Programmed Cell Deaths and Potential Crosstalk With Blood–Brain Barrier Dysfunction After Hemorrhagic Stroke
 Yuanjian Fang1†
Yuanjian Fang1†  Shiqi Gao1†
Shiqi Gao1†  Xiaoyu Wang1
Xiaoyu Wang1  Yang Cao1
Yang Cao1  Jianan Lu1
Jianan Lu1  Sheng Chen1
Sheng Chen1  Cameron Lenahan2,3,4
Cameron Lenahan2,3,4  John H. Zhang2,4,5,6
John H. Zhang2,4,5,6  Anwen Shao1*
Anwen Shao1*  Jianmin Zhang1,7,8*
Jianmin Zhang1,7,8*- 1Department of Neurosurgery, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 2Department of Physiology and Pharmacology, Loma Linda University School of Medicine, Loma Linda, CA, United States
- 3Burrell College of Osteopathic Medicine, Las Cruces, NM, United States
- 4Center for Neuroscience Research, Loma Linda University School of Medicine, Loma Linda, CA, United States
- 5Department of Anesthesiology, Loma Linda University School of Medicine, Loma Linda, CA, United States
- 6Department of Neurosurgery, Loma Linda University School of Medicine, Loma Linda, CA, United States
- 7Brain Research Institute, Zhejiang University, Hangzhou, China
- 8Collaborative Innovation Center for Brain Science, Zhejiang University, Hangzhou, China
Hemorrhagic stroke is a life-threatening neurological disease characterized by high mortality and morbidity. Various pathophysiological responses are initiated after blood enters the interstitial space of the brain, compressing the brain tissue and thus causing cell death. Recently, three new programmed cell deaths (PCDs), necroptosis, pyroptosis, and ferroptosis, were also found to be important contributors in the pathophysiology of hemorrhagic stroke. Additionally, blood–brain barrier (BBB) dysfunction plays a crucial role in the pathophysiology of hemorrhagic stroke. The primary insult following BBB dysfunction may disrupt the tight junctions (TJs), transporters, transcytosis, and leukocyte adhesion molecule expression, which may lead to brain edema, ionic homeostasis disruption, altered signaling, and immune infiltration, consequently causing neuronal cell death. This review article summarizes recent advances in our knowledge of the mechanisms regarding these new PCDs and reviews their contributions in hemorrhagic stroke and potential crosstalk in BBB dysfunction. Numerous studies revealed that necroptosis, pyroptosis, and ferroptosis participate in cell death after subarachnoid hemorrhage (SAH) and intracerebral hemorrhage (ICH). Endothelial dysfunction caused by these three PCDs may be the critical factor during BBB damage. Also, several signaling pathways were involved in PCDs and BBB dysfunction. These new PCDs (necroptosis, pyroptosis, ferroptosis), as well as BBB dysfunction, each play a critical role after hemorrhagic stroke. A better understanding of the interrelationship among them might provide us with better therapeutic targets for the treatment of hemorrhagic stroke.
Introduction
Hemorrhagic stroke, including intracerebral hemorrhage (ICH) and subarachnoid hemorrhage (SAH), is an important public health concern with high morbidity and mortality worldwide (Scimemi, 2018; Fang et al., 2019). ICH and SAH have annual incidence rates of nearly 20 and 10 cases per 100,000, respectively, but with some regional variations (de Rooij et al., 2007; van Asch et al., 2010). Despite significant improvements in the prevention and clinical treatment of ICH and SAH, the global incidence, as well as related mortality and morbidity rates, has increased over the past several decades (Qureshi et al., 2009; Turan et al., 2016; Macdonald and Schweizer, 2017). As reported, the 5-year survival rate of patients with hemorrhagic stroke is less than 50%. Most survivors of hemorrhagic stroke have a reduced quality of life and may require more time and resources during hospitalization and rehabilitation (Gustavsson et al., 2011). Recently, numerous studies have focused on the pathological mechanisms after hemorrhagic stroke, with the intention of seeking potential therapeutic targets to guide treatment (Grysiewicz et al., 2008). However, because of the complexity of its mechanism and the limitations of translational studies, the benefits have been limited (Wilkinson et al., 2018). Hemorrhagic stroke occurs when a weakened vessel ruptures, allowing blood to traverse the broken blood–brain barrier (BBB) into the brain tissue, or fissures, which leads to a mass effect that increases the intracranial pressure and decreases the cerebral blood flow (Topkoru et al., 2017; Tao et al., 2019). Thus, the deficiency of ATP and increased blood toxic substances initiate a cascade of pathophysiological changes, such as depolarization, excitotoxicity, cell edema, inflammatory responses, oxidative stress, ionic homeostasis, and secondary BBB disruption (Zille et al., 2017; Fang et al., 2018; Marbacher et al., 2018; Shao A. et al., 2019; Shao Z. et al., 2019; Wan et al., 2019). These pathophysiological changes may lead to various forms of cell death, including apoptosis, necrosis, necroptosis, autosis, ferroptosis, pyroptosis, parthanatos, and cyclophilin D necrosis, which are also categorized under programmed cell deaths (PCDs; Fuchs and Steller, 2015; Sekerdag et al., 2018). Increasing efforts have been deployed to study these PCDs and their related pathways to discover potential therapeutic targets that will provide neuroprotection after hemorrhagic stroke (Leist and Jaattela, 2001).
Pathological changes in BBB function are widely implicated in the pathophysiological changes after hemorrhagic stroke (Daneman and Prat, 2015; Lublinsky et al., 2019; Ma et al., 2020). Recent studies have proposed that endothelial cells (ECs) do not work alone in the maintenance of the BBB and that astrocytes, pericytes, neurons, and microglia also contribute and are collectively known as the neurovascular unit (NVU). The death of these cells may directly or indirectly result in dysfunction of the BBB (Jiang et al., 2018). Additionally, BBB disruption may further aggravate brain edema, ionic homeostasis disruption, altered signaling, and immune infiltration, consequently causing cell death (Daneman and Prat, 2015).
Given the significant role of PCDs and BBB dysfunction in the pathophysiological mechanisms after hemorrhagic stroke, this review article focuses on the mechanism and recent studies of three PCDs (necroptosis, pyroptosis, and ferroptosis) after hemorrhagic stroke. In addition, we will also discuss, summarize, and hypothesize that the crosstalk between these PCDs and BBB dysfunction after hemorrhagic stroke may lead to the development of novel therapeutic approaches in the future.
The Mechanism of Necroptosis
Necroptosis is a cell death similar to necrosis in terms of morphological features, such as being triggered by death ligands or intracellular stimuli and being executed in a caspase-independent manner (Liu et al., 2018). Morphologically, necroptosis is distinguished from apoptosis by the presence of clusters of dying cells, which may be indicated by early destruction of membrane integrity, cell and organelle swelling, cytoplasmic granulation, chromatin fragmentation, and cell lysis (Zhang et al., 2017). Ligands, such as tumor necrosis factor (TNF), TNF-related apoptosis-inducing ligand, and the intracellular stimuli, such as DNA-dependent activator of interferon regulatory factors (which may act as a cytoplasmic viral RNA sensor) and protein kinase R, can initiate necroptosis (Linkermann and Green, 2014). Furthermore, necroptosis can also be triggered by interferons and Toll-like receptor signaling (Zhang et al., 2017). The receptor-interacting kinase 3 (RIPK3) and its substrate, the pseudokinase mixed lineage kinase domain-like protein (MLKL), execute the core component of necroptosis without caspase participation (Zhang et al., 2009; Sun et al., 2012). Generally, there are several signaling pathways involved in necroptosis, including TNFα, TNF receptor (TNFR1)-related signaling pathway, TRAIL, and other factors associated with the apoptosis signaling pathway, as well as the RIPK3 mitochondrial reactive oxygen species (ROS) metabolic pathway and the zVAD-mediated PKC/mitogen-activated protein kinase (MAPK)/AP1-related signaling pathway (Jin and El-Deiry, 2006; Vanden Berghe et al., 2007; Wu et al., 2011; Liu et al., 2018).
The first step of necroptosis is the formation of the necrosome (Zhang et al., 2017; Figure 1). Take the thoroughly studied TNFα–TNFR1-related signaling pathway as an example. TNF α activates TNFR1 through the extracellular domain and subsequently triggers the trimerization of TNFR1, which recruits several proteins to form complex I at the plasma membrane. Complex I comprised several components, such as TNFα receptor-associated death domain (interact with RIPK1), RIPK1 (core protein), TNFR-associated factor 2 (TRAF2), TRAF5, cellular inhibitor of apoptosis 1 (cIAP1), and cIAP2 (mediates the ubiquitination of RIPK1; Micheau and Tschopp, 2003). Ubiquitination of RIPK1 generates binding sites for TAB2/3 (transforming growth factor beta–activated kinase 1 (TAK1) and NEMO [the regulatory subunit of the IκB kinase (IKK) complex], which leads to further recruitment and activation of TAK1 and IKKα/β. Then, activated IKKα/β phosphorylated IκB and leads to nuclear factor κB release and MAPK activation, promoting cell survival (Bertrand et al., 2008; Hayden and Ghosh, 2014). Conversely, RIPK1 loses its default prosurvival function when it is deubiquitinated by the induction of deubiquitinases, such as cylindromatosis and A20 (Wertz et al., 2004; Moquin et al., 2013). Then, the deubiquitinated RIPK1 can bind to FAS-associated protein with a death domain and recruit procaspase 8 to form complex II within the cytoplasm. Apoptosis occurs when caspase 8 is activated. However, necroptosis occurs when caspase 8 is inhibited (Oberst et al., 2011). In the necrosome, RIPK1 can phosphorylate the RIPK3 to execute the following necroptosis (Zhang et al., 2009).
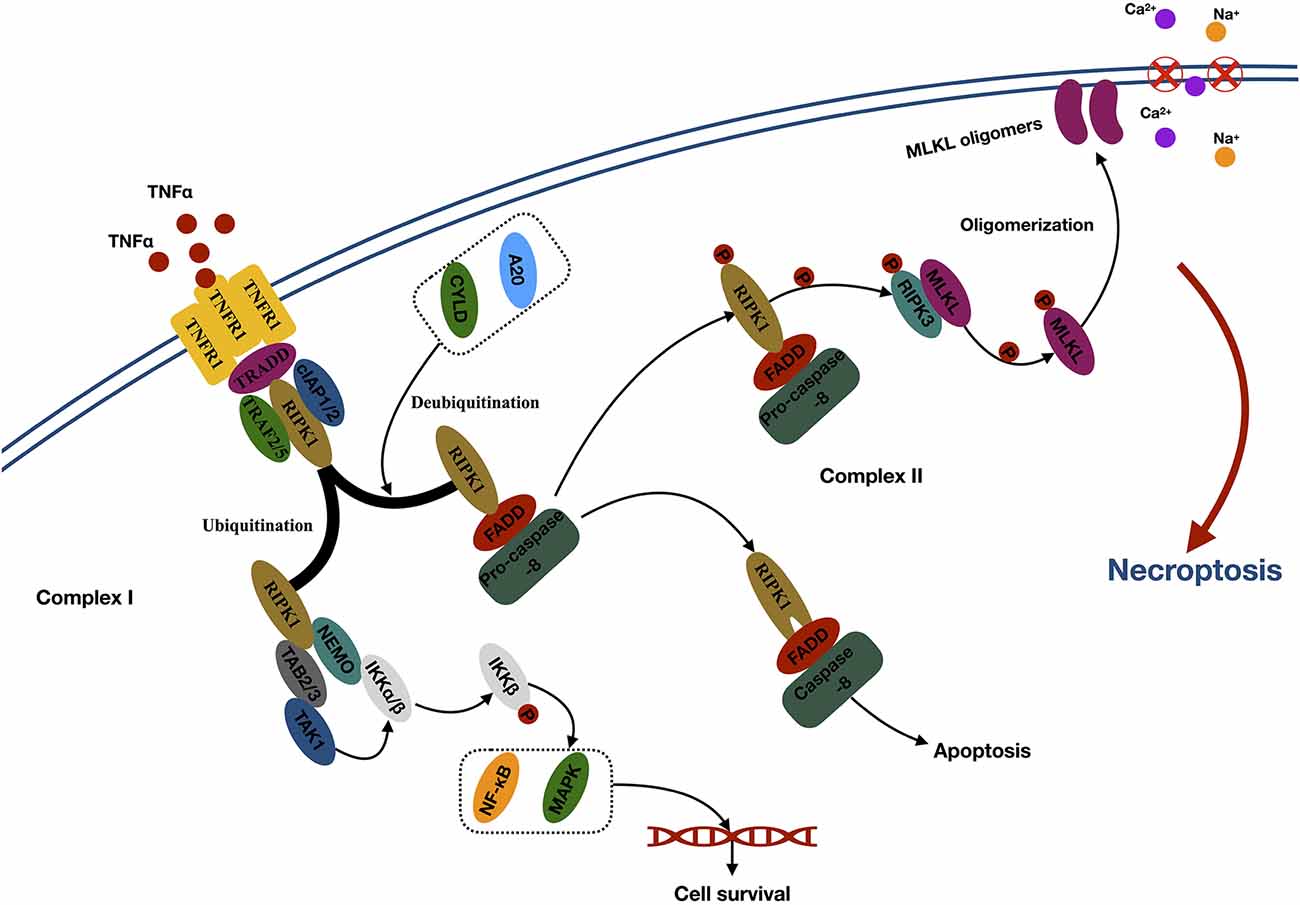
Figure 1. Mechanisms of necroptosis. Abbreviations: TNFα, tumor necrosis factor; TNFR1, tumor necrosis factor receptor type 1; TRADD, TNFα receptor-associated death domain; TRAF2, TNFR-associated factor 2; cIAP1, cellular inhibitor of apoptosis 1; RIPK1, receptor-interacting kinase 1; CYLD, cylindromatosis; FADD, Fas-associated death domain; RIPK3, receptor-interacting kinase 3; MLKL, mixed lineage kinase domain-like protein; NEMO, NF-κB essential modulator; TAB2/3, TAK1-binding 2/3; TAK1, TGF-β–activated kinase 1; IKKα/β, IκB kinase α/β; NF-κb, nuclear factor κB; MAPK, mitogen-activated protein kinase.
