Tracking the Origin of Austrian Human Brucellosis Cases Using Whole Genome Sequencing
 Justine Schaeffer1,2*
Justine Schaeffer1,2*  Sandra Revilla-Fernández3
Sandra Revilla-Fernández3  Erwin Hofer3
Erwin Hofer3  Romana Posch3 Anna Stoeger1
Romana Posch3 Anna Stoeger1  Christoph Leth3
Christoph Leth3  Friedrich Schmoll3 Vesna Djordjevic4
Friedrich Schmoll3 Vesna Djordjevic4  Brankica Lakicevic4 Kazimir Matovic5
Brankica Lakicevic4 Kazimir Matovic5  Peter Hufnagl1 Alexander Indra1
Peter Hufnagl1 Alexander Indra1  Franz Allerberger1
Franz Allerberger1  Werner Ruppitsch1
Werner Ruppitsch1- 1Institute for Medical Microbiology and Hygiene, Austrian Agency for Health and Food Safety (AGES), Vienna, Austria
- 2EUPHEM Fellowship, European Centre for Disease Prevention and Control (ECDC), Stockholm, Sweden
- 3Institute for Veterinary Disease Control Mödling, Austrian Agency for Health and Food Safety (AGES), Mödling, Austria
- 4Department of Microbiology and Molecular Biology, Institute of Meat Hygiene and Technology, Belgrade, Serbia
- 5Department for Laboratory Diagnostic, Veterinary Specialized Institute, Kraljevo, Serbia
Brucellosis is a zoonotic disease caused by Brucella spp. and a major concern for livestock. Most human cases are caused by B. melitensis and clinical presentation is usually a mild febrile illness. However, treatment failure is frequent and more severe complications can occur. In Austria, every human brucellosis is investigated to determine whether it was imported from endemic areas or is the sign of an undetected autochthonous transmission. For this study, 21 B. melitensis strains isolated in Austria between 2005 and 2019 were collected, 17 strains from 15 different patients and four strains from cattle. Whole genome sequencing combined with core-genome MLST analysis was used to characterize these strains. A cluster of seven isolates from 2018 (three human and four cattle isolates) was identified, with fewer than two allelic differences. They corresponded to the only Austrian B. melitensis outbreak that happened over the past 15 years. The other 12 Austrian brucellosis cases were single cases, and geographical origins were available for 8/12. Genomic data was used to locate probable geographical origins and compared with the results of the epidemiological investigations. Austrian strains were compared with 67 published B. melitensis sequences available on NCBI. The result of genomic analysis matched for 7/8 cases with documented conclusion of the epidemiological investigation. Genome analysis also pointed to the geographical origin for three of the four cases with missing epidemiological data. Strains from six cases were grouped together (<40 allelic differences) with 4/6 cases imported from the Balkans. Additional B. melitensis isolates from Serbian animals were analyzed and grouped with this branch, suggesting frequent importation from Balkan countries to Austria. Overall, this study highlights the specificities of human brucellosis in Austria. It also underlines the value of whole genome sequencing as a tool to investigate brucellosis cases, allowing to identify and investigate outbreaks but also to support epidemiological investigation of imported cases. However, the reliability of such methods depends on the number of strains for comparison, which can be challenging in low incidence countries. Increasing the availability of published sequences with documented geographical origins would help establishing genomic-based methods for investigating brucellosis cases.
Introduction
Brucellosis is a zoonotic disease caused by Brucella spp. Four species are responsible for most human brucellosis cases, and each one is usually associated with a different animal reservoir: B. melitensis (sheep and goat), B. abortus (cow), B. suis (pig), and B. canis (dog) (1). B. melitensis is the predominant species causing human brucellosis, and is associated with more severe disease (2). In sheep and goat, brucellosis is usually a subacute or chronic infection of the reproductive system, but can result in abortions in pregnant females (2). These abortions make brucellosis a leading cause of economic losses in the livestock sector (3). B. melitensis can be transmitted to humans via direct contact with infected animals or consumption of contaminated food (mainly dairy products) (4). Clinical presentation is usually an unspecific mild febrile illness that can be difficult to diagnose (5). Failure of early treatment often leads to bacterial spread and disease progression in the genitourinary, neurological, pulmonary, or cardiovascular systems (6). The most serious complication is endocarditis, reported in 2% of cases (2). Human brucellosis treatment is tedious, the recommended protocol consisting of two combined antibiotics for at least 6 weeks (7). Even with antibiotic treatment, the disease may persist as relapse, chronic localized infection or delayed convalescence (2).
Brucellosis is one of the world's most widespread zoonosis, with an estimated incidence of 500,000 cases per year (3). It is considered a re-emerging disease, highly spread by traveling and globalization (8). In Europe, brucellosis is rare in both cattle and humans (9). Most EU countries have eradicated bovine and ovine/caprine brucellosis, preventing food-borne outbreaks. However, over 380 cases of human brucellosis, mainly B. melitensis infections, were reported in the EU in 2017 and more than 60% required hospitalization (4). In cattle, Austria is officially brucellosis-free (OBF), but a few human cases are still reported every year (10). The incidence is lower than the EU average, with 0.05 cases/100,000 inhabitants. To maintain this OBF status, every human brucellosis is reported and investigated to identify the source of infection. In the past years most investigations found only imported cases, until a local outbreak was identified in 2018.
As B. melitensis infections in Austria are rare and usually imported, collecting as many details as possible is essential to better understand patterns of transmission. This includes epidemiological information on patient's history and also microbiological data on bacterial strains. Whole genome sequencing (WGS) allows to study the entire genetic information of the strain and is therefore highly discriminatory (11). In species with low genetic diversity such as B. melitensis, it is the easiest way to accurately type isolates which is highly valuable to identify the source of infection. Additional data, such as the identification of antimicrobial resistance (AMR) genes, can also be extracted from WGS data. Molecular surveillance of B. melitensis allows to investigate precisely every case, human and animal.
In this study, WGS was used to characterize Austrian B. melitensis isolates. The sequences of the isolates were compared with published genomes of strains from various countries. Similarities between the Austrian and published sequences were estimated using classical multi-locus sequence typing (MLST), but also stable core genome MLST (cgMLST) (12). This analysis provided information on the clustering of the Austrian isolates and comparison with each other but also with published sequences from various geographical areas. Additional data on the possible origins of the strains would allow further study of the patterns of B. melitensis importation to Austria.
Materials and Methods
Isolates
Between 2005 and 2019, 21 B. melitensis strains were received by the Austrian National Reference Center (NRC) for brucellosis for confirmation and species identification. Among these strains, 17 were human clinical isolates (blood or pus from an epidural abscess) and four were isolated from cattle (Table 1). Clinical, epidemiological and microbiological data used in this study fell within the legal mandate given to the NRC by the Austrian Ministry of Health and did not require additional ethical approval. Informed consent was not required because of the retrospective design of this study.
