Targeting Amyloidogenic Processing of APP in Alzheimer’s Disease
 Jing Zhao
Jing Zhao Xinyue Liu1
Xinyue Liu1 - 1Center for Biotechnology and Interdisciplinary Studies, Rensselaer Polytechnic Institute, Troy, NY, United States
- 2Geriatric Research Education Clinical Center, Edith Nourse Rogers Memorial Veterans Hospital, Bedford, MA, United States
- 3Department of Pharmacology and Experimental Therapeutics, School of Medicine, Boston University, Boston, MA, United States
- 4Department of Chemistry, New York University, New York, NY, United States
- 5Department of Biological Sciences, Rensselaer Polytechnic Institute, Troy, NY, United States
- 6Department of Chemistry and Chemical Biology, Rensselaer Polytechnic Institute, Troy, NY, United States
Alzheimer’s disease (AD) is the most common type of senile dementia, characterized by neurofibrillary tangle and amyloid plaque in brain pathology. Major efforts in AD drug were devoted to the interference with the production and accumulation of amyloid-β peptide (Aβ), which plays a causal role in the pathogenesis of AD. Aβ is generated from amyloid precursor protein (APP), by consecutive cleavage by β-secretase and γ-secretase. Therefore, β-secretase and γ-secretase inhibition have been the focus for AD drug discovery efforts for amyloid reduction. Here, we review β-secretase inhibitors and γ-secretase inhibitors/modulators, and their efficacies in clinical trials. In addition, we discussed the novel concept of specifically targeting the γ-secretase substrate APP. Targeting amyloidogenic processing of APP is still a fundamentally sound strategy to develop disease-modifying AD therapies and recent advance in γ-secretase/APP complex structure provides new opportunities in designing selective inhibitors/modulators for AD.
Introduction
Alzheimer’s disease (AD) is a progressive and incurable neurodegenerative disorder, characterized by progressive and irreversible loss of memory. AD is the leading cause of senile dementia. The number of people age 65 and older living with AD’s dementia in the United States is projected to grow from 5.8 million in 2020 to 13.8 million by 2050 (Alzheimer’s_Association, 2020). Cognitive deficits caused by AD, such as progressive memory loss, difficulty in communication and movement disorder, significantly compromise the patients’ quality of life, leading to hospitalization and eventually death due to complications. AD has been recognized as one of the most difficult medical problems with hefty economic burden (Wimo et al., 2010). Total medical expenses for Alzheimer’s or other dementias in the United States are projected to be $305 billion in 2020 (Alzheimer’s_Association, 2020). The cost of AD is likely to skyrocket in the near future, due to rising ageing population, increasing mortality relative to other disease and the absence of a disease-modifying drug. Therefore, there is a major unmet medical need for disease-modifying therapies for AD.
Approved drugs for AD include acetylcholinesterase inhibitors (Aricept®, Exelon®, Razadyne®) and NMDA-antagonist memantine (Namenda®). However, these drugs are not disease-modifying, only improving symptoms without slowing down or stopping AD from progressing. In the last 30 years, a large number of drug candidates have entered clinical development but no new drug for AD has been approved since memantine in 2003. The majority of AD drug discovery focused on inhibiting the amyloid-β peptide (Aβ) production from the amyloidogenic processing of APP.
Amyloid Cascade Hypothesis and Amyloidogenic Processing of APP
In the past 30 years, the amyloid hypothesis has been extensively tested and amyloid has been the most compelling therapeutic target for AD. Despite ongoing debates about this hypothesis in light of recent failures of anti-amyloid-based clinical trials, new evidences continue to emerge to support the idea that an imbalance between production and clearance of Aβ peptides is the initiating event of AD pathogenic processes. Abnormal accumulation of amyloid eventually leads to formation of senile plaques and neurofibrillary tangles, two pathological hallmarks of AD. Aβ aggregates were found to be toxic both in vitro and in vivo. Numerous studies have shown that Aβ aggregates, especially soluble oligomers, impair both synaptic function and structure (Kokubo et al., 2005; Wilcox et al., 2011). Injection of soluble Aβ42 oligomers directly isolated form AD cerebral cortex into healthy rats leads to impaired memory (Selkoe et al., 2016). In addition, accumulation of Aβ oligomers can not only trigger AD-type tau hyperphosphorylation and cause neurotic dystrophy (Jin et al., 2011; Stancu et al., 2014; Jacobs et al., 2018), but also activate neuroinflammation (Park et al., 2018; Henstridge et al., 2019). Apolipoprotein E4, the greatest genetic risk factor for late-onset AD, impairs Aβ clearance and promotes Aβ accumulation in the brain (Carter et al., 2001; Wildsmith et al., 2013). Along with tau, Aβ might be transmissible through the Aβ contaminants in cadaver-derived human growth hormone for the treatment of Creutzfeldt-Jakob disease (Duyckaerts et al., 2018). From human genetics, dominant mutations causing early-onset familial AD reside either in APP or presenilin (catalytic sub-unit of γ-secretase), which alter the proteolytic processing of APP in ways either elevating the Aβ42/Aβ40 ratio or increasing the self-aggregation propensity of resultant Aβ peptides (De Jonghe, 2001; Selkoe, 2001; Chen et al., 2014). Duplication of the APP gene in Down’s syndrome leads to Aβ deposits in the teens, and almost invariably leads to AD at an early age (Lejeune et al., 1959; Head et al., 2012). Interestingly, three DS patients with partial trisomy that excludes the APP gene did not develop dementia (Korbel et al., 2009; Doran et al., 2016). The human genetics of DS strikingly demonstrates that increasing Aβ dosage (APP duplication) causes dementia, while normalized Aβ dosage in DS (partial trisomy without APP duplication) prevents dementia (albeit the sample size = 3 is low), affirming that amyloid reduction is a fundamentally sound strategy for disease-modifying treatment of AD. The failures of anti-amyloid clinical trials in recent years can be attributed to giving the therapy too late to the patients, poor clinical trial design, heterogeneity of the trial patient population etc.
Aβ is a small peptide generated by proteolytic processing of APP (Figure 1A), a type-I transmembrane protein with a large extracellular domain. APP is transported to the plasma membrane through the endoplasmic reticulum -Golgi secretory pathway. The majority of APP is processed via the non-amyloidogenic pathway at the plasma membrane (Figure 1A). In the non-amyloidogenic pathway, APP is cleaved by α-secretase within the Aβ domain between Lys16 and Leu17, producing a soluble N-terminal fragment (APPs α) and a membrane-bound C-terminal fragment, C83, which can be further cleaved by γ-secretases and generates a soluble extracellular p3 peptide, thus precluding the formation of intact Aβ (Figure 1A; Anderson et al., 1991; Sisodia, 1992; Wilson et al., 1999). Unlike the non-amyloidogenic pathway, APP is internalized and delivered to endosomes in the amyloidogenic pathway (Bu, 2009). During amyloidogenic APP processing, APP is cleaved by β-secretase (BACE1, β-site APP-cleaving enzyme 1), generating a soluble N-terminal fragment (APPsβ) and a membrane-bound C-terminal fragment (C99) (Vassar, 2004; Vassar et al., 1999). Within the membrane, C99 is subsequently cleaved by an enzymatic complex known as γ-secretase, releasing a cytoplasmic polypeptide termed AICD (APP intracellular domain) at the luminal side and Aβ peptides (Thinakaran and Koo, 2008) at the other side of the membrane. AICD is transferred to the nucleus, where it functions as a transcriptional factor (Berridge, 2010), whereas the Aβ peptides are secreted into the extracellular space when the endosome recycles to cell surface. γ-Secretase cleaves APP at variable sites within the transmembrane domain, generating Aβ peptides ranging in length from 38 to 43 residues (Selkoe and Wolfe, 2007). Among different Aβ species, Aβ42 and Aβ43 are highly self-aggregating, while Aβ40 and shorter peptides are relatively benign (Burdick et al., 1992). Aβ42 and Aβ40 are the two common Aβ species in the human brain and the increased Aβ42/Aβ40 ratio is a common biochemical feature in the early-onset familial AD (FAD) caused by mutations in APP and presenilin. Aβ42 aggregates rapidly into neurotoxic oligomers, leading to fibrils and plaques. It has been proposed that Aβ oligomers are more toxic than fibrils, therefore it may play a more important role than amyloid plaque in AD progression. Aberrant process of APP by β-secretase and γ-secretase may result in imbalance between production and clearance of Aβ peptides, leading to toxic oligomers, fibrils and senile plaques. Interestingly, pathogenic mutations in presenilin were found to destabilize γ-secretase-APP interactions and thus enhance the production of longer Aβ peptides (Chévez-Gutiérrez et al., 2012; Veugelen et al., 2016; Szaruga et al., 2017). These finding points to enhancing the stability of γ-secretase-Aβn complex as a potential therapeutic approach for AD.
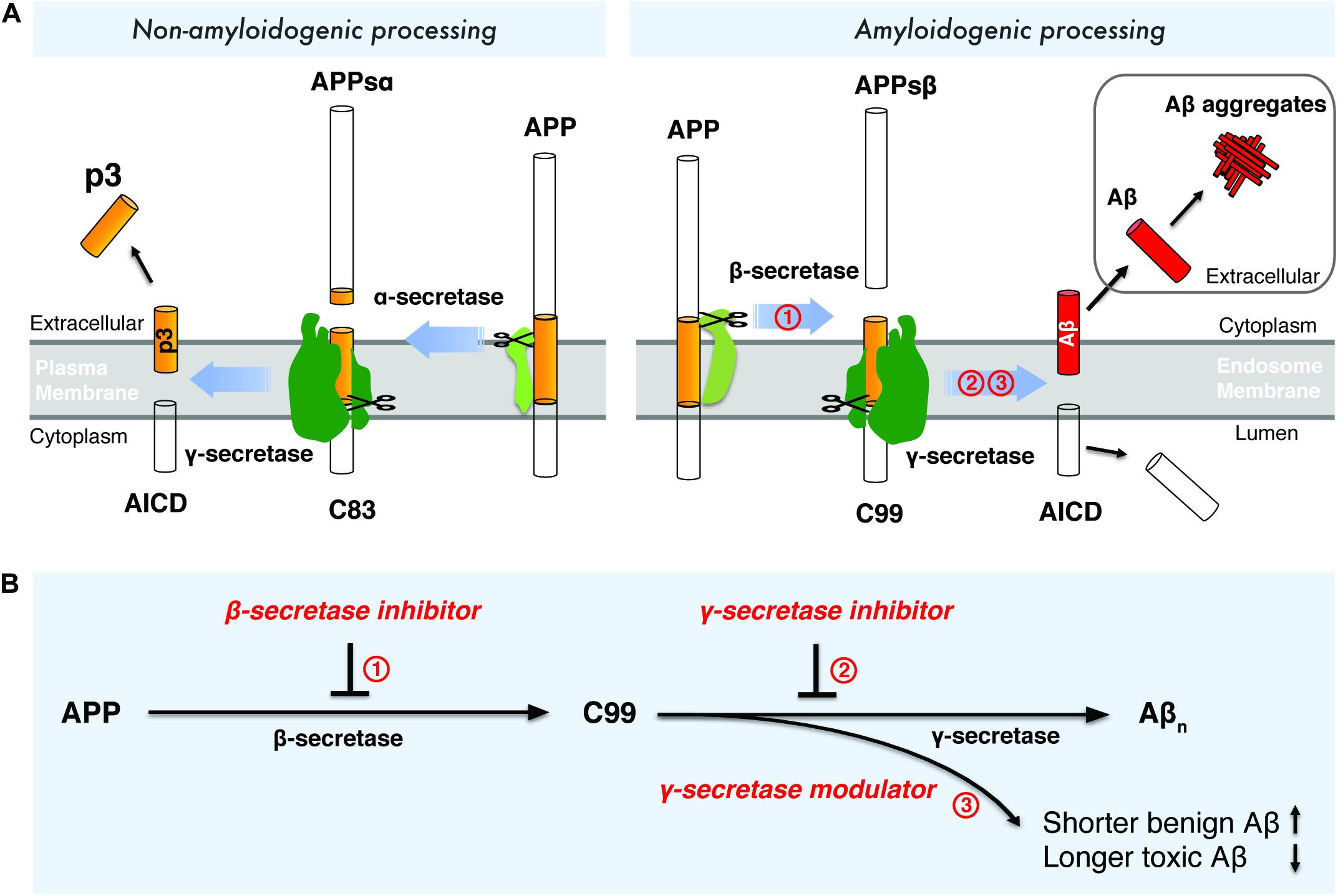
Figure 1. Drugdiscovery strategies targeting the amyloidogenic processing of APP. (A) Amyloidogenic and non-amyloidogenic processing of APP. (B) Drug discovery strategies targeting β-secretase and γ-secretase.
Drug Discovery Targeting Amyloidogenic Processing of APP
Interference with the amyloidogenic processing of APP has been a major strategy to modulate Aβ production. As two crucial enzymes catalyzing the intramembrane proteolysis of APP, β-secretase and γ-secretase have been the most prominent targets for AD drug discovery. In the past two decades, numerous β-secretase inhibitors and γ-secretase inhibitors/modulators were discovered to inhibit or modulate the amyloidogenic processing of APP, either causing the reduced production of total Aβn or shifting the production of Aβ to shorter and more benign Aβ species (Figure 1B).
Drug Discovery Targeting β-Secretase
β-Secretase
β-secretase, also named as BACE1 (β-site APP-cleaving enzyme 1), was identified in 1999 (Fairbanks et al., 1999; Hussain et al., 1999; Vassar et al., 1999; Yan et al., 1999; Lin et al., 2000) as an APP-cleaving aspartyl protease. BACE1 is the principal neuronal protease for generating C99 from APP, which leads to subsequent Aβ generation by γ-secretase. Importantly, BACE1 shedding of APP is a prerequisite for γ-secretase cleavage within the transmembrane domain of APP for Aβ production. Secretion of Aβ peptides is abolished in cultures of BACE1-deficient embryonic cortical neurons (Cai et al., 2001). Naturally, BACE1 inhibition has been a widely pursued therapeutic target for amyloid reduction.