The second step of necroptosis is processing (Figure 1). The activated RIPK3 binds to and induces phosphorylation of cytoplasmic MLKL. Phosphorylation leads to the oligomerization of MLKL and membrane translocation of the MLKL oligomers, which disrupts the integrity and increases permeability of the membrane, therefore inducing internal flow of Ca2+ or Na+ ions, finally leading to cell death (Wang et al., 2014).
Necroptosis in Hemorrhagic Stroke
Although the phenomenon of necroptosis has been extensively investigated in the pathophysiological mechanisms of other diseases, there are relatively few studies regarding necroptosis and hemorrhagic stroke (Table 1). It is known that necroptosis can be blocked by a small, specific molecular compound, known as necrostatin 1 (nec-1), by targeting and binding with RIPK1 (Galluzzi et al., 2017). Numerous studies have attempted to inhibit necroptosis by using nec-1 to reduce cell death and improve neurological function, thereby indirectly demonstrating the existence of necroptosis in the pathophysiological development after hemorrhagic stroke (Laird et al., 2008; King et al., 2014; Majmundar et al., 2016). Administration of nec-1 and the irreversible pan-caspase inhibitor, z-VAD, in astrocytic cells revealed that only nec-1 could significantly reduce hemin-induced astrocytic death, suggesting that caspase-independent necroptosis may mediate astrocytic death after hemorrhagic injury in an in vitro model. In addition, the proinflammatory effect of hemin relied on the enhanced astrocytic necroptosis after ICH (Laird et al., 2008). Meanwhile, nec-1 presented the capacity to reduce hematoma volume and neurovascular injury, while improving neurological outcomes after ICH in mice (King et al., 2014; Majmundar et al., 2016). However, they did not reveal detailed information pertaining to the cell types and mechanisms of necroptosis in their studies.
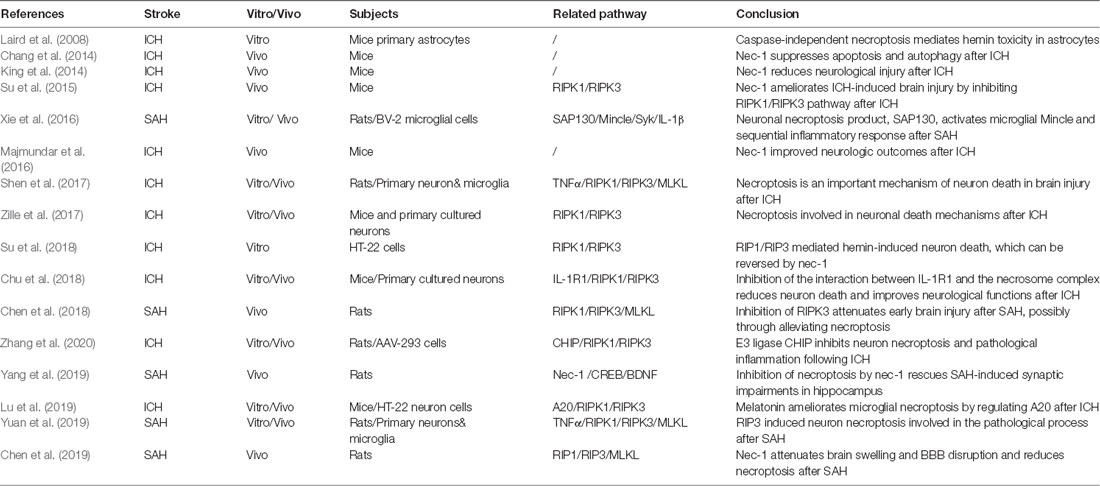
Table 1. Latest research of necroptosis in hemorrhagic stroke.
It should be mentioned that nec-1 administration may also suppress the apoptotic and autophagic pathways after ICH and exert a neuroprotective effect (Chang et al., 2014). Necrostatin 1 treatment increased Bcl-2 expression and decreased cleaved caspase 3 levels, as well as the beclin 1/Bcl-2 ratio at 24 and 72 h after ICH (Chang et al., 2014). Thus, the following study attempts to identify the interaction between the key pathway in necroptosis (RIPK1/RIPK3) and nec-1. It has been found that RIPK1 and RIPK3 were significantly decreased, with reduced necrotic cell death (no detailed cell type was shown), under the treatment of nec-1 in mice after ICH, further suggesting that nec-1 inhibited necroptosis after ICH (Su et al., 2015).
The RIPK1/RIPK3 pathway was shown to play an important role in hemin-induced cell death (Zille et al., 2017; Su et al., 2018). RIPK1 and RIPK3 mRNA levels and phospho-RIPK1 increased after the primary cortical neuron had been treated with hemin (Zille et al., 2017). Necrostatin 1 or RIPK3 siRNA dramatically attenuated hemin-induced cell death and ROS accumulation in the HT-22 neuron cell line (Su et al., 2018). Furthermore, the neuronal necroptosis can be diminished if mutations are present at the serine kinase phosphorylation site of RIPK1, further supporting the significance of RIPK1 phosphorylation in necroptosis (Shen et al., 2017).
Meanwhile, the RIPK1/RIPK3-mediated necroptosis pathway was also involved in the alterations of pathophysiology after SAH (Chen et al., 2018, 2019; Yuan et al., 2019). The RIPK3 protein level increased in the rat brain and peaked at 24 h after SAH. Inhibition of RIPK3 by genetic or pharmacological treatment attenuated the brain injury in rats after SAH and neuronal necroptosis induced by oxygen hemoglobin (OxyHb; Chen et al., 2018, 2019; Yuan et al., 2019). Besides, pretreatment of nec-1 in rats after SAH demonstrated that nec-1 could also prevent BBB dysfunction by reducing the degradation of TJ proteins (occludin, claudin-5, and ZO-1), by increasing active matrix metalloproteinase 9 (MMP-9), and by reducing neuroinflammation via reduction of proinflammatory cytokines, such as interleukin 1β (IL-1β), IL-6, and TNFα after SAH (Chen et al., 2019). These findings seemingly indicated that nec-1 attenuated astrocytic and endothelial cell necroptosis. Additionally, it has been reported that the neuroprotective effect of nec-1 was also connected with another pathway. Necrostatin 1 rescues SAH-induced synaptic impairments and neuronal death in the hippocampus of rats via the cAMP-responsive element-binding proteins (CREB)/brain-derived neurotrophic factor (BDNF) pathway (Yang et al., 2019). cAMP-responsive element-binding protein and BDNF play an important role in synaptic plasticity, which contributes to memory processing (Seoane et al., 2011). Necrostatin 1 can reverse the decreased protein level of CREB and BDNF in rats after SAH (Yang et al., 2019).
Recently, several upstream regulators of necroptosis were found in hemorrhagic stroke (Shen et al., 2017; Chu et al., 2018). Necroptosis of primary cultured neurons could be induced by supernatant medium derived from microglia treated with OxyHb, but could be countered by a TNFα inhibitor, indicating that the TNFα–TNFR1-related signaling pathway participates in this process (Shen et al., 2017). Besides, hemin triggers neuronal necroptosis through promotion of IL-1 receptor 1 (IL-1R1) and RIPK complex formation. Inhibition of IL-1R1 can prevent necrosome and RIPK1/RIPK3 pathway activation, suggesting that the IL-1R1/RIPK1/RIPK3 pathway was also an important signaling pathway involved in necroptosis (Chu et al., 2018).
Downstream in the RIPK1/RIPK3 pathway, there are several regulators of necroptosis found in hemorrhagic stroke, such as carboxyl terminus of Hsp70-interacting protein (CHIP) and A20 (Lu et al., 2019; Zhang et al., 2020). CHIP is an E3 ligase that mediates ubiquitylation and negatively regulates the protein level of RIPK1 and RIPk3 (Seo et al., 2016). The expression of CHIP increased in the perihematomal region in rats after ICH. Overexpression of CHIP exerts neuroprotective effects by regulating the RIPK1/RIPK3 necroptosis pathway in neurons and therefore attenuating ICH-associated cerebral inflammation (Zhang et al., 2020). Additionally, the deubiquitylating enzyme, A20, was shown to inhibit RIP3 activity and reduce microglial necroptosis after ICH, in vitro and in vivo. Moreover, the A20 was identified as a novel target of melatonin, which upregulates A20 activity and suppresses necroptosis following ICH (Lu et al., 2019). On the other hand, it was found that the neuronal necroptosis product, SAP130, activates microglial macrophage-inducible C-type lectin (Mincle) and the sequential inflammatory response by mediating Mincle/Syk/IL-1b signaling in the ipsilateral hemisphere that had been subjected to SAH (Xie et al., 2017).
In conclusion, necroptosis is initiated after hemorrhagic stroke or hemin treatment in different cell types, such as astrocytes, neurons, and microglia. Inhibition of necroptosis by administration of nec-1 appears to be a potential target for the treatment of hemorrhagic stroke. However, the detailed mechanisms of necroptosis should be further investigated, not only in vivo but also in vitro, with particular emphasis regarding ECs due to the numerous studies demonstrating that inhibition of necroptosis can protect the BBB after hemorrhagic stroke.
The Mechanism of Ferroptosis
Ferroptosis is a form of cell death characterized by the accumulation of intracellular iron and lipid ROS. The primary morphologic manifestations of ferroptosis include cell volume shrinkage and increased mitochondrial membrane density (Yu et al., 2017). The ferroptosis can be roughly divided into two segments: the core pathway of ferroptosis and iron metabolism (Xie et al., 2016; Figure 2).
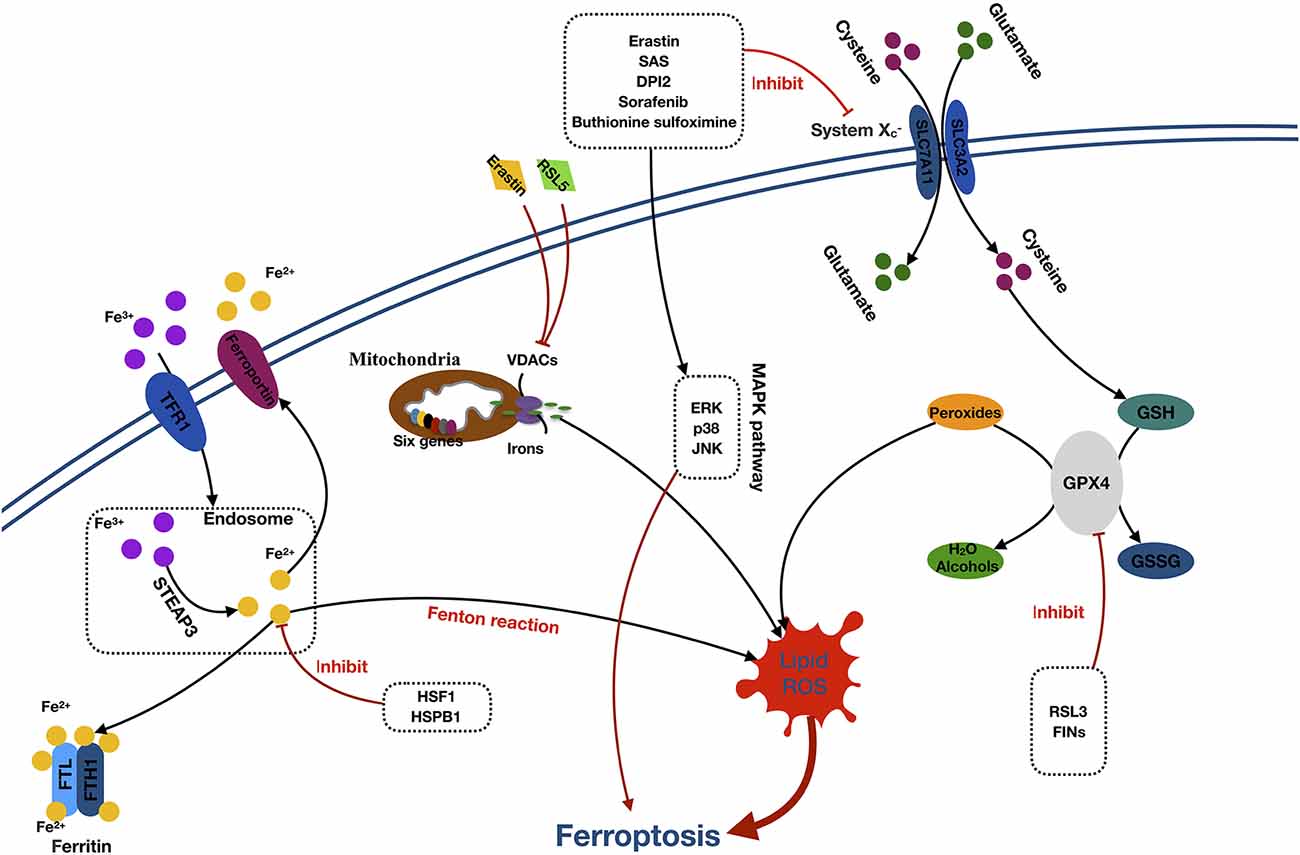
Figure 2. Mechanisms of ferroptosis. Abbreviations: TFR1, transferrin receptor 1; STEAP3, six-transmembrane epithelial antigen of prostate 3; FTL, ferritin light chain; FTH1, ferritin heavy chain 1; HSF1, heat shock factor 1; HSPB1, (HSF1)-heat-shock protein B1; RSL5, Ras selective lethal 5; VDACs, voltage-dependent anion channels; SAS, sulfasalazine; DPI2, diphenyleneiodonium 2; ERK, extracellular signal–regulated kinase; JNK, c-jun–N-terminal kinase; MAPK pathway, mitogen-activated protein kinase pathway; SLC7A11, solute carrier family 7 member 11; SLC3A2, solute carrier family 3 member 2; GSH, glutathione; GPX4, glutathione peroxidase 4; GSSG, oxidized glutathione; RL3, ribonuclease 1; FINS, fasting serum insulin.
