 Esha Sehanobish
Esha Sehanobish Mohammad Asad
Mohammad Asad Mali Barbi
Mali Barbi Steven A. Porcelli1,2
Steven A. Porcelli1,2- 1Department of Medicine, Albert Einstein College of Medicine, Bronx, NY, United States
- 2Department of Microbiology and Immunology, Albert Einstein College of Medicine, Bronx, NY, United States
Non-steroidal Anti-inflammatory drugs (NSAID)-exacerbated respiratory disease (N-ERD) is characterized by nasal polyposis, chronic rhinosinusitis, adult-onset asthma and hypersensitive reactions to cyclooxygenase-1 (COX-1) inhibitors. Among the available treatments for this disease, a combination of endoscopic sinus surgery followed by aspirin desensitization and aspirin maintenance therapy has been an effective approach. Studies have shown that long-term aspirin maintenance therapy can reduce the rate of nasal polyp recurrence in patients with N-ERD. However, the exact mechanism by which aspirin can both trigger and suppress airway disease in N-ERD remains poorly understood. In this review, we summarize current knowledge of aspirin effects in N-ERD, cardiovascular disease, and cancer, and consider potential mechanistic pathways accounting for the effects of aspirin in N-ERD.
Introduction
NSAID Exacerbated Respiratory Disease (N-ERD) is characterized by nasal polyp formation, asthma, and hypersensitivity to all cyclooxygenase-1 (COX-1) inhibitors, which are commonly used non-steroidal anti-inflammatory drugs (NSAIDs). COX-1 catalyzes the production of prostaglandins, thromboxanes and prostacyclins from arachidonic acid, and NSAIDs act by blocking the ability of COX-1 to initiate the biosynthesis of these mediators by catalyzing oxidation of arachidonic acid (1). The earliest known description of N-ERD in the medical literature can be traced to a 1922 article by Widal et al. (2) Over four decades later, Samter and Beers provided a more systematic characterization of the combination of aspirin-intolerance with nasal polyps and asthma, referred to initially as Samter’s triad (3, 4). Subsequently, this became known as aspirin exacerbated respiratory disease (AERD), and more recently as N-ERD to more accurately reflect its association with hypersensitivity to all drugs that inhibit COX-1 (5, 6). Clinically, N-ERD is characterized by an adult onset of severe nasal congestion followed by chronic rhinosinusitis, and eventually by the development of nasal polyps. Asthma is frequently although not universally present in N-ERD (7). Nasal polyps, which are benign growths in the paranasal sinuses that can obstruct airflow leading to difficulty in breathing and loss of olfactory sense, are a key feature of N-ERD, and the severity of sinonasal symptoms due to nasal polyps is often correlated with the severity of asthma symptoms (8). In addition, endoscopic sinus surgery decreases or abolishes reactions to aspirin in most N-ERD patients, and improves long term responses to aspirin desensitization and maintenance treatment (9, 10).
In N-ERD, the underlying inflammatory process of the upper and lower respiratory systems begins and occurs independently of the NSAID consumption. However, intake of COX-1 inhibitors triggers symptoms resembling an allergic reaction or an asthma attack (11). Around 15% of patients with N-ERD may be unaware of their hypersensitivity to these drugs (12). Surveillance studies indicate that N-ERD is not a rare condition. From one study by Rajan et al. it was found that approximately 7% of all adult asthmatics have N-ERD (13). The prevalence of N-ERD is higher in severe asthma patients (15%) followed by patients with nasal polyps (10%) (13). N-ERD patients have greater morbidity than aspirin tolerant asthma patients, as characterized by more frequent corticosteroid bursts, increased hospitalizations and emergency visits, and lower baseline forced expiratory volume in 1 second (FEV1) (12, 14).
Among treatment approaches that have shown benefit in N-ERD, aspirin desensitization followed by a continuous long-term aspirin administration has yielded significant improvement in the clinical course of many patients (15–19). This treatment reduces asthma and sinonasal symptoms, daily use of corticosteroids, ER visits or hospitalization due to asthma, and sinus infections (20, 21). Desensitization and aspirin maintenance therapy reduces and delays the occurrence of nasal polyps in more than 70% of N-ERD patients (22, 23). Even though the beneficial effects of aspirin desensitization and maintenance therapy are now accepted by many practitioners, the mechanisms, by which aspirin leads to the suppression of clinical symptoms and to polyp prevention are not well understood. In this review we focus on pathways by which aspirin can exert anti-inflammatory effects and prevent the recurrence of the nasal polyps in N-ERD.
Immunological Features of N-ERD
N-ERD is characterized by increased numbers of eosinophils and mast cells, upregulation of type-2 (T2) pro-inflammatory cytokines, and abnormalities in the production of cysteinyl leukotrienes and prostaglandins (24–29). Hypersensitivity reactions are caused by inhibition of COX-1 by the NSAIDs. Even though increases in several inflammatory mediators such as cysteinyl leukotrienes and prostaglandins are associated with N-ERD, the exact events that result in the initiation and the progression of the disease are not yet known.
Involvement of Leukotrienes and Their Receptors in N-ERD
5-lipoxygenase (5-LO) mediated oxidation of arachidonic acid produces cysteinyl leukotrienes (cysLTs) and leukotriene B4 (LTB4).
CysLTs
Leukotrienes are a family of inflammatory lipid mediators that are synthesized from arachidonic acid by eosinophils, mast cells, macrophages, neutrophils and basophils (30–33). Leukotrienes C4 (LTC4), D4 (LTD4) and E4 (LTE4) are collectively known as cysLTs due to the presence of a cysteine residue. In neutrophils, monocytes, eosinophils, mast cells, and basophils, arachidonic acid is oxidized by 5-lipoxygenase (5-LO) to produce an unstable metabolite, leukotriene A4 (LTA4) (34). In monocytes, mast cells, eosinophils, and basophils, LTA4 upon conjugation with glutathione is then converted to LTC4 by leukotriene C4 synthase (LTC4S) (35–37). LTC4 is then extracellularly converted to LTD4 and its stable metabolite, LTE4 (38–40). LTC4, LTD4 and LTE4 are potent bronchoconstrictors in human and in other species (41–44). LTD4 functions via its interaction with a G protein-coupled receptor, cysteinyl leukotriene receptor 1 (CysLT1R), which has a lower affinity for LTC4 and LTE4. In addition, LTD4 and LTE4 have a similar affinity to another leukotriene receptor, cysteinyl leukotriene receptor 2 (CysLT2R), also a G-protein coupled receptor. There may also be other less investigated cysLT receptors that could be involved in the action of these lipid mediators (45, 46).
The over-production of cysteinyl leukotrienes is a characteristic feature of N-ERD (26, 35). Urinary levels of LTE4 are elevated at baseline and increase several fold after aspirin challenge in N-ERD patients compared to aspirin-tolerant asthma patients (47, 48). Several possible mechanisms have been proposed to account for the overproduction of cysLTs. For example, eosinophils and mast cells can produce cysLTs, raising the possibility that the overproduction of cysLTs is a result of the higher numbers or an increased activation of these cells in respiratory tissue of N-ERD patients (24, 25). The two main enzymes involved in the production of these lipid mediators are 5-LO and LTC4S. In case of biopsies from N-ERD patients, the percentages of mast cells and eosinophils staining positive for 5-LO was much higher than in the biopsies from the aspirin tolerant patients (36, 49). Platelets too can express LTC4S, and upon adhering to leukocytes they are capable of using the leukocyte-derived LTA4 as a substrate for conversion to LTC4. In this regard, it is interesting that N-ERD patients had a higher percentage of platelet-adherent leukocytes than aspirin-tolerant asthma patients (35). Another reason for the overproduction of cysLTs is related to the decrease in the prostaglandin E2 (PGE2) levels in nasal tissue samples including nasal polyps. Low PGE2 levels may contribute to the shift of the arachidonic acid metabolism pathway towards leukotriene synthesis. PGE2 inhibits cysLT production in people with N-ERD via the inhibition of 5-LO (50, 51). Therefore, the overexpression of cysLTs in N-ERD is likely a result of an increased activity of 5-LO and LTC4S, and an increased availability of the LTC4S due to the increase of platelet-adherent leukocytes (52).
Overexpression of cysteinyl leukotriene receptor CysLT1R in nasal biopsies of N-ERD patients has also been reported (53). The mRNA and the protein expression of CysLT1R can be increased by interleukin-13 (IL-13) and inteleukin-4 (IL-4), as observed in human monocytes and monocyte-derived macrophages (54). Both of these cytokines have an increased expression in N-ERD, suggesting that they could act as important drivers of pathology in N-ERD at least in part by augmenting cysLT receptor expression. Potentially, genetic polymorphisms in cysLT receptor genes could contribute to this mechanism of disease pathogenesis, although such polymorphisms have not yet been described in N-ERD. Little is currently known about the involvement of CysLT2R in N-ERD.
LTB4
5-LO catalyzes the conversion of arachidonic acid to LTA4 (34). LTA4 can then be converted either to cysLTs as mentioned in the above section or can be converted to LTB4 by LTA4 hydrolase in neutrophils and eosinophils (55). LTB4 is a pro-inflammatory lipid mediator that functions as chemoattractant for eosinophils, monocytes, macrophages, and neutrophils (56–58). LTB4 can activate leukocytes via its interaction with a G-protein-coupled surface receptor BLT1 (59). LTB4 concentrations are elevated in the bronchoalveolar lavage fluid from patients with severe asthma, with eosinophils being the major source of LTB4 production (60). In N-ERD, along with an increase in the urinary level of the cysLT, LTE4, there is an increase in the urinary levels of an LTB4 metabolite during reactions to COX-1 inhibitions (61).
Involvement of Prostaglandins and Their Receptors in N-ERD
Prostaglandins are lipid molecules derived from arachidonic acid by the action of the COX enzymes via endoperoxide intermediates such as prostaglandin G2 (PGG2) and H2 (PGH2). The latter leads to the production of the five main bioactive prostaglandins, prostaglandins D2 (PGD2), E2 (PGE2), F2α (PGF2α), prostacyclin (PGI2) and thromboxane A2 (TXA2) (62). Three of these (PGD2, PGE2 and TXA2) have been implicated in the pathogenesis of N-ERD.
PGD2
PGD2 is the main product of COX-derived intermediates in mast cells (63). It is also produced by eosinophils, although the lower expressions of the terminal PGD2 synthase gene compared to mast cells makes eosinophils a smaller source of this prostaglandin (64). PGD2 performs a wide range of functions through its interaction with its receptors: the thromboxane prostanoid (TP) receptors, and D-prostanoid (DP) receptors DP1 and DP2. The DP2 receptor is also known as chemoattractant receptor-homologous molecule expressed on TH2 cells (CRTH2). Upon binding to the TP receptor, PGD2 can function as a bronchoconstrictor and can also function as a potent chemoattractant for eosinophils and basophils via its interaction with CRTH2 (65–67). PGD2 can mediate vasorelaxation, inhibition of platelet aggregation, and bronchodilation primarily through stimulation of the DP1 receptor (68), and its signaling through this receptor also exerts an anti-inflammatory role in allergic inflammation (69). However, the functions of the DP1 and DP2 receptors in allergic inflammation are considered to be antagonistic (70), with DP1 stimulation resulting in mostly anti-inflammatory effects such as inhibition of cell migration, vasodilation, eosinophil apoptosis (71), whereas DP2 triggers pro-inflammatory effects by upregulating type 2 cytokines in Th2 cells (72, 73).
N-ERD is characterized by an overexpression of PGD2, as shown by the higher concentration of PGD2 in the sputum of N-ERD patients compared to aspirin tolerant asthma patients (74). N-ERD patients who have higher baseline levels of PGD2-metabolite in urine experience more severe clinical reactions to aspirin (75, 76), and the inability to tolerate aspirin desensitization has been attributed to the high levels of urinary PGD-metabolite (75). There are various mechanisms responsible for the high expression of PGD2 in N-ERD. Nasal polyps of N-ERD patients have a high concentration of mast cells and eosinophils which both express hematopoietic prostaglandin D synthase required for the synthesis of the PGD2 (64, 77, 78). Nasal polyp tissue and bronchial mucosa of asthmatic patients have been found to be rich in the cytokine thymic stromal lymphopoietin (TSLP), which can stimulate PGD2 production by mast cells (79). Nasal polyps have increased expression of TSLP compared to healthy nasal tissues (80–82), and Buchheit et al. showed that the levels of TSLP were significantly higher in the nasal polyps of N-ERD patients compared to nasal polyps of aspirin-tolerant asthma patients (64). Additionally in N-ERD patients, the levels of PGD2 paradoxically increase in plasma and urine after aspirin challenge, and correlate with the severity of the reaction to aspirin (64, 76, 83, 84). This is counterintuitive, considering that aspirin inhibits COX enzymes that represent a key step in prostaglandin synthesis, and could indicate an alternate pathway for PGD2 synthesis. Alternatively, this elevation of PGD2 could reflect effects of aspirin on targets other than COX enzymes, or indirect effects resulting from increased levels of leukotrienes (85).
PGE2
In contrast to PGD2, the levels of PGE2 in N-ERD are greatly reduced both in peripheral blood cells and nasal tissue samples, including nasal polyps (86, 87). There are 4 subtypes of the E-prostanoid (EP) receptor to which PGE2 binds, designated as EP1, EP2, EP3 and EP4 (88, 89). PGE2 can function both as a pro- and an anti-inflammatory mediator. PGE2 may exhibit opposing functions via interaction with different EP receptors based on the cell type and the location (90). The pro-inflammatory effects of PGE2 is usually observed in various inflammatory conditions such as arthritis, inflammatory bowel disease, and also in different types of cancers (91–94).However, PGE2 exerts anti-inflammatory and bronchoprotective effects in the airways by suppressing allergen induced-inflammatory responses (95, 96). In N-ERD, PGE2 functions as an anti-inflammatory mediator as it prevents both airway obstruction and the increase in the urinary LTE4 associated with aspirin challenge in N-ERD (50, 97). PGE2 exerts its anti-inflammatory effects in the airways through the EP2 and EP4 receptors by activating protein kinase A (PKA). PGE2 binds to EP2 and EP4 receptors that activate adenylate cyclase. Next adenylate cyclase increases the levels of cellular cyclic AMP. This in turn activates PKA (98). PKA then phosphorylates 5-LO, thus directly inhibiting the catalytic activity of 5-LO and regulating the leukotriene synthesis by working as a brake for the production of cysLT (51). PGE2 signaling through its EP2 receptor blocks mast cell degranulation, limits eosinophil migration, and inhibits the allergen-stimulated release of mast cell-derived inflammatory mediators including PGD2 in the airways of asthma patients, providing another possible mechanism for its bronchoprotective action (99, 100).