BACE1 is a type-I membrane protein with 501 amino acid residues related to the pepsin family. It is localized within acidic subcellular compartments of the secretory pathway, primarily the Golgi apparatus and endosomes. As shown in Figure 2A. BACE1 has an N-terminal signal sequence (residues 1–21), a pro-peptide domain (residues 22–45), a large luminal catalytic domain (residues 46-451), a single transmembrane domain (residues 452-483), and a short cytoplasmic domain (residues 484-501) (Hussain et al., 1999; Benjannet et al., 2001). In addition, BACE1 has several N-linked glycosylation sites and six cysteine residues that form three intramolecular disulfide bonds, C216-C420, C278-C443, and C330-C380. The N-terminal signal sequence and pro-peptide domain were removed post-translationally, so the mature BACE1 sequence begins at residue Glu46 (Creemers et al., 2001). In the luminal catalytic domain for the β-site cleavage of APP, BACE1 contains two motifs, DTGS (residues 93–96) and DSGT (residues 289–292), which contain the two highly conserved catalytic aspartates (Hussain et al., 1999; Bennett et al., 2000). The crystal structure of the catalytic domain (residues 56-446) of BACE1 with an inhibitor was first published by Hong et al. in 2000 (PDB code: 1FKN) (Hong et al., 2000). Like other aspartic proteases, the substrate binding cleft was located between the N- and C-terminal lobes of BACE1, together with a β-hairpin loop which forms the flap region. The flap opens to allow the substrate to enter and then closes down on the substrate during catalysis and reopens to release the hydrolyzed products. As shown in Figure 2B, the two conserved aspartate, D93 and D289, are located at the groove between the N- and C-terminal lobes, partially covered by the “flap” (residues 130-135) (Hong et al., 2000). A later substrate-free (apo) structure of BACE1 (PDB code: 1W50) showed a water molecule located between D93 and D289, which is likely involved in nucleophilic attack for peptide hydrolysis and important binding site for inhibitors (Patel et al., 2004; Ghosh and Osswald, 2014).
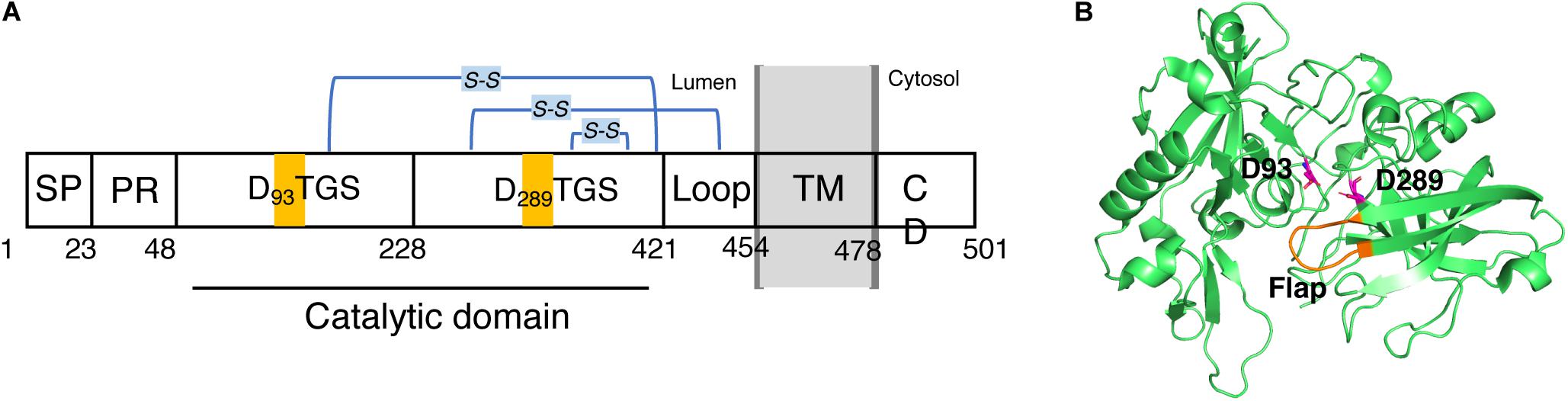
Figure 2. Schematic view of the domains of BACE1 and structure of its catalytic domain. (A) BACE1 is a membrane protein consists of (from N- to C- terminal) a signal peptide (SP, 1–22), a proline rich domain (PR, 23–47), a large luminal catalytic domain (48-420), a loop (421-453), a transmembrane domain (TM, 454-477), and a cytoplasmic domain (CD, 478-501). (B) 3D structure of the catalytic domain. Two conserved catalytic aspartic acids, D93 and D289, are highlighted.
BACE1 cleavage is highly specific and cleaves APP only at the β-secretase sites of Asp1 and Glu11 of Aβ (Vassar et al., 1999). As expected, BACE1 cleaves the Swedish APP mutant 10 to 100-fold more efficiently than wild-type APP. BACE1 appeared to be an ideal target for drug discovery since its complete inhibition should shut down the amyloidogenic APP processing.
Drug Discovery Targeting BACE1
The first generation of BACE1 inhibitors are peptidomimetic transition-state analogs, which were designed to be accommodated within the substrate-binding cleft of BACE1. In 2000, Hong et al. developed an 8-residue peptide OM99-2 utilizing the template of the β-cleavage site of Swedish APP, where the scissile peptide bond was replaced by a Leu-Ala hydroxyethylene transition-state isostere (Figure 3B; Ghosh et al., 2000). The crystal structure of BACE1 and OM99-2 complex (Figure 3A) provided coveted molecular insight into the ligand/enzyme interaction and significantly advanced the development of BACE1 inhibitors (Hong et al., 2000). Catalytic aspartates D93 and D289 (see Figure 3A, in green) are located at the center of the substrate binding pocket. There are four hydrogen bonds between the catalytic aspartates and the transition state isostere hydroxyl, and ten hydrogen bonds between the binding cleft and flap to OM99-2 backbone. As shown in Figure 3A, the “flap” (in yellow) closes over the top of the cleft upon the binding of the inhibitor. Futher investigation led to the design of inhibitor OM00-3 by the replacement of the P2’ Ala in OM99-2 with a Val, which achived a five-fold enhancement of inhibitory potency (Hong et al., 2002). Additional peptidomimetic inhibitors were developed, including KMI-008, KMI-420, and KMI-429 by Kiso’s group (Shuto et al., 2003; Kimura et al., 2005; Asai et al., 2006), GSK188909 (Hussain et al., 2007), and compound 11 (Iserloh et al., 2008), etc. However, the big molecular size precludes the application of peptidomimetic BACE1 inhibitors in vivo, due to their short half-life, deficiency in crossing the BBB, and low oral availability. Therefore, later generations of BACE1 inhibitors are mostly non-peptidic small molecules.
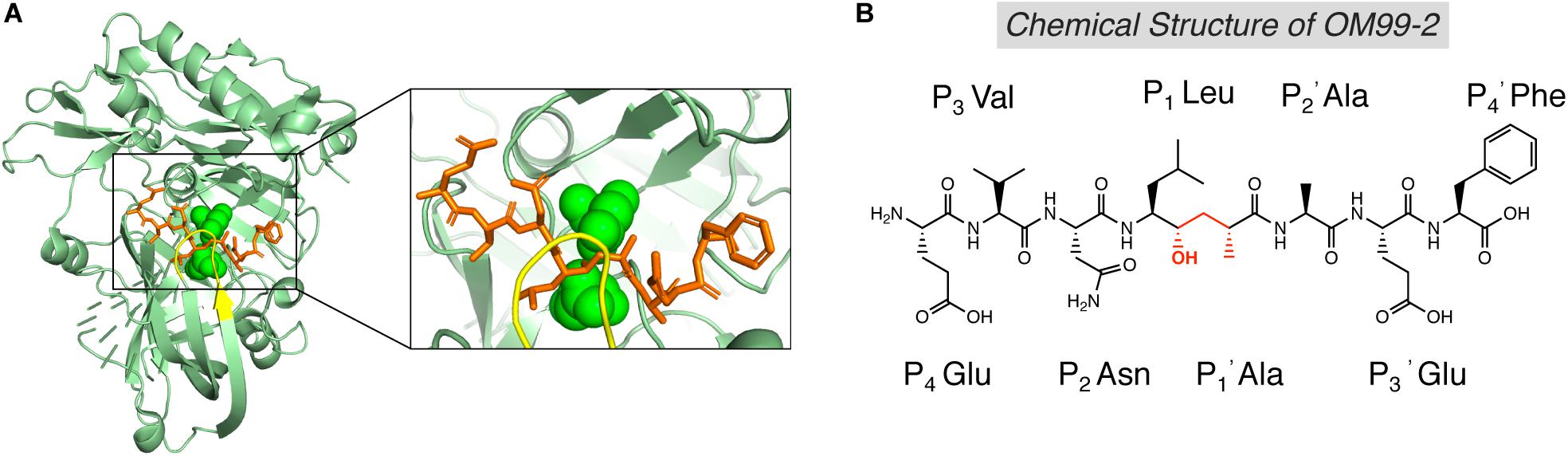
Figure 3. Structure of BACE1 complexed with peptidomimetic inhibitor OM99-2. (A) Crystal structure of BACE1-OM99-2 complex (PDB code: 1FKN). The ligand binding region on BACE1 was magnified on the right. Inhibitor OM99-2 was shown as sticks in orange. Two catalytic aspartic acid residues were shown in green as sphere. The “flap” region was shown in yellow. (B) Chemical structure of OM99-2. The hydroxyethylene transition-state isostere was indicated in red.
High-throughput screening (HTS) and fragment-based drug discovery (FBDD) are two major methods employed to explore non-peptidyl small molecule BACE1 inhibitors. In contrast to traditional HTS, which uses libraries of drug-like compounds (typically 300–500 Da), FBDD screens libraries of small fragments (typically < 300 Da), allowing more thorough exploration of the chemical space and the identification of weakly binding chemical fragments. Small fragment hits can then be linked or extended into unique multi-fragment scaffold (Erlanson, 2012). Screening of 8000 fragments by Lilly Research Laboratories produced two promising fragment hits, amino-benzothiazine and amino-thiadiazine (May et al., 2011). Evolution of the fragment based on crystal structure produced compound LY2811376 [(S)-4-(2,4-difluoro-5-pyrimidin-5-yl-phenyl)-4-methyl-5,6-dihydro- 4H-[1,3]thiazin-2-ylamine]. LY2811376 was the first orally available, non-peptidic small-molecule BACE1 inhibitor, showing satisfactory pharmacokinetic and pharmacodynamic properties in preclinical animal models and in humans (May et al., 2011). The chemical structure of LY2811376 and its complex with BACE1 were shown in Figure 4. LY2811376 binds to the active site of BACE1 and forms an optimal H-bonds network with the catalytic aspartates (May et al., 2011). Despite the encouraging results in preclinical animal models, LY2811376 shows toxicity in long-term studies. Other fragment-based compounds, including AZD3839 from Astra-Zeneca (Jeppsson et al., 2012) and NB-360 from Novartis (Neumann et al., 2015), also showed great potential in reducing brain Aβ levels in preclinical studies.
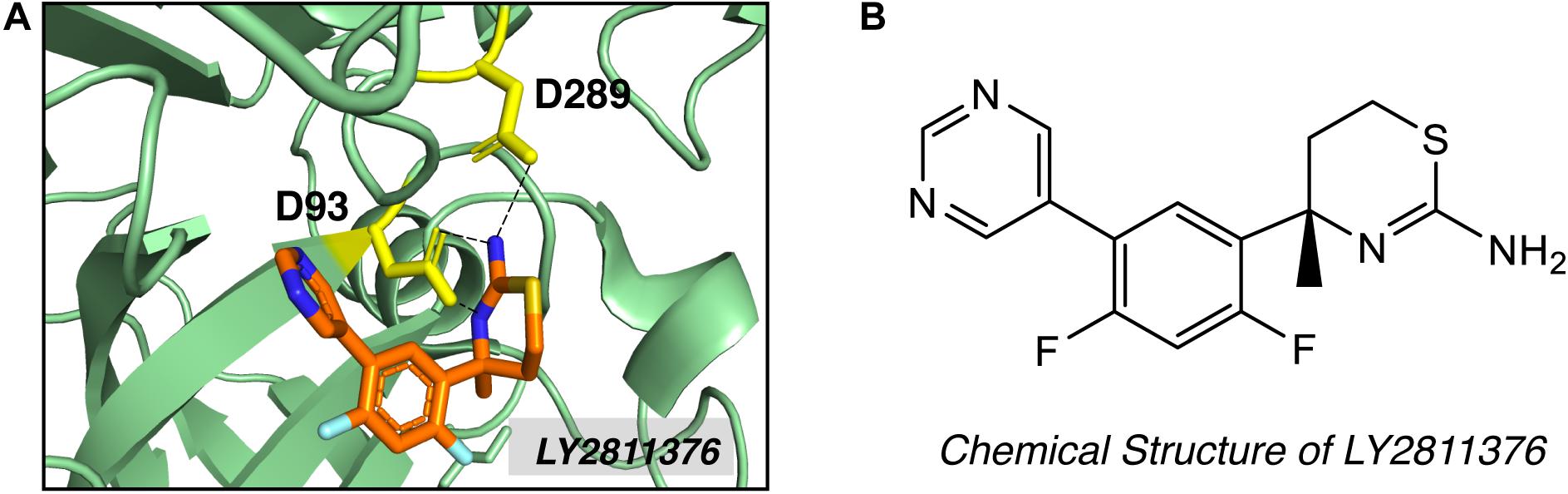
Figure 4. Structure of BACE1 complexed with small molecue inhibitor LY2811376. (A) LY2811376 binds to the active site of BACE1 (PDB code: 4YBI). Inhibitor LY2811376 was shown as sticks colored with orange for carbon, blue for nitrogen, light blue for fluorine and light-orange for sulfur. The side chain of two catalytic aspartate (D93 and D289) were shown in yellow as sticks. Hydrogen bonds between aspartate side chain and LY2811376 were shown in dotted black lines. (B) Chemical structure of LY2811376.