The core component of ferroptosis includes lipid ROS accumulation due to the inhibition of System and glutathione peroxidase (GPX4). According to the different inhibition targets, the reducers can be divided into two classes. The first class includes erastin, sulfasalazine, DPI-sorafenib, and buthionine sulfoximine, which inhibits the depletion of System and glutathione (GSH; Xie et al., 2016). The second class comprised Ras-selective lethal 3 compound (RSL3), as well as DPI family members, which inactivate GPX4 (Yang et al., 2014). System is a membrane Na+-dependent cysteine–glutamate exchange transporter (with a ratio of 1:1) that is composed of a light-chain subunit (xCT, SLC7A11) and a heavy-chain subunit (CD98hc, SLC3A2). It is critical for maintaining redox homeostasis by reducing the intracellular cysteine that is required for the synthesis of GSH, a major antioxidant. When cells are cultured in glutamate, a substance causing neuronal hyperexcitability (Yang et al., 2019), the intracellular GSH content will decrease, and ferroptosis will be induced. Cells stimulated with class 1 ferroptosis inducers will usually have a significantly reduced GSH level (Yang and Stockwell, 2016). GSH peroxidase was a specific downstream site of GSH. GSH peroxidase uses two GSH molecules as electron donors to reduce H2O2, as well as other common small-molecule and complex lipid peroxides via decomposition of H2O2 into water or corresponding alcohols (Ursini et al., 1995). The class 2 ferroptosis inducers, such as RSL3 and FINs, directly inhibit GPX4 activity, contributing to the intracellular accumulation of lipid peroxides and subsequent ferroptosis (Yang et al., 2014).
The detailed role of iron in ferroptosis remains unclear. However, it was considered that the participation of iron is necessary for ferroptosis in oxidative reactions (Dixon et al., 2012; Gao et al., 2015). Free Fe3+ induces ferroptosis via importation into cells through the membrane protein transferrin receptor 1 and then stored in the endosome where Fe3+ is converted into Fe2+ by STEAP3. Subsequently, Fe2+ is transported by the divalent metal transporter 1 out of the endosome. The cytoplasmic Fe2+ is then transferred to three locations. Some is stored in ferritin, an iron storage protein complex, which includes a ferritin light chain and ferritin heavy chain 1. Some is transported out of the cell by the transmembrane protein, ferroportin. The rest is utilized in the Fenton reaction to create lipid ROS, which is an important source of ROS (Dixon and Stockwell, 2014; Xie et al., 2016; Wu et al., 2019). Moreover, erastin and RSL5 can alter the ion selectivity of the voltage-dependent anion channels, preventing the bidirectional movement of cations within the mitochondria, causing mitochondrial dysfunction and oxidant release (Yagoda et al., 2007; Yang and Stockwell, 2008).
Additionally, activation of the MAPK pathway and heat shock factor 1 (HSF1)–heat shock protein B1 (HSPB1) pathway also plays a significant role in ferroptosis. Inhibition of the MAPKs family, such as extracellular signal regulated kinase (ERK), p38, and c-Jun NH2-terminal kinase (JNK) can significantly attenuate the erastin-induced cell death, which indicates the potential role of MAPKs in ferroptosis (Yagoda et al., 2007; Yu et al., 2015). Furthermore, research has proven that HSF1 and HSPB1 can inhibit cell ferroptosis by decreasing the concentrations of iron and lipid ROS in cells (Sun et al., 2015).
Ferroptosis in Hemorrhagic Stroke
Ferroptosis was first reported in cell death during tumor and embryonic development (Dixon et al., 2012; Jiang et al., 2015; Sun et al., 2015). However, few studies have reported this form of cell death in hemorrhagic stroke (Table 2). Morphologically, the mitochondria of ferroptotic neurons appeared shrunken, with decreased cytomembrane mitochondria at 3 and 6 days after ICH in mice. After several days, the swollen mitochondrial count appears to stabilize and is sustained until 28 days (Li et al., 2018). Interestingly, another study failed to find the shrunken mitochondria in primary neurons subjected to hemin stimulation (Zille et al., 2017). Regarding the molecular mechanism of ferroptosis, most studies attempt to confirm the presence of ferroptosis and the effects of ferroptosis inhibition via modulation of downstream pathways. To date, several ferroptosis inhibitors have been found. Accordingly, these inhibitors are primarily categorized as either antioxidants or iron chelators. The antioxidants include ferrostatin 1 (fer-1), liproxstatin 1 (lip-1), cycloheximide, N-acetylcysteine (NAC), Trolox, and U0126 (Dixon et al., 2012; Cao and Dixon, 2016; Zille et al., 2017), whereas the iron chelators include deferoxamine (DFO), ciclopirox (CPO), and dihydroxybenzoic acid (DHB; Okauchi et al., 2010; Karuppagounder et al., 2016).
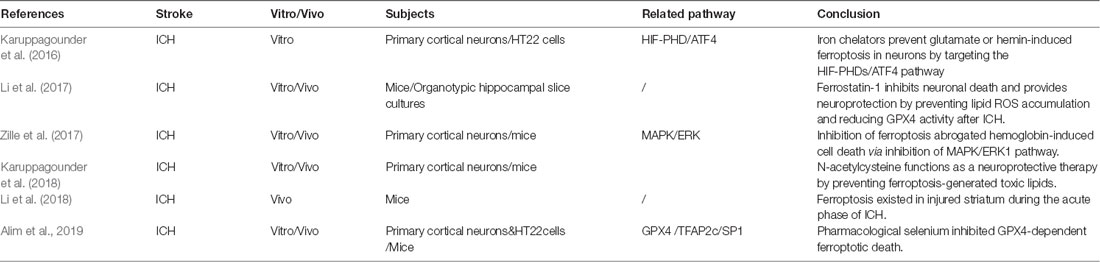
Table 2. Latest research of ferroptosis in hemorrhagic stroke.
It was found that the neuron death and iron deposition, induced by hemoglobin in organotypic hippocampal slice cultures and primary cortical neurons, can be attenuated by administration of fer-1, or other ferroptosis inhibitors (Li et al., 2017; Zille et al., 2017). Ferrostatin 1 prevents Hb-induced GPX4 deficits and lipid ROS accumulation and attenuates the injury volume and neurological deficits after ICH in mice (Li et al., 2017). The phospho-ERK1/2 level was significantly elevated at 6 and 24 h after ICH in mice and blocked by U0126 (an inhibitor of MAPK), suggesting the involvement of the MAPK/ERK pathway in the regulation of ferroptosis (Zille et al., 2017). Regarding the downstream production of ferroptosis, cyclooxygenase 2 (COX-2), an enzyme encoded by the PTGS-2 gene, was found to be significantly increased within the first 3 days after ICH in mice and can be inhibited by fer-1 (Li et al., 2017). By using spectrometric analysis, the following study also proved that COX-dependent lipid species participate in the facilitation of ferroptosis (Karuppagounder et al., 2018).
N-acetylcysteine is a clinically approved cysteine prodrug, which modulates redox reactions by mediating activity of the transporter and the cysteine/glutamate ratio. Moreover, recent data suggest that NAC can inhibit ferroptosis in vitro and in vivo (Zille et al., 2017; Karuppagounder et al., 2018). N-acetylcysteine attenuates hemin-/hemoglobin-induced cell death in primary cortical neuronal cells and improved functional recovery after ICH in mice (Zille et al., 2017; Karuppagounder et al., 2018). In this study, arachidonate 5-lipoxygenase (ALOX5)–derived reactive lipid species was revealed to contribute significantly in hemin-induced ferroptosis. The NAC-mediated neuroprotective effect was executed by increasing GSH levels and subsequently neutralizing nuclear ALOX5-derived lipid species after ICH (Karuppagounder et al., 2018). Additionally, this study also found that the COX-2–derived species, PGE2 and NAC, could synergize to prevent hemin-induced ferroptosis in vitro and improve functional recovery after ICH (Karuppagounder et al., 2018). However, this result seems to be in contrast of the detrimental effect of COX-2 after ICH, as mentioned in the previous study (Li et al., 2017).
A recent study revealed that iron chelators (DFO, CPO, DHB) inhibit glutamate- or hemin-induced ferroptosis in neurons by targeting the hypoxia-inducible factor prolyl hydroxylase domain enzymes (HIF-PHDs), a family of iron-dependent enzymes necessary for ATF4-dependent prodeath transcription. However, this process was identified independently to the inhibition of the Fenton reaction (Karuppagounder et al., 2016). ATF4 is a leucine zipper transcription factor that is activated in the ferroptosis-induced transcriptional responses found in neurons and cancer cells. Neurons with a germline deletion of ATF4 had resistance to the homocysteic acid–induced ferroptosis (Lange et al., 2008; Karuppagounder et al., 2016; Chen et al., 2017). This evidence suggests that the ATF4-dependent pathway may have an important role in ferroptosis after ICH.
The GPX4 homeostasis is a critical component of ferroptotic cell death after ICH. However, little is known about the expression levels of GPX4 and other selenoproteins after ferroptotic initiation. The latest study found an increased expression of several selenium-containing antioxidant enzymes, including GPX4, thioredoxin reductase 1, GPX3, and selenoprotein P in vivo and in vitro (Alim et al., 2019). The neuron-specific expression of GPX4 after ICH functions as a protective factor to avoid neuronal loss from ferroptosis-induced oxidative damage. However, it seems to be an insufficient response in preventing cell death as a result of ferroptotic insults (Karuppagounder et al., 2018; Alim et al., 2019). Interestingly, this study found that selenium augments the transcriptional response involving GPX4 via coordinated activation of the transcription factors, TFAP2c and Sp1, to protect the neurons from ferroptotic death and improve functional recovery after ICH in mice (Alim et al., 2019).
It should be mentioned that there have not been any studies that investigate the role of ferroptosis in the pathophysiology after SAH. Our laboratory has found that lip-1 can reduce the characteristic shrunken mitochondria in ipsilateral cortical neurons after SAH in mice. Liproxstatin 1 participated in attenuation of active lipid ROS by reducing the production of malondialdehyde and 4-hydroxynonenal (4-HNE) and by increasing the level of GSH and the activity GPX. Furthermore, lip-1 downregulated acyl-CoA synthetase long-chain family member 4 and COX-2, but also reduced the activation of microglia and inflammatory response (data unpublished).
In conclusion, ferroptosis has a critical role in the cell death of neurons. However, there have not been any studies conducted that focus on other cell types. After hemorrhagic stroke, the iron metabolic disorders and ROS accumulation both existed in the endothelium, microglia, and astrocytes. In addition, the BBB dysfunction and inflammation were also shown to be associated with the ferroptosis inhibitors (Zhang Z. et al., 2018; Zhang Y. H. et al., 2018). Ferroptosis may also occur in these cells after hemorrhagic stroke. Further studies should expand their research beyond the role of ferroptosis in neuronal death.
The Mechanism of Pyroptosis
Pyroptosis is a proinflammatory form of cell death (Bergsbaken et al., 2009; Chen et al., 2018). Morphologically, pyroptotic cell death possesses features of both necrosis and apoptosis. The morphological changes include necrosis-like cell membrane rupture, pore formation, cellular swelling, proinflammatory intracellular content release, as well as apoptosis-like nuclear condensation and DNA fragmentation. In contrast to apoptosis, pyroptotic cells have an integral mitochondrion and “balloon-shaped” vesicle formation, but no cytochrome c release (Xu et al., 2018; Jia et al., 2019). Molecularly, pyroptosis is characteristic in gasdermin-mediated cell death (Shi et al., 2017). Normally, the gasdermin is activated by two caspase-dependent pathways, including canonical caspase 1 and the noncanonical caspase 4/5/11 pathway (Jia et al., 2019; Figure 3).
