 D. Channe Gowda
D. Channe Gowda Xianzhu Wu
Xianzhu Wu- Department of Biochemistry and Molecular Biology, The Pennsylvania State University College of Medicine, Hershey, PA, United States
Malaria caused by the Plasmodium family of parasites, especially P.falciparum and P. vivax, is a major health problem in many countries in the tropical and subtropical regions of the world. The disease presents a wide array of systemic clinical conditions and several life-threatening organ pathologies, including the dreaded cerebral malaria. Like many other infectious diseases, malaria is an inflammatory response-driven disease, and positive outcomes to infection depend on finely tuned regulation of immune responses that efficiently clear parasites and allow protective immunity to develop. Immune responses initiated by the innate immune system in response to parasites play key roles both in protective immunity development and pathogenesis. Initial pro-inflammatory responses are essential for clearing infection by promoting appropriate cell-mediated and humoral immunity. However, elevated and prolonged pro-inflammatory responses owing to inappropriate cellular programming contribute to disease conditions. A comprehensive knowledge of the molecular and cellular mechanisms that initiate immune responses and how these responses contribute to protective immunity development or pathogenesis is important for developing effective therapeutics and/or a vaccine. Historically, in efforts to develop a vaccine, immunity to malaria was extensively studied in the context of identifying protective humoral responses, targeting proteins involved in parasite invasion or clearance. The innate immune response was thought to be non-specific. However, during the past two decades, there has been a significant progress in understanding the molecular and cellular mechanisms of host-parasite interactions and the associated signaling in immune responses to malaria. Malaria infection occurs at two stages, initially in the liver through the bite of a mosquito, carrying sporozoites, and subsequently, in the blood through the invasion of red blood cells by merozoites released from the infected hepatocytes. Soon after infection, both the liver and blood stage parasites are sensed by various receptors of the host innate immune system resulting in the activation of signaling pathways and production of cytokines and chemokines. These immune responses play crucial roles in clearing parasites and regulating adaptive immunity. Here, we summarize the knowledge on molecular mechanisms that underlie the innate immune responses to malaria infection.
Background
Malaria is a widespread infectious disease that is prevalent in most tropical regions of the world (1–4). About half of the world population is at risk of contracting malaria. World Health Organization has reported an estimated 216 million malaria clinical cases and about 445,000 deaths during 2016 (1). Besides huge health burden and mortality, malaria morbidity is a substantial hindrance to socio-economic development in endemic areas due to the loss of man power (5, 6). The disease is caused by protozoan parasites of the genus Plasmodium. Five parasite species infect humans that include Plasmodium falciparum, P. vivax, P. malariae, P. ovale, and P. knowlesi (7–9). However, most malaria infections are caused by P. falciparum and P. vivax, and infections by the other three species are relatively rare. Several parasite species, including P. berghei, P. yoelii, P. chabaudi, and P. vinckei, infect rodents, but not humans (10). Different strains of rodent parasites used in laboratories show distinct growth rates, and infected mice exhibit a range of immunological and pathological conditions, resembling a wide spectrum of pathophysiologic conditions of human malaria infection. As such, mice infected with different parasite strains are useful models to study distinct systemic and organ-specific clinical conditions of human malaria.
Malaria is a highly complex disease that displays a wide variety of pathological conditions. At the early stages, malaria infection presents a number of systemic clinical conditions, including the characteristic periodic fever, chills, headache, dizziness, malaise, abdominal discomfort, nausea, and muscle and joint aches (11, 12). As the infection progresses and parasite biomass increases, pathogenic processes follow, resulting in severe anemia, blood acidosis, splenomegaly and hepatomegaly, acute respiratory distress syndrome, and several other clinical conditions. In the case of P. falciparum, the infected red blood cells (IRBCs) bind to certain cell surface proteins of vascular endothelia, including CD36, intracellular adhesion molecule 1 (ICAM-1), vascular adhesion molecule 1 (VCAM-1), and endothelial protein receptor (EPCR) (4, 13–20). These binding events allow parasites to sequester in organs, such as brain, lungs, liver, intestine, dermal tissues, and placenta, thereby avoiding splenic clearance. Parasite sequestration contributes to single and multiorgan fatal pathologic conditions, including cerebral malaria, and renal, liver and lung dysfunction and failure. In pregnant women, P. falciparum sequesters in the placenta through the binding of IRBCs to chondroitin 4-sulfate in the intervillous space and on the syncytiotrophoblast cell surface (21–23). This process contributes to pregnancy-associated malaria, characterized by a number of clinical conditions, including low birth weight, abortion, and death in the baby and the mother (24). The binding of IRBCs to the endothelial cell surface proteins in the microvasculature of vital organs and chondroitin 4-sulfate in the placenta is mediated by a family of antigenically variant parasite proteins encoded by about 60 var genes (15, 16, 25). These proteins are collectively called P. falciparum erythrocyte membrane protein 1 (PfEMP1). PfEMP1 confers virulence to P. falciparum through IRBC binding to endothelial cells in various organs contributing to microvascular plugging and hypoxia. In addition, IRBC-binding and parasite accumulation amplify inflammatory responses locally, leading to immune cell infiltration, endothelial damage, and organ dysfunction and failure. P. vivax lacks PfEMP1 ortholog (26) and thus, P. vivax is less virulent compared to P. falciparum. However, in recent years, P. vivax is becoming increasingly virulent, causing severe malaria in significant numbers (11, 27–30). Although, the underlying reasons in P. vivax causing severe illnesses remain unclear, drug resistance and changes in genetic makeup appear to be significant factors (31, 32).
Innate immune responses that are initiated in response to malaria infection play key roles both in the development of protective immunity and pathogenesis (14, 33–38). Early pro-inflammatory responses regulate antiparasitic Th1 development and promote effector cell function for efficiently clearing infections. Usually, as infection progresses, pro-inflammatory responses are gradually downregulated with parallel increase in anti-inflammatory responses (39). Generally, this leads to Th2 development, resulting in balanced pro-/anti-inflammatory and Th1/Th2 responses and resistance against pathogenesis (35, 39–42). However, this is not always the case. Depending upon the host-parasite interaction dynamics, factors such as parasite sequestration and alterations in host genetics, pro-inflammatory responses may be overly upregulated, resulting in systemic and organ-related severe illnesses.
During the past two decades, there has been a substantial progress in our understanding of the host receptors that sense parasite factors involved in inducing immune responses and the associated signaling pathways (43–47). A substantial progress has also been made in our understanding of parasite immunostimulatory factors that are the targets of host receptors (43–47). However, much remained to be learned on the spectrum of molecular and cellular processes and immune responses that are initiated upon parasite sensing. Development of protective immunity to malaria requires repetitive infection over a period of time (48, 49). Information is limited as to whether and how the innate immune responses contribute to the inefficient development of protective immunity to malaria. In addition to signaling initiated by specific sensing of parasite factors, signals induced by processes such as phagocytosis and cytoadherence also contribute; information is also limited as to whether and to what extent these signaling contribute to the overall innate immune responses. In this review, we are providing an overview of the available information on the molecular and cellular mechanisms of parasite-host interactions that involved in innate immune responses to malaria.
Malaria Infection
Malaria infection begins with the entry of sporozoite form of parasites when infected mosquitoes inject saliva during blood meal (7, 46, 50–52). A substantial number of injected sporozoites is unable to enter the blood stream and stuck in the dermis, and these parasites are removed likely by the resident macrophages (Mφs). Those sporozoites that enter the blood stream target liver, where they exclusively infect hepatocytes. In hepatocytes, parasites reside inside parasitophorous vacuole formed during invasion and develop into merozoites over a period of 7–10 days in humans and 2 days in rodents, vastly expanding hepatocyte size. Upon parasite maturation, each infected hepatocyte releases 10,000–30,000 merozoites into the blood stream (46, 50–52). This period of cell cycles represents the first stage of malaria infection, referred to as the liver stage or the tissue stage infection. The merozoites released from the matured infected hepatocytes are called exo-erythrocytic merozoites. These merozoites do not infect hepatocytes, but instead exclusively invade red blood cells, and reside inside parasitophorous vacuole. Soon after invasion, parasites appear morphologically like rings inside IRBCs. Parasites then develop into early and late trophozoites, and finally undergo schizogony to form differentiated merozoites, which are released into the blood stream. Each matured erythrocytic stage schizont releases 8–24 merozoites (53) (Table 1), which can invade red blood cells. The parasite developmental process inside red blood cells occurs over a period of 24–72 h, depending on the Plasmodium species (Table 1). This cycle of invading red blood cells and parasite growth is called the blood stage infection. The repetitive erythrocytic cell cycles result in the exponential growth of parasites and if the growth is unchecked, most red blood cells are consumed, resulting in severe anemia and pathologies, and death. However, soon after infection, the innate immune system detects parasites at both the liver and blood stages, and responds by inducing pro-inflammatory cytokines and chemokines. The cytokines prime phagocytes for an efficient uptake and clearance, while chemokines help recruit effector cells to sites, where parasites are sequestered or accumulated, for effective infection clearance.
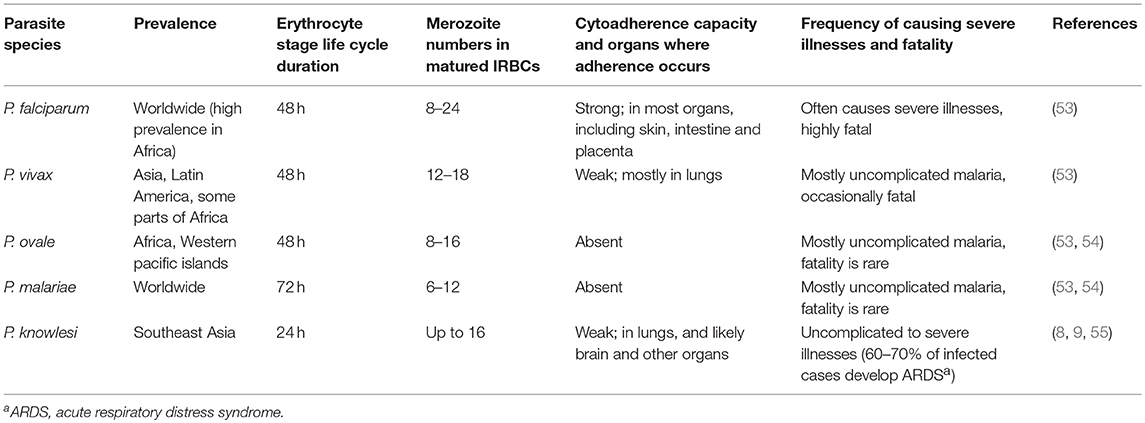
Table 1. Prevalence and features of human blood stage malaria parasites.
Pathogen Sensing Mechanisms
Hosts respond to various infections by sensing certain evolutionarily conserved molecules (signature structures) of pathogens called pathogen-associated molecular patterns (PAMPs); these include bacterial LPS and peptidoglycan, fungal glucans, and microbial DNA and RNA, and even self-DNA under certain pathologic conditions (56–61). Host detects PAMPs through receptors called pathogen-recognition receptors (PRRs). Host also detects certain endogenous factors released during infection and thus can induce danger signaling. These factors are called danger-associated molecular patterns (DAMPs). Examples of DAMPs include high mobility box 1 (HMGB1), HSP70, SP100 family of proteins, and degraded hyaluronic acid (62–65). The innate immune system is equipped with a wide range of PAMP- and DAMP-recognizing PRRs. After recognition of PAMPs and DAMPs by PRRs, the innate immune cells are activated through the initiation of specific signaling pathways, producing cytokines and chemokines. PRRs are present at various cellular locations, including outer surface of plasma membrane, luminal surface of endosomal membrane, outer membrane of mitochondria, and cytosol (59, 60, 66). Prominent among transmembrane PRRs are toll-like receptors (TLRs) (56–59, 61); others transmembrane PRRs include, c-type lectin receptors, such as mannose- and galactose-binding proteins, and scavenger receptors, such as CD36, CD204, and MARCO (67). Examples of cytosolic sensors include dectin-1 that binds fungal β-1,3-glucan, cyclic GMP-AMP synthase (cGAS, senses dsDNA), retinoic acid-inducible gene-I (RIG-I, respond to viral RNA), RIG-I-like receptors (RLRs), such as melanoma differentiation-associated gene 5 (MDA5) that respond to dsRNA, nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs, senses bacterial peptidoglycan), and absent in melanoma 2 (AIM2, respond to dsDNA). Malaria parasites are sensed by several receptors, leading to cell activation and immune responses (see below). Several excellent reviewers that comprehensively discuss various host-pathogen interactions and signaling mechanisms are available (56–60, 62–66, 68–70).
Malarial PAMPs and Host PRRs
A notable feature of malaria infection is that the liver stage is clinically completely silent, that is, the host does not exhibit any symptoms of malaria (71–73). All the malaria clinical conditions and fatal illnesses are manifested during the blood stage infection (74). At both stages of infection, the host detects parasites immediately after infection and initiates innate immune responses. These responses are geared toward clearing the infection and shaping the development of protective adaptive immunity (14, 34, 44, 71–78). However, the complex parasite-host interaction dynamics are not always in favor of achieving this goal, but instead often result in dysregulated immune responses and uncontrolled parasite growth, leading to pathogenesis. Understanding the malaria parasite-host interaction dynamics that shape the parasite-specific immunity and the molecular and cellular processes that contribute to pathogenesis is important for developing suitable treatment strategies. Below, we summarize the advancements that have been made in identifying the malaria PAMPs, the cognate host PRRs, and the signaling events that lead to innate immune responses.