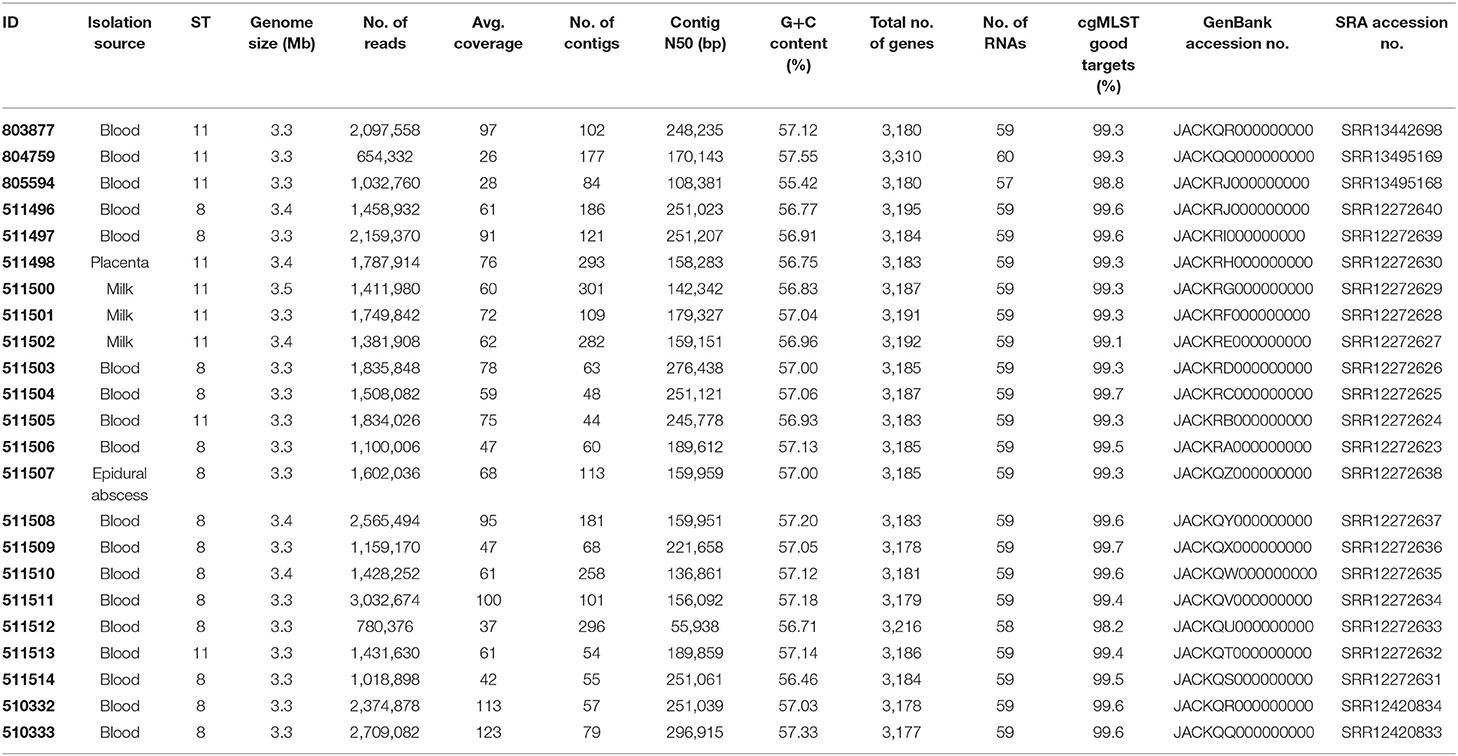
Table 1. Sequencing data, quality data, and accession numbers of the B. melitensis isolate genomes.
Whole Genome Sequencing
For WGS genomic DNA isolation, library preparation, sequencing run, assembly, and contig filtering were performed as described previously (13). Briefly, high-molecular-weight DNA was isolated from a culture using MagAttract HMW DNA Kit (QIAGEN, Hilden, Germany), following the manufacturers' protocol for Gram-negative bacteria. Library preparation to obtain ready-to-sequence libraries was done with a NexteraXT kit (Illumina, Inc., San Diego, CA, USA). Paired-end sequencing (2 × 300 bp) was performed using a MiSeq system (Illumina, Inc.) and raw reads were de novo assembled into a draft genome using SPAdes (version 3.11.1) (14). Contigs were filtered for a minimum coverage of 5 and minimum length of 200 bp. Sequencing generated 654,332–3,032,674 reads and a mean coverage of 26 to 123-fold (Table 1). Assemblies were analyzed with the NCBI prokaryotic genome automatic annotation pipeline (Table 1).
Sequence Analysis
SeqSphere + (Ridom GmbH, Münster, Germany) was used for isolate characterization. Two publicly available typing schemes available on the SeqSphere database, the 9 loci B. melitensis MLST scheme (15) and the B. melitensis cgMLST scheme (2,704 targets in core genome and 360 targets in accessory genome) were used (12). Minimum Spanning Tree (MST) and Neighbor Joining Tree (NJT) were made with SeqSphere.
Sixty-seven published B. melitensis sequences were obtained from GenBank or the Sequence Read Archive (SRA, Supplementary Table 1) (16). Accessing the National Center for Biotechnology Information (NCBI) from Seqsphere, 63 assembled complete genomes of B. melitensis were found. The 51 belonging to ST8 (49 strains) and ST11 (2 strains) were included. As more ST11 sequences were needed for our analysis, our search was extended for scaffold and contig assemblies. Thirteen ST11 published sequences with documented origin could be added. Finally, some ST11 Italian strains were added because of the geographic proximity of Austria and Italy. From the Janowicz et al. study (12), one sheep isolate from a 2015–2016 outbreak in Molise and two human isolates from northern Italy (Piedmont, 2015 and 2017) were added. For these three isolates, read files were downloaded from SRA and assembled using SPAdes.
Single nucleotide polymorphism (SNP) analysis of sequences was performed with CSI Phylogeny 1.4, available from the Center for Genomic Epidemiology (17), using the strain 16M as a reference genome (NC_003317.1 and NC_003318.1). Dendrograms were created from distance matrix (of SNP analysis or cgMLST) with R (version 3.6.1) using packages msa and dendextend.