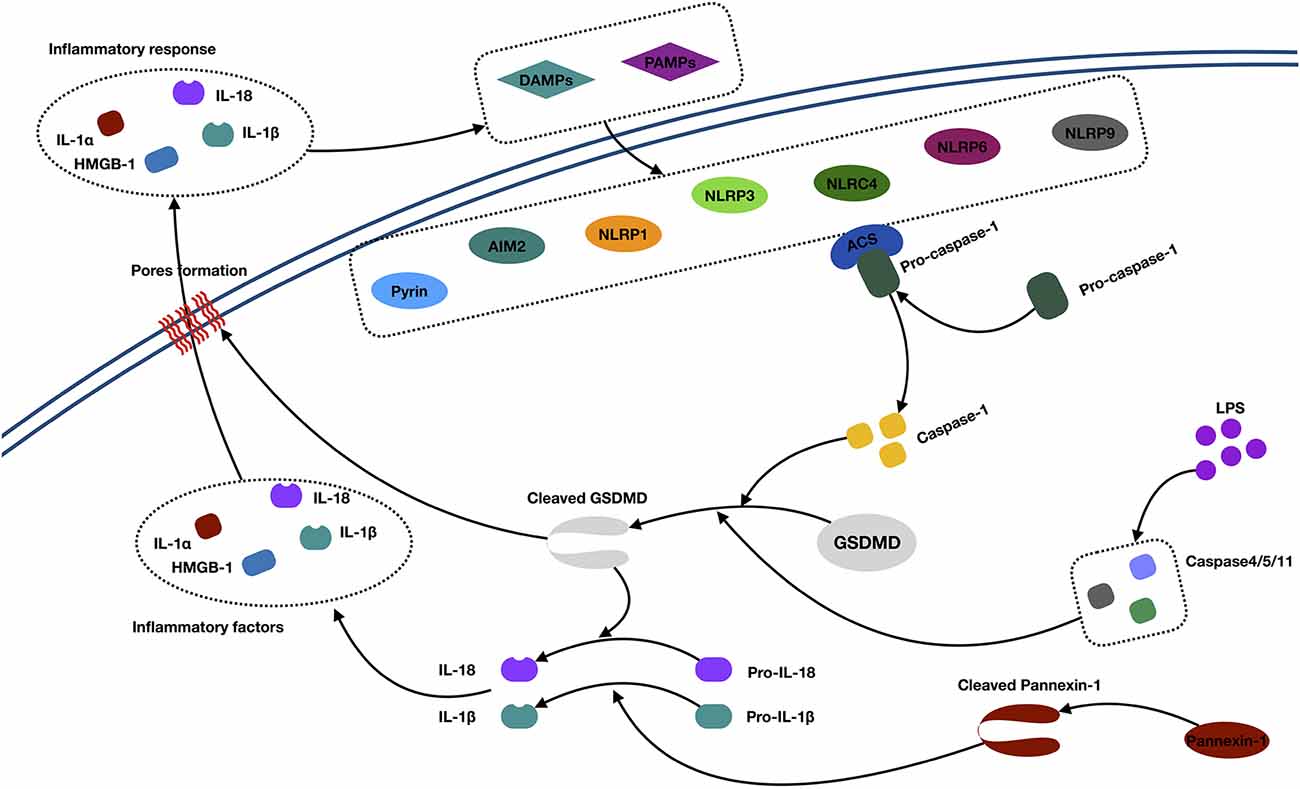
Figure 3. Mechanisms of pyroptosis. Abbreviations: IL-1α/1β/18, interleukin 1α/1β/18; DAMPs, danger-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; AIM2, absent in melanoma 2; NLRP, (NOD)-like receptor protein; ACS, andacyl-CoA synthetase; GSDMD, Gasdermin D; HMGB1, high-mobility group box 1; LPS, lipopolysaccharide.
In the canonical caspase 1 pathway, inflammasomes (also called pyroptosomes) assemble in the cytosol after recognizing pathogen-associated molecular patterns and danger-associated molecular patterns released by dying cells and some proinflammatory cytokines (Lamkanfi and Dixit, 2014). Inflammasomes are multimeric protein complexes, which are composed of the nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family (NLRP1, NLRP3, NLRC4, NLRP6, and NLRP9), PYHIN protein families [absent in melanoma 2 (AIM2)], or pyrin proteins (Zheng et al., 2011; Sekerdag et al., 2018). Typically, an NLR consists of three parts: C-terminal leucine-rich repeats, which are responsible for ligand recognition and autoinhibition, a central NOD (NACHT), which activates the signaling complex, and an N-terminal caspase activation and recruitment domain (CARD) or pyrin domain (PYD), which mediates homotypic protein–protein interactions (Martinon et al., 2002; Aachoui et al., 2013). The AIM2 contains a DNA-binding HIN-200 domain and PYD. The pyrin protein consists of the PYD, a zinc finger domain (bBOX), a coiled coil domain, and/or a B30.2/SPRY domain (Jia et al., 2019). The signaling domains, such as the PYD and CARD, both recruit the apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC). This subsequently activates pro–caspase 1 to generate active caspase 1 (Aachoui et al., 2013). Then, the activated caspase 1 cleaves the gasdermin D (GSDMD) and triggers oligomerization of the GSDMD-N domain in the cell, which finally forms the pores and releases cellular contents, such as IL-1α and HMGB1. Besides, the active caspase 1 is also responsible for processing and maturing IL-1β/18, which cause inflammation (Dinarello, 2011; Xu et al., 2018).
In the noncanonical caspase 4/5/11 pathway, caspase 4/5/11 was activated by a direct interaction with cytosolic lipopolysaccharide. Next, activated caspase 4/5/11 directly initiates the cleavage of GSDMD and triggers pyroptosis. Meanwhile, active caspase 4/5/11 can initiate pannexin 1 cleavage and K+ efflux, indirectly facilitating the release of mature IL-1β (Kayagaki et al., 2011; Cheng et al., 2017).
Pyroptosis in Hemorrhagic Stroke
Pyroptosis, unlike other types of PCDs, cannot be detected morphologically. Thus, the existing research is limited to the molecular level (Table 3). It was shown that the NLRP3 protein is upregulated after ICH and SAH and peaked at 24 h, along with elevation of inflammatory factors, IL-1β and IL-18 (Chen et al., 2013; Ma et al., 2014; Feng et al., 2015; Dong et al., 2016). Meanwhile, the significant upregulation of caspase 1 was detected at 3 h and peaked at 24–72 h after ICH (Ma et al., 2014; Feng et al., 2015). Inhibition of caspase 1 by Ac-YVAD-CMK could significantly decrease brain injury presentation, as evidenced by improved neurological functions, and amelioration of brain edema after ICH (Wu et al., 2010; Lin et al., 2018). The neuroprotection was associated with decreased expression of IL-1β, JNK, and MMP-9 and inhibition of ZO-1 degradation (Wu et al., 2010). The Ac-YVAD-CMK administration reduced M1-type microglia polarization and increased the number of M2-type cells around the hematoma (Lin et al., 2018). This characteristic polarization of microglia was also reported to reduce the poststroke neuroinflammatory damage in an ischemic stroke study (Xu et al., 2019).
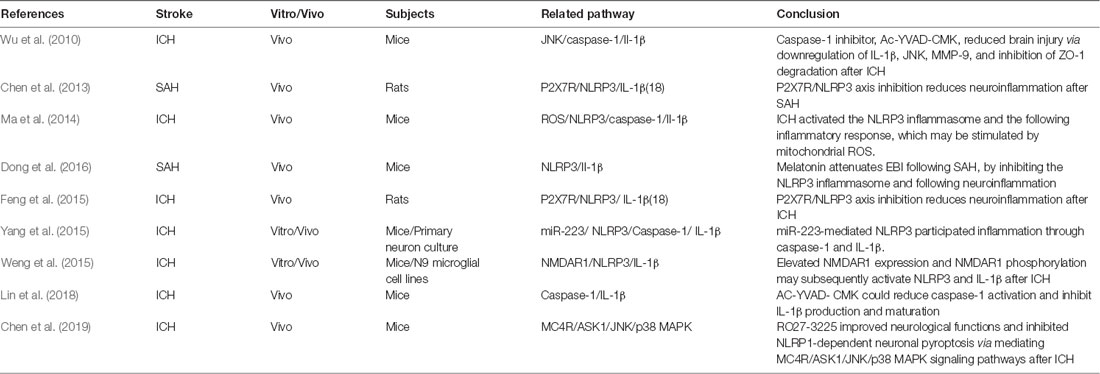
Table 3. Latest research of pyroptosis in hemorrhagic stroke.
The NLRP1 inflammasome is the first member that has been characterized among the NLR family. It has been reported to be primarily expressed in neurons and glial cells (Abulafia et al., 2009; Tan et al., 2014). The melanocortin 4 receptor (MC4R) is a seven-transmembrane G-protein–coupled receptor that could be activated by neuropeptide α-MSH, which exerts anti-inflammatory and neuroprotective effects after traumatic brain injury and cerebral ischemia (Forslin Aronsson et al., 2006; Yang et al., 2014). Treatment with the MC4R agonist, RO27-3225, successfully attenuated NLRP1-dependent neuron pyroptosis (including the cleaved caspase 1 and IL-1β level) after ICH in mice (Chen et al., 2019). Furthermore, RO27-3225 also reduced the expression of p-ASK1, JNK, and p-p38 mitogen-activated protein kinase (p38 MAPK) after ICH. This study presented a hypothesis that RO27-3225–mediated neuronal pyroptosis suppression may be regulated by the activation of MC4R and the inhibition of ASK1/JNK/p38 MAPK signaling pathways (Chen et al., 2019).
NLRP3 is another member of the NLR family reported in the central nervous system (CNS), with expression primarily located within the microglia and endothelium (Ma et al., 2014; Yang et al., 2014; Lin et al., 2018). Previous studies investigated several upstream regulators of NLRP3 involved in ICH pathophysiology. The ATP-gated transmembrane cation channel purinergic 2X7 receptor (P2X7R) is the key regulator in upstream activation of the NLRP3 inflammasome (Di Virgilio, 2007). P2X7R was activated after ICH and SAH in vivo. Inhibition of P2X7R activity can reduce NLRP3, IL-1β, and IL-18 from microglia (Chen et al., 2013; Feng et al., 2015). It was also postulated that peroxynitrite (ONOO−) could be involved in the P2X7R-regulated NLRP3 inflammasome formation after ICH (Feng et al., 2015).
Another study found evidence that microRNA-223 was a potential negative regulator of NLRP3 formation by using TargetScan (a searching program for potential regulator microRNA; Yang et al., 2015). The NLRP3 mRNA has conserved miR-223 binding sites in its 3′ UTR, which could be used to regulate the expression of NLRP3. Inhibition of NLRP3 by miR-223 reduces erythrocyte lysis-induced microglial inflammation and neuronal injury after ICH in mice (Yang et al., 2015). It was also found that the miR-223 levels decreased after erythrocyte lysis stimulation was decreased in microglia and ICH in mice (Yang et al., 2015). Meanwhile, N-methyl-D-aspartic acid receptor 1 (NMDAR1) was also suggested to play an important role in NLRP3 regulation after ICH in vivo and in vitro (Weng et al., 2015). The expression and phosphorylation of NMDAR1 were significantly increased after ICH and in cultured microglial cells treated with hemin (Weng et al., 2015). Furthermore, they demonstrated that an NMDAR1 inhibitor (MK801) attenuated hemin-induced activation of microglia, subsequently leading to a decrease in NLRP3 and Il- 1β microglia production (Weng et al., 2015).
Although several studies have attempted to investigate the expression of inflammasomes and upstream regulators of canonical inflammasomes after ICH, few have explored the key protein, GSDMD, downstream of the inflammasome and the noncanonical inflammasome. As we know, pyroptosis is a GSDMD-related cell death process, where two inflammasome signaling pathways converge at the GSDMD and are executed following pore formation (Jia et al., 2019). In addition to the canonical-inflammasome activation, caspase 4/5/11 also participates in IL-1/IL-18 processing and cell death (Jia et al., 2019). We propose that future studies should investigate the role that GSDMD and caspase 4/5/11 signaling pathways have in hemorrhagic stroke.
Mechanisms of BBB Dysfunction Secondary to Hemorrhagic Stroke
The mechanism of BBB dysfunction after hemorrhagic is a complex process as shown in Figure 4. ICH and SAH are two categories of hemorrhagic stroke that occur in different intracerebral regions. ICH is mainly secondary to arteriolosclerosis induced by hypertension, coagulopathy, and vascular malformation rupture, whereas SAH is mainly caused by the rupture of an intracranial aneurysm (Gross et al., 2019; Zhang et al., 2019) Even with different etiologies, ICH and SAH are initially caused by vascular disruption in different intracerebral sites, which can be defined as the first phase of BBB disruption. This initial hemorrhage may result in early phases of cell death because of physical injury, brain edema, and increased intracranial pressure. At this stage, the death of cells comprising the NVU may significantly contribute to the induction of secondary BBB dysfunction (Hawkins and Davis, 2005; Knowland et al., 2014; Zhang et al., 2019). Currently, a normal BBB is no longer just a physical “barrier,” but a dynamic and metabolic interface, based on the novel structural foundation known as the NVU (Tso and Macdonald, 2014; Zou et al., 2017). This structure includes components, such as ECs and their linking TJs, pericytes, astrocytic end-feet, neurons, and extracellular matrix (ECM) components (Tso and Macdonald, 2014; Erdo and Krajcsi, 2019). These components interact with each other to form a highly connected entirety, which has a critical function in regulating the homeostasis of water and electrolytes, immune cell trafficking, transporting necessary nutrients, and preventing the entry of compounds into the brain (Abbott et al., 2010; Hladky and Barrand, 2016; Erdo and Krajcsi, 2019).
