodium bisulfite facelift is the SssI methyltransferase assay. In its new incarnation, the enzymatic regional methylation assay (ERMA), genomic DNA is treated with sodium bisulfite prior to amplification of a particular region of interest with non-discriminating primers containing flanking GATC sites. These tetranucleotide sequences are required to standardize DNA quantity in this assay, as they are dam sites that accept methyl groups from dam methyltransferase. To quantify DNA methylation, E. coli cytosine methyltransferase SssI was used to specifically methylate the cytosine in all of the CpG dinucleotides that remained after sodium bisulfite treatment, using 3H-labeled S-adenosyl-L-methionine (SAM) as a methyl donor. For the aforementioned standardization step, 14C-labeled SAM was incubated along with dam methyltransferase so the total number of amplicons could be visualized (Galm et al., 2002).
In 2001, the working draft of the Human Genome was published in special issues of Nature and Science (Pennisi, 2001). Later that year, Human chromosome 20 was fully sequenced, the third chromosome to be completed in the Human Genome Project. This year was also an important year for DNA methylation and epigenetics too, because it was here that a new phrase entered the vocabulary of the scientific community: The Methylome (Feinberg, 2001).
The post-genome era
By the beginning of the twenty-first century, a great deal of the epigenetic landscape had been explored. While the role and mechanism of gene regulation via DNA methylation was well understood, the gene-specific methods described above helped bring these ideas to the context of complex diseases states, especially tumorigenesis (Jones and Laird, 1999). However, very little was known about the genome-wide distribution of 5-methylcytosine until robust array precipitation methods were devised.
Comparative Methylation Profiling Using Microarray Technology
Throughout the 1990s, the development of DNA microarray technology was responsible for a revolution in functional genomics, paving the way for high-throughput analysis of single nucleotide polymorphisms and other genomic variants (Southern et al., 1999). With the help of these novel tools, the three traditional lines of attack on the DNA methylation landscape; immunoprecipitation, endonuclease digestion, and sodium bisulfite treatment, would each receive a post-genome era transformation (Figure 2). These three DNA methylation differentiation and isolation methods have been the principal approaches used to compare the DNA methylation patterns between samples over the last decade. In the microarray assays discussed here, the underlying principle is the same in each: methylated and unmethylated fragments of the genome are separated and analyzed. Hybridization to a microarray of known probes allows for quantification and identification of areas of the genome that are methylated or unmethylated. All of the microarray-based techniques discussed here are listed in Table 4 and are based on one of the three approaches described below and in Figure 2.
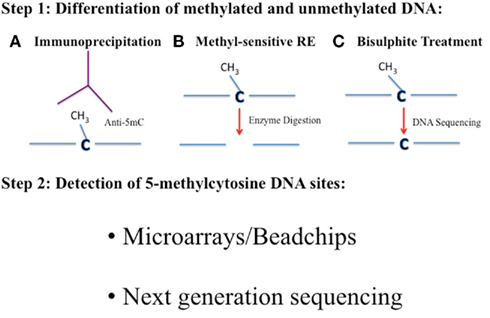
Figure 2. The three main current approaches for DNA methylation analysis of genomes. The analyses of DNA methylation patterns across a genome at varying degrees of resolution involves three main approaches. In Step 1 methylated and unmethylated cytosines need to be distinguished. This can be achieved by using methods A, B, or C. (A) Immunoprecipitation with an antibody against 5mC (Anti-5mC)/methyl-binding protein or precipitation with specific methyl-binding proteins. (B) Digestion of DNA with methyl-sensitive restriction enzymes (RE) that cleave methylated and unmethylated cytosines differently. (C) Bisulfite treatment of DNA will convert unmethylated cytosines to uracil, which are “read” as Ts when PCR amplified and sequenced. Methylated cytosines remain as cytosines when sequenced. The 5mC sites can then be identified in Step 2 by either using a microarray or beadchip platform or by next-generation sequencing.
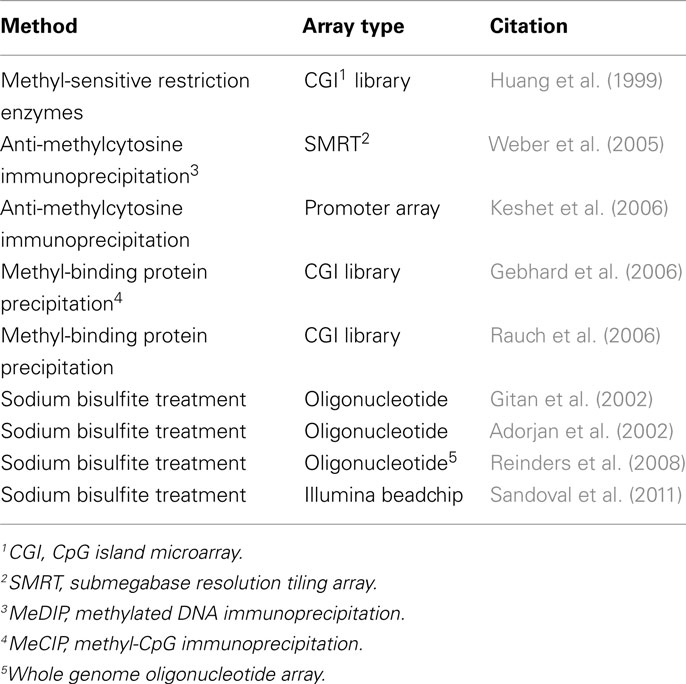
Table 4. Applications of microarray technology to differential DNA methylation analyses.
Endonuclease Digestion
Differential methylation hybridization (DMH) was the first array-based method for genome-wide screening of hypermethylated-CpG islands in tumor cells (Huang et al., 1999). This early array was only able to asses about 300 CpG islands at a time, and suffered from major sequence bias. Genomic DNA was first sheared with a methylation insensitive restriction enzyme, MseI. Having a restriction recognition site at TTAA, MseI was unlikely to interfere with any CpG islands. After the ligation of linkers to the end of each DNA fragment, half of the pool was treated with methylation-sensitive BstUI. As a result, the methylated fragments, and those only treated with MseI remained intact, and only these could be amplified via PCR, with primers specific to the linkers. These amplicons were differentially labeled and co-hybridized to a CpG island array. As for any array-based method, the analysis is limited to the number of genomic elements represented on the array. The array, used to determine the methylation status of CpG islands in breast cancer cells, was constructed from a physical library of CpG islands generated from a novel column separation strategy (Cross et al., 1994). Shortly after it was introduced, DMH was used to detect specific methylation profiles in breast and ovarian cancer cells (Yan et al., 2000; Ahluwalia et al., 2001). Following on from this, the same group improved on this method further (Yan et al., 2001; Chen et al., 2003).
In 2005, the Promoter-associated methylated DNA amplification DNA chip was introduced. Contrary to the use of a second wave of restriction enzymes, the restriction endonuclease McrBC has been used to fractionate methylated regions of DNA. In a protocol pioneered by Nouzova et al. (2004), DNA is treated with MseI, and the fragments are ligated to linkers, in concurrence to the previous methods. However, the fragments are then divided into two pools: one to be treated with McrBC, while the other is not. Unlike the other restriction enzymes discussed so far, McrBC only cuts at methylated sequences. After PCR, both pools are differentially labeled with Cy3 and Cy5 fluorescent dyes and co-hybridized to a CpG island array. In the Methylscope platform (Ordway et al., 2006), DNA fragments are prepared in a similar way, but the DNA is randomly sheared in the first step.