Data Availability
Raw sequence reads and assemblies were deposited in the SRA and in the GenBank database as project PRJNA647424. Biosamples were assigned accession nos. SAMN15586354 to SAMN15586371, SAMN15774184, SAMN15774185, SAMN17320939, SAMN17393919, and SAMN17393920. SRA accessions no. of raw sequences and GenBank accession no. assigned to assemblies are indicated in Table 1.
Results
Samples and WGS
Twenty-one strains were analyzed in this study including four strains isolated from cattle (three from raw milk samples and one from a placenta tissue sample) and 17 strains isolated from 15 different patients (Table 2). These brucellosis cases were reported between 2005 and 2019. Two patients had two isolates each (511509/511510 and 511496/511497). For 13 cases clinical data were available in the register of mandatorily reportable diseases. These patients were mostly adults (12/13) and male (9/13). This is in accordance with usual brucellosis patterns of contamination with adult men being predominantly affected (4). The clinical presentation was mostly fever and fatigue. The delay between date of onset and date of microbiological investigation (or reception date in Table 2) was between eight and 34 days (mean: 24 days; median: 18 days). However, given the low specificity of symptoms, date of onset can be subject to recall bias. In 11/15 cases epidemiological investigation documented imported etiology with the suspected country of infection.
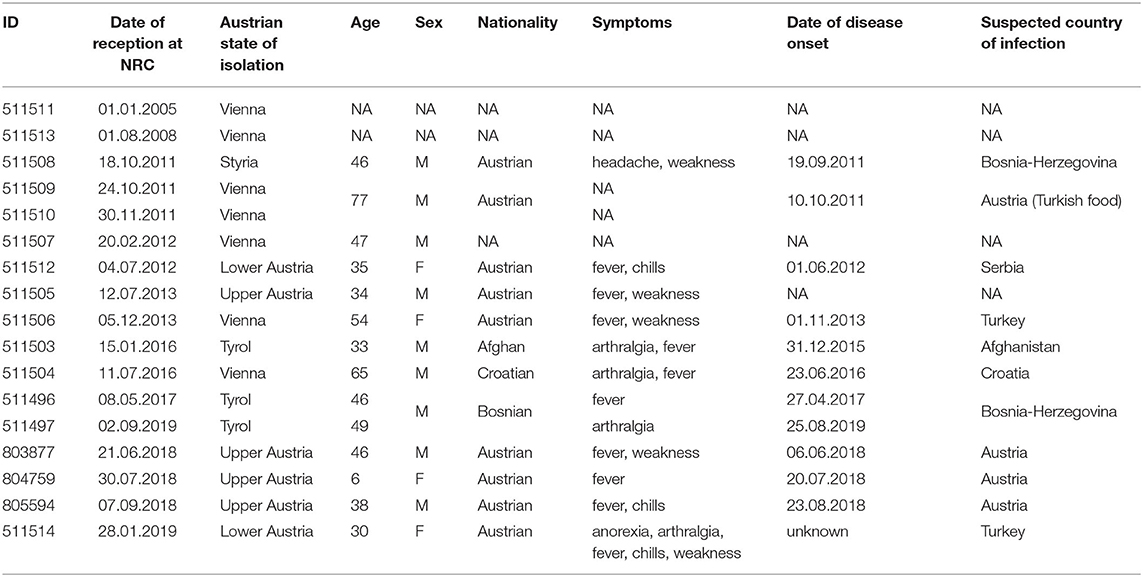
Table 2. Patient and epidemiological data corresponding to the B. melitensis isolates.
Draft genomes obtained by de novo assembly consisted of 44–186 contigs. The NCBI Prokaryotic Genome Automatic Annotation Pipeline identified 3,177–3,310 genes, 2,964–3,008 coding sequences, 57–60 RNAs, 133–161 pseudogenes, 4 rRNA genes and 51−52 tRNA genes (Table 1). Sequences were analyzed for AMR genes using the CARD database (18). Assemblies from all the isolates contained B. suis mprF, which confers AMR to defensins. No other strict hit for resistance genes was found in any of the isolates.
Comparison of Austrian Strains
To compare B. melitensis isolates with each other, MLST typing was first used, extracting sequence types (ST) from WGS data (Table 1). The 21 strains belonged to 2 STs: ST8 (12 strains) and ST11 (9 strains). Previously isolated ST11 B. melitensis strains were of European origin, whereas ST8 strains had been isolated all over the world. Obtaining only 2 different ST for 21 strains isolated between 2005 and 2019 underlines the low discriminatory power of MLST for B. melitensis typing and the need for better alternatives. Therefore, a genome wide approach such as cgMLST seemed to be more appropriate.
The Janowicz cgMLST scheme was used to compare the Austrian B. melitensis isolates (Figures 1A,B) (12). Isolates segregated in two groups separated by a minimum of 1,302 allelic differences which corresponded to ST8 and ST11 strains (Figure 1A). Among ST11 isolates, the four cattle strains and three clinical isolates clustered together. They corresponded to an Austrian outbreak detected at a dairy farm in May 2018 (19). Following an epidemic of miscarriages, B. melitensis was isolated from aborted bovine material (isolate 511498) and milk (isolates 511500, 511501, and 511502). Two veterinarians and the farmer's child were hospitalized and diagnosed with brucellosis (isolates 803877, 804759, and 805594). The genetic similarity between the cattle and human isolates (<2 allelic differences) confirmed that they all belonged to the same outbreak. It proved the first zoonotic transmission of B. melitensis in Austria in more than 15 years. During the outbreak investigation, three additional cases were identified using serology. Patient isolate 511513 from 2008 had only 27 allelic differences to the outbreak cluster of 2018, indicating that the strain responsible for the 2018 outbreak might have been circulating undetected for a longer time (Figure 1B).