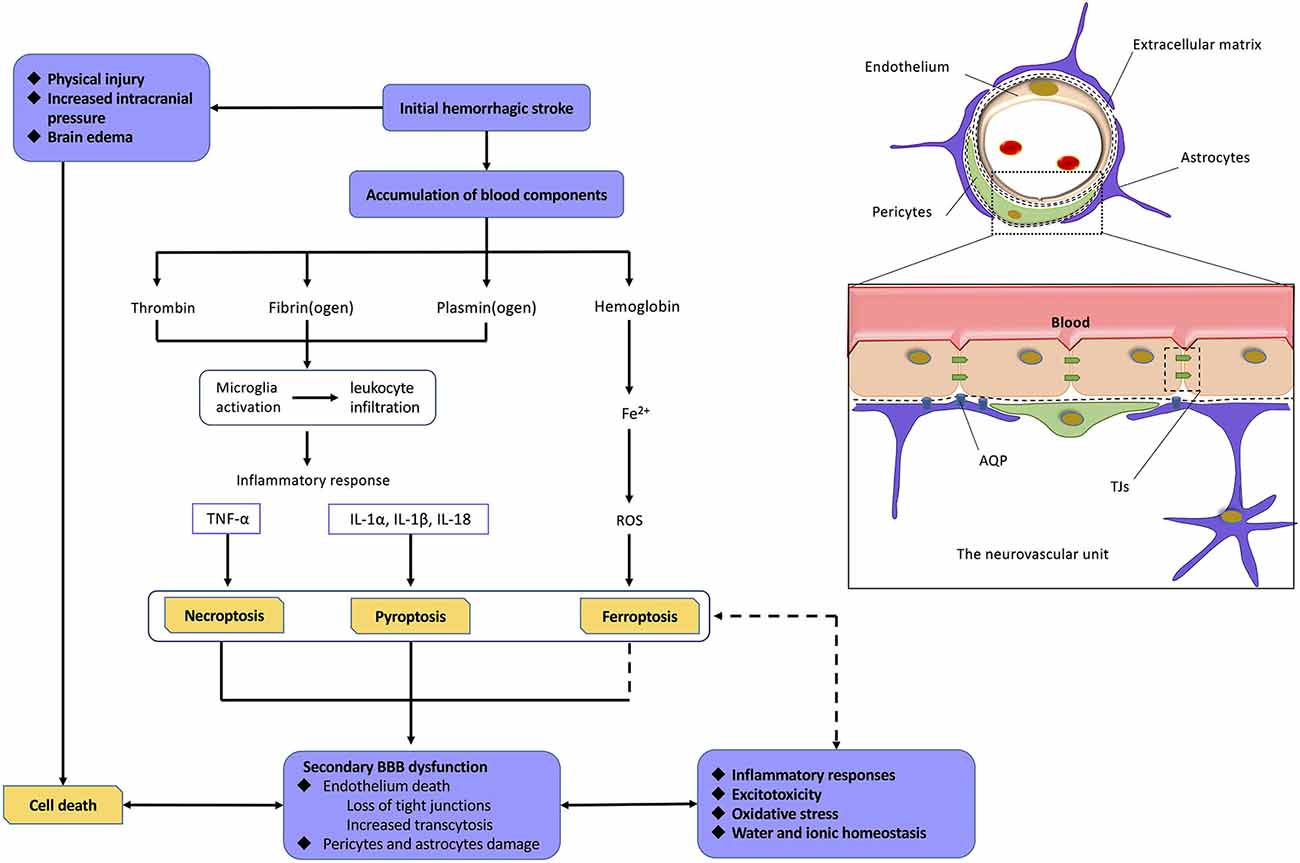
Figure 4. Relationship among hemorrhagic stroke, programmed cell deaths, and blood–brain barrier (BBB) dysfunction. Normal neurovascular units (NVUs) are composed of intact endothelial cells (ECs), pericytes, astrocytic endfeet, and extracellular matrix (ECM) components. ECs, with their tight junctions (TJs), are the most important components. On the one hand, initial hemorrhagic stroke directly causes cell death in central nervous system (CNS) as a result of physical injury, intracranial pressure, and brain edema. On the other hand, abundant blood components can also mediate programmed cell deaths (PCDs) via proinflammatory response, reactive oxygen species (ROS), and so on. The death of cell, especially ECs, leads to severe damage on BBB. As a result, BBB dysfunction induces a variety of pathophysiological processes including inflammatory responses, excitotoxicity, oxidative stress, water, and ionic homeostasis, which finally mediate more extensive PCDs. Abbreviations: TNFα, tumor necrosis factor; IL-1α/1β/18, interleukin 1α/1β/18.
After hemorrhagic stroke, the blood components initiate the process of cell excitotoxicity, cell edema, oxidative stress, and neuroinflammation, which further disrupt the BBB. These neurotoxic blood–derived factors include thrombin, fibrin, and erythrocyte components (Keep et al., 2014; Tso and Macdonald, 2014; Zhu et al., 2019). Thrombin is the cascade product of prothrombin during hemostasis after hemorrhagic stroke. The binding of thrombin to protease activated receptor 1 induces secondary BBB disruption through phosphorylating Src kinases and activating microglia (Möller et al., 2006; Liu et al., 2010). Fibrin cleaved from fibrinogen has been reported to play a potential role in neuroinflammation and microglial activation (Lim-Hing and Rincon, 2017). Iron, degraded from hemoglobin, is one of most important factors among various components for hemorrhagic stroke–induced BBB hyperpermeability (Hua et al., 2007; Gomes et al., 2014), which is supported by ICH/SAH studies revealing alleviated brain edema with administration of the iron chelator, deferoxamine (Lee et al., 2010; Okauchi et al., 2010; Yu et al., 2014), or a heme oxygenase (HO) inhibitor (Wagner et al., 2000; Han et al., 2018). HO plays a critical role in the degradation of heme and the release of free ferrous iron (Kamat et al., 2019). Importantly, iron has been shown to mediate BBB dysfunction mainly through generating ROS, which can directly induce degradation of the endothelium and activate signaling pathways (Mori et al., 2001; Fraser, 2011). Meanwhile, the influx of albumin accompanied by disequilibrium of water and ion homeostasis leads to cerebral edema (Lehmann et al., 2013; Di Napoli et al., 2018).
It is widely accepted that the inflammatory response occurs when resident microglia of the brain are activated within the early stages of hemorrhagic stroke. This is due to stimuli, such as neurotoxic factor accumulation and reduced blood flow, which then causes the release of proinflammatory cytokines (Ge et al., 2018; Chen et al., 2019). Among other CNS diseases, such as cerebral ischemia, Alzheimer disease, or multiple sclerosis, the mechanisms of leukocytic infiltration into the brain and the immune response with the complement system have been widely studied (Jiang et al., 2018; Sweeney et al., 2018; Ma et al., 2019; Shen et al., 2019). DL-3-n-Butylphthalide, a synthetic compound that has been approved for the treatment of ischemic stroke in China, has been shown to inhibit neurovascular inflammation via downregulation of intercellular adhesion molecule 1 (ICAM-1; Yang et al., 2019). Despite a slight lag, it was recently revealed that these cytokines may also upregulate adhesion molecules, including ICAM-1, vascular adhesion molecule 1, and vascular adhesion protein 1 after hemorrhagic stroke (Ma et al., 2011; Xu et al., 2015; Cheng et al., 2018; Gris et al., 2019). These recruited leukocytes further accumulate and transmigrate across the endothelium, finally releasing an abundance of cytokines and chemokines (Cheng et al., 2018; Gris et al., 2019). Cytokines mediating various signaling pathways are an important part of the inflammatory processes after hemorrhagic stroke. For example, TNFα and IL-1β are both found to mediate cell function, including endothelial cell death, followed by BBB hyperpermeability (King et al., 2013; Yang et al., 2014; Liu et al., 2019). Chemokines, such as monocyte chemoattractant protein 1, not only can mediate leukocyte chemotaxis, but also can directly damage TJs. These both result in BBB dysfunction (Niwa et al., 2016; Yang et al., 2016). Meanwhile, more recent studies have focused on the immune response, with an emphasis on lymphocytic infiltration, as they contribute to brain recovery through hematoma resolution, neurogenesis, and axonal regeneration after ICH. Regulatory T cells were shown to mediate anti-inflammatory responses and improve neurological function in a chronic subdural hematoma rat model (Frost et al., 2019; Quan et al., 2019). These studies may provide emerging therapeutic targets for ICH (Shao A. et al., 2019). It should be noted that MMPs, which are primarily released by neutrophils, are also thoroughly studied potent proteinases and play a critical role in the increase of endothelial permeability via disruption of TJs, ECM degradation, and increased transcytosis of the endothelium (Abilleira et al., 2003; Qi et al., 2016; Liu et al., 2019). Recently, MMP-9 was also found to mediate endothelial permeability through the activity of von Willebrand factor and the initial recruitment of inflammatory leukocytes (Askenase and Sansing, 2016).
Crosstalk Between BBB Dysfunction and PCDs in Hemorrhagic Stroke
Cell death, which occurs over an extended period following hemorrhagic stroke, not only can be directly induced by physical injury, but also indirectly mediated by pathophysiological process associated with BBB dysfunction mentioned above (Keep et al., 2014). The death of NVU cells further exacerbates BBB damage. While the three PCDs (necroptosis, ferroptosis, and pyroptosis) have distinct morphological, biochemical, and genetic characteristics, they have been shown to mutually participate in BBB dysfunction. The endothelium remains the primary target of study because of their paramount significance in the BBB (Figure 4; Zille et al., 2017; Chen et al., 2018, 2019; Zhao et al., 2018).
A significant amount of proinflammatory cytokines are increased during these three processes, such as TNFα, IL-1β, and IL-6 (Ge et al., 2018; Zhang Z. et al., 2018; Chen et al., 2019). As described above, upregulation of these cytokines will significantly induce microglial activation and increase BBB permeability, either directly or indirectly. To support this, several animal experiments have demonstrated this hyperpermeability of BBB via albumin extravasation or an Evans blue dye extravasation assay, and this pathological change can be reversed by specific inhibitors of necroptosis, ferroptosis, and pyroptosis (Chen et al., 2017; Ge et al., 2018; Zhang Z. et al., 2018; Yuan et al., 2019). More recently, research found that necroptosis occurred in the endothelium and is initiated by interactions between TNFα secreted by M1-type microglia and TNFR1 on the endothelium (Chen et al., 2019). The significant decrease in the levels of occludin and claudin-5 (major transmembrane TJs) and the increase in MMP-9 levels and activity can both be reversed by nec-1 after SAH (Chen et al., 2019). It implies that there is a high degree of correlation between necroptosis and BBB dysfunction after SAH. Similarly, pyroptosis of brain microvascular ECs was shown in a TBI model and was induced by GSDMD. BBB function and neurological outcomes improved when the process of pyroptosis was inhibited (Chen et al., 2018). According to the discovery above, it seems reasonable to speculate that ferroptosis also occurred in the endothelium after stroke events. In an ICH model, GPX4 reduced oxidative stress and thus inhibited ferroptosis to recover the function of the BBB (Zhang Z. et al., 2018). The inflammatory and innate immune responses are among the most important factors in BBB regulation. Recently, a series of experiments on aged mice revealed severe dysregulation of immunity and an excessive inflammatory response (Freitas et al., 2019; Shen et al., 2019; Zhang et al., 2019). As hemorrhagic stroke is a disease with an increased incidence in elderly populations, we should further investigate the effect of these changes on BBB dysfunction and PCDs.
Currently, research pertaining to ferroptosis and BBB dysfunction is very limited. However, according to the mechanisms of BBB dysfunction and ferroptosis discussed above, the role of ferroptosis on BBB damage warrants more attention. We have mentioned above with sufficient evidence that iron, when degraded from hemoglobin, has a critically important role in BBB hyperpermeability after hemorrhagic stroke, and decreasing iron content through various methods significantly reversed the brain edema (Wagner et al., 2000; Hua et al., 2007; Gomes et al., 2014; Yu et al., 2014). The key point of this problem lies in the poor understanding regarding the exact mechanism of iron overload-induced BBB hyperpermeability. Oxidative stress seems to be a promising candidate (Fraser, 2011). Interestingly, the mechanism of ferroptosis has not been fully clarified. However, iron and lipid peroxidation products have been demonstrated to be indispensable initiators of critical death signals (Yang and Stockwell, 2016; Stockwell et al., 2017). Moreover, studies had shown increased deleterious effects of lipid peroxidation products, such as HO-1, on free radical scavenging after ICH (Chang et al., 2014). Therefore, there are theoretical bases to prove the role of ferroptosis in BBB dysfunction.
In addition to the mechanisms of endothelial cell destruction being elucidated to some extent, these three PCDs may have some potential mechanisms relevant to other BBB structures and signaling pathways based on current research findings. Astrocytes are an important component of the BBB basic structure; they regulate electrolyte and water balance via an abundance of AQP-4 and gap junctions in the end-feet (Zlokovic, 2008). In the condition of depleted intracellular GSH, astrocytes undergo caspase-independent cell death. This process is highly relevant to the secretion of TNFα from human astrocytes and can be inhibited by nec-1 (Laird et al., 2008). It suggests that the studies regarding the exact mechanisms of necroptosis and nec-1 in astrocytes, as well as other BBB structures, strengthen the understanding of BBB dysfunction after hemorrhagic stroke.
Interestingly, there may also be some intersection between the three PCDs. Inducible GPX4 alleviates ferroptosis and pyroptosis in bacterial infection, whereas degradation of GSH enhances necroptosis and ferroptosis in human triple-negative breast cancer cells, suggesting a potential pathway involving pyroptosis and necroptosis (Zille et al., 2017; Zhu et al., 2019). Lastly, these three patterns of death may share common causes, which are not limited to oxidative stress. A logical plan of action would be to find shared targets of different PCDs to inhibit, therefore improving the efficacy of neurological recovery after hemorrhagic stroke.
Conclusion
A growing number of studies show that PCDs are involved in hemorrhagic stroke. This review article summarizes the role of necroptosis, ferroptosis, and pyroptosis after hemorrhagic stroke, as well as their relationship with BBB dysfunction. Although most consequences of BBB dysfunction are detrimental, one potential beneficial result is that the entire brain would benefit if a novel therapeutic target was uncovered. Further studies exploring the mechanisms of these three forms of cell death and the crosstalk with BBB dysfunction will assist in the search for promising interventional targets of hemorrhagic stroke. Furthermore, more studies should broaden their scope to include research on cell types other than neurons.