Comprehensive high-throughput arrays for relative methylation (CHARM) is another platform for array-based methylation analysis (Irizarry et al., 2008). The workflow is based on some of the methods already mentioned in this section, and works to eliminate the disadvantages of each. CHARM was conceived while Irizarry and colleagues were comparing three already established methods for analyzing DNA methylation: methylated DNA immunoprecipitation (MeDIP), HELP, and fractionation by McrBC. The HELP assay (HpaII tiny fragment enrichment by ligation-mediated PCR) is based again on the use of two sets of restriction enzymes, but the fragments are amplified via ligation-mediated PCR and hybridized to a custom microarray along with separate fluorochromes (Khulan et al., 2006). In the 2008 paper, Irizarry points out significant flaws with each of the array-based methods in use. MeDIP (also discussed below), was shown to have a significant bias to CpG islands, HELP had incomplete genomic coverage and McrBC fractionation displayed location imprecision. After the rigorous comparison of these methods, the second half of the paper discusses how a new platform of original array design strategies and statistical procedures involving genome-weighted averages from larger genomic areas was capable of countering these limitations (Irizarry et al., 2008).
Immunoprecipitation
Differentiation between methylated DNA and non-methylated DNA using anti-methyl antibodies has been discussed already. However, with the requirement to enrich methylated DNA prior to microarray hybridization, immunological separation techniques became relevant again.
In 2005, MeDIP was used to immunocapture methylated cytosines with an antibody specific for methylated cytosines for array hybridization (Weber et al., 2005). Prior to immunoprecipitation, genomic DNA was randomly fragmented via sonication or enzyme restriction. Immunocaptured DNA and control genomic DNA were both labeled with Cy5 and Cy3 fluorescent dyes, producing a ratio of green fluorescence to red fluorescence which would be indicative of the relative levels of hypermethylation or hypomethylation. In the 2005 paper, Weber and colleagues used a submegabase resolution tiling (SMRT) array consisting of 32,433 overlapping BAC clones spanning the entire genome (Ishkanian et al., 2004; Weber et al., 2005). Independently, Keshet et al. (2006) devised a similar array-based approach: methyl-DNA immunoprecipitation (MDIP). They found that tumor specific methylated genes are found in clusters on chromosomes, and shared many structural and functional features. This reinforced the hypothesis that tumorigenesis arises as a result of de novo mechanisms. One of the major drawbacks of MDIP and MeDIP is their inability to pinpoint DNA methylation changes at a single base-pair resolution (Beck and Rakyan, 2008). However, some argue that since neighboring CpG islands spanning up to 1000 bp are co-methylated in healthy cells, there is no need for methylation analyses with single base-pair resolutions (Eckhardt et al., 2006). In 2006 MDIP was responsible for producing the first complete Methylome: Arabidopsis thaliana (Zhang et al., 2006). Although much smaller than the mammalian genome, the map of the plant’s methylome represents an important milestone in epigenetics, while the data produced was of interest in itself. It was found that one third of expressed genes contained DNA methylation in their transcribed regions, and these regions were still highly expressed and constitutionally active.
Methyl-CpG immunoprecipitation (MeCIP) is another immunoprecipitation assay similar to MeDIP in terms of the techniques used and its applications, but a recombinant protein complex with similar properties to an anti-methylcytosine antibody is used (Gebhard et al., 2006). Epigenetic gene silencing via DNA methylation is caused by steric hindrance, resulting from methylated DNA recruiting methyl-binding domain proteins (MBDs; Thu et al., 2010). In the 2006 paper, Gebhard and colleagues introduce a recombinant protein made up of MBD2 combined with the Fc tail of a human Ig1 with very high affinity to single-stranded methylated DNA, stronger than that of the MeDIP and MDIP methods. Also, it is possible to separate DNA fragments into fractions of increasing methylation density by eluting with a salt gradient (Schilling and Rehli, 2007). This approach allows for the quantification of tissue specific methylation differences for a wide range of DNA methylation densities.
Another protein complex, MBD3LI bound to MBD2, has also been shown to have a high affinity to methylated DNA (Rauch and Pfeifer, 2005). Methylated-CpG island recovery assay (MIRA) separates fragmented DNA by incubation with a matrix containing glutathione-S-transferase-MBD2b in the presence of methyl-CpG-binding domain protein 3-like-1, which increases the affinity of MBD2b when paired. CpG island methylation can be detected using PCR or array-based methods (Rauch et al., 2006, 2009).
Bisulfite Treatment
In 2002, the principles of DHM were expanded to the use of novel methylation-specific oligonucleotide arrays (Adorjan et al., 2002; Gitan et al., 2002). This time, DNA was prepared for hybridization by bisulfite modification and PCR amplification to convert unmethylated cytosines to thymidine, allowing the epigenetic modification to be detected via traditional hybridization methods. This approach has the potential to detect methylated-CpG islands at a single-base resolution, but the global conversion of cytosines to thymidines results in a reduction in sequence complexity, making it difficult to design enough unique probes to scale up to a genome-wide level (Beck and Rakyan, 2008). Although it is possible to design probes taken from amplified bisulfite treated DNA, novel approaches have been devised. For small, methylation-rich genomes, a method called bisulfite methylation profiling (BiMP) can be employed (Reinders et al., 2008). The entire genome of A. thaliana was amplified using a technique utilizing random tetranucleotides primers reducing the amplification bias usually associated with bisulfite treated DNA. The BiMP data from the paper was compared to the MDIP results cited earlier (Zhang et al., 2006). Data from both studies were in concordance, although the former exhibited profiles of considerably higher resolution than the latter (Reinders et al., 2008). As a result, BiMP is more likely to pick up specific, localized changes in DNA methylation patterns that could prove elusive to detection via MDIP.
The Illumina Beadchip technology, while technically different from the types of arrays discussed above, do fall under the microarray category. Illumina Infinium has been applied to the DNA methylation analysis. Fryer et al. (2011) examined DNA methylation patterns at 27,578 CpG sites using the Infinium HumanMethylation27K in cord blood samples and correlated to homocysteine levels and birth weight. More recently, Illumina have launched the Infinium HumanMethylation450Karray which allows the analysis of >450,000 DNA methylation sites (Sandoval et al., 2011) with up to 12 samples at a time. This is by far the most high-throughput comprehensive method available for whole genome DNA methylation analysis outside of the next-generation sequencing methods described below.
The use of microarrays for DNA methylation analysis proves to be a versatile strategy for probing the methylome. Before hybridization, methylated DNA can be purified by a number of different strategies, each with their own unique merits. The strengths and weaknesses of most of these methods have been systematically evaluated in Laird (2010). DNA methylation microarrays provide cheap and accessible genome-wide insights to the DNA methylation status of a sample, or even a large number of samples. However, as with the Human Genome Project, there needs to be a trend toward a gold standard: a perfect assay. Although none exists at time of writing, only sequencing-based assays have the potential to provide such a detailed look at the enigmatic methylome, i.e., at single-base resolution.