BACE1 Inhibitors in Clinical Trials
BACE1 inhibitors effectively reduced brain and CSF Aβ levels in both animal studies and human clinical trials. In the past two decades, many potent BACE inhibitors have been developed, but only a small portion entered the clinical trials. Selectivity over other aspartic protease (BACE2, pepsin, renin, cathepsin D, and cathepsin E) and blood-brain barrier (BBB) permeability are major hurdles. The first Phase I clinical trial on a BACE1 inhibitor, CTS-21166, was conducted by CoMentis in 2008. CTS-21166 passes BBB, with high oral bio-availability and selectivity of BACE1 over other proteases. Results from clinical studies indicated a dose-dependent reduction of plasma Aβ for an extended period of time, with up to 80% inhibition at the highest dosage (Gabrielle, 2008). Merck conducted phase I clinical trial on their inhibitor MK-8931 (Verubecestat) in 2012, which is well-tolerated and demonstrates a profound (up to 94%) reduction in CSF Aβ (Forman et al., 2012). MK-8931 is the first BACE1 inhibitor to advance to phase II/III clinical trial in patients with mild to moderate AD. It was discontinued in 2017 (Table 1), due to a lack of clinical benefit in cognition (Egan et al., 2018). Subsequent phase III clinical trials of MK-8931 in patients with prodromal AD was also discontinued in 2018, because it was deemed unlikely to exhibit a positive benefit/risk ratio (Merck, 2018). Several other promising BACE1 inhibitors in late state clinical trials also reported disappointing results, including LY2886721 (Eli Lilly), AZD3839 (AstraZeneca), atabecestat (JNJ-54,861,911, Janssen), and lanabecestat (AZD3293, LY3314814, AstraZeneca and Eli Lilly) (Table 1).
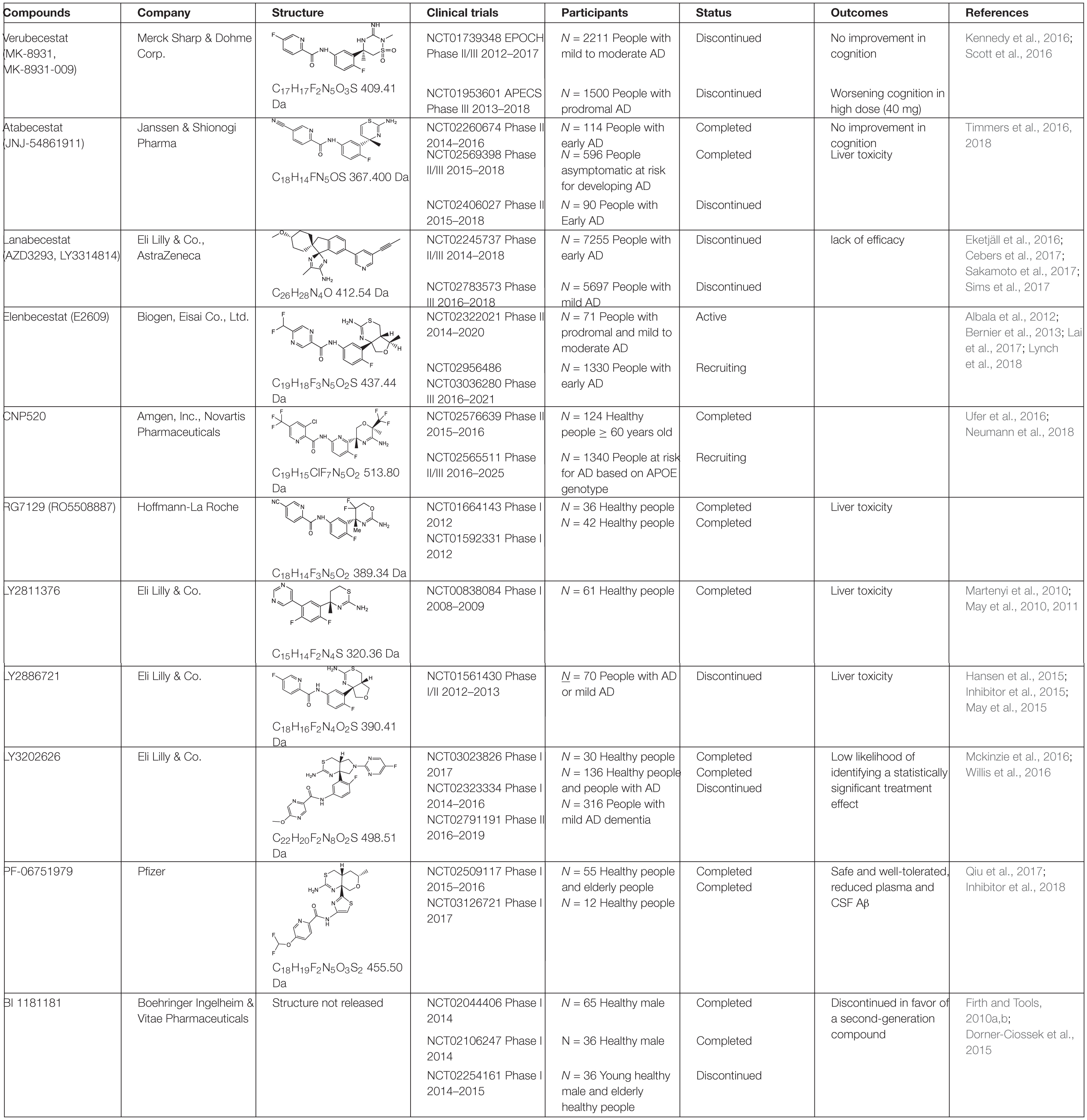
Table 1. BACE1 inhibitors in clinical trials.
Drug Discovery Targeting γ-Secretase
γ-Secretase
γ-secretase is a membrane protein complex composed of four essential subunits, with presenilin-1 (PS1) or presenilin-2 (PS2) as the catalytic subunit, nicastrin (Nct), anterior pharynx-defective 1 (Aph-1), and presenilin enhancer 2 (Pen-2) (De Strooper, 2003; Wolfe and Kopan, 2004; Wolfe, 2006). PS1 undergoes autocleavage through its endopeptidase activity to produce a N-terminal fragment (NTF) and a C-terminal fragment (CTF) (Thinakaran et al., 1996), each of which contributes a conserved catalytic aspartate, D257 and D385, respectively (Figure 5A). Nct is believed to play a role in substrate recognition and its initial docking (Shah et al., 2005). Aph-1 stabilizes the complex and Pen-2 is required for maturation of γ-secretase (Takasugi et al., 2003). γ-secretase cleaves type I transmembrane proteins and has more than 90 reported substrates (Haapasalo and Kovacs, 2011; Jurisch-Yaksi et al., 2013), of which APP and Notch are the most well-characterized. After the cleavage of APP by BACE1, a C-terminal 99-residue fragment of APP (C99) is generated and subsequently cleaved by γ-secretase. The initial cleavage of γ-secretase produces a 48-residue (Aβ48) or 49-redidue (Aβ49) amyloid peptide, while at the same time the intracellular domains (AICD) are liberated into the cytoplasm (Figure 5B; De Strooper et al., 1998; Wolfe et al., 1999). Subsequent cleavage of Aβ49 by the C-terminal peptidase activity of γ-secretase leads to the generation of Aβ46, Aβ43, and Aβ40, while cleavage of Aβ48 results in Aβ45, Aβ42, and Aβ38 (Golde et al., 1992; Sano et al., 2009). Among Aβ peptides with different lengths, Aβ42 and Aβ43 are most prone to aggregation, while Aβ40 and shorter peptides are relatively benign (Burdick et al., 1992).
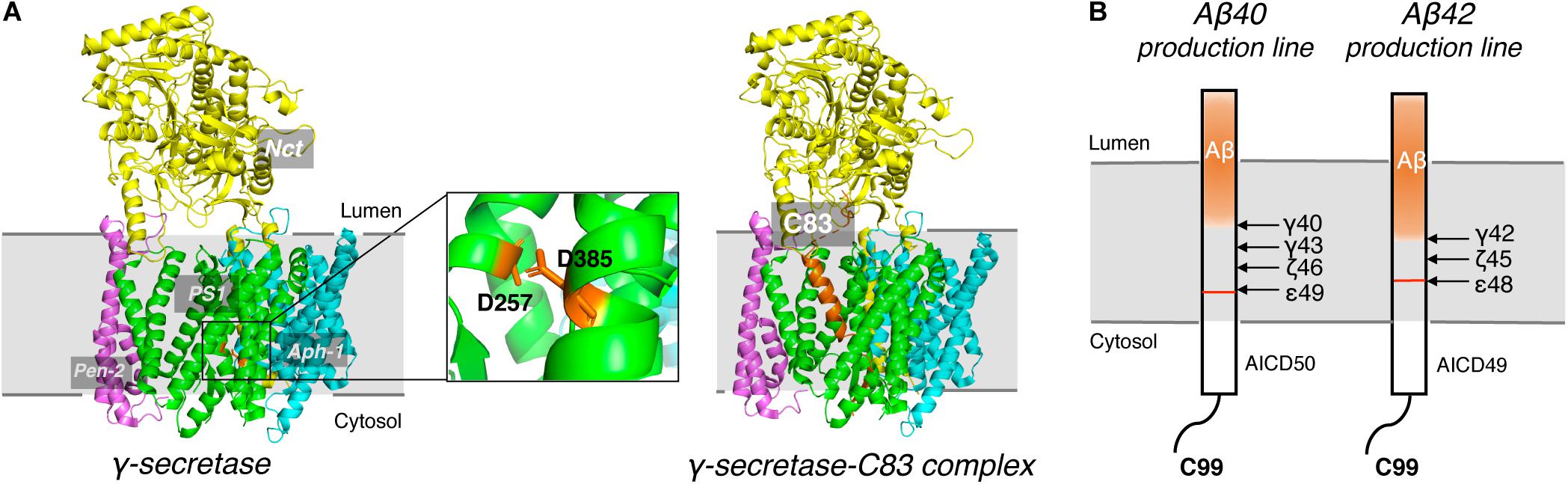
Figure 5. Cryo-EM Structure of γ-secretase and progressive cleavage of APP transmembrane domain. (A) The cryo-EM structure of γ-secretase apo (left) and complexed with substrate C83 (right). Four subunits, Nct, PS1, Pen-2 and Aph-1, were indicated. Two conserved catalytic aspartates, D257 and D385, were shown as sticks in orange in the magnified graph. The APP substrate C83 was shown as cartoon in orange in the complex. (B) Schematic diagram of the progressive cleavage sites of γ-secretase on substrate C99.
Drug Discovery Targeting γ-Secretase
γ-Secretase cleaves within APP transmembrane domain (APPTM) of C99 to release Aβ, which aggregates to form neurotoxic oligomers and fibrils. Thus, γ-secretase is an obvious drug target for reducing amyloid load. Efforts have been devoted to develop γ-secretase targeting compounds, γ-secretase inhibitors (GSIs) and later γ-secretase modulators (GSMs).
Early studies on γ-secretase targeting compounds have shown an increase of the levels of substrates C99 and C83, and a reduction of the levels of the γ-secretase cleavage products Aβ and p3 in APP-transfected cells (Higaki et al., 1995, 1999; Klafki et al., 1996). Those compounds were named as GSIs, among which dipeptide aldehydes (such as MG-132 and MDL-28170) were firstly reported (Higaki et al., 1995; Klafki et al., 1996). At the same time, substrate-based GSIs were designed, and these compounds show pharmacological effects through occupying the binding site of APP on γ-secretase. The substrate-based GSIs (Figure 6) includes: (1) transition-state analog inhibitor of aspartyl proteases, such as hydroxyethylene L-685,458, which was identified to target PS1 NTF and CTF (Shearman et al., 2000); (2) helical peptide (Das et al., 2003), which was found to directly bind to the PS1 NTF/CTF interface (Kornilova et al., 2005); (3) Chemically modified compounds based on hits from high-throughput screening, such as DAPT (Dovey et al., 2001), for which PS1 was identified as the direct target (Morohashi et al., 2006; Bai et al., 2015). The complex structure of presenilin homolog PSH bound to a hydroxyethylene derivative L-682,679 was shown in Figure 7. PSH is composed of 9 transmembrane helices (TMs). L-682,679 was found to bind in the cleft surrounded by TMs 2, 6, 7, 8, and 9. The two catalytic aspartates D162 and D220 were separated by the phenol group in the amide end of L-682679 (Dang et al., 2015).