The reduced expression and function of PGE2 in N-ERD could be attributed to a combination of factors. One of the enzymes responsible for the synthesis of PGE2 is COX-2, and expression of the mRNA for this enzyme is markedly reduced in nasal polyps of N-ERD patients compared to polyps and normal mucosa of aspirin-tolerant asthma patients (101). There is also diminished expression of EP2 receptors on mast cells and nasal fibroblasts of N-ERD patients (102, 103). Since PGE2 exerts its anti-inflammatory activities mainly through EP2, a reduced expression of the latter could decrease the ability of PGE2 to perform its anti-inflammatory actions in N-ERD patients. The reduction in PGE2 observed in N-ERD may also account at least in part for the increased baseline levels of PGD2 that are generally observed in N-ERD. Consistent with this, an elevated PGD2/PGE2 ratio in the nasal polyps of patients with chronic rhinosinusitis is often indicative of N-ERD (104). In addition, a reduction in the level of PGE2 in the nasal polyps of N-ERD patients may remove the normal brake on cysLT production, leading to the increased levels of leukotrienes (89). Consistent with the proposed important role of PGE2 deficiency in N-ERD, studies have shown inhaled PGE2 to serve as a bronchodilator in N-ERD patients (50, 105).
TXA2
TXA2 is the main COX-product derived from platelets (106). It is a potent unstable vasoconstrictor and hydrolyzes to an inactive but stable form, thromboxane B2 (TXB2) (107). TXA2 is a pro-inflammatory prostanoid involved in platelet aggregation and activation, and facilitates leukocyte recruitment (108, 109). Studies in animal models show that TXA2 can function as a potent bronchoconstrictor and mediates its effects through interaction with the TP receptors (110–112). Similar to PGD2, an overexpression of TXA2 is also associated with the pathogenesis of N-ERD. The basal urinary level of the stable thromboxane metabolite (TX-M) is found to be higher in N-ERD patients espcially those who are unable to tolerate aspirin desensitization as a treatment (75). The exact mechanism for the overexpression of TXA2 is not known.
Involvement of Lipoxins in N-ERD
In 1984, Serhan et al. first described a new set of oxygenated derivatives of arachidonic acid isolated from human leukocytes that were different from the other eicosanoids as they contained a conjugated tetraene structure (113). In a follow-up study, these trihydroxytetraenes, generated from the interactions of the 5- and 15-lipoxygenase (5-LO and 15-LO) pathways of human leukocytes, were designated lipoxins and considered to be a newly recognized series of arachidonic acid metabolites. The two main products were designated lipoxin A4 and lipoxin B4 (114). There are two major pathways for lipoxin biosynthesis. One of these involves platelet-leukocyte interactions in which leukocyte-produced LTA4 is converted to lipoxin A4 and lipoxin B4, by platelet 12-lipoxygenase (115). The other pathway involves the action of 15-LO on arachidonic acid to produce 15(S)-hydroxyeicosatetraenoic acid (15(S)-HETE) which is then used as a substrate by 5-LO to produce lipoxins (116). 5-LO is involved in the production of both leukotrienes and lipoxins, and is thus an integral part of both pro- and anti-inflammatory pathways.
Lipoxins are produced by a variety of cells, including airway epithelium, platelets and eosinophils, and they perform functions different than CysLTs (117, 118). Unlike CysLTs which mainly function as bronchoconstrictors, lipoxins exert anti-inflammatory properties and inhibit bronchoconstriction (119). Lipoxins inhibit eosinophilic and neutrophilic migration dampening the airway hyperreactivity and allergic airway inflammation (120–122). The anti-inflammatory effects of lipoxins are mediated by interaction with a high-affinity, G-protein-coupled receptor called ALX/FPR2 (lipoxin receptor (ALX)/N-formyl peptide receptor (FPR)-2) (120, 123–125). These lipid mediators bind to ALX/FPR2 present on T and B cells, and regulate B and T cell-mediated responses during resolution of inflammation (126, 127). ALX/FPR2 receptors are also found on natural killer and innate lymphoid type 2 cells, and lipoxin A4 inhibits inflammatory responses by these cells (128, 129). Apart from this, lipoxin A4 also binds to the CysLT1R with equal affinity as LTD4 and acts as an antagonist by regulating the action of LTD4 as well as leukocyte trafficking (130).
A characteristic feature of N-ERD is the diminished lipoxin levels, although the mechanism accounting for this remains unclear (117). At baseline, N-ERD patients have a lower level of lipoxins in plasma and blood leukocytes as compared to aspirin tolerant asthmatic patients (131, 132). The levels of lipoxins also correlate directly with the severity of asthma. Studies comparing N-ERD and aspirin tolerant asthma patients suggest a link between aspirin sensitivity and the ability to generate lipoxins (117). This may potentially involve altered metabolism of the lipoxin precursor 15(S)-HETE (133, 134). The low levels of lipoxins in N-ERD could be due to preferential conversion of 15(S)-HETE into 15-oxo-Eicosatetraenoic acid (15-oxo-ETE) rather than lipoxins (135). Another possible cause for reduced lipoxins in N-ERD could be the low levels of PGE2, since PGE2 is capable of switching lipid mediator biosynthesis from LTB4, a proinflammatory product of 5-LO, to the production of 15-LO product, lipoxin A4 (136). Additionally, there is a reduced expression of the ALX/FPR2 receptor genes in the peripheral blood of severe asthma patients, which included NERD patients, likely leading to an overall reduction in lipoxin function (137).
Another aspect of lipoxin biosynthesis that may be relevant to N-ERD is the finding that aspirin has different effects on the COX-1 and COX-2 enzymes, which leads to generation of bioactive anti-inflammatory tetraene eicosanoids known as aspirin-triggered 15-epi-lipoxins (138). Aspirin covalently modifies both COX-1 and COX-2 by acetylating their active site serine residues (139). Acetylation of COX-1 results in the complete inhibition of its activity of generating prostaglandins from arachidonic acid (1, 140). In contrast, the acetylation of a serine residue in COX-2 results in a switch from a prostaglandin-producing COX-2 into an enzyme that converts arachidonic acid to the R stereoisomer of 15-HETE (i.e., 15(R)-HETE) (138, 140). 15(R)-HETE is then metabolized in a transcellular manner by 5-LO in leukocytes to produce 15-epi-lipoxin. A defect in this aspirin-dependent pathway in N-ERD patients might be another mechanism contributing to their deficiency in lipoxins.
Treatment of N-ERD
There are several treatments that benefit N-ERD patients, including leukotriene modifiers, corticosteroids, anti-immunoglobulin E (IgE) monoclonal antibody, endoscopic sinus surgery, and aspirin desensitization and maintenance therapy (141). In this section we will focus on the beneficial effects of aspirin treatment in N-ERD and discuss possible mechanisms for its action.
Aspirin Desensitization and Maintenance Therapy
Although aspirin acts as a potent and rapid trigger of respiratory symptoms in N-ERD patients, extensive clinical experience shows that desensitization can be achieved in many patients and reinforced by continuous aspirin maintenance therapy. One of the earliest reports on aspirin benefits in the treatment of N-ERD was conducted in 1980 by Stevenson et al. They showed that aspirin-sensitive patients were able to continue on a daily dose of aspirin with subsequent symptom improvement and a decrease in daily corticosteroid dose (142). Since then, various short and long-term studies have been conducted that corroborate the benefits of aspirin desensitization followed by aspirin maintenance therapy in N-ERD (22, 143, 144). Aspirin desensitization in N-ERD involves a gradual administration of aspirin to the patient in a controlled setting. Available protocols suggest administration of 40 mg, 80, mg, 160, mg, and 325 mg every 60 to 90 minutes, until the patient is able to tolerate a dose of 325 mg of aspirin. Following aspirin desensitization, aspirin maintenance therapy is initiated, commonly with twice daily dose of 650 mg followed by tapering to 325 mg twice daily (145, 146). Clinically, N-ERD patients treated with aspirin maintenance regimens experience a reduction in their daily need for maintenance corticosteroid, improvement in clinical symptoms of asthma, and experience a significant reduction in ER visits or hospitalization due to asthma (16, 20, 147, 148). Endoscopic sinus surgery enhances the response to aspirin treatment, and can even create responses to aspirin maintenance in cases that previously failed aspirin treatment (9, 149). The effects of NSAIDs other than aspirin in alleviating the symptoms of N-ERD have not been extensively studied. At present, a combination of endoscopic sinus surgery followed by aspirin desensitization and aspirin maintenance therapy is widely used to treat N-ERD patients.
Potential Mechanism of Aspirin Action
The underlying mechanism of aspirin’s anti-inflammatory action in N-ERD is not completely understood. Long-term aspirin treatment has a beneficial effect in a wide range of diseases. Its therapeutic effects are well-established in cardiovascular disease, thalassemia, and various types of cancers with particular relevance to prevention of colorectal cancers (150–152). Table 1 summarizes the function of aspirin in different diseases. There is a suggestion that the continuous blocking of the COX-activity is generally associated with aspirin treatment benefits (18, 53, 176). Since aspirin is a powerful inhibitor of COX-1, long-term aspirin action has resulted in reduced levels of prostaglandin D2 (75, 177). However, there are difficulties in explaining the beneficial effects of aspirin maintenance therapy in N-ERD by solely considering known effects on the arachidonic acid pathway. For example, a potential pathway for alleviating the symptoms of N-ERD should include lowering urinary levels of LTE4 as a measure of cysLTs and increasing the levels of PGE2. However, high doses of aspirin treatment of N-ERD patients for eight weeks was found to either not change or increase urinary LTE4 levels (75, 177), and also induced a further reduction in urinary PGE-metabolite levels. Thus, aspirin therapy appears to reduce nasal symptoms in N-ERD patients without correcting any of the known defects in PGE2 and cysLT production. These findings imply involvement of other pathways that may be unrelated to direct COX-1/2 inhibition and the modulation of products of the cyclooxygenase and lipoxygenase pathways. Some of the possible alternative mechanisms are summarized in Figure 1 and are discussed in the sections that follow.
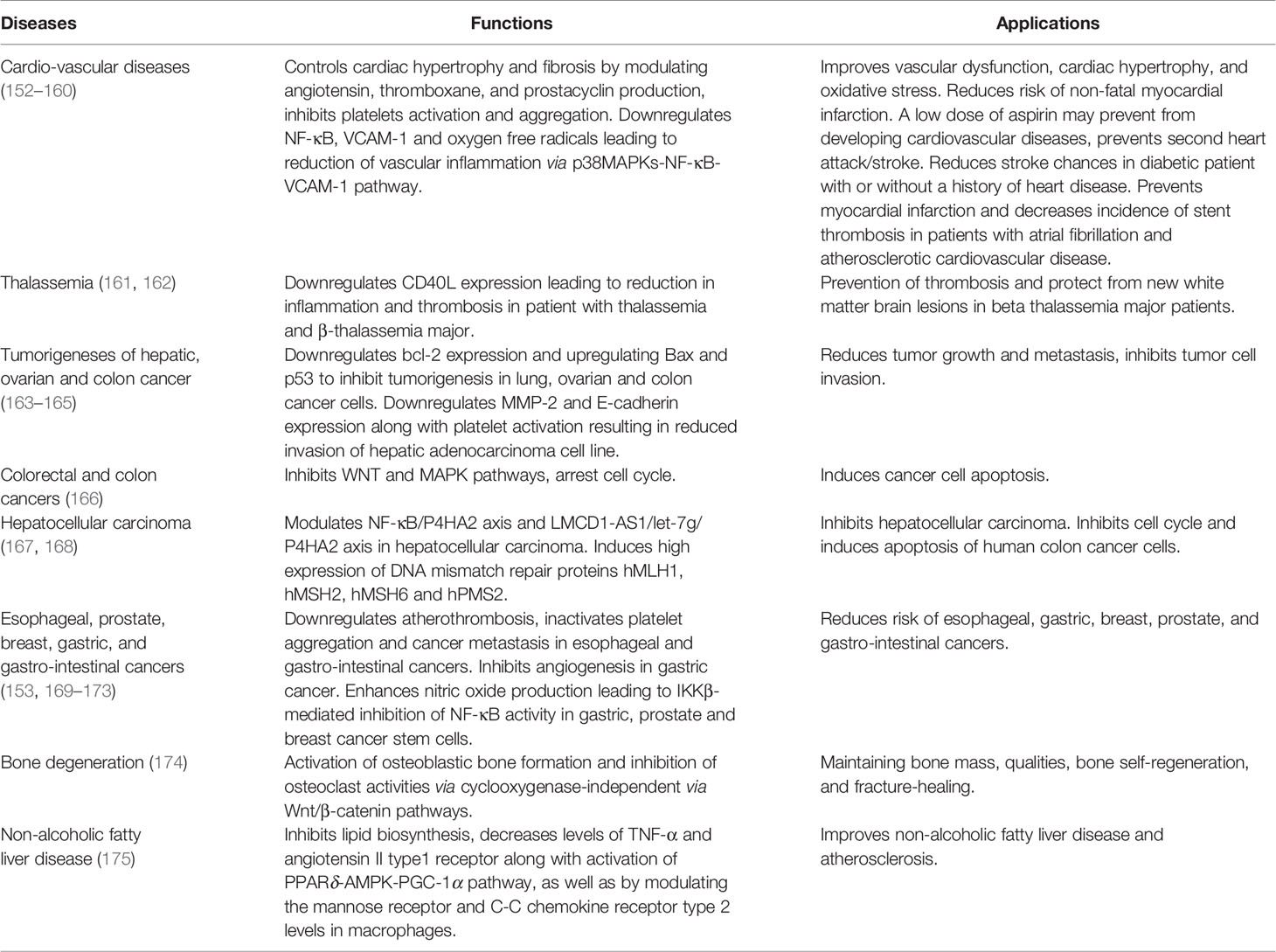
Table 1 Effect of Aspirin on various diseases.