Sequencing-Based Approaches
Sanger Sequencing
Although the bisulfite reaction itself has been adapted and applied to the conventional genetic techniques aforementioned, the technology has taken a novel route while genome sequencing platforms have improved as the Human Genome Project progressed over the subsequent years. At the moment, it is possible to directly sequence the human genome with sophisticated technology; technology that is starting to be applied to the field of DNA methylation. First, it is worth revisiting how sequencing-based DNA methylation analyses have evolved over the past two decades.
The original bisulfite sequencing protocol from Frommer et al. (1992) suffers from several difficulties. For example, relatively large quantities of genomic DNA are needed for a full profile, limiting its proficiencies in a genome-wide perspective. In 1994, the same lab (Clark et al., 1994), integrated a PCR amplification step to increase the assay’s sensitivity by 104 fold. The old protocol also required DNA to be denatured in order to expose the individual bases to bisulfite treatment. Some workarounds have been devised to counter this, but according to a review by Oakeley (1999), the best approach at the time was to denature DNA in solution with NaOH, then mix with molten agarose. Cooling the agarose locks the DNA in the denatured conformation, allowing subsequent reactions to be performed on the agarose block (Olek et al., 1996).
In the pilot study of the Human Epigenome Project, Rakyan et al. (2004) aimed to profile the DNA methylation patterns of the human major histocompatibility complex (MHC) located on chromosome six. This region of the genome was selected as it is associated with more diseases than any other region of the genome, and also it’s the most polymorphic area of the genome, so complete sequencing and annotation from the Human Genome was readily available for the study. This sequencing method was innovative as it did not require a sub-cloning step, but utilized a novel high-throughput method of direct sequencing of PCR products. An algorithm described by Lewin et al. (2004) allows for quantitative analysis of DNA methylation from four-dye electropherogram data obtained from direct sequencing. Although such data was previously used in the human genome project and for analyzing single-base SNPs (Qiu et al., 2003), earlier applications of bisulfite treated PCR were impeded by unique technical difficulties. The new software, called epigenetic sequencing methylation analysis software (ESME), corrects for incomplete bisulfite conversion, performs quality control tests on data, and maps methylation positions to the reference sequence. This approach was also used in a related paper reporting the DNA methylation profiles of human chromosomes 6, 20, and 22 (Eckhardt et al., 2006).
Reduced representation bisulfite sequencing (RRBS) is a random sequencing-based method for analyzing and comparing DNA methylation patterns on a genome scale (Meissner et al., 2005). Size selected BglII fragments of a whole genome were fixed with ligation linkers and denatured. Bisulfite treatment of these fragments yielded single-stranded DNA, as complementarity between both strands was lost when methylated cytosines were converted to uracil residues. Converted fragments were amplified via PCR and cloned into plasmid vectors for sequencing. Comparison of the bisulfite treated DNA sequence to a reference sequence allows the operator to pinpoint which cytosines have been methylated, as those are the only ones to remain cytosines at this stage. Thymidines that align to cytosines during this comparison stage represent cytosines that were once unmethylated. RRBS has the obvious advantage over PCR-based bisulfite sequencing methods in generating a reproducible library of a small, defined area of a genome. This makes RRBS suitable for comparative methylation studies across different tissue or cell types.
Next-Generation Sequencing
Up until 2005, most whole genome DNA sequencing strategies were based on the cloning of fragments into bacterial vectors, followed by amplification and Sanger sequencing via chain terminating fluorescent signaling, visualized with capillary electrophoresis (Prober et al., 1987). In recent years, however, a new parallel sequencing method was developed that did not require a sub-cloning step. It involves a previously established method for genotyping known as pyrosequencing, which incidentally has also been applied to the analysis of gene-specific/local DNA methylation patterns (Tost and Gut, 2007). The emulsion based PCR method, described by Margulies et al. (2005), utilizes a pyrosequencing protocol optimized for picoliter-scale volumes in the high density picoliter “reactors” formed by the emulsion droplets. In 2007, this massively parallel sequencing system (commercialized as Roche 454 FLX) was employed for bisulfite sequencing (Taylor et al., 2007). The pilot study showed robustness and superiority of this approach by analyzing methylation in 25 gene-related CpG rich regions from over 40 primary cell lines. During the process, specific four-nucleotide tags were added to the 5′end of each primer, so each amplicon could be individually indexed, pooled, and manipulated (Taylor et al., 2007).
Further advances in next-generation sequencing including the Illumina/Solexa Genome Analyzer and the Applied Biosystems SOLiD™ System (reviewed in Mardis, 2008) has meant that going forward, most genome-wide DNA methylation protocols will feature some form of next-generation sequencing. The current gold standard is to carry out whole genome bisulfite sequencing of target samples where a reference genome is available. However, the costs for such an approach are still not trivial and adaptations of methods to produce a representation of genome-wide DNA methylation have been developed. Table 5 describes some of the current options for DNA methylation analysis in combination with next-generation sequencing and are described below. It is worth noting that similarly to microarray analysis, for next-generation sequencing, the three principle approaches still employ sodium bisulfite treatment, immunoprecipitation, and the utilization of methyl-sensitive restriction enzymes (Figure 2).

Table 5. Approaches for DNA methylation analyses by next-generation sequencing.
Whole Genome Sequencing-Bisulfite Treatment
An entire DNA methylome can be assessed at a single nucleotide resolution with sodium bisulfite treatment followed by whole genome sequencing. This approach has been taken to generate a DNA methylation map of A. thaliana (Cokus et al., 2008). Unlike previous genome-wide approaches, this allowed for the sensitive measurement of cytosine-methylation across the genome with sequence specific contexts. When compared to array-based methods, the authors reported the discovery of new methylation sites in previously inaccessible areas of the genome. A whole genome approach was also recently applied to mammalian cells. The first human DNA Methylome in embryonic and fetal cells at single-base resolution was recently published (Lister et al., 2009), identifying a significant proportion of non-CG methylation. Additional single-base resolution human methylomes continue to be published (Maunakea et al., 2010) highlighting the importance of intragenic DNA methylation in the regulation of gene expression. Thus, the elusive Human DNA Methylome is more complex than previously thought.
The whole genome approach is the most desirable with unlimited resources, but realistically for a lot of laboratories this is not an approach that can be taken for the analysis of numerous samples. A more cost effective approach is to reduce the complexity of the genome in order to reduce the amount of sequencing required per sample. The methods described below are some examples of how this can be done.
Methylated DNA Immunoprecipitation Sequencing
The methylated DNA immunoprecipitation sequencing (MeDIP-seq) approach incorporates the anti-methylcytosine antibody described earlier. Briefly, methylated DNA is immunoprecipitated using the antibody against 5-methylcytosine and sequenced (Maunakea et al., 2010; Table 5). The portion of DNA that is immunoprecipitated represents the methylated portion of DNA and is identified by comparison to the reference genome.
Methyl-Binding Domain Isolated Genome Sequencing
Methyl-binding domain isolated genome sequencing (MBDiGs) uses recombinant MBD and MBD2 proteins to enrich methyl-rich DNA fragments from a pool of sonicated genomic DNA (Serre et al., 2009). According to the review by Hirst and Mara (2010), MBDiGS is preferable over MeDIP-seq because a gradient in salt concentrations can be used to elute DNA fragments at different rates depending on their methylation status.