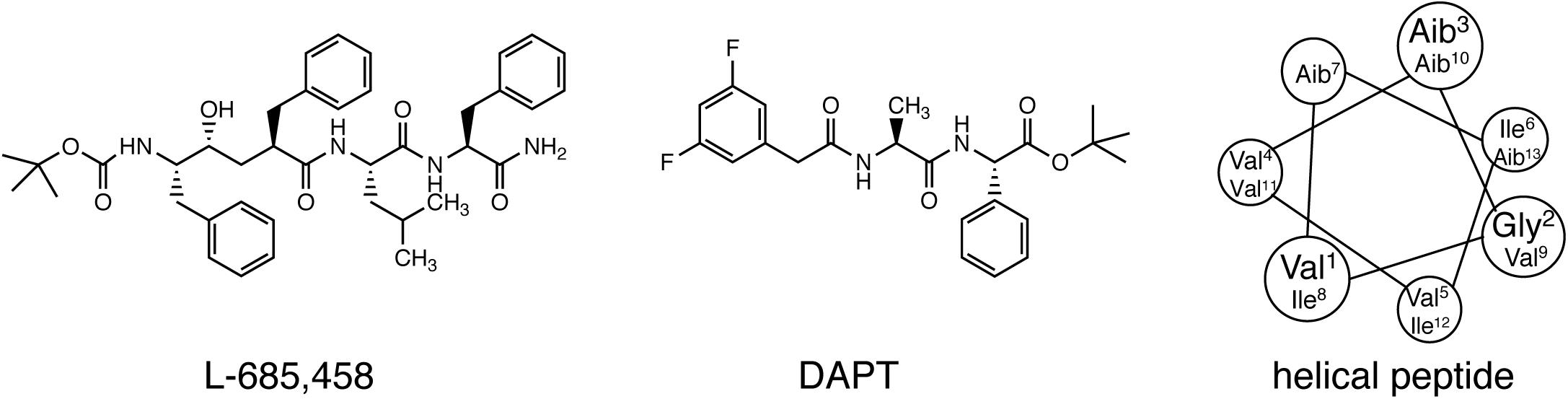
Figure 6. Chemical structures of representative compounds in three classes of substrate-based GSIs. From left to right: transition-state analog inhibitor (L-685,458), chemically modified compounds based on hits from high-throughput screening (DAPT), and helical peptide.
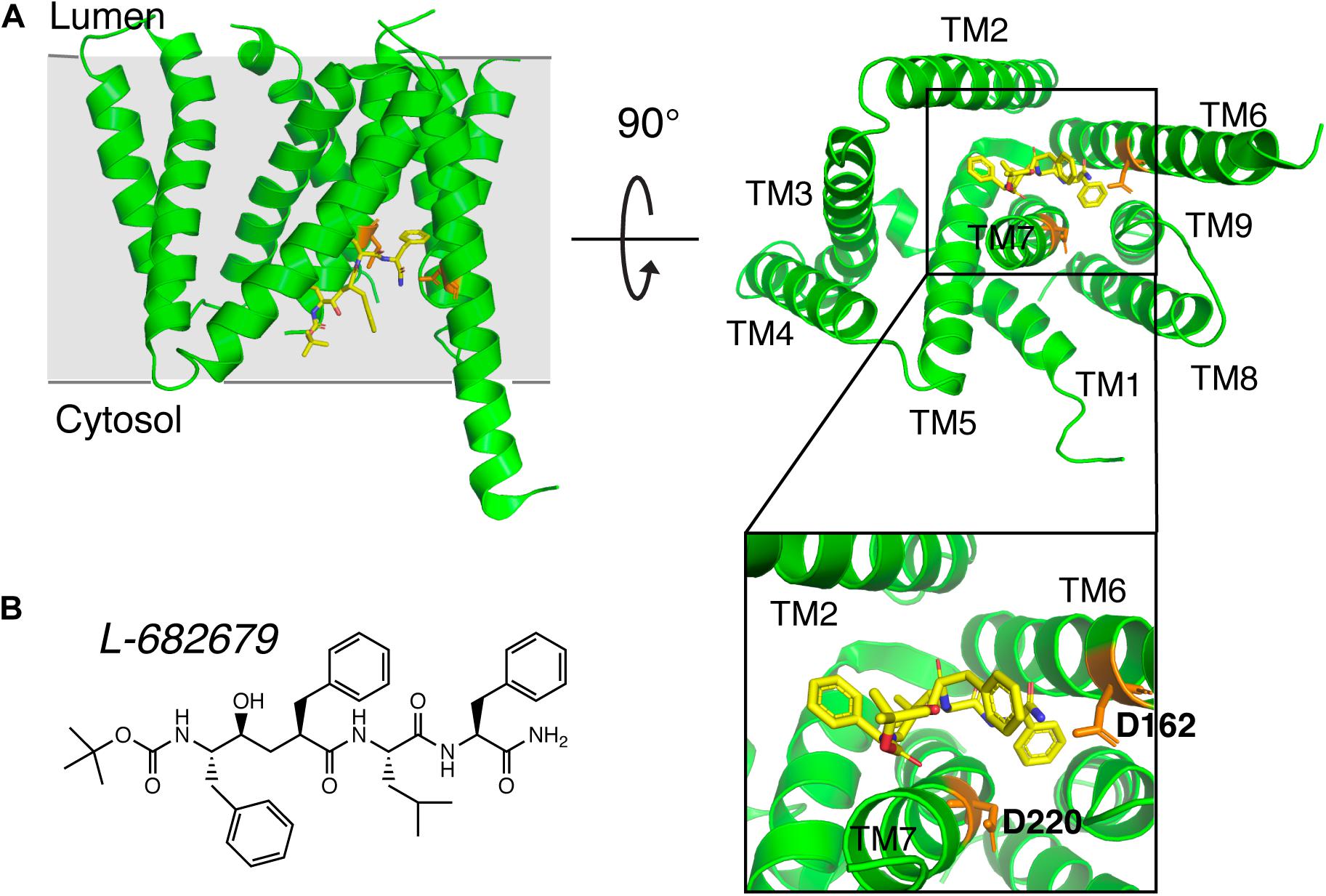
Figure 7. The complex structure of presenilin homolog PSH bound to a hydroxyethylene derivative L-682679. (A) Structure of PSH bound to L-682679. The two catalytic aspartates D162 and D220 were shown in orange. L-682679 was shown in stick model with yellow for carbon, blue for nitrogen, red for oxygen (Dang et al., 2015). (B) Chemical structure of L-682679.
Additional non-peptidyl GSIs were developed, e.g., modification of DAPT led to a much more potent compound LY-411,575, which was further modified to be LY-450,139 (semagacestat, Table 2), a compound that was advanced to phase III clinical trials. However, the phase III clinical trial revealed severe gastrointestinal toxicity and skin cancer, which were probably due to impairment of Notch signaling.
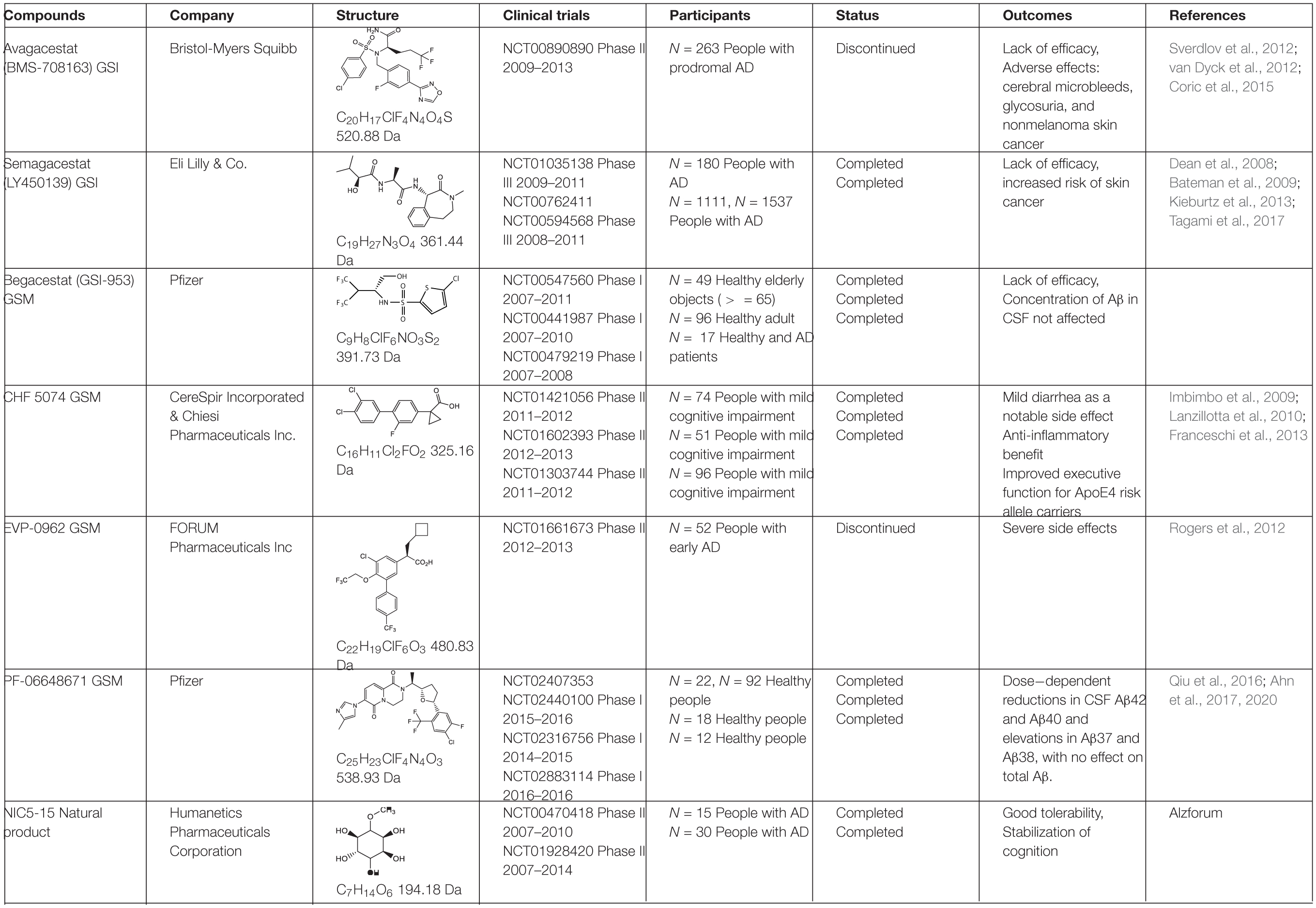
Table 2. γ-secretase inhibitors/modulators in clinical trials.
To avoid the toxicity of GSIs, extensive efforts have been devoted toward selective γ-secretase inhibitors/γ-secretase modulators (GSMs) (Weggen et al., 2001). Some GSMs interact with γ-secretase through the allosteric binding site, and therefore do not interfere with the normal γ-secretase processing of other physiological substrates, such as Notch (called “Notch-sparing GSMs”). The first generation of GSMs is a subset of non-steroidal anti-inflammatory agents (NSAIDs), including ibuprofen, sulindac and indomethacin. These NSAIDs interact with PS at the luminal side and increase the distance between the N- and C- termini of PS, thus direct the cleavage sites toward Aβ38 instead of Aβ42, without altering total Aβ production (Lleó et al., 2004; Takeo et al., 2014). The second generation of GSMs include NSAID-derived carboxylic acid analogs (Beher et al., 2004; Hall et al., 2010) and non-NSAID GSMs (Huang et al., 2012; Sun et al., 2012). The second-generation GSMs have better pharmacokinetic properties, and several of them entered clinical trials, including CHF 5074, EVP-0962, and PF-06648671 (Table 2). Natural products were also screened and characterized for their GSM activities, such as dihydroergocristine (DHEC) (Lei et al., 2015), curcumin (Goozee et al., 2016), and luteolin (Wang et al., 2016). However, the mechanism of how GSMs shift Aβ production is poorly understood, partially because the complexity of the structure of γ-secretase. γ-Secretase contains 4 subunits and at least 18 transmembrane domains, structural information on this high molecular weight complex is limited until recently. Moreover, the binding sites of GSMs are still not understood. GSMs fenofibrate and tarenflurbil were initially reported to modulate γ-secretase cleavage by binding to the substrate APP (Kukar et al., 2008), but the specificity of this binding is questionable (Beel et al., 2009). Other GSMs have been reported to bind presenilin (Crump et al., 2011; Ohki et al., 2011).
γ-Secretase Inhibitors/Modulators in Clinical Trials
The most potent GSI semagacestat (LY450139) failed in Phase III clinical trials with more than 2600 patients from 31 countries, due to worsening cognition and increased risk of skin cancer in test patients comparing to placebo group (Kieburtz et al., 2013; Tagami et al., 2017). The side effects are likely due to the impaired Notch signaling. Focus was then shifted toward “Notch-sparing” GSMs, which shift the Aβ production to shorter and benign peptides without affecting the total Aβ production. However, avagacestat (BMS-708163), which was claimed to be a Notch-sparing GSI, also failed in a Phase II clinical trial (Table 2) with similar side effects to those found with semagacestat (Sverdlov et al., 2012; van Dyck et al., 2012; Coric et al., 2015). Later a lack of Notch selectivity was reported for avagacestat, which may contribute to the failure of clinical trial (Crump et al., 2012). Two more GSMs entered clinical trials. EVP-0962, entered phase II clinical trial in 2012 but no results have ever been reported. NIC5-15, a naturally occurring sugar alcohol found in plants and fruits, also entered phase II clinical trial without follow-up studies.
Discussion
All clinical trials of BACE1 inhibitors and GSIs/GSMs failed, raising the question about causal role of Aβ in AD pathogenesis, and the validity of amyloid cascade hypothesis in general. However, the amyloid hypothesis refers to a continuum of pathological processes at different stages of disease lasting 15–20 years before symptoms appear (Rowe et al., 2010). It is widely accepted that amyloid biomarkers (detected by reduced Aβ42 in cerebrospinal fluid (CSF) and/or positive brain amyloid PET imaging) represent the earliest evidence of AD neuropathologic change currently detectable in humans (Donohue et al., 2014; Young et al., 2014; Xiong et al., 2016). A causative role of Aβ in AD pathogenesis is supported by strong evidences, including genetic of FAD (De Jonghe, 2001; Selkoe, 2001; Chen et al., 2014) and Down’s syndrome (Lejeune et al., 1959; Head et al., 2012), toxicity of Aβ aggregates (Selkoe et al., 2016), Aβ activation of neuron inflammation (Eng et al., 2004) and Aβ potentiation of tau pathology (Jin et al., 2011). Very recently, increased Aβ42/40 ratio was shown to drive tau pathology in 3-dimensional human AD neural cell culture models (Kwak et al., 2020). These studies support the causal upstream role for Aβ in the pathogenesis of AD.