Liver Stage Parasite Sensing
Since the liver stage malaria infection is clinically silent, it has long been thought that parasites inside hepatocytes grow undetected by the innate immune system. However, recent studies in P. berghei- and P. yoelii-infected mice show that, although parasites inside hepatocytes are shielded from recognition by Mφs and dendritic cells (DCs), the growing parasites are recognized by the cytosolic PRRs of hepatocytes, initiating antiparasitic type I IFN response (79, 80). The parasites in infected hepatocytes are detected through the interaction of parasite RNA with RIG-I family of proteins homolog called melanoma differentiation-associated protein 5 (MDA5), but not by RIG-I, leading to the activation of MDA5-MAVS-IRF3/IRF7 signaling axis and downstream production of type I IFN (79, 80) (Figure 1). This signaling event results in an array of type I IFN receptor (IFNαR)-mediated innate immune responses, which include: (i) expression of interferon-stimulated genes by hepatocytes; (ii) production of chemokines by hepatocytes, and chemotaxis-mediated recruitment of Mφs, neutrophils and lymphocytes to the proximity of infected hepatocytes; (iii) production of IFN-γ and chemokines by NK and NKT cells, which are abundantly present in the liver; (iv) infiltration of NK and NKT cells to the liver; (v) CD1d-resitrcted elimination of infected hepatocytes by NKT cells (79, 80). Since parasites reside inside parasitophorous vacuole, it appears that parasite RNA is exported to the cytosol, but not to phagolysosomes. It is unlikely that cytosolic RNA can enter endosomes and moreover hepatocytes may not express significant levels of TLR7. Thus, it appears that cytosolic sensors are the only PRRs that interact with parasite factors in infected hepatocytes. These findings represent a significant advancement in our understanding of parasite recognition mechanisms involved in innate immune responses to the liver stage malaria infection. However, it remains unclear if parasite DNA is exported to the cytosol, where it can be sensed by cytosolic nucleic acid sensors. DNA is a prominent immunostimulatory PAMP of the blood stage malaria parasites and the AT-rich stem loops of parasite DNA can induce the production of type I IFN through sensing in the cytosol (44, 60). If the liver stage parasite DNA has no role in type I IFN response, then DNA is either not exported to the cytosol of hepatocytes or non-stimulatory.
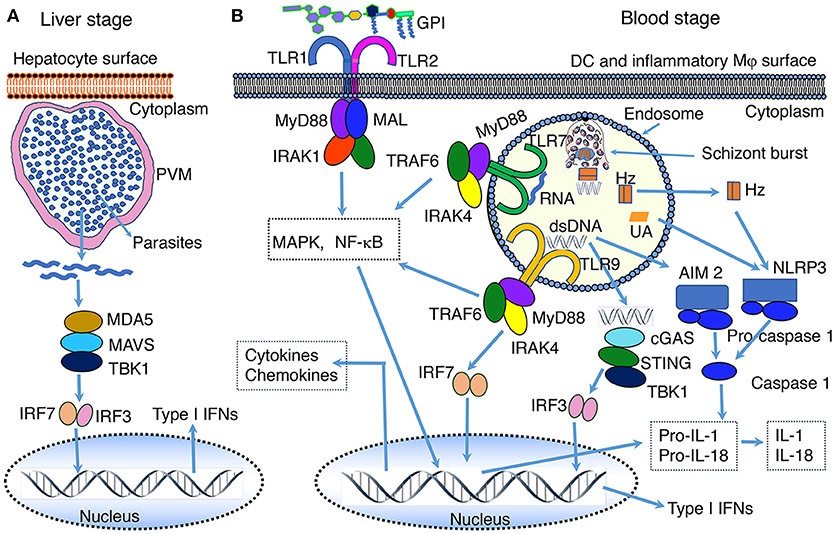
Figure 1. PAMP-PRR interaction-induced signaling pathways. (A) RNA of the liver stage parasites growing inside hepatocytes is recognized by MDA5 leading to the activation of MAVS-TBK1-IRF3/IRF7 signaling and downstream production of type I IFNs. (B) At the blood stage infection, parasite DNA, RNA and GPI interact with, respectively, TLR9, TLR7, and TLR2, leading to the activation of primarily MAPK and NF-κB signaling pathways and downstream cytokine and chemokine responses. In the cytosol, similar to the liver stage parasite RNA, the blood stage parasite RNA is sensed by MDA5 leading to the activation of MAVS-TBK1-IRF3/IRF7 signaling (see A). However, this signaling seems induce the expression of SOCS1, which downregulate RNA-TLR7-induced type I IFN production (81). Parasite DNA in the cytosol is sensed by cGAS, resulting in the activation of STING-TBK1-IRF3 signaling and type I IFN response. Parasite DNA also activates AIM2 inflammasome, which cleaves pro-caspase 1 to activate caspase 1. Hemozoin (Hz) and uric acid (UA) induce danger signaling, activating NLRP3 inflammasome and the cleavage of pro-caspase 1 to activate caspase 1. Parasites have also been reported to activate NLRP12 inflammasome through unidentified interaction, leading to the cleavage of pro-caspase 1 to activate caspase 1 (44, 82). It appears that microparticles released from IRBCs and heme produced during infection activate TLR4 signaling (83, 84). Ligands bind to TLR4 homodimer through the cooperation of accessory proteins CD14 and MD2, leading to MAPK, NF-κB and TRIF-TBK1-IRF3 signaling. Note: the diagram depicts a simplified version of indicated signaling pathways and additional details can be found in review articles (44, 56–70). The abbreviations are defined in footnote in page 1.
Blood Stage Parasite Sensing
As mentioned earlier, the blood stage infection accounts for all the pathological conditions of malaria (74). In infected non-immune people, P. falciparum parasites multiply rapidly through the release of large numbers of merozoites by matured schizonts to the blood circulation every 48 h (53, 85). Some of the released merozoites invade erythrocytes and the reminders become dead and are likely targeted by Mφs and DCs. In addition, the burst of schizont stage erythrocytes releases large amounts of parasite's digestive vacuoles containing hemozoin and other waste products. The blood stage parasites grow synchronously by adapting to daily rhythm of systemic TNF-α production and the level of glucose in the blood (86). Thus, parasites develop at similar rates, releasing merozoites, digestive vacuoles and other contents more or less at the same time point (86). This leads to peak concentrations of parasite stimulatory components, to which the innate immune system potently responds and induces the production of pro-inflammatory cytokines and chemokines at high levels. This periodic elicitation of strong inflammatory responses within a narrow window of time period after each erythrocytic cell cycle is responsible for the characteristic periodicity of malaria paroxysms, including periodic recurrent of peak levels of fever, chills, headache, shock, and malaise. Several malaria parasite PAMPs have been identified, and the mechanisms by which they are sensed and the associated signaling pathways and immune responses have been delineated (44, 46, 73, 87). This body of information represents a substantial progress in our understanding of malaria innate immunity. Below we describe the current knowledge on malaria parasite PAMP and host PRR interaction mechanisms involved in innate immune responses.
Malarial PAMPs
GPI
P. falciparum glycosylphosphatidylinositol (GPI) is the first factor that was identified as a malaria parasite PAMP (88). Structurally, parasite GPI comprises a heterogenous group of molecules consisting of triacylated phosphatidylinositol linked to the glucosamine moiety of glycan having four mannose residues and a glucosamine residue (89–91). The structural heterogeneity of malaria GPI is due to the variation in length and level of unsaturation in acyl residues present at different positions of the phosphatidylinositol moiety. This structural heterogeneity in the lipid moieties has no bearing on immune response-inducing activity of GPI. This is evident from the fact that the sn-2 lyso GPI, obtained by the removal of acyl moiety at the sn-2 position of parasite GPI, could efficiently induce cytokine responses similar to the unmodified parasite GPI. The biosynthesis of GPI is essential for the survival of parasites as GPI anchors several proteins of merozoites to the plasma membrane that are involved in erythrocyte invasion (92–94). In the absence of GPI anchoring, these proteins are not expressed on the surface and hence merozoites cannot invade erythrocytes. Malaria parasites synthesize GPI in several folds excess over the actual amounts needed for anchoring proteins to the surface of merozoites and thus, significant amounts of GPI remain not linked to proteins (90). The GPI molecules that are not linked to proteins are exposed on the cell surface and thus are likely targeted by the innate immune system.
The identification that parasite GPI is a malaria inflammatory response-inducing pathogenicity factor was based on the observation that the purified parasite GPI could induce strong pro-inflammatory responses by Mφs (88). When administered to mice, GPI induced symptoms that resembled the systemic clinical conditions of malaria, including pyrexia, cachexia, hypoglycemia, and TNF-α-induced sepsis. In several subsequent studies, GPI was shown to induce a wide range of immune responses, including the production of TNF-α and IL-1 by Mφs, expression of nitric oxide synthase by Mφs and endothelial cells, and the expression of ICAM-1, VCAM-1, and E-selectin by leukocytes and endothelial cells through the activation of several signaling events (95–97). Consistent with the property of GPI in inducing malaria-like symptoms, immunization of mice with a synthetic glycan portion of GPI produced anti-GPI antibodies, and immunized mice infected with P. berghei ANKA, an experimental cerebral malaria model, were protected from cerebral malaria (98). Further, the presence of anti-GPI antibodies in people in malaria endemic was associated with a significant protective immunity against malaria illnesses (99, 100).
Subsequent studies have shown that P. falciparum GPI activates Mφs through the induction of an outside-in signaling, without binding to plasma membrane or being internalized by the cells, but instead by recognition through weak interactions on the cell surface (101). The intact structure of GPI is essential for bioactivity as neither the carbohydrate moiety nor the triacylated phosphatidylinositol lipid portion can induce immune responses. Further studies have shown that the parasite GPI induced production of pro-inflammatory cytokines by Mφs occurs through recognition mainly by TLR2-TLR1 heterodimer and to a much lesser extent by TLR4 (102–105); Figure 1 and Table 2. Sensing of GPI by TLR2-TLR1 leads to the activation of ERK, p38, JNK MAPK, and NF-κB signaling pathways, which differentially contribute to the production of various inflammatory mediators (103). Thus, malaria parasite GPI is mainly a TLR2-activating PAMP.
Table 2. Innate sensing of malaria parasites and signaling mechanisms.
DNA
A large body of accumulated data over the past two decades demonstrates that microbial (bacteria, viruses, and parasites) DNA, and self-DNA in some pathological situations, function as PAMP and are recognized by TLR9 in endosomes and by DNA sensors in the cytosol (56–60). In the case of malaria, the first demonstration that TLR9 senses malaria parasites and induces immune responses was by Pinchyangukul et al. (123). They showed that a soluble component in the schizont extract of P. falciparum that was heat labile and precipitable with ammonium sulfate (the description agrees with the active component being protein-DNA complex) induces cytokine and chemokine responses by human plasmacytoid DCs (pDCs) and mouse DCs through the activation of TLR9-MyD88 signaling pathway. Subsequently, it was shown that the TLR9 signaling-inducing malaria factor is DNA (107, 108, 124). TLR9 specifically recognizes the unmethylated CpG motifs of DNA (68, 69). The genomic DNA of P. falciparum and P. vivax contain, respectively, ~300 and ~2,000 CpG motifs (109). The higher content of CpG motifs is likely responsible for the strong fever-inducing ability of P. vivax compared to P. falciparum (125).
Malaria parasite DNA enters the endosomes of the innate immune cells, such as Mφs and DCs, through phagocytic uptake of IRBCs, merozoites, the nuclear material of parasites, DNA-protein-hemozoin complex, and DNA-containing immune complexes (107, 108, 124, 126). The endosomes are then fused to lysosomes to form phagolysosomes. In the acidic environment of phagolysosomes, DNA is released and recognized by TLR9, leading to the activation of MAPK and NF-κB signaling pathways and cytokine and chemokine responses (Figure 1 and Table 2). In addition to TLR9, parasite DNA is recognized by cytosolic DNA sensors upon the release of phagolysosomal contents into the cytosol. In the cytosol, several distinct cytosolic sensors can potentially recognize DNA (Figure 1). Thus far, it has been demonstrated that two cytosolic PRRs sense parasite DNA: (i) cGAS sensing dsDNA and inducing the activation of STING-TBK1-IRF3 signaling and downstream production of type I IFNs (44, 110), and (ii) AIM2 recognizing dsDNA, resulting in the activation of inflammasome and caspase 1. The activated caspase 1 converts pro forms of IL-1 and IL-18 into active IL-1 and IL-18. Robust production of IL-1 and IL-18 in response to malaria infection requires, in addition to inflammasome signaling, TLR (mainly TLR9 and TLR7) mediated production of pro-IL-1 and pro-IL-18. P. falciparum genomic DNA contains >80% AT nucleotides. The AT-rich motifs of parasite DNA form loop structures, which have been shown to induce type I IFN response through STING-TBK1-IRF3 signaling (109). In some viruses, the AT-rich loop motifs of DNA are transcribed by RNA polymerase III to form double-stranded RNA containing 5′-triphosphate that induces type I IFNs through RIG-I-MAVS-IRF3 signaling (110). However, in the case of P. falciparum DNA, pol III-dependent recognition of AT-rich motifs of DNA seems to be not involved in cytosolic sensing as deficiency in pol III had no effect on immune responses to the parasite DNA (109).
RNA
The innate immune system senses RNA of both the liver and the blood stage parasites. In the liver stage, as described in the section Liver Stage Parasite Sensing above, RNA is recognized exclusively in the cytosol by MDA5 (Figure 1) (79, 80). In contrast, in the blood stage infection, mouse parasite RNA is recognized by TLR7 in phagolysosomes of DCs, leading to type I IFN production (Figure 1). In fact, studies in a mouse model of P. chabaudi-infection showed that type I IFN production is the earliest cytokine response (24 h postinfection) during the blood stage malaria infection (127). Parasite RNA enters endosomes of Mφs and DCs upon the uptake of parasites/parasite components, inducing type I IFN response through TLR7 signaling (81, 106, 128). Type I IFNs thus produced prominently contribute to the upregulation of pro-inflammatory cytokines, such as IFN-γ and IL-12, during the early stages of infection (106). RNA of the blood stage mouse parasites is also sensed by cytosolic MDA5, leading to type I IFN production (81).