Methyl-Sensitive Restriction Enzyme Sequencing
Methyl-sensitive restriction enzyme sequencing (MRE-seq), as its name suggests, involves methylation-sensitive restriction enzymes (Maunakea et al., 2010). Genomic DNA samples are digested with the restriction enzymes and the subsequent DNA fragments are size selected and sequenced. Differential DNA methylation may be identified by comparison of the fragments sequenced between samples and site specific information is identified by comparison to a reference genome. This method analyses a different portion of the genome compared to MeDIP-seq and therefore, they can be viewed as complimentary approaches.
Modified Methylation-Specific Digital Karyotyping
Modified methylation-specific digital karyotyping (MMSDK) or MSDK-seq (Li et al., 2009) is similar to MRE-seq in that a methyl-sensitive restriction enzyme is employed but includes additional steps that reduce the amount of sequencing required. Rather than sequencing sections of the genome, specific regions of the genome can be identified from their short sequence tags. This significantly reduces the amount of sequencing and in turn reduces the costs of this approach.
Comparison of Contemporary DNA Methylation Methods
In this review, we aim to detail the development and evolution of these analytical methods over time with respect to advancements made in genetics, nucleotide biochemistry, and DNA sequencing technology. As a result, many of the methods discussed are obsolete today and have been replaced by more recent technologies. However, some of the techniques described in the latter part of this review have subtle strengths and weaknesses, and careful judgment should be employed in adopting any of these methods in a research laboratory. There are many recent reviews that compare most recent methods of DNA methylation analysis as mentioned below.
Laird (2010) list the features and source of bias for various sequencing and microarray-based techniques including CpG ambiguity, fragment size bias, cross-hybridization bias. All of the methods that involve sodium bisulfite treatment, they argue, are subject to incomplete bisulfite conversion bias. Thu et al. (2010), also compare the strengths and weaknesses of each method, with a special focus on techniques based on immunoprecipitation. In more detail, a paper by Harris et al. (2010) quantitatively compares the sequencing-based methods MethylC-seq, RRBS, MeDIP-seq and MBD-seq (2010). Due to the nature of their processes, the two bisulfite-based methods yield data with single base-pair resolution, augmented with the capacity to quantify methylation levels. At a reduced coverage, the enrichment methods both have a lower cost-per-CpG in a genome-wide context, but not allowing precise quantification of methylation levels on a genome-wide scale. It appears that none of the currently available methods are without their flaws but bisulfite treated whole genome sequencing offers complete genome coverage at single-base resolution and is currently the method of choice for genome-wide DNA methylation analysis where costs are not prohibitive.
Future of Methylome Analysis
The Next-generation sequencing approaches for DNA methylation analysis will dominate for the moment. However, the methods discussed here cannot detect non-cytosine related methylation reactions, i.e., N6-methyladenine nor 5-hydroxymethylcytosine and therefore, more sophisticated methods are required than currently on offer. Newer sequencing technologies such as single-molecule real-time (SMRT) sequencing (Flusberg et al., 2010) can directly detect all known DNA methylation reactions without the need for bisulfite treatment and is likely to take over from next-generation sequencing in the very near future.
The analysis of 5-hydroxymethylcytosine (5hmC) via HPLC based methods has been discussed briefly. Recently, two novel approaches have been described to discern the genomic distribution of 5hmC (Pastor et al., 2011). The first, called GLIB involves the glucosylation, periodate oxidation, and biotinylation of 5mhC. Biotin molecules can be added to newly formed aldehyde groups on pretreated 5hmC. A glucose moiety is added to 5hmC via a glucosyltransferase enzyme, which has its vicinal hydroxyl groups converted to aldehydes via treatment with sodium periodate. We have previously discussed at length how sodium bisulfite treatment of 5mC does not result in a conversion in a similar time-frame to unmethylated cytosine. However, treatment of 5hmC yields another molecule: 5-methylenesulfonate. For the second method discussed in the article, Pastor et al. (2011) have succeeded in selectively isolating biotinylated 5hmC and sodium bisulfite converted 5hmC using streptavidin and anti-5-methylenesulfonate, respectively. In another recent publication, Kinney et al. (2011) exploited an isoschizomer pair of restriction enzymes, MspI and HpaII, that can differentiate between 5hmC and its glucosylated form. Coupled with qPCR, the team found that ES and brain cell genomic DNA contains a considerable amount of 5hmC, and identified novel loci containing 5hmC in both mouse ES and human brain DNA (Kinney et al., 2011).
Conclusion
The major developments in the methodologies for profiling and fingerprinting the human methylome have followed a clear progression toward innovative sequencing techniques at a single base-pair resolution. As this technology improves, the cost of genome-wide sequencing will decrease, resulting in a new wave of DNA methylation data as more labs become fully immersed in the field. The bioinformatic tools will continue to improve in order to accurately analyze the vast datasets that will no doubt be generated in the coming years. The precedent for this has already been set through the Human Genome Project. Earlier this year, the International Human Epigenome Consortium was launched, aiming to map 1000 Epigenome by 2020 (IHEC, 2010). This is by no means an easy task, but if we see as many technical advances in the field in the next 10 years as we have in the previous decade, it is a challenge we are more than capable of facing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adorjan, P., Distler, J., Lipscher, E., Model, F., Muller, J., Pelet, C., Braun, A., Florl, A. R., Gutig, D., Grabs, G., Howe, A., Kursar, M., Lesche, R., Leu, E., Lewin, A., Maier, S., Muller, V., Otto, T., Scholz, C., Schulz, W. A., Seifert, H.-H., Schwope, I., Ziebarth, H., Berlin, K., Piepenbrock, C., and Olek, A. (2002). Tumour class prediction and discovery by microarray-based DNA methylation analysis. Nucleic Acids Res. 30, e21.
Adouard, V., Dante, R., Niveleau, A., Delain, E., Revet, B., and Ehrlich, M. (1985). The accessibility of 5-methylcytosine to specific antibodies in double-stranded DNA of Xanthomonas phage XP12. Eur. J. Biochem. 152, 115–121.
Ahluwalia, A., Yan, P., Bigsby, R., Hurteau, J., Jung, S., Huang, T., and Nephew, K. (2001). DNA methylation and ovarian cancer I. Analysis of CpG island hypermethylation in human ovarian cancer using differential methylation hybridization. Gynecol. Oncol. 82, 261–268.
Annan, R., Kresbach, G., Giese, R., and Vouros, P. (1989). Trace detection of modified DNA bases via moving-belt liquid chromatography-mass spectrometry using electrophoric derivatization and negative chemical ionization. J. Chromatogr. 465, 285–296.
Beck, S., and Rakyan, V. (2008). The methylome: approaches for global DNA methylation profiling. Trends Genet. 24, 231–237.
Bestor, T., Hellewell, S., and Ingram, V. (1984). Differentiation of two mouse cell lines is associated with hypomethylation of their genomes. Mol. Cell. Biol. 4, 1800–1806.
Bianco, T., Hussey, D., and Dobrovic, A. (1999). Methylation-sensitive, single-strand conformation analysis (MS-SSCA): a rapid method to screen for and analyze methylation. Hum. Mutat. 14, 289–293.
Bird, A., and Southern, E. (1978). Use of restriction enzymes to study eukaryotic DNA methylation: I. The methylation patterns in ribosomal DNA from Xenopus laevis. J. Mol. Biol. 118, 27–47.
Casadesús, J., and Low, D. (2006). Epigenetic gene regulation in the bacterial world. Microbiol. Mol. Biol. Rev. 70, 830–856.
