 C. I. Edvard Smith
C. I. Edvard Smith Jan A. Burger
Jan A. Burger- 1Department of Laboratory Medicine, Karolinska Institutet (KI), Huddinge, Sweden
- 2Department of Leukemia, University of Texas MD Anderson Cancer Center, Houston, TX, United States
Since the first clinical report in 2013, inhibitors of the intracellular kinase BTK (BTKi) have profoundly altered the treatment paradigm of B cell malignancies, replacing chemotherapy with targeted agents in patients with chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), and Waldenström’s macroglobulinemia. There are over 20 BTKi, both irreversible and reversible, in clinical development. While loss-of-function (LoF) mutations in the BTK gene cause the immunodeficiency X-linked agammaglobulinemia, neither inherited, nor somatic BTK driver mutations are known. Instead, BTKi-sensitive malignancies are addicted to BTK. BTK is activated by upstream surface receptors, especially the B cell receptor (BCR) but also by chemokine receptors, and adhesion molecules regulating B cell homing. Consequently, BTKi therapy abrogates BCR-driven proliferation and the tissue homing capacity of the malignant cells, which are being redistributed into peripheral blood. BTKi resistance can develop over time, especially in MCL and high-risk CLL patients. Frequently, resistance mutations affect the BTKi binding-site, cysteine 481, thereby reducing drug binding. Less common are gain-of-function (GoF) mutations in downstream signaling components, including phospholipase Cγ2 (PLCγ2). In a subset of patients, mechanisms outside of the BCR pathway, related e.g. to resistance to apoptosis were described. BCR signaling depends on many proteins including SYK, BTK, PI3K; still based on the resistance pattern, BTKi therapy only selects GoF alterations in the NF-κB arm, whereas an inhibitor of the p110δ subunit of PI3K instead selects resistance mutations in the RAS-MAP kinase pathway. BTK and PLCγ2 resistance mutations highlight BTK’s non-redundant role in BCR-mediated NF-κB activation. Of note, mutations affecting BTK tend to generate clone sizes larger than alterations in PLCγ2. This infers that BTK signaling may go beyond the PLCγ2-regulated NF-κB and NFAT arms. Collectively, when comparing the primary and acquired mutation spectrum in BTKi-sensitive malignancies with the phenotype of the corresponding germline alterations, we find that certain observations do not readily fit with the existing models of BCR signaling.
Introduction
The sequence of the BTK gene was reported in 1993 by two independent research teams, as a result of a hunt for the genetic cause of X-linked agammaglobulinemia, XLA (1, 2), and a search for novel tyrosine kinases (3). Given that patients with XLA are essentially devoid of mature B lymphocytes it was apparent already prior to the gene cloning that if it were possible to inhibit the product of the gene mutated in XLA, such drugs could become highly useful for targeting B cells.
However, it was not until 2007 that a compound was selected for clinical development, the first-in-class BTKi ibrutinib (4–6). Patients with diverse B cell malignancies were enrolled in the initial Phase 1 ibrutinib trial, which reported about particularly high response rates in patients with CLL and MCL (7), and from then on, the development of BTKi has been no less than astounding.
Focusing on CLL, the first ibrutinib trial demonstrated high response rates (8), and very durable effects in treatment naïve patients (7-year progression-free survival 83%), while relapsed/refractory patients had shorter remissions (7-year progression-free survival 34%) (9). Subsequently, a series of randomized clinical trials demonstrated major improvement in progression-free survival (PFS) in CLL patients (untreated or previously treated) receiving ibrutinib (alone or with CD20 antibodies), as compared with previous standard therapies, most of them chemotherapy-based (10–13). Based on these data, ibrutinib and other BTK inhibitors have become the preferred CLL treatment to date. A similar development has taken place in Waldenström’s macroglobulinemia, WM (14).
At least 22 BTKi are in clinical development (15) and sales have already placed BTKi among the most commonly sold cancer drugs worldwide (16).
While the first approved drugs, ibrutinib, acalabrutinib and zanubrutinib, bind irreversibly to a cysteine in the catalytic domain, there are now several compounds, which tether reversibly. Many of these are investigated as treatment of autoimmune and inflammatory disorders (15). They also are explored as a treatment for patients with resistance mutations to irreversible inhibitors (17, 18). The concept of irreversible kinase inhibitors was relatively novel around the time when BTKi were first generated (19). A covalent bond is formed between the BTKi, at the electrophilic site, and its target where the cysteine 481 serves as the corresponding nucleophile. Since only few kinases carry a corresponding cysteine residue this likely limits off-target side effects (15, 20–22). A more detailed description of the interaction between various BTKi and the catalytic domain of BTK is found in an accompanying paper (23).
B Cell Malignancies Responding to BTKi Depend on BCR Signaling
To date, the responsiveness to BTKi has been studied in all major B cell malignancies and the emerging pattern is that sensitive malignancies are dependent on BCR signaling for their growth and survival. For example, in CLL, the importance of the BCR pathway was already emphasized prior to the clinical use of BTKi (24), whereas in mantle cell lymphoma (MCL) and Waldenström’s macroglobulinemia (WM) the dependence of the malignant B cells on BCR and/or BTK was mostly discovered later (25, 26).
In CLL, it was reported as early as in 1972 (27) that the specificity of the monoclonal surface immunoglobulins (IG) was not random. In the 1990s, it was noted that CLL cells express a restricted repertoire of IG heavy variable (IGHV) genes (28–30). During that time, it became obvious that the IG gene repertoire of CLL is non-random and, moreover, that a major fraction of CLL patients carried IGHV somatic hypermutations (SHM) (31). Interestingly, the SHM status of the IGHV genes is a reliable predictor for patient survival, as reported in two seminal publications by Stevenson, Chiorazzi, and colleagues (32, 33). Patients with mutated IGHV genes (‘IG-mutated’ CLL, M-CLL; 50-60% of cases) generally have more indolent disease, whereas those with unmutated IGHV genes (‘IG-unmutated’ CLL, U-CLL; 30-40% of cases) typically have more rapidly progressive disease and respond poorly to chemotherapy-based treatment (32, 33). Importantly, BTKi therapy overcomes the negative prognostic impact of U-CLL, based on longer follow-up of M-CLL and U-CLL patients treated with ibrutinib (34). Moreover, stereotyped BCR, defined as the existence of (quasi)identical BCR in different CLL patients has been another discovery highlighting the importance of the BCR and common antigens in CLL pathogenesis (35). Over the last years it was established that a large fraction of CLL, at least 41%, expressed stereotyped BCR, which is remarkable considering the extremely low probability of randomly finding identical VH CDR3 sequences in different people (36, 37). Also in MCL there is a bias in the VH usage (38) demonstrating the involvement of the BCR in these lymphomas.
As described in the section “Recurrent Primary Mutations in B Cell Malignancies Responding to BTKi”, BCR-mediated signaling differs in certain other BTKi-sensitive malignancies, such as WM. Here, the formation of a constitutively active intracellular signaling hub is the driving force rather than stereotypic IG at the membrane.
Not All B-Cell Malignancies Respond to BTKi
In the activated B-cell-like (ABC) subtype of diffuse large B-cell lymphoma (DLBCL) an RNA interference genetic screen revealed that BTK is essential for the survival of lymphoma cells (39) with wild-type Caspase recruitment domain family, member 11 (CARD11). Thus, while DLBCL tumors belonging to the group of germinal center B cell-like (GCB) are unresponsive to BTKi, the ABC subtype can be sensitive (40), although only subsets respond, and responses generally are short. Similarly, only few patients with follicular lymphoma responded transiently (7). Furthermore, when CLL is affected by Richter’s transformation additional genetic changes cause the tumor to become unresponsive to BTKi (41, 42). Thus, unless the tumor is dependent on BCR signaling sensitivity to BTKi is not expected.
The BCR Signaling Pathway and the Critical Role of NF-κB
In classical experiments in which the BCR was genetically disabled in adult mice it was demonstrated that B-cells lacking this receptor could not survive (43), and this contributed to the idea of ‘tonic signaling’. Later this concept was also recapitulated for distinct signaling components of the BCR (44). Particularly in B cells, NF-κB serves as a survival factor and tonic signaling achieves sufficient NF-κB activity to prevent apoptotic events from occurring (45). This was recently corroborated at the single-cell level, by combining transcriptome profiling and chromatin mapping in BTKi-treated CLL patients. A sharp decrease of NF-κB binding was found, followed by the acquisition of a quiescence-like gene signature (46). Subsequently there was reduced chromatin accessibility at binding sites of the transcription factors regulated by NF-κB, such as PU.1. This is of interest, since mutation of the PU.1 binding site in the BTK promoter causes XLA (47–49), and, furthermore, because BTK uses a positive autoregulatory feedback mechanism to stimulate transcription from its own promoter via NF-κB (50).
Resistance Mutations Predominantly Affect BTK
In 2014, the first BTKi resistance mutations were reported. They were identified in CLL patients receiving ibrutinib treatment (51, 52). They fell into two categories: [1] point mutations that disable covalent binding of ibrutinib to BTK, by replacing the ibrutinib-target, cysteine 481 (C481) in the kinase domain (Figures 1 and 2), with a different amino acid, and [2] mutations causing constitutive activation of PLCγ2, the downstream substrate of BTK (variations altering PLCγ2 are described in detail in the next section). BTK mutations mainly originate from single base changes yielding substitution not only to serine, but also to arginine, phenylalanine, tryptophan or tyrosine (53). Predictions suggested that glycine substitutions also may occur (54), and indeed, later, glycine substitution of C481 was found in patients (55).
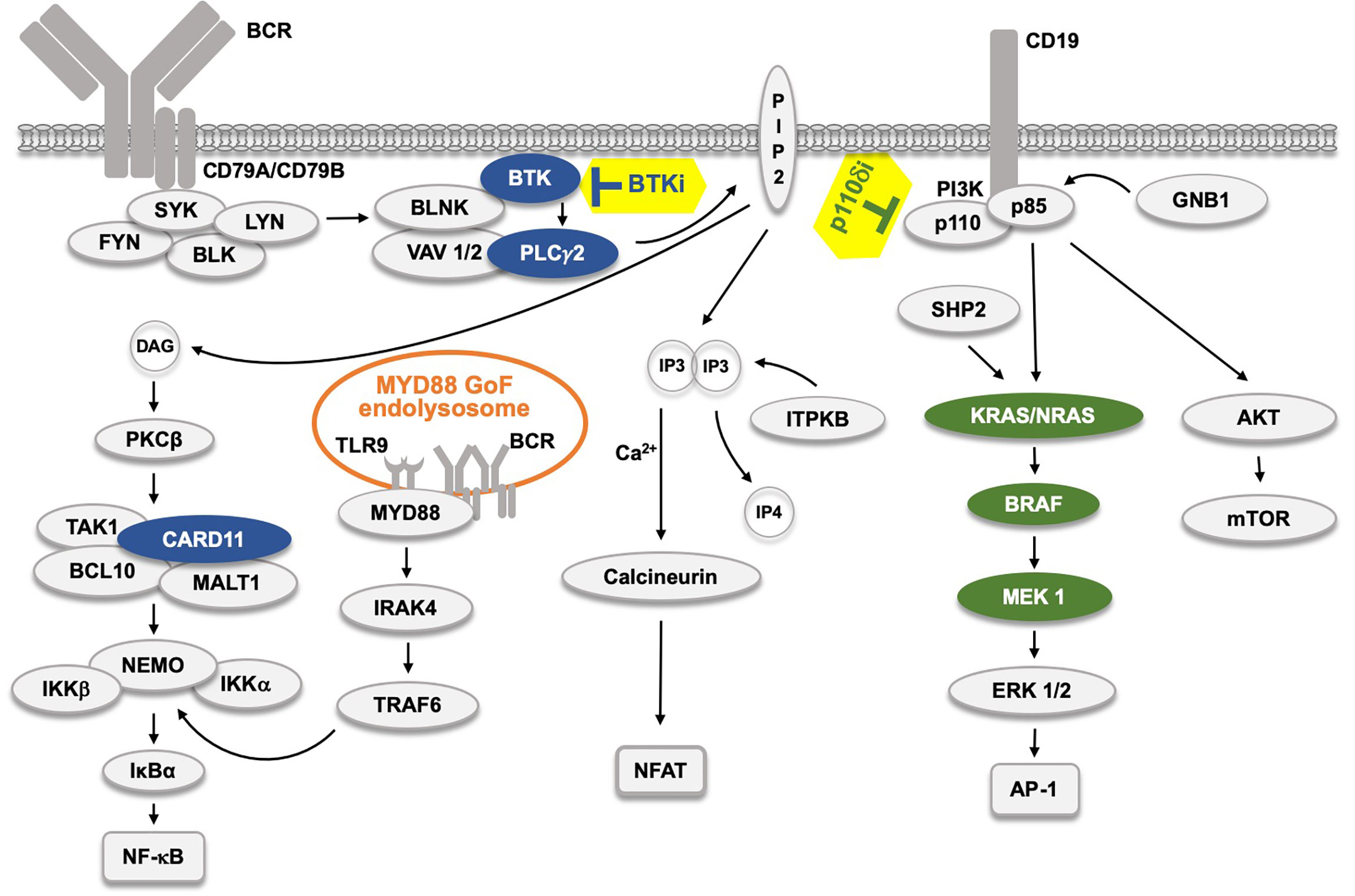
Figure 1 The B cell receptor (BCR) signaling pathway and the effect of kinase inhibitors. The BCR pathway trifurcates into, from left, NF-κB, NFAT and AP-1 signaling. The AP-1 pathway is believed to be connected to the BCR through LYN, SYK and ZAP70 (not depicted). Filled protein symbols correspond to Gain-of-Function (GoF) drug resistance mutations. BTK inhibitors (BTKi) and p110δ inhibitors (p110δi) are marked with yellow background. The MYD88 L265P GoF mutation found in 95% of WM patients, and also in ABC-type DLBCL, and in some CLL patients induces an activated endolysosome containing TLR9 and the BCR.  proteins affected by BTKi,
proteins affected by BTKi,  proteins affected by p110δi.