While it is clear that RNA induced TLR7 signaling plays an important role in early type I IFN production in mouse malaria, it remains unclear whether or to what extent RNA of human parasites is able to induce type I IFN response. This is because, although RNA of human malaria parasite P. falciparum has been reported to induce cytokine responses through TLR7 recognition, the reported activity appears to be very low (106). Further studies are needed to determine whether or to what extent P. falciparum RNA is immunostimulatory.
Malarial Danger-associated Molecular Patterns (DAMPs)
Hemozoin
Hemozoin is a hydrophobic, crystalline insoluble polymer of heme formed during the digestion of hemoglobin by parasites in the digestive vacuoles to use the released amino acid as a food source (45, 129–131). In parasites, hemozoin is associated with lipids and proteins, and is released into the blood circulation when merozoites are egressed from matured schizonts. Hemozoin by itself is an inert material and appears to have no specific receptor for recognition, but it influences the innate immune responses to malaria parasites in several ways; reviewed in Olivier et al. (45). Studies have reported that hemozoin is a carrier of malaria parasite DNA into endosomes for TLR9 recognition (107). Although how and where parasite DNA associates with hemozoin have not been specifically studied in vivo, there exist several possibilities. Malaria merozoites egressed from the matured schizonts have a half-life of <5 min (132), and many merozoites cannot invade red blood cells within this short period time. These uninvaded merozoites are likely to be lysed, releasing DNA that may complex with hemozoin via the associated proteins. Additionally, Mφs and neutrophils undergo apoptotic death after ingesting IRBCs (133), releasing the degraded parasite materials, including DNA, RNA, hemozoin and other components. The released DNA can complex with hemozoin. However, hemozoin is not obligatory for the entry of parasite DNA into endosomes. The parasite nuclear material, DNA containing immune complexes, merozoites, and IRBCs are also taken up by phagocytic cells (107, 108, 124, 126). It is difficult to quantify in vivo the extent to which DNA enters endosomes through phagocytic uptake of infected erythrocytes, merozoites and nuclear materials compared to hemozoin-bound DNA in inducing immune responses.
Since hemozoin is an inert, indigestible material, it curtails the ability of Mφs to induce innate immune responses to malaria. Upon uptake of IRBCs or hemozoin, Mφs become immunosuppressive and dysfunctional due to damages caused by oxidative burst (134). Monocytes and Mφs that ingest parasite IRBCs undergo apoptotic death with little or negligible release of cytokines, although cytokines are expressed to certain extent (133). Phagocytic internalization of large amounts of whole parasites and non-digestible hemozoin renders Mφs nonfunctional because of phagolysosomal acidification and apoptotic death (133, 135). Hemozoin is known to inhibit the differentiation and maturation of human monocyte-derived DCs as well (136, 137). However, certain subsets of inflammatory Mφs, such as spleen red pulp Mφs, CD169+ Mφs, and CD16+ monocytes produce cytokines in response to malaria parasites (126–128). It is not known whether hemozoin significantly alters the capacity of these cells to produce cytokines and chemokines.
The effect of hemozoin on DC function remains unclear, reviewed in Wykes and Good (138). A previous study has shown that hemozoin-internalized DCs localized in T cell areas of the spleen in P. chabaudi-infected mice and that T cell activated by these DCs lacked effector function (139). These results suggest that hemozoin considerably compromises DC function. However, in a later study, DCs efficiently produced DNA-TLR9 signaling-induced cytokines in response to DNA bound to natural hemozoin and thus it was suggested that hemozoin enhances the activity of DNA by facilitating its entry to endosomes (107). It remains unclear whether this is the case in infected host. On the other hand, it has been shown that subsequent to the uptake of parasite components and the activation of TLR7 and TLR9 signaling, hemozoin destabilizes phagolysosomal membrane, leading to the release of nucleic acid and hemozoin into the cytosol (111). The released parasite components are sensed by cytosolic PRRs, leading to several signaling pathways (44, 81, 111–113), including: (i) cGAS-STING-TBK1-IRF3 signaling by dsDNA; (ii) MDA5-MAVS-TBK1-IRF3 signaling by RNA; (iii) STING signaling mediated by AT-rich motifs through sensing by an unidentified receptor; (iv) AIM2 inflammasome signaling induced by parasite DNA, and (v) NLRP12 inflammasome activation via an unidentified mechanism (82). In addition, hemozoin also activates NLRP3 inflammasome (45, 111–113); Figure 1 and Table 2. The activation of NLRP3 inflammasome by malarial hemozoin is mediated through the activation of Lyn and Syk tyrosine kinases (45). Synthetic hemozoin also activates Lyn and Syk signaling and, depending on the morphology and particle size, induces distinct immune response. It appears that hemozoin has no specific PRR, but it induces a danger signal (112, 113); hence, hemozoin is a DAPM. While nucleic acid-TLR-mediated signaling results in the synthesis of large amounts of pro-IL-1, inflammasome signaling and activation of caspase 1 lead to the cleavage of pro-IL-1 to the active, fever-inducing IL-1. Thus, hemozoin plays a significant role in the production of IL-1, contributing to fever induction in infected people. Besides hemozoin, parasite biomass, and purified merozoites that are devoid of hemozoin can potentially activate inflammasome and caspase 1 as it is known that even inert materials such as alum, asbestos, silica, uric acid crystals and cholesterol particles, activate NLRP3 inflammasome (45).
Uric Acid
Uric acid is the final oxidation product of purine metabolism and is released in large amounts by dying cells. Under physiological conditions, uric acid exists as a monoionic urate and forms insoluble monourate crystal (140). Uric acid is an important antioxidant in plasma and can induce protective anti-inflammatory responses in vascular and other diseases (141, 142). However, excessive formation of uric acid promotes pathogenic conditions, such as severe and chronic inflammatory arthritis, gout, and certain metabolic syndromes (141, 142). The pathogenic role of uric acid is due to the activation of NLRP3 inflammasome, which results in caspase 1 activation and conversion of pro-IL-1 into active IL-1 (114–116); Figure 1 and Table 2. During malaria, a large number of parasite-ingested Mφs, neutrophils and other immune cells die, which likely release high levels of uric acid. Importantly, a significant amount of uric acid is formed during purine nucleotide metabolism by parasites and accumulates as a precipitate in IRBCs (143). Large amounts of hypoxanthine also accumulate in parasite IRBCs. The uric acid precipitate and hypoxanthine are released to the blood upon schizont burst. In the blood stream, hypoxanthine is oxidized to uric acid. In agreement with its pathogenic role, high levels of uric acid are found in the blood circulation of patients having severe malaria (144).
Phagocytosis- and Adherence-Induced Signaling
Phagocytic uptake of parasite IRBCs, merozoites, hemozoin, and immune complexes by Mφs, neutrophils and DCs is an important process during malaria infection. Studies have shown that phagocytosis generally activates Src/Syk family of nonreceptor kinases, leading to the activation of a wide range of signaling pathways, including PKC, RAS-ERK, , NF-κB signaling (145–147). These signaling events can integrate into TLR and inflammasome signaling, thereby modulating immune responses (148). Phagocytosis of malaria parasites and hemozoin suppresses immune function of Mφs and monocytes due to phagolysosomal acidification (133, 135). However, phagocytosis by DCs and inflammatory Mφs likely induces Src/Syk-mediated signaling, contributing to immune responses. The signaling induced by CD36-mediated phagocytosis and IRBC adherence in malaria immune responses are described below.
CD36 Signaling
CD36 is a multifunctional class B scavenger receptor that binds diverse types of ligands and pathogens, facilitating internalization by cells (67, 118, 149). Many cell types, including Mφs, DCs, platelets, and endothelial cells, express CD36. CD36-mediated phagocytosis of pathogens and pathogenic molecules activates Src/Syk kinases, leading to the activation of ERK, p38 and Jun members of the MAPK signaling pathways and NF-κB (67, 118, 119, 150) (Figure 2). Signals of these pathways synergize with TLR and inflammasome signaling, contributing to innate immune responses (119, 156). In malaria, CD36 plays several roles, including (i) mediating the sequestration of P. falciparum in vascular capillaries through the binding of adhesive PfEMP1 expressed on the surface of IRBCs, (ii) phagocytic uptake of parasites, and (iii) enhancing innate immune responses (120, 157–162). Studies have shown that CD36-induced MAPK signaling contributes to the production of TNF-α in mouse malaria infection and modulates parasite GPI-induced cytokine responses in mouse Mφs and human blood DCs (120, 161, 163). In P. falciparum endemic regions, single nucleotide polymorphisms in the Cd36 gene have been linked to protection from cerebral and other severe malaria (164, 165). A more recent study in P. yoelii-infected mice demonstrated that CD36 significantly contributes to cytokine responses by innate immune cells, and the upregulation of MHCII expression, phagocytic activity, Th1 responses and expression of complement and Fc receptors (117). Overall, these responses lead to decreased parasite burden in infected host. It is possible that, similar to CD36, other scavenger receptors, such as MARCO modulate innate immunity to malaria to a certain extent.
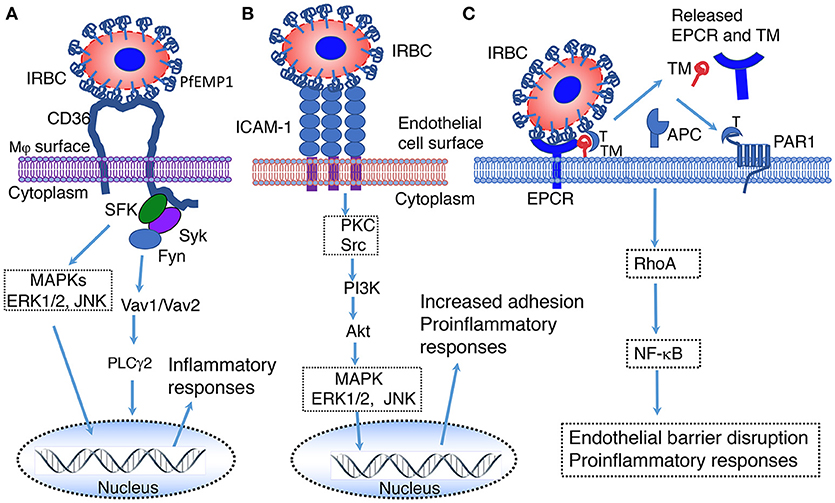
Figure 2. Illustration of predicted pathways activated by the binding of PfEMP 1 expressed on the IRBC surface to CD36 (A), ICAM-1 (B) and EPCR (C). (A) Interaction of PfEMP1 on the surface of IRBC with CD36 on the surface of macrophages. SFK, Src family of protein tyrosine kinases; Syk, spleen tyrosine kinase; Fyn, proto-oncogene tyrosine protein kinase Fyn; MAPK, mitogen-activated protein kinase; ERK1/2, extracellular signal-regulated kinase 1 and 2; JNK, c-Jun N-terminal kinase; Vav1/Vav2, Vav family of guanine nucleotide exchange factors; PLCγ2, phospholipase Cγ2 (151). (B) Interaction of PfEMP1 on the surface of IRBC with ICAM-1 on the surface of endothelial cells surface. PKC, protein kinase C; Src, proto-oncogene tyrosine-protein kinase Src; PI3K, Phosphatidyl inositol 3-kinase; Akt, RAC-alpha serine/threonine-protein kinase (152). (C) Interaction of EPCR on the surface of endothelial cells with PfEMP1 on the surface of IRBCs (153–155). EPCR, endothelial protein C receptor; TM, thrombomodulin; T, thrombin; APC, Activated protein C; PAR1, protease-activated receptor 1; RhoA, Ras homolog gene family, member A GTAase.
EPCR Signaling
Endothelial protein C receptor (EPCR), which is also known as activated protein C (APC) receptor, is a transmembrane glycoprotein in vasculatures that binds protein C and promotes thrombin-thrombomodulin complex-mediated protein C activation (166). Under normal conditions, thrombomodulin on the endothelial surface binds thrombin and activates protein C to APC; this process is strongly promoted by EPCR. APC is detached from EPCR and inactivates blood coagulation factors Va and VIIIa, thereby exerting anticoagulant effect. APC binding to EPCR confers cytoprotective roles, such as antiapoptotic, anti-inflammatory and barrier stabilization responses (153–155). In malaria, EPCR binds certain members of PfEMP1 expressed on the surface of IRBCs and contributes to severe malaria (17, 167–169). Although the downstream events of EPCR-PfEMP1 binding-mediated signaling remain to be understood, it appears that the binding results in the loss of EPCR and thrombomodulin from the endothelial cell surface by shedding, leading to decreased protein C activation and compromised EPCR-mediated protection (Figure 2). This may reduce anti-inflammatory responses and increase thrombin production, blood coagulation, pro-inflammatory responses, endothelial cell apoptosis, leading to the loss of barrier function. The cumulative effects of these responses may contribute to endothelial cell damage, promoting cerebral and other severe malaria illnesses.