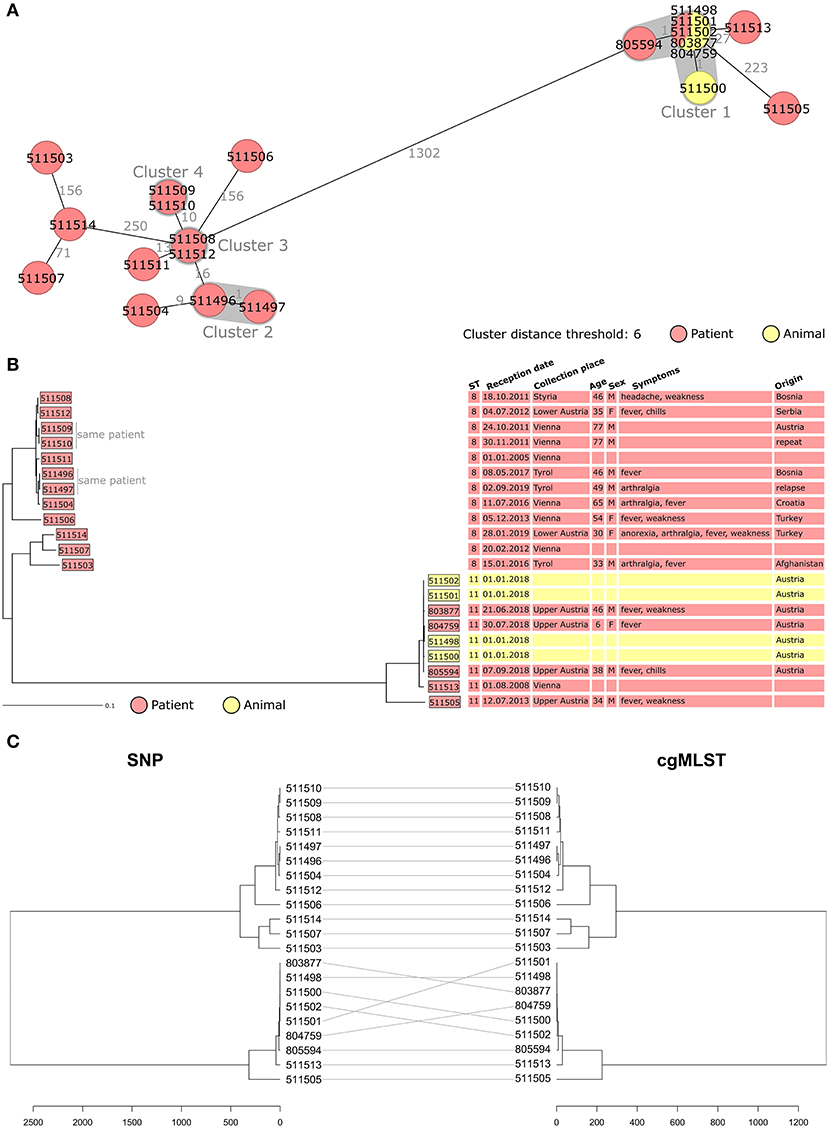
Figure 1. (A,B) Minimum spanning tree [MST, (A)] and Neighbor Joining Tree [NJT, (B)] of B. melitensis isolates obtained in Austria between 2005 and 2019 (N = 21). MST and NJT were computed using the number of allelic differences in stable B. melitensis cgMLST. Colors indicate the type of sample from which strain was isolated. (A) Isolate IDs are indicated on the nodes. Distance between the isolates (in number of allelic differences) are indicated on the connecting lines. Clusters with fewer than 6 allelic differences are shaded in dark gray and named (cluster 1, cluster 2, etc.). (B) Scale bar indicates the percentage of allelic differences between the isolates. Columns on the right provide additional information on isolates: ST, sample information and patient data. (C) Dendrogram computed from the distance matrix of single nucleotide polymorphism analysis (left) and cgMLST analysis (right). Gray lines connect isolates in both trees. Scale bar indicates the number of differences between the isolates, in nucleotides (left) or allelic differences (right).
Among the 12 isolates corresponding to ST8, eight (from six human cases) grouped together with fewer than 40 allelic differences. Among them, two clusters were identified (cluster 2 and cluster 3, Figure 1A) and corresponded to repeated sampling from the same patient. For cluster 3, the second isolate was collected 1 month after the first one to test for treatment efficacy and was identical in cgMLST. For cluster 2, the two isolates were obtained 2 years apart (2017 and 2019). After being cured of brucellosis in 2017 this patient developed a second episode of brucellosis in 2019. WGS analysis of the 2019 isolate showed only one allelic difference to the 2017 strain.
To assess whether the discriminatory power of cgMLST was sufficient to efficiently compare B. melitensis isolates, SNP analysis was performed. The alignments obtained with SNP and cgMLST (Figure 1C) were compared. Both analyses showed identical clustering of the Austrian isolates. This result confirmed that cgMLST was powerful enough for B. melitensis strain typing, and it was therefore used for the rest of the study.
Comparison With Published Sequences
To investigate the divergence between the Austrian B. melitensis strains and the epidemiological relevance, it was decided to compare them with published sequences. Forty-nine ST8 strains and 18 ST11 strains were included in the analysis (Supplementary Table 1). Published sequences corresponding to ST8 strains were more abundant, as ST8 is a common ST. ST8 and ST11 isolates grouped separately, with a minimum of 1,281 allelic differences (Figure 2). Two groups of Austrian isolates (one ST8 group and one ST11 group) were identified.