proteins affected by p110δi.
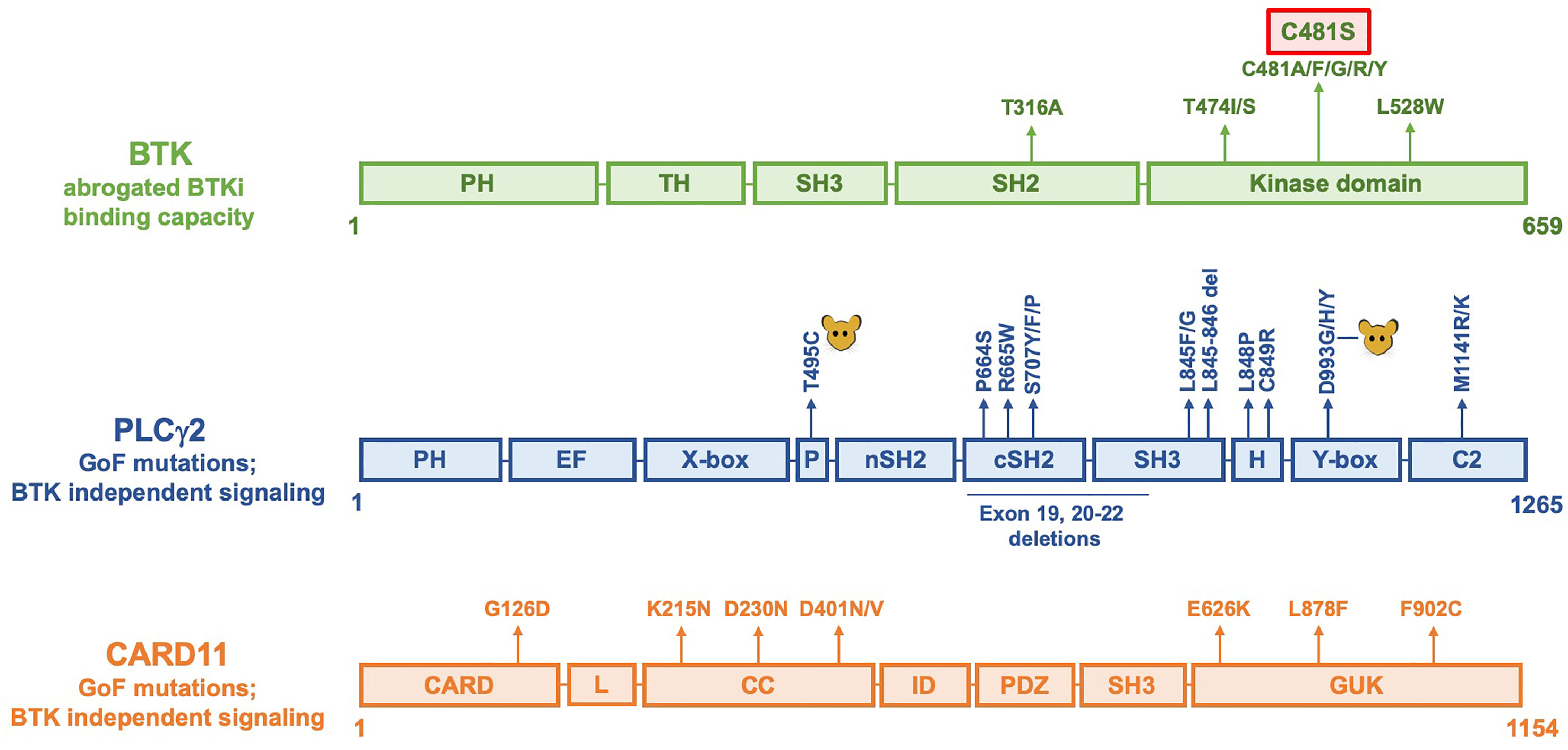
Figure 2 Linear representation of BTK, PLCγ2 and CARD11 molecules showing amino acid replacements causing resistance to BTKi. The C481S replacement is the most common alteration and is marked with a red box. In PLCγ2, alterations induced by mutagenesis in mice are indicated as  . Not all reported alterations are listed, only recurrent mutations have been included.
. Not all reported alterations are listed, only recurrent mutations have been included.
The C481S, and also the experimentally generated, C481T, threonine substitution were shown to maintain kinase activity (51, 52, 54), whereas the other substitutions are only weakly active (glycine) or kinase-dead (arginine, phenylalanine, tryptophan, tyrosine) (54). To this end, knock-in mice carrying serine 481 in their germline have a phenotype essentially identical to wildtype animals (56). Thus, this demonstrates a normal function of BTK harboring a serine at the 481 position.
The observation that in vivo-selection of kinase-dead mutants occurs, may be unexpected; the underlying mechanism has been discussed elsewhere (54). The C481 mutants are not exclusively seen during ibrutinib treatment, they are also found in patients treated with other irreversible BTKi that target C481, namely acalabrutinib (57) and zanubrutinib (58). Furthermore, resistance mutations not only appear in CLL, but also in MCL (59), marginal zone leukemia (MZL) (60) and Waldenström’s macroglobulinemia (14, 61).
A second site with amino acid substitutions affects the gatekeeper residue, threonine 474 (Figure 2). These replacements are more rarely seen during BTKi therapy, with only two exchanges reported to date, namely to isoleucine or to serine (53). However, the gatekeeper residue is the predominantly substituted amino acid in patients developing resistance against most other kinase-inhibitors, such as those directed against ABL in chronic myeloid leukemia and against EGFR in lung cancer [reviewed in (62)].
In a recent study, mutations affecting T474 and those yielding the C481S replacement were combined (18). Surprisingly, certain double mutants demonstrated super-resistance tolerating more than 16-fold the therapeutic BTKi concentration. Even more unexpected was the fact that C481S combined with T474A restored sensitivity to ibrutinib.
As depicted in Figure 2, rare recurrent replacements have also been reported affecting the leucine 528 in the catalytic domain of BTK (53, 58, 63) as well as threonine 316 in the SH2 domain (64, 65).
Furthermore, apart from the BCR-signaling components which are affected, there are additional resistance mutations. Those located to the short arm of chromosome 8 have been suggested to impair apoptosis by causing haploinsufficiency for the TRAIL receptor (61, 66), whereas other mutations affect additional genes (61, 67). Del(8p) was found in a sizable number of ibrutinib-resistant CLL cases, resulting in deletion and haploinsufficiency of tumor necrosis factor-related apoptosis-inducing ligand-receptor (TRAIL-R) (66, 68). This was associated with a decrease in the TRAIL-R mRNA and protein levels which corresponded to the size of the del(8p)-harboring clone. Interestingly, a potential indirect mechanism that would link TRAIL resistance to positive selection by ibrutinib therapy is suggested by the fact that TRAIL concentrations are higher in circulating blood as compared with the lymph node environment. Ibrutinib therapy is known to mobilize CLL cells from the lymph nodes into to the peripheral blood. Hence, haploinsufficiency of TRAIL-R could provide a survival advantage for CLL cells by making such CLL cells less sensitivity to cell death in the periphery, once released from the lymph node and exposed to higher levels of TRAIL.
BTK’s best known role in the BCR signaling pathway is to phosphorylate tyrosine residues in PLCγ2 (69, 70). GoF mutations affecting PLCγ2 therefore obviate the need for posttranscriptional phosphorylation, and, hence, BTK becomes redundant when malignant B cells acquire this mode of resistance to BTKi.
PLCγ2 Resistance Mutations for BTKi
Two different amino acid substitutions, which caused PLCγ2 BTKi resistance mutations, arginine 665 to tryptophan and leucine 845 to phenylalanine, were presented in the first report on BTKi resistance (51). They were classified as GoF (51, 71), and, in line with this, activating germline mutations in the PLCG2 gene located in chromosome 16q23.3, with dominant inheritance, were published already two years earlier (72). Not all PLCG2 mutations found causing BTKi resistance have yet been reported to also induce PLCγ2-associated antibody deficiency with immune dysregulation (APLAID), or without autoinflammation (PLAID), but many do. The most recurrent mutations are depicted in Figure 2. Even prior to the identification of constitutively active PLCγ2 in humans in 2012, GoF mutations affecting PLCγ2 were observed in mice following N-ethyl-N-nitrosourea mutagenesis (73). They occur in the same domains, which are affected by both BTKi resistance and germline PLCG2 mutations in humans. The first reported alteration in mice caused an aspartate 993 to glycine replacement in the C-terminal part of the catalytic domain, known as the Y-box (73). Later a mutation was identified in the split pleckstrin homology (PH) domain yielding a tyrosine 495 to cysteine substitution (74). Both mutant strains develop severe spontaneous inflammation and autoimmunity (73–75).
PLCγ2 is a large molecule with a M.W. of 147,870 Da, i.e. more than twice the size of BTK. It consists of 10 domains as depicted in Figure 2. The activation state of the mutated PLCγ2 has been suggested to be a form of pre-activation, rather than constitutive activation (76, 77). However, constitutive activation does not necessarily mean full activation, and, hypothetically, if the constitutive activation is too strong, the affected cell, or its host, may not survive long-term.
CARD11 (CARMA1) Resistance Mutations for BTKi
CARD11, also known as caspase recruitment domain-containing membrane-associated guanylate kinase protein-1 (CARMA1), is an adaptor protein, which forms an integral part of BCR signaling (Figures 1 and 2). CARD11 is autoinhibited in its natural state, but upon activation the latch domain is released, and the protein forms a complex with B-cell lymphoma/leukemia 10 (BCL-10) and mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1), generating the CBM scaffold, which can oligomerize (78). The activation is initiated by PKCβ, which phosphorylates a series of serine sites in the CARD11 inhibitory domain, the initial modification necessary for the assembly of the CBM complex (79, 80). The serine phosphorylation alters CARD 11 into an open conformation accessible for BCL10-MALT1 binding.
CARD11 mutations in tumors were first reported in DLBCL in 2008 and transfer of the identified CARD11 coiled-coil domain mutants into lymphoma cell lines resulted in constitutive NF-κB activation (81). As depicted in Figure 2, the leucine 878 to phenylalanine resistance mutation to BTKi has been found in CLL, MCL as well as in WM and several more in MCL (82, 83). CARD11 is located downstream of BTK in the BCR pathway, but upstream of NF-κB, whose signaling is crucial for B-cell survival. In a set of follicular lymphomas treated with ibrutinib, CARD11 mutations were significantly associated with resistance (84).