Cedar, H., Solage, A., Glaser, G., and Razin, A. (1979). Direct detection of methylated cytosine in DNA by use of the restriction enzyme MspI. Nucleic Acids Res. 6, 2125–2132.
Chen, C., Chen, H., Hsiau, T., Hsiau, A., Shi, H., Brock, G., Wei, S., Caldwell, C., Yan, P., and Huang, T. (2003). Methylation target array for rapid analysis of CpG island hypermethylation in multiple tissue genomes. Am. J. Pathol. 163, 37–45.
Clark, S., Harrison, J., Paul, C., and Frommer, M. (1994). High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 22, 2990–2997.
Cokus, S., Feng, S., Zhang, X., Chen, Z., Merriman, B., Haudenschild, C. D., Pradhan, S., Nelson, S. F., Pellegrini, M., and Jacobsen, S. E. (2008). Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–219.
Cross, S., Charlton, J., Nan, X., and Bird, A. (1994). Purification of CpG islands using a methylated DNA binding column. Nat. Genet. 6, 236–244.
Doskocil, J., and Sorm, F. (1962). Distribution of 5-methylcytosine in pyrimidine sequences of deoxyribonucleic acids. Biochim. Biophys. Acta 55, 953–959.
Durbin, R. M., Abecasis, G. R., Altshuler, D. L., Auton, A., Brooks, L. D., Gibbs, R. A., Hurles, M. E., McVean, G. A., 1000 Genomes Project Consortium. (2010). A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073.
Duthie, S., Narayanan, S., Blum, S., Pirie, L., and Brand, G. (2000). Folate deficiency in vitro induces uracil misincorporation and DNA hypomethylation and inhibits DNA excision repair in immortalized normal human colon epithelial cells. Nutr. Cancer 37, 245–251.
Eckhardt, F., Lewin, J., Cortese, R., Rakyan, V. K., Attwood, J., Burger, M., Burton, J., Cox, T. V., Davies, R., Down, T. A., Haefliger, C., Horton, R., Howe, K., Jackson, D. K., Kunde, J., Koenig, C., Liddle, J., Niblett, D., Otto, T., Pettett, R., Seemann, S., Thompson, C., West, T., Rogers, J., Olek, A., Berlin, K., and Beck, S. (2006). DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 38, 1378–1385.
Feil, R., Charlton, J., Bird, A., Walter, J., and Reik, W. (1994). Methylation analysis on individual chromosomes: improved protocol for bisulphite genomic sequencing. Nucleic Acids Res. 22, 695–696.
Flusberg, B., Webster, D., Lee, J., Travers, K., Olivares, E., Clark, T., Korlach, J., and Turner, S. (2010). Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7, 461–465.
Frigola, J., Ribas, M., Risques, R., and Peinado, M. (2002). Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS). Nucleic Acids Res. 30, e28.
Frommer, M., McDonald, L., Millar, D. S., Collis, C., Watt, F., Grigg, G., Molloy, P., and Paul, C. (1992). A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strand. Proc. Natl. Acad. Sci. U.S.A. 89, 1827–1831.
Fryer, A., Emes, R., Ismail, K., Haworth, K., Mein, C., Carroll, W., and Farrell, W. (2011). Quantitative, high-resolution epigenetic profiling of CpG loci identifies associations with cord blood plasma homocysteine and birth weight in humans. Epigenetics 6, 86–94.
Galm, O., Rountree, M., Bachman, K., Jair, K., Baylin, S., and Herman, J. (2002). Enzymatic regional methylation assay: a novel method to quantify regional CpG methylation density. Genome Res. 12, 153–157.
Gama-Sosa, M., Midgett, R., Slagel, V., Githens, S., Kuo, K., Gehrke, C., and Ehrlich, M. (1983). Tissue-specific differences in DNA methylation in various mammals. Biochim. Biophys. Acta 740, 212–219.
Gebhard, C., Schwarzfischer, L., Pham, T., Schilling, E., Klug, M., Andreesen, R., and Rehli, M. (2006). Genome-wide profiling of CpG methylation identifies novel targets of aberrant hypermethylation in myeloid leukemia. Cancer Res. 66, 6118–6128.
Gitan, R., Shi, H., Chen, C., Yan, P., and Huang, T.-M. (2002). Methylation-specific oligonucleotide microarray: a new potential for high-throughput methylation analysis. Genome Res. 12, 158–164.
Gomes, J., and Chang, C. (1983). Reverse-phase high-performance liquid chromatography of chemically modified DNA. Anal. Biochem. 129, 387–391.
Gonzalgo, M., and Jones, P. (1997). Rapid quantitation of methylationdifferences at specific sites using methylation-sensitive single nucleotide primer extension (Ms-SNuPE). Nucleic Acids Res. 25, 2529–2531.
Gonzalgo, M., Liang, G., Spruck, C. III, Zingg, J., Rideout, W. III, and Jones, P. (1997). Identification and characterization of differentially methylated regions of genomic DNA by methylation-sensitive arbitrarily primed PCR1. Cancer Res. 57, 594–599.
Harris, R. A., Wang, T., Coarfa, C., Nagarajan, R. P., Hong, C., Downey, S. L., Johnson, B. E., Fouse, S. D., Delaney, A., Zhao, Y., Oishen, A., Ballinger, T., Zhou, X., Forsberg, K. J., Gu, J., Echipare, L., O’Geen, H., Lister, R., Pelizzola, M., Xi, Y., Epstein, C. B., Bernstein, B. E., Hawkins, R. D., Ren, B., Chung, W.-Y., Gu, H., Bock, C., Gnirke, A., Zhang, M. Q., Haussler, D., Ecker, J. R., Li, W., Farnham, P. J., Waterland, R. A., Meissner, A., Marra, M. A., Hirst, M., Milosavljevic, A., and Costello, J. F. (2010). Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotechnol. 28, 1097–1105.
Hatada, I., Hayashizaki, Y., Hirotsune, S., Komatsubara, H., and Mukai, T. (1991). A genomic scanning method for higher organisms using restriction sites as landmarks. Proc. Natl. Acad. Sci. U.S.A. 88, 9523–9527.
Hayashizaki, Y., Hirosune, S., Okazaki, Y., Hatada, I., Shibata, H., Kawai, J., Hirose, K., Watanabe, S., Fushiki, S., Wada, S., Sugimoto, T., Kobayakawa, K., Kawara, T., Katsuki, M., Shibuya, T., and Mukai, T. (1993). Restriction landmark genomic scanning method and its various applications. Electrophoresis 14, 251–258.
Hayatsu, H., Wataya, Y., Kai, K., and Iida, S. (1970). Reaction of sodium bisulfite with uracil, cytosine, and their derivatives. Biochemistry 9, 2858–2865.
Herman, J., Graff, J., Myöhänen, S., Nelkin, B., and Baylin, S. (1996). Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. U.S.A. 93, 9821–9826.
Hirst, M., and Mara, M. (2010). Next generation sequencing based approaches to epigenomics. Brief. Funct. Genomics 9, 455–465.
Holliday, R., and Pugh, J. (1975). DNA modification mechanisms and gene activity during development. Science 187, 226–232.
Huang, T., Perry, M., and Laux, D. (1999). Methylation profiling of CpG islands in human breast cancer cells. Hum. Mol. Genet. 8, 459–470.
Internation Human Epigenome Consortium (IHEC). (2010). Available at: http://www.ihec-epigenomes.org/ [accessed May 16, 2011].