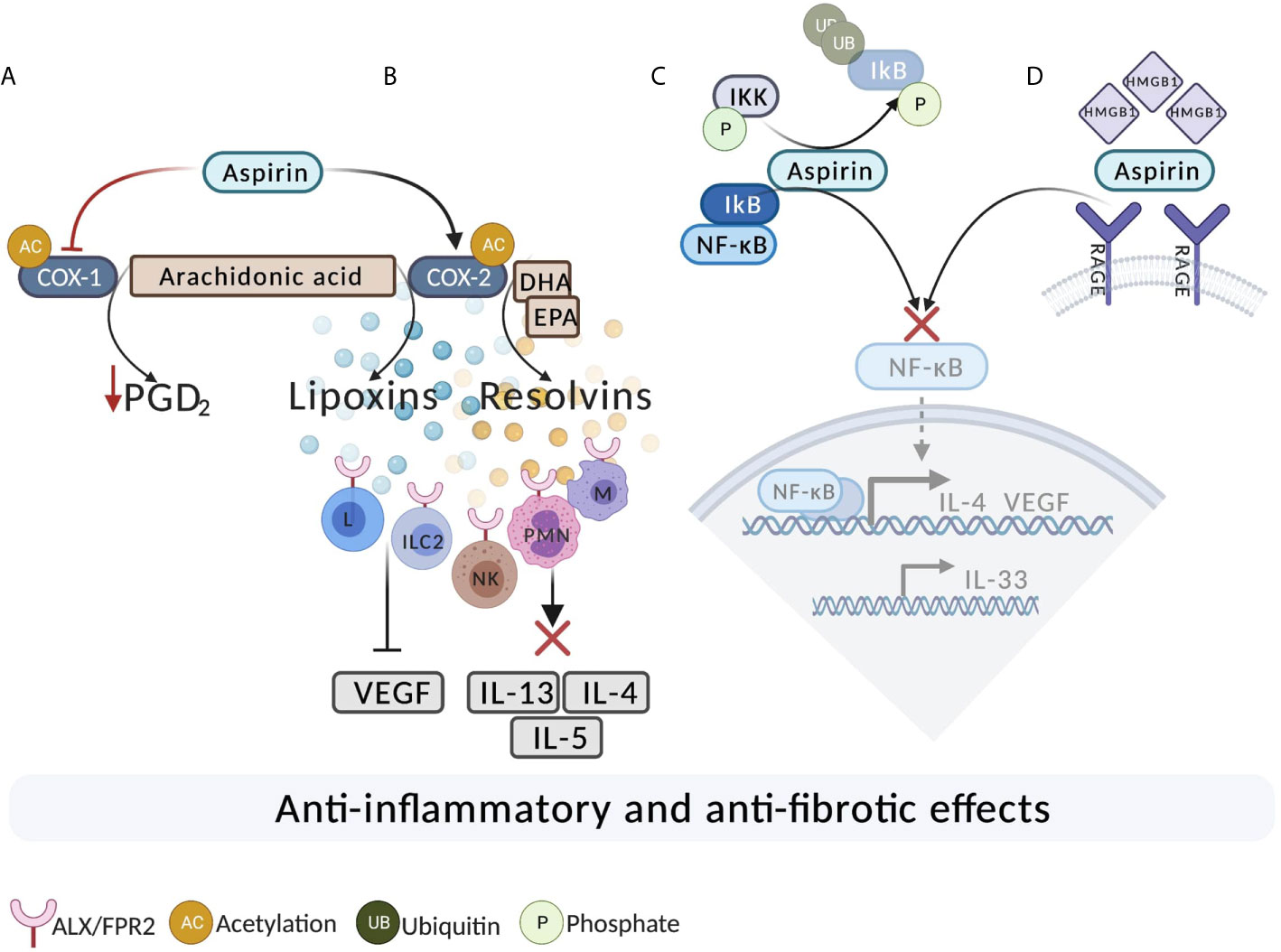
Figure 1 The potential mechanisms of aspirin action in N-ERD. (A) Aspirin acetylates and completely inhibits COX-1. As a result of long-term, high-dose aspirin treatment, there is a reduction in the level of PGD2. (B) Aspirin acetylates COX-2. Acetylation of COX-2 results in the production of 15(R)-HETE, leading to generation of 15-epi-lipoxins from arachidonic acid and resolvins from DHA and EPA. Lipoxins and resolvins bind to ALX/FPR2 on ILC2, T, B, and NK cells leading to decreased inflammation. Lipoxins also exert antifibrotic effects by reducing the expression of VEGF. Resolvins act on ALX/FPR2 on macrophages and PMN and promote the resolution of the allergic reaction. (C) Aspirin may inhibit the activation of NF-κB. Aspirin potentially binds to IKK-β (IκB Kinase) and inhibits the degradation of IκB, resulting in inhibition of the NF-κB pathway. (D) Inhibition of high mobility group box 1 protein (HMGB1). Aspirin directly binds and inhibits HMGB1 and subsequently leads to the inhibition of the downstream pro-inflammatory activating signaling pathway via NF-ĸB. DHA, Docosahexaenoic acid; EPA, Eicosapentaenoic acid; ILC2, innate lymphoid type 2 cells; M, macrophage; L, lymphocyte; NK. Natural Killer; PMN, Polymorphonuclear; UB, ubiquitin; P, Phosphate.
Aspirin-Triggered Lipoxins and Inhibition of Growth Factors
Lipoxins and 15-epi-lipoxin reduce symptoms of severe asthma, airway inflammation and airway hyper-responsiveness (119, 120, 178, 179). For example, lipoxin A4 blocks airway hyper-responsiveness and pulmonary inflammation by decreasing leukocytes and mediators such as interleukin-5 (IL-5) and interleukin-13 (IL-13) (120). 15(S)-HETE is considered to function both as a pro- and an anti-inflammatory molecule and is a precursor to lipoxins and 15-epi-lipoxin. Kowalski et al. showed for the first time with cultured epithelial cells from nasal polyps, a significant increase in 15(R)-HETE generation following aspirin exposure was observed only in cells derived from N-ERD patients (87). This was closely followed by another study which showed a similar observation in peripheral blood leukocytes where higher levels of 15(R)-HETE were generated in N-ERD patients upon aspirin exposure, when compared to the aspirin tolerant asthma patients (132). In addition, a recent report showed that N-ERD patients with higher baseline 15(S)-HETE plasma levels showed a greater improvement in respiratory symptoms and pulmonary function during treatment with aspirin (134). Thus, aspirin treatment benefit in N-ERD patients may be associated with the ability to convert 15(R)-HETE to 15-epi-lipoxin by 5-lipoxygenase.
Lipoxins, including aspirin-triggered lipoxins, may potentially exert antifibrotic effects by reducing the expression of vascular endothelial growth factor (VEGF). VEGF causes angiogenesis and drives the proliferation and survival of the epithelial cells in chronic rhinosinusitis (180, 181). An increased level of VEGF is also found in the nasal lavage of chronic rhinosinusitis patients with nasal polyposis (181). The increased PGD2 levels in nasal polyps of patients with chronic rhinosinusitis has been identified as a dominant factor in inducing the production of VEGF via the DP receptors, and an increased level of VEGF has been documented in nasal polyps of N-ERD patients (24, 182, 183). In N-ERD, overexpression of PGD2 may explain the increase in VEGF, and suppression of VEGF production could be a potential mechanism, by which nasal polyp recurrence is reduced in N-ERD. While the effect of aspirin on VEGF expression levels has not been studied in N-ERD, its effect in other disorders has been documented. For example, aspirin inhibits tumor angiogenesis and cell proliferation by reducing the expression of VEGF (184, 185). Lipoxin A4 suppresses tumor growth by inhibiting the VEGF production in a hepatocarcinoma cell line (186). Even though the direct effects of aspirin to reduce the levels of VEGF has not been studied in N-ERD, it could be a possible mechanism of aspirin action.
Production of Resolvins and Aspirin-Triggered Resolvins
The enzymatic oxygenation of arachidonic acid, an omega-6 fatty acid, produces both anti- and pro-inflammatory lipid mediators. On the other hand, metabolism of the omega-3 fatty acids such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) mainly produces pro-resolving mediators such as resolvins of the E-series and D-series and protectins. Resolvin D1 (RvD1) and resolvin E1 (RvE1) promote resolution of inflammation in allergic airway disease, and have also been implicated in resolution of inflammation in tumors (187, 188). Resolvin D2 (RvD2) and resolvin D3 (RvD3) are also known to have potent inflammation resolving activities (188, 189). DHA is converted to the D-series resolvins via the sequential oxygenation initiated by 15-LO and followed by 5-LO via 17S-H(p)DHA (17(S)-hydroperoxy-docosahexaenoic acid). On the other hand, aspirin acetylated COX-2 and 5-LO converts DHA to aspirin triggered resolvins via 17R-H(p)DHA (17(R)-hydroperoxy-docosahexaenoic acid) (190). Along with the D-series resolvins, their aspirin-triggered counterparts also function as anti-inflammatory and pro-resolving factors in tumors and allergic airway inflammation by reducing polymorphonuclear leukocyte infiltration (189, 191).
RvD1 and its aspirin-triggered 17R epimer (AT-RvD1) promote resolution in allergic airway inflammation by decreasing eosinophil counts and proinflammatory mediators, and by increasing the clearance of allergens from the bronchial tree (192). These molecules can also reduce the levels of Th2 cytokines IL-4 and IL-13 in the lung, as measured in bronchoalveolar lavage fluids (192). Both RvD1 and aspirin-triggered RvD1 exert their pro-resolving effects via the ALX/FPR2 receptors that are also used by lipoxins to exert their anti-inflammatory actions (193, 194). While the effects of resolvins in N-ERD have not been studied extensively, indirect evidence of their relevance is suggested by the finding that a diet high in omega-3 fatty acids can improve N-ERD-associated symptoms, and this is associated with increased levels of RvD1 and reduced urinary levels of pro-inflammatory LTE4 and PGD-metabolite (195). Enhanced production of resolvins upon aspirin treatment should thus be examined further as a possible mechanism of aspirin benefits in N-ERD.
Aspirin Effects on Inflammatory Cytokine Levels
Cytokines play an integral part in allergic inflammation. IL-4 for example, is involved in the progression of several allergic conditions (196, 197). In N-ERD, overexpression of IL-4 in the nasal and sinus mucosa of the nasal polyps contributes to the pathogenesis of the disease (198). IL-4 can strongly and selectively upregulate the expression of the LTC4S mRNA and proteins (199). It can also upregulate the m-RNA and protein expression of CysLT1R (an LTD4 receptor) in human monocytes and monocyte-derived macrophages, thus also contributing to the pathogenesis of allergic disease and asthma by modulating the responsiveness to LTD4 (54). A possible mechanism is the activation of the transcription factor signal transducer and activator of transcription 6 (STAT6) by IL-4 (200). At the transcriptional level, the expression of CysLT1R and LTC4S can be induced by IL-4 through a STAT6 response element located in the promoter region of CysLT1R and LTC4S (201, 202).
Prolonged aspirin use reduces the level of IL-4 in sputum samples of N-ERD patients (18, 144, 203, 204). The mechanism by which aspirin reduces IL-4 levels in N-ERD is not well understood, but one possibility could be that this occurs at the level of IL-4 transcription as observed in an in vitro analysis where aspirin (1 mM) was effective in inhibiting gene expression of IL-4 in activated CD4+ T cells (198, 204). This aspirin-induced reduction in the levels of IL-4 transcription can lead to beneficial downstream effects. For example, an aspirin-induced reduction in the level of IL-4 can prevent the activation of STAT6 as seen in in vitro studies in human PBMCs (205). In case of N-ERD, two studies have shown that aspirin can inhibit the IL-4-STAT6 axis, thus resulting in therapeutic benefits associated with aspirin treatment by reducing the level of CysLT1R and LTC4S (202, 206).
Regulation of the NF-κB Pathway
Nuclear factor kappa light chain enhancer of activated B cells (NF-κB) is a protein complex that plays an important role in promoting inflammation, cell proliferation and survival. It is present in complex with inhibitor of nuclear factor κB (IκB), an inhibitory protein. The IκB Kinase (IKK) is an enzyme complex consisting of two catalytic subunits involving kinases (IKK-α and IKK-β) and a regulatory subunit (IKK-γ). Upon activation, IKK phosphorylates IκB resulting in the degradation of the protein complex and translocation of NF-κB to the nucleus. After its own activation, NF-κB can activate the transcription of various pro-inflammatory cytokines, chemokines and adhesion molecules. In cancer, a deregulated NF-κB pathway promotes tumor cell survival, proliferation, migration, invasion, angiogenesis, and resistance to therapy (169).
Increased expression of NF-κB is associated with nasal polyposis (207). The involvement of the NF-κB signaling pathway has been well documented in chronic airway diseases such as asthma by activation and translocation of NF-κB via the phosphorylation of IKK-β (208, 209). Interleukin-25 (IL-25) induces myofibroblast differentiation, extracellular matrix production and matrix metalloprotease expression in nasal fibroblasts via the NF- κB signaling pathway, thus aiding the process of the tissue growth and leading to nasal polyposis in chronic rhinosinusitis (210). IL-25 is also upregulated in N-ERD but, to our knowledge, no direct association of the NF-κB pathway has been established in case of N-ERD. An elevated level of the ligand for Receptor Activator of NF- κB (RANK-L) was observed in the tissue homogenates from nasal polyps of N-ERD patients (211). An increased expression of RANK-L could result in downstream activation of NF- κB, however this will have to be investigated further to determine the exact role of NF- κB in N-ERD.
There is evidence that, besides inhibiting the cyclooxygenase-prostaglandin axis, aspirin also mediates anti-tumor and anti-inflammatory effects through inhibiting NF-ĸB (212). There are various ways by which aspirin can inhibit NF-κB, which may include aspirin binding to the kinase IKK-β to reduce its accessibility to ATP and prevent phosphorylation of IκB (212, 213). Stabilization of IκB in this way has been suggested as a possible mechanism of inhibition of allergic airway inflammation by aspirin (192). Resolvin D1 and aspirin triggered resolvin D1 can reduce airway eosinophilia by decreasing IκB-α degradation, leading to a reduced activation of NF-ĸB (192). Therefore aspirin can eventually reduce the activation of NF-ĸB via an indirect mechanism involving resolvins, although the exact mechanism remains unknown at this time. Association of NF-κB with nasal polyp formation in N-ERD needs further investigation as a potential target for anti-inflammatory aspirin action.
Inhibition of High Mobility Group Box 1 Protein
High mobility group box 1 or HMGB1 is a non-histone, chromatin binding protein that belongs to the family of damage-associated molecular patterns and plays an important part in inflammation. HMGB1 is involved in cancer angiogenesis and in cancer progression and metastasis development (214, 215). It exerts its pro-inflammatory and pro-angiogenic activity via its interaction with various toll-like receptors but most importantly toll-like receptor 4 and via receptor for advanced glycation end products (RAGE) (216, 217). These interactions then activate downstream signaling pathways such as MAPK and NF-ĸB which in turn releases pro-inflammatory cytokines such as tumor necrosis factor-α, interleukin-1,-6,-8, and VEGF (218). Apart from cancer, HMGB1 is also involved in asthma pathogenesis as HMGB1 and its receptor RAGE are overexpressed in the sputum of severe asthma patients (219). The protein-receptor pair is also overexpressed in the nasal tissues of those diagnosed with eosinophilic chronic rhinosinusitis with nasal polyps (220, 221). Interleukin-33 (IL-33) is a an inducer of mast cell activation and innate type 2 immunity and is present in increased levels in the nasal polyp tissues of patients with N-ERD (222). A recent study in mice showed that stimulation with LTC4 upregulates the expression of surface HMGB1 (223). HMGB1 can then signal through RAGE resulting in the amplification of CysLT2R-mediated platelet activation which can lead to an increase in IL-33 levels.
Due to its pro-inflammatory effects, HMGB1 is an important drug target and a potential biomarker for various neurodegenerative diseases and cancers. It has been suggested that aspirin is capable of inhibiting HMGB1-mediated tumor progression (224). Even though the exact mechanism of HMGB1 inhibition by aspirin is not fully known, aspirin may function by directly binding to HMGB1 and suppressing its proinflammatory functions even at low doses (225). HMGB1 activates VEGF, downstream signaling of NF-ĸB pathway, and IL-33. Inhibition of HMGB1 by aspirin can therefore lead to a downregulation of these pathways, although a clinical relevance of these potential actions in N-ERD needs to be further investigated.
Epigenetic Changes
Aspirin’s effectiveness in treatment of cancer may be related to epigenetic changes, such as histone acetylation. Histone acetylation involves histone acetyltransferase and histone deacetylase enzymes (226), and the activities of these enzymes can be regulated by aspirin (163). In fact, inhibition of histone deacetylase in general is now considered to be a possible therapeutic approach for colorectal and other cancers by regulating epigenetic changes (227–229). Aspirin in particular was shown to modulate epigenetic changes in colon cancer by inhibiting the activity of histone deacetylase which leads to restoration of histone 3 lysine 27 acetylation, eventually suppressing levels of tumor necrosis factor-α and interleukin-6 (230).