Inherited mutations in CARD11 are associated with different diseases. Thus, heterozygous dominant-negative variants of CARD11 result in combined immunodeficiency with, or without atopy, high IgE levels and other autoimmune features [reviewed in (85, 86)]. Germline GoF CARD11 mutations instead cause B cell expansion with NF-κB and T cell anergy (BENTA), accompanied by elevated NF-κB activity and hyperproliferative B cells (87). Very recently, more than two thousand CARD11 variants were generated by multiplexed, CRISPR-mediated, saturation genome editing, functionally scored and classified (88). A single LoF mutation affecting BCL-10 has been identified, whereas none has been reported in MALT1, and GoF variants were not published for any of them (89).
Collectively, this clearly demonstrates that CARD11 is a complex molecule with a multitude of activities. Importantly, together with PLCγ2 it is the only protein in which GoF mutations have been repeatedly found to cause resistance to BTKi.
Lack of BTKi Resistance Mutations in the PI3K-RAS and Calcineurin Arms of the BCR Pathway
The B cell malignancies that respond well to BTKi, especially CLL, are particularly dependent on BCR pathway activation for growth and survival (24). This goes beyond tonic BCR signaling that all B cells need for survival, and is induced, presumably by activating conformational BCR properties or tissue auto-antigens in the supporting microenvironment of secondary lymphatic organs (i.e. spleen and lymph nodes). Activating mutations upstream of BTK will be inhibited by BTKi, whereas downstream GoF alterations would be insensitive. Based on this, it could be hypothesized that any component downstream of BTK in this signaling pathway could induce unresponsiveness to BTKi when constitutively active. Of note, the signaling downstream of PLCγ2 diverges into diacylglycerol (DAG)- and inositol 1,4,5-trisphosphate (IP3)-dependent pathways and it is therefore conceivable that GoF mutations would need to affect both of these arms. However, this is not the case in B-cell malignancies, since to date resistance mutations have only been reported in the DAG pathway (Figure 1).
Hence, after the initial phase, in which PLCγ2 generates diacyl glycerol (DAG) and inositol 3-phosphate (IP3), BCR signaling bifurcates into the DAG and IP3 pathways, and subsequently trifurcates since DAG activates RAS (not depicted) and NF-κB (Figure 1). The BCR connection to RAS is likely mainly through CD19 and PI3K. Hence, LYN, SYK and ZAP-70 have been shown to phosphorylate the YXXM motif in CD19, to which PI3K can tether (90–92). Moreover, generation of phosphatidylinositol (3–5)-trisphosphate (PIP3) by PI3K is essential for the activity of and membrane tethering of BTK (93–95). While resistance mechanisms affecting the PI3K-AKT-FOXO3a axis, without involving genetic alterations, may occur (96–98), including transcriptional reprogramming in MCL (99), it is still highly unexpected that GoF substitutions affecting the corresponding signal transducers would be absent.
Interestingly, GoF resistance mutations to BTKi are confined to the protein kinase Cβ (PKCβ) arm, although variations in PKCβ itself do not seem to cause resistance. Concerning the RAS-ERK pathway, in which BTKi resistance mutations were not reported, primary mutations causing RAS GoF are the hallmark of numerous forms of neoplasms (100). They have been found in CLL but seem unrelated to BTKi treatment (64). Furthermore, nor have any resistance variants been accounted for in the calcineurin arm (Figure 1). It should be mentioned that a cysteine to phenylalanine substitution at amino acid 873 in inositol-trisphosphate 3-kinase B (ITPKB) has been reported under ibrutinib-treatment (68). However, this alteration was identified in a patient with Richter’s transformation and variations are also known in DLCBL. Lack of ITPKB is manifested at the transition from immature to transitional B-cells (101). Thus, while this mutation potentially could affect the calcineurin arm, there is no clear evidence that it is related to BTKi resistance.
This does not in any way mean that the RAS and calcineurin pathways are unimportant for B-cells; it could merely reflect that they are redundant in the leukemias and lymphomas, which are sensitive to treatment with BTKi. In conclusion, acquisition of GoF mutations seems to be rare among BTK downstream signaling components. As described in the next section, the lack of such GoF mutations in transformed B-cells is even more surprising, since they do occur in malignant T-cells.
PKCβ and VAV1 GoF Variants Are Drivers in Adult T Cell Malignancy
Adult T cell leukemia/lymphoma (ATL) is a highly malignant, retrovirus-induced cancer caused by HTLV-1. It is endemic in certain parts of Japan, the Caribbean and Africa (102). In a study of 426 ATL patients, activating mutations were found in a large proportion (103). These variants are depicted in yellow with a T in Figure 3. Similar to what has been observed for BTKi unresponsiveness in B cell malignancies, GoF mutations affecting PLCγ were also identified in ATL. These alterations were found in PLCγ1, which is the isoform expressed in T-cells, whereas in B-cells the corresponding form is PLCγ2. Furthermore, acquired GoF variants were observed in CARD11, as also noted in some patients treated with BTKi.
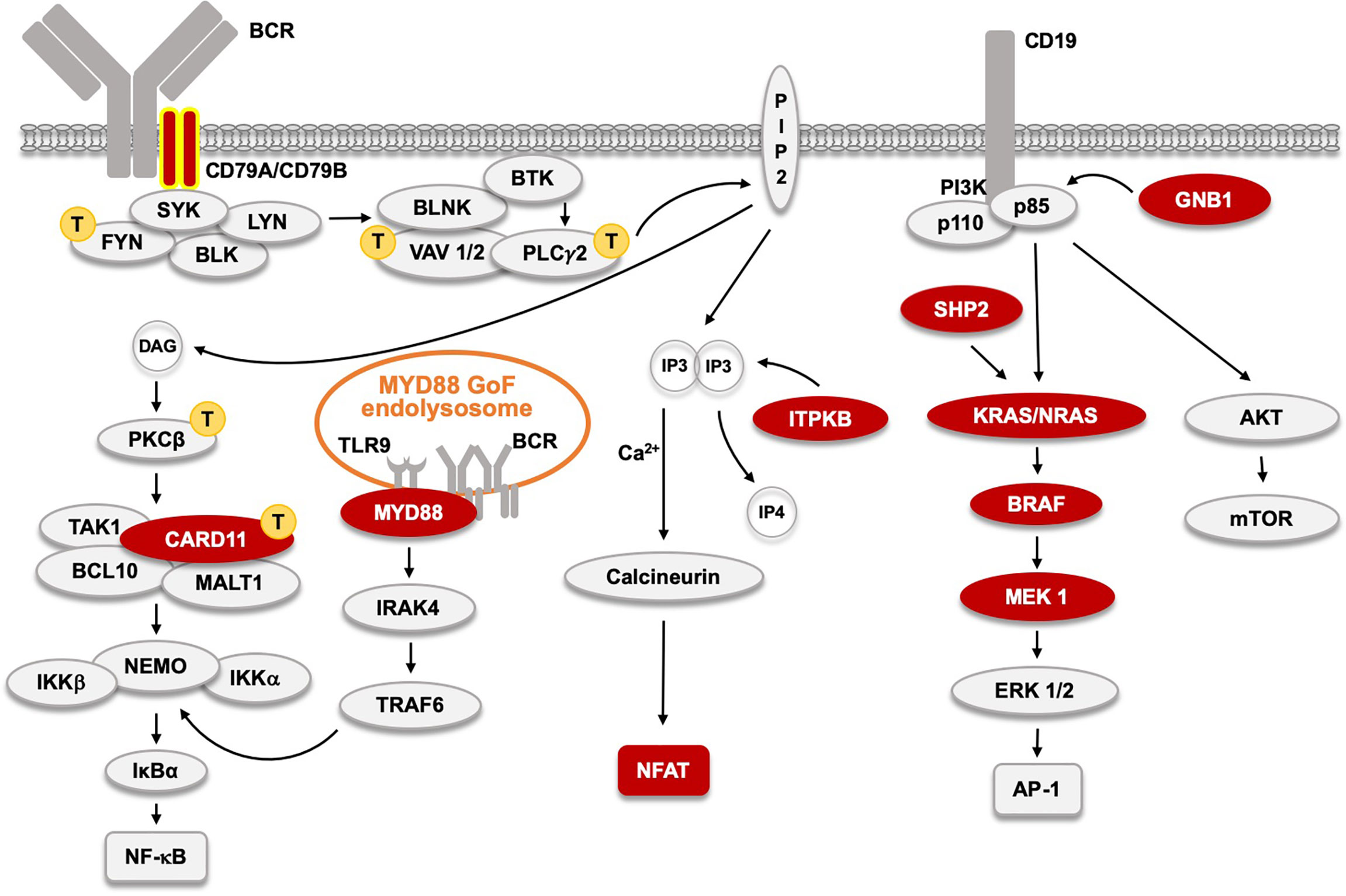
Figure 3 The B cell receptor (BCR) signaling pathway and acquired, primary, driver Gain-of-Function (GoF) mutations found in CLL and other BTKi-sensitive malignancies  . CD79A and B GoF, found in WM and ABC-type DLBCL, and very rare in CLL, are labeled with yellow contour. Signaling proteins marked with
. CD79A and B GoF, found in WM and ABC-type DLBCL, and very rare in CLL, are labeled with yellow contour. Signaling proteins marked with  indicate those affected by primary GoF mutations in adult T cell leukemia as reported (103). The MYD88 L265P GoF mutation in 95% of WM patients, and also in ABC-type DLBCL, and in some CLL patients induces an activated endolysosome containing TLR9 and the BCR.
indicate those affected by primary GoF mutations in adult T cell leukemia as reported (103). The MYD88 L265P GoF mutation in 95% of WM patients, and also in ABC-type DLBCL, and in some CLL patients induces an activated endolysosome containing TLR9 and the BCR.
However, a number of the GoF mutations found in ATLs affected proteins for which variants were never reported to cause resistance to BTKi, namely PKCβ, FYN and VAV1 (Figure 3). Regarding FYN, activating SRC-family kinases are located upstream of BTK, and hence alterations in FYN, LYN or BLK would not be expected to cause resistance to BTKi.
ATL is the first tumor in which recurrent GoF alterations in the PRKCB gene have been described (103). This was unexpected, in particular for the reason that PKCθ is known to the most important isoform in T-cell receptor signaling (104). More than 90% of the mutations affecting PKCβ in ATL were confined to a conserved region of the catalytic domain. In contrast, PKC LoF mutations have been identified in many solid tumors. The PKC family contains 9 genes, and in a comprehensive study, 46/554 of the identified PKC mutations were selected for functional analysis (105). The absolute majority was found to cause LoF and none was activating.
While heritable LoF or GoF mutations affecting PKCβ have not been reported in humans (Figure 4), inactivation of the corresponding gene in the mouse germline causes a phenotype similar to BTK deficiency (106). As mentioned, GoF mutations affecting PKCβ frequently occur in ATL. It is therefore likely that spontaneous GoF mutations in the PRKCB gene, would have occurred multiple times in B-CLL, but seemingly without providing any survival advantage for the affected cell. Identifying the reason for this conundrum would be of great interest to further our understanding of BCR-signaling, including in CLL.