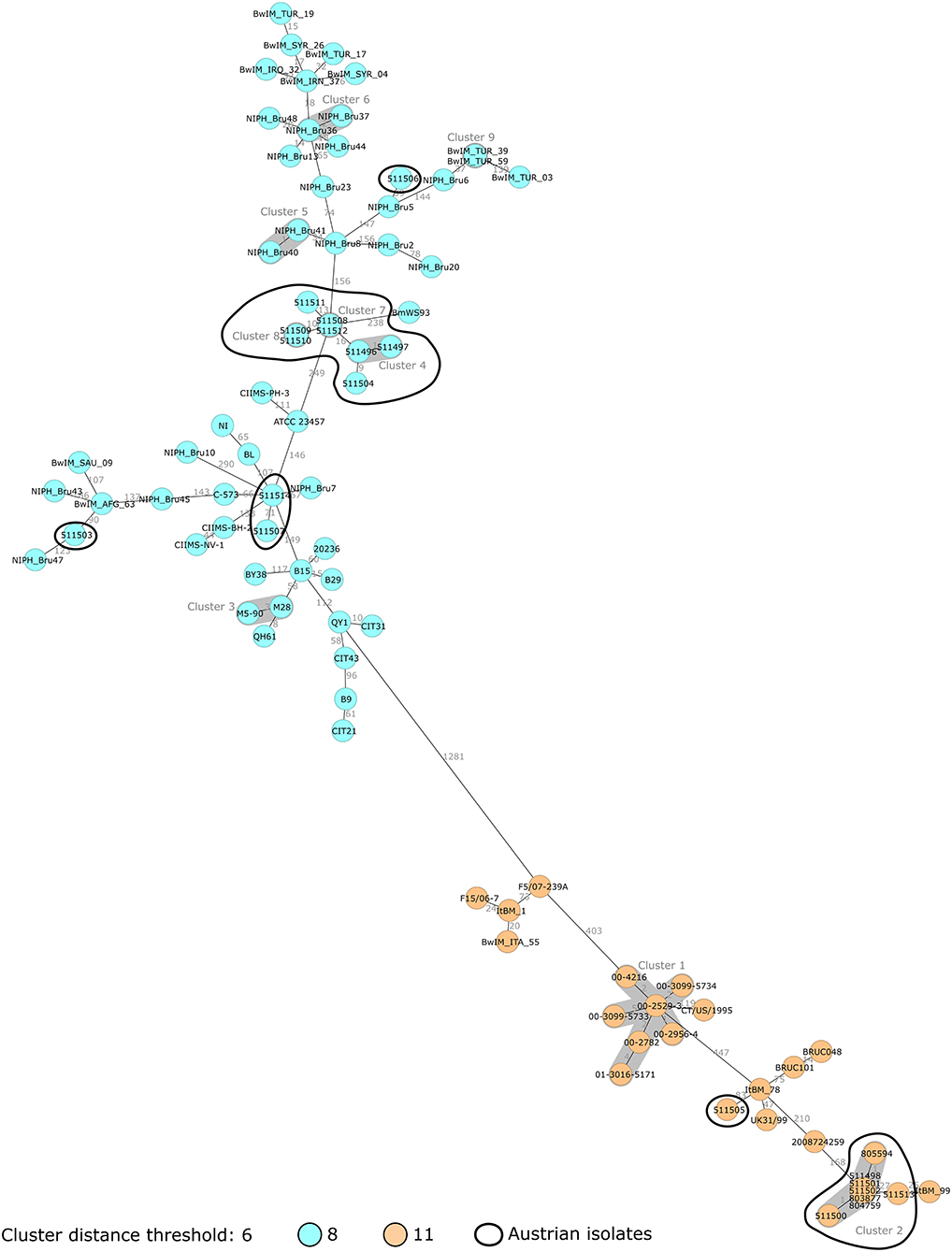
Figure 2. Minimum spanning tree (MST) of B. melitensis isolates obtained in Austria between 2005 and 2019 (N = 21, circled in black) and publicly available sequences (N = 67). MST was computed using the number of allelic differences in stable B. melitensis cgMLST. Isolate IDs are indicated on the nodes. Distance between the isolates (in number of allelic differences) are indicated on the connecting lines. Nodes are colored according to isolate ST. Clusters with fewer than 6 allelic differences are shaded in dark gray and named (cluster 1, cluster 2, etc.).
In Figure 3, Austrian isolates and published sequences were compared using a NJT and colored depending on the reported region of origin. Isolate 511506 grouped with Turkish strains, and epidemiological investigation concluded that it was imported from Turkey (with documented exposure to raw cheese in Turkey). This isolate showed concordance between epidemiological investigations and genomic clustering. Isolate 511503 corresponded to an Afghan patient with reported travel from Afghanistan but no documented exposure (neither contact with animals nor consumption of raw food). This isolate grouped with other strains from central Asia supporting the hypothesis of having been imported from Afghanistan. Isolates 511507 and 511514 grouped with strains from Russia and Georgia, which corresponded to an eastern Mediterranean region clade. As only two NCBI strains belonged to this branch, no global conclusion could be drawn. However, for isolate 511514, the epidemiological investigation highlighted exposure to raw milk in both Turkey and Austria. The genetic data indicates Turkey as the more likely source of infection than Austria.
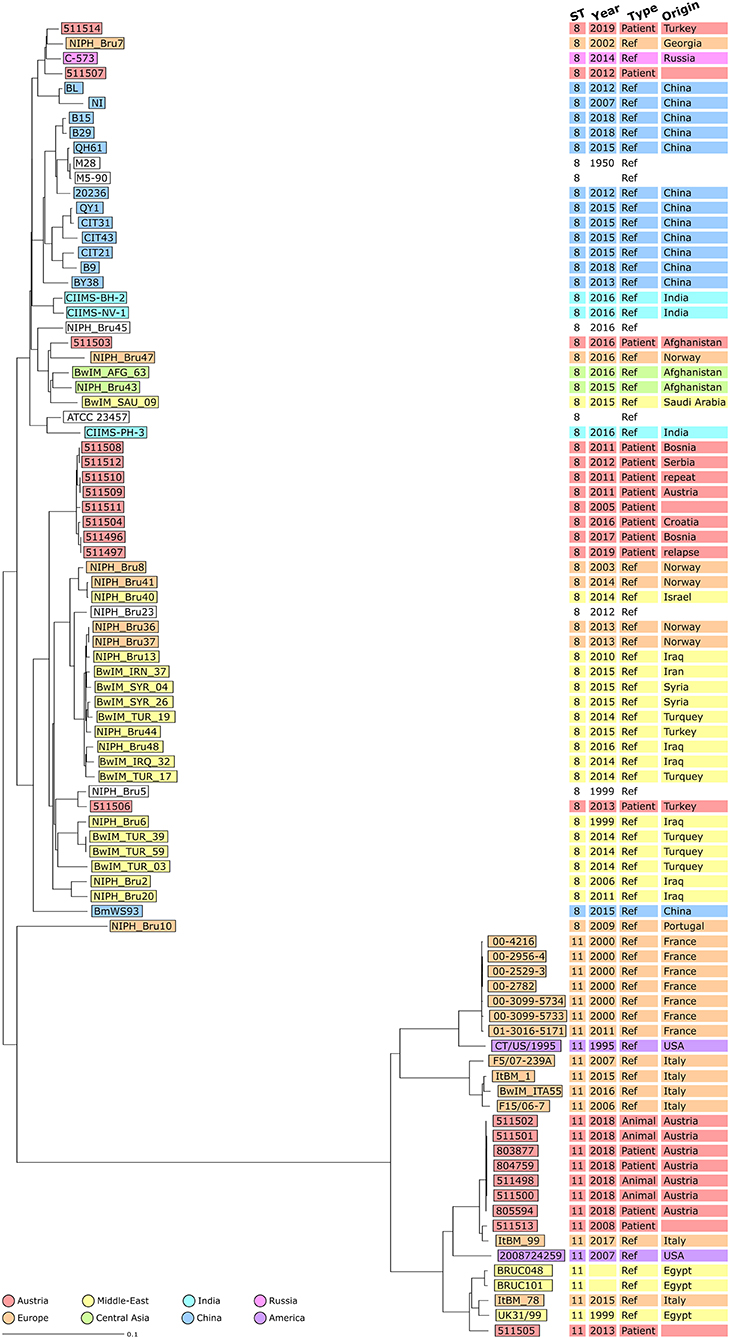
Figure 3. Neighbor Joining Tree (NJT) of B. melitensis isolates obtained in Austria between 2005 and 2019 (N = 21) and publicly available sequences (N = 67). NJT was computed using the number of allelic differences in stable B. melitensis cgMLST. Scale bar indicates the percentage of allelic differences between the isolates. Colors indicate isolate geographical region of origin. Columns on the right provide additional information on isolates: ST, collection date of the sample, type of sample used for isolation and country of infection.