Irizarry, R. A., Ladd-Acosta, C., Carvalho, B., Wu, H., Brandenburg, S. A., Jeddeloh, J. A., Wen, B., and Feinberg, A. P. (2008). Comprehensive high-throughput arrays for relative methylation (CHARM). Genome Res. 18, 780–790.
Ishkanian, A., Malloff, C. A., Watson, S. K., DeLeeuw, R. J., Chi, B., Coe, B. P., Snijders, A., Albertson, D. G., Pinkel, D., Marra, M. A., Ling, V., MacAulay, C., and Lam, W. L. (2004). A tiling resolution DNA microarray with complete coverage of the human genome. Nat. Genet. 36, 299–303.
Kaiser, J. (2008). DNA sequencing – a plan to capture human diversity in 1000 genomes. Science 319, 395.
Kawai, J., Hirotsune, S., Hirose, K., Fushiki, S., Watanabe, S., and Hayashizaki, Y. (1993). Methylation profiles of genomic DNA of mouse developmental brain detected by restriction landmark genomic scanning (RLGS) method. Nucleic Acids Res. 21, 5604–5608.
Keshet, I., Schlesinger, Y., Farkash, S., Rand, E., Hecht, M., Segal, E., Pikarski, E., Young, R. A., Niveleau, A., Cedar, H., and Simon, I. (2006). Evidence for an instructive mechanism of de novo methylation in cancer cells. Nat. Genet. 38, 149–153.
Khulan, B., Thompson, R., Ye, K., Fazzari, M., Suzuki, M., Stasiek, E., Figueroa, M., Glass, J., Chen, Q., Montagna, C., Hatchwell, E., Selzer, R. R., Richmond, T. A., Green, R. D., Maniatis Melnick, A., and Greally, J. M. (2006). Comparative isoschizomer profiling of cytosine methylation: the HELP assay. Genome Res. 16, 1046–1055.
Kinney, S., Chin, H., Vaisvilla, R., Bitinaite, J., Zheng, Y., Estève, P., Feng, S., Stroud, H., Jacobsen, S., and Pradhan, S. (2011). Tissue-specific distribution and dynamic changes of 5-hydroxymethylcytosine in mammalian genomes. J. Biol. Chem. 286, 24685–24683.
Kriaucionis, S., and Heintz, N. (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930.
Kuo, K., McCune, R., Gehrke, C., Midgett, R., and Ehrlich, M. (1980). Quantitative reversed-phase high performance liquid chromatographic determination of major and modified deoxyribonucleosides in DNA. Nucleic Acids Res. 8, 4763–4776.
Kuppuswamy, M., Hoffmann, J., Kasper, C., Spitzer, S., Groce, S., and Bajaj, S. P. (1991). Single nucleotide primer extension to detect genetic diseases: experimental application to hemophilia B (factor IX) and cystic fibrosis genes. Proc. Natl. Acad. Sci. U.S.A. 88, 1143–1147.
Laird, C., Pleasant, N., Clark, A., Sneeden, J., Hassan, K., Manley, N., Vary, J., Morgan, T., Hansen, R., and Stöger, R. (2004). Hairpin-bisulfite PCR: assessing epigenetic methylation patterns on complementary strands of individual DNA molecules. Proc. Natl. Acad. Sci. U.S.A. 101, 204–209.
Laird, P. (2010). Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 11, 191–203.
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., Funke, R., Gage, D., Harris, K., Heaford, A., Howland, J., Kann, L., Lehoczky, J., LeVine, R., McEwan, P., McKernan, K., Meldrim, J., Mesirov, J. P., Miranda, C., Morris, W., Naylor, J., Raymond, C., Rosetti, M., Santos, R., Sheridan, A., Sougnez, C., Stange-Thomann, N., Stojanovic, N., Subramanian, A., Wyman, D., Rogers, J., Sulston, J., Ainscough, R., Beck, S., Bentley, D., Burton, J., Clee, C., Carter, N., Coulson, A., Deadman, R., Deloukas, P., Dunham, A., Dunham, I., Durbin, R., French, L., Grafham, D., Gregory, S., Hubbard, T., Humphray, S., Hunt, A., Jones, M., Lloyd, C., McMurray, A., Matthews, L., Mercer, S., Milne, S., Mullikin, J. C., Mungall, A., Plumb, R., Ross, M., Shownkeen, R., Sims, S., Waterston, R. H., Wilson, R. K., Hillier, L. W., McPherson, J. D., Marra, M. A., Mardis, E. R., Fulton, L. A., Chinwalla, A. T., Pepin, K. H., Gish, W. R., Chissoe, S. L., Wendl, M. C., Delehaunty, K. D., Miner, T. L., Delehaunty, A., Kramer, J. B., Cook, L. L., Fulton, R. S., Johnson, D. L., Minx, P. J., Clifton, S. W., Hawkins, T., Branscomb, E., Predki, P., Richardson, P., Wenning, S., Slezak, T., Doggett, N., Cheng, J. F., Olsen, A., Lucas, S., Elkin, C., Uberbacher, E., Frazier, M., Gibbs, R. A., Muzny, D. M., Scherer, S. E., Bouck, J. B., Sodergren, E. J., Worley, K. C., Rives, C. M., Gorrell, J. H., Metzker, M. L., Naylor, S. L., Kucherlapati, R. S., Nelson, D. L., Weinstock, G. M., Sakaki, Y., Fujiyama, A., Hattori, M., Yada, T., Toyoda, A., Itoh, T., Kawagoe, C., Watanabe, H., Totoki, Y., Taylor, T., Weissenbach, J., Heilig, R., Saurin, W., Artiguenave, F., Brottier, P., Bruls, T., Pelletier, E., Robert, C., Wincker, P., Smith, D. R., Doucette-Stamm, L., Rubenfield, M., Weinstock, K., Lee, H. M., Dubois, J., Rosenthal, A., Platzer, M., Nyakatura, G., Taudien, S., Rump, A., Yang, H., Yu, J., Wang, J., Huang, G., Gu, J., Hood, L., Rowen, L., Madan, A., Qin, S., Davis, R. W., Federspiel, N. A., Abola, A. P., Proctor, M. J., Myers, R. M., Schmutz, J., Dickson, M., Grimwood, J., Cox, D. R., Olson, M. V., Kaul, R., Raymond, C., Shimizu, N., Kawasaki, K., Minoshima, S., Evans, G. A., Athanasiou, M., Schultz, R., Roe, B. A., Chen, F., Pan, H., Ramser, J., Lehrach, H., Reinhardt, R., McCombie, W. R., de la Bastide, M., Dedhia, N., Blöcker, H., Hornischer, K., Nordsiek, G., Agarwala, R., Aravind, L., Bailey, J. A., Bateman, A., Batzoglou, S., Birney, E., Bork, P., Brown, D. G., Burge, C. B., Cerutti, L., Chen, H. C., Church, D., Clamp, M., Copley, R. R., Doerks, T., Eddy, S. R., Eichler, E. E., Furey, T. S., Galagan, J., Gilbert, J. G., Harmon, C., Hayashizaki, Y., Haussler, D., Hermjakob, H., Hokamp, K., Jang, W., Johnson, L. S., Jones, T. A., Kasif, S., Kaspryzk, A., Kennedy, S., Kent, W. J., Kitts, P., Koonin, E. V., Korf, I., Kulp, D., Lancet, D., Lowe, T. M., McLysaght, A., Mikkelsen, T., Moran, J. V., Mulder, N., Pollara, V. J., Ponting, C. P., Schuler, G., Schultz, J., Slater, G., Smit, A. F., Stupka, E., Szustakowski, J., Thierry-Mieg, D., Thierry-Mieg, J., Wagner, L., Wallis, J., Wheeler, R., Williams, A., Wolf, Y. I., Wolfe, K. H., Yang, S. P., Yeh, R. F., Collins, F., Guyer, M. S., Peterson, J., Felsenfeld, A., Wetterstrand, K. A., Patrinos, A., Morgan, M. J., de Jong, P., Catanese, J. J., Osoegawa, K., Shizuya, H., Choi, S., Chen, Y. J., International Human Genome Sequencing Consortium. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921.