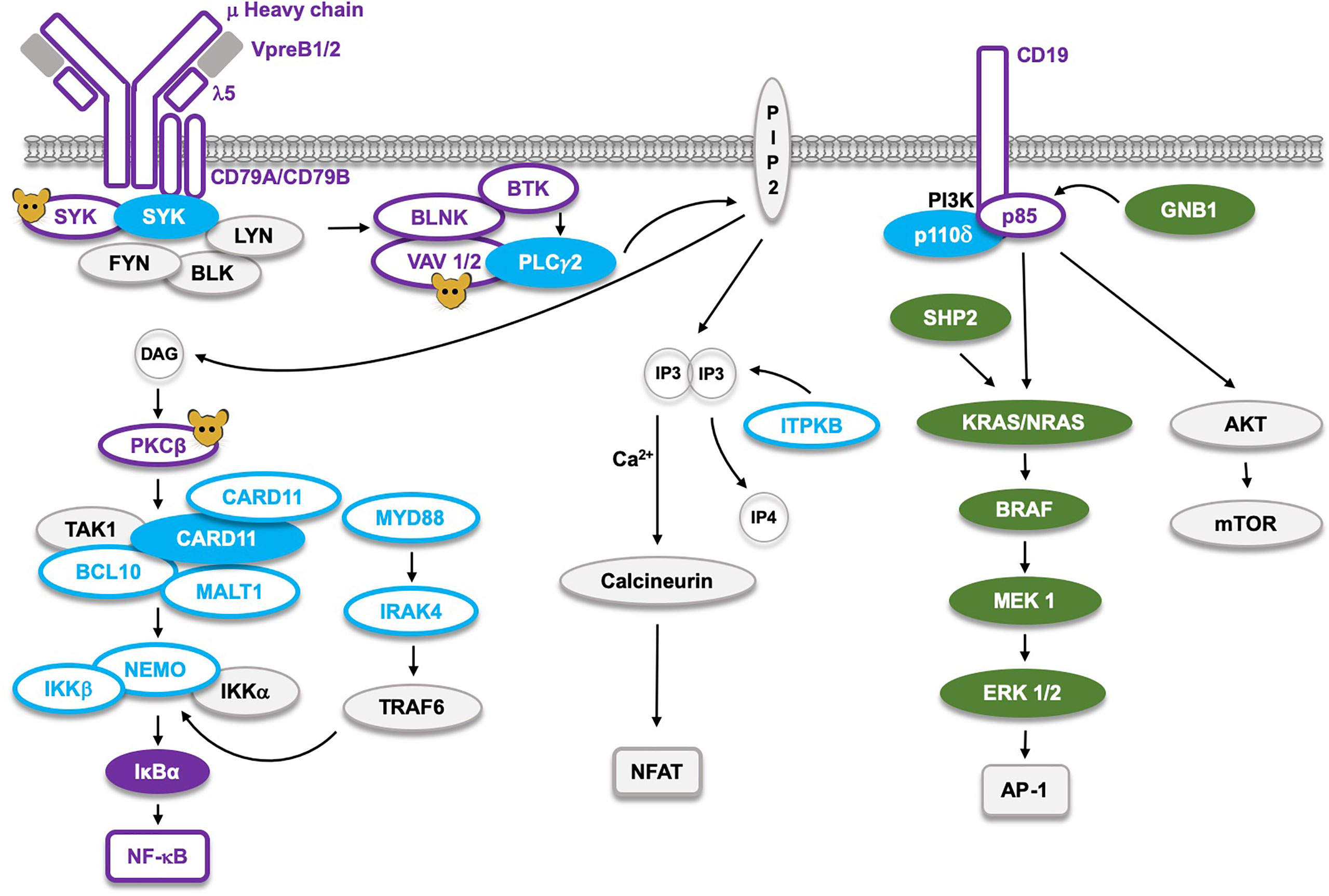
Figure 4 The B cell receptor (BCR) signaling pathway and the corresponding germline mutations found in man, or only generated in mice (molecules marked with ). Proteins marked as  represent Loss-of-Function (LoF) mutations with defects limited to B cells, whereas the LoF mutations marked as
represent Loss-of-Function (LoF) mutations with defects limited to B cells, whereas the LoF mutations marked as  cause defects not limited to B cells. The corresponding filled symbols mark Gain-of-Function (GoF). CARD11 and SYK, which are the only proteins affected by either GoF or LoF, appear as two molecules. LoF mutations in the NF-κB pathway cause B cell defects, whereas those affecting AP-1 signaling result in GoF RASopathies marked as . The single LoF mutation in the NFAT pathway affects the ITPKB gene causing severe combined immunodeficiency (SCID), primarily influencing development of the T cell lineage.
cause defects not limited to B cells. The corresponding filled symbols mark Gain-of-Function (GoF). CARD11 and SYK, which are the only proteins affected by either GoF or LoF, appear as two molecules. LoF mutations in the NF-κB pathway cause B cell defects, whereas those affecting AP-1 signaling result in GoF RASopathies marked as . The single LoF mutation in the NFAT pathway affects the ITPKB gene causing severe combined immunodeficiency (SCID), primarily influencing development of the T cell lineage.
The VAV guanine nucleotide exchange factor is, similar to PKCβ, positioned downstream of BTK, and could therefore potentially cause resistance to BTKi. However, alterations in this gene family have so far not been reported, suggesting that, if they at all exist, they must be very rare. In contrast, VAV1 GoF mutations clustering into hotspots in the acidic, zinc-finger and SH3 domains were found in ATL (103), which demonstrates that they do occur and are selected for. They disable an autoinhibitory mechanism and are considered to cause constitutive activation. Mice with inactivation of the genes encoding VAV1- 2- and -3 in the germline (Figure 4) fail to produce significant numbers of recirculating follicular or marginal zone B cells (107). Similar to the situation for PKCβ, it would be interesting to understand why there is an absence of detectable VAV1 GoF variants in CLL, since they do appear in ATL.
The Origin of the Resistance Mutation Affects Leukemic Clone Size and Variant Allele Frequencies
The analysis of the size of resistance clones has demonstrated interesting differences. Although there is considerable heterogeneity among patients, mutations affecting BTK itself show higher allele frequencies than those affecting PLCγ2 or CARD11, and the resulting clone size is often also larger. It is common that several resistance variants exist in the same patient, such as C481S combined with C481Y, or C481S combined with a GoF mutation in the PLCG2 gene, for example. It has also been found that BTK C481S mutants can occur in the same patient and result from different changes in the TGC codon for cysteine, to AGC or to TCC, both of which encode serine (108). While in this case it was possible to prove different origins because of sequence differences, the identical mutation in which the TGC cysteine codon is altered to the same codon for serine, could also occur in different CLL clones as recurring events. Collectively this demonstrates that resistant clones in the same patient can arise repeatedly during the evolution of the malignant B cell clone, and that the frequency by which they occur likely is underestimated. Since resistance mutations are random events, the highest number of mutational events is expected to exist at the time when the tumor burden is the largest (62), which often is prior to therapy.
To this end, careful dissection of the evolution of CLL under the treatment with BTKi established that resistance mutants were present already prior to therapy (66). This was predicted based on calculation of the leukemia cell burden and other parameters at the onset of treatment (109). During ibrutinib therapy it was estimated that 1.7% of blood CLL cells and 2.7% of tissue CLL cells die per day, 3-5 times higher rate than without treatment (110). This suggests that the ibrutinib-induced reduced tissue disease burden is primarily caused by CLL cell death and to a lesser degree due to CLL cell egress from nodal compartments.
These findings do, however, not explain the reason for that resistance mutations affecting BTK are more frequent and yield a larger clone size than those affecting PLCγ2 or CARD11. Owing to that mutations affecting PLCγ2 and CARD11 are known to occur in multiple sites along the molecules, the mere lack of residues suitable for replacement cannot be the underlying mechanism. Current knowledge (Figure 1) suggests a crucial role for PLCγ2 in the generation of diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), which subsequently activate PKCβ, and intracellular calcium-signaling, respectively. However, it is possible that it is not sufficient to only activate PLCγ2, but that BTK has yet another role beyond DAG- and IP3-mediated signaling.
Recurrent Primary Mutations in B Cell Malignancies Responding to BTKi
While the focus of this review is on resistance mutations to BTKi, it is of interest to compare these alterations with those found as recurrent primary mutations. Genetic alterations are the basis for tumorigenesis, including leukemogenesis. How neoplasms evolve with regard to their mutation spectrum over the course of disease and therapy is key to the understanding of cancer biology. Interestingly, the primary mutations differ substantially among BTKi sensitive tumors.
Hence, in CLL the majority of the primary driver mutations are unrelated to BCR signaling proteins with alterations affecting the MIR15A and MIR16-1, ATM, BIRC3, NOTCH, SF3B1, and TP53 genes being highly prevalent (111–113). In contrast, in WM, the genetic changes mainly affect the BCR pathway, with 95% of patients carrying the MYD88 leucine 265 to proline substitution, L265P (114, 115). Mutations affecting the G-protein coupled receptor CXCR4 are also prevalent in WM, but not in CLL (115). However, MYD88 mutations are overrepresented in CLL (112, 113) and they are more often clonal than subclonal (116).
Even if MYD88 is not part of the classical BCR pathway it activates canonical NF-κB signaling (117). MYD88 is an adaptor protein mediating signals from Toll-like and interleukin 1 receptors. It forms a supramolecular complex, known as the Myddosome (118). As depicted in Figures 1 and 3, in both ABC-type DLBCL and WM, a MYD88-TLR9-BCR (My-T-BCR) complex is generated at the endolysosome, while this is not characteristic of MCL (116). My-T-BCR connects MYD88 to IgM, PLCγ2, the CARD11-BCL10-MALT1 complex and to mTOR, and the L265P substitution in MYD88 is crucial for generating such a constitutively active signaling hub. In animal experimental model systems TLR9 acts together with BTK to induce autoimmunity (119). Remarkably, primary GoF mutations affecting components downstream of MYD88, such as IRAK4 have not been described in WM.
In WM, gain-of-function mutations instead affect CD79B (Igβ), which is an essential upstream signaling module of the BCR complex (115),. In the ABC subtype of DLBCL GoF alterations mainly also affect CD79B, but they are found in CD79A, too (116). In CLL, GoF mutations in CD79A and B exist, but are very rare (112, 113).
In CLL, primary GoF, driver mutations are found in components of not only the NF-κB but also of the RAS-MAPK pathway, as depicted in Figure 3 (112, 113, 120). The very recent report demonstrating that resistance mutations to inhibitors of p110δ selectively affects the MAPK pathway in CLL patients is highly interesting (121). It shows that also this arm of the BCR pathway can be affected by resistance mutations (Figure 1), although they never appear after BTKi treatment. The existence of such resistance mutations in CLL upon p110δi therapy suggests that the PI3K arm of BCR signaling primarily affects RAS-MAPK signaling. Furthermore, these findings suggest that combined treatment with BTKi and p110δi may give synergistic effects, and such studies have already been performed (122, 123).
Finally, the third arm of BCR signaling is also affected in CLL. This pathway is formed by PLCγ2-generated inositol 1,4,5-trisphosphate (IP3) activating the phosphatase calcineurin, which subsequently de-phosphorylates the transcriptional regulator nuclear factor of activated T-cells, NFAT (Figures 1 and 3). De-phosphorylation of NFAT results in nuclear translocation and enhanced nuclear presence of NFAT is observed in CLL (124). The underlying mechanism involves hypomethylation of the NFATC1 promoter (125), and the degree of NFAT activation has been reported to predict the clinical outcome in CLL (126).
Proteins involved in the adhesion and migration of leukemia and lymphoma cells have demonstrated their importance in both CLL and MCL (127, 128). However, with the exception of CXCR4 in WM (115), the corresponding activating mutations have not been identified. Notably, in both CLL and MCL adhesion molecules, including CXCR4, are affected by BTKi treatment (127, 129, 130). In WM somatic CXCR4 mutations were reported to activate AKT and ERK (131).
Phenotype of Germline, Loss-of-Function Mutations Affecting Components of the BCR Pathway
Mutations in leukemia and lymphoma, which cause resistance to treatment with kinase inhibitors, mainly are GoF. Importantly, only a subset of these proteins is also frequently affected by primary GoF mutations in B cell malignancies (Figures 1 and 3). For NF-κB signaling these are CD79A, CD79B, CARD11 and in particular MYD88. A likely explanation for this selectivity is that many of the molecules in this pathway simply are not susceptible to GoF activity through a mutational event. This seems to be the case for e. g. BTK and the adaptor BLNK for which constitutively activating variants are lacking. Conversely, as depicted in Figure 4, the corresponding LoF mutations cause very severe B cell deficiency (1, 132, 133).
Recently (Figure 4), inherited GoF mutations were observed in the SYK gene (134). They cause a B cell defect combined with a multiorgan, inflammatory disorder and patients do not develop CLL, but, instead, an increased frequency of DLBCL. While having driver activity, primary SYK mutations are very rare among DLBCL patients (135, 136), but are increased in many cancer cell lines (137). Moreover, in peripheral T cell lymphoma the BTK-related ITK kinase, upon a translocation event, forms a fusion protein with SYK (138), which induces STAT3 phosphorylation and CD69 expression (139).