The ST11 strains formed a different branch compared to other STs (Figure 3). The seven isolates from the 2018 Austrian outbreak clustered together. Strain 511505 belonged to a branch including three strains from Egypt and an Italian strain of unknown epidemiological status. This suggested that both the Austrian case corresponding to isolate 511505 and also the Italian case corresponding to isolate ItBM_78, were imported from Egypt. Strain 511513 was closer to an Italian clinical strain from Turin (25 allelic differences) as well as the 2018 Austrian outbreak strains (27 allelic differences).
The group of eight ST8 Austrian strains that was identified previously did not cluster with other strains from the NCBI (Figure 3). Among these patient isolates, four out of six cases were suspected to have been infected in Balkan countries. The two patients corresponding to isolates 511508 and 511512 had documented exposure to raw food (unpasteurized milk in Bosnia and raw cheese in Serbia) supporting the conclusion of the epidemiological investigation that these cases were imported. However, for isolates 511496/511497 and 511504, patients had traveled to Bosnia and Croatia but had no documented exposure to animals or raw food. For isolates 511509/511510, the patient did not report travel to endemic countries, but consumed food imported from Turkey in Austria. Altogether, on the assumption of an importation route from the Balkans to Austria, some of these cases would require further investigation.
To test the hypothesis that these six cases were imported from the Balkans, additional B. melitensis strains from these countries were requested. Serbia provided two strains isolated from sheep and goats which were included in the analysis (Figure 4). They also grouped with the eight Austrian isolates. This finding supported the hypothesis that these eight Austrian strains, including those from patients with no reported exposure to the Balkans, might have been imported from this region. If this conclusion was in accordance with the epidemiological investigation for isolates 511496/511497 and 511504, it is not the case for isolate 511509/511510. In this case, the assumed contamination via imported Turkish food was not supported by the WGS analysis. Overall, these results also highlight that a large proportion of Austrian brucellosis cases were linked to the Balkans.
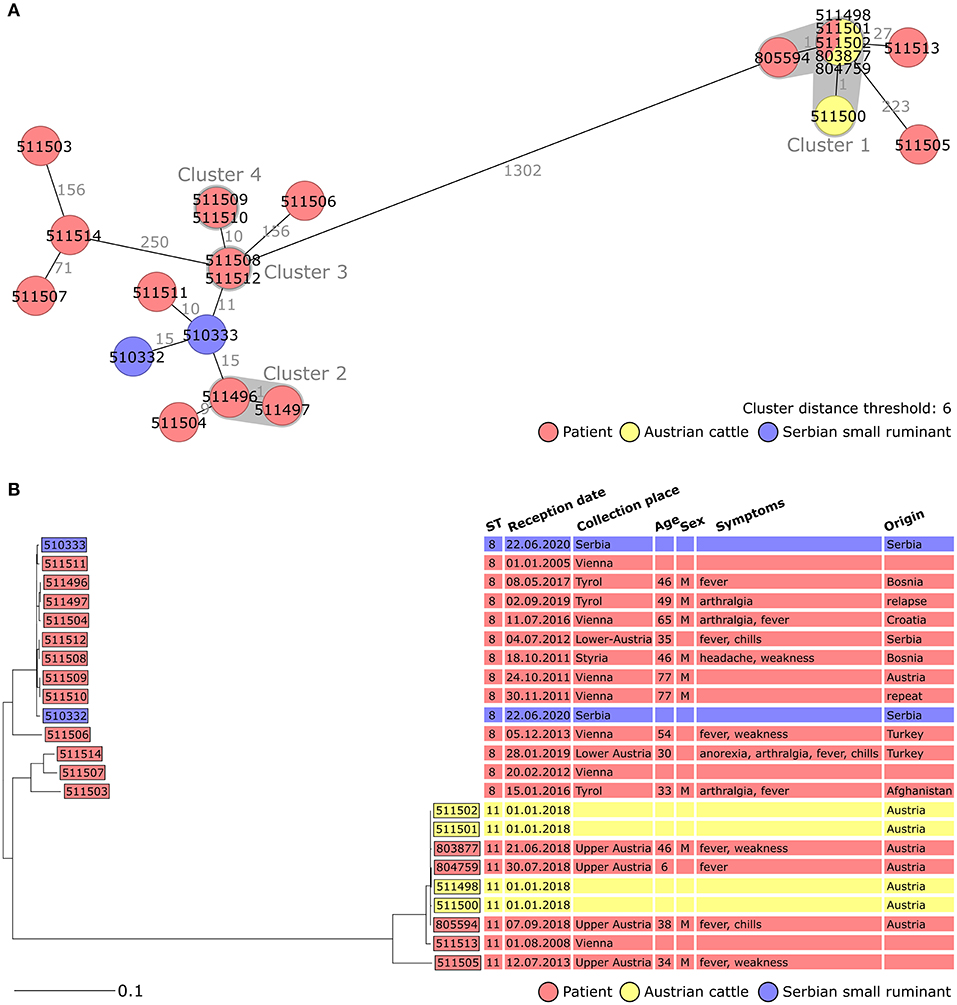
Figure 4. Minimum spanning tree [MST, (A)] and Neighbor Joining Tree [NJT, (B)] of B. melitensis isolates obtained in Austria between 2005 and 2019 (N = 21, red and yellow) and isolates obtained from Serbian animals (N = 2, blue). MST and NJT were computed using the number of allelic differences in stable B. melitensis cgMLST. (A) Isolate IDs are indicated on the nodes. Distance between the isolates (in number of allelic differences) are indicated on the connecting lines. Clusters with fewer than 6 allelic differences are shaded in dark gray and named (cluster 1, cluster 2, etc.). (B) Scale bar indicates the percentage of allelic differences between the isolates. Columns on the right provide additional information on isolates: ST, sample information and patient data.
Discussion
Although the incidence of human brucellosis in Austria is very low, with fewer than 10 cases per year, the individual burden of the disease makes it a relevant public health problem (10). In 2014, an exploratory nationwide cross-sectional seroprevalence survey in 526 healthy adults was conducted and all individuals tested negative for antibodies against B. melitensis/B. abortus (20). A solid surveillance system tracks brucellosis cases in both humans and animals and every case has to be reported and confirmed by the NRC for brucellosis. Human cases are investigated to find the source of infection and prevent further cases. Identifying local outbreaks and segregating them from imported cases is of high importance to maintain Austria's OBF status.