Epigenetic changes such as hyper and hypo-methylation of genes mainly associated with the arachidonic acid pathway have been observed in N-ERD (231). In the nasal polyp samples of N-ERD patients, Cheong et al. observed that the following genes were hypomethylated: the PGDS gene encoding prostaglandin D synthase, the ALOX5AP gene encoding 5-LO activation protein, and the LTB4R gene encoding the leukotriene B4 receptor, while the PTGES gene encoding prostaglandin E synthase was hypermethylated (231). Laidlaw et al. also observed epigenetic changes in the form of histone acetylation at the PTGER2 promoter that dysregulates the EP2 receptor expression in the nasal fibroblast of N-ERD patients (103). Whether or not these epigenetic changes could function as possible targets for aspirin therapy in N-ERD is still unknown.
Conclusion
Therapeutic Implications
Aspirin desensitization and maintenance therapy following endoscopic sinus surgery is an effective treatment for N-ERD patients that reduces the recurrence of nasal polyps. Aspirin and its use as an anti-inflammatory agent have been widely studied in cancer, but the exact mechanism of its actions in N-ERD is not completely understood. It is also unexplained why some N-ERD patients fail aspirin desensitization and maintenance therapy. In this review we summarized the potential pathways of aspirin action based on the available knowledge of aspirin’s effectiveness on eicosanoids, growth factors, pro- and anti-inflammatory pathways, and cytokines. Exploring these mechanisms may highlight the diversity of aspirin action. Mechanistic studies may help in developing new and more effective treatments for N-ERD using aspirin and other agents. Information on aspirin actions may help identify N-ERD patients who are appropriate candidates for this effective and affordable treatment.
Limitations and Future Directions
The mechanism of aspirin action to relieve the symptoms of N-ERD is not fully understood. There are various avenues for further investigation of aspirin actions in N-ERD. A major emphasis has been primarily on the arachidonic acid pathway and its regulation by aspirin in N-ERD. Aspirin exerts its anti-inflammatory effects in other diseases, for example in cancer, by several other pathways. Limited information is available for some of these pathways such as production of resolvins and modulation of epigenetic changes in N-ERD. Further studies are needed to determine the role of these pathways in N-ERD. The role of NF-κB, VEGF, HMGB1, and RAGE in N-ERD needs to be better defined. Long-term studies with an emphasis on molecular pathways would provide a robust understanding of the disease and pathways that could serve as therapeutic targets.
Author Contributions
ES, MA, and EJ were involved in conceptualizing the review. All authors contributed to the drafting and editing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grant R21AI146804 from the National Institutes of Health (NIH)/National institute of Allergy and Infectious diseases (NIAID). Portions of this work were supported by the generous contribution of Hiram and Jeanne Gray Endowment Fund. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Conflict of Interest
EJ served on the Advisory Board for GSK, Sanofi/Regeneron, and Novartis/Genentech; she is a consultant for GSK and has research support from AstraZeneca and from Cumberland Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Loll PJ, Picot D, Garavito RM. The Structural Basis of Aspirin Activity Inferred From the Crystal Structure of Inactivated Prostaglandin H2 Synthase. Nat Struct Biol (1995) 2(8):637–43. doi: 10.1038/nsb0895-637
2. Widal F, Abrami P, Lermoyez J. First Complete Description of the Aspirin Idiosyncrasy-Asthma-Nasal Polyposis Syndrome (Plus Urticaria)–1922 (With a Note on Aspirin Desensitization). J Asthma (1987) 24(5):297–300.
3. Samter M, Beers RF Jr. Intolerance to Aspirin. Clinical Studies and Consideration of its Pathogenesis. Ann Intern Med (1968) 68(5):975–83. doi: 10.7326/0003-4819-68-5-975
4. Samter M, Beers RF Jr. Concerning the Nature of Intolerance to Aspirin. J Allergy (1967) 40(5):281–93. doi: 10.1016/0021-8707(67)90076-7
5. Kowalski ML, Asero R, Bavbek S, Blanca M, Blanca-Lopez N, Bochenek G, et al. Classification and Practical Approach to the Diagnosis and Management of Hypersensitivity to Nonsteroidal Anti-Inflammatory Drugs. Allergy (2013) 68(10):1219–32. doi: 10.1111/all.12260
6. Kowalski ML, Agache I, Bavbek S, Bakirtas A, Blanca M, Bochenek G, et al. Diagnosis and Management of NSAID-Exacerbated Respiratory Disease (N-ERD)-a EAACI Position Paper. Allergy (2019) 74(1):28–39. doi: 10.1111/all.13599
7. Szczeklik A, Nizankowska E. Clinical Features and Diagnosis of Aspirin Induced Asthma. Thorax (2000) 55(Suppl 2):S42–4. doi: 10.1136/thorax.55.suppl_2.s42
8. Slavin RG. Nasal Polyps and Sinusitis. JAMA (1997) 278(22):1849–54. doi: 10.1001/jama.278.22.1849
9. Shah SJ, Abuzeid WM, Ponduri A, Pelletier T, Ren Z, Keskin T, et al. Endoscopic Sinus Surgery Improves Aspirin Treatment Response in Aspirin-Exacerbated Respiratory Disease Patients. Int Forum Allergy Rhinol (2019) 9(12):1401–8. doi: 10.1002/alr.22418
10. Jerschow E, Edin ML, Chi Y, Hurst B, Abuzeid WM, Akbar NA, et al. Sinus Surgery is Associated With a Decrease in Aspirin-Induced Reaction Severity in Patients With Aspirin Exacerbated Respiratory Disease. J Allergy Clin Immunol Pract (2019) 7(5):1580–88. doi: 10.1016/j.jaip.2018.12.014
11. Sanchez-Borges M, Caballero-Fonseca F, Capriles-Hulett A. Cofactors and Comorbidities in Patients With Aspirin/NSAID Hypersensitivity. Allergol Immunopathol (Madr) (2017) 45(6):573–78. doi: 10.1016/j.aller.2016.08.010
12. Szczeklik A, Nizankowska E, Duplaga M. Natural History of Aspirin-Induced Asthma. AIANE Investigators. European Network on Aspirin-Induced Asthma. Eur Respir J (2000) 16(3):432–6. doi: 10.1034/j.1399-3003.2000.016003432.x
13. Rajan JP, Wineinger NE, Stevenson DD, White AA. Prevalence of Aspirin-Exacerbated Respiratory Disease Among Asthmatic Patients: A Meta-Analysis of the Literature. J Allergy Clin Immunol (2015) 135(3):676–81 e1. doi: 10.1016/j.jaci.2014.08.020
14. Mascia K, Haselkorn T, Deniz YM, Miller DP, Bleecker ER, Borish L, et al. Aspirin Sensitivity and Severity of Asthma: Evidence for Irreversible Airway Obstruction in Patients With Severe or Difficult-to-Treat Asthma. J Allergy Clin Immunol (2005) 116(5):970–5. doi: 10.1016/j.jaci.2005.08.035
15. Berges-Gimeno MP, Simon RA, Stevenson DD. Early Effects of Aspirin Desensitization Treatment in Asthmatic Patients With Aspirin-Exacerbated Respiratory Disease. Ann Allergy Asthma Immunol (2003) 90(3):338–41. doi: 10.1016/S1081-1206(10)61803-0
16. Berges-Gimeno MP, Simon RA, Stevenson DD. Long-Term Treatment With Aspirin Desensitization in Asthmatic Patients With Aspirin-Exacerbated Respiratory Disease. J Allergy Clin Immunol (2003) 111(1):180–6. doi: 10.1067/mai.2003.7
17. Lee JY, Simon RA, Stevenson DD. Selection of Aspirin Dosages for Aspirin Desensitization Treatment in Patients With Aspirin-Exacerbated Respiratory Disease. J Allergy Clin Immunol (2007) 119(1):157–64. doi: 10.1016/j.jaci.2006.09.011
18. Katial RK, Strand M, Prasertsuntarasai T, Leung R, Zheng W, Alam R. The Effect of Aspirin Desensitization on Novel Biomarkers in Aspirin-Exacerbated Respiratory Diseases. J Allergy Clin Immunol (2010) 126(4):738–44. doi: 10.1016/j.jaci.2010.06.036
19. Stevenson DD, Hankammer MA, Mathison DA, Christiansen SC, Simon RA. Aspirin Desensitization Treatment of Aspirin-Sensitive Patients With Rhinosinusitis-Asthma: Long-Term Outcomes. J Allergy Clin Immunol (1996) 98(4):751–8. doi: 10.1016/s0091-6749(96)70123-9
20. Walters KM, Waldram JD, Woessner KM, White AA. Long-Term Clinical Outcomes of Aspirin Desensitization With Continuous Daily Aspirin Therapy in Aspirin-Exacerbated Respiratory Disease. Am J Rhinol Allergy (2018) 32(4):280–86. doi: 10.1177/1945892418770260
21. Rozsasi A, Polzehl D, Deutschle T, Smith E, Wiesmiller K, Riechelmann H, et al. Long-Term Treatment With Aspirin Desensitization: A Prospective Clinical Trial Comparing 100 and 300 Mg Aspirin Daily. Allergy (2008) 63(9):1228–34. doi: 10.1111/j.1398-9995.2008.01658.x
22. Swierczynska-Krepa M, Sanak M, Bochenek G, Strek P, Cmiel A, Gielicz A, et al. Aspirin Desensitization in Patients With Aspirin-Induced and Aspirin-Tolerant Asthma: A Double-Blind Study. J Allergy Clin Immunol (2014) 134(4):883–90. doi: 10.1016/j.jaci.2014.02.041
23. Laidlaw TM. Clinical Updates in Aspirin-Exacerbated Respiratory Disease. Allergy Asthma Proc (2019) 40(1):4–6. doi: 10.2500/aap.2019.40.4188
24. Payne SC, Early SB, Huyett P, Han JK, Borish L, Steinke JW. Evidence for Distinct Histologic Profile of Nasal Polyps With and Without Eosinophilia. Laryngoscope (2011) 121(10):2262–7. doi: 10.1002/lary.21969
25. Park HS, Nahm DH, Park K, Suh KS, Yim HE. Immunohistochemical Characterization of Cellular Infiltrate in Nasal Polyp From Aspirin-Sensitive Asthmatic Patients. Ann Allergy Asthma Immunol (1998) 81(3):219–24. doi: 10.1016/s1081-1206(10)62815-3
26. Sladek K, Szczeklik A. Cysteinyl Leukotrienes Overproduction and Mast Cell Activation in Aspirin-Provoked Bronchospasm in Asthma. Eur Respir J (1993) 6(3):391–9.
27. Fischer AR, Rosenberg MA, Lilly CM, Callery JC, Rubin P, Cohn J, et al. Direct Evidence for a Role of the Mast Cell in the Nasal Response to Aspirin in Aspirin-Sensitive Asthma. J Allergy Clin Immunol (1994) 94(6 Pt 1):1046–56. doi: 10.1016/0091-6749(94)90123-6
28. Bachert C, Wagenmann M, Hauser U, Rudack C. IL-5 Synthesis is Upregulated in Human Nasal Polyp Tissue. J Allergy Clin Immunol (1997) 99(6 Pt 1):837–42. doi: 10.1016/s0091-6749(97)80019-x
29. Hamilos DL, Leung DY, Wood R, Cunningham L, Bean DK, Yasruel Z, et al. Evidence for Distinct Cytokine Expression in Allergic Versus Nonallergic Chronic Sinusitis. J Allergy Clin Immunol (1995) 96(4):537–44. doi: 10.1016/s0091-6749(95)70298-9
30. Ochensberger B, Tassera L, Bifrare D, Rihs S, Dahinden CA. Regulation of Cytokine Expression and Leukotriene Formation in Human Basophils by Growth Factors, Chemokines and Chemotactic Agonists. Eur J Immunol (1999) 29(1):11–22. doi: 10.1002/(SICI)1521-4141(199901)29:01<11::AID-IMMU11>3.0.CO;2-B
31. Schleimer RP, MacGlashan DW Jr., Peters SP, Pinckard RN, Adkinson NF Jr., Lichtenstein LM. Characterization of Inflammatory Mediator Release From Purified Human Lung Mast Cells. Am Rev Respir Dis (1986) 133(4):614–7. doi: 10.1164/arrd.1986.133.4.614
32. Williams JD, Czop JK, Austen KF. Release of Leukotrienes by Human Monocytes on Stimulation of Their Phagocytic Receptor for Particulate Activators. J Immunol (1984) 132(6):3034–40.