In Figure 4, components of the BCR causing hereditary disease are highlighted. In the NF-κB pathway LoF mutations predominantly induce defects in B cell development. This is true for all the components of the pre-BCR and several of the downstream signaling proteins. BTK is highly conserved during evolution (140) and mutations in the BTK gene are the most frequent, since this gene is located on the X-chromosome making boys particularly susceptible (1, 133, 141). Single LoF mutations affecting the SRC-family kinases, BLK, FYN and LYN do not cause B cell deficiency, while a combined triple-knockout in mice does (142). Whereas LoF alterations for PLCγ2 have not been described in humans, activating mutations cause inherited hyperinflammation as depicted in Figure 2.
LoF mutations affecting VAV or PKCβ proteins have not been identified in the germline of humans, likely because more than a single gene needs to be inactivated. SYK LoF is lethal in mice but has not been observed in humans. In addition to an endothelial abnormality, it causes a severe B-lineage defect in mice (143, 144). Thus, it belongs to the group of proteins whose genes have been inactivated in mice, but for which LoF variants were never reported in humans (marked with a mouse symbol in Figure 4).
Mutations in components of the CARD11-BCL10-MALT1 complex affect the development of the B cell lineage but also give rise to other phenotypes (86, 89). CARD11 is so far unique in that both GoF and LoF germline mutations have been identified in humans, with LoF causing a combined immunodeficiency affecting both B- and T-cells (86, 89, 145). The inhibitory IκBα can be affected with GoF variations, whereas NF-κB only shows LoF and these cause B cell deficiency (145). LoF affecting MYD88 mainly affects innate immunity and patients surviving bacterial infections early in life develop adaptive immunity protecting them from subsequent infectious episodes (145, 146).
Inherited variations in the calcineurin-NFAT pathway have not been reported. However, LoF mutations in the inositol-trisphosphate 3-kinase B gene, ITPKB, cause severe combined immunodeficiency (Figure 4). While B cells secondarily are influenced by this loss, the defect primarily affects T cells (147). Moreover, GoF alterations in this gene belong to the group of primary mutations found in CLL (Figure 3).
Germline mutations in the RAS-MAPK pathway (Figure 4) cause RASopathies, many of which belong to the Noonan syndrome (148). RASopathies are characterized by unusual facial features, congenital heart disease, growth and skeletal abnormalities, intellectual disability and predisposition to neoplastic development. Of note, the overlap between primary mutations in CLL (Figure 3) and those providing resistance (Figure 1) is so far only partial. Thus, activating mutations affecting SHP2 and GNB1 exist as primary alterations in CLL and cause RASopathy, but have not as yet been found to induce resistance to p110δi. Inherited GoF mutations affecting p110δi cause a combined immunodeficiency and hyperinflammatory state (149), whereas LoF defects of its partner p85 result in a primary B cell defect (145).
Collectively this demonstrates that there is an interesting, but complex, interplay between signaling proteins, with often major phenotypic differences between germline and acquired primary mutations in B-lineage cells. Moreover, the patterns that are being revealed through mutational analysis of both primary and drug-induced resistance provide important insights into leukemogenesis.
Of further note are the RAS-MAPK pathway mutations found in CLL, since they clearly exemplify that the phenotype of germline mutations and those found in tumors do not necessarily overlap. It can be anticipated that every single gene likely is mutated as a rare event in every tumor type (but not in every patient). Because cells with an acquired driver mutation will be selected for, this experiment of nature, suggests that continued sequencing of tumor genomes eventually will reveal every key component in tumor biology. Because of the dependence on additional genomic alterations for the selection of cells having acquired a potential driver mutation, revealing the interplay is a formidable task. Drug treatment and the selection of resistance mutations will add another layer of information, which may aid in this process.
Concluding Remarks
The common linear representation of the BCR-signaling pathway, which trifurcates into the NF-κB, NFAT and AP-1 arms, does not fully explain the observed pattern of resistance mutations in patients treated with BTKi. Although GoF mutations affecting both PLCγ2 and CARD11 are compatible with such a scheme, the lack of PKCβ and VAV GoF variants is not. Furthermore, resistance mutations in the NFAT or the AP-1 arms have not been observed in patients treated with BTKi.
Similarly, in WM, why is there such a predominance of primary driver mutations generating an activated form of MYD88? Why are there no examples of primary GoF driver mutations affecting proteins downstream of MYD88, such as IRAK4? This indicates that MYD88 has some unique properties apart from activating IRAK1, IRAK4 and TRAF6 mediated signaling. Alternatively, these components may not be susceptible to GoF.
Inherited SYK GoF mutations frequently cause DLBCL, whereas inherited GoF mutations of CD79A and B were not yet reported. In contrast, CD79A and B primary mutations in tumors are found in CLL, WM as well as in DLBCL, whereas primary SYK mutations are very rare, calling into question a strict linear signaling relationship.
A conundrum has been the mechanism underlying the variant allele frequencies among resistant clones. Thus, replacement of cysteine 481 is the most frequent alteration and the corresponding clones are usually also the largest. This observation suggests a gradual reduction of the biological activity of individual proteins reflecting their position in the descending BCR pathway. The recent finding that the arms of the BCR pathway are differentially affected by BTKi and p110δi, is compatible with this idea and also has therapeutic implications in favor of combination therapies.
Hence, although the NF-κB, NFAT and AP-1 downstream signaling arms from the BCR appear in the form of textbook information, the resistance patterns observed in patients treated with BTKi and p110δi reveal that there is more to learn. For example, precisely how the BCR is connected to PI3K signaling needs further investigation as well as the role of PI3K-regulated AKT and mTOR. This is key for the basic understanding of the BCR multicomponent signalosome, and also for providing optimal therapeutic solutions, since combination therapies may prevent resistance from developing.
Author Contributions
CS determined the scope and focus of the review and drafted the manuscript and generated the figures. JB wrote part of the manuscript and provided critical feedback. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Swedish Cancer Society, Stockholm County Council (ALF-project) and Swedish Medical Research Council. This work was supported in part by the generous philanthropic contributions to The University of Texas MD Anderson Cancer Center Moon Shots Program™, the CLL Global Research Foundation, and the MD Anderson Cancer Support Grant CA016672.
Conflict of Interest
JB reports honoraria from consulting/an advisory role with Janssen; research funding from Gilead, Astra Zeneca, BeiGene, and Pharmacyclics LLC, an AbbVie Company; travel, accommodations, or other expenses from Gilead and Janssen.
CS declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Vetrie D, Vořechovský I, Sideras P, Holland J, Davies A, Flinter F, et al. The Gene Involved in X-Linked Agammaglobulinaemia Is a Member of the SRC Family of Protein-Tyrosine Kinases. Nature (1993) 361(6409):226–33. doi: 10.1038/361226a0
2. Smith CIE. From Identification of the BTK Kinase to Effective Management of Leukemia. Oncogene (2017a) 36(15):2045–53. doi: 10.1038/onc.2016.343
3. Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, et al. Deficient Expression of a B Cell Cytoplasmic Tyrosine Kinase in Human X-Linked Agammaglobulinemia. Cell (1993) 72(2):279–90. doi: 10.1016/0092-8674(93)90667-f
4. Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, et al. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem (2007) 2:58–61. doi: 10.1002/cmdc.200600221
5. Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton Tyrosine Kinase Inhibitor PCI-32765 Blocks B-Cell Activation and Is Efficacious in Models of Autoimmune Disease and B-Cell Malignancy. Proc Natl Acad Sci USA (2010) 107(29):13075–80. doi: 10.1073/pnas.1004594107
6. Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s Tyrosine Kinase in B Cell Malignancies. Nat Rev Cancer (2014) 14(4):219–32. doi: 10.1038/nrc3702
7. Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B. Et AlBruton Tyrosine Kinase Inhibitor Ibrutinib (PCI-32765) Has Significant Activity in Patients With Relapsed/Refractory B-Cell Malignancies. J Clin Oncol (2013) 31:88–94. doi: 10.1200/JCO.2012.42.7906
8. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK With Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med (2013) 369(1):32–42. doi: 10.1056/NEJMoa1215637
9. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum K, et al. Ibrutinib Treatment for First-Line and Relapsed/Refractory Chronic Lymphocytic Leukemia: Final Analysis of the Pivotal Phase IB/II PCYC-1102 Study. Clin Cancer Res (2020) 26(15):3918–27. doi: 10.1158/1078-0432.CCR-19-2856
10. Byrd JC, Brown JR, O’Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib Versus Ofatumumab in Previously Treated Chronic Lymphoid Leukemia. N Engl J Med (2014) 371(3):213–23. doi: 10.1056/NEJMoa1400376
11. Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, et al. Ibrutinib as Initial Therapy for Patients With Chronic Lymphocytic Leukemia. N Engl J Med (2015) 373(25):2425–37. doi: 10.1056/NEJMoa1509388
12. Woyach JA, Ruppert AS, Heerema NA, Zhao W, Booth AM, Ding W, et al. Ibrutinib Regimens Versus Chemoimmunotherapy in Older Patients With Untreated CLL. N Engl J Med (2018) 379(26):2517–28. doi: 10.1056/NEJMoa1812836
13. Shanafelt TD, Wang XV, Kay NE, Hanson CA, O’Brien S, Barrientos J, et al. Ibrutinib-Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N Engl J Med (2019) 381(5):432–43. doi: 10.1056/NEJMoa1817073
14. Castillo JJ, Treon SP. Management of Waldenström Macroglobulinemia in 2020. Hematol Am Soc Hematol Educ Program (2020) 1:372–9. doi: 10.1182/hematology.2020000121
15. Estupiñan HY, Berglöf A, Zain R, Smith CIE. Comparative Analysis of BTK Inhibitors and Mechanisms Underlying Adverse Effects. Front Cell Dev Biol (2021a) 9:630942. doi: 10.3389/fcell.2021.630942
16. Wen T, Wang J, Shi Y, Qian H, Liu P. Inhibitors Targeting Bruton’s Tyrosine Kinase in Cancers: Drug Development Advances. Leukemia. (2021) 35(2):312–32. doi: 10.1038/s41375-020-01072-6
17. Reiff SD, Mantel R, Smith LL, Greene JT, Muhowski EM, Fabian CA, et al. The BTK Inhibitor ARQ 531 Targets Ibrutinib-Resistant CLL and Richter Transformation. Cancer Discovery (2018) 8(10):1300–15. doi: 10.1158/2159-8290.CD-17-1409
18. Estupiñán HY, Wang Q, Berglöf A, Schaafsma GCP, Shi Y, Zhou L, et al. BTK Gatekeeper Residue Variation Combined With Cysteine 481 Substitution Causes Super-Resistance to Irreversible Inhibitors Acalabrutinib, Ibrutinib and Zanubrutinib. Leukemia (2021b) 35(5):1317–29. doi: 10.1038/s41375-021-01123-6
19. Garuti L, Roberti M, Bottegoni G. Irreversible Protein Kinase Inhibitors. Curr Med Chem (2011) 18(20):2981–94. doi: 10.2174/092986711796391705
20. Berglöf A, Hamasy A, Meinke S, Palma M, Krstic A, Månsson R, et al. Targets for Ibrutinib Beyond B Cell Malignancies. Scand J Immunol (2015) 82(3):208–17. doi: 10.1111/sji.12333
21. Brown JR. How I Treat CLL Patients With Ibrutinib. Blood (2018) 131(4):379–86. doi: 10.1182/blood-2017-08-764712
22. Thompson PA, Burger JA. Bruton’s Tyrosine Kinase Inhibitors: First and Second Generation Agents for Patients With Chronic Lymphocytic Leukemia (CLL). Expert Opin Investig Drugs (2018) 27(1):31–42. doi: 10.1080/13543784.2018.1404027
23. Zain R, Vihinen M. Structure-Function Relationships of Covalent and Non-Covalent BTK Inhibitors. Front Immunol (2021).