In this study, WGS was used to characterize 21 B. melitensis strains isolated in Austria since 2005. WGS data were searched for AMR genes and only one gene conferring AMR to defensins (B. suis mprF) was found. This gene usually belongs to the resistome of Brucella spp and was identified in over 99% of B. melitensis WGS from NCBI. Testing AMR in B. melitensis is rarely included in surveillance, because it requires bacterial culture in a BLS3 laboratory. Using WGS data to screen for AMR genes could be a convenient alternative, although it is not as informative as proper phenotypical AMR testing. Among the 21 Austrian isolates, 17 strains were isolated from 15 different patients. Two patients had repeated sampling, 1 month apart for the first case and 2 years apart for the second. In both cases, the two strains were similar, reducing the possibility of two independent infections. For the first patient, the fact that bacteria could still be isolated after 1 month of treatment illustrated the potential for B. melitensis persistence. For the second case, the second episode of brucellosis was concluded to be a relapse, with the bacteria persisting in the organism for 2 years.
Isolate typing was performed using cgMLST. One major cluster was identified including three human and four cattle isolates. They corresponded to an autochthonous outbreak identified in 2018. WGS had enabled detection of this outbreak, showing that three patients diagnosed over a period of 3 months were infected by the same B. melitensis strain (1 allelic difference). It underlines how useful genetic comparison of strains (using standard MLVA-typing or more recently WGS) can be to support outbreak investigations in real time (19, 21). A retrospective study in Portugal even identified likely “missed outbreaks” using WGS, as isolates with no documented links clustered (22). When environmental samples are available, comparing isolates using genetic techniques can also confirm the source of infection. It was the case in the 2018 Austrian outbreak, where B. melitensis isolates from cattle placenta and milk clustered with the three clinical isolates. For such investigations, it is worth implementing molecular techniques with the highest possible discriminatory power because they provide a higher probability that two isolates, with similar genetic profiles, are indeed related.
With the exception of the three cases from the 2018 outbreak mentioned above, the other 12 human cases included in this study were single cases. For 8/12, the conclusion of the epidemiological investigation regarding the source of infection was documented. However, for the other 4 cases, epidemiological data were incomplete and no data on whether or not the cases were imported was available. Therefore, it was decided to use the genomic data to identify the most likely geographical origins of the cases. This could have been achieved using environmental or animal samples, as done in a study from Germany where they collected B. melitensis strains from Turkish sheep (23). They were able to link these animal isolates to clinical isolates from travelers returning from Turkey or Turkish immigrants, implying that these cases were indeed imported from Turkey. However, neither food nor animal samples were collected for any of the 12 Austrian patients. The next option was to compare Austrian B. melitensis strains with each other, analyzing how they clustered together. Previous studies used this method, but they had access to a large number of isolates (11, 12). Among the 21 Austrian isolates, one branch with a high proportion of cases imported from the Balkans was identified, but the low number of cases prevented any strong conclusions.
To counterbalance the limitations due to the low sample size, publically available sequences were included in the analysis. This method has proven useful to indicate the geographical origin of brucellosis cases in previous studies, using either MLVA-typing or WGS (24–26). As shown in the study from Sacchini et al., (27) which compared MLST, MLVA and cgMLST to analyze B. melitensis isolates, higher throughput techniques give results with higher discriminatory power (27). In our WGS-based study, the clustering of seven Austrian isolates with published sequences corroborated the country of infection found by the epidemiological investigation. For the four remaining patients it allowed us to suggest a plausible origin; the confidence of this conclusion depends on the number of publically available sequences. This is one of the biggest limitations of this method: its accuracy depends strongly on the number of published sequences with reliable metadata (11). Increasing the pool of available B. melitensis sequences and better data concerning geographical origins would allow WGS-typing to better support epidemiological investigations.
One of the most interesting findings of this study was the identification of a group of isolates originating in the Balkans. The group included cases imported from Balkan countries and also two animal isolates provided by Serbia. This finding was quite unusual among EU countries. Previous studies from Belgium, Germany, Norway and Sweden have highlighted a high proportion of imported cases originating from the eastern Mediterranean region (11, 25–27). Such differences might rely on the geopolitical situation of Austria which has tighter bonds with Eastern Europe and Balkan countries compared to other EU member states. Enhanced international collaboration between Austrian health authorities and their Balkan counterparts would allow a better understanding of these patterns of importation. It could also suggest targeted control measures to reduce the incidence of brucellosis, both in Austria and in the Balkans.
In conclusion, this study highlights several specificities of brucellosis in Austria. It relied on WGS, which is a well-adapted tool for B. melitensis typing thanks to its high discriminatory power. WGS has proven valuable to identify and investigate outbreaks. In addition, comparing large batches of sequences could help identify possible geographical origins of the strains. For countries with low brucellosis incidence, such techniques require an external data source. More publicly available sequences with documented origin need to be available in order to increase the reliability of this method. Overall, this study underlines the usefulness of implementing WGS for routine B. melitensis surveillance, in both human and animal health. It also advocates publishing B. melitensis sequences with relevant metadata thus increasing the pool of available genomic data.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author Contributions
JS and WR planned and conducted the study. SR-F, EH, RP, CL, FS, PH, and AI provided the Austrian strains and the corresponding patient data. AS sequenced the isolates. VD, BL, and KM provided the Serbian isolates. FS and FA provided funding. JS wrote the manuscript and all co-authors provided feedback on the draft. All authors contributed to the article and approved the submitted version.
Funding
JS was supported by a grant from the European Public Health Microbiology Training Program (EUPHEM), European Center for Disease Prevention and Control (specific grant agreement number 1 ECD.7550 implementing ECDC/GRANT/2017/003).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as ass potential conflict of interest.
Acknowledgments
We thank the staff of the Brucella National Reference Center for isolate collection. We thank Loredana Ingrosso for her feedback throughout the project and on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2021.635547/full#supplementary-material
References
1. Hull NC, Schumaker BA. Comparisons of brucellosis between human and veterinary medicine. Infect. Ecol. Epidemiol. (2018) 8:1500846. doi: 10.1080/20008686.2018.1500846
3. Pappas G, Papadimitriou P, Akritidis N, Christou L, Tsianos EV. The new global map of human brucellosis. Lancet Infect. Dis. (2006) 6:91–9. doi: 10.1016/S1473-3099(06)70382-6
5. de Figueiredo P, Ficht TA, Rice-Ficht A, Rossetti CA, Adams LG. Pathogenesis and immunobiology of brucellosis: review of Brucella-host interactions. Am. J. Pathol. (2015) 185:1505–17. doi: 10.1016/j.ajpath.2015.03.003
6. Buzgan T, Karahocagil MK, Irmak H, Baran AI, Karsen H, Evirgen O, Akdeniz H. Clinical manifestations and complications in 1028 cases of brucellosis: a retrospective evaluation and review of the literature. Int. J. Infect. Dis. (2010) 14:e469–78. doi: 10.1016/j.ijid.2009.06.031
7. Ariza J, Bosilkovski M, Cascio A, Colmenero JD, Corbel MJ, Falagas ME, Memish ZA, et al. Perspectives for the treatment of brucellosis in the 21st century: the Ioannina recommendations. PLoS Med. (2007) 4:e317. doi: 10.1371/journal.pmed.0040317
8. Pappas G, Akritidis N, Bosilkovski M, Tsianos E. Brucellosis. N. Engl. J. Med. (2005) 352:2325–36. doi: 10.1056/NEJMra050570
9. EFSA and ECDC. The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2017. EFSA J. (2018) 16:e05500. doi: 10.2903/j.efsa.2018.5500
10. Much P, Arrouas M, Herzog U. Zoonoses and Zoonotic Agents in Austria - Report 2018. Austrian Agency for Health and Food Safety (AGES) (2018).