ICAM-1 Signaling
ICAM-1 mediates the sequestration of P. falciparum in the brain through the binding of PfEMP1 on the surface of IRBCs to endothelial cells (15, 16, 25). The sequestration of parasites in vascular capillaries of the brain induces inflammatory responses, resulting in the infiltration of cytotoxic effector cells and endothelial barrier damage. These processes have been implicated in the development of cerebral malaria, but the experimental evidence is controversial. An earlier study has reported that interaction with ICAM-1 contributes to increased serum TNF-α in cerebral malaria model of P. berghei ANKA-infected mice and that ICAM-1 deficient mice survive >15 days compared to 6–8 days in wild type (WT) mice (170). However, a later study reported that ICAM-1 is dispensable for cerebral malaria pathogenesis (171). NK cells cross-talk with myeloid cells through LFA-1 binding to ICAM-1, producing IFN-γ (172). Similarly, T cells are also known to interact with endothelial cells through LFA-1 mediated binding to ICAM-1, producing IFN-γ. Studies have shown that, upon binding to its ligands, ICAM-1 is activated by phosphorylation, inducing Src-PI3K-Akt, , Rho and MAPK signaling (121, 122) (Figure 2). It is likely that these signals contribute to immune responses to malaria; thus far this aspect has not been examined.
Innate Immune Responses to Malaria
Innate Immune Responses at the Liver Stage Infection
In malaria, like in most pathogenic infections, the innate immune system functions as the first line of defense by controlling parasite growth and regulating the development of adaptive immunity (14, 75–77). As discussed in section Liver Stage Parasite Sensing, during the liver stage malaria infection, parasite-infected hepatocytes produce type I IFNs through cytosolic sensing of RNA. This cytokine response contributes to the killing of parasite-infected hepatocytes by NKT cells, exposing parasite components. The antigen-presenting cells, primarily DCs and inflammatory Mφs, can potentially recognize the exposed parasite factors. In addition, sporozoites injected by infected mosquitoes that could not enter the blood circulation remain in dermis and die. In addition, some sporozoites that enter blood circulation may not invade hepatocytes and die. These dead parasites are likely sampled by DCs and inflammatory Mφs, leading to TLR- and inflammasome-mediated immune responses. The activated antigen-presenting cells have potential to modulate immune responses to the blood stage infection. However, because the parasite load in liver in natural infections is very low, the innate immune responses are likely to be also very low. Thus, the responses produced by antigen presenting cells against liver stage parasites may not exert a significant influence on immune responses induced by the blood stage parasites. However, in hyper endemic areas, repetitive infections in humans may induce immune tolerance in antigen-presenting cells that may modulate immunity to the blood stage infection to a certain extent.
Innate Immune Responses at the Blood Stage Infection
During the blood stage infection, unlike the liver stage, parasites grow exponentially through repetitive erythrocytic cycles, rapidly accumulating the biomass. This leads to an efficient induction of innate immune responses. Early during the blood stage infection, DCs and Mφs are key first responders of the innate immune system. However, as noted above, Mφs upon internalization of infected erythrocytes, merozoites or hemozoin become immunosuppressive and dysfunctional (133–137); unable to secrete cytokines and chemokines. It appears that the primary role of Mφs during the early stages of blood stage malaria infection is to control parasite growth through phagocytic clearance. In contrast to Mφs, DCs efficiently produce cytokines and chemokines in response malaria parasites, and effectively interact with cells of the innate and adaptive immune system. Thus, DCs play key roles in the initiation and regulation of innate and adaptive immunity to malaria. Also, it should be noted that some subsets of Mφs having certain features of DCs, such as CD11c+ spleen red pulp Mφs and CD169+ inflammatory Mφs, produce cytokines (127, 133, 173).
As noted in section RNA, during the blood stage malaria infection, type I IFNs are the earliest (24 h postinfection) cytokines produced through the activation of TLR7-MyD88 and IRF7 signaling (174). Early type I IFN response is produced primarily by the spleen red pulp Mφs and pDCs in P. chabaudi-infected mice (106, 127). Two recent studies by using lethal malaria model of P. yoelii YM infection, in which parasites grow rapidly to attain peak parasitemia of ~80% by 6 days postinfection, have also showed TLR7-dependent peak levels of type I IFN production at 24–36 h postinfection (81, 128). This early cytokine response is mediated through the coordination of TLR7- and cytosolic sensing mechanisms: TLR7-MyD88-IRF7 signaling in endosomes, and DNA-cGAS-STING-TBK1-IRF3/IRF7 and RNA-MDA5-MAVS-TBK1-IRF3 signaling in the cytosol (81). It has been found that SOCS1 expressed in response to cytosolic nucleic acid sensor-mediated signaling significantly downregulates TLR7-mediated type I IFN response. As such, deficiency in SOCS1 results in markedly high levels of type I IFNs, providing resistance against high parasite burden-dependent lethality (81). Additionally, it has been shown that CD169+ Mφs activated through STING-mediated signaling migrate to bone marrow, where they interact with pDCs to induce type I IFN production through TLR7-MyD88 signaling (128). Interaction of classical DCs with pDCs is also important in pDCs producing cytokine responses to malaria parasites (38, 108). It appears that Mφs and neutrophils that internalize parasites undergo pyroptosis (111), exposing parasite components, which are taken up by classical DCs and CD169+ Mφs, and activated through STING-mediated cytosolic signaling. The early type I IFN response triggers the infiltration of immune cells to the blood that may subsequently localize to the spleen. In contrast, in P. berghei ANKA-infected mice, relatively high level of IFN-α was seen at 4 days postinfection with low or negligible IFN-α production during 1–3 days postinfection (175). Collectively, the above results indicate that different strains of malaria parasites differentially induce type I IFN. That is, the earliest type I IFN production (24–36 h postinfection) by P. chabaudi and P. yoelii is mediated mainly through parasite RNA-induced TLR7 signaling (81, 106, 128), whereas such RNA-mediated early type I IFN response is not readily apparent in P. berghei ANKA (175). The production of type I IFNs at later stages of P. berghei ANKA infection likely involves a different mechanism. In other studies, cGAS sensing of the AT-rich motifs of P. falciparum DNA in the cytosol could induce type I IFN production through the STING-TBK1-IRF3/IRF7 signaling pathway, independent of TLR9-MyD88 signaling (109, 110). This could be the mechanism through which P. berghei ANKA induces significant levels of type I IFN at 4 days pi (175).
Early type I IFN response to malaria infection contributes to the suppression of antiparasitic immunity and promotes cerebral and other severe malaria illnesses under certain situations. This is evident from the observations that deficiency in IFNαR in P. yoelii YM infection resulted in increased IFN-γ levels, significantly decreased parasitemia and resistant to parasite burden-dependent death (128). Also, in P. berghei ANKA-infected mice, which produce low levels of early type I IFN response but produce a significant amount at a somewhat later stage (4 days postinfection), deficiency in IFNαR or treatment with anti-IFNαR1 antibody resulted in increased IFN-γ production, increased number of IFN-γ-positive NK and CD4+ T cells in the spleen, and significantly decreased parasitemia and protection from cerebral malaria (175, 176). From these results, it is evident that type I IFNs suppress the production of IFN-γ and thus anti-parasitic function under certain conditions. By contrast, in a different situation, high levels of type I IFN production at early stages of P. yoelii YM infection promoted antiparasitic immunity. In this case, blocking of SOCS1, a suppressor of cytokine signaling 1, expression resulted in high levels of type I IFN production, leading to increased IFN-γ production and reduced parasitemia, protecting mice from parasite burden-dependent death (81). Consistent with the observations of the latter study (81), daily treatment of P. berghei ANKA-infected mice with recombinant IFN-α, which to a certain extent resembles a situation of high levels of type I IFN production at early stage of infection, also significantly increased IFN-γ production by splenic CD8+ T cells, markedly reducing parasitemia and preventing cerebral malaria (177). The results of these two studies suggest that type I IFNs promote IFN-γ-dependent anti-parasitic immunity, providing resistance against severe malaria. On the other hand, treatment of P. berghei ANKA-infected mice with recombinant IFN-β resulted in reduced TNF-α and IFN-γ production, decreased expression of ICAM-1 and CXCL9 in the brain, reduced CXCR3 expression by T cells and T cell infiltration to the brain, thereby significantly preventing cerebral malaria and increasing mice survival (178). Thus, type I IFNs play contrasting roles in malaria in a context-dependent manner and also dependent on type I IFN isomeric composition.
Compared to the results of two studies outlined above (175, 176), wherein deficiency in IFNαR contributed to increased IFN-γ response and decreased parasitemia in P. berghei ANKA-infected mice, Palomo et al. observed contrasting immune responses (179), although in all these studies mice were protected from cerebral malaria. Palomo et al. found that, compared to infected WT mice, P. berghei ANKA-infected mice deficient in IFNαR had similar parasitemia, reduced expression of IFN-γ and granzyme B by T cells, decreased expression of T cell-attracting chemokine CXCL9, and low infiltration of CXCR3+-expressing CD8 T cells to the brain. A notable difference in these studies is that the latter study used GFP knock-in transgenic parasite clone, which appeared to have slower growth rate than the WT parasites used in the former study. These differences may alter the dynamics of immune responses. Nevertheless, the results agree with the notion that type I IFNs play distinct roles under different malaria conditions.
Overall, the available data indicate that type I IFNs play disparate roles in malaria infection depending on the timing and amount of production, relative levels of IFN-α, IFN-β and perhaps other isomers, and compositions of cellular and cytokine milieu during the progression of infection, and parasite strains. A recent review provides a comprehensive and up-to-date account on the role and contrasting effects of type I IFNs in malaria (180). Type I IFNs are pleiotropic cytokines and as such, they induce a wide range of effects on innate and adaptive immune cells during various pathogenic infections, contributing to either protection against infection or pathogenesis (181–183). These differential effects are likely dependent on the levels of type I IFNs; low levels at early stages of infection mediate cell-mediated immunity, but high levels cause immunosuppression (182). Thus, it is not surprising that type I IFNs produced during malaria infection also induce a wide range of cellular effects involving a complex interplay between various cell types.
In addition to producing type I IFNs, DCs produce a wide range of pro-inflammatory cytokines, including TNF-α, IL-12, and IL-6, and chemokines, such as CXCL1, CXCL2, CCL2, CCL5, CXCL9, and CXCL10 in response to malaria parasites, and play crucial roles in malaria immunity and pathogenesis (34, 38, 108, 184–186). Type I IFNs prime DCs for efficient cytokine and chemokine production and activate NK, NKT, γδ T, and T cells to induce IFN-γ and other inflammatory responses (182). IL-12 produced by DCs activates NK cells to induce the production of IFN-γ, which promotes Th1 and effector T cell responses (187). The augmented production of IFN-γ contributes to an efficient parasitemia control by priming Mφs and neutrophils for increased phagocytic activity and thus parasite clearance (188, 189). IFN-γ also contributes to cerebral and other severe malaria clinical conditions under certain situations, such as parasite sequestering in vital organs (187, 188, 190). Chemokines, on the other hand, promote the recruitment of immune cells to mount effective cell-mediated anti-parasitic effects (191). However, these responses also contribute to severe pathology (37, 192, 193). By and large, the initial innate immune responses are mainly aimed at controlling parasite growth by potentiating antiparasitic cell-mediated immunity. However, since pro-inflammatory responses contribute to pathogenesis (14, 33–38), as infection progresses, the function of DCs switches from pro-inflammatory and Th1-inducing to anti-inflammatory and Th2-inducing phenotypes (39, 40, 194). Eventually, balanced pro- and anti-inflammatory and Th1/Th2 responses prevent pathogenesis and promote protective humoral immunity to malaria (39–42); imbalanced responses contribute to pathogenesis. Thus, innate immune responses contribute to either protective immunity or pathogenesis in a context dependent manner.
TLR-MYD88 Signaling Prominently Contributes to Protective Immunity and Pathogenesis
DNA and RNA play prominent roles in mouse malaria immunity and pathogenesis; under certain conditions TLR2 and TLR4 also play important roles. As such, studies in various mouse models have demonstrated that TLR9, TLR7, TLR4, and TLR2 play important roles in malaria immunity and cerebral, placental and other severe malaria pathology (43, 44, 81, 102, 103, 106–108, 124, 128, 174, 195–201). Gene polymorphism studies in endemic areas that assessed the role of TLRs in malaria have linked TLR9, TLR4, and TLR2 to either susceptibility or resistance to malaria (202–214). While the involvement of TLR9, TLR4, and TLR2 in malaria immunity/pathology is evident from studies in both mouse malaria models and in humans (43, 106, 195, 197, 198, 200–203, 205, 210), thus far none of the studies in endemic areas have revealed the association of TLR7 with human malaria immunity/pathology. It is unclear whether studies in endemic areas that have assessed association of TLRs have investigated the role of TLR7 in malaria susceptibility/resistance. However, since P. falciparum RNA appears to have very low immunostimulatory activity, it is possible that TLR7 has either minor or no role in human malaria. Accumulated evidence indicates that TLRs prominently contribute malaria immunity and pathology, whereas other signaling mechanisms such as inflammasome and adherence-mediated signaling may play minor roles (172, 215).
The contribution of pro-inflammatory responses produced by the innate immune system to protective immunity and pathogenesis is primarily context dependent. In non-cytoadherent parasites, such as P. yoelii 17XNL strain and P. chabaudi that exhibit slow growth rates during the initial phase of infection, pro-inflammatory responses are protective by facilitating anti-parasitic effector function and cell-mediated immunity. In contrast, in the case of cytoadherent parasites, such as P. berghei ANKA that sequesters in brain, lungs, liver and adipose tissues, and P. berghei NK65 that sequesters in lungs and liver, pro-inflammatory responses are pathogenic by promoting cytotoxicity in effector cells, which cause organ damage. In this regard, the observations made in mouse models parallel those of vivax and falciparum malaria in humans. In P. falciparum infection, strong pro-inflammatory responses contribute to an effective control of parasitemia which otherwise results in fulminant infection (41, 42). However, despite this beneficial effect, pro-inflammatory responses promote effector cell function, which contributes to organ damage and severe illnesses. In contrast, in the case of P. vivax, which is relatively slow growing and does not cytoadhere except at low levels in lungs and the placenta (125), despite causing a number of clinical conditions, fatal illnesses are relatively rare compared to P. falciparum.