24. Burger JA, Chiorazzi N. B Cell Receptor Signaling in Chronic Lymphocytic Leukemia. Trends Immunol (2013) 34(12):592–601. doi: 10.1016/j.it.2013.07.002
25. Saba NS, Liu D, Herman SE, Underbayev C, Tian X, Behrend D, et al. Pathogenic Role of B-Cell Receptor Signaling and Canonical NF-κB Activation in Mantle Cell Lymphoma. Blood (2016) 128(1):82–92. doi: 10.1182/blood-2015-11-681460
26. Argyropoulos KV, Vogel R, Ziegler C, Altan-Bonnet G, Velardi E, Calafiore M, et al. Clonal B Cells in Waldenström’s Macroglobulinemia Exhibit Functional Features of Chronic Active B-Cell Receptor Signaling. Leukemia (2016) 30:1116–25. doi: 10.1038/leu.2016.8
27. Preud’homme JL, Seligmann M. Anti-Human Immunoglobulin G Activity of Membrane-Bound Monoclonal Immunoglobulin M in Lymphoproliferative Disorders. Proc Natl Acad Sci USA (1972) 69(8):2132–5. doi: 10.1073/pnas.69.8.2132
28. Schroeder HW Jr, Dighiero G. The Pathogenesis of Chronic Lymphocytic Leukemia: Analysis of the Antibody Repertoire. Immunol Today (1994) 15(6):288–94. doi: 10.1016/0167-5699(94)90009-4
29. Hashimoto S, Dono M, Wakai M, Allen SL, Lichtman SM, Schulman P, et al. Somatic Diversification and Selection of Immunoglobulin Heavy and Light Chain Variable Region Genes in Igg+ CD5+ Chronic Lymphocytic Leukemia B Cells. J Exp Med (1995) 181(4):1507–17. doi: 10.1084/jem.181.4.1507
30. Johnson TA, Rassenti LZ, Kipps TJ. Ig VH1 Genes Expressed in B Cell Chronic Lymphocytic Leukemia Exhibit Distinctive Molecular Features. J Immunol (1997) 158(1):235–46.
31. Fais F, Ghiotto F, Hashimoto S, Sellars B, Valetto A, Allen SL, et al. Chronic Lymphocytic Leukemia B Cells Express Restricted Sets of Mutated and Unmutated Antigen Receptors. J Clin Invest (1998) 102(8):1515–25. doi: 10.1172/JCI3009
32. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) Genes are Associated With a More Aggressive Form of Chronic Lymphocytic Leukemia. Blood (1999) 94(6):1848–54. doi: 10.1182/blood.V94.6.1848
33. Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, et al. Ig V Gene Mutation Status and CD38 Expression as Novel Prognostic Indicators in Chronic Lymphocytic Leukemia. Blood (1999) 94(6):1840–7. doi: 10.1182/blood.V94.6.1840.418k06_1840_1847
34. Burger JA, Barr PM, Robak T, Owen C, Ghia P, Tedeschi A, et al. Long-Term Efficacy and Safety of First-Line Ibrutinib Treatment for Patients With CLL/SLL: 5 Years of Follow-Up From the Phase 3 RESONATE-2 Study. Leukemia (2020) 34(3):787–98. doi: 10.1038/s41375-019-0602-x
35. Stamatopoulos K, Agathangelidis A, Rosenquist R, Ghia P. Antigen Receptor Stereotypy in Chronic Lymphocytic Leukemia. Leukemia (2017) 31(2):282–91. doi: 10.1038/leu.2016.322
36. Briney B, Inderbitzin A, Joyce C, Burton DR. Commonality Despite Exceptional Diversity in the Baseline Human Antibody Repertoire. Nature (2019) 566(7744):393–7. doi: 10.1038/s41586-019-0879-y
37. Agathangelidis A, Chatzidimitriou A, Gemenetzi K, Giudicelli V, Karypidou M, Plevova K, et al. Higher-Order Connections Between Stereotyped Subsets: Implications for Improved Patient Classification in CLL. Blood. (2021) 137(10):1365–76. doi: 10.1182/blood.2020007039
38. Kienle D, Kröber A, Katzenberger T, Ott G, Leupolt E, Barth TF, et al. VH Mutation Status and VDJ Rearrangement Structure in Mantle Cell Lymphoma: Correlation With Genomic Aberrations, Clinical Characteristics, and Outcome. Blood (2003) 102(8):3003–9. doi: 10.1182/blood-2003-05-1383
39. Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic Active B−Cell-Receptor Signalling in Diffuse Large B−Cell Lymphoma. Nature (2010) 463(7277):88–92. doi: 10.1038/nature08638
40. Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B Cell Receptor Signaling With Ibrutinib in Diffuse Large B Cell Lymphoma. Nat Med (2015) 21(8):922–6. doi: 10.1038/nm.3884
41. Klintman J, Appleby N, Stamatopoulos B, Ridout K, Eyre TA, Robbe P, et al. Genomic and Transcriptomic Correlates of Richter’s Transformation in Chronic Lymphocytic Leukemia. Blood In press (2021) 137(20):2800–16. doi: 10.1182/blood.2020005650
42. Petrackova A, Turcsanyi P, Papajik T, Kriegova E. Revisiting Richter Transformation in the Era of Novel CLL Agents. Blood Rev (2021) 12:100824. doi: 10.1016/j.blre.2021.100824
43. Lam KP, Kühn R, Rajewsky K. In Vivo Ablation of Surface Immunoglobulin on Mature B Cells by Inducible Gene Targeting Results in Rapid Cell Death. Cell. (1997) 90(6):1073–83. doi: 10.1016/s0092-8674(00)80373-6
44. Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K. Survival of Resting Mature B Lymphocytes Depends on BCR Signaling Via the Igα/β Heterodimer. Cell (2004) 117(6):787–800. doi: 10.1016/j.cell.2004.05.014
45. Pasparakis M, Schmidt-Supprian M, Rajewsky K. Ikappab Kinase Signaling Is Essential for Maintenance of Mature B Cells. J Exp Med (2002) 196(6):743–52. doi: 10.1084/jem.20020907
46. Rendeiro AF, Krausgruber T, Fortelny N, Zhao F, Penz T, Farlik M, et al. Chromatin Mapping and Single-Cell Immune Profiling Define the Temporal Dynamics of Ibrutinib Response in CLL. Nat Commun (2020) 11(1):577. doi: 10.1038/s41467-019-14081-6
47. Müller S, Sideras P, Smith CI, Xanthopoulos KG. Cell Specific Expression of Human Bruton’s Agammaglobulinemia Tyrosine Kinase Gene (Btk) Is Regulated by Sp1- and Spi-1/PU.1-Family Members. Oncogene (1996) 13(9):1955–64.
48. Holinski-Feder E, Weiss M, Brandau O, Jedele KB, Nore B, Bäckesjö CM, et al. Mutation Screening of the BTK Gene in 56 Families With X-Linked Agammaglobulinemia (XLA): 47 Unique Mutations Without Correlation to Clinical Course. Pediatrics (1998) 101(2):276–84. doi: 10.1542/peds.101.2.276
49. Bravo García-Morato M, Del Pino Molina L, Torres Canizales JM, Del Rosal Rabes T, Méndez Echevarría A, González Martínez B, et al. A Mutation in the Promoter Region of BTK Causes Atypical XLA. Heliyon (2020) 6(9):e04914. doi: 10.1016/j.heliyon.2020.e04914
50. Yu L, Mohamed AJ, Simonson OE, Vargas L, Blomberg KEM, Björkstrand B, et al. Proteasome-Dependent Autoregulation of Bruton Tyrosine Kinase (Btk) Promoter Via NF-κB. Blood (2008) 111(9):4617–26. doi: 10.1182/blood-2007-10-121137
51. Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance Mechanisms for the Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. N Engl J Med (2014) 370(24):2286–94. doi: 10.1056/NEJMoa1400029
52. Furman RR, Cheng S, Lu P, Setty M, Perez AR, Guo A, et al. Ibrutinib Resistance in Chronic Lymphocytic Leukemia. N Engl J Med (2014) 370(24):2352–4. doi: 10.1056/NEJMc1402716
53. Maddocks KJ, Ruppert AS, Lozanski G, Heerema NA, Zhao W, Abruzzo L, et al. Etiology of Ibrutinib Therapy Discontinuation and Outcomes in Patients With Chronic Lymphocytic Leukemia. JAMA Oncol (2015) 1(1):80–7. doi: 10.1001/jamaoncol.2014.218
54. Hamasy A, Wang Q, Blomberg KE, Mohammad DK, Yu L, Vihinen M, et al. Substitution Scanning Identifies a Novel, Catalytically Active Ibrutinib-Resistant BTK Cysteine 481 to Threonine (C481T) Variant. Leukemia (2017) 31(1):177–85. doi: 10.1038/leu.2016.153
55. Quinquenel A, Fornecker L-M, Letestu R, Ysebaert L, Fleury C, Lazarian G, et al. Prevalence of BTK and PLCG2 Mutations in a Real−Life CLL Cohort Still on Ibrutinib After Three Years: A FILO Group Study. Blood (2019) 134(7):641–4. doi: 10.1182/blood.2019000854
56. Estupiñan HY, Bouderlique T, He C, Berglöf A, Gupta D, Peña-Perez L, et al. Novel Mouse Model Resistant to Irreversible BTK Inhibitors: A Tool Identifying New Therapeutic Targets and Side Effects. Blood Adv (2020) 4(11):2439–50. doi: 10.1182/bloodadvances.2019001319
57. Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med (2016) 374(4):323–32. doi: 10.1056/NEJMoa1509981
58. Handunnetti SM, Tang CPS, Nguyen T, Zhou X, Thompson E, Sun H, et al. BTK Leu528Trp - a Potential Secondary Resistance Mechanism Specific for Patients With Chronic Lymphocytic Leukemia Treated With the Next Generation BTK Inhibitor Zanubrutinib. Blood (2019) 134(Supplement 1):170. doi: 10.1182/blood-2019-125488
59. Chiron D, Di Liberto M, Martin P, Huang X, Sharman J, Blecua P, et al. Cell-Cycle Reprogramming for PI3K Inhibition Overrides a Relapse-Specific C481S BTK Mutation Revealed by Longitudinal Functional Genomics in Mantle Cell Lymphoma. Cancer Discovery (2014) 4(9):1022–35. doi: 10.1158/2159-8290.CD-14-0098