Lewin, J., Schmitt, A. O., Adorjan, P., Hildmann, T., and Piepenbrock, C. (2004). Quantitative DNA methylation analysis based on four-dye trace data from direct sequencing of PCR amplificates. Bioinformatics 20, 3005–3012.
Li, E., Beard, C., and Jaenisch, R. (1993). Role for DNA methylation in genomic imprinting. Nature 366, 362–365.
Li, J., Gao, F., Li, N., Li, S., Yin, G., Tian, G., Jia, S., Wang, K., Zhang, X., Yang, H., Nielson, A., and Bolund, L. (2009). An improved method for genome wide DNA methylation profiling correlated to transcription and genomic instability in two breast cancer cell lines. BMC Genomics 10, 223. doi:10.1186/1471-2164-10-223
Liang, G., Gonzalgo, M., Salem, C., and Jones, P. (2002). Identification of DNA methylation differences during tumorigenesis by methylation-sensitive arbitrarily primed polymerase chain reaction. Methods 27, 150–155.
Lister, R., Pelizzola, M., Dowen, R., Hawkins, R., Hon, G., Tonti-Filippini, J., Nery, J., Lee, L., Ye, Z., Ngo, Q.-M., Edsall, L., Antosiewicz-Bourget, J., Stewart, R., Ruotti, V., Millar, A., Thomson, J., Ren, B., and Ecker, J. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322.
Mardis, E. (2008). Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 9, 387–402.
Margulies, M., Egholm, M., Altman, W. E., Attiya, S., Bader, J. S., Bemben, L. A., Berka, J., Braverman, M. S., Chen, Y.-J., Chen, Z., Dewell, S. B., Du, L., Fierro, J. M., Gomes, X. V., Godwin, B. C., He, W., Helgesen, S., Ho, C. H., Irzyk, G. P., Jando, S. C., Alenquer, M. L. I., Jarvie, T. P., Jirage, K. B., Kim, J.-B., Knight, J. R., Lanza, J. R., Leamon, J. H., Lefkowitz, S. M., Lei, M., Li, J., Lohman, K. L., Lu, H., Makhijani, V. B., McDade, K. E., McKenna, M. P., Myers, E. W., Nickerson, E., Nobile, J. R., Plant, R., Puc, B. P., Ronan, M. T., Roth, G. T., Sarkis, G. J., Simons, J. F., Simpson, J. W., Srinivasan, M., Tartaro, K. R., Tomasz, A., Vogt, K. A., Volkmer, G. A., Wang, S. H., Wang, Y., Weiner, M. P., Yu, P., Richard, F., Begley, R. F., and Rothberg, J. M. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380.
Maunakea, A., Nagarajan, R., Bilenky, M., Ballinger, T., D’Souza, C., Fouse, S., Johnson, B., Hong, C., Nielsen, C., Zhao, Y., Turecki, G., Delaney, A., Varhol, R., Thiessen, N., Shchors, K., Heine, V., Rowitch, D., Xing, X., Fiore, C., Schillebeechx, M., Jones, S., Haussler, D., Marra, M., Hirst, M., Wang, T., and Costello, J. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257.
Meissner, A., Gnirke, A., Bell, G. W., Ramsahoye, B., Lander, E. S., and Jaenisch, R. (2005). Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33, 5868–5877.
Meselson, M., Yuan, R., and Heywood, J. (1972). Restriction and Modification of DNA. Annu. Rev. Biochem. 41, 447–466.
Monk, M., Boubelik, M., and Lehnert, S. (1987). Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development 99, 371–382.
Nouzova, M., Holtan, N., Oshiro, M., Isett, R. B., Munoz-Rodriguez, J. F., List, A. F., Narro, M. L., Miller, S. J., Merchant, N. C., and Futscher, B. W. (2004). Epigenomic changes during leukemia cell differentiation: analysis of histone acetylation and cytosine methylation using CpG island microarrays. J. Pharmacol. Exp. Ther. 311, 968–981.
Oakeley, E. (1999). DNA methylation analysis: a review of current methodologies. Pharmacol. Ther. 84, 389–400.
Oakeley, E., Podestà, A., and Jost, J. (1997). Developmental changes in DNA methylation of the two tobacco pollen nuclei during maturation. Proc. Natl. Acad. Sci. U.S.A. 94, 11721–11725.
Olek, A., Oswald, J., and Walter, J. (1996). A modified and improved method for bisulphite based cytosine methylation analysis. Nucleic Acids Res. 24, 5064–5066.
Ordway, J., Bedell, J., Citek, R., Nunberg, A., Garrido, A., Kendall, R., Stevens, J., Cao, D., Doerge, R., Korshunova, Y., Holemon, H., McPherson, J., Lakey, N., Leon, J., Martienssen, R., and Jeddeloh, J. (2006). Comprehensive DNA methylation profiling in a human cancer genome identifies novel epigenetic targets. Carcinogenesis 27, 2409–2423.
Pastor, W., Pape, U., Huang, Y., Henderson, H., Lister, R., Ko, M., McLoughlin, E., Brudno, Y., Mahapatra, S., Kapranov, P., Tahiliani, M., Daley, G., Liu, X., Ecker, J., Milos, P., Agarwal, S., and Rao, A. (2011). Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 473, 394–397.
Patel, C., and Gopinathan, K. (1987). Determination of trace amounts of 5-methylcytosine in DNA by reverse-phase high-performance liquid chromatography. Anal. Biochem. 164, 164–169.
Pfeifer, G., Steigerwald, S., Mueller, P., Wold, B., and Riggs, A. (1989). Genomic sequencing and methylation analysis by ligation mediated PCR. Science 246, 810–881.
Poduslo, S., Dean, M., Kolch, U., and O’Brien, S. (1991). Detecting high-resolution polymorphisms in human coding loci by combining PCR and single-strand conformation polymorphism (SSCP) analysis. Am. J. Hum. Genet. 49, 106–111.
Prober, J., Trainor, G., Dam, R., Hobbs, R., Robertson, C., Zagursky, R., Cocuzza, J. A., and Baumeister, K. (1987). A system for rapid DNA sequencing with fluorescent chain-terminating dideoxynucleotides. Science 238, 336–341.
Qiu, P., Soder, G., Sanfiorenzo, V., Wang, L., Greene, J., Fritz, M., and Cai, X. (2003). Quantification of single nucleotide polymorphisms by automated DNA sequencing. Biochem. Biophys. Res. Commun. 309, 331–338.