Reducing Aβ42 by modulating β- or γ-secretase activity may inhibit subsequent neurodegenerative changes in the brain. It is clear that Aβ alone is not sufficient for cognitive dysfunction, but it may play a crucial role in potentiating downstream tauopathy and neuroinflammation, converting a cognitively unimpaired preclinical AD subject to a patient with MCI and finally with dementia. With recent advances in ultra-sensitive quantification of pathological proteins in human plasma samples, levels of phosphorylated Tau protein (pTau181) were used to distinguish plasma samples from AD vs. control subjects (Janelidze et al., 2020; Thijssen et al., 2020). For the first time, both Tau and pTau181 can be measured in plasma to predict brain Tau load and neurodegeneration, for monitoring the efficacy of β- or γ-secretase inhibitors/modulators in clinical trials. Existing β-/γ-secretase inhibitors/modulators can then be tested in sub-cohorts selected by AT(N) biomarker.
One plausible explanation for negative clinical results is that BACE1 inhibitors and GSIs/GSMs were given too late, during the irreversible phase of the disease (MCI/prodromal AD, mild-to-moderate AD), and patients need to be treated at earlier stage of AD to prevent neurodegeneration. The phase III trial of BACE1 inhibitor verubecestat by Merck in patients with prodromal AD (MCI) was discontinued in 2018, similar to the phase II/III trial of atabecestat by Janssen.
In addition, side effects and toxicity account for a number of failed trials, such as the inhibition of Notch signaling by GSIs. Recent studies on BACE1 knockout mice suggested that BACE1 inhibitors may disrupt the axonal organization in the hippocampus, and impair synaptic plasticity, leading to defects in learning and memory (Ou-Yang et al., 2018; Lombardo et al., 2019). Inhibition of γ-secretase cleavage can lead to the accumulation of C99 fragment (Lauritzen et al., 2019a), which was correlated to the acceleration of early neurodegenerative process in AD (Lauritzen et al., 2012; Lauritzen et al., 2019b).
An obstacle for the development of selective GSMs is the lack of information about the structural characteristics of γ-secretase-substrate complex. Recent cryo-EM structure of γ-secretase bound to the C83 fragment and Notch substrate (Yang et al., 2019; Zhou et al., 2019) started to fill this crucial knowledge gap. An α-helical to β-strand transition was observed at the C-terminal of APPTM, forming an anti-parallel, intermolecular β-sheet with two induced β-strands from presenilin (Figure 8). The unwinding of C-terminal transmembrane helix exposes the initial ε-cleavage sites (T48, L49) to interact with γ-secretase (Zhou et al., 2019). These are in agreement with earlier Raman and NMR spectroscopic studies carried out on APPTM and PSHs in solution, which showed that substrate binding is coupled with helical unwinding to prime the substrate for peptide bond hydrolysis (Brown et al., 2018; Clemente et al., 2018).
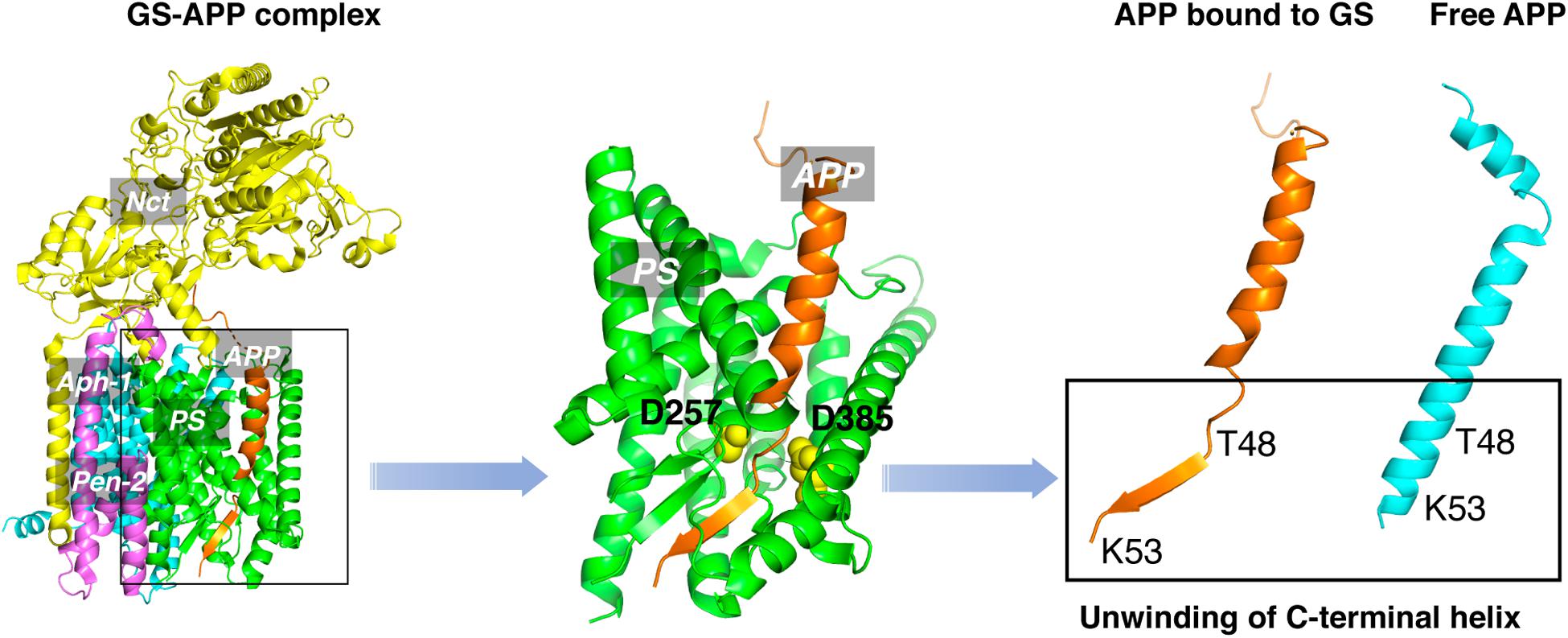
Figure 8. The unwinding model of γ-secretase-APP interaction.
This unwinding model of the substrate in γ-secretase-APP interaction suggests that targeting APP, especially the C-terminal of APPTM, could be a new strategy in selective amyloid reduction. A substrate-specific inhibitor is not expected to affect the γ-secretase cleavage of other physiological substrates, the assembly of the γ-secretase complex, or any presenilin function (Saura et al., 2004; Wines-Samuelson et al., 2010; Watanabe et al., 2012; Barthet et al., 2013), thereby sparing the side effects associated with broad-spectrum γ-secretase inhibitors. A novel compound C1 was found to interact with the C-terminal juxtamembrane lysines of APP and inhibit the γ-secretase production of Aβ both in vitro and in cell (Zhao et al., 2020). This study provides the first in vitro evidence that targeting the C-terminal juxtamembrane lysines is sufficient for reducing Aβ production.
Besides cleavage by the well-known α-, β-, and γ-secretases, APP can be alternatively cleaved by meprin-β, legumain, rhomboid-like protein-4 (RHBDL4), caspases, η-secretase (MT-MMPs) at different sites, resulting in different APP fragments (reviewed by García-González et al., 2019). These emerging proteinases in APP metabolism opened more possibilities for the intervention of amyloidogenic APP processing in AD. For example, cleavage of APP by meprin-β can generate a N-terminally truncated Aβ (Aβ2–X), which is more prone to aggregation than Aβ40; while the inhibitor of meprin-β, fetuin-A, was reduced in CSF of AD patients (Puchades et al., 2003), indicating a potentially pathogenic role of enhanced meprin-β activity in AD. Further investigations are needed to link the new proteinases to AD pathogenesis, which will offer novel insights and more molecular targets in AD drug discovery.
AD is a complex disease and therefore drugs with a single molecular target may not be sufficient for reversing the progression of AD. Thus, multi-target molecules able to inhibit both APP processing and tau pathology, or neuroinflammation etc., may be a promising approach for the treatment of AD. Emerging multi-targeted molecules are developed based on natural products and their derivatives, such as curcumin, berberine, and epigallocatechin gallate (EGCG) (Shan et al., 2011; Di Martino et al., 2016). Multiple AD-related clinical trials were carried out for EGCG, which is a polyphenolic flavonoid extracted from green tea. EGCG has been proposed to not only inhibit Aβ misfolding and tau aggregation in vitro but also increase α-secretase cleavage of APP and improve inflammatory in APP/PS1 transgenic mouse models (Obregon et al., 2006; Ahmed et al., 2017; Guéroux et al., 2017; Ettcheto et al., 2020). While the outcome of clinical trials on EGCG is currently inconclusive, more trials are ongoing and it represents an intriguing multi-target strategy in AD treatment.
Summary
Drug discovery targeting Aβ-producing enzymes is one of the most important strategies to develop disease modifying therapeutics for AD. Exploration of “Notch-sparing” GSMs and inhibitors targeting the substrate APP are two promising future directions. Beyond obvious significance in AD drug discovery, investigation of the γ-secretase and BACE inhibitors will not only provide chemical probes to study fundamental enzymatic mechanisms, but also help settling the debates on whether Aβ is indeed a molecular driver in AD pathogenesis.
Author Contributions
JZ and CW conceived and wrote the manuscript. XL, WX, and YZ edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the Warren Alpert Foundation (to CW) and NIH grant R21-NS109926 (to CW).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahmed, R., Vanschouwen, B., Jafari, N., Ni, X., Ortega, J., and Melacini, G. (2017). Molecular mechanism for the (-)-Epigallocatechin gallate-induced toxic to nontoxic remodeling of Aβ oligomers. J. Am. Chem. Soc. 139, 13720–13734. doi: 10.1021/jacs.7b05012
Ahn, J. E., Carrieri, C., Dela Cruz, F., Fullerton, T., Hajos-Korcsok, E., He, P., et al. (2020). Pharmacokinetic and pharmacodynamic effects of a γ-secretase modulator, PF-06648671, on CSF amyloid-β peptides in randomized Phase I studies. Clin. Pharmacol. Ther. 107, 211–220. doi: 10.1002/cpt.1570
Ahn, J. E., Liu, R., Trapa, P., Wood, K. M., Hajos-Korcsok, E., Fullerton, T., et al. (2017). Pharmacokinetic/Pharmacodynamic (Pk/Pd) effects of Pf-06648671, a novel gamma secretase modulator (GSM), following single and multiple dose administration in healthy volunteers. Alzheimer’s Dement. 13:601. doi: 10.1016/j.jalz.2017.07.244
Albala, B., Kaplow, J. M., Lai, R., Matijevic, M., Aluri, J., and Satlin, A. (2012). CSF amyloid lowering in human volunteers after 14 days’ oral administration of the novel BACE1 inhibitor E2609. Alzheimer’s Dement. 8:S743. doi: 10.1016/j.jalz.2013.08.023
Alzheimer’s_Association (2020). Alzheimer’s Association 2020 Facts and Figures Report. Available online at: https://www.alz.org/alzheimers-dementia/facts-figures (accessed July 24, 2020).
Anderson, J. P., Esch, F. S., Keim, P. S., Sambamurti, K., Lieberburg, I., and Robakis, N. K. (1991). Exact cleavage site of Alzheimer amyloid precursor in neuronal PC-12 cells. Neurosci. Lett. 128, 126–128. doi: 10.1016/0304-3940(91)90775-O
Asai, M., Hattori, C., Iwata, N., Saido, T. C., Sasagawa, N., Szabó, B., et al. (2006). The novel β-secretase inhibitor KMI-429 reduces amyloid β peptide production in amyloid precursor protein transgenic and wild-type mice. J. Neurochem. 96, 533–540. doi: 10.1111/j.1471-4159.2005.03576.x
Bai, X. C., Rajendra, E., Yang, G., Shi, Y., and Scheres, S. H. W. (2015). Sampling the conformational space of the catalytic subunit of human γ-secretase. elife 4:e11182. doi: 10.7554/eLife.11182
Barthet, G., Dunys, J., Shao, Z., Xuan, Z., Ren, Y., Xu, J., et al. (2013). Presenilin mediates neuroprotective functions of ephrinB and brain-derived neurotrophic factor and regulates ligand-induced internalization and metabolism of EphB2 and TrkB receptors. Neurobiol. Aging 34, 499–510. doi: 10.1016/j.neurobiolaging.2012.02.024
Bateman, R. J., Siemers, E. R., Mawuenyega, K. G., Wen, G., Browning, K. R., Sigurdson, W. C., et al. (2009). A γ -secretase inhibitor decreases amyloid-β production in the central nervous system. Ann. Neurol. 66, 48–54. doi: 10.1002/ana.21623
Beel, A. J., Barrett, P., Schnier, P. D., Hitchcock, S. A., Bagal, D., Sanders, C. R., et al. (2009). Nonspecificity of binding of gamma-secretase modulators to the amyloid precursor protein. Biochemistry 48, 11837–11839. doi: 10.1021/bi901839d
Beher, D., Clarke, E. E., Wrigley, J. D. J., Martin, A. C. L., Nadin, A., Churcher, I., et al. (2004). Selected non-steroidal anti-inflammatory drugs and their derivatives target γ-secretase at a novel site: evidence for an allosteric mechanism. J. Biol. Chem. 279, 43419–43426. doi: 10.1074/jbc.M404937200
Benjannet, S., Elagoz, A., Wickham, L., Mamarbachi, M., Munzer, J. S., Basak, A., et al. (2001). Post-translational processing of β-secretase (β -amyloid-converting enzyme) and its ectodomain shedding: the pro- and transmembrane/cytosolic domains affect its cellular activity and amyloid-β production. J. Biol. Chem. 276, 10879–10887. doi: 10.1074/jbc.M009899200
Bennett, B. D., Denis, P., Haniu, M., Teplow, D. B., Kahn, S., Louis, J. C., et al. (2000). A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer’s β-secretase. J. Biol. Chem. 275, 37712–37717. doi: 10.1074/jbc.M005339200
Bernier, F., Sato, Y., Matijevic, M., Desmond, H., McGrath, S., Burns, L., et al. (2013). Clinical study of E2609, a novel BACE1 inhibitor, demonstrates target engagement and inhibition of BACE1 activity in CSF. Alzheimer’s Dement. 9:886. doi: 10.1016/j.jalz.2013.08.244
Berridge, M. J. (2010). Calcium hypothesis of Alzheimer’s disease. Pflugers Arch. Eur. J. Physiol. 459, 441–449. doi: 10.1007/s00424-009-0736-1
Brown, M. C., Abdine, A., Chavez, J., Schaffner, A., Torres-Arancivia, C., Lada, B., et al. (2018). Unwinding of the substrate transmembrane helix in intramembrane proteolysis. Biophys. J. 114, 1579–1589. doi: 10.1016/j.bpj.2018.01.043
Bu, G. (2009). Apolipoprotein e and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344. doi: 10.1038/nrn2620
Burdick, D., Soreghan, B., Kwon, M., Kosmoski, J., Knauer, M., Henschen, A., et al. (1992). Assembly and aggregation properties of synthetic Alzheimer’s A4/β amyloid peptide analogs. J. Biol. Chem. 267, 546–554.