60. Epperla N, Shana’ah AY, Jones D, Christian BA, Ayyappan S, Maddocks K, et al. Resistance Mechanism for Ibrutinib in Marginal Zone Lymphoma. Blood Adv (2019) 3(4):500–2. doi: 10.1182/bloodadvances.2018029058
61. Jiménez C, Chan GG, Xu L, Tsakmaklis N, Kofides A, Demos MG, et al. Genomic Evolution of Ibrutinib-Resistant Clones in Waldenström Macroglobulinaemia. Br J Haematol (2020) 189(6):1165–70. doi: 10.1111/bjh.16463
62. Smith CIE. Enigmas in Tumor Resistance to Kinase Inhibitors and Calculation of the Drug Resistance Index for Cancer (DRIC). Semin Cancer Biol (2017b) 45:36–49. doi: 10.1016/j.semcancer.2016.11.008
63. Kanagal-Shamanna R, Jain P, Patel KP, Routbort M, Bueso-Ramos C, Alhalouli T, et al. Targeted Multigene Deep Sequencing of Bruton Tyrosine Kinase Inhibitor-Resistant Chronic Lymphocytic Leukemia With Disease Progression and Richter Transformation. Cancer (2019) 125(4):559–74. doi: 10.1002/cncr.31831
64. Kadri S, Lee J, Fitzpatrick C, Galanina N, Sukhanova M, Venkataraman G, et al. Clonal Evolution Underlying Leukemia Progression and Richter Transformation in Patients with Ibrutinib-relapsed CLL. Blood Adv (2017) 1(12):715–27. doi: 10.1182/bloodadvances.2016003632
65. Sharma S, Galanina N, Guo A, Lee J, Kadri S, Van Slambrouck C, et al. Identification of a Structurally Novel BTK Mutation That Drives Ibrutinib Resistance in CLL. Oncotarget (2016) 7(42):68833–41. doi: 10.18632/oncotarget.11932
66. Burger JA, Landau DA, Taylor-Weiner A, Bozic I, Zhang H, Sarosiek K, et al. Clonal Evolution in Patients With Chronic Lymphocytic Leukaemia Developing Resistance to BTK Inhibition. Nat Commun (2016) 7:11589. doi: 10.1038/ncomms11589
67. Gángó A, Alpár D, Galik B, Marosvári D, Kiss R, Fésüs V, et al. Dissection of Subclonal Evolution by Temporal Mutation Profiling in Chronic Lymphocytic Leukemia Patients Treated With Ibrutinib. Int J Cancer (2020) 146(1):85–93. doi: 10.1002/ijc.32502
68. Landau DA, Sun C, Rosebrock D, Herman SEM, Fein J, Sivina M, et al. The Evolutionary Landscape of Chronic Lymphocytic Leukemia Treated With Ibrutinib Targeted Therapy. Nat Commun (2017) 8(1):2185. doi: 10.1038/s41467-017-02329-y
69. Takata M, Kurosaki T. A Role for Bruton’s Tyrosine Kinase in B Cell Antigen Receptor-Mediated Activation of Phospholipase C-Gamma 2. J Exp Med (1996) 184(1):31–40. doi: 10.1084/jem.184.1.31
70. Watanabe D, Hashimoto S, Ishiai M, Matsushita M, Baba Y, Kishimoto T, et al. Four Tyrosine Residues in Phospholipase C-Gamma 2, Identified as Btk-Dependent Phosphorylation Sites, Are Required for B Cell Antigen Receptor-Coupled Calcium Signaling. J Biol Chem (2001) 276(42):38595–601. doi: 10.1074/jbc.M103675200
71. Liu TM, Woyach JA, Zhong Y, Lozanski A, Lozanski G, Dong S, et al. Hypermorphic Mutation of Phospholipase C, γ2 Acquired in Ibrutinib-Resistant CLL Confers BTK Independency Upon B-Cell Receptor Activation. Blood (2015) 126(1):61–8. doi: 10.1182/blood-2015-02-626846
72. Zhou Q, Lee GS, Brady J, Datta S, Katan M, Sheikh A, et al. A Hypermorphic Missense Mutation in PLCG2, Encoding Phospholipase Cγ2, Causes a Dominantly Inherited Autoinflammatory Disease With Immunodeficiency. Am J Hum Genet (2012) 91(4):713–20. doi: 10.1016/j.ajhg.2012.08.006
73. Yu P, Constien R, Dear N, Katan M, Hanke P, Bunney TD, et al. Autoimmunity and Inflammation Due to a Gain-of-Function Mutation in Phospholipase Cγ2 That Specifically Increases External Ca2+ Entry. Immunity (2005) 22(4):451–65. doi: 10.1016/j.immuni.2005.01.018
74. Everett KL, Bunney TD, Yoon Y, Rodrigues-Lima F, Harris R, Driscoll PC, et al. Characterization of Phospholipase C Gamma Enzymes With Gain-of-Function Mutations. J Biol Chem (2009) 284(34):23083–93. doi: 10.1074/jbc.M109.019265
75. Abe K, Fuchs H, Boersma A, Hans W, Yu P, Kalaydjiev S, et al. A Novel N-Ethyl-N-Nitrosourea-Induced Mutation in Phospholipase Cγ2 Causes Inflammatory Arthritis, Metabolic Defects, and Male Infertility In Vitro in a Murine Model. Arthritis Rheumatol (2011) 63(5):1301–11. doi: 10.1002/art.30280
76. Walliser C, Tron K, Clauss K, Gutman O, Kobitski AY, Retlich M, et al. Rac-Mediated Stimulation of Phospholipase Cγ2 Amplifies B Cell Receptor-Induced Calcium Signaling. J Biol Chem (2015) 290(28):17056–72. doi: 10.1074/jbc.M115.645739
77. Wist M, Meier L, Gutman O, Haas J, Endres S, Zhou Y, et al. Noncatalytic Bruton’s Tyrosine Kinase Activates Plcγ2 Variants Mediating Ibrutinib Resistance in Human Chronic Lymphocytic Leukemia Cells. J Biol Chem (2020) 295(17):5717–36. doi: 10.1074/jbc.RA119.011946
78. Rawlings DJ, Sommer K, Moreno-García ME. The CARMA1 Signalosome Links the Signalling Machinery of Adaptive and Innate Immunity in Lymphocytes. Nat Rev Immunol (2006) 6(11):799–812. doi: 10.1038/nri1944
79. Matsumoto R, Wang D, Blonska M, Li H, Kobayashi M, Pappu B, et al. Phosphorylation of CARMA1 Plays a Critical Role in T Cell Receptor-Mediated NF-κB Activation. Immunity (2005) 23:575–85. doi: 10.1016/j.immuni.2005.10.007
80. Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno- Garcia ME, Ovechkina YL, et al. Phosphorylation of the CARMA1 Linker Controls NF-κB Activation. Immunity (2005) 23:561–74. doi: 10.1016/j.immuni.2005.09.014
81. Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, et al. Molecular Subtypes of Diffuse Large B-Cell Lymphoma Arise by Distinct Genetic Pathways. Proc Natl Acad Sci USA (2008) 105(36):13520–5. doi: 10.1073/pnas.0804295105
82. Wu C, de Miranda NF, Chen L, Wasik AM, Mansouri L, Jurczak W, et al. Genetic Heterogeneity in Primary and Relapsed Mantle Cell Lymphomas: Impact of Recurrent CARD11 Mutations. Oncotarget (2016) 7(25):38180–90. doi: 10.18632/oncotarget.9500
83. Xu L, Tsakmaklis N, Yang G, Chen JG, Liu X, Demos M, et al. Acquired Mutations Associated With Ibrutinib Resistance in Waldenström Macroglobulinemia. Blood (2017) 129(18):2519–25. doi: 10.1182/blood-2017-01-761726
84. Bartlett NL, Costello BA, LaPlant BR, Ansell SM, Kuruvilla JG, Reeder CB, et al. Single-Agent Ibrutinib in Relapsed or Refractory Follicular Lymphoma: A Phase 2 Consortium Trial. Blood (2018) 131(2):182–90. doi: 10.182/blood-2017-09-804641
85. Béziat V, Jouanguy E, Puel A. Dominant Negative CARD11 Mutations: Beyond Atopy. J Allergy Clin Immunol (2019) 143:1345–7. doi: 10.1016/j.jaci.2018.12.1006
86. Lu HY, Sharma M, Sharma AA, Lacson A, Szpurko A, Luider J, et al. Mechanistic Understanding of the Combined Immunodeficiency in Complete Human CARD11 Deficiency. J Allergy Clin Immunol (2021) 21)00612-6:S0091–6749. doi: 10.1016/j.jaci.2021.04.006
87. Snow AL, Xiao W, Stinson JR, Lu W, Chaigne-Delalande B, Zheng L, et al. Congenital B Cell Lymphocytosis Explained by Novel Germline CARD11 Mutations. J Exp Med (2012) 209(12):2247–61. doi: 10.1084/jem.20120831
88. Meitlis I, Allenspach EJ, Bauman BM, Phan IQ, Dabbah G, Schmitt EG, et al. Multiplexed Functional Assessment of Genetic Variants in CARD11. Am J Hum Genet (2020) 107(6):1029–43. doi: 10.1016/j.ajhg.2020.10.015
89. Lu HY, Bauman BM, Arjunaraja S, Dorjbal B, Milner JD, Snow AL, et al. The CBM-Opathies - A Rapidly Expanding Spectrum of Human Inborn Errors of Immunity Caused by Mutations in the CARD11-BCL10-MALT1 Complex. Front Immunol (2018) 9:2078. doi: 10.3389/fimmu.2018.02078
90. Roifman CM, Ke S. CD19 is a Substrate of the Antigen Receptor-Associated Protein Tyrosine Kinase in Human B Cells. Biochem Biophys Res Commun (1993) 194(1):222–5. doi: 10.1006/bbrc.1993.1807
91. Tuveson DA, Carter RH, Soltoff SP, Fearon DT. CD19 of B Cells as a Surrogate Kinase Insert Region to Bind Phosphatidylinositol 3-Kinase. Science (1993) 260(5110):986–9. doi: 10.1126/science.7684160
92. Sadras T, Martin M, Kume K, Robinson ME, Saravanakumar S, Lenz G, et al. Developmental Partitioning of SYK and ZAP70 Prevents Autoimmunity and Cancer. Mol Cell (2021) 2100264-1:S1097–2765. doi: 10.1016/j.molcel.2021.03.043
93. Salim K, Bottomley MJ, Querfurth E, Zvelebil MJ, Gout I, Scaife R, et al. Distinct Specificity in the Recognition of Phosphoinositides by the Pleckstrin Homology Domains of Dynamin and Bruton’s Tyrosine Kinase. EMBO J (1996) 15(22):6241–50. doi: 10.1002/j.1460-2075.1996.tb01014.x
94. Buhl AM, Cambier JC. Phosphorylation of CD19 Y484 and Y515, and Linked Activation of Phosphatidylinositol 3-Kinase, Are Required for B Cell Antigen Receptor-Mediated Activation of Bruton’s Tyrosine Kinase. J Immunol (1999) 1162(8):4438–46.