11. Georgi E, Walter MC, Pfalzgraf M-T, Northoff BH, Holdt LM, Scholz HC, et al. Whole genome sequencing of Brucella melitensis isolated from 57 patients in Germany reveals high diversity in strains from Middle East. PLoS ONE. (2017) 12:e0175425. doi: 10.1371/journal.pone.0175425
12. Janowicz A, De Massis F, Ancora M, Cammà C, Patavino C, Battisti A, Prior K, et al. Core genome multilocus sequence typing and single nucleotide polymorphism analysis in the epidemiology of Brucella melitensis infections. J. Clin. Microbiol. (2018) 56:e00517-18. doi: 10.1128/JCM.00517-18
13. Lepuschitz S, Sorschag S, Springer B, Allerberger F, Ruppitsch W. Draft genome sequence of carbapenemase-producing Serratia marcescens isolated from a patient with chronic obstructive pulmonary disease. Genome Announc. (2017) 5:e01288-17. doi: 10.1128/genomeA.01288-17
14. Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. (2013) 20:714–37. doi: 10.1089/cmb.2013.0084
15. Whatmore AM, Perrett LL, MacMillan AP. Characterisation of the genetic diversity of Brucella by multilocus sequencing. BMC Microbiol. (2007) 7:34. doi: 10.1186/1471-2180-7-34
16. NCBI. NCBI Genome. Brucella Melitensis Genome Tree Report. (2019). Available online at: https://www.ncbi.nlm.nih.gov/genome/genomes/943 (accessed January 5, 2021).
17. Kaas RS, Leekitcharoenphon P, Aarestrup FM, Lund O. Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS ONE. (2014) 9:e104984. doi: 10.1371/journal.pone.0104984
18. Alcock BP, Raphenya AR, Lau TTY, Tsang KK, Bouchard M, Edalatmand A, et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. (2020) 48:D517–25. doi: 10.1093/nar/gkz935
19. Blaschitz M, Hufnagl P, Stöger A, Felsberger G, Schmid D, Allerberger F, et al. Use of whole genome sequencing in an outbreak investigation of bovine brucellosis due to Brucella melitensis, Austria, 2018. Int. J. Infect. Dis. (2019) 79:84. doi: 10.1016/j.ijid.2018.11.213
20. Tobudic S, Nedomansky K, Poeppl W, Müller M, Faas A, Mooseder G, et al. Seroprevalence for Coxiella burnetii, Francisella tularensis, Brucella abortus and Brucella melitensis in Austrian adults: a cross-sectional survey among military personnel and civilians. Ticks Tick Borne Dis. (2014) 5:315–7. doi: 10.1016/j.ttbdis.2013.12.007
21. Arapović J, Špičić S, Ostojić M, Duvnjak S, Arapović M, Nikolić J, et al. Epidemiological, clinical and molecular characterization of human brucellosis in Bosnia and Herzegovina - an ongoing brucellosis outbreak. Acta Med. Acad. (2018) 47:50–60. doi: 10.5644/ama2006-124.214
22. Pelerito A, Nunes A, Núncio MS, Gomes JP. Genome-scale approach to study the genetic relatedness among Brucella melitensis strains. PLoS ONE. (2020) 15:e0229863. doi: 10.1371/journal.pone.0229863
23. Gwida M, Neubauer H, Ilhan Z, Schmoock G, Melzer F, Nöckler K, et al. Cross-border molecular tracing of brucellosis in Europe. Comp. Immunol. Microbiol. Infect. Dis. (2012) 35:181–5. doi: 10.1016/j.cimid.2011.12.012
24. Muchowski JK, Koylass MS, Dainty AC, Stack JA, Perrett L, Whatmore AM, et al. Using molecular tools to identify the geographical origin of a case of human brucellosis. Epidemiol. Infect. (2015) 143:3110–3. doi: 10.1017/S0950268814003896
25. Hanot Mambres D, Boarbi S, Michel P, Bouker N, Escobar-Calle L, Desqueper D, et al. Imported human brucellosis in Belgium: bio and molecular typing of bacterial isolates, 1996-2015. PLoS ONE. (2017) 12:e0174756. doi: 10.1371/journal.pone.0174756
26. Johansen TB, Scheffer L, Jensen VK, Bohlin J, Feruglio SL. Whole-genome sequencing and antimicrobial resistance in Brucella melitensis from a Norwegian perspective. Sci. Rep. (2018) 8:8538. doi: 10.1038/s41598-018-26906-3
Keywords: brucellosis, Brucella melitensis, whole genome sequencing, core genome multilocus sequence typing, imported case
Citation: Schaeffer J, Revilla-Fernández S, Hofer E, Posch R, Stoeger A, Leth C, Schmoll F, Djordjevic V, Lakicevic B, Matovic K, Hufnagl P, Indra A, Allerberger F and Ruppitsch W (2021) Tracking the Origin of Austrian Human Brucellosis Cases Using Whole Genome Sequencing. Front. Med. 8:635547. doi: 10.3389/fmed.2021.635547
Received: 03 December 2020; Accepted: 04 February 2021;
Published: 24 February 2021.
Edited by:
Jacques Xavier Godfroid, Arctic University of Norway, NorwayReviewed by:
Gamal Wareth, Friedrich-Loeffler-Institute, GermanyHolger C. Scholz, Institut für Mikrobiologie der Bundeswehr, Germany
Copyright © 2021 Schaeffer, Revilla-Fernández, Hofer, Posch, Stoeger, Leth, Schmoll, Djordjevic, Lakicevic, Matovic, Hufnagl, Indra, Allerberger and Ruppitsch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Justine Schaeffer, justine.schaeffer@ages.at; schaeffer.justine.91@gmail.com