Author Contributions
All the authors participated in analyzing and discussing the literature, commenting on, and approving the manuscript. AS and JZ supervised the research, led the discussion, wrote and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was funded by the China Postdoctoral Science Foundation (2017M612010), the National Natural Science Foundation of China (81701144) and Fundamental Research Funds for the Central Universities (2019QNA7038).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aachoui, Y., Sagulenko, V., Miao, E. A., and Stacey, K. J. (2013). Inflammasome-mediated pyroptotic and apoptotic cell death and defense against infection. Curr. Opin. Microbiol. 16, 319–326. doi: 10.1016/j.mib.2013.04.004
Abbott, N. J., Patabendige, A. A., Dolman, D. E., Yusof, S. R., and Begley, D. J. (2010). Structure and function of the blood-brain barrier. Neurobiol. Dis. 37, 13–25. doi: 10.1016/j.nbd.2009.07.030
Abilleira, S., Montaner, J., Molina, C. A., Monasterio, J., Castillo, J., and Alvarez-Sabin, J. (2003). Matrix metalloproteinase-9 concentration after spontaneous intracerebral hemorrhage. J. Neurosurg. 99, 65–70. doi: 10.3171/jns.2003.99.1.0065
Abulafia, D. P., de Rivero Vaccari, J. P., Lozano, J. D., Lotocki, G., Keane, R. W., and Dietrich, W. D. (2009). Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J. Cereb. Blood Flow Metab. 29, 534–544. doi: 10.1038/jcbfm.2008.143
Alim, I., Caulfield, J. T., Chen, Y., Swarup, V., Geschwind, D. H., Ivanova, E., et al. (2019). Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177, 1262.e25–1279.e25. doi: 10.1016/j.cell.2019.03.032
Askenase, M. H., and Sansing, L. H. (2016). Stages of the inflammatory response in pathology and tissue repair after intracerebral hemorrhage. Semin. Neurol. 36, 288–297. doi: 10.1055/s-0036-1582132
Bergsbaken, T., Fink, S. L., and Cookson, B. T. (2009). Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109. doi: 10.1038/nrmicro2070
Bertrand, M. J., Milutinovic, S., Dickson, K. M., Ho, W. C., Boudreault, A., Durkin, J., et al. (2008). cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700. doi: 10.1016/j.molcel.2008.05.014
Cao, J. Y., and Dixon, S. J. (2016). Mechanisms of ferroptosis. Cell. Mol. Life Sci. 73, 2195–2209. doi: 10.1007/s00018-016-2194-1
Chang, C. F., Cho, S., and Wang, J. (2014). (−)-Epicatechin protects hemorrhagic brain via synergistic Nrf2 pathways. Ann. Clin. Transl. Neurol. 1, 258–271. doi: 10.1002/acn3.54
Chang, P., Dong, W., Zhang, M., Wang, Z., Wang, Y., Wang, T., et al. (2014). Anti-necroptosis chemical necrostatin-1 can also suppress apoptotic and autophagic pathway to exert neuroprotective effect in mice intracerebral hemorrhage model. J. Mol. Neurosci. 52, 242–249. doi: 10.1007/s12031-013-0132-3
Chen, A. Q., Fang, Z., Chen, X. L., Yang, S., Zhou, Y. F., Mao, L., et al. (2019). Microglia-derived TNF-α mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke. Cell Death Dis. 10:487. doi: 10.1038/s41419-019-1716-9
Chen, D., Fan, Z., Rauh, M., Buchfelder, M., Eyupoglu, I. Y., and Savaskan, N. (2017). ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 36, 5593–5608. doi: 10.1038/onc.2017.146
Chen, J., Jin, H., Xu, H., Peng, Y., Jie, L., Xu, D., et al. (2019). The neuroprotective effects of necrostatin-1 on subarachnoid hemorrhage in rats are possibly mediated by preventing blood-brain barrier disruption and RIP3-mediated necroptosis. Cell Transplant. 28, 1358–1372. doi: 10.1177/0963689719867285
Chen, S., Ma, Q., Krafft, P. R., Hu, Q., Rolland, W. II., Sherchan, P., et al. (2013). P2X7R/cryopyrin inflammasome axis inhibition reduces neuroinflammation after SAH. Neurobiol. Dis. 58, 296–307. doi: 10.1016/j.nbd.2013.06.011
Chen, S., Mei, S., Luo, Y., Wu, H., Zhang, J., and Zhu, J. (2018). Gasdermin family: a promising therapeutic target for stroke. Transl. Stroke Res. 9, 555–563. doi: 10.1007/s12975-018-0666-3
Chen, T., Pan, H., Li, J., Xu, H., Jin, H., Qian, C., et al. (2018). Inhibiting of RIPK3 attenuates early brain injury following subarachnoid hemorrhage: possibly through alleviating necroptosis. Biomed. Pharmacother. 107, 563–570. doi: 10.1016/j.biopha.2018.08.056
Chen, F., Su, X., Lin, Z., Lin, Y., Yu, L., Cai, J., et al. (2017). Necrostatin-1 attenuates early brain injury after subarachnoid hemorrhage in rats by inhibiting necroptosis. Neuropsychiatr. Dis. Treat. 13, 1771–1782. doi: 10.2147/NDT.S140801
Chen, S., Zuo, Y., Huang, L., Sherchan, P., Zhang, J., Yu, Z., et al. (2019). The MC4 receptor agonist RO27–3225 inhibits NLRP1-dependent neuronal pyroptosis via the ASK1/JNK/p38 MAPK pathway in a mouse model of intracerebral haemorrhage. Br. J. Pharmacol. 176, 1341–1356. doi: 10.1111/bph.14639
Cheng, K. T., Xiong, S., Ye, Z., Hong, Z., Di, A., Tsang, K. M., et al. (2017). Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J. Clin. Invest. 127, 4124–4135. doi: 10.1172/jci94495
Cheng, Y., Zan, J., Song, Y., Yang, G., Shang, H., and Zhao, W. (2018). Evaluation of intestinal injury, inflammatory response and oxidative stress following intracerebral hemorrhage in mice. Int. J. Mol. Med. 42, 2120–2128. doi: 10.3892/ijmm.2018.3755
Chu, X., Wu, X., Feng, H., Zhao, H., Tan, Y., Wang, L., et al. (2018). Coupling between interleukin-1R1 and necrosome complex involves in hemin-induced neuronal necroptosis after intracranial hemorrhage. Stroke 49, 2473–2482. doi: 10.1161/strokeaha.117.019253
Daneman, R., and Prat, A. (2015). The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 7:a020412. doi: 10.1101/cshperspect.a020412
de Rooij, N. K., Linn, F. H., van der Plas, J. A., Algra, A., and Rinkel, G. J. (2007). Incidence of subarachnoid haemorrhage: a systematic review with emphasis on region, age, gender, and time trends. J. Neurol. Neurosurg. Psychiatry 78, 1365–1372. doi: 10.1136/jnnp.2007.117655
Di Napoli, M., Slevin, M., Popa-Wagner, A., Singh, P., Lattanzi, S., and Divani, A. A. (2018). Monomeric C-reactive protein and cerebral hemorrhage: from bench to bedside. Front. Immunol. 9:1921. doi: 10.3389/fimmu.2018.01921
Di Virgilio, F. (2007). Liaisons dangereuses: P2X7 and the inflammasome. Trends Pharmacol. Sci. 28, 465–472. doi: 10.1016/j.tips.2007.07.002
Dinarello, C. A. (2011). A clinical perspective of IL-1β as the gatekeeper of inflammation. Eur. J. Immunol. 41, 1203–1217. doi: 10.1002/eji.201141550
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Dixon, S. J., and Stockwell, B. R. (2014). The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10, 9–17. doi: 10.1038/nchembio.1416
Dong, Y., Fan, C., Hu, W., Jiang, S., Ma, Z., Yan, X., et al. (2016). Melatonin attenuated early brain injury induced by subarachnoid hemorrhage via regulating NLRP3 inflammasome and apoptosis signaling. J. Pineal Res. 60, 253–262. doi: 10.1111/jpi.12300
Erdo, F., and Krajcsi, P. (2019). Age-related functional and expressional changes in efflux pathways at the blood-brain barrier. Front. Aging Neurosci. 11:196. doi: 10.3389/fnagi.2019.00196
Fang, Y., Chen, S., Reis, C., and Zhang, J. (2018). The role of autophagy in subarachnoid hemorrhage: an update. Curr. Neuropharmacol. 16, 1255–1266. doi: 10.2174/1570159x15666170406142631
Fang, Y., Shao, Y., Lu, J., Dong, X., Zhao, X., Zhang, J., et al. (2019). The effectiveness of lumbar cerebrospinal fluid drainage in aneurysmal subarachnoid hemorrhage with different bleeding amounts. Neurosurg. Rev. doi: 10.1007/s10143-019-01116-1 [Epub ahead of print].
Feng, L., Chen, Y., Ding, R., Fu, Z., Yang, S., Deng, X., et al. (2015). P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: involvement of peroxynitrite. J. Neuroinflammation 12:190. doi: 10.1186/s12974-015-0409-2
Forslin Aronsson, S., Spulber, S., Popescu, L. M., Winblad, B., Post, C., Oprica, M., et al. (2006). α-Melanocyte-stimulating hormone is neuroprotective in rat global cerebral ischemia. Neuropeptides 40, 65–75. doi: 10.1016/j.npep.2005.10.006
Fraser, P. A. (2011). The role of free radical generation in increasing cerebrovascular permeability. Free Radic. Biol. Med. 51, 967–977. doi: 10.1016/j.freeradbiomed.2011.06.003
Freitas, G. R. R., da Luz Fernandes, M., Agena, F., Jaluul, O., Silva, S. C., Lemos, F. B. C., et al. (2019). Aging and end stage renal disease cause a decrease in absolute circulating lymphocyte counts with a shift to a memory profile and diverge in treg population. Aging Dis. 10, 49–61. doi: 10.14336/ad.2018.0318
Frost, P. S., Barros-Aragão, F., da Silva, R. T., Venancio, A., Matias, I., Lyra E Silva, N. M., et al. (2019). Neonatal infection leads to increased susceptibility to Aβ oligomer-induced brain inflammation, synapse loss and cognitive impairment in mice. Cell Death Dis. 10:323. doi: 10.1038/s41419-019-1529-x
Fuchs, Y., and Steller, H. (2015). Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 16, 329–344. doi: 10.1038/nrm3999
Galluzzi, L., Kepp, O., Chan, F. K., and Kroemer, G. (2017). Necroptosis: mechanisms and relevance to disease. Annu. Rev. Pathol. 12, 103–130. doi: 10.1146/annurev-pathol-052016-100247
Gao, M., Monian, P., Quadri, N., Ramasamy, R., and Jiang, X. (2015). Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 59, 298–308. doi: 10.1016/j.molcel.2015.06.011
Ge, X., Li, W., Huang, S., Yin, Z., Xu, X., Chen, F., et al. (2018). The pathological role of NLRs and AIM2 inflammasome-mediated pyroptosis in damaged blood-brain barrier after traumatic brain injury. Brain Res. 1697, 10–20. doi: 10.1016/j.brainres.2018.06.008
Gomes, J. A., Selim, M., Cotleur, A., Hussain, M. S., Toth, G., Koffman, L., et al. (2014). Brain iron metabolism and brain injury following subarachnoid hemorrhage: iCeFISH-pilot (CSF iron in SAH). Neurocrit. Care 21, 285–293. doi: 10.1007/s12028-014-9977-8
Gris, T., Laplante, P., Thebault, P., Cayrol, R., Najjar, A., Joannette-Pilon, B., et al. (2019). Innate immunity activation in the early brain injury period following subarachnoid hemorrhage. J. Neuroinflammation 16:253. doi: 10.1186/s12974-019-1629-7
Gross, B. A., Jankowitz, B. T., and Friedlander, R. M. (2019). Cerebral intraparenchymal hemorrhage: a review. JAMA 321, 1295–1303. doi: 10.1001/jama.2019.2413
Grysiewicz, R. A., Thomas, K., and Pandey, D. K. (2008). Epidemiology of ischemic and hemorrhagic stroke: incidence, prevalence, mortality, and risk factors. Neurol. Clin. 26, 871–895. doi: 10.1016/j.ncl.2008.07.003
Gustavsson, A., Svensson, M., Jacobi, F., Allgulander, C., Alonso, J., Beghi, E., et al. (2011). Cost of disorders of the brain in Europe 2010. Eur. Neuropsychopharmacol. 21, 718–779. doi: 10.1016/j.euroneuro.2011.08.008
Han, Y.-W., Liu, X.-J., Zhao, Y., and Li, X.-M. (2018). Role of Oleanolic acid in maintaining BBB integrity by targeting p38MAPK/VEGF/Src signaling pathway in rat model of subarachnoid hemorrhage. Eur. J. Pharmacol. 839, 12–20. doi: 10.1016/j.ejphar.2018.09.018
Hawkins, B. T., and Davis, T. P. (2005). The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 57, 173–185. doi: 10.1124/pr.57.2.4
Hayden, M. S., and Ghosh, S. (2014). Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 26, 253–266. doi: 10.1016/j.smim.2014.05.004
Hladky, S. B., and Barrand, M. A. (2016). Fluid and ion transfer across the blood-brain and blood-cerebrospinal fluid barriers; a comparative account of mechanisms and roles. Fluids Barriers CNS 13:19. doi: 10.1186/s12987-016-0040-3
Hua, Y., Keep, R. F., Hoff, J. T., and Xi, G. (2007). Brain injury after intracerebral hemorrhage: the role of thrombin and iron. Stroke 38, 759–762. doi: 10.1161/01.str.0000247868.97078.10
Jia, C., Chen, H., Zhang, J., Zhou, K., Zhuge, Y., Niu, C., et al. (2019). Role of pyroptosis in cardiovascular diseases. Int. Immunopharmacol. 67, 311–318. doi: 10.7150/ijbs.33568
Jiang, X., Andjelkovic, A. V., Zhu, L., Yang, T., Bennett, M. V. L., Chen, J., et al. (2018). Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 163–164, 144–171. doi: 10.1016/j.pneurobio.2017.10.001
Jiang, L., Kon, N., Li, T., Wang, S. J., Su, T., Hibshoosh, H., et al. (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. doi: 10.1038/nature14344
Jin, Z., and El-Deiry, W. S. (2006). Distinct signaling pathways in TRAIL- versus tumor necrosis factor-induced apoptosis. Mol. Cell. Biol. 26, 8136–8148. doi: 10.1128/mcb.00257-06
Kamat, P. K., Ahmad, A. S., and Doré, S. (2019). Carbon monoxide attenuates vasospasm and improves neurobehavioral function after subarachnoid hemorrhage. Arch. Biochem. Biophys. 676:108117. doi: 10.1016/j.abb.2019.108117
Karuppagounder, S. S., Alim, I., Khim, S. J., Bourassa, M. W., Sleiman, S. F., John, R., et al. (2016). Therapeutic targeting of oxygen-sensing prolyl hydroxylases abrogates ATF4-dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci. Transl. Med. 8:328ra29. doi: 10.1126/scitranslmed.aac6008
Karuppagounder, S. S., Alin, L., Chen, Y., Brand, D., Bourassa, M. W., Dietrich, K., et al. (2018). N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann. Neurol. 84, 854–872. doi: 10.1002/ana.25356
Kayagaki, N., Warming, S., Lamkanfi, M., Vande Walle, L., Louie, S., Dong, J., et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. doi: 10.1038/nature10558