Concluding Remarks
Malaria continues to be a major global health problem. In many endemic areas, parasites are becoming increasingly resistant to the currently widely used artemisinin-based combination drugs, which have been very effective in treating infection. Therefore, new drugs or other treatment strategies are urgently needed. Mass vaccination is the best strategy to prevent malaria. However, despite huge efforts during the past decades in many laboratories around the world, obtaining an efficacious vaccine suitable for mass vaccination remains challenging. It is thought that better understanding of molecular and cellular processes involved in the development of protective immunity and those that contribute to severe pathogenesis will be useful in designing an effective vaccine. During the past decades, a significant progress has been made in identifying the parasite factors and host receptors involved in the activation of the innate immune system and the associated cell signaling pathways. Significant progress has also been made in understanding how cellular activation and immune responses initiated upon parasite-host interactions influence subsequent development of antiparasitic immunity and contribute to severe pathogenesis.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants AI41139 and AI104844 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, and by the financial support from the Department of Biochemistry and Molecular Biology, The Penn State University College of Medicine.
Abbreviations
IRBCs, infected red blood cells; DC, dendritic cell; Mφ, macrophage; NK cells, natural killer cells; NKT cells, NK T cells; GPI, glycosylphosphatidylinositol; PfEMP1, P. falciparum erythrocyte membrane protein 1; ICAM-1, intracellular adhesion molecule 1; VCAM-1, vascular adhesion molecule 1; LFA-1, lymphocyte function-associated antigen-1; EPCR, endothelial protein receptor; Th, helper T cells; IFNαR, type I IFN-α receptor; PAMPs, pathogen-associated molecular patterns; DAMPs, danger-associated molecular patterns; PRRs, pathogen recognition receptors; TLR, toll-like receptor; MyD88, myeloid differentiation factor 88; MAL, MyD88 adapter-like; IRAK, IL-1 receptor-associated kinase; TRAF6, tumor necrosis factor receptor-associated factor 6; TRIF, TIR (Toll/IL-1 receptor) domain-containing adaptor inducing IFN-β; TRAM, TRIF-related adaptor molecule; MAPK, mitogen-activated protein kinases; ERK, extracellular signal-regulated kinases; JNK, c-Jun N-terminal kinases; NF-κB, nuclear factor κB; TBK1, TRAF family member-associated NF-κB activator (TANK)-binding kinase 1; cGAS, cyclic GMP-AMP synthase; STING, stimulator of IFN genes; MDA5, melanoma differentiation-associated gene 5; MVAS, mitochondrial antiviral-signaling protein; IRF, IFN regulatory factor; AIM2, absent in melanoma 2; NOD, nucleotide-binding oligomerization domain; NLR, (NOD)-like receptor; LLR, leucine-rich repeat kinase-like protein; NLRP, NOD- LRR- and pyrin domain-containing protein.
References
1. World Health Organization. World Malaria Report Geneva. (2017). Available online at: http://www.who.int/malaria/publications/world-malaria-report-2017/en/
2. Battle KE, Guerra CA, Golding N, Duda KA, Cameron E, Howes RE, et al. Global database of matched Plasmodium falciparum and P. vivax incidence and prevalence records from 1985-2013. Sci Data (2015) 2:150012. doi: 10.1038/sdata.2015.12
3. Howes RE, Battle KE, Mendis KN, Smith DL, Cibulskis RE, Baird JK, et al. Global epidemiology of Plasmodium vivax. Am J Trop Med Hyg. (2016) 95(Suppl. 6):15–34. doi: 10.4269/ajtmh.16-0141
4. Rogerson SJ, Desai M, Mayor A, Sicuri E, Taylor SM, van Eijk AM. Burden, pathology, and costs of malaria in pregnancy: new developments for an old problem. Lancet Infect Dis. (2018) 18:e107–18. doi: 10.1016/S1473-3099(18)30066-5
5. Gallup JL, Sachs JD. The economic burden of malaria. Am J Trop Med Hyg. (2001) 64(Suppl. 1–2):85–96. doi: 10.4269/ajtmh.2001.64.85
6. Chima RI, Goodman CA, Mills A. The economic impact of malaria in Africa: a critical review of the evidence. Health Policy (2003) 63:17–36. doi: 10.1016/S0168-8510(02)00036-2
7. Phillips MA, Burrows JN, Manyando C, van Huijsduijnen RH, Van Voorhis WC, Wells TNC. Malaria. Nat Rev Dis Primers (2017) 3:17050. doi: 10.1038/nrdp.2017.50
8. Singh B, Daneshvar C. Human infections and detection of Plasmodium knowlesi. Clinical Microbiol Rev. (2013) 26:165–84. doi: 10.1128/CMR.00079-12
9. Lee K-S, Cox-Singh J, Singh B. Morphological features and differential counts of Plasmodium knowlesi parasites in naturally acquired human infections. Malaria J. (2009) 8:73. doi: 10.1186/1475-2875-8-73
10. Otto TD, Bohme U, Jackson AP, Hunt M, Franke-Fayard B, Hoeijmakers WA, et al. A comprehensive evaluation of rodent malaria parasite genomes and gene expression. BMC Biol. (2014) 12:86. doi: 10.1186/s12915-014-0086-0
11. Anstey NM, Russell B, Yeo TW, Price RN. The pathophysiology of vivax malaria. Trends Parasitol. (2009) 25:220–7. doi: 10.1016/j.pt.2009.02.003
12. Autino B, Corbett Y, Castelli F, Taramelli D. Pathogenesis of malaria in tissues and blood. Mediterr J Hematol Infect Dis. (2012) 4:e2012061. doi: 10.4084/mjhid.2012.061
13. Storm J, Craig AG. Pathogenesis of cerebral malaria–inflammation and cytoadherence. Front Cellular Infect Microbiol. (2014) 4:100. doi: 10.3389/fcimb.2014.00100
14. Deroost K, Pham T-T, Opdenakker G, Van den Steen PE. The immunological balance between host and parasite in malaria. FEMS Microbiol Rev. (2015) 40:208–57. doi: 10.1093/femsre/fuv046
15. Wahlgren M, Goel S, Akhouri RR. Variant surface antigens of Plasmodium falciparum and their roles in severe malaria. Nat Rev Microbiol. (2017) 15:479–91. doi: 10.1038/nrmicro.2017.47
16. Smith JD, Rowe JA, Higgins MK, Lavstsen T. Malaria's deadly grip: cytoadhesion of Plasmodium falciparum-infected erythrocytes. Cellular Microbiol. (2013) 15:1976–83. doi: 10.1111/cmi.12183
17. Bernabeu M, Smith JD. EPCR and malaria severity: the center of a perfect storm. Trends Parasitol. (2017) 33:295–308. doi: 10.1016/j.pt.2016.11.004
18. Sharma L, Shukla G. Placental malaria: a new insight into the pathophysiology. Front Med. (2017) 4:117. doi: 10.3389/fmed.2017.00117
19. Coban C, Lee MSJ, Ishii KJ. Tissue-specific immunopathology during malaria infection. Nat Rev Immunol. (2018) 18:266–78. doi: 10.1038/nri.2017.138
20. Lee MSJ, Coban C. Unforeseen pathologies caused by malaria. Int Immunol. (2018) 30:121–9. doi: 10.1093/intimm/dxx076
21. Fried M, Duffy PE. Adherence of Plasmodium falciparum to chondroitin sulfate A in the human placenta. Science (1996) 272:1502–4. doi: 10.1126/science.272.5267.1502
22. Nunes MC, Scherf A. Plasmodium falciparum during pregnancy: a puzzling parasite tissue adhesion tropism. Parasitology (2007) 134(Pt 13):1863–9. doi: 10.1017/S0031182007000133
23. Goel S, Gowda DC. How specific is Plasmodium falciparum adherence to chondroitin 4-sulfate? Trends Parasitol. (2011) 27:375–81. doi: 10.1016/j.pt.2011.03.005
24. Fried M, Duffy PE. Malaria during pregnancy. Cold Spring Harb Perspect Med. (2017) 7:a025551. doi: 10.1101/cshperspect.a025551
25. Hviid L, Jensen AT. PfEMP1 - a parasite protein family of key importance in Plasmodium falciparum malaria immunity and pathogenesis. Adv Parasitol. (2015) 88:51–84. doi: 10.1016/bs.apar.2015.02.004
26. Totino PR, Lopes SC. Insights into the cytoadherence phenomenon of Plasmodium vivax: the putative role of phosphatidylserine. Front Immunol. (2017) 8:1148. doi: 10.3389/fimmu.2017.01148
27. Kochar DK, Saxena V, Singh N, Kochar SK, Kumar SV, Das A. Plasmodium vivax malaria. Emerg Infect Dis. (2005) 11:132–4. doi: 10.3201/eid1101.040519
28. Genton B, D'Acremont V, Rare L, Baea K, Reeder JC, Alpers MP, et al. Plasmodium vivax and mixed infections are associated with severe malaria in children: a prospective cohort study from Papua New Guinea. PLoS Med. (2008) 5:e127. doi: 10.1371/journal.pmed.0050127
29. Baird JK. Evidence and implications of mortality associated with acute Plasmodium vivax malaria. Clin Microbiol Rev. (2013) 26:36–57. doi: 10.1128/CMR.00074-12
30. Naing C, Whittaker MA, Wai VN, Mak JW. Is Plasmodium vivax malaria a severe malaria?: a systematic review and meta-analysis. PLoS Neglect Trop Dis. (2014) 8:e3071. doi: 10.1371/journal.pntd.0003071
31. Price RN, Douglas NM, Anstey NM. New developments in Plasmodium vivax malaria: severe disease and the rise of chloroquine resistance. Curr Opin Infect Dis. (2009) 22:430–5. doi: 10.1097/QCO.0b013e32832f14c1
32. Lima-Junior Jda C, Pratt-Riccio LR. Major histocompatibility complex and malaria: focus on Plasmodium vivax infection. Front Immunol. (2016) 7:13. doi: 10.3389/fimmu.2016.00013
33. Angulo I, Fresno M. Cytokines in the pathogenesis of and protection against malaria. Clin Diagn Lab Immunol. (2002) 9:1145–52. doi: 10.1128/CDLI.9.6.1145-1152.2002
34. Stevenson MM, Riley EM. Innate immunity to malaria. Nat Rev Immunol. (2004) 4:169–80. doi: 10.1038/nri1311
35. Schofield L, Grau GE. Immunological processes in malaria pathogenesis. Nat Rev Immunol. (2005) 5:722–35. doi: 10.1038/nri1686
36. Clark IA, Alleva LM, Budd AC, Cowden WB. Understanding the role of inflammatory cytokines in malaria and related diseases. Travel Med Infect Dis. (2008) 6:67–81. doi: 10.1016/j.tmaid.2007.07.002
37. Dunst J, Kamena F, Matuschewski K. Cytokines and chemokines in cerebral malaria pathogenesis. Front Cellular Infect Microbiol. (2017) 7:324. doi: 10.3389/fcimb.2017.00324
38. Gotz A, Tang MS, Ty MC, Arama C, Ongoiba A, Doumtabe D, et al. Atypical activation of dendritic cells by Plasmodium falciparum. Proc Natl Acad Sci USA. (2017) 114:E10568–77. doi: 10.1073/pnas.1708383114
39. Perry JA, Olver CS, Burnett RC, Avery AC. Cutting edge: the acquisition of TLR tolerance during malaria infection impacts T cell activation. J Immunol. (2005) 174:5921–5. doi: 10.4049/jimmunol.174.10.5921
40. Riley EM, Wahl S, Perkins DJ, Schofield L. Regulating immunity to malaria. Parasite Immunol. (2006) 28:35–49. doi: 10.1111/j.1365-3024.2006.00775.x
41. Goncalves RM, Scopel KK, Bastos MS, Ferreira MU. Cytokine balance in human malaria: does Plasmodium vivax elicit more inflammatory responses than Plasmodium falciparum? PLoS ONE (2012) 7:e44394. doi: 10.1371/journal.pone.0044394
42. Goncalves RM, Lima NF, Ferreira MU. Parasite virulence, co-infections and cytokine balance in malaria. Pathogens Global Health (2014) 108:173–8. doi: 10.1179/2047773214Y.0000000139
43. Eriksson E, Sampaio N, Schofield L. Toll-like receptors and malaria–sensing and susceptibility. J Trop Dis. (2013) 2:126. doi: 10.4172/2329-891X.1000126
44. Gazzinelli RT, Kalantari P, Fitzgerald KA, Golenbock DT. Innate sensing of malaria parasites. Nat Rev Immunol. (2014) 14:744–57. doi: 10.1038/nri3742
45. Olivier M, Van Den Ham K, Shio MT, Kassa FA, Fougeray S. Malarial pigment hemozoin and the innate inflammatory response. Front Immunol. (2014) 5:25. doi: 10.3389/fimmu.2014.00025
46. Liehl P, Meireles P, Albuquerque IS, Pinkevych M, Baptista F, Mota MM, et al. Innate immunity induced by Plasmodium liver infection inhibits malaria reinfections. Infect Immun. (2015) 83:1172–80. doi: 10.1128/IAI.02796-14
47. Kalantari P. The emerging role of pattern recognition receptors in the pathogenesis of malaria. Vaccines (2018) 6:1–15. doi: 10.3390/vaccines6010013
48. Struik SS, Riley EM. Does malaria suffer from lack of memory? Immunol Rev. (2004) 201:268–90. doi: 10.1111/j.0105-2896.2004.00181.x
49. Langhorne J, Ndungu FM, Sponaas AM, Marsh K. Immunity to malaria: more questions than answers. Nat Immunol. (2008) 9:725–32. doi: 10.1038/ni.f.205
50. Aly AS, Vaughan AM, Kappe SH. Malaria parasite development in the mosquito and infection of the mammalian host. Ann Rev Microbiol. (2009) 63:195–221. doi: 10.1146/annurev.micro.091208.073403
51. Vaughan AM, Aly AS, Kappe SH. Malaria parasite pre-erythrocytic stage infection: gliding and hiding. Cell Host Microbe (2008) 4:209–18. doi: 10.1016/j.chom.2008.08.010
52. Bertolino P, Bowen DG. Malaria and the liver: immunological hide-and-seek or subversion of immunity from within? Front Microbiol. (2015) 6:41. doi: 10.3389/fmicb.2015.00041