95. Nore BF, Vargas L, Mohamed AJ, Brandén LJ, Bäckesjö CM, Islam TC, et al. Redistribution of Bruton’s Tyrosine Kinase by Activation of Phosphatidylinositol 3-Kinase and Rho-Family Gtpases. Eur J Immunol (2000) 30(1):145–54. doi: 10.1002/1521-4141(200001)30:1<145::AID-IMMU145>3.0.CO;2-0
96. Kapoor I, Li Y, Sharma A, Zhu H, Bodo J, Xu W, et al. Resistance to BTK Inhibition by Ibrutinib Can be Overcome by Preventing FOXO3a Nuclear Export and PI3K/AKT Activation in B-Cell Lymphoid Malignancies. Cell Death Dis (2019) 10(12):924. doi: 10.1038/s41419-019-2158-0
97. Seda V, Vojackova E, Ondrisova L, Kostalova L, Sharma S, Loja T, et al. Foxo1-GAB1 Axis Regulates Homing Capacity and Tonic AKT Activity in Chronic Lymphocytic Leukemia. Blood (2021) 2020008101. doi: 10.1182/blood.2020008101
98. Zhao X, Lwin T, Silva A, Shah B, Tao J, Fang B, et al. Unification of De Novo and Acquired Ibrutinib Resistance in Mantle Cell Lymphoma. Nat Commun (2017) 8:14920. doi: 10.1038/ncomms14920
99. Zhao X, Wang MY, Jiang H, Lwin T, Park PM, Gao J, et al. Transcriptional Programming Drives Ibrutinib-Resistance Evolution in Mantle Cell Lymphoma. Cell Rep (2021) 34(11):108870. doi: 10.1016/j.celrep.2021.108870
100. Prior IA, Hood FE, Hartley JL. The Frequency of Ras Mutations in Cancer. Cancer Res (2020) 80(14):2969–74. doi: 10.1158/0008-5472.CAN-19-3682
101. Sauer K, Cooke MP. Regulation of Immune Cell Development Through Soluble Inositol-1,3,4,5-Tetrakisphosphate. Nat Rev Immunol (2010) 10(4):257–71. doi: 10.1038/nri2745
102. Ishitsuka K, Tamura K. Human T-Cell Leukaemia Virus Type I and Adult T-Cell Leukaemia-Lymphoma. Lancet Oncol (2014) 15(11):e517–26. doi: 10.1016/S1470-2045(14)70202-5
103. Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, et al. Integrated Molecular Analysis of Adult T Cell Leukemia/Lymphoma. Nat Genet (2015) 47(11):1304–15. doi: 10.1038/ng.3415
104. Vallabhapurapu S, Karin M. Regulation and Function of NF-κB Transcription Factors in the Immune System. Annu Rev Immunol (2009) 27:693–733. doi: 10.1146/annurev.immunol.021908.132641
105. Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, et al. Cancer-Associated Protein Kinase C Mutations Reveal Kinase’s Role as Tumor Suppressor. Cell (2015) 160(3):489–502. doi: 10.1016/j.cell.2015.01.001
106. Leitges M, Schmedt C, Guinamard R, Davoust J, Schaal S, Stabel S, et al. Immunodeficiency in Protein Kinase Cbeta-Deficient Mice. Science (1996) 273(5276):788–91. doi: 10.1126/science.273.5276.788
107. Vigorito E, Gambardella L, Colucci F, McAdam S, Turner M. Vav Proteins Regulate Peripheral B-Cell Survival. Blood (2005) 106(7):2391–8. doi: 10.1182/blood-2004-12-4894
108. Ahn IE, Underbayev C, Albitar A, Herman SE, Tian X, Maric I, et al. Clonal Evolution Leading to Ibrutinib Resistance in Chronic Lymphocytic Leukemia. Blood (2017) 129(11):1469–79. doi: 10.1182/blood-2016-06-719294
109. Komarova NL, Burger JA, Wodarz D. Evolution of Ibrutinib Resistance in Chronic Lymphocytic Leukemia (CLL). Proc Natl Acad Sci USA (2014) 111(38):13906–11. doi: 10.1073/pnas.1409362111
110. Wodarz D, Garg N, Komarova NL, Benjamini O, Keating MJ, Wierda WG, et al. Kinetics of CLL Cells in Tissues and Blood During Therapy With the BTK Inhibitor Ibrutinib. Blood (2014) 123(26):4132–5. doi: 10.1182/blood-2014-02-554220
111. Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. Mir-15 and Mir-16 Induce Apoptosis by Targeting BCL2. Proc Natl Acad Sci USA (2005) 102(39):13944–9. doi: 10.1073/pnas.0506654102
112. Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations Driving CLL and Their Evolution in Progression and Relapse. Nature (2015) 526(7574):525–30. doi: 10.1038/nature15395
113. Puente XS, Beà S, Valdés-Mas R, Villamor N, Gutiérrez-Abril J, Martín-Subero JI, et al. Non-Coding Recurrent Mutations in Chronic Lymphocytic Leukaemia. Nature (2015) 526(7574):519–24. doi: 10.1038/nature14666
114. Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P Somatic Mutation in Waldenström’s Macroglobulinemia. N Engl J Med (2012) 367(9):826–33. doi: 10.1056/NEJMoa1200710
115. Treon SP, Xu L, Guerrera ML, Jimenez C, Hunter ZR, Liu X, et al. Genomic Landscape of Waldenström Macroglobulinemia and Its Impact on Treatment Strategies. J Clin Oncol (2020) 38(11):1198–208. doi: 10.1200/JCO.19.02314
116. Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and Impact of Subclonal Mutations in Chronic Lymphocytic Leukemia. Cell (2013) 152(4):714–26. doi: 10.1016/j.cell.2013.01.019
117. Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, et al. A Multiprotein Supercomplex Controlling Oncogenic Signalling in Lymphoma. Nature (2018) 560(7718):387–91. doi: 10.1038/s41586-018-0290-0
118. Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM, et al. An Oligomeric Signaling Platform Formed by the Toll-Like Receptor Signal Transducers Myd88 and IRAK-4. J Biol Chem (2009) 284(37):25404–11. doi: 10.1074/jbc.M109.022392
119. Rip J, de Bruijn MJW, Appelman MK, Pal Singh S, Hendriks RW, Corneth OBJ. Toll-Like Receptor Signaling Drives Btk-Mediated Autoimmune Disease. Front Immunol (2019) 10:95. doi: 10.3389/fimmu.2019.00095
120. Giménez N, Martínez-Trillos A, Montraveta A, Lopez-Guerra M, Rosich L, Nadeu F, et al. Mutations in the RAS-BRAF-MAPK-ERK Pathway Define a Specific Subgroup of Patients With Adverse Clinical Features and Provide New Therapeutic Options in Chronic Lymphocytic Leukemia. Haematologica (2019) 104(3):576–86. doi: 10.3324/haematol.2018.196931
121. Murali I, Kasar S, Naeem A, Tyekucheva S, Khalsa JK, Thrash EM, et al. Activation of the MAPK Pathway Mediates Resistance to PI3K Inhibitors in Chronic Lymphocytic Leukemia (Cll). Blood (2021) 2020006765. doi: 10.1182/blood.2020006765
122. Davids MS, Kim HT, Nicotra A, Savell A, Francoeur K, Hellman JM, et al. Umbralisib in Combination With Ibrutinib in Patients With Relapsed or Refractory Chronic Lymphocytic Leukaemia or Mantle Cell Lymphoma: A Multicentre Phase 1-1b Study. Blood Cancer Research Partnership of the Leukemia and Lymphoma Society. Lancet Haematol (2019) 6(1):e38–47. doi: 10.1016/S2352-3026(18)30196-0
123. Mato AR, Ghosh N, Schuster SJ, Lamanna N, Pagel JM, Flinn IW, et al. Phase 2 Study of the Safety and Efficacy of Umbralisib in Patients With CLL Who are Intolerant to BTK or PI3Kδ Inhibitor Therapy. Blood press (2021) 137(20):2817–26. doi: 10.1182/blood.2020007376
124. Schuh K, Avots A, Tony HP, Serfling E, Kneitz C. Nuclear NF-Atp is a Hallmark of Unstimulated B Cells From B-CLL Patients. Leuk Lymphoma (1996) 23(5-6):583–92. doi: 10.3109/10428199609054868
125. Wolf C, Garding A, Filarsky K, Bahlo J, Robrecht S, Becker N, et al. NFATC1 Activation by DNA Hypomethylation in Chronic Lymphocytic Leukemia Correlates With Clinical Staging and Can be Inhibited by Ibrutinib. Int J Cancer (2018) 142(2):322–33. doi: 10.1002/ijc.31057
126. Le Roy C, Deglesne PA, Chevallier N, Beitar T, Eclache V, Quettier M, et al. The Degree of BCR and NFAT Activation Predicts Clinical Outcomes in Chronic Lymphocytic Leukemia. Blood (2012) 120(2):356–65. doi: 10.1182/blood-2011-12-397158
127. Chang BY, Francesco M, De Rooij MF, Magadala P, Steggerda SM, Huang MM, et al. Egress of CD19(+)CD5(+) Cells Into Peripheral Blood Following Treatment With the Bruton Tyrosine Kinase Inhibitor Ibrutinib in Mantle Cell Lymphoma Patients. Blood (2013) 122(14):2412–24. doi: 10.1182/blood-2013-02-482125
128. de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, et al. The Clinically Active BTK Inhibitor PCI-32765 Targets B-Cell Receptor- and Chemokine-Controlled Adhesion and Migration in Chronic Lymphocytic Leukemia. Blood (2012) 119(11):2590–4. doi: 10.1182/blood-2011-11-390989
129. Chen SS, Chang BY, Chang S, Tong T, Ham S, Sherry B, et al. Expression, Signaling and Function in Chronic Lymphocytic Leukemia. Leukemia (2016) 30(4):833–43. doi: 10.1038/leu.2015.316
130. Pavlasova G, Borsky M, Seda V, Cerna K, Osickova J, Doubek M, et al. Ibrutinib Inhibits CD20 Upregulation on CLL B Cells Mediated by the CXCR4/SDF-1 Axis. Blood (2016) 128(12):1609–13. doi: 10.1182/blood-2016-04-709519
131. Cao Y, Hunter ZR, Liu X, Xu L, Yang G, Chen J, et al. The WHIM-Like CXCR4(S338X) Somatic Mutation Activates AKT and ERK, and Promotes Resistance to Ibrutinib and Other Agents Used in the Treatment of Waldenstrom’s Macroglobulinemia. Leukemia (2015) 29(1):169–76. doi: 10.1038/leu.2014.187
132. Della Mina E, Guérin A, Tangye SG. Molecular Requirements for Human Lymphopoiesis as Defined by Inborn Errors of Immunity. Stem Cells (2021) 39(4):389–402. doi: 10.1002/stem.3327
133. Berglöf A, Turunen JJ, Gissberg O, Bestas B, Blomberg KE, Smith CIE. Agammaglobulinemia: Causative Mutations and Their Implications for Novel Therapies. Expert Rev Clin Immunol (2013) 9(12):1205–21. doi: 10.1586/1744666X.2013.850030
134. Wang L, Aschenbrenner D, Zeng Z, Cao X, Mayr D, Mehta M, et al. Gain-of-Function Variants in SYK Cause Immune Dysregulation and Systemic Inflammation in Humans and Mice. Nat Genet (2021) 53(4):500–10. doi: 10.1038/s41588-021-00803-4
135. Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell (2017) 171(2):481–494.e15. doi: 10.1016/j.cell.2017.09.027
136. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med (2018) 378(15):1396–407. doi: 10.1056/NEJMoa1801445
137. Ruhe JE, Streit S, Hart S, Wong CH, Specht K, Knyazev P, et al. Genetic Alterations in the Tyrosine Kinase Transcriptome of Human Cancer Cell Lines. Cancer Res (2007) 67(23):11368–76. doi: 10.1158/0008-5472.CAN-07-2703
138. Streubel B, Vinatzer U, Willheim M, Raderer M, Chott A. Novel T(5;9)(Q33;Q22) Fuses ITK to SYK in Unspecified Peripheral T-Cell Lymphoma. Leukemia. (2006) 20(2):313–8. doi: 10.1038/sj.leu.2404045
139. Fathi NN, Mohammad DK, Görgens A, Andaloussi SE, Zain R, Nore BF, et al. Translocation-Generated ITK-FER and ITK-SYK Fusions Induce STAT3 Phosphorylation and CD69 Expression. Biochem Biophys Res Commun (2018) 504(4):749–52. doi: 10.1016/j.bbrc.2018.09.019
140. Ortutay C, Nore BF, Vihinen M, Smith CIE. Phylogeny of Tec Family Kinases: Identification of a Pre-Metzoan Origin of Btk, Bmx, Itk, Tec, Txk and the Btk Regulator SH3BP5. Adv Genet (2008) 64:51–80. doi: 10.1016/S0065-2660(08)00803-1
141. Väliaho J, Smith CI, Vihinen M. Btkbase: The Mutation Database for X-Linked Agammaglobulinemia. Hum Mutat (2006) 27(12):1209–17. doi: 10.1002/humu.20410
142. Saijo K, Schmedt C, Su IH, Karasuyama H, Lowell CA, Reth M, et al. Essential Role of Src-Family Protein Tyrosine Kinases in NF-Kappab Activation During B Cell Development. Nat Immunol (2003) 24(3):274–9. doi: 10.1038/ni893
143. Cheng AM, Rowley B, Pao W, Hayday A, Bolen JB, Pawson T. Syk Tyrosine Kinase Required for Mouse Viability and B-Cell Development. Nature (1995) 378(6554):303–6. doi: 10.1038/378303a0
144. Turner M, Mee PJ, Costello PS, Williams O, Price AA, Duddy LP, et al. Perinatal Lethality and Blocked B-Cell Development in Mice Lacking the Tyrosine Kinase Syk. Nature (1995) 378(6554):298–302. doi: 10.1038/378298a0
145. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
146. von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, et al. Pyogenic Bacterial Infections in Humans With Myd88 Deficiency. Science (2008) 321(5889):691–6. doi: 10.1126/science.1158298
147. Almutairi A, Wallace JG, Jaber F, Alosaimi MF, Jones J, Sallam MTH, et al. Severe Combined Immunodeficiency Caused by Inositol-Trisphosphate 3-Kinase B (ITPKB) Deficiency. J Allergy Clin Immunol (2020) 145(6):1696–1699.e6. doi: 10.1016/j.jaci.2020.01.014
148. Tajan M, Paccoud R, Branka S, Edouard T, Yart A. The Rasopathy Family: Consequences of Germline Activation of the RAS/MAPK Pathway. Endocr Rev (2018) 39(5):676–700. doi: 10.1210/er.2017-00232
Keywords: Btk, ibrutinib, B cell receptor, chronic lymphocytic leukemia, mantle cell lymphoma, Waldenström’s macroglobulinemia, resistance mutation, CARD11
Citation: Smith CIE and Burger JA (2021) Resistance Mutations to BTK Inhibitors Originate From the NF-κB but Not From the PI3K-RAS-MAPK Arm of the B Cell Receptor Signaling Pathway. Front. Immunol. 12:689472. doi: 10.3389/fimmu.2021.689472
Received: 31 March 2021; Accepted: 05 May 2021;
Published: 10 June 2021.
Edited by:
Laura Mandik-Nayak, Lankenau Institute for Medical Research, United StatesReviewed by:
Marek Mraz, Central European Institute of Technology (CEITEC), CzechiaMarcel Spaargaren, Academic Medical Center, Netherlands
Copyright © 2021 Smith and Burger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: C. I. Edvard Smith, edvard.smith@ki.se