Keep, R. F., Zhou, N., Xiang, J., Andjelkovic, A. V., Hua, Y., and Xi, G. (2014). Vascular disruption and blood-brain barrier dysfunction in intracerebral hemorrhage. Fluids Barriers CNS 11:18. doi: 10.1186/2045-8118-11-18
King, M. D., Alleyne, C. H. Jr., and Dhandapani, K. M. (2013). TNF-alpha receptor antagonist, R-7050, improves neurological outcomes following intracerebral hemorrhage in mice. Neurosci. Lett. 542, 92–96. doi: 10.1016/j.neulet.2013.02.051
King, M. D., Whitaker-Lea, W. A., Campbell, J. M., Alleyne, C. H. Jr., and Dhandapani, K. M. (2014). Necrostatin-1 reduces neurovascular injury after intracerebral hemorrhage. Int. J. Cell Biol. 2014:495817. doi: 10.1155/2014/495817
Knowland, D., Arac, A., Sekiguchi, K. J., Hsu, M., Lutz, S. E., Perrino, J., et al. (2014). Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 82, 603–617. doi: 10.1016/j.neuron.2014.03.003
Laird, M. D., Wakade, C., Alleyne, C. H. Jr., and Dhandapani, K. M. (2008). Hemin-induced necroptosis involves glutathione depletion in mouse astrocytes. Free Radic. Biol. Med. 45, 1103–1114. doi: 10.1016/j.freeradbiomed.2008.07.003
Lamkanfi, M., and Dixit, V. M. (2014). Mechanisms and functions of inflammasomes. Cell 157, 1013–1022. doi: 10.1016/j.cell.2014.04.007
Lange, P. S., Chavez, J. C., Pinto, J. T., Coppola, G., Sun, C. W., Townes, T. M., et al. (2008). ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J. Exp. Med. 205, 1227–1242. doi: 10.1084/jem.20071460
Lee, J.-Y., Keep, R. F., He, Y., Sagher, O., Hua, Y., and Xi, G. (2010). Hemoglobin and iron handling in brain after subarachnoid hemorrhage and the effect of deferoxamine on early brain injury. J. Cereb. Blood Flow Metab. 30, 1793–1803. doi: 10.1038/jcbfm.2010.137
Lehmann, L., Bendel, S., Uehlinger, D. E., Takala, J., Schafer, M., Reinert, M., et al. (2013). Randomized, double-blind trial of the effect of fluid composition on electrolyte, acid-base, and fluid homeostasis in patients early after subarachnoid hemorrhage. Neurocrit. Care 18, 5–12. doi: 10.1007/s12028-012-9764-3
Leist, M., and Jaattela, M. (2001). Four deaths and a funeral: from caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2, 589–598. doi: 10.1038/35085008
Li, Q., Han, X., Lan, X., Gao, Y., Wan, J., Durham, F., et al. (2017). Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2:e90777. doi: 10.1172/jci.insight.90777
Li, Q., Weiland, A., Chen, X., Lan, X., Han, X., Durham, F., et al. (2018). Ultrastructural characteristics of neuronal death and white matter injury in mouse brain tissues after intracerebral hemorrhage: coexistence of ferroptosis, autophagy, and necrosis. Front. Neurol. 9:581. doi: 10.3389/fneur.2018.00581
Lim-Hing, K., and Rincon, F. (2017). Secondary hematoma expansion and perihemorrhagic edema after intracerebral hemorrhage: from bench work to practical aspects. Front. Neurol. 8:74. doi: 10.3389/fneur.2017.00074
Lin, X., Ye, H., Siaw-Debrah, F., Pan, S., He, Z., Ni, H., et al. (2018). AC-YVAD-CMK inhibits pyroptosis and improves functional outcome after intracerebral hemorrhage. Biomed. Res. Int. 2018:3706047. doi: 10.1155/2018/3706047
Linkermann, A., and Green, D. R. (2014). Necroptosis. N. Engl. J. Med. 370, 455–465. doi: 10.1056/NEJMra1310050
Liu, D. Z., Ander, B. P., Xu, H., Shen, Y., Kaur, P., Deng, W., et al. (2010). Blood-brain barrier breakdown and repair by Src after thrombin-induced injury. Ann. Neurol. 67, 526–533. doi: 10.1002/ana.21924
Liu, Y., Luo, H., Wang, L., Li, C., Liu, L., Huang, L., et al. (2019). Increased serum matrix metalloproteinase-9 levels are associated with anti-jo1 but not anti-MDA5 in myositis patients. Aging Dis. 10, 746–755. doi: 10.14336/ad.2018.1120
Liu, Z., Wang, X., Jiang, K., Ji, X., Zhang, Y. A., and Chen, Z. (2019). TNFα-induced up-regulation of Ascl2 affects the differentiation and proliferation of neural stem cells. Aging Dis. 10, 1207–1220. doi: 10.14336/ad.2018.1028
Liu, C., Zhang, K., Shen, H., Yao, X., Sun, Q., and Chen, G. (2018). Necroptosis: a novel manner of cell death, associated with stroke (Review). Int. J. Mol. Med. 41, 624–630. doi: 10.3892/ijmm.2017.3279
Lu, J., Sun, Z., Fang, Y., Zheng, J., Xu, S., Xu, W., et al. (2019). Melatonin suppresses microglial necroptosis by regulating deubiquitinating enzyme A20 after intracerebral hemorrhage. Front. Immunol. 10:1360. doi: 10.3389/fimmu.2019.01360
Lublinsky, S., Major, S., Kola, V., Horst, V., Santos, E., Platz, J., et al. (2019). Early blood-brain barrier dysfunction predicts neurological outcome following aneurysmal subarachnoid hemorrhage. EBioMedicine 43, 460–472. doi: 10.1016/j.ebiom.2019.04.054
Möller, T., Weinstein, J. R., and Hanisch, U.-K. (2006). Activation of microglial cells by thrombin: past, present, and future. Semin. Thromb. Hemost. 32, 69–76. doi: 10.1055/s-2006-939556
Ma, Q., Chen, S., Hu, Q., Feng, H., Zhang, J. H., and Tang, J. (2014). NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann. Neurol. 75, 209–319. doi: 10.1002/ana.24070
Ma, Y., Liu, Y., Zhang, Z., and Yang, G. Y. (2019). Significance of complement system in ischemic stroke: a comprehensive review. Aging Dis. 10, 429–462. doi: 10.14336/ad.2019.0119
Ma, Q., Manaenko, A., Khatibi, N. H., Chen, W., Zhang, J. H., and Tang, J. (2011). Vascular adhesion protein-1 inhibition provides antiinflammatory protection after an intracerebral hemorrhagic stroke in mice. J. Cereb. Blood Flow Metab. 31, 881–893. doi: 10.1038/jcbfm.2010.167
Ma, F., Zhang, X., and Yin, K. J. (2020). MicroRNAs in central nervous system diseases: a prospective role in regulating blood-brain barrier integrity. Exp. Neurol. 323:113094. doi: 10.1016/j.expneurol.2019.113094
Macdonald, R. L., and Schweizer, T. A. (2017). Spontaneous subarachnoid haemorrhage. Lancet 389, 655–666. doi: 10.1016/S0140-6736(16)30668-7
Majmundar, N., Kim, B., and Prestigiacomo, C. J. (2016). Necroptosis pathway in treatment of intracerebral hemorrhage: novel therapeutic target. World Neurosurg. 89, 716–717. doi: 10.1016/j.wneu.2016.03.071
Marbacher, S., Gruter, B., Schopf, S., Croci, D., Nevzati, E., D’Alonzo, D., et al. (2018). Systematic review of in vivo animal models of subarachnoid hemorrhage: species, standard parameters, and outcomes. Transl. Stroke Res. 10, 250–258. doi: 10.1007/s12975-018-0657-4
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 10, 417–426. doi: 10.1016/s1097-2765(02)00599-3
Micheau, O., and Tschopp, J. (2003). Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190. doi: 10.1016/s0092-8674(03)00521-x
Moquin, D. M., McQuade, T., and Chan, F. K. (2013). CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One 8:e76841. doi: 10.1371/journal.pone.0076841
Mori, T., Nagata, K., Town, T., Tan, J., Matsui, T., and Asano, T. (2001). Intracisternal increase of superoxide anion production in a canine subarachnoid hemorrhage model. Stroke 32, 636–642. doi: 10.1161/01.str.32.3.636
Niwa, A., Osuka, K., Nakura, T., Matsuo, N., Watabe, T., and Takayasu, M. (2016). Interleukin-6, MCP-1, IP-10, and MIG are sequentially expressed in cerebrospinal fluid after subarachnoid hemorrhage. J. Neuroinflammation 13:217. doi: 10.1186/s12974-016-0675-7
Oberst, A., Dillon, C. P., Weinlich, R., McCormick, L. L., Fitzgerald, P., Pop, C., et al. (2011). Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367. doi: 10.1038/nature09852
Okauchi, M., Hua, Y., Keep, R. F., Morgenstern, L. B., Schallert, T., and Xi, G. (2010). Deferoxamine treatment for intracerebral hemorrhage in aged rats: therapeutic time window and optimal duration. Stroke 41, 375–382. doi: 10.1161/strokeaha.109.569830
Qi, Y. X., Zhang, X. H., Wang, Y. Q., Pang, Y. Z., Zhang, Z. B., Zhang, T. L., et al. (2016). Expression of MMP-1, -2, and -8 in longissimus dorsi muscle and their relationship with meat quality traits in cattle. Genet. Mol. Res. 15:15017593. doi: 10.4238/gmr.15017593
Quan, W., Zhang, Z., Li, P., Tian, Q., Huang, J., Qian, Y., et al. (2019). Role of regulatory T cells in atorvastatin induced absorption of chronic subdural hematoma in rats. Aging Dis. 10, 992–1002. doi: 10.14336/ad.2018.0926
Qureshi, A. I., Mendelow, A. D., and Hanley, D. F. (2009). Intracerebral haemorrhage. Lancet 373, 1632–1644. doi: 10.1016/S0140-6736(09)60371-8
Scimemi, A. (2018). Astrocytes and the warning signs of intracerebral hemorrhagic stroke. Neural Plast. 2018:7301623. doi: 10.1155/2018/7301623
Sekerdag, E., Solaroglu, I., and Gursoy-Ozdemir, Y. (2018). Cell death mechanisms in stroke and novel molecular and cellular treatment options. Curr. Neuropharmacol. 16, 1396–1415. doi: 10.2174/1570159x16666180302115544
Seo, J., Lee, E. W., Sung, H., Seong, D., Dondelinger, Y., Shin, J., et al. (2016). CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat. Cell Biol. 18, 291–302. doi: 10.1038/ncb3314
Seoane, A., Tinsley, C. J., and Brown, M. W. (2011). Interfering with perirhinal brain-derived neurotrophic factor expression impairs recognition memory in rats. Hippocampus 21, 121–126. doi: 10.1002/hipo.20763
Shao, Z., Tu, S., and Shao, A. (2019). Pathophysiological mechanisms and potential therapeutic targets in intracerebral hemorrhage. Front. Pharmacol. 10:1079. doi: 10.3389/fphar.2019.01079
Shao, A., Zhu, Z., Li, L., Zhang, S., and Zhang, J. (2019). Emerging therapeutic targets associated with the immune system in patients with intracerebral haemorrhage (ICH): from mechanisms to translation. EBioMedicine 45, 615–623. doi: 10.1016/j.ebiom.2019.06.012
Shen, F., Jiang, L., Han, F., Degos, V., Chen, S., and Su, H. (2019). Increased inflammatory response in old mice is associated with more severe neuronal injury at the acute stage of ischemic stroke. Aging Dis. 10, 12–22. doi: 10.14336/AD.2018.0205
Shen, H., Liu, C., Zhang, D., Yao, X., Zhang, K., Li, H., et al. (2017). Role for RIP1 in mediating necroptosis in experimental intracerebral hemorrhage model both in vivo and in vitro. Cell Death Dis. 8:e2641. doi: 10.1038/cddis.2017.58
Shi, J., Gao, W., and Shao, F. (2017). Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 42, 245–254. doi: 10.1016/j.tibs.2016.10.004
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Sun, X., Ou, Z., Xie, M., Kang, R., Fan, Y., Niu, X., et al. (2015). HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 34, 5617–5625. doi: 10.1038/onc.2015.32
Su, X., Wang, H., Kang, D., Zhu, J., Sun, Q., Li, T., et al. (2015). Necrostatin-1 ameliorates intracerebral hemorrhage-induced brain injury in mice through inhibiting RIP1/RIP3 pathway. Neurochem. Res. 40, 643–650. doi: 10.1007/s11064-014-1510-0
Su, X., Wang, H., Lin, Y., and Chen, F. (2018). RIP1 and RIP3 mediate hemin-induced cell death in HT22 hippocampal neuronal cells. Neuropsychiatr. Dis. Treat. 14, 3111–3119. doi: 10.2147/ndt.s181074
Sun, L., Wang, H., Wang, Z., He, S., Chen, S., Liao, D., et al. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. doi: 10.1016/j.cell.2011.11.031
Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2018). Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150. doi: 10.1038/nrneurol.2017.188
Tan, M. S., Tan, L., Jiang, T., Zhu, X. C., Wang, H. F., Jia, C. D., et al. (2014). Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis. 5:e1382. doi: 10.1038/cddis.2014.348
Tao, X., Yang, W., Zhu, S., Que, R., Liu, C., Fan, T., et al. (2019). Models of poststroke depression and assessments of core depressive symptoms in rodents: how to choose? Exp. Neurol. 322:113060. doi: 10.1016/j.expneurol.2019.113060
Topkoru, B., Egemen, E., Solaroglu, I., and Zhang, J. H. (2017). Early brain injury or vasospasm? an overview of common mechanisms. Expert Rev. Cardiovasc. Ther. 18, 1424–1429. doi: 10.2174/1389450117666160905112923
Tso, M. K., and Macdonald, R. L. (2014). Subarachnoid hemorrhage: a review of experimental studies on the microcirculation and the neurovascular unit. Transl. Stroke Res. 5, 174–189. doi: 10.1007/s12975-014-0323-4
Turan, N., Heider, R. A., Zaharieva, D., Ahmad, F. U., Barrow, D. L., and Pradilla, G. (2016). Sex differences in the formation of intracranial aneurysms and incidence and outcome of subarachnoid hemorrhage: review of experimental and human studies. Transl. Stroke Res. 7, 12–19. doi: 10.1007/s12975-015-0434-6
Ursini, F., Maiorino, M., Brigelius-Flohé, R., Aumann, K. D., Roveri, A., Schomburg, D., et al. (1995). Diversity of glutathione peroxidases. Methods Enzymol. 252, 38–53. doi: 10.1016/0076-6879(95)52007-4
van Asch, C. J., Luitse, M. J., Rinkel, G. J., van der Tweel, I., Algra, A., and Klijn, C. J. (2010). Incidence, case fatality, and functional outcome of intracerebral haemorrhage over time, according to age, sex, and ethnic origin: a systematic review and meta-analysis. Lancet Neurol. 9, 167–176. doi: 10.1016/s1474-4422(09)70340-0
Vanden Berghe, T., Declercq, W., and Vandenabeele, P. (2007). NADPH oxidases: new players in TNF-induced necrotic cell death. Mol. Cell 26, 769–771. doi: 10.1016/j.molcel.2007.06.002
Wagner, K. R., Hua, Y., de Courten-Myers, G. M., Broderick, J. P., Nishimura, R. N., Lu, S. Y., et al. (2000). Tin-mesoporphyrin, a potent heme oxygenase inhibitor, for treatment of intracerebral hemorrhage: in vivo and in vitro studies. Cell. Mol. Biol. 46, 597–608.