Cai, H., Wang, Y., McCarthy, D., Wen, H., Borchelt, D. R., Price, D. L., et al. (2001). BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat. Neurosci. 4, 233–234. doi: 10.1038/85064
Carter, D. B., Dunn, E., McKinley, D. D., Stratman, N. C., Boyle, T. P., Kuiper, S. L., et al. (2001). Human apolipoprotein E4 accelerates β-amyloid deposition in APPsw transgenic mouse brain. Ann. Neurol. 50, 468–475. doi: 10.1002/ana.1134
Cebers, G., Alexander, R. C., Haeberlein, S. B., Han, D., Goldwater, R., Ereshefsky, L., et al. (2017). AZD3293: pharmacokinetic and pharmacodynamic effects in healthy subjects and patients with Alzheimer’s disease. J. Alzheimer’s Dis. 55, 1039–1053. doi: 10.3233/JAD-160701
Chen, W., Gamache, E., Rosenman, D. J., Xie, J., Lopez, M. M., Li, Y. M., et al. (2014). Familial Alzheimer’s mutations within APPTM increase Aβ42 production by enhancing accessibility of ε-cleavage site. Nat. Commun. 5:3037. doi: 10.1038/ncomms4037
Chévez-Gutiérrez, L., Bammens, L., Benilova, I., Vandersteen, A., Benurwar, M., Borgers, M., et al. (2012). The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 31, 2261–2274. doi: 10.1038/emboj.2012.79
Clemente, N., Abdine, A., Ubarretxena-Belandia, I., and Wang, C. (2018). Coupled transmembrane substrate docking and helical unwinding in intramembrane proteolysis of amyloid precursor Protein. Sci. Rep. 8:12411. doi: 10.1038/s41598-018-30015-6
Coric, V., Salloway, S., Van Dyck, C. H., Dubois, B., Andreasen, N., Brody, M., et al. (2015). Targeting prodromal Alzheimer disease with avagacestat: a randomized clinical trial. JAMA Neurol. 72, 1324–1333. doi: 10.1001/jamaneurol.2015.0607
Creemers, J. W. M., Dominguez, D. I., Plets, E., Serneels, L., Taylor, N. A., Multhaup, G., et al. (2001). Processing of β-secretase by furin and other members of the proprotein convertase family. J. Biol. Chem. 276, 4211–4217. doi: 10.1074/jbc.M006947200
Crump, C. J., Castro, S. V., Wang, F., Pozdnyakov, N., Ballard, T. E., Sisodia, S. S., et al. (2012). BMS-708,163 targets presenilin and lacks notch-sparing activity. Biochemistry 51, 7209–7211. doi: 10.1021/bi301137h
Crump, C. J., Fish, B. A., Castro, S. V., Chau, D. M., Gertsik, N., Ahn, K., et al. (2011). Piperidine acetic acid based γ-secretase modulators directly bind to presenilin-1. ACS Chem. Neurosci. 2, 705–710. doi: 10.1021/cn200098p
Dang, S., Wu, S., Wang, J., Li, H., Huang, M., He, W., et al. (2015). Cleavage of amyloid precursor protein by an archaeal presenilin homologue PSH. Proc. Natl. Acad. Sci. U.S.A. 112, 3344–3349. doi: 10.1073/pnas.1502150112
Das, C., Berezovska, O., Diehl, T. S., Genet, C., Buldyrev, I., Tsai, J. Y., et al. (2003). Designed helical peptides inhibit an intramembrane protease. J. Am. Chem. Soc. 125, 11794–11795. doi: 10.1021/ja037131v
De Jonghe, C. (2001). Pathogenic APP mutations near the gamma-secretase cleavage site differentially affect Abeta secretion and APP C-terminal fragment stability. Hum. Mol. Genet. 10, 1665–1671. doi: 10.1093/hmg/10.16.1665
De Strooper, B. (2003). Aph-1, Pen-2, and nicastrin with presenilin generate an active γ-secretase complex. Neuron 38, 9–12. doi: 10.1109/TASL.2012.2197612
De Strooper, B., Saftig, P., Craessaerts, K., Vanderstichele, H., Guhde, G., Annaert, W., et al. (1998). Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390. doi: 10.1038/34910
Dean, R. A., Farlow, M. R., Galvin, J. E., Peskind, E. R., and Quinn, J. F. (2008). Phase 2 safety trial targeting amyloid NL production with a γ-secretase inhibitor in Alzheimer disease. Arch. Neurol. 65, 1031–1038.
Di Martino, R. M. C., De Simone, A., Andrisano, V., Bisignano, P., Bisi, A., Gobbi, S., et al. (2016). Versatility of the curcumin scaffold: discovery of potent and balanced dual BACE-1 and GSK-3β inhibitors. J. Med. Chem. 59, 531–544. doi: 10.1021/acs.jmedchem.5b00894
Donohue, M. C., Jacqmin-Gadda, H., Le Goff, M., Thomas, R. G., Raman, R., Gamst, A. C., et al. (2014). Estimating long-term multivariate progression from short-term data. Alzheimer’s Dement. 10, S400–S410. doi: 10.1016/j.jalz.2013.10.003
Doran, E., Small, G. W., Lott, I. T., Kim, R., Barrio, J. R., Keator, D., et al. (2016). Down syndrome, partial trisomy 21, and absence of Alzheimer’s disease: the role of APP. J. Alzheimer’s Dis. 56, 459–470. doi: 10.3233/jad-160836
Dorner-Ciossek, C., Hobson, S., Fuchs, K., Sauer, A., Bauer, M., Morales-Ramos, A., et al. (2015). Pharmacological characterization of the new bace1 inhibitor bi 1181181. Alzheimer’s Dement. 11:477. doi: 10.1016/j.jalz.2015.06.529
Dovey, H. F., John, V., Anderson, J. P., Chen, L. Z., De Saint Andrieu, P., Fang, L. Y., et al. (2001). Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J. Neurochem. 76, 173–181. doi: 10.1046/j.1471-4159.2001.00012.x
Duyckaerts, C., Sazdovitch, V., Ando, K., Seilhean, D., Privat, N., Yilmaz, Z., et al. (2018). Neuropathology of iatrogenic Creutzfeldt–Jakob disease and immunoassay of French cadaver-sourced growth hormone batches suggest possible transmission of tauopathy and long incubation periods for the transmission of Abeta pathology. Acta Neuropathol. 135, 201–212. doi: 10.1007/s00401-017-1791-x
Egan, M. F., Kost, J., Tariot, P. N., Aisen, P. S., Cummings, J. L., Vellas, B., et al. (2018). Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 378, 1691–1703. doi: 10.1056/NEJMoa1706441
Eketjäll, S., Janson, J., Kaspersson, K., Bogstedt, A., Jeppsson, F., Fälting, J., et al. (2016). AZD3293: a novel, orally active BACE1 inhibitor with high potency and permeability and markedly slow Off-rate kinetics. J. Alzheimer’s Dis. 50, 1109–1123. doi: 10.3233/JAD-150834
Eng, J. A., Frosch, M. P., Choi, K., Rebeck, G. W., and Greenberg, S. M. (2004). Clinical manifestations of cerebral amyloid angiopathy – related inflammation. Ann. Neurol. 55, 250–256. doi: 10.1002/ana.10810
Erlanson, D. A. (2012). Introduction to fragment-based drug discovery. Top. Curr. Chem. 317, 1–32. doi: 10.1007/128-2011-180
Ettcheto, M., Cano, A., Manzine, P. R., Busquets, O., Verdaguer, E., Castro-Torres, R. D., et al. (2020). Epigallocatechin-3-Gallate (EGCG) improves cognitive deficits aggravated by an obesogenic diet through modulation of unfolded protein response in APPswe/PS1dE9 Mice. Mol. Neurobiol. 57, 1814–1827. doi: 10.1007/s12035-019-01849-6
Fairbanks, M., Leone, J., Emmons, T., Drong, R., Slightom, J., and Winterrowd, G. (1999). Purification and cloning of amyloid precursor protein β-secretase from human brian. Nature 705, 537–540.
Firth, I. E., and Tools, M. H. (2010b). Single oral doses of the novel BACE inhibitor BI 1181181 significantly reduce concentrations of cerebrospinal fluid amyloid-beta peptides in healthy subjects. JALZ 11:740. doi: 10.1016/j.jalz.2015.06.1655
Firth, I. E., and Tools, M. H. (2010a). Pharmacokinetics, pharmacodynamics, and safety of the novel bace inhibitor BI1181181 after oral administration of single ascending doses in healthy subjects. JALZ 11, 740–741. doi: 10.1016/j.jalz.2015.06.1656
Forman, M., Tseng, J., Palcza, J., Leempoels, J., Ramael, S., Krishna, G., et al. (2012). The novel BACE inhibitor MK-8931 dramatically lowers CSF Abeta peptides in healthy subjects: results from a rising single dose study. Neurology 78:PL02.004-PL02.004. doi: 10.1212/WNL.78.1_MeetingAbstracts.PL02.004
Franceschi, M., Casula, D., Bottini, G., Giardino, L., Scarpini, E., Norris, D., et al. (2013). CHF5074 reduces biomarkers of neuroinflammation in patients with mild cognitive impairment: a 12-week, double-blind, placebo- controlled study. Curr. Alzheimer Res. 10, 742–753. doi: 10.2174/13892037113149990144
Gabrielle, S. (2008). Keystone Drug News: coMentis BACE Inhibitor Debuts. Available online at: http://www.alzforum.org/new/detail.asp?id=1790 (accessed April 5, 2018).
García-González, L., Pilat, D., Baranger, K., and Rivera, S. (2019). Emerging alternative proteinases in APP metabolism and alzheimer’s disease pathogenesis: a focus on MT1-MMP and MT5-MMP. Front. Aging Neurosci. 11:244. doi: 10.3389/fnagi.2019.00244
Ghosh, A. K., and Osswald, H. L. (2014). BACE1 (β-Secretase) inhibitors for the treatment of Alzheimer’s disease arun. Chem. Soc. Rev. 43, 6765–6813. doi: 10.1039/c3cs60460h.BACE1
Ghosh, A. K., Shin, D., Downs, D., Koelsch, G., Lin, X., Ermolieff, J., et al. (2000). Design of potent inhibitors for human brain memapsin 2 (β-secretase) [1]. J. Am. Chem. Soc. 122, 3522–3523. doi: 10.1021/ja000300g
Golde, T. E., Estus, S., Younkin, L. H., Selkoe, D. J., Steven, G., Golde, T. E., et al. (1992). Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science 255, 728–730. doi: 10.1126/science.1738847
Goozee, K. G., Shah, T. M., Sohrabi, H. R., Rainey-Smith, S. R., Brown, B., Verdile, G., et al. (2016). Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br. J. Nutr. 115, 449–465. doi: 10.1017/S0007114515004687
Guéroux, M., Fleau, C., Slozeck, M., Laguerre, M., and Pianet, I. (2017). Epigallocatechin 3-Gallate as an inhibitor of Tau phosphorylation and aggregation: a molecular and structural insight. J. Prev. Alzheimer’s Dis. 4, 218–225. doi: 10.14283/jpad.2017.35
Haapasalo, A., and Kovacs, D. M. (2011). The many substrates of presenilin/γ-secretase. J. Alzheimer’s Dis. 25, 3–28. doi: 10.3233/JAD-2011-101065.The
Hall, A., Elliott, R. L., Giblin, G. M. P., Hussain, I., Musgrave, J., Naylor, A., et al. (2010). Piperidine-derived γ-secretase modulators. Bioorgan. Med. Chem. Lett. 20, 1306–1311. doi: 10.1016/j.bmcl.2009.08.072
Hansen, M. M., Jarmer, D. J., Arslantas, E., Debaillie, A. C., Frederick, A. L., Harding, M., et al. (2015). Synthesis of BACE inhibitor LY2886721. Part II. isoxazolidines as precursors to chiral aminothiazines, selective peptide coupling, and a controlled reactive crystallization. Organ. Process Res. Dev. 19:150828123638003. doi: 10.1021/op500327t
Head, E., Powell, D., Gold, B. T., and Schmitt, F. A. (2012). Alzheimer’s disease in down syndrome. Eur. J. Neurodegener. Dis. 1, 353–364.