33. Weller PF, Lee CW, Foster DW, Corey EJ, Austen KF, Lewis RA. Generation and Metabolism of 5-Lipoxygenase Pathway Leukotrienes by Human Eosinophils: Predominant Production of Leukotriene C4. Proc Natl Acad Sci USA (1983) 80(24):7626–30. doi: 10.1073/pnas.80.24.7626
34. Reid GK, Kargman S, Vickers PJ, Mancini JA, Leveille C, Ethier D, et al. Correlation Between Expression of 5-Lipoxygenase-Activating Protein, 5-Lipoxygenase, and Cellular Leukotriene Synthesis. J Biol Chem (1990) 265(32):19818–23. doi: 10.1016/S0021-9258(17)45446-9
35. Laidlaw TM, Kidder MS, Bhattacharyya N, Xing W, Shen S, Milne GL, et al. Cysteinyl Leukotriene Overproduction in Aspirin-Exacerbated Respiratory Disease Is Driven by Platelet-Adherent Leukocytes. Blood (2012) 119(16):3790–8. doi: 10.1182/blood-2011-10-384826
36. Cowburn AS, Sladek K, Soja J, Adamek L, Nizankowska E, Szczeklik A, et al. Overexpression of Leukotriene C4 Synthase in Bronchial Biopsies From Patients With Aspirin-Intolerant Asthma. J Clin Invest (1998) 101(4):834–46. doi: 10.1172/JCI620
37. Lam BK, Penrose JF, Freeman GJ, Austen KF. Expression Cloning of a cDNA for Human Leukotriene C4 Synthase, An Integral Membrane Protein Conjugating Reduced Glutathione to Leukotriene A4. Proc Natl Acad Sci USA (1994) 91(16):7663–7. doi: 10.1073/pnas.91.16.7663
38. Peters-Golden M, Brock TG. 5-Lipoxygenase and FLAP. Prostaglandins Leukot Essent Fatty Acids (2003) 69(2-3):99–109. doi: 10.1016/s0952-3278(03)00070-x
39. Shimizu T, Izumi T, Seyama Y, Tadokoro K, Radmark O, Samuelsson B. Characterization of Leukotriene A4 Synthase From Murine Mast Cells: Evidence for Its Identity to Arachidonate 5-Lipoxygenase. Proc Natl Acad Sci USA (1986) 83(12):4175–9. doi: 10.1073/pnas.83.12.4175
40. Borgeat P, Hamberg M, Samuelsson B. Transformation of Arachidonic Acid and Homo-Gamma-Linolenic Acid by Rabbit Polymorphonuclear Leukocytes. Monohydroxy Acids From Novel Lipoxygenases. J Biol Chem (1976) 251(24):7816–20. doi: 10.1016/S0021-9258(19)57008-9
41. Dahlen SE, Hedqvist P, Hammarstrom S, Samuelsson B. Leukotrienes are Potent Constrictors of Human Bronchi. Nature (1980) 288(5790):484–6. doi: 10.1038/288484a0
42. Smedegard G, Hedqvist P, Dahlen SE, Revenas B, Hammarstrom S, Samuelsson B. Leukotriene C4 Affects Pulmonary and Cardiovascular Dynamics in Monkey. Nature (1982) 295(5847):327–9. doi: 10.1038/295327a0
43. Smith LJ, Kern R, Patterson R, Krell RD, Bernstein PR. Mechanism of Leukotriene D4-induced Bronchoconstriction in Normal Subject. J Allergy Clin Immunol (1987) 80(3 Pt 1):340–7. doi: 10.1016/0091-6749(87)90040-6
44. Davidson AB, Lee TH, Scanlon PD, Solway J, McFadden ER Jr., Ingram RH Jr., et al. Bronchoconstrictor Effects of Leukotriene E4 in Normal and Asthmatic Subjects. Am Rev Respir Dis (1987) 135(2):333–7. doi: 10.1164/arrd.1987.135.2.333
45. Maekawa A, Kanaoka Y, Xing W, Austen KF. Functional Recognition of a Distinct Receptor Preferential for Leukotriene E4 in Mice Lacking the Cysteinyl Leukotriene 1 and 2 Receptors. Proc Natl Acad Sci USA (2008) 105(43):16695–700. doi: 10.1073/pnas.0808993105
46. Paruchuri S, Tashimo H, Feng C, Maekawa A, Xing W, Jiang Y, et al. Leukotriene E4-Induced Pulmonary Inflammation is Mediated by the P2Y12 Receptor. J Exp Med (2009) 206(11):2543–55. doi: 10.1084/jem.20091240
47. Christie PE, Tagari P, Ford-Hutchinson AW, Charlesson S, Chee P, Arm JP, et al. Urinary Leukotriene E4 Concentrations Increase After Aspirin Challenge in Aspirin-Sensitive Asthmatic Subjects. Am Rev Respir Dis (1991) 143(5 Pt 1):1025–9. doi: 10.1164/ajrccm/143.5_Pt_1.1025
48. Mastalerz L, Januszek R, Kaszuba M, Wojcik K, Celejewska-Wojcik N, Gielicz A, et al. Aspirin Provocation Increases 8-iso-PGE2 in Exhaled Breath Condensate of Aspirin-Hypersensitive Asthmatics. Prostaglandins Other Lipid Mediat (2015) 121(Pt B):163–9. doi: 10.1016/j.prostaglandins.2015.07.001
49. Nasser SM, Pfister R, Christie PE, Sousa AR, Barker J, Schmitz-Schumann M, et al. Inflammatory Cell Populations in Bronchial Biopsies From Aspirin-Sensitive Asthmatic Subjects. Am J Respir Crit Care Med (1996) 153(1):90–6. doi: 10.1164/ajrccm.153.1.8542168
50. Sestini P, Armetti L, Gambaro G, Pieroni MG, Refini RM, Sala A, et al. Inhaled PGE2 Prevents Aspirin-Induced Bronchoconstriction and Urinary LTE4 Excretion in Aspirin-Sensitive Asthma. Am J Respir Crit Care Med (1996) 153(2):572–5. doi: 10.1164/ajrccm.153.2.8564100
51. Luo M, Jones SM, Phare SM, Coffey MJ, Peters-Golden M, Brock TG. Protein Kinase A Inhibits Leukotriene Synthesis by Phosphorylation of 5-Lipoxygenase on Serine 523. J Biol Chem (2004) 279(40):41512–20. doi: 10.1074/jbc.M312568200
52. Laidlaw TM, Cahill KN, Cardet JC, Murphy K, Cui J, Dioneda B, et al. A Trial of Type 12 Purinergic (P2Y12) Receptor Inhibition With Prasugrel Identifies a Potentially Distinct Endotype of Patients With Aspirin-Exacerbated Respiratory Disease. J Allergy Clin Immunol (2019) 143(1):316–24 e7. doi: 10.1016/j.jaci.2018.06.001
53. Sousa AR, Parikh A, Scadding G, Corrigan CJ, Lee TH. Leukotriene-Receptor Expression on Nasal Mucosal Inflammatory Cells in Aspirin-Sensitive Rhinosinusitis. N Engl J Med (2002) 347(19):1493–9. doi: 10.1056/NEJMoa013508
54. Thivierge M, Stankova J, Rola-Pleszczynski M. Il-13 and IL-4 Up-Regulate Cysteinyl Leukotriene 1 Receptor Expression in Human Monocytes and Macrophages. J Immunol (2001) 167(5):2855–60. doi: 10.4049/jimmunol.167.5.2855
55. Lewis RA, Austen KF, Soberman RJ. Leukotrienes and Other Products of the 5-Lipoxygenase Pathway. Biochemistry and Relation to Pathobiology in Human Diseases. N Engl J Med (1990) 323(10):645–55. doi: 10.1056/NEJM199009063231006
56. Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. Leukotriene B, a Potent Chemokinetic and Aggregating Substance Released From Polymorphonuclear Leukocytes. Nature (1980) 286(5770):264–5. doi: 10.1038/286264a0
57. Serhan CN, Prescott SM. The Scent of a Phagocyte: Advances on Leukotriene B(4) Receptors. J Exp Med (2000) 192(3):F5–8. doi: 10.1084/jem.192.3.f5
58. Huang WW, Garcia-Zepeda EA, Sauty A, Oettgen HC, Rothenberg ME, Luster AD. Molecular and Biological Characterization of the Murine Leukotriene B4 Receptor Expressed on Eosinophils. J Exp Med (1998) 188(6):1063–74. doi: 10.1084/jem.188.6.1063
59. Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. A G-Protein-Coupled Receptor for Leukotriene B4 That Mediates Chemotaxis. Nature (1997) 387(6633):620–4. doi: 10.1038/42506
60. Pal K, Feng X, Steinke JW, Burdick MD, Shim YM, Sung SS, et al. Leukotriene A4 Hydrolase Activation and Leukotriene B4 Production by Eosinophils in Severe Asthma. Am J Respir Cell Mol Biol (2019) 60(4):413–19. doi: 10.1165/rcmb.2018-0175OC
61. Mita H, Higashi N, Taniguchi M, Higashi A, Akiyama K. Increase in Urinary Leukotriene B4 Glucuronide Concentration in Patients With Aspirin-Intolerant Asthma After Intravenous Aspirin Challenge. Clin Exp Allergy (2004) 34(8):1262–9. doi: 10.1111/j.1365-2222.2004.02034.x
62. Ricciotti E, FitzGerald GA. Prostaglandins and Inflammation. Arterioscler Thromb Vasc Biol (2011) 31(5):986–1000. doi: 10.1161/ATVBAHA.110.207449
63. Mita H, Endoh S, Kudoh M, Kawagishi Y, Kobayashi M, Taniguchi M, et al. Possible Involvement of Mast-Cell Activation in Aspirin Provocation of Aspirin-Induced Asthma. Allergy (2001) 56(11):1061–7. doi: 10.1111/j.1398-9995.2001.00913.x
64. Buchheit KM, Cahill KN, Katz HR, Murphy KC, Feng C, Lee-Sarwar K, et al. Thymic Stromal Lymphopoietin Controls Prostaglandin D2 Generation in Patients With Aspirin-Exacerbated Respiratory Disease. J Allergy Clin Immunol (2016) 137(5):1566–76 e5. doi: 10.1016/j.jaci.2015.10.020
65. Hardy CC, Robinson C, Tattersfield AE, Holgate ST. The Bronchoconstrictor Effect of Inhaled Prostaglandin D2 in Normal and Asthmatic Men. N Engl J Med (1984) 311(4):209–13. doi: 10.1056/NEJM198407263110401
66. Johnston SL, Freezer NJ, Ritter W, O’Toole S, Howarth PH. Prostaglandin D2-induced Bronchoconstriction Is Mediated Only in Part by the Thromboxane Prostanoid Receptor. Eur Respir J (1995) 8(3):411–5. doi: 10.1183/09031936.95.08030411
67. Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, et al. Prostaglandin D2 Selectively Induces Chemotaxis in T Helper Type 2 Cells, Eosinophils, and Basophils Via Seven-Transmembrane Receptor CRTH2. J Exp Med (2001) 193(2):255–61. doi: 10.1084/jem.193.2.255
68. Giles H, Leff P, Bolofo ML, Kelly MG, Robertson AD. The Classification of Prostaglandin DP-Receptors in Platelets and Vasculature Using BW A868C, A Novel, Selective and Potent Competitive Antagonist. Br J Pharmacol (1989) 96(2):291–300. doi: 10.1111/j.1476-5381.1989.tb11816.x
69. Nakamura T, Maeda S, Horiguchi K, Maehara T, Aritake K, Choi BI, et al. PGD2 Deficiency Exacerbates Food Antigen-Induced Mast Cell Hyperplasia. Nat Commun (2015) 6:7514. doi: 10.1038/ncomms8514
70. Kupczyk M, Kuna P. Targeting the PGD2/CRTH2/DP1 Signaling Pathway in Asthma and Allergic Disease: Current Status and Future Perspectives. Drugs (2017) 77(12):1281–94. doi: 10.1007/s40265-017-0777-2
71. Gervais FG, Cruz RP, Chateauneuf A, Gale S, Sawyer N, Nantel F, et al. Selective Modulation of Chemokinesis, Degranulation, and Apoptosis in Eosinophils Through the PGD2 Receptors CRTH2 and DP. J Allergy Clin Immunol (2001) 108(6):982–8. doi: 10.1067/mai.2001.119919
72. Xue L, Gyles SL, Wettey FR, Gazi L, Townsend E, Hunter MG, et al. Prostaglandin D2 Causes Preferential Induction of Proinflammatory Th2 Cytokine Production Through an Action on Chemoattractant Receptor-Like Molecule Expressed on Th2 Cells. J Immunol (2005) 175(10):6531–6. doi: 10.4049/jimmunol.175.10.6531
73. Spik I, Brenuchon C, Angeli V, Staumont D, Fleury S, Capron M, et al. Activation of the Prostaglandin D2 Receptor DP2/CRTH2 Increases Allergic Inflammation in Mouse. J Immunol (2005) 174(6):3703–8. doi: 10.4049/jimmunol.174.6.3703
74. Higashi N, Taniguchi M, Mita H, Osame M, Akiyama K. A Comparative Study of Eicosanoid Concentrations in Sputum and Urine in Patients With Aspirin-Intolerant Asthma. Clin Exp Allergy (2002) 32(10):1484–90. doi: 10.1046/j.1365-2745.2002.01507.x
75. Cahill KN, Bensko JC, Boyce JA, Laidlaw TM. Prostaglandin D(2): A Dominant Mediator of Aspirin-Exacerbated Respiratory Disease. J Allergy Clin Immunol (2015) 135(1):245–52. doi: 10.1016/j.jaci.2014.07.031
76. Jerschow E, Ren Z, Hudes G, Sanak M, Morales E, Schuster V, et al. Utility of Low-Dose Oral Aspirin Challenges for Diagnosis of Aspirin-Exacerbated Respiratory Disease. Ann Allergy Asthma Immunol (2016) 116(4):321–28 e1. doi: 10.1016/j.anai.2015.12.026
77. Kanaoka Y, Ago H, Inagaki E, Nanayama T, Miyano M, Kikuno R, et al. Cloning and Crystal Structure of Hematopoietic Prostaglandin D Synthase. Cell (1997) 90(6):1085–95. doi: 10.1016/s0092-8674(00)80374-8
78. Luna-Gomes T, Magalhaes KG, Mesquita-Santos FP, Bakker-Abreu I, Samico RF, Molinaro R, et al. Eosinophils as a Novel Cell Source of Prostaglandin D2: Autocrine Role in Allergic Inflammation. J Immunol (2011) 187(12):6518–26. doi: 10.4049/jimmunol.1101806
79. Ying S, O’Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, et al. Thymic Stromal Lymphopoietin Expression is Increased in Asthmatic Airways and Correlates With Expression of Th2-Attracting Chemokines and Disease Severity. J Immunol (2005) 174(12):8183–90. doi: 10.4049/jimmunol.174.12.8183
80. Kimura S, Pawankar R, Mori S, Nonaka M, Masuno S, Yagi T, et al. Increased Expression and Role of Thymic Stromal Lymphopoietin in Nasal Polyposis. Allergy Asthma Immunol Res (2011) 3(3):186–93. doi: 10.4168/aair.2011.3.3.186
81. Nagarkar DR, Poposki JA, Tan BK, Comeau MR, Peters AT, Hulse KE, et al. Thymic Stromal Lymphopoietin Activity Is Increased in Nasal Polyps of Patients With Chronic Rhinosinusitis. J Allergy Clin Immunol (2013) 132(3):593–600 e12. doi: 10.1016/j.jaci.2013.04.005
82. Liu T, Li TL, Zhao F, Xie C, Liu AM, Chen X, et al. Role of Thymic Stromal Lymphopoietin in the Pathogenesis of Nasal Polyposis. Am J Med Sci (2011) 341(1):40–7. doi: 10.1097/MAJ.0b013e3181f20489