53. Bruce-Chwatt LJ. Essential Malariology. London, UK: William Heinemann Medical Books Ltd. (1985).
54. Roucher C, Rogier C, Sokhna C, Tall A, Trape JF. A 20-year longitudinal study of Plasmodium ovale and Plasmodium malariae prevalence and morbidity in a West African population. PLoS ONE (2014) 9:e87169. doi: 10.1371/journal.pone.0087169
55. Van den Steen PE, Deroost K, Deckers J, Van Herck E, Struyf Opdenakker G. Pathogenesis of malaria-associated acute respiratory distress syndrome. Trends Parasitol. (2013) 29:346–58. doi: 10.1016/j.pt.2013.04.006
56. Beutler BA. TLRs and innate immunity. Blood (2009) 113:1399–407. doi: 10.1182/blood-2008-07-019307
57. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
58. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Ann Rev Immunol. (2015) 33:257–90. doi: 10.1146/annurev-immunol-032414-112240
59. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
60. Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Ann Rev Immunol. (2014) 32:461–88. doi: 10.1146/annurev-immunol-032713-120156
61. Vijay K. Toll-like receptors in immunity and inflammatory diseases: past, present, and future. Intl Immunopharmacol. (2018) 59:391–412. doi: 10.1016/j.intimp.2018.03.002
62. Rubartelli A, Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. (2007) 28:429–36. doi: 10.1016/j.it.2007.08.004
63. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. (2012) 249:158–75. doi: 10.1111/j.1600-065X.2012.01146.x
64. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity (2013) 38:209–23. doi: 10.1016/j.immuni.2013.02.003
65. Venereau E, Ceriotti C, Bianchi ME. DAMPs from cell death to new life. Front Immunol. (2015) 6:422. doi: 10.3389/fimmu.2015.00422
66. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity (2011) 34:637–50. doi: 10.1016/j.immuni.2011.05.006
67. Freeman SA, Grinstein S. Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol Rev. (2014) 262:193–215. doi: 10.1111/imr.12212
68. Lamphier MS, Sirois CM, Verma A, Golenbock DT, Latz E. TLR9 and the recognition of self and non-self nucleic acids. Ann N Y Acad Sci. (2006) 1082:31–43. doi: 10.1196/annals.1348.005
69. Ohto U, Shimizu T. Structural aspects of nucleic acid-sensing Toll-like receptors. Biophys Rev. (2016) 8:33–43. doi: 10.1007/s12551-015-0187-1
70. Franchi L, McDonald C, Kanneganti TD, Amer A, Núñez G. Nucleotide-binding oligomerization domain-like receptors: intracellular pattern recognition molecules for pathogen detection and host defense. J Immunol. (2006) 177:3507–13. doi: 10.4049/jimmunol.177.6.3507
71. Silvie O, Amino R, Hafalla JC. Tissue-specific cellular immune responses to malaria pre-erythrocytic stages. Curr Opin Microbiol. (2017) 40:160–7. doi: 10.1016/j.mib.2017.12.001
72. Holz LE, Fernandez-Ruiz D, Heath WR. Protective immunity to liver-stage malaria. Clin Transl Immunol. (2016) 5:e105. doi: 10.1038/cti.2016.60
73. Gätz A, Ty M, Chora AF, Zuzarte-Luís V, Mota MM, Rodriguez A. Innate immunity to malaria. In: Malaria: Immune Response to Infection and Vaccination, eds Mota MM and Rodriguez A (Cham: Springer International Publishing AG) (2017). p. 3–25.
74. Yazdani SS, Mukherjee P, Chauhan VS, Chitnis CE. Immune responses to asexual blood-stages of malaria parasites. Curr Mol Med. (2006) 6:187–203. doi: 10.2174/156652406776055212
75. Smith TG, Ayi K, Serghides L, McAllister CD, Kain KC. Innate immunity to malaria caused by Plasmodium falciparum. Clin Invest Med. (2002) 25:262–72.
76. Urban BC, Ing R, Stevenson MM. Early interactions between blood-stage Plasmodium parasites and the immune system. Curr Top Microbiol Immunol. (2005) 297:25–70. doi: 10.1007/3-540-29967-X_2
77. Walther M, Woodruff J, Edele F, Jeffries D, Tongren JE, King E, et al. Innate immune responses to human malaria: heterogeneous cytokine responses to blood-stage Plasmodium falciparum correlate with parasitological and clinical outcomes. J Immunol. (2006) 177:5736–45. doi: 10.4049/jimmunol.177.8.5736
78. Stevenson MM, Ing R, Berretta F, Miu J. Regulating the adaptive immune response to blood-stage malaria: role of dendritic cells and CD4+Foxp3+ regulatory T cells. Int J Biol Sci. (2011) 7:1311–22. doi: 10.7150/ijbs.7.1311
79. Liehl P, Zuzarte-Luis V, Chan J, Zillinger T, Baptista F, Carapau D, et al. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat Med. (2014) 20:47–53. doi: 10.1038/nm.3424
80. Miller JL, Sack BK, Baldwin M, Vaughan AM, Kappe SH. Interferon-mediated innate immune responses against malaria parasite liver stages. Cell Rep. (2014) 7:436–47. doi: 10.1016/j.celrep.2014.03.018
81. Yu X, Cai B, Wang M, Tan P, Ding X, Wu J, et al. Cross-regulation of two type I interferon signaling pathways in plasmacytoid dendritic cells controls anti-malaria immunity and host mortality. Immunity (2016) 45:1093–107. doi: 10.1016/j.immuni.2016.10.001
82. Ataide MA, Andrade WA, Zamboni DS, Wang D, do Carmo Souza M, Franklin BS, et al. Malaria-induced NLRP12/NLRP3-dependent caspase-1 activation mediates inflammation and hypersensitivity to bacterial superinfection. PLoS Pathog. (2014) 10:e1003885. doi: 10.1371/journal.ppat.1003885
83. Couper KN, Barnes T, Hafalla JC, Combes V, Ryffel B, Secher T, et al. Parasite-derived plasma microparticles contribute significantly to malaria infection-induced inflammation through potent macrophage stimulation. PLoS Pathog. (2010) 6:e1000744. doi: 10.1371/journal.ppat.1000744
84. Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, et al. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem. (2007) 282:20221–9. doi: 10.1074/jbc.M610737200
85. Cowman AF, Healer J, Marapana D, Marsh K. Malaria: biology and disease. Cell (2016) 167:610–24. doi: 10.1016/j.cell.2016.07.055
86. Hirako IC, Assis PA, Hojo-Souza NS, Reed G, Nakaya H, Golenbock DT, et al. Daily rhythms of TNF-α expression and food intake regulate synchrony of Plasmodium stages with the host circadian cycle. Cell Host Microbe (2018) 23:796–808.e6. doi: 10.1016/j.chom.2018.04.016
87. Schreibelt G, Tel J, Sliepen KH, Benitez-Ribas D, Figdor CG, Adema GJ, et al. Toll-like receptor expression and function in human dendritic cell subsets: implications for dendritic cell-based anti-cancer immunotherapy. Cancer Immunol Immunother. (2010) 59:1573–82. doi: 10.1007/s00262-010-0833-1
88. Schofield L, Hackett F. Signal transduction in host cells by a glycosylphosphatidylinositol toxin of malaria parasites. J Exp Med. (1993)177:145–53. doi: 10.1084/jem.177.1.145
89. Gowda DC. TLR-mediated cell signaling by malaria GPIs. Trends Parasitol. (2007) 23:596–604. doi: 10.1016/j.pt.2007.09.003
90. Naik RS, Branch OH, Woods AS, Vijaykumar M, Perkins DJ, Nahlen BL, et al. Glycosylphosphatidylinositol anchors of Plasmodium falciparum: molecular characterization and naturally elicited antibody response that may provide immunity to malaria pathogenesis. J Exp Med. (2000) 192:1563–76. doi: 10.1084/jem.192.11.1563
91. Gerold P, Jung N, Azzouz N, Freiberg N, Kobe S, Schwarz RT. Biosynthesis of glycosylphosphatidylinositols of Plasmodium falciparum in a cell-free incubation system: inositol acylation is needed for mannosylation of glycosylphosphatidylinositols. Biochem J. (1999) 344(Pt 3):731–8. doi: 10.1042/bj3440731
92. Gowda DC, Gupta P, Davidson EA. Glycosylphosphatidylinositol anchors represent the major carbohydrate modification in proteins of intraerythrocytic stage Plasmodium falciparum. J Biol Chem. (1997) 272:6428–39. doi: 10.1074/jbc.272.10.6428
93. Gilson PR, Nebl T, Vukcevic D, Moritz RL, Sargeant T, Speed TP, et al. Identification and stoichiometry of glycosylphosphatidylinositol-anchored membrane proteins of the human malaria parasite Plasmodium falciparum. Mol Cell Proteomics (2006) 5:1286–99. doi: 10.1074/mcp.M600035-MCP200
94. Sanders PR, Gilson PR, Cantin GT, Greenbaum DC, Nebl T, Carucci DJ, et al. Distinct protein classes including novel merozoite surface antigens in Raft-like membranes of Plasmodium falciparum. J Biol Chem. (2005) 280:40169–76. doi: 10.1074/jbc.M509631200
95. Schofield L, Novakovic S, Gerold P, Schwarz RT, McConville MJ, Tachado SD. Glycosylphosphatidylinositol toxin of Plasmodium up-regulates intercellular adhesion molecule-1, vascular cell adhesion molecule-1, and E-selectin expression in vascular endothelial cells and increases leukocyte and parasite cytoadherence via tyrosine kinase-dependent signal transduction. J Immunol. (1996) 156:1886–96.
96. Tachado SD, Gerold P, McConville MJ, Baldwin T, Quilici D, Schwarz RT, et al. Glycosylphosphatidylinositol toxin of Plasmodium induces nitric oxide synthase expression in macrophages and vascular endothelial cells by a protein tyrosine kinase-dependent and protein kinase C-dependent signaling pathway. J Immunol. (1996) 156:1897–907.