Wan, J., Ren, H., and Wang, J. (2019). Iron toxicity, lipid peroxidation and ferroptosis after intracerebral haemorrhage. Stroke Vasc. Neurol. 4, 93–95. doi: 10.1136/svn-2018-000205
Wang, H., Sun, L., Su, L., Rizo, J., Liu, L., Wang, L. F., et al. (2014). Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146. doi: 10.1016/j.molcel.2014.03.003
Weng, X., Tan, Y., Chu, X., Wu, X. F., Liu, R., Tian, Y., et al. (2015). N-methyl-D-aspartic acid receptor 1 (NMDAR1) aggravates secondary inflammatory damage induced by hemin-NLRP3 pathway after intracerebral hemorrhage. Chin. J. Traumatol. 18, 254–258. doi: 10.1016/j.cjtee.2015.11.010
Wertz, I. E., O’Rourke, K. M., Zhou, H., Eby, M., Aravind, L., Seshagiri, S., et al. (2004). De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430, 694–699. doi: 10.1038/nature02794
Wilkinson, D. A., Pandey, A. S., Thompson, B. G., Keep, R. F., Hua, Y., and Xi, G. (2018). Injury mechanisms in acute intracerebral hemorrhage. Neuropharmacology 134, 240–248. doi: 10.1016/j.neuropharm.2017.09.033
Wu, B., Ma, Q., Khatibi, N., Chen, W., Sozen, T., Cheng, O., et al. (2010). Ac-YVAD-CMK decreases blood-brain barrier degradation by inhibiting caspase-1 activation of interleukin-1β in intracerebral hemorrhage mouse model. Transl. Stroke Res. 1, 57–64. doi: 10.1007/s12975-009-0002-z
Wu, Y., Song, J., Wang, Y., Wang, X., Culmsee, C., and Zhu, C. (2019). The potential role of ferroptosis in neonatal brain injury. Front. Neurosci. 13:115. doi: 10.3389/fnins.2019.00115
Wu, Y. T., Tan, H. L., Huang, Q., Sun, X. J., Zhu, X., and Shen, H. M. (2011). zVAD-induced necroptosis in L929 cells depends on autocrine production of TNFα mediated by the PKC-MAPKs-AP-1 pathway. Cell Death Differ. 18, 26–37. doi: 10.1038/cdd.2010.72
Xie, Y., Guo, H., Wang, L., Xu, L., Zhang, X., Yu, L., et al. (2017). Human albumin attenuates excessive innate immunity via inhibition of microglial Mincle/Syk signaling in subarachnoid hemorrhage. Brain Behav. Immun. 60, 346–360. doi: 10.1016/j.bbi.2016.11.004
Xie, Y., Hou, W., Song, X., Yu, Y., Huang, J., Sun, X., et al. (2016). Ferroptosis: process and function. Cell Death Differ. 23, 369–379. doi: 10.1038/cdd.2015.158
Xu, H., Testai, F. D., Valyi-Nagy, T., N Pavuluri, M., Zhai, F., Nanegrungsunk, D., et al. (2015). VAP-1 blockade prevents subarachnoid hemorrhage-associated cerebrovascular dilating dysfunction via repression of a neutrophil recruitment-related mechanism. Brain Res. 1603, 141–149. doi: 10.1016/j.brainres.2015.01.047
Xu, P., Zhang, X., Liu, Q., Xie, Y., Shi, X., Chen, J., et al. (2019). Microglial TREM-1 receptor mediates neuroinflammatory injury via interaction with SYK in experimental ischemic stroke. Cell Death Dis. 10:555. doi: 10.1038/s41419-019-1777-9
Xu, Y. J., Zheng, L., Hu, Y. W., and Wang, Q. (2018). Pyroptosis and its relationship to atherosclerosis. Clin. Chim. Acta 476, 28–37. doi: 10.1016/j.cca.2017.11.005
Yagoda, N., von Rechenberg, M., Zaganjor, E., Bauer, A. J., Yang, W. S., Fridman, D. J., et al. (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868. doi: 10.1038/nature05859
Yang, C. S., Guo, A., Li, Y., Shi, K., Shi, F. D., and Li, M. (2019). Dl-3-n-butylphthalide reduces neurovascular inflammation and ischemic brain injury in mice. Aging Dis. 10, 964–976. doi: 10.14336/ad.2019.0608
Yang, Q., Huang, Z., Luo, Y., Zheng, F., Hu, Y., Liu, H., et al. (2019). Inhibition of Nwd1 activity attenuates neuronal hyperexcitability and GluN2B phosphorylation in the hippocampus. EBioMedicine 47, 470–483. doi: 10.1016/j.ebiom.2019.08.050
Yang, C., Li, T., Xue, H., Wang, L., Deng, L., Xie, Y., et al. (2019). Inhibition of necroptosis rescues SAH-induced synaptic impairments in hippocampus via CREB-BDNF pathway. Front. Neurosci. 12:990. doi: 10.3389/fnins.2018.00990
Yang, Z., Liu, Y., Yuan, F., Li, Z., Huang, S., Shen, H., et al. (2014). Sinomenine inhibits microglia activation and attenuates brain injury in intracerebral hemorrhage. Mol. Immunol. 60, 109–114. doi: 10.1016/j.molimm.2014.03.005
Yang, W. S., and Stockwell, B. R. (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245. doi: 10.1016/j.chembiol.2008.02.010
Yang, W. S., and Stockwell, B. R. (2016). Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 26, 165–176. doi: 10.1016/j.tcb.2015.10.014
Yang, W. S., SriRamaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi: 10.1016/j.cell.2013.12.010
Yang, F., Wang, Z., Wei, X., Han, H., Meng, X., Zhang, Y., et al. (2014). NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J. Cereb. Blood Flow Metab. 34, 660–667. doi: 10.1038/jcbfm.2013.242
Yang, Z., Wang, J., Yu, Y., and Li, Z. (2016). Gene silencing of MCP-1 prevents microglial activation and inflammatory injury after intracerebral hemorrhage. Int. Immunopharmacol. 33, 18–23. doi: 10.1016/j.intimp.2016.01.016
Yang, Z., Zhong, L., Xian, R., and Yuan, B. (2015). MicroRNA-223 regulates inflammation and brain injury via feedback to NLRP3 inflammasome after intracerebral hemorrhage. Mol. Immunol. 65, 267–276. doi: 10.1016/j.molimm.2014.12.018
Yu, H., Guo, P., Xie, X., Wang, Y., and Chen, G. (2017). Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell. Mol. Med. 21, 648–657. doi: 10.1111/jcmm.13008
Yu, Y., Lin, Z., Yin, Y., and Zhao, J. (2014). The ferric iron chelator 2,2’-dipyridyl attenuates basilar artery vasospasm and improves neurological function after subarachnoid hemorrhage in rabbits. Neurol. Sci. 35, 1413–1419. doi: 10.1007/s10072-014-1730-8
Yu, Y., Xie, Y., Cao, L., Yang, L., Yang, M., Lotze, M. T., et al. (2015). The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell. Oncol. 2:e1054549. doi: 10.1080/23723556.2015.1054549
Yuan, S., Yu, Z., Zhang, Z., Zhang, J., Zhang, P., Li, X., et al. (2019). RIP3 participates in early brain injury after experimental subarachnoid hemorrhage in rats by inducing necroptosis. Neurobiol. Dis. 129, 144–158. doi: 10.1016/j.nbd.2019.05.004
Zhang, S., Hu, Z. W., Luo, H. Y., Mao, C. Y., Tang, M. B., Li, Y. S., et al. (2020). AAV/BBB-mediated gene transfer of CHIP attenuates brain injury following experimental intracerebral hemorrhage. Transl. Stroke Res. 11, 296–309. doi: 10.1007/s12975-019-00715-w
Zhang, C., Jiang, M., Wang, W. Q., Zhao, S. J., Yin, Y. X., Mi, Q. J., et al. (2019). Selective mGluR1 negative allosteric modulator reduces blood-brain barrier permeability and cerebral edema after experimental subarachnoid hemorrhage. Transl. Stroke Res. doi: 10.1007/s12975-019-00758-z [Epub ahead of print].
Zhang, D. W., Shao, J., Lin, J., Zhang, N., Lu, B. J., Lin, S. C., et al. (2009). RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336. doi: 10.1126/science.1172308
Zhang, S., Tang, M. B., Luo, H. Y., Shi, C. H., and Xu, Y. M. (2017). Necroptosis in neurodegenerative diseases: a potential therapeutic target. Cell Death Dis. 8:e2905. doi: 10.1038/cddis.2017.286
Zhang, J., Wang, Y., Aili, A., Sun, X., Pang, X., Ge, Q., et al. (2019). Th1 biased progressive autoimmunity in aged aire-deficient mice accelerated thymic epithelial cell senescence. Aging Dis. 10, 497–509. doi: 10.14336/ad.2018.0608
Zhang, Y. H., Wang, D. W., Xu, S. F., Zhang, S., Fan, Y. G., Yang, Y. Y., et al. (2018). α-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 14, 535–548. doi: 10.1016/j.redox.2017.11.001
Zhang, Z., Wu, Y., Yuan, S., Zhang, P., Zhang, J., Li, H., et al. (2018). Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res. 1701, 112–125. doi: 10.1016/j.brainres.2018.09.012
Zhao, H., Chen, Y., and Feng, H. (2018). P2X7 receptor-associated programmed cell death in the pathophysiology of hemorrhagic stroke. Curr. Neuropharmacol. 16, 1282–1295. doi: 10.2174/1570159x16666180516094500
Zheng, Y., Gardner, S. E., and Clarke, M. C. (2011). Cell death, damage-associated molecular patterns, and sterile inflammation in cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 31, 2781–2786. doi: 10.1161/atvbaha.111.224907
Zhu, H., Santo, A., Jia, Z., and Robert Li, Y. (2019). GPx4 in bacterial infection and polymicrobial sepsis: involvement of ferroptosis and pyroptosis. React. Oxyg. Species Apex 7, 154–160. doi: 10.20455/ros.2019.835
Zhu, S., Wei, X., Yang, X., Huang, Z., Chang, Z., Xie, F., et al. (2019). Plasma lipoprotein-associated phospholipase A2 and superoxide dismutase are independent predicators of cognitive impairment in cerebral small vessel disease patients: diagnosis and assessment. Aging Dis. 10, 834–846. doi: 10.14336/ad.2019.0304
Zille, M., Karuppagounder, S. S., Chen, Y., Gough, P. J., Bertin, J., Finger, J., et al. (2017). Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke 48, 1033–1043. doi: 10.1161/strokeaha.116.015609
Zlokovic, B. V. (2008). The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57, 178–201. doi: 10.1016/j.neuron.2008.01.003
Keywords: programmed cell death, necroptosis, ferroptosis, pyroptosis, blood–brain barrier dysfunction, crosstalk
Citation: Fang Y, Gao S, Wang X, Cao Y, Lu J, Chen S, Lenahan C, Zhang JH, Shao A and Zhang J (2020) Programmed Cell Deaths and Potential Crosstalk With Blood–Brain Barrier Dysfunction After Hemorrhagic Stroke. Front. Cell. Neurosci. 14:68. doi: 10.3389/fncel.2020.00068
Received: 23 December 2019; Accepted: 06 March 2020;
Published: 03 April 2020.
Edited by:
Dennis Qing Wang, Zhujiang Hospital, Southern Medical University, ChinaReviewed by:
Xiaoya Gao, Southern Medical University, ChinaStefano Angiari, Trinity College Dublin, Ireland
Copyright © 2020 Fang, Gao, Wang, Cao, Lu, Chen, Lenahan, Zhang, Shao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anwen Shao, anwenshao@sina.com; 21118116@zju.edu.cn; Jianmin Zhang, zjm135@zju.edu.cn
† These authors have contributed equally to this work