83. Bochenek G, Nagraba K, Nizankowska E, Szczeklik A. A Controlled Study of 9alpha,11beta-PGF2 (a Prostaglandin D2 Metabolite) in Plasma and Urine of Patients With Bronchial Asthma and Healthy Controls After Aspirin Challenge. J Allergy Clin Immunol (2003) 111(4):743–9. doi: 10.1067/mai.2003.1387
84. Higashi N, Mita H, Ono E, Fukutomi Y, Yamaguchi H, Kajiwara K, et al. Profile of Eicosanoid Generation in Aspirin-Intolerant Asthma and Anaphylaxis Assessed by New Biomarkers. J Allergy Clin Immunol (2010) 125(5):1084–91 e6. doi: 10.1016/j.jaci.2009.12.977
85. Paruchuri S, Jiang Y, Feng C, Francis SA, Plutzky J, Boyce JA. Leukotriene E4 Activates Peroxisome Proliferator-Activated Receptor Gamma and Induces Prostaglandin D2 Generation by Human Mast Cells. J Biol Chem (2008) 283(24):16477–87. doi: 10.1074/jbc.M705822200
86. Schmid M, Gode U, Schafer D, Wigand ME. Arachidonic Acid Metabolism in Nasal Tissue and Peripheral Blood Cells in Aspirin Intolerant Asthmatics. Acta Otolaryngol (1999) 119(2):277–80. doi: 10.1080/00016489950181819
87. Kowalski ML, Pawliczak R, Wozniak J, Siuda K, Poniatowska M, Iwaszkiewicz J, et al. Differential Metabolism of Arachidonic Acid in Nasal Polyp Epithelial Cells Cultured From Aspirin-Sensitive and Aspirin-Tolerant Patients. Am J Respir Crit Care Med (2000) 161(2 Pt 1):391–8. doi: 10.1164/ajrccm.161.2.9902034
88. Abramovitz M, Adam M, Boie Y, Carriere M, Denis D, Godbout C, et al. The Utilization of Recombinant Prostanoid Receptors to Determine the Affinities and Selectivities of Prostaglandins and Related Analogs. Biochim Biophys Acta (2000) 1483(2):285–93. doi: 10.1016/s1388-1981(99)00164-x
89. Machado-Carvalho L, Roca-Ferrer J, Picado C. Prostaglandin E2 Receptors in Asthma and in Chronic Rhinosinusitis/Nasal Polyps With and Without Aspirin Hypersensitivity. Respir Res (2014) 15:100. doi: 10.1186/s12931-014-0100-7
90. O’Callaghan G, Houston A. Prostaglandin E2 and the EP Receptors in Malignancy: Possible Therapeutic Targets? Br J Pharmacol (2015) 172(22):5239–50. doi: 10.1111/bph.13331
91. Sheibanie AF, Yen JH, Khayrullina T, Emig F, Zhang M, Tuma R, et al. The Proinflammatory Effect of Prostaglandin E2 in Experimental Inflammatory Bowel Disease is Mediated Through the IL-23–>IL-17 Axis. J Immunol (2007) 178(12):8138–47. doi: 10.4049/jimmunol.178.12.8138
92. Sheibanie AF, Khayrullina T, Safadi FF, Ganea D. Prostaglandin E2 Exacerbates Collagen-Induced Arthritis in Mice Through the Inflammatory Interleukin-23/Interleukin-17 Axis. Arthritis Rheum (2007) 56(8):2608–19. doi: 10.1002/art.22794
93. Kawamori T, Uchiya N, Sugimura T, Wakabayashi K. Enhancement of Colon Carcinogenesis By Prostaglandin E2 Administration. Carcinogenesis (2003) 24(5):985–90. doi: 10.1093/carcin/bgg033
94. Wang D, Buchanan FG, Wang H, Dey SK, DuBois RN. Prostaglandin E2 Enhances Intestinal Adenoma Growth Via Activation of the Ras-Mitogen-Activated Protein Kinase Cascade. Cancer Res (2005) 65(5):1822–9. doi: 10.1158/0008-5472.CAN-04-3671
95. Gauvreau GM, Watson RM, O’Byrne PM. Protective Effects of Inhaled PGE2 on Allergen-Induced Airway Responses and Airway Inflammation. Am J Respir Crit Care Med (1999) 159(1):31–6. doi: 10.1164/ajrccm.159.1.9804030
96. Lundequist A, Nallamshetty SN, Xing W, Feng C, Laidlaw TM, Uematsu S, et al. Prostaglandin E(2) Exerts Homeostatic Regulation of Pulmonary Vascular Remodeling in Allergic Airway Inflammation. J Immunol (2010) 184(1):433–41. doi: 10.4049/jimmunol.0902835
97. Rusznak M, Peebles RS Jr. Prostaglandin E2 in NSAID-Exacerbated Respiratory Disease: Protection Against Cysteinyl Leukotrienes and Group 2 Innate Lymphoid Cells. Curr Opin Allergy Clin Immunol (2019) 19(1):38–45. doi: 10.1097/ACI.0000000000000498
98. Negishi M, Sugimoto Y, Ichikawa A. Molecular Mechanisms of Diverse Actions of Prostanoid Receptors. Biochim Biophys Acta (1995) 1259(1):109–19. doi: 10.1016/0005-2760(95)00146-4
99. Hartert TV, Dworski RT, Mellen BG, Oates JA, Murray JJ, Sheller JR. Prostaglandin E(2) Decreases Allergen-Stimulated Release of Prostaglandin D(2) in Airways of Subjects With Asthma. Am J Respir Crit Care Med (2000) 162(2 Pt 1):637–40. doi: 10.1164/ajrccm.162.2.9904038
100. Sturm EM, Schratl P, Schuligoi R, Konya V, Sturm GJ, Lippe IT, et al. Prostaglandin E2 Inhibits Eosinophil Trafficking Through E-Prostanoid 2 Receptors. J Immunol (2008) 181(10):7273–83. doi: 10.4049/jimmunol.181.10.7273
101. Picado C, Fernandez-Morata JC, Juan M, Roca-Ferrer J, Fuentes M, Xaubet A, et al. Cyclooxygenase-2 mRNA Is Downexpressed in Nasal Polyps From Aspirin-Sensitive Asthmatics. Am J Respir Crit Care Med (1999) 160(1):291–6. doi: 10.1164/ajrccm.160.1.9808048
102. Ying S, Meng Q, Scadding G, Parikh A, Corrigan CJ, Lee TH. Aspirin-Sensitive Rhinosinusitis Is Associated With Reduced E-Prostanoid 2 Receptor Expression on Nasal Mucosal Inflammatory Cells. J Allergy Clin Immunol (2006) 117(2):312–8. doi: 10.1016/j.jaci.2005.10.037
103. Cahill KN, Raby BA, Zhou X, Guo F, Thibault D, Baccarelli A, et al. Impaired E Prostanoid2 Expression and Resistance to Prostaglandin E2 in Nasal Polyp Fibroblasts From Subjects With Aspirin-Exacerbated Respiratory Disease. Am J Respir Cell Mol Biol (2016) 54(1):34–40. doi: 10.1165/rcmb.2014-0486OC
104. Yoshimura T, Yoshikawa M, Otori N, Haruna S, Moriyama H. Correlation Between the Prostaglandin D(2)/E(2) Ratio in Nasal Polyps and the Recalcitrant Pathophysiology of Chronic Rhinosinusitis Associated With Bronchial Asthma. Allergol Int (2008) 57(4):429–36. doi: 10.2332/allergolint.o-08-545
105. Szczeklik A, Mastalerz L, Nizankowska E, Cmiel A. Protective and Bronchodilator Effects of Prostaglandin E and Salbutamol in Aspirin-Induced Asthma. Am J Respir Crit Care Med (1996) 153(2):567–71. doi: 10.1164/ajrccm.153.2.8564099
106. Takami M, Tsukada W. Correlative Alteration of Thromboxane A2 With Antigen-Induced Bronchoconstriction and the Role of Platelets as a Source of TXA2 Synthesis in Guinea Pigs: Effect of DP-1904, an Inhibitor of Thromboxane Synthetase. Pharmacol Res (1998) 38(2):133–9. doi: 10.1006/phrs.1998.0345
107. McDonald JW, Ali M, Morgan E, Townsend ER, Cooper JD. Thromboxane Synthesis by Sources Other Than Platelets in Association With Complement-Induced Pulmonary Leukostasis and Pulmonary Hypertension in Sheep. Circ Res (1983) 52(1):1–6. doi: 10.1161/01.res.52.1.1
108. Hamberg M, Svensson J, Samuelsson B. Thromboxanes: A New Group of Biologically Active Compounds Derived From Prostaglandin Endoperoxides. Proc Natl Acad Sci USA (1975) 72(8):2994–8. doi: 10.1073/pnas.72.8.2994
109. Ishizuka T, Suzuki K, Kawakami M, Hidaka T, Matsuki Y, Nakamura H. Thromboxane A2 Receptor Blockade Suppresses Intercellular Adhesion Molecule-1 Expression By Stimulated Vascular Endothelial Cells. Eur J Pharmacol (1996) 312(3):367–77. doi: 10.1016/0014-2999(96)00478-5
110. Paul BZ, Jin J, Kunapuli SP. Molecular Mechanism of Thromboxane A(2)-Induced Platelet Aggregation. Essential Role for p2t(ac) and Alpha(2a) Receptors. J Biol Chem (1999) 274(41):29108–14. doi: 10.1074/jbc.274.41.29108
111. Bureau MF, De Clerck F, Lefort J, Arreto CD, Vargaftig BB. Thromboxane A2 Accounts for Bronchoconstriction But Not for Platelet Sequestration and Microvascular Albumin Exchanges Induced by fMLP in the Guinea Pig Lung. J Pharmacol Exp Ther (1992) 260(2):832–40.
112. Ueno A, Tanaka K, Katori M. Possible Involvement of Thromboxane in Bronchoconstrictive and Hypertensive Effects of LTC4 and LTD4 in Guinea Pigs. Prostaglandins (1982) 23(6):865–80. doi: 10.1016/0090-6980(82)90130-7
113. Serhan CN, Hamberg M, Samuelsson B. Trihydroxytetraenes: A Novel Series of Compounds Formed From Arachidonic Acid in Human Leukocytes. Biochem Biophys Res Commun (1984) 118(3):943–9. doi: 10.1016/0006-291x(84)91486-4
114. Serhan CN, Hamberg M, Samuelsson B. Lipoxins: Novel Series of Biologically Active Compounds Formed From Arachidonic Acid in Human Leukocytes. Proc Natl Acad Sci USA (1984) 81(17):5335–9. doi: 10.1073/pnas.81.17.5335
115. Serhan CN, Sheppard KA. Lipoxin Formation During Human Neutrophil-Platelet Interactions. Evidence for the Transformation of Leukotriene A4 by Platelet 12-Lipoxygenase In Vitro. J Clin Invest (1990) 85(3):772–80. doi: 10.1172/JCI114503
116. Levy BD, Romano M, Chapman HA, Reilly JJ, Drazen J, Serhan CN. Human Alveolar Macrophages Have 15-Lipoxygenase and Generate 15(s)-hydroxy-5,8,11-cis-13-trans-eicosatetraenoic Acid and Lipoxins. J Clin Invest (1993) 92(3):1572–9. doi: 10.1172/JCI116738
117. Celik GE, Erkekol FO, Misirligil Z, Melli M. Lipoxin A4 Levels in Asthma: Relation With Disease Severity and Aspirin Sensitivity. Clin Exp Allergy (2007) 37(10):1494–501. doi: 10.1111/j.1365-2222.2007.02806.x
118. Serhan CN, Hirsch U, Palmblad J, Samuelsson B. Formation of Lipoxin A by Granulocytes From Eosinophilic Donors. FEBS Lett (1987) 217(2):242–6. doi: 10.1016/0014-5793(87)80671-3
119. Christie PE, Spur BW, Lee TH. The Effects of Lipoxin A4 on Airway Responses in Asthmatic Subjects. Am Rev Respir Dis (1992) 145(6):1281–4. doi: 10.1164/ajrccm/145.6.1281
120. evy BD, De Sanctis GT, Devchand PR, Kim E, Ackerman K, Schmidt BA, et al. Multi-Pronged Inhibition of Airway Hyper-Responsiveness and Inflammation by Lipoxin a(4). Nat Med (2002) 8(9):1018–23. doi: 10.1038/nm748
121. Colgan SP, Serhan CN, Parkos CA, Delp-Archer C, Madara JL. Lipoxin A4 Modulates Transmigration of Human Neutrophils Across Intestinal Epithelial Monolayers. J Clin Invest (1993) 92(1):75–82. doi: 10.1172/JCI116601
122. Lee TH, Lympany P, Crea AE, Spur BW. Inhibition of Leukotriene B4-induced Neutrophil Migration by Lipoxin A4: Structure-Function Relationships. Biochem Biophys Res Commun (1991) 180(3):1416–21. doi: 10.1016/s0006-291x(05)81354-3
123. Fiore S, Maddox JF, Perez HD, Serhan CN. Identification of a Human cDNA Encoding a Functional High Affinity Lipoxin A4 Receptor. J Exp Med (1994) 180(1):253–60. doi: 10.1084/jem.180.1.253
124. Takano T, Fiore S, Maddox JF, Brady HR, Petasis NA, Serhan CN. Aspirin-Triggered 15-Epi-Lipoxin A4 (LXA4) and LXA4 Stable Analogues Are Potent Inhibitors of Acute Inflammation: Evidence for Anti-Inflammatory Receptors. J Exp Med (1997) 185(9):1693–704. doi: 10.1084/jem.185.9.1693
125. Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, et al. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the Formyl Peptide Receptor (Fpr) Family. Pharmacol Rev (2009) 61(2):119–61. doi: 10.1124/pr.109.001578
126. Ramon S, Bancos S, Serhan CN, Phipps RP. Lipoxin A(4) Modulates Adaptive Immunity by Decreasing Memory B-Cell Responses Via an ALX/FPR2-dependent Mechanism. Eur J Immunol (2014) 44(2):357–69. doi: 10.1002/eji.201343316
127. Ariel A, Chiang N, Arita M, Petasis NA, Serhan CN. Aspirin-Triggered Lipoxin A4 and B4 Analogs Block Extracellular Signal-Regulated Kinase-Dependent TNF-Alpha Secretion From Human T Cells. J Immunol (2003) 170(12):6266–72. doi: 10.4049/jimmunol.170.12.6266
128. Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, et al. Lipoxin A4 Regulates Natural Killer Cell and Type 2 Innate Lymphoid Cell Activation in Asthma. Sci Transl Med (2013) 5(174):174ra26. doi: 10.1126/scitranslmed.3004812
129. Lu X, Fu H, Han F, Fang Y, Xu J, Zhang L, et al. Lipoxin A4 Regulates PM2.5-induced Severe Allergic Asthma in Mice Via the Th1/Th2 Balance of Group 2 Innate Lymphoid Cells. J Thorac Dis (2018) 10(3):1449–59. doi: 10.21037/jtd.2018.03.02
130. Gronert K, Martinsson-Niskanen T, Ravasi S, Chiang N, Serhan CN. Selectivity of Recombinant Human Leukotriene D(4), Leukotriene B(4), and Lipoxin A(4) Receptors With Aspirin-Triggered 15-epi-LXA(4) and Regulation of Vascular and Inflammatory Responses. Am J Pathol (2001) 158(1):3–9. doi: 10.1016/S0002-9440(10)63937-5
131. Sanak M, Levy BD, Clish CB, Chiang N, Gronert K, Mastalerz L, et al. Aspirin-Tolerant Asthmatics Generate More Lipoxins Than Aspirin-Intolerant Asthmatics. Eur Respir J (2000) 16(1):44–9. doi: 10.1034/j.1399-3003.2000.16a08.x
132. Kowalski ML, Ptasinska A, Bienkiewicz B, Pawliczak R, DuBuske L. Differential Effects of Aspirin and Misoprostol on 15-Hydroxyeicosatetraenoic Acid Generation By Leukocytes From Aspirin-Sensitive Asthmatic Patients. J Allergy Clin Immunol (2003) 112(3):505–12. doi: 10.1016/s0091-6749(03)01716-0