97. Tachado SD, Gerold P, Schwarz R, Novakovic S, McConville M, Schofield L. Signal transduction in macrophages by glycosylphosphatidylinositols of Plasmodium, Trypanosoma, and Leishmania: activation of protein tyrosine kinases and protein kinase C by inositolglycan and diacylglycerol moieties. Proc Natl Acad Sci USA. (1997) 94:4022–7. doi: 10.1073/pnas.94.8.4022
98. Schofield L, Hewitt MC, Evans K, Siomos MA, Seeberger PH. Synthetic GPI as a candidate anti-toxic vaccine in a model of malaria. Nature (2002) 418:785–9. doi: 10.1038/nature00937
99. Naik RS, Krishnegowda G, Gowda DC. Glucosamine inhibits inositol acylation of the glycosylphosphatidylinositol anchors in intraerythrocytic Plasmodium falciparum. J Biol Chem. (2003) 278:2036–42. doi: 10.1074/jbc.M208976200
100. Franca CT, Li Wai Suen CSN, Carmagnac A, Lin E, Kiniboro B, Siba P, et al. IgG antibodies to synthetic GPI are biomarkers of immune-status to both Plasmodium falciparum and Plasmodium vivax malaria in young children. Malar J. (2017) 16:386. doi: 10.1186/s12936-017-2042-2
101. Vijaykumar M, Naik RS, Gowda DC. Plasmodium falciparum glycosylphosphatidylinositol-induced TNF-α secretion by macrophages is mediated without membrane insertion or endocytosis. J Biol Chem. (2001) 276:6909–12. doi: 10.1074/jbc.C100007200
102. Krishnegowda G, Hajjar AM, Zhu J, Douglass EJ, Uematsu S, Akira S, et al. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J Biol Chem. (2005) 280:8606–16. doi: 10.1074/jbc.M413541200
103. Zhu J, Krishnegowda G, Gowda DC. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: the requirement of extracellular signal-regulated kinase, p38, c-Jun N-terminal kinase and NF-κB pathways for the expression of proinflammatory cytokines and nitric oxide. J Biol Chem. (2005) 280:8617–27. doi: 10.1074/jbc.M413539200
104. Lu Z, Serghides L, Patel SN, Degousee N, Rubin BB, Krishnegowda G, et al. Disruption of JNK2 decreases the cytokine response to Plasmodium falciparum glycosylphosphatidylinositol In vitro and confers protection in a cerebral malaria model. J Immunol. (2006) 177:6344–52. doi: 10.4049/jimmunol.177.9.6344
105. Zhu J, Wu X, Goel S, Gowda NM, Kumar S, Krishnegowda G, et al. MAPK-activated protein kinase 2 differentially regulates Plasmodium falciparum glycosylphosphatidylinositol-induced production of tumor necrosis factor-α and interleukin-12 in macrophages. J Biol Chem. (2009) 284:15750–61. doi: 10.1074/jbc.M901111200
106. Baccarella A, Fontana MF, Chen EC, Kim CC. Toll-like receptor 7 mediates early innate immune responses to malaria. Infect Immun. (2013) 81:4431–42. doi: 10.1128/IAI.00923-13
107. Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, et al. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci USA. (2007) 104:1919–24. doi: 10.1073/pnas.0608745104
108. Wu X, Gowda NM, Kumar S, Gowda DC. Protein-DNA complex is the exclusive malaria parasite component that activates dendritic cells and triggers innate immune responses. J Immunol. (2010) 184:4338–48. doi: 10.4049/jimmunol.0903824
109. Sharma S, DeOliveira RB, Kalantari P, Parroche P, Goutagny N, Jiang Z, et al. Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity (2011) 35:194–207. doi: 10.1016/j.immuni.2011.05.016
110. Gallego-Marin C, Schrum JE, Andrade WA, Shaffer SA, Giraldo LF, Lasso AM, et al. Cyclic GMP-AMP synthase is the cytosolic sensor of Plasmodium falciparum genomic DNA and activates type I IFN in malaria. J Immunol. (2018) 200:768–74. doi: 10.4049/jimmunol.1701048
111. Kalantari P, DeOliveira RB, Chan J, Corbett Y, Rathinam V, Stutz A, et al. Dual engagement of the NLRP3 and AIM2 inflammasomes by plasmodium-derived hemozoin and DNA during malaria. Cell Rep. (2014) 6:196–210. doi: 10.1016/j.celrep.2013.12.014
112. Shio MT, Eisenbarth SC, Savaria M, Vinet AF, Bellemare MJ, Harder KW, et al. Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog. (2009) 5:e1000559. doi: 10.1371/annotation/abca067d-b82b-4de6-93c5-0fcc38e3df05
113. Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, et al. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS ONE (2009) 4:e6510. doi: 10.1371/journal.pone.0006510
114. Griffith JW, Sun T, McIntosh MT, Bucala R. Pure Hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J Immunol. (2009) 183:5208–20. doi: 10.4049/jimmunol.0713552
115. Braga TT, Forni MF, Correa-Costa M, Ramos RN, Barbuto JA, Branco P, et al. Soluble uric acid activates the NLRP3 inflammasome. Sci Rep. (2017) 7:39884. doi: 10.1038/srep39884
116. Romero CA, Remor A, Latini A, De Paul AL, Torres AI, Mukdsi JH. Uric acid activates NRLP3 inflammasome in an in-vivo model of epithelial to mesenchymal transition in the kidney. J Mol Histol. (2017) 48:209–18. doi: 10.1007/s10735-017-9720-9
117. Thylur RP, Wu X, Gowda NM, Punnath K, Neelgund SE, Febbraio M, et al. CD36 receptor regulates malaria-induced immune responses primarily at early blood stage infection contributing to parasitemia control and resistance to mortality. J Biol Chem. (2017) 292:9394–408. doi: 10.1074/jbc.M117.781294
118. Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. (2009) 2:re3. doi: 10.1126/scisignal.272re3
119. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. (2010) 11:155–61. doi: 10.1038/ni.1836
120. Erdman LK, Cosio G, Helmers AJ, Gowda DC, Grinstein S, Kain KC. CD36 and TLR interactions in inflammation and phagocytosis: implications for malaria. J Immunol. (2009) 183:6452–9. doi: 10.4049/jimmunol.0901374
121. Liu G, Place AT, Chen Z, Brovkovych VM, Vogel SM, Muller WA, et al. ICAM-1-activated Src and eNOS signaling increase endothelial cell surface PECAM-1 adhesivity and neutrophil transmigration. Blood (2012) 120:1942–52. doi: 10.1182/blood-2011-12-397430
122. Ramos TN, Bullard DC, Barnum SR. ICAM-1: isoforms and phenotypes. J Immunol. (2014) 192:4469–74. doi: 10.4049/jimmunol.1400135
123. Pichyangkul S, Yongvanitchit K, Kum-arb U, Hemmi H, Akira S, Krieg AM, et al. Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a Toll-like receptor 9-dependent pathway. J Immunol. (2004) 172:4926–33. doi: 10.4049/jimmunol.172.8.4926
124. Gowda NM, Wu X, Gowda DC. The nucleosome (histone-DNA complex) is the TLR9-specific immunostimulatory component of Plasmodium falciparum that activates DCs. PLoS ONE (2011) 6:e20398. doi: 10.1371/journal.pone.0020398
125. Anstey NM, Douglas NM, Poespoprodjo JR, Price RN. Plasmodium vivax: clinical spectrum, risk factors and pathogenesis. Adv Parasitol. (2012) 80:151–201. doi: 10.1016/B978-0-12-397900-1.00003-7
126. Hirako IC, Gallego-Marin C, Ataide MA, Andrade WA, Gravina H, Rocha BC, et al. DNA-containing immunocomplexes promote inflammasome assembly and release of pyrogenic cytokines by CD14+CD16+CD64highCD32low inflammatory monocytes from malaria patients. MBio (2015) 6:e01605–15. doi: 10.1128/mBio.01605-15
127. Kim CC, Nelson CS, Wilson EB, Hou B, DeFranco AL, DeRisi JL. Splenic red pulp macrophages produce type I interferons as early sentinels of malaria infection but are dispensable for control. PloS ONE (2012) 7:e48126. doi: 10.1371/journal.pone.0048126
128. Spaulding E, Fooksman D, Moore JM, Saidi A, Feintuch CM, Reizis B, et al. STING-licensed macrophages prime type I IFN production by plasmacytoid dendritic cells in the bone marrow during severe Plasmodium yoelii malaria. PLoS Pathog. (2016) 12:e1005975. doi: 10.1371/journal.ppat.1005975
129. Pagola S, Stephens PW, Bohle DS, Kosar AD, Madsen SK. The structure of malaria pigment β-haematin. Nature (2000) 404:307–10. doi: 10.1038/35005132
130. Jani D, Nagarkatti R, Beatty W, Angel R, Slebodnick C, Andersen J, et al. HDP—a novel heme detoxification protein from the malaria parasite. PLoS Pathog. (2008) 4:e1000053. doi: 10.1371/journal.ppat.1000053
131. Arese P, Schwarzer E. Malarial pigment (haemozoin): a very active 'inert' substance. Ann Trop Med Parasitol. (1997) 91:501–16. doi: 10.1080/00034983.1997.11813168
132. Boyle MJ, Wilson DW, Richards JS, Riglar DT, Tetteh KK, Conway DJ, et al. Isolation of viable Plasmodium falciparum merozoites to define erythrocyte invasion events and advance vaccine and drug development. Proc Natl Acad Sci USA. (2010) 107:14378–83. doi: 10.1073/pnas.1009198107
133. Wu X, Gowda NM, Gowda DC. Phagosomal acidification prevents macrophage inflammatory cytokine production to malaria, and dendritic cells are the major source at the early stages of infection: IMPLICATION FOR MALARIA PROTECTIVE IMMUNITY DEVELOPMENT. J Biol Chem. (2015) 290:23135–47. doi: 10.1074/jbc.M115.671065
134. Schwarzer E, Turrini F, Ulliers D, Giribaldi G, Ginsburg H, Arese P. Impairment of macrophage functions after ingestion of Plasmodium falciparum-infected erythrocytes or isolated malarial pigment. J Exp Med. (1992) 176:1033–41. doi: 10.1084/jem.176.4.1033
135. Schwarzer E, Bellomo G, Giribaldi G, Ulliers D, Arese P. Phagocytosis of malarial pigment haemozoin by human monocytes: a confocal microscopy study. Parasitology (2001) 123(Pt 2):125–31. doi: 10.1017/S0031182001008216
136. Skorokhod OA, Alessio M, Mordmuller B, Arese P, Schwarzer E. Hemozoin (malarial pigment) inhibits differentiation and maturation of human monocyte-derived dendritic cells: a peroxisome proliferator-activated receptor-gamma-mediated effect. J Immunol. (2004) 173:4066–74. doi: 10.4049/jimmunol.173.6.4066
137. Schwarzer E, Alessio M, Ulliers D, Arese P. Phagocytosis of the malarial pigment, hemozoin, impairs expression of major histocompatibility complex class II antigen, CD54, and CD11c in human monocytes. Infect Immun. (1998) 66:1601–6.
138. Wykes MN, Good MF. What really happens to dendritic cells during malaria? Nat Rev Microbiol. (2008) 6:864–70. doi: 10.1038/nrmicro1988
139. Millington OR, Di Lorenzo C, Phillips RS, Garside P, Brewer JM. Suppression of adaptive immunity to heterologous antigens during Plasmodium infection through hemozoin-induced failure of dendritic cell function. J Biol. (2006) 5:5. doi: 10.1186/jbiol34
140. Martillo MA, Nazzal L, Crittenden DB. The crystallization of monosodium urate. Curr Rheumatol Rep. (2014) 16:400. doi: 10.1007/s11926-013-0400-9
141. Jin M, Yang F, Yang I, Yin Y, Luo JJ, Wang H, et al. Uric acid, hyperuricemia and vascular diseases. Front Biosci. (2012) 17:656–69. doi: 10.2741/3950
142. El Ridi R, Tallima H. Physiological functions and pathogenic potential of uric acid: a review. J Adv Res. (2017) 8:487–93. doi: 10.1016/j.jare.2017.03.003
143. Gallego-Delgado J, Ty M, Orengo JM, van de Hoef D, Rodriguez A. A surprising role for uric acid: the inflammatory malaria response. Curr Rheumatol Rep. (2014)16:401. doi: 10.1007/s11926-013-0401-8
144. Lopera-Mesa TM, Mita-Mendoza NK, van de Hoef DL, Doumbia S, Konate D, Doumbouya M, et al. Plasma uric acid levels correlate with inflammation and disease severity in Malian children with Plasmodium falciparum malaria. PLoS ONE (2012) 7:e46424. doi: 10.1371/journal.pone.0046424
145. Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu Rev Immunol. (2002) 20:825–52. doi: 10.1146/annurev.immunol.20.103001.114744
146. Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. (2010) 10:387–402. doi: 10.1038/nri2765
147. Lowell CA. Src-family and Syk kinases in activating and inhibitory pathways in innate immune cells: signaling cross talk. Cold Spring Harb Perspect Biol. (2011) 3:a002352. doi: 10.1101/cshperspect.a002352
148. Underhill DM, Gantner B. Integration of Toll-like receptor and phagocytic signaling for tailored immunity. Microbes Infect. (2004) 6:1368–73. doi: 10.1016/j.micinf.2004.08.016
149. Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. (2001) 108:785–91. doi: 10.1172/JCI14006
150. Cabrera A, Neculai D, Kain KC. CD36 and malaria: friends or foes? A decade of data provides some answers. Trends Parasitol. (2014) 30:436–44. doi: 10.1016/j.pt.2014.07.006
151. Silverstein RL. Disabling the platelet's brakes to promote thrombosis. Blood (2015) 125:2591–3. doi: 10.1182/blood-2015-03-630822
152. Dragoni S, Hudson N, Kenny BA, Burgoyne T, McKenzie JA, Gill Y et al. Endothelial MAPKs direct ICAM-1 signaling to divergent inflammatory functions. J Immunol. (2017) 198:4074–85. doi: 10.4049/jimmunol.1600823
153. Mohan Rao LV, Esmon CT, Pendurthi UR. Endothelial cell protein C receptor: a multi-liganded and multi-functional receptor. Blood (2014) 124:1553–62. doi: 10.1182/blood-2014-05-578328
154. Pendurthi UR, Rao LVM. Endothelial cell protein C receptor-dependent signaling. Curr Opin Hematol. (2018) 25:219–26. doi: 10.1097/MOH.0000000000000416
155. van der Poll T. The endothelial protein C receptor and malaria. Blood (2013) 122:624–5. doi: 10.1182/blood-2013-06-508531
156. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. (2013) 14:812–20. doi: 10.1038/ni.2639
157. Pasloske BL, Howard RJ. Malaria, the red cell, and the endothelium. Annu Rev Med. (1994) 45:283–95. doi: 10.1146/annurev.med.45.1.283
158. Day KP, Hayward RE, Smith D, Culvenor JG. CD36-dependent adhesion and knob expression of the transmission stages of Plasmodium falciparum is stage specific. Mol Biochem Parasitol. (1998) 93:167–77. doi: 10.1016/S0166-6851(98)00040-1