Rakyan, V., Hildmann, T., Novik, K. L., Lewin, J., Tost, J., Cox, A. V., Andrews, T. D., Howe, K. L., Otto, T., Olek, A., Fischer, J., Gut, I. G., Berlin, K., and Beck, S. (2004). DNA methylation profiling of the human major histocompatibility complex: a pilot study for the human epigenome project. PLoS Biol. 2, e405. doi:10.1371/journal.pbio.0020405
Rauch, T., Hongwei, L., Xiwei, W., and Pfeifer, G. (2006). MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 66, 7939–7947.
Rauch, T., and Pfeifer, G. (2005). Methylated-CpG island recovery assay: a new technique for the rapid detection of methylated-CpG islands in cancer. Lab. Invest. 85, 1172–1180.
Rauch, T., Wu, X., Zhong, X., Riggs, A., and Pfeifer, G. (2009). A human B cell methylome at 100-base pair resolution. Proc. Natl. Acad. Sci. U.S.A. 106, 671–678.
Razin, A., and Ceder, H. (1991). DNA methylation and gene expression. Microbiol. Mol. Biol. Rev. 55, 451–458.
Reinders, J., Vivier, D., Theiler, G., Chollet, D., Descombes, P., and Paszkowski, J. (2008). Genome-wide, high-resolution DNA methylation profiling using bisulfite-mediated cytosine conversion. Genome Res. 18, 469–476.
Riggs, A. (1975). X inactivation, differentiation, and DNA methylation. Cytogenet. Cell Genet. 14, 9–25.
Sadri, R., and Hornsby, P. (1996). Rapid analysis of DNA methylation using new restriction enzyme sites created by bisulfite modification. Nucleic Acids Res. 24, 5058–5059.
Sandoval, J., Heyn, H. A., Moran, S., Serra-Musach, J., Pujana, M. A., Bibikova, M., and Esteller, M. (2011). Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 6, 692–702.
Santos, F., Hendrich, B., Reik, W., and Dean, W. (2002). Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev. Biol. 241, 172–182.
Schilling, E., and Rehli, M. (2007). Global, comparative analysis of tissue-specific promoter CpG methylation. Genomics 90, 314–323.
Serre, D., Lee, B., and Ting, A. (2009). MBD-isolated genome sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 38, 391–399.
Singer-Sam, J., Grant, M., LeBon, J., Okuyama, K., Chapman, V., Monk, M., and Riggs, A. (1990). Use of HpaII-polymerase chain reaction assay to study DNA methylation in the Pgk-1 CpG island of mouse embryos at the time of X-chromosome inactivation. Mol. Cell Biol. 10, 4987–4498.
Smith, J., Arber, W., and Kühnlein, U. (1973). Host specificity of DNA produced by Escherichia coli: XIV. The role of nucleotide methylation in in vivo B-specific modification. J. Mol. Biol. 63, 1–8.
Southern, E. (1975). Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 98, 503–517.
Southern, E., Mir, K., and Schchepinov, M. (1999). Molecular interactions on microarrays. Nat. Genet. 21, 5–9.
Suzuki, H., Itoh, F., Toyota, M., Kikuchi, T., Kakiuchi, H., Hinoda, Y., and Imai, K. (2000). Quantitative DNA methylation analysis by fluorescent polymerase chain reaction single strand conformation polymorphism using an automated DNA sequencer. Electrophoresis 21, 904–908.
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., Agarwal, S., Iyer, L. M., Liu, D. R., Aravind, L., and Rao, A. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935.
Taylor, K., Kramer, R., Davis, J., Guo, J., Duff, D., Xu, D., Caldwell, C., and Shi, H. (2007). Ultradeep bisulfite sequencing analysis of DNA methylation patterns in multiple gene promoters by 454 sequencing. Cancer Res. 67, 8511–8518.
Thu, K., Pikor, L., Kennet, J., Alvarez, C., and Lam, W. (2010). Methylation analysis by DNA immunoprecipitation. J. Cell. Physiol. 222, 522–531.
Tost, J., and Gut, I. G. (2007). DNA methylation analysis by pyrosequencing. Nat. Protoc. 2, 2265–2275.
Vischer, E., and Chargaff, E. (1948). The composition of the pentose nucleic acids of yeast and pancreas. J. Biol. Chem. 176, 715–734.
Wagner, I., and Capesius, I. (1981). Determination of 5-methylcytosine from plant DNA by high-performance liquid chromatography. Biochim. Biophys. Acta 654, 52–56.
Wang, Y., Gehrke, W., and Ehrlich, M. (1980). Comparison of bisulfite modification of 5-methyldeoxycytidine and deoxycytidine residues. Nucleic Acids Res. 8, 4777–4790.
Weber, M., Davies, J., Wittig, D., Oakeley, E., Haase, M., Lam, W., and Schübeler, D. (2005). Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 37, 853–862.
Welsh, J., and McClelland, M. (1990). Fingerprinting genomes using PCR with arbitrary primers. Nucleic Acids Res. 18, 7213–7218
Wilton, S., Lim, L., Dye, D., and Laing, N. (1997). Bandstab: a PCR based alternative to cloning PCR products. Biotechniques 22, 642–645.
Wittwer, C., Reed, G., Gundry, C., Vandersteen, J., and Pryor, R. (2003). High-resolution genotyping by amplicon melting analysis using LCGreen. Clin. Chem. 49, 853–860.
Wojdacz, T. K., and Dobrovic, A. (2007). Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res. 35, e41.
Wojdacz, T. K., Dobrovic, A., and Hansen, L. L. (2008). Methylation-sensitive high-resolution melting. Nat. Protoc. 3, 1903–1908.
Wu, J., Issa, J., Herman, J., Bassett, D., Nelkin, B., and Baylin, S. (1993). Expression of an exogenous eukaryotic DNA methyltransferase gene induces transformation of NIH 3T3 cells. Proc. Natl. Acad. Sci. U.S.A. 90, 8891–8895.
Wyatt, G. (1950). The purine and pyrimidine composition of deoxypentose nucleic acids. Biochem. J. 48, 584–590.
Xiong, Z., and Liard, P. (1997). COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acid Res. 25, 2532–2534.
Yan, P., Chen, C., and Shi, H. (2001). Dissecting complex epigenetic alterations in breast cancer using CpG island microarrays. Cancer Res. 61, 8375–8380.
Yan, P., Perry, M., Laux, D., Asare, A., Caldwell, C., and Huang, T. H.-M. (2000). CpG island arrays: an application toward deciphering epigenetic signatures of breast cancer. Clin. Cancer Res. 6, 1432–1438.
Keywords: DNA, methylation, bisulfite, sequencing, methods
Citation: Harrison A and Parle-McDermott A (2011) DNA methylation: a timeline of methods and applications. Front. Gene. 2:74. doi: 10.3389/fgene.2011.00074
Received: 07 July 2011; Accepted: 04 October 2011;
Published online: 25 October 2011.
Edited by:
Jill Ann McKay, Newcastle University, UKReviewed by:
Jafar Sharif, RIKEN, JapanRavi Goyal, Loma Linda University, USA
Lisa Jane Coneyworth, Nottingham University, UK
Copyright: © 2011 Harrison and Parle-McDermott. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Anne Parle-McDermott, Nutritional Genomics Group, School of Biotechnology, Dublin City University, Glasnevin, Dublin 9, Ireland. e-mail: anne.parle-mcdermott@dcu.ie