133. Levy BD, Bonnans C, Silverman ES, Palmer LJ, Marigowda G, Israel E, et al. Diminished Lipoxin Biosynthesis in Severe Asthma. Am J Respir Crit Care Med (2005) 172(7):824–30. doi: 10.1164/rccm.200410-1413OC
134. Jerschow E, Edin ML, Pelletier T, Abuzeid WM, Akbar NA, Gibber M, et al. Plasma 15-Hydroxyeicosatetraenoic Acid Predicts Treatment Outcomes in Aspirin-Exacerbated Respiratory Disease. J Allergy Clin Immunol Pract (2017) 5(4):998–1007.e2. doi: 10.1016/j.jaip.2016.11.021
135. Stevens WW, Staudacher AG, Hulse KE, Carter RG, Winter DR, Abdala-Valencia H, et al. Activation of the 15-Lipoxygenase Pathway in Aspirin-Exacerbated Respiratory Disease. J Allergy Clin Immunol (2021) 147(2):600–12. doi: 10.1016/j.jaci.2020.04.031
136. Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid Mediator Class Switching During Acute Inflammation: Signals in Resolution. Nat Immunol (2001) 2(7):612–9. doi: 10.1038/89759
137. Planaguma A, Kazani S, Marigowda G, Haworth O, Mariani TJ, Israel E, et al. Airway Lipoxin A4 Generation and Lipoxin A4 Receptor Expression Are Decreased in Severe Asthma. Am J Respir Crit Care Med (2008) 178(6):574–82. doi: 10.1164/rccm.200801-061OC
138. Claria J, Serhan CN. Aspirin Triggers Previously Undescribed Bioactive Eicosanoids by Human Endothelial Cell-Leukocyte Interactions. Proc Natl Acad Sci USA (1995) 92(21):9475–9. doi: 10.1073/pnas.92.21.9475
139. Meade EA, Smith WL, DeWitt DL. Differential Inhibition of Prostaglandin Endoperoxide Synthase (Cyclooxygenase) Isozymes by Aspirin and Other non-Steroidal Anti-Inflammatory Drugs. J Biol Chem (1993) 268(9):6610–4. doi: 10.1016/S0021-9258(18)53294-4
140. Lecomte M, Laneuville O, Ji C, DeWitt DL, Smith WL. Acetylation of Human Prostaglandin Endoperoxide Synthase-2 (cyclooxygenase-2) by Aspirin. J Biol Chem (1994) 269(18):13207–15. doi: 10.1016/S0021-9258(17)36820-5
141. Li KL, Lee AY, Abuzeid WM. Aspirin Exacerbated Respiratory Disease: Epidemiology, Pathophysiology, and Management. Med Sci (Basel) (2019) 7(3). doi: 10.3390/medsci7030045
142. Stevenson DD, Simon RA, Mathison DA. Aspirin-Sensitive Asthma: Tolerance to Aspirin After Positive Oral Aspirin Challenges. J Allergy Clin Immunol (1980) 66(1):82–8. doi: 10.1016/0091-6749(80)90143-8
143. Stevenson DD, Pleskow WW, Simon RA, Mathison DA, Lumry WR, Schatz M, et al. Aspirin-Sensitive Rhinosinusitis Asthma: A Double-Blind Crossover Study of Treatment With Aspirin. J Allergy Clin Immunol (1984) 73(4):500–7. doi: 10.1016/0091-6749(84)90361-0
144. Esmaeilzadeh H, Nabavi M, Aryan Z, Arshi S, Bemanian MH, Fallahpour M, et al. Aspirin Desensitization for Patients With Aspirin-Exacerbated Respiratory Disease: A Randomized Double-Blind Placebo-Controlled Trial. Clin Immunol (2015) 160(2):349–57. doi: 10.1016/j.clim.2015.05.012
145. Macy E, Bernstein JA, Castells MC, Gawchik SM, Lee TH, Settipane RA, et al. Aspirin Challenge and Desensitization for Aspirin-Exacerbated Respiratory Disease: A Practice Paper. Ann Allergy Asthma Immunol (2007) 98(2):172–4. doi: 10.1016/S1081-1206(10)60692-8
146. Stevens WW, Jerschow E, Baptist AP, Borish L, Bosso JV, Buchheit KM, et al. The Role of Aspirin Desensitization Followed by Oral Aspirin Therapy in Managing Patients With Aspirin-Exacerbated Respiratory Disease: A Work Group Report From the Rhinitis, Rhinosinusitis and Ocular Allergy Committee of the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol (2021) 147(3):827–44. doi: 10.1016/j.jaci.2020.10.043
147. Chiu JT. Improvement in Aspirin-Sensitive Asthmatic Subjects After Rapid Aspirin Desensitization and Aspirin Maintenance (ADAM) Treatment. J Allergy Clin Immunol (1983) 71(6):560–7. doi: 10.1016/0091-6749(83)90437-2
148. Sweet JM, Stevenson DD, Simon RA, Mathison DA. Long-Term Effects of Aspirin Desensitization–Treatment for Aspirin-Sensitive Rhinosinusitis-Asthma. J Allergy Clin Immunol (1990) 85(1 Pt 1):59–65. doi: 10.1016/0091-6749(90)90222-p
149. Woessner KM, White AA. Evidence-Based Approach to Aspirin Desensitization in Aspirin-Exacerbated Respiratory Disease. J Allergy Clin Immunol (2014) 133(1):286–7.e1-9. doi: 10.1016/j.jaci.2013.11.016
150. Flossmann E, Rothwell PM, British Doctors Aspirin T. The UKTIAAT. Effect of Aspirin on Long-Term Risk of Colorectal Cancer: Consistent Evidence From Randomised and Observational Studies. Lancet (2007) 369(9573):1603–13. doi: 10.1016/S0140-6736(07)60747-8
151. Cuzick J, Otto F, Baron JA, Brown PH, Burn J, Greenwald P, et al. Aspirin and Non-Steroidal Anti-Inflammatory Drugs for Cancer Prevention: An International Consensus Statement. Lancet Oncol (2009) 10(5):501–7. doi: 10.1016/S1470-2045(09)70035-X
152. Capodanno D, Angiolillo DJ. Aspirin for Primary Prevention of Cardiovascular Disease. Lancet (2018) 392(10152):988–90. doi: 10.1016/S0140-6736(18)31990-1
153. Wu YL, Fu SL, Zhang YP, Qiao MM, Chen Y. Cyclooxygenase-2 Inhibitors Suppress Angiogenesis and Growth of Gastric Cancer Xenografts. BioMed Pharmacother (2005) 59 Suppl 2:S289–92. doi: 10.1016/s0753-3322(05)80048-4
154. Capodanno D, Angiolillo DJ. Aspirin for Primary Cardiovascular Risk Prevention and Beyond in Diabetes Mellitus. Circulation (2016) 134(20):1579–94. doi: 10.1161/CIRCULATIONAHA.116.023164
155. Cheng G, Shan XF, Wang XL, Dong WW, Li Z, Liu XH, et al. Endothelial Damage Effects of Circulating Microparticles From Patients With Stable Angina are Reduced by Aspirin Through ERK/p38 Mapks Pathways. Cardiovasc Ther (2017) 35(4). doi: 10.1111/1755-5922.12273
156. Guirguis-Blake JM, Evans CV, Senger CA, O’Connor EA, Whitlock EP. Aspirin for the Primary Prevention of Cardiovascular Events: A Systematic Evidence Review for the U.S. Preventive Services Task Force. Ann Intern Med (2016) 164(12):804–13. doi: 10.7326/M15-2113
157. Peters AT, Mutharasan RK. Aspirin for Prevention of Cardiovascular Disease. JAMA (2020) 323(7):676. doi: 10.1001/jama.2019.18425
158. Rocca B, Patrono C. Aspirin in the Primary Prevention of Cardiovascular Disease in Diabetes Mellitus: A New Perspective. Diabetes Res Clin Pract (2020) 160:108008. doi: 10.1016/j.diabres.2020.108008
159. Luo CF, Mo P, Li GQ, Liu SM. Aspirin-Omitted Dual Antithrombotic Therapy in Nonvalvular Atrial Fibrillation Patients Presenting With Acute Coronary Syndrome or Undergoing Percutaneous Coronary Intervention: Results of a Meta-Analysis. Eur Heart J Cardiovasc Pharmacother (2020) 7(3):218–224. doi: 10.1093/ehjcvp/pvaa016
160. Thobani A, Dhindsa DS, DeMoss BD, Raad M, Sandesara PB, Sperling LS, et al. Usefulness of Aspirin for Primary Prevention of Atherosclerotic Cardiovascular Disease. Am J Cardiol (2019) 124(11):1785–89. doi: 10.1016/j.amjcard.2019.08.040
161. Morsilli O, Guerriero R, Lulli V, Maffei L, Pasquini L, Pulcinelli FM, et al. Platelet and Megakaryocyte CD40L Expression in Beta-Thalassemic Patients. Thromb Res (2020) 189:108–11. doi: 10.1016/j.thromres.2020.02.026
162. Karimi M, Haghpanah S, Pishdad P, Zahedi Z, Parand S, Safaei S. Frequency of Silent Brain Lesions and Aspirin Protection Evaluation Over 3 Years Follow-Up in Beta Thalassemia Patients. Ann Hematol (2019) 98(10):2267–71. doi: 10.1007/s00277-019-03765-0
163. Passacquale G, Phinikaridou A, Warboys C, Cooper M, Lavin B, Alfieri A, et al. Aspirin-Induced Histone Acetylation in Endothelial Cells Enhances Synthesis of the Secreted Isoform of Netrin-1 Thus Inhibiting Monocyte Vascular Infiltration. Br J Pharmacol (2015) 172(14):3548–64. doi: 10.1111/bph.13144
164. Zhang Z, Chen F, Shang L. Advances in Antitumor Effects of Nsaids. Cancer Manag Res (2018) 10:4631–40. doi: 10.2147/CMAR.S175212
165. Jiang MC, Liao CF, Lee PH. Aspirin Inhibits Matrix Metalloproteinase-2 Activity, Increases E-cadherin Production, and Inhibits In Vitro Invasion of Tumor Cells. Biochem Biophys Res Commun (2001) 282(3):671–7. doi: 10.1006/bbrc.2001.4637
166. Bagheri M, Tabatabae Far MA, Mirzaei H, Ghasemi F. Evaluation of Antitumor Effects of Aspirin and LGK974 Drugs on Cellular Signaling Pathways, Cell Cycle and Apoptosis in Colorectal Cancer Cell Lines Compared to Oxaliplatin Drug. Fundam Clin Pharmacol (2020) 34(1):51–64. doi: 10.1111/fcp.12492
167. Wang T, Fu X, Jin T, Zhang L, Liu B, Wu Y, et al. Aspirin Targets P4HA2 Through Inhibiting NF-kappaB and LMCD1-AS1/let-7g to Inhibit Tumour Growth and Collagen Deposition in Hepatocellular Carcinoma. EBioMedicine (2019) 45:168–80. doi: 10.1016/j.ebiom.2019.06.048
168. Goel A, Chang DK, Ricciardiello L, Gasche C, Boland CR. A Novel Mechanism for Aspirin-Mediated Growth Inhibition of Human Colon Cancer Cells. Clin Cancer Res (2003) 9(1):383–90.
169. Kastrati I, Delgado-Rivera L, Georgieva G, Thatcher GR, Frasor J. Synthesis and Characterization of an Aspirin-fumarate Prodrug That Inhibits Nfkappab Activity and Breast Cancer Stem Cells. J Vis Exp (2017) 119). doi: 10.3791/54798
170. Gelosa P, Castiglioni L, Camera M, Sironi L. Repurposing of Drugs Approved for Cardiovascular Diseases: Opportunity or Mirage? Biochem Pharmacol (2020) 177:113895. doi: 10.1016/j.bcp.2020.113895
171. Patrignani P, Patrono C. Aspirin, Platelet Inhibition and Cancer Prevention. Platelets (2018) 29(8):779–85. doi: 10.1080/09537104.2018.1492105
172. Shi C, Zhang N, Feng Y, Cao J, Chen X, Liu B. Aspirin Inhibits IKK-Beta-Mediated Prostate Cancer Cell Invasion by Targeting Matrix Metalloproteinase-9 and Urokinase-Type Plasminogen Activator. Cell Physiol Biochem (2017) 41(4):1313–24. doi: 10.1159/000464434
173. Chattopadhyay M, Goswami S, Rodes DB, Kodela R, Velazquez CA, Boring D, et al. NO-Releasing NSAIDs Suppress NF-KappaB Signaling In Vitro and In Vivo Through s-Nitrosylation. Cancer Lett (2010) 298(2):204–11. doi: 10.1016/j.canlet.2010.07.006
174. Xie Y, Pan M, Gao Y, Zhang L, Ge W, Tang P. Dose-Dependent Roles of Aspirin and Other Non-Steroidal Anti-Inflammatory Drugs in Abnormal Bone Remodeling and Skeletal Regeneration. Cell Biosci (2019) 9:103. doi: 10.1186/s13578-019-0369-9
175. Han YM, Lee YJ, Jang YN, Kim HM, Seo HS, Jung TW, et al. Aspirin Improves Nonalcoholic Fatty Liver Disease and Atherosclerosis Through Regulation of the PPARdelta-AMPK-PGC-1alpha Pathway in Dyslipidemic Conditions. BioMed Res Int (2020) 2020:7806860. doi: 10.1155/2020/7806860
176. Nasser SM, Patel M, Bell GS, Lee TH. The Effect of Aspirin Desensitization on Urinary Leukotriene E4 Concentrations in Aspirin-Sensitive Asthma. Am J Respir Crit Care Med (1995) 151(5):1326–30. doi: 10.1164/ajrccm.151.5.7735581
177. Cahill KN, Cui J, Kothari P, Murphy K, Raby BA, Singer J, et al. Unique Effect of Aspirin Therapy on Biomarkers in Aspirin-Exacerbated Respiratory Disease. A Prospective Trial. Am J Respir Crit Care Med (2019) 200(6):704–11. doi: 10.1164/rccm.201809-1755OC
178. Bonnans C, Vachier I, Chavis C, Godard P, Bousquet J, Chanez P. Lipoxins are Potential Endogenous Antiinflammatory Mediators in Asthma. Am J Respir Crit Care Med (2002) 165(11):1531–5. doi: 10.1164/rccm.200201-053OC
179. Levy BD, Lukacs NW, Berlin AA, Schmidt B, Guilford WJ, Serhan CN, et al. Lipoxin A4 Stable Analogs Reduce Allergic Airway Responses Via Mechanisms Distinct From CysLT1 Receptor Antagonism. FASEB J (2007) 21(14):3877–84. doi: 10.1096/fj.07-8653com
180. Gosepath J, Brieger J, Lehr HA, Mann WJ. Expression, Localization, and Significance of Vascular Permeability/Vascular Endothelial Growth Factor in Nasal Polyps. Am J Rhinol (2005) 19(1):7–13. doi: 10.1177/194589240501900102
181. Lee HS, Myers A, Kim J. Vascular Endothelial Growth Factor Drives Autocrine Epithelial Cell Proliferation and Survival in Chronic Rhinosinusitis With Nasal Polyposis. Am J Respir Crit Care Med (2009) 180(11):1056–67. doi: 10.1164/rccm.200905-0740OC
182. Fruth K, Zhu C, Schramek E, Angermair J, Kassem W, Haxel BR, et al. Vascular Endothelial Growth Factor Expression in Nasal Polyps of Aspirin-Intolerant Patients. Arch Otolaryngol Head Neck Surg (2012) 138(3):286–93. doi: 10.1001/archoto.2011.1474
183. Kanai K, Okano M, Fujiwara T, Kariya S, Haruna T, Omichi R, et al. Effect of Prostaglandin D2 on VEGF Release by Nasal Polyp Fibroblasts. Allergol Int (2016) 65(4):414–19. doi: 10.1016/j.alit.2016.03.003