159. Weatherall DJ, Miller LH, Baruch DI, Marsh K, Doumbo OK, Casals-Pascual C, et al. Malaria and the red cell. Hematol Am Soc Hematol Educ Program (2002) 2002:35–57. doi: 10.1182/asheducation-2002.1.35
160. Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, et al. CD36 is a sensor of diacylglycerides. Nature (2005) 433:523–7. doi: 10.1038/nature03253
161. Patel SN, Lu Z, Ayi K, Serghides L, Gowda DC, Kain KC. Disruption of CD36 impairs cytokine response to Plasmodium falciparum glycosylphosphatidylinositol and confers susceptibility to severe and fatal malaria in vivo. J Immunol. (2007) 178:3954–61. doi: 10.4049/jimmunol.178.6.3954
162. Urban BC, Willcox N, Roberts DJ. A role for CD36 in the regulation of dendritic cell function. Proc Natl Acad Sci USA. (2001) 98:8750–5. doi: 10.1073/pnas.151028698
163. Gowda NM, Wu X, Kumar S, Febbraio M, Gowda DC. CD36 contributes to malaria parasite-induced pro-inflammatory cytokine production and NK and T cell activation by dendritic cells. PLoS ONE (2013) 8:e77604. doi: 10.1371/journal.pone.0077604
164. Pain A, Urban BC, Kai O, Casals-Pascual C, Shafi J, Marsh K, et al. A non-sense mutation in Cd36 gene is associated with protection from severe malaria. Lancet (2001) 357:1502–3. doi: 10.1016/S0140-6736(00)04662-6
165. Omi K, Ohashi J, Patarapotikul J, Hananantachai H, Naka I, Looareesuwan S, et al. CD36 polymorphism is associated with protection from cerebral malaria. Am J Hum Genet. (2003) 72:364–74. doi: 10.1086/346091
166. Van de Wouwer MV, Collen D, Conway EM. Thrombomodulin-protein C-EPCR system: integrated to regulate coagulation and inflammation. Arterioscler Thromb Vasc Biol. (2004) 24:1374–83. doi: 10.1161/01.ATV.0000134298.25489.92
167. Turner L, Lavstsen T, Berger SS, Wang CW, Petersen JE, Avril M, et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature (2013) 498:502–5. doi: 10.1038/nature12216
168. Avril M, Bernabeu M, Benjamin M, Brazier AJ, Smith JD. Interaction between endothelial protein C receptor and intercellular adhesion molecule 1 to mediate binding of Plasmodium falciparum-infected erythrocytes to endothelial cells. MBio (2016) 7:e00615–16. doi: 10.1128/mBio.00615-16
169. Tuikue Ndam N, Moussiliou A, Lavstsen T, Kamaliddin C, Jensen AT, Mama A, et al. Parasites causing cerebral falciparum malaria bind multiple endothelial receptors and express EPCR and ICAM-1-binding PfEMP1. J Infect Dis. (2017) 215:1918–25. doi: 10.1093/infdis/jix230
170. Favre N, Da Laperousaz C, Ryffel B, Weiss NA, Imhof BA, Rudin W, et al. Role of ICAM-1 (CD54) in the development of murine cerebral malaria. Microbes Infect. (1999) 1:961–8. doi: 10.1016/S1286-4579(99)80513-9
171. Ramos TN, Bullard DC, Darley MM, McDonald K, Crawford DF, Barnum SR. Experimental cerebral malaria develops independently of endothelial expression of intercellular adhesion molecule-1 (icam-1). J Biol Chem. (2013) 288:10962–6. doi: 10.1074/jbc.C113.457028
172. Baratin M, Roetynck S, Pouvelle B, Lemmers C, Viebig NK, Johansson S, et al. Dissection of the role of PfEMP1 and ICAM-1 in the sensing of Plasmodium falciparum-infected erythrocytes by natural killer cells. PLoS ONE (2007) 2:e228. doi: 10.1371/journal.pone.0000228
173. Gupta P, Lai SM, Sheng J, Tetlak P, Balachander A, Claser C, et al. Tissue-resident CD169+ macrophages form a crucial front line against Plasmodium infection. Cell Rep. (2016) 16:1749–61. doi: 10.1016/j.celrep.2016.07.010
174. Baccarella A, Huang BW, Fontana MF, Kim CC. Loss of Toll-like receptor 7 alters cytokine production and protects against experimental cerebral malaria. Malar J. (2014) 13:354. doi: 10.1186/1475-2875-13-354
175. Haque A, Best SE, Montes de Oca M, James KR, Ammerdorffer A, Edwards CL, et al. Type I IFN signaling in CD8− DCs impairs Th1-dependent malaria immunity. J Clin Invest. (2014) 124:2483–96. doi: 10.1172/JCI70698
176. Haque A, Best SE, Ammerdorffer A, Desbarrieres L, de Oca MM, Amante FH, et al. Type I interferons suppress CD4+ T-cell-dependent parasite control during blood-stage Plasmodium infection. Eur J Immunol. (2011) 41:2688–98. doi: 10.1002/eji.201141539
177. Vigario AM, Belnoue E, Gruner AC, Mauduit M, Kayibanda M, Deschemin JC, et al. Recombinant human IFN-α inhibits cerebral malaria and reduces parasite burden in mice. J Immunol. (2007) 178:6416–25. doi: 10.4049/jimmunol.178.10.6416
178. Morrell CN, Srivastava K, Swaim A, Lee MT, Chen J, Nagineni C, et al. Beta interferon suppresses the development of experimental cerebral malaria. Infect Immun. (2011) 79:1750–8. doi: 10.1128/IAI.00810-10
179. Palomo J, Fauconnier M, Coquard L, Gilles M, Meme S, Szeremeta F, et al. Type I interferons contribute to experimental cerebral malaria development in response to sporozoite or blood-stage Plasmodium berghei ANKA. Eur J Immunol. (2013) 43:2683–95. doi: 10.1002/eji.201343327
180. Sebina I, Haque A. Effects of type I interferons in malaria. Immunology (2018). 155:176–85. doi: 10.1111/imm.12971
181. Trinchieri G. Type I interferon: friend or foe? J Exp Med. (2010) 207:2053–63. doi: 10.1084/jem.20101664
182. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
183. Moreira-Teixeira L, Mayer-Barber K, Sher A, O'Garra A. Type I interferons in tuberculosis: foe and occasionally friend. J Exp Med. (2018) 215:1273–85. doi: 10.1084/jem.20180325
184. Stevenson MM, Urban BC. Antigen presentation and dendritic cell biology in malaria. Parasite Immunol. (2006) 28:5–14. doi: 10.1111/j.1365-3024.2006.00772.x
185. Todryk SM, Urban BC. Dendritic cells in Plasmodium infection. Future Microbiol. (2008) 3:279–86. doi: 10.2217/17460913.3.3.279
186. Amorim KN, Chagas DC, Sulczewski FB, Boscardin SB. Dendritic cells and their multiple roles during malaria infection. J Immunol Res. (2016) 2016:2926436. doi: 10.1155/2016/2926436
187. Walsh KP, Mills KH. Dendritic cells and other innate determinants of T helper cell polarisation. Trends Immunol. (2013) 34:521–30. doi: 10.1016/j.it.2013.07.006
188. King T, Lamb T. Interferon-γ: the Jekyll and Hyde of malaria. PLoS Pathog. (2015) 11:e1005118. doi: 10.1371/journal.ppat.1005118
189. Ing R, Stevenson MM. Dendritic cell and NK cell reciprocal cross talk promotes gamma interferon-dependent immunity to blood-stage Plasmodium chabaudi AS infection in mice. Infect Immun. (2009) 77:770–82. doi: 10.1128/IAI.00994-08
190. D'ombrain MC, Robinson LJ, Stanisic DI, Taraika J, Bernard N, Michon P, et al. Association of early interferon-γ production with immunity to clinical malaria: a longitudinal study among Papua New Guinean children. Clin Infect Dis. (2008) 47:1380–7. doi: 10.1086/592971
191. McGovern KE, Wilson EH. Role of chemokines and trafficking of immune cells in parasitic infections. Curr Immunol Rev. (2013) 9:157–68. doi: 10.2174/1573395509666131217000000
192. Rocha BC, Marques PE, Leoratti FMS, Junqueira C, Pereira DB, Antonelli L, et al. Type I interferon transcriptional signature in neutrophils and low-density granulocytes are associated with tissue damage in malaria. Cell Rep. (2015) 13:2829–41. doi: 10.1016/j.celrep.2015.11.055
193. Ioannidis LJ, Nie CQ, Hansen DS. The role of chemokines in severe malaria: more than meets the eye. Parasitology (2014) 141:602–13. doi: 10.1017/S0031182013001984
194. Sponaas AM, Cadman ET, Voisine C, Harrison V, Boonstra A, O'Garra A, et al. Malaria infection changes the ability of splenic dendritic cell populations to stimulate antigen-specific T cells. J Exp Med. (2006) 203:1427–33. doi: 10.1084/jem.20052450
195. Gowda NM, Wu X, Gowda DC. TLR9 and MyD88 are crucial for the development of protective immunity to malaria. J Immunol. (2012) 188:5073–85. doi: 10.4049/jimmunol.1102143
196. Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, et al. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J Exp Med. (2005) 201:19–25. doi: 10.1084/jem.20041836
197. Coban C, Ishii KJ, Uematsu S, Arisue N, Sato S, Yamamoto M, et al. Pathological role of Toll-like receptor signaling in cerebral malaria. Int Immunol. (2007) 19:67–79. doi: 10.1093/intimm/dxl123
198. Franklin BS, Ishizaka ST, Lamphier M, Gusovsky F, Hansen H, Rose J, et al. Therapeutical targeting of nucleic acid-sensing Toll-like receptors prevents experimental cerebral malaria. Proc Natl Acad Sci USA. (2011) 108:3689–94. doi: 10.1073/pnas.1015406108
199. Zhang Y, Zhu X, Feng Y, Pang W, Qi Z, Cui L, et al. TLR4 and TLR9 signals stimulate protective immunity against blood-stage Plasmodium yoelii infection in mice. Exp Parasitol. (2016) 170:73–81. doi: 10.1016/j.exppara.2016.09.003
200. Griffith JW, O'Connor C, Bernard K, Town T, Goldstein DR, Bucala R. Toll-like receptor modulation of murine cerebral malaria is dependent on the genetic background of the host. J Infect Dis. (2007) 196:1553–64. doi: 10.1086/522865
201. Barboza R, Lima FA, Reis AS, Murillo OJ, Peixoto EPM, Bandeira CL, et al. TLR4-mediated placental pathology and pregnancy outcome in experimental malaria. Sci Rep. (2017) 7:8623. doi: 10.1038/s41598-017-08299-x
202. Mockenhaupt FP, Cramer JP, Hamann L, Stegemann MS, Eckert J, Oh NR, et al. Toll-like receptor (TLR) polymorphisms in African children: common TLR-4 variants predispose to severe malaria. Proc Natl Acad Sci USA. (2006) 103:177–82. doi: 10.1073/pnas.0506803102
203. Esposito S, Molteni CG, Zampiero A, Baggi E, Lavizzari A, Semino M, et al. Role of polymorphisms of toll-like receptor (TLR) 4, TLR9, toll-interleukin 1 receptor domain containing adaptor protein (TIRAP) and FCGR2A genes in malaria susceptibility and severity in Burundian children. Malar J. (2012) 11:196. doi: 10.1186/1475-2875-11-196
204. Mockenhaupt FP, Hamann L, von Gaertner C, Bedu-Addo G, von Kleinsorgen C, Schumann RR, et al. Common polymorphisms of toll-like receptors 4 and 9 are associated with the clinical manifestation of malaria during pregnancy. J Infect Dis. (2006) 194:184–8. doi: 10.1086/505152
205. Leoratti FM, Farias L, Alves FP, Suarez-Mutis MC, Coura JR, Kalil J, et al. Variants in the toll-like receptor signaling pathway and clinical outcomes of malaria. J Infect Dis. (2008) 198:772–80. doi: 10.1086/590440
206. Khor CC, Chapman SJ, Vannberg FO, Dunne A, Murphy C, Ling EY, et al. A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat Genet. (2007) 39:523–8. doi: 10.1038/ng1976
207. Crompton PD, Mircetic M, Weiss G, Baughman A, Huang CY, Topham DJ, et al. The TLR9 ligand CpG promotes the acquisition of Plasmodium falciparum-specific memory B cells in malaria-naive individuals. J Immunol. (2009) 82:3318–26. doi: 10.4049/jimmunol.0803596
208. Munde EO, Okeyo WA, Anyona SB, Raballah E, Konah S, Okumu W, et al. Polymorphisms in Fc gamma receptor (FcγRIIIA-176 F/V) and Toll-like receptor (TLR9 [-1237 T/C]) are associated with protection against severe malarial anemia and changes in circulating IFN-γ. Infect Immun. (2012) 80:4435–43. doi: 10.1128/IAI.00945-12
209. Sawian CE, Lourembam SD, Banerjee A, Baruah S. Polymorphisms and expression of TLR4 and 9 in malaria in two ethnic groups of Assam, northeast India. Innate Immun. (2013) 19:174–83. doi: 10.1177/1753425912455675
210. Greene JA, Moormann AM, Vulule J, Bockarie MJ, Zimmerman PA, Kazura JW. Toll-like receptor polymorphisms in malaria-endemic populations. Malar J. (2009) 8:50. doi: 10.1186/1475-2875-8-50
211. Greene JA, Sam-Agudu N, John CC, Opoka RO, Zimmerman PA, Kazura JW. Toll-like receptor polymorphisms and cerebral malaria: TLR2 Δ22 polymorphism is associated with protection from cerebral malaria in a case control study. Malar J. (2012) 11:47. doi: 10.1186/1475-2875-11-47
212. Omar AH, Yasunami M, Yamazaki A, Shibata H, Ofori MF, Akanmori BD, et al. Toll-like receptor 9 (TLR9) polymorphism associated with symptomatic malaria: a cohort study. Malar J. (2012) 11:168. doi: 10.1186/1475-2875-11-168
213. Sam-Agudu NA, Greene JA, Opoka RO, Kazura JW, Boivin MJ, Zimmerman PA, et al. TLR9 polymorphisms are associated with altered IFN-γ levels in children with cerebral malaria. Am J Trop Med Hyg. (2010) 82:548–55. doi: 10.4269/ajtmh.2010.09-0467
214. Manning L, Cutts J, Stanisic DI, Laman M, Carmagnac A, Allen S, et al. A Toll-like receptor-1 variant and its characteristic cellular phenotype is associated with severe malaria in Papua New Guinean children. Genes Immun. (2016) 17:52–9. doi: 10.1038/gene.2015.50
Keywords: malaria, immunostimulatory factors, host receptors, signaling mechanisms, innate immune responses, protective immunity, pathogenesis
Citation: Gowda DC and Wu X (2018) Parasite Recognition and Signaling Mechanisms in Innate Immune Responses to Malaria. Front. Immunol. 9:3006. doi: 10.3389/fimmu.2018.03006
Received: 30 August 2018; Accepted: 05 December 2018;
Published: 19 December 2018.
Edited by:
Ashraful Haque, QIMR Berghofer Medical Research Institute, AustraliaReviewed by:
Dolores Correa, Instituto Nacional de Pediatria, MexicoKylie Renee James, Wellcome Trust Sanger Institute (WT), United Kingdom
Copyright © 2018 Gowda and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: D. Channe Gowda, gowda@psu.edu