Henstridge, C. M., Hyman, B. T., and Spires-Jones, T. L. (2019). Beyond the neuron–cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 20, 94–108. doi: 10.1038/s41583-018-0113-1
Higaki, J., Quon, D., Zhong, Z., and Cordell, B. (1995). Inhibition of β-amyloid formation identifies proteolytic precursors and subcellular site of catabolism. Neuron 14, 651–659. doi: 10.1016/0896-6273(95)90322-4
Higaki, J. N., Chakravarty, S., Bryant, C. M., Cowart, L. R., Harden, P., Scardina, J. M., et al. (1999). A combinatorial approach to the identification of dipeptide aldehyde inhibitors of β-amyloid production. J. Med. Chem. 42, 3889–3898. doi: 10.1021/jm990009f
Hong, L., Koelsch, G., Lin, X., Wu, S., Terzyan, S., Ghosh, A. K., et al. (2000). Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science 290, 150–153. doi: 10.1126/science.290.5489.150
Hong, L., Turner, R. T., Koelsch, G., Shin, D., Ghosh, A. K., and Tang, J. (2002). Crystal structure of memapsin 2 (β-secretase) in complex with an inhibitor OM00-3. Biochemistry 41, 10963–10967. doi: 10.1021/bi026232n
Huang, X., Zhou, W., Liu, X., Li, H., Sun, G., Mandal, M., et al. (2012). Synthesis and SAR studies of fused oxadiazines as γ-secretase modulators for treatment of Alzheimer’s disease. ACS Med. Chem. Lett. 3, 931–935. doi: 10.1021/ml300209g
Hussain, I., Hawkins, J., Harrison, D., Hille, C., Wayne, G., Cutler, L., et al. (2007). Oral administration of a potent and selective non-peptidic BACE-1 inhibitor decreases β-cleavage of amyloid precursor protein and amyloid-β production in vivo. J. Neurochem. 100, 802–809. doi: 10.1111/j.1471-4159.2006.04260.x
Hussain, I., Powell, D., Howlett, D. R., Tew, D. G., Meek, T. D., Chapman, C., et al. (1999). Identification of a novel aspartic protease (Asp 2) as β-secretase. Mol. Cell. Neurosci. 14, 419–427. doi: 10.1006/mcne.1999.0811
Imbimbo, B. P., Hutter-Paier, B., Villetti, G., Facchinetti, F., Cenacchi, V., Volta, R., et al. (2009). CHF5074, a novel γ-secretase modulator, attenuates brain β-amyloid pathology and learning deficit in a mouse model of Alzheimer’s disease. Br. J. Pharmacol. 156, 982–993. doi: 10.1111/j.1476-5381.2008.00097.x
Inhibitor, B., Hypopigmentation, L., Neill, B. T. O., Beck, E. M., Butler, C. R., Nolan, C. E., et al. (2018). Design and synthesis of clinical candidate PF-06751979: a potent, brain penetrant, β-Site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitor lacking hypopigmentation. J. Med. Chem. 61, 4476–4504. doi: 10.1021/acs.jmedchem.8b00246
Inhibitor, B., Part, L. Y., Asymmetric, I. A., Kolis, S. P., Hansen, M. M., Arslantas, E., et al. (2015). Synthesis of BACE inhibitor LY2886721. Part I. An asymmetric nitrone cycloaddition strategy. Org. Process Res. Dev. 19, 1203–1213. doi: 10.1021/op500351q
Iserloh, U., Pan, J., Stamford, A. W., Kennedy, M. E., Zhang, Q., Zhang, L., et al. (2008). Discovery of an orally efficaceous 4-phenoxypyrrolidine-based BACE-1 inhibitor. Bioorganic Med. Chem. Lett. 18, 418–422. doi: 10.1016/j.bmcl.2007.10.053
Jacobs, H. I. L., Hedden, T., Schultz, A. P., Sepulcre, J., Perea, R. D., Amariglio, R. E., et al. (2018). Structural tract alterations predict downstream tau accumulation in amyloid-positive older individuals. Nat. Neurosci. 21, 424–431. doi: 10.1038/s41593-018-0070-z
Janelidze, S., Mattsson, N., Palmqvist, S., Smith, R., Beach, T. G., Serrano, G. E., et al. (2020). Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, and longitudinal progression to Alzheimer’s dementia. Nat. Med 26, 379–386. doi: 10.1038/s41591-020-0755-1
Jeppsson, F., Eketjäll, S., Janson, J., Karlström, S., Gustavsson, S., Olsson, L. L., et al. (2012). Discovery of AZD3839, a potent and selective BACE1 inhibitor clinical candidate for the treatment of alzheimer disease. J. Biol. Chem. 287, 41245–41257. doi: 10.1074/jbc.M112.409110
Jin, M., Shepardson, N., Yang, T., Chen, G., Walsh, D., and Selkoe, D. J. (2011). Soluble amyloid -protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 5819–5824. doi: 10.1073/pnas.1017033108
Jurisch-Yaksi, N., Sannerud, R., and Annaert, W. (2013). A fast growing spectrum of biological functions of γ-secretase in development and disease. Biochim. Biophys. Acta Biomembr. 1828, 2815–2827. doi: 10.1016/j.bbamem.2013.04.016
Kennedy, M. E., Stamford, A. W., Chen, X., Cox, K., Cumming, J. N., Dockendorf, M. F., et al. (2016). The BACE1 inhibitor verubecestat (MK-8931) reduces CNS b-Amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 8:363ra150. doi: 10.1126/scitranslmed.aad9704
Kieburtz, K., He, F., Sun, X., Thomas, R. G., Ph, D., and Aisen, P. S. (2013). A Phase 3 trial of semagacestat for treatment of Alzheimer’s disease. 369, 341–350. doi: 10.1056/NEJMoa1210951
Kimura, T., Shuto, D., Hamada, Y., Igawa, N., Kasai, S., Liu, P., et al. (2005). Design and synthesis of highly active Alzheimer’s β-secretase (BACE1) inhibitors, KMI-420 and KMI-429, with enhanced chemical stability. Bioorganic Med. Chem. Lett. 15, 211–215. doi: 10.1016/j.bmcl.2004.09.090
Klafki, H. W., Abramowski, D., Swoboda, R., Paganetti, P. A., and Staufenbiel, M. (1996). The carboxyl termini of β-amyloid peptides 1-40 and 1-42 are generated by distinct γ-secretase activities. J. Biol. Chem. 271, 28655–28659. doi: 10.1074/jbc.271.45.28655
Kokubo, H., Kayed, R., Glabe, C. G., and Yamaguchi, H. (2005). Soluble Aβ oligomers ultrastructurally localize to cell processes and might be related to synaptic dysfunction in Alzheimer’s disease brain. Brain Res. 1031, 222–228. doi: 10.1016/j.brainres.2004.10.041
Korbel, J. O., Tirosh-Wagner, T., Urban, A. E., Chen, X.-N., Kasowski, M., Dai, L., et al. (2009). The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc. Natl. Acad. Sci. U.S.A. 106, 12031–12036. doi: 10.1073/pnas.0813248106
Kornilova, A. Y., Bihel, F., Das, C., and Wolfe, M. S. (2005). The initial substrate-binding site of γ-secretase is located on presenilin near the active site. Proc. Natl. Acad. Sci. U.S.A. 102, 3230–3235. doi: 10.1073/pnas.0407640102
Kukar, T. L., Ladd, T. B., Bann, M. A., Fraering, P. C., Narlawar, R., Maharvi, G. M., et al. (2008). Substrate-targeting γ-secretase modulators. Nature 453, 925–929. doi: 10.1038/nature07055
Kwak, S. S., Washicosky, K. J., Brand, E., von Maydell, D., Aronson, J., Kim, S., et al. (2020). Amyloid-β42/40 ratio drives tau pathology in 3D human neural cell culture models of Alzheimer’s disease. Nat. Commun. 11:1377.
Lai, R. Y., Darpo, B., Dayal, S., Hall, N., Chang, M.-K., Albala, B., et al. (2017). Elenbecestat, a novel oral bace inhibitor, has no clinically meaningful effect on Qtc interval up to a supratherapeutic dose of 200 Mg. Alzheimer’s Dement. 13, 250–251. doi: 10.1016/j.jalz.2017.06.110
Lanzillotta, A., Sarnico, I., Benarese, M., Branca, C., Baiguera, C., Hutter-Paier, B., et al. (2010). The γ-secretase modulator CHF5074 reduces the accumulation of native hyperphosphorylated Tau in a transgenic mouse model of Alzheimer’s disease. J. Mol. Neurosci. 45, 22–31. doi: 10.1007/s12031-010-9482-2
Lauritzen, I., Bécot, A., Bourgeois, A., Pardossi-Piquard, R., Biferi, M. G., Barkats, M., et al. (2019a). Targeting γ-secretase triggers the selective enrichment of oligomeric APP-CTFs in brain extracellular vesicles from Alzheimer cell and mouse models. Transl. Neurodegener. 8:35. doi: 10.1186/s40035-019-0176-6
Lauritzen, I., Pardossi-Piquard, R., Bauer, C., Brigham, E., Abraham, J. D., Ranaldi, S., et al. (2012). The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J. Neurosci. 32, 16243–16255. doi: 10.1523/JNEUROSCI.2775-12.2012
Lauritzen, I., Pardossi-Piquard, R., Bourgeois, A., Bécot, A., and Checler, F. (2019b). Does intraneuronal accumulation of carboxyl-terminal fragments of the amyloid precursor protein trigger early neurotoxicity in Alzheimer’s Disease? Curr. Alzheimer Res. 16, 453–457. doi: 10.2174/1567205016666190325092841
Lei, X., Yu, J., Niu, Q., Liu, J., Fraering, P. C., and Wu, F. (2015). The FDA-approved natural product dihydroergocristine reduces the production of the Alzheimer’s disease amyloid-β peptides. Sci. Rep. 5:16541. doi: 10.1038/srep16541
Lejeune, J., Gautier, M., and Turpin, R. (1959). Study of the somatic chromosomes of nine mongoloid children. C. R. Hebd. Seances Acad. Sci. 248, 1721–1722.
Lin, X., Koelsch, G., Wu, S., Downs, D., Dashti, A., and Tang, J. (2000). Human aspartic protease memapsin 2 cleaves the beta -secretase site of beta -amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 97, 1456–1460. doi: 10.1073/pnas.97.4.1456
Lleó, A., Berezovska, O., Herl, L., Raju, S., Deng, A., Bacskai, B. J., et al. (2004). Nonsteroidal anti-inflammatory drugs lower Aβ42 and change presenilin 1 conformation. Nat. Med. 10, 1065–1066. doi: 10.1038/nm1112
Lombardo, S., Chiacchiaretta, M., Tarr, A., Kim, W. H., Cao, T., Sigal, G., et al. (2019). BACE1 partial deletion induces synaptic plasticity deficit in adult mice. Sci. Rep. 9:19877. doi: 10.1038/s41598-019-56329-7
Lynch, S. Y., Kaplow, J., Zhao, J., Dhadda, S., Luthman, J., and Albala, B. (2018). Elenbecestat, E2609, a bace inhibitor: results from a Phase-2 study in subjects with mild cognitive impairment and mild-to-moderate dementia due to Alzheimer’S disease. Alzheimer’s Dement. 14:1623. doi: 10.1016/j.jalz.2018.07.213
Martenyi, F., Lowe, S., Dean, R. A., Monk, S. A., Gonzales, C. R., Friedrich, S., et al. (2010). Central and peripheral pharmacokinetic and pharmacodynamic effects of the & beta;-site APP cleavage enzyme (BACE1) inhibitor LY2811376 In humans. Alzheimer’s Dement. 6:e48. doi: 10.1016/j.jalz.2010.08.148
May, P. C., Boggs, L. N., Yang, Z., Lindstrom, T., Calligaro, D., Citron, M., et al. (2010). Central and peripheral pharmacodynamic effects of BACE1 inhibition following oral administration of LY2811376 to PDAPP mice and beagle dog. Alzheimer’s Dement. 6, S590–S591. doi: 10.1016/j.jalz.2010.05.2010
May, P. C., Dean, R. A., Lowe, S. L., Martenyi, F., Sheehan, S. M., Boggs, L. N., et al. (2011). Robust central reduction of amyloid- in humans with an orally available, non-peptidic -secretase inhibitor. J. Neurosci. 31, 16507–16516. doi: 10.1523/JNEUROSCI.3647-11.2011
May, P. C., Willis, B. A., Lowe, S. L., Dean, R. A., Monk, S. A., Cocke, P. J., et al. (2015). The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. J. Neurosci. 35, 1199–1210. doi: 10.1523/JNEUROSCI.4129-14.2015
Mckinzie, D. L., May, P. C., Boggs, L. N., Yang, Z., Brier, R. A., Monk, S. A., et al. (2016). Nonclinical pharmacological characterization of The BACE1 inhibitor LY3202626. Alzheimer’s Dement. 12, 432–433. doi: 10.1016/j.jalz.2016.06.828
Merck (2018). Merck Announces Discontinuation of APECS Study Evaluating Verubecestat (MK-8931) for the Treatment of People with Prodromal Alzheimer’s Disease. Available online at: https://investors.merck.com/news/press-release-details/2018/Merck-Announces-Discontinuation-of-APECS-Study-Evaluating-Verubecestat-MK-8931-for-the-Treatment-of-People-with-Prodromal-Alzheimers-Disease/default.aspx (accessed February 13, 2018).
Morohashi, Y., Kan, T., Tominari, Y., Fuwa, H., Okamura, Y., Watanabe, N., et al. (2006). C-terminal fragment of presenilin is the molecular target of a dipeptidic γ-secretase-specific inhibitor DAPT (N-[N-(3,5-difluorophenacetyl)-L- alanyl]-S-phenylglycine t-butyl ester). J. Biol. Chem. 281, 14670–14676. doi: 10.1074/jbc.M513012200
Neumann, U., Rueeger, H., MacHauer, R., Veenstra, S. J., Lueoend, R. M., Tintelnot-Blomley, M., et al. (2015). A novel BACE inhibitor NB-360 shows a superior pharmacological profile and robust reduction of amyloid-β and neuroinflammation in APP transgenic mice. Mol. Neurodegener. 10, 1–15. doi: 10.1186/s13024-015-0033-8
Neumann, U., Ufer, M., Jacobson, L. H., Rouzade−Dominguez, M., Huledal, G., Kolly, C., et al. (2018). The BACE−1 inhibitor CNP520 for prevention trials in Alzheimer’s disease. EMBO Mol. Med. 10:e9316. doi: 10.15252/emmm.201809316
Obregon, D. F., Rezai-Zadeh, K., Bai, Y., Sun, N., Hou, H., Ehrhart, J., et al. (2006). ADAM10 activation is required for green tea (-)-epigallocatechin-3-gallate- induced α-secretase cleavage of amyloid precursor protein. J. Biol. Chem. 281, 16419–16427. doi: 10.1074/jbc.M600617200
Ohki, Y., Higo, T., Uemura, K., Shimada, N., Osawa, S., Berezovska, O., et al. (2011). Phenylpiperidine-type γ-secretase modulators target the transmembrane domain 1 of presenilin 1. EMBO J. 30, 4815–4824. doi: 10.1038/emboj.2011.372
Ou-Yang, M. H., Kurz, J. E., Nomura, T., Popovic, J., Rajapaksha, T. W., Dong, H., et al. (2018). Axonal organization defects in the hippocampus of adult conditional BACE1 knockout mice. Sci. Transl. Med. 10:eaao5620. doi: 10.1126/scitranslmed.aao5620
Park, J., Wetzel, I., Marriott, I., Dréau, D., D’Avanzo, C., Kim, D. Y., et al. (2018). A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 21, 941–951. doi: 10.1038/s41593-018-0175-4
Patel, S., Vuillard, L., Cleasby, A., Murray, C. W., and Yon, J. (2004). Apo and inhibitor complex structures of BACE (β-secretase). J. Mol. Biol. 343, 407–416. doi: 10.1016/j.jmb.2004.08.018
Puchades, M., Hansson, S. F., Nilsson, C. L., Andreasen, N., Blennow, K., and Davidsson, P. (2003). Proteomic studies of potential cerebrospinal fluid protein markers for Alzheimer’s disease. Mol. Brain Res. 118, 140–146. doi: 10.1016/j.molbrainres.2003.08.005
Qiu, R., Liu, R., He, P., Wills, A., Tankisheva, E., Alvey, C., et al. (2017). Tolerability, and pharmacokinetic and pharmacodynamic effects of PF-06751979, a potent and selective oral BACE1 inhibitor: results from Phase1 single and multiple ascending dose studies in healthy young and older volunteers. Alzheimer’s Dement. J. Alzheimer’s Assoc. 13, 575–576. doi: 10.1016/j.jalz.2017.07.196
Qiu, R., Liu, R., Wills, A., He, P., Leurent, C., Hajos-korcsok, E., et al. (2016). PF-06648671-A novel gamma secretase modulator: safety, tolerability, pharmacokinetics, and effects on plasma amyloid-β Levels following single oral ascending doses in healthy volunteers. Alzheimer’s Dement. 12, 611–612. doi: 10.1016/j.jalz.2016.06.1213
Rogers, K., Felsenstein, K. M., Hrdlicka, L., Tu, Z., Albayya, F., Lee, W., et al. (2012). Modulation of γ -secretase by EVP-0015962 reduces amyloid deposition and behavioral deficits in Tg2576 mice. Mol. Neurodegen. 7:61. doi: 10.1186/1750-1326-7-61
Rowe, C. C., Ellis, K. A., Rimajova, M., Bourgeat, P., Pike, K. E., Jones, G., et al. (2010). Amyloid imaging results from the Australian imaging, biomarkers and lifestyle (AIBL) study of aging. Neurobiol. Aging 31, 1275–1283. doi: 10.1016/j.neurobiolaging.2010.04.007
Sakamoto, K., Matsuki, S., Matsuguma, K., Yoshihara, T., Uchida, N., Azuma, F., et al. (2017). BACE1 inhibitor lanabecestat (AZD3293) in a Phase 1 study of healthy Japanese subjects: pharmacokinetics and effects on plasma and cerebrospinal fluid Aβ peptides. J. Clin. Pharmacol. 57, 1460–1471. doi: 10.1002/jcph.950
Sano, Y., Morishima-Kawashima, M., Funamoto, S., Ihara, Y., Nagashima, Y., Ishihara, S., et al. (2009). γ-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of -Carboxyl terminal fragment. J. Neurosci. 29, 13042–13052. doi: 10.1523/jneurosci.2362-09.2009
Saura, C. A., Choi, S. Y., Beglopoulos, V., Malkani, S., Zhang, D., Rao, B. S. S., et al. (2004). Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42, 23–36. doi: 10.1016/S0896-6273(04)00182-5
Scott, J. D., Li, S. W., Brunskill, A. P. J., Chen, X., Cox, K., Cumming, J. N., et al. (2016). Discovery of the 3-Imino-1,2,4-thiadiazinane 1,1-dioxide derivative verubecestat (MK-8931)-A β-site amyloid precursor protein cleaving enzyme 1 inhibitor for the treatment of Alzheimer’s disease. J. Med. Chem. 59, 10435–10450. doi: 10.1021/acs.jmedchem.6b00307
Selkoe, D. J., Hardy, J., Sciences, B., Hu, N.-W., Nicoll, A. J., Zhang, D., et al. (2016). Amyloid β-protein dimers isolated directly from Alzheimer brains impair synaptic plasticity and memory. Nat. Med. 7:3374. doi: 10.1038/nm1782.Amyloid
Selkoe, D. J., and Wolfe, M. S. (2007). Presenilin: running with scissors in the membrane. Cell 131, 215–221. doi: 10.1016/j.cell.2007.10.012
Shah, S., Lee, S. F., Tabuchi, K., Hao, Y. H., Yu, C., LaPlant, Q., et al. (2005). Nicastrin functions as a γ-secretase-substrate receptor. Cell 122, 435–447. doi: 10.1016/j.cell.2005.05.022
Shan, W. J., Huang, L., Zhou, Q., Meng, F. C., and Li, X. S. (2011). Synthesis, biological evaluation of 9-N-substituted berberine derivatives as multi-functional agents of antioxidant, inhibitors of acetylcholinesterase, butyrylcholinesterase and amyloid-β aggregation. Eur. J. Med. Chem. 46, 5885–5893. doi: 10.1016/j.ejmech.2011.09.051
Shearman, M. S., Beher, D., Clarke, E. E., Lewis, H. D., Harrison, T., Hunt, P., et al. (2000). L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid β-protein precursor γ-secretase activity. Biochemistry 39, 8698–8704. doi: 10.1021/bi0005456
Shuto, D., Kasai, S., Kimura, T., Liu, P., Hidaka, K., Hamada, T., et al. (2003). KMI-008, a novel β-Secretase inhibitor containing a hydroxymethylcarbonyl isostere as a transition-state mimic: design and synthesis of substrate-based octapeptides. Bioorganic Med. Chem. Lett. 13, 4273–4276. doi: 10.1016/j.bmcl.2003.09.053
Sims, J. R., Selzler, K. J., Downing, A. M., Willis, B. A., Aluise, C. D., Zimmer, J., et al. (2017). Development review of the BACE1 inhibitor lanabecestat (AZD3293/LY3314814). J. Prev. Alzheimer’s Dis. 4, 247–254. doi: 10.14283/jpad.2017.38
Sisodia, S. S. (1992). Secretion of the β−amyloid precursor protein. Ann. N. Y. Acad. Sci. 674, 53–57. doi: 10.1111/j.1749-6632.1992.tb27476.
Stancu, I.-C., Vasconcelos, B., Terwel, D., and Dewachter, I. (2014). Models of beta-amyloid induced Tau-pathology: the long and folded road to understand the mechanism Molecular Neurodegeneration. Mol. Neurodegener. 9:51. doi: 10.1186/1750-1326-9-51
Sun, Z. Y., Asberom, T., Bara, T., Bennett, C., Burnett, D., Chu, I., et al. (2012). Cyclic hydroxyamidines as amide isosteres: discovery of oxadiazolines and oxadiazines as potent and highly efficacious γ-secretase modulators in vivo. J. Med. Chem. 55, 489–502. doi: 10.1021/jm201407j
Sverdlov, O., Berman, R. M., Castaneda, L., Li, H., Albright, C. F., Wong, O., et al. (2012). Multicenter, randomized, double-blind, placebo-controlled, single-ascending dose study of the oral γ-secretase inhibitor BMS-708163 (Avagacestat): tolerability profile, pharmacokinetic parameters, and pharmacodynamic markers. Clin. Ther. 34, 654–667. doi: 10.1016/j.clinthera.2012.01.022
Szaruga, M., Munteanu, B., Lismont, S., Veugelen, S., Horré, K., Mercken, M., et al. (2017). Alzheimer’s-causing mutations shift Aβ length by Destabilizing γ-secretase-Aβn interactions. Cell 170:443–456.e14. doi: 10.1016/j.cell.2017.07.004
Tagami, S., Yanagida, K., Kodama, T. S., Takami, M., Mizuta, N., Oyama, H., et al. (2017). Semagacestat is a pseudo-inhibitor of γ-secretase. Cell Rep. 21, 259–273. doi: 10.1016/j.celrep.2017.09.032
Takasugi, N., Tomita, T., Hayashi, I., Tsuruoka, M., Niimura, M., Takahashi, Y., et al. (2003). The role of presenilin cofactors in the gamma-secretase complex. Nature 422, 438–441. doi: 10.1038/nature01506
Takeo, K., Tanimura, S., Shinoda, T., Osawa, S., Zahariev, I. K., Takegami, N., et al. (2014). Allosteric regulation of -secretase activity by a phenylimidazole-type -secretase modulator. Proc. Natl. Acad. Sci. U.S.A. 111, 10544–10549. doi: 10.1073/pnas.1402171111
Thijssen, E. H., La Joie, R., Wolf, A., Strom, A., Wang, P., Iaccarino, L., et al. (2020). Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat. Med. 26, 387–397.
Thinakaran, G., Borchelt, D. R., Lee, M. K., Slunt, H. H., Spitzer, L., Kim, G., et al. (1996). Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17, 181–190. doi: 10.1016/S0896-6273(00)80291-3
Thinakaran, G., and Koo, E. H. (2008). Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619. doi: 10.1074/jbc.R800019200
Timmers, M., Streffer, J. R., Russu, A., Tominaga, Y., Shimizu, H., Shiraishi, A., et al. (2018). Pharmacodynamics of atabecestat (JNJ-54861911), an oral BACE1 inhibitor in patients with early Alzheimer’s disease: randomized, double-blind, placebo-controlled study. Alzheimer’s Res. Ther. 10, 1–18. doi: 10.1186/s13195-018-0415-6
Timmers, M., Van Broeck, B., Ramael, S., Slemmon, J., De Waepenaert, K., Russu, A., et al. (2016). Profiling the dynamics of CSF and plasma Aβ reduction after treatment with JNJ-54861911, a potent oral BACE inhibitor. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2, 202–212. doi: 10.1016/j.trci.2016.08.001
Ufer, M., Rouzade-Dominguez, M.-L., Huledal, G., Pezous, N., Avrameas, A., David, O., et al. (2016). Results from a first-in-human study with the bace inhibitor Cnp520. Alzheimer’s Dement. 12:200. doi: 10.1016/j.jalz.2016.06.351
van Dyck, C. H., Brody, M., Salloway, S., Rollin, L., Pilcher, G., Berman, R. M., et al. (2012). Safety and tolerability of the γ-secretase inhibitor avagacestat in a Phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 69:1430. doi: 10.1001/archneurol.2012.2194
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Rogers, G., et al. (1999). β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. doi: 10.1126/science.286.5440.735
Veugelen, S., Saito, T., Saido, T. C., Chávez-Gutiérrez, L., and De Strooper, B. (2016). Familial Alzheimer’s disease mutations in presenilin generate amyloidogenic Aβ peptide seeds. Neuron 90, 410–416. doi: 10.1016/j.neuron.2016.03.010
Wang, H., Wang, H., Cheng, H., and Che, Z. (2016). Ameliorating effect of luteolin on memory impairment in an Alzheimer’s disease model. Mol. Med. Rep. 13, 4215–4220. doi: 10.3892/mmr.2016.5052
Watanabe, H., Xia, D., Kanekiyo, T., Kelleher, R. J., and Shen, J. (2012). Familial frontotemporal dementia-associated presenilin-1 c.548G>T mutation causes decreased mRNA expression and reduced presenilin function in knock-in mice. J. Neurosci. 32, 5085–5096. doi: 10.1523/jneurosci.0317-12.2012
Weggen, S., Eriksen, J. L., Das, P., Sagi, S. A., Wang, R., Pietrzik, C. U., et al. (2001). A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414, 212–216. doi: 10.1038/35102591
Wilcox, K. C., Lacor, P. N., Pitt, J., and Klein, W. L. (2011). Aβ oligomer-induced synapse degeneration in Alzheimer’s disease. Cell. Mol. Neurobiol. 31, 939–948. doi: 10.1007/s10571-011-9691-4
Wildsmith, K. R., Holley, M., Savage, J. C., Skerrett, R., and Landreth, G. E. (2013). Evidence for impaired amyloid β clearance in Alzheimer’s disease. Alzheimer’s Res. Ther. 5:33. doi: 10.1186/alzrt187
Willis, B. A., Lowe, S. L., Daugherty, L. L., Dean, R. A., English, B., Ereshefsky, L., et al. (2016). Pharmacokinetics, pharmacodynamics, safety, and tolerability of Ly3202626, a novel Bace1 inhibitor, in healthy subjects and patients with Alzheimer’S disease. Alzheimer’s Dement. 12:418. doi: 10.1016/j.jalz.2016.06.791
Wilson, C. A., Doms, R. W., and Lee, V. M. Y. (1999). Intracellular APP processing and Aβ production in Alzheimer disease. J. Neuropathol. Exp. Neurol. 58, 787–794. doi: 10.