184. Zhang X, Wang Z, Wang Z, Zhang Y, Jia Q, Wu L, et al. Impact of Acetylsalicylic Acid on Tumor Angiogenesis and Lymphangiogenesis Through Inhibition of VEGF Signaling in a Murine Sarcoma Model. Oncol Rep (2013) 29(5):1907–13. doi: 10.3892/or.2013.2339
185. Ding JH, Yuan LY, Huang RB, Chen GA. Aspirin Inhibits Proliferation and Induces Apoptosis of Multiple Myeloma Cells Through Regulation of Bcl-2 and Bax and Suppression of VEGF. Eur J Haematol (2014) 93(4):329–39. doi: 10.1111/ejh.12352
186. Chen Y, Hao H, He S, Cai L, Li Y, Hu S, et al. Lipoxin A4 and its Analogue Suppress the Tumor Growth of Transplanted H22 in Mice: The Role of Antiangiogenesis. Mol Cancer Ther (2010) 9(8):2164–74. doi: 10.1158/1535-7163.MCT-10-0173
187. Levy BD. Resolvin D1 and Resolvin E1 Promote the Resolution of Allergic Airway Inflammation Via Shared and Distinct Molecular Counter-Regulatory Pathways. Front Immunol (2012) 3:390. doi: 10.3389/fimmu.2012.00390
188. Sulciner ML, Serhan CN, Gilligan MM, Mudge DK, Chang J, Gartung A, et al. Resolvins Suppress Tumor Growth and Enhance Cancer Therapy. J Exp Med (2018) 215(1):115–40. doi: 10.1084/jem.20170681
189. Dalli J, Winkler JW, Colas RA, Arnardottir H, Cheng CY, Chiang N, et al. Resolvin D3 and Aspirin-Triggered Resolvin D3 are Potent Immunoresolvents. Chem Biol (2013) 20(2):188–201. doi: 10.1016/j.chembiol.2012.11.010
190. Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, et al. Resolvins: A Family of Bioactive Products of Omega-3 Fatty Acid Transformation Circuits Initiated By Aspirin Treatment That Counter Proinflammation Signals. J Exp Med (2002) 196(8):1025–37. doi: 10.1084/jem.20020760
191. Sun YP, Oh SF, Uddin J, Yang R, Gotlinger K, Campbell E, et al. Resolvin D1 and its Aspirin-Triggered 17R Epimer. Stereochemical Assignments, Anti-Inflammatory Properties, and Enzymatic Inactivation. J Biol Chem (2007) 282(13):9323–34. doi: 10.1074/jbc.M609212200
192. Rogerio AP, Haworth O, Croze R, Oh SF, Uddin M, Carlo T, et al. Resolvin D1 and Aspirin-Triggered Resolvin D1 Promote Resolution of Allergic Airways Responses. J Immunol (2012) 189(4):1983–91. doi: 10.4049/jimmunol.1101665
193. Krishnamoorthy S, Recchiuti A, Chiang N, Yacoubian S, Lee CH, Yang R, et al. Resolvin D1 Binds Human Phagocytes With Evidence for Proresolving Receptors. Proc Natl Acad Sci USA (2010) 107(4):1660–5. doi: 10.1073/pnas.0907342107
194. Norling LV, Dalli J, Flower RJ, Serhan CN, Perretti M. Resolvin D1 Limits Polymorphonuclear Leukocyte Recruitment to Inflammatory Loci: Receptor-Dependent Actions. Arterioscler Thromb Vasc Biol (2012) 32(8):1970–8. doi: 10.1161/ATVBAHA.112.249508
195. Schneider TR, Johns CB, Palumbo ML, Murphy KC, Cahill KN, Laidlaw TM. Dietary Fatty Acid Modification for the Treatment of Aspirin-Exacerbated Respiratory Disease: A Prospective Pilot Trial. J Allergy Clin Immunol Pract (2018) 6(3):825–31. doi: 10.1016/j.jaip.2017.10.011
196. Cohn L, Homer RJ, Marinov A, Rankin J, Bottomly K. Induction of Airway Mucus Production by T Helper 2 (Th2) Cells: A Critical Role for Interleukin 4 in Cell Recruitment But Not Mucus Production. J Exp Med (1997) 186(10):1737–47. doi: 10.1084/jem.186.10.1737
197. Corry DB, Folkesson HG, Warnock ML, Erle DJ, Matthay MA, Wiener-Kronish JP, et al. Interleukin 4, But Not Interleukin 5 or Eosinophils, is Required in a Murine Model of Acute Airway Hyperreactivity. J Exp Med (1996) 183(1):109–17. doi: 10.1084/jem.183.1.109
198. Steinke JW, Payne SC, Borish L. Interleukin-4 in the Generation of the AERD Phenotype: Implications for Molecular Mechanisms Driving Therapeutic Benefit of Aspirin Desensitization. J Allergy (Cairo) (2012) 2012:182090. doi: 10.1155/2012/182090
199. Hsieh FH, Lam BK, Penrose JF, Austen KF, Boyce JA. T Helper Cell Type 2 Cytokines Coordinately Regulate Immunoglobulin E-dependent Cysteinyl Leukotriene Production by Human Cord Blood-Derived Mast Cells: Profound Induction of Leukotriene C(4) Synthase Expression by Interleukin 4. J Exp Med (2001) 193(1):123–33. doi: 10.1084/jem.193.1.123
200. Chen XH, Patel BK, Wang LM, Frankel M, Ellmore N, Flavell RA, et al. Jak1 Expression is Required for Mediating interleukin-4-induced Tyrosine Phosphorylation of Insulin Receptor Substrate and Stat6 Signaling Molecules. J Biol Chem (1997) 272(10):6556–60. doi: 10.1074/jbc.272.10.6556
201. Woszczek G, Pawliczak R, Qi HY, Nagineni S, Alsaaty S, Logun C, et al. Functional Characterization of Human Cysteinyl Leukotriene 1 Receptor Gene Structure. J Immunol (2005) 175(8):5152–9. doi: 10.4049/jimmunol.175.8.5152
202. Steinke JW, Culp JA, Kropf E, Borish L. Modulation by Aspirin of Nuclear Phospho-Signal Transducer and Activator of Transcription 6 Expression: Possible Role in Therapeutic Benefit Associated With Aspirin Desensitization. J Allergy Clin Immunol (2009) 124(4):724–30 e4. doi: 10.1016/j.jaci.2009.07.031
203. Goodwin JS, Ceuppens JL. Effect of Nonsteroidal Antiinflammatory Drugs on Immune Function. Semin Arthritis Rheum (1983) 13(1 Suppl 1):134–43. doi: 10.1016/0049-0172(83)90033-1
204. Cianferoni A, Schroeder JT, Kim J, Schmidt JW, Lichtenstein LM, Georas SN, et al. Selective Inhibition of Interleukin-4 Gene Expression in Human T Cells by Aspirin. Blood (2001) 97(6):1742–9. doi: 10.1182/blood.v97.6.1742
205. Perez GM, Melo M, Keegan AD, Zamorano J. Aspirin and Salicylates Inhibit the IL-4- and IL-13-Induced Activation of STAT6. J Immunol (2002) 168(3):1428–34. doi: 10.4049/jimmunol.168.3.1428
206. Katial RK, Martucci M, Burnett T, Faino A, Finkas L, Liu S, et al. Nonsteroidal Anti-Inflammatory-Induced Inhibition of Signal Transducer and Activator of Transcription 6 (STAT-6) Phosphorylation in Aspirin-Exacerbated Respiratory Disease. J Allergy Clin Immunol (2016) 138(2):579–85. doi: 10.1016/j.jaci.2015.11.038
207. Valera FC, Queiroz R, Scrideli C, Tone LG, Anselmo-Lima WT. Expression of Transcription Factors NF-kappaB and AP-1 in Nasal Polyposis. Clin Exp Allergy (2008) 38(4):579–85. doi: 10.1111/j.1365-2222.2007.02929.x
208. Hart LA, Krishnan VL, Adcock IM, Barnes PJ, Chung KF. Activation and Localization of Transcription Factor, Nuclear factor-kappaB, in Asthma. Am J Respir Crit Care Med (1998) 158(5 Pt 1):1585–92. doi: 10.1164/ajrccm.158.5.9706116
209. Gagliardo R, Chanez P, Mathieu M, Bruno A, Costanzo G, Gougat C, et al. Persistent Activation of Nuclear Factor-Kappab Signaling Pathway in Severe Uncontrolled Asthma. Am J Respir Crit Care Med (2003) 168(10):1190–8. doi: 10.1164/rccm.200205-479OC
210. Park SK, Jin YD, Park YK, Yeon SH, Xu J, Han RN, et al. Il-25-induced Activation of Nasal Fibroblast and Its Association With the Remodeling of Chronic Rhinosinusitis With Nasal Polyposis. PloS One (2017) 12(8):e0181806. doi: 10.1371/journal.pone.0181806
211. Ogasawara N, Poposki JA, Klingler AI, Tan BK, Hulse KE, Stevens WW, et al. Role of RANK-L as a Potential Inducer of ILC2-Mediated Type 2 Inflammation in Chronic Rhinosinusitis With Nasal Polyps. Mucosal Immunol (2020) 13(1):86–95. doi: 10.1038/s41385-019-0215-8
212. Yin MJ, Yamamoto Y, Gaynor RB. The Anti-Inflammatory Agents Aspirin and Salicylate Inhibit the Activity of I(kappa)B Kinase-Beta. Nature (1998) 396(6706):77–80. doi: 10.1038/23948
213. Kopp E, Ghosh S. Inhibition of NF-Kappa B by Sodium Salicylate and Aspirin. Science (1994) 265(5174):956–9. doi: 10.1126/science.8052854
214. Chung HW, Lim JB. High-Mobility Group Box-1 Contributes Tumor Angiogenesis Under Interleukin-8 Mediation During Gastric Cancer Progression. Cancer Sci (2017) 108(8):1594–601. doi: 10.1111/cas.13288
215. Tang D, Kang R, Zeh HJ 3rd, Lotze MT. High-Mobility Group Box 1 and Cancer. Biochim Biophys Acta (2010) 1799(1-2):131–40. doi: 10.1016/j.bbagrm.2009.11.014
216. Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, et al. High Mobility Group 1 Protein (HMG-1) Stimulates Proinflammatory Cytokine Synthesis in Human Monocytes. J Exp Med (2000) 192(4):565–70. doi: 10.1084/jem.192.4.565
217. Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A Critical Cysteine is Required for HMGB1 Binding to Toll-like Receptor 4 and Activation of Macrophage Cytokine Release. Proc Natl Acad Sci USA (2010) 107(26):11942–7. doi: 10.1073/pnas.1003893107
218. Lin Q, Yang XP, Fang D, Ren X, Zhou H, Fang J, et al. High-Mobility Group Box-1 Mediates Toll-Like Receptor 4-Dependent Angiogenesis. Arterioscler Thromb Vasc Biol (2011) 31(5):1024–32. doi: 10.1161/ATVBAHA.111.224048
219. Watanabe T, Asai K, Fujimoto H, Tanaka H, Kanazawa H, Hirata K. Increased Levels of HMGB-1 and Endogenous Secretory RAGE in Induced Sputum From Asthmatic Patients. Respir Med (2011) 105(4):519–25. doi: 10.1016/j.rmed.2010.10.016
220. Hong SM, Cho JS, Um JY, Shin JM, Park IH, Lee SH, et al. Increased Expression of High-Mobility Group Protein B1 in Chronic Rhinosinusitis. Am J Rhinol Allergy (2013) 27(4):278–82. doi: 10.2500/ajra.2013.27.3909
221. Dzaman K, Szczepanski MJ, Molinska-Glura M, Krzeski A, Zagor M. Expression of the Receptor for Advanced Glycation End Products, a Target for High Mobility Group Box 1 Protein, and its Role in Chronic Recalcitrant Rhinosinusitis With Nasal Polyps. Arch Immunol Ther Exp (Warsz) (2015) 63(3):223–30. doi: 10.1007/s00005-014-0325-7
222. Liu T, Kanaoka Y, Barrett NA, Feng C, Garofalo D, Lai J, et al. Aspirin-Exacerbated Respiratory Disease Involves a Cysteinyl Leukotriene-Driven IL-33-Mediated Mast Cell Activation Pathway. J Immunol (2015) 195(8):3537–45. doi: 10.4049/jimmunol.1500905
223. Liu T, Barrett NA, Kanaoka Y, Buchheit K, Laidlaw TM, Garofalo D, et al. Cysteinyl Leukotriene Receptor 2 Drives Lung Immunopathology Through a Platelet and High Mobility Box 1-Dependent Mechanism. Mucosal Immunol (2019) 12(3):679–90. doi: 10.1038/s41385-019-0134-8
224. Yang H, Pellegrini L, Napolitano A, Giorgi C, Jube S, Preti A, et al. Aspirin Delays Mesothelioma Growth by Inhibiting HMGB1-Mediated Tumor Progression. Cell Death Dis (2015) 6:e1786. doi: 10.1038/cddis.2015.153
225. Choi HW, Tian M, Song F, Venereau E, Preti A, Park SW, et al. Aspirin’s Active Metabolite Salicylic Acid Targets High Mobility Group Box 1 to Modulate Inflammatory Responses. Mol Med (2015) 21:526–35. doi: 10.2119/molmed.2015.00148
226. Villagra A, Sotomayor EM, Seto E. Histone Deacetylases and the Immunological Network: Implications in Cancer and Inflammation. Oncogene (2010) 29(2):157–73. doi: 10.1038/onc.2009.334
227. Lee HY, Tsai AC, Chen MC, Shen PJ, Cheng YC, Kuo CC, et al. Azaindolylsulfonamides, With a More Selective Inhibitory Effect on Histone Deacetylase 6 Activity, Exhibit Antitumor Activity in Colorectal Cancer HCT116 Cells. J Med Chem (2014) 57(10):4009–22. doi: 10.1021/jm401899x
228. Ishikawa S, Hayashi H, Kinoshita K, Abe M, Kuroki H, Tokunaga R, et al. Statins Inhibit Tumor Progression Via an Enhancer of Zeste Homolog 2-Mediated Epigenetic Alteration in Colorectal Cancer. Int J Cancer (2014) 135(11):2528–36. doi: 10.1002/ijc.28672
229. Li Y, Seto E. Hdacs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med (2016) 6(10). doi: 10.1101/cshperspect.a026831
230. Guo Y, Liu Y, Zhang C, Su ZY, Li W, Huang MT, et al. The Epigenetic Effects of Aspirin: The Modification of Histone H3 Lysine 27 Acetylation in the Prevention of Colon Carcinogenesis in Azoxymethane- and Dextran Sulfate Sodium-Treated CF-1 Mice. Carcinogenesis (2016) 37(6):616–24. doi: 10.1093/carcin/bgw042
Keywords: N-ERD, aspirin, COX-1 inhibitor, NSAIDs, nasal polyps, prostaglandins, leukotrienes, lipoxins
Citation: Sehanobish E, Asad M, Barbi M, Porcelli SA and Jerschow E (2021) Aspirin Actions in Treatment of NSAID-Exacerbated Respiratory Disease. Front. Immunol. 12:695815. doi: 10.3389/fimmu.2021.695815
Received: 15 April 2021; Accepted: 07 June 2021;
Published: 25 June 2021.
Edited by:
Beate E. Kehrel, University Hospital Münster, GermanyReviewed by:
Abed N. Azab, Ben-Gurion University of the Negev, IsraelToshiyuki Murai, Osaka University, Japan
Copyright © 2021 Sehanobish, Asad, Barbi, Porcelli and Jerschow. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elina Jerschow, ejerscho@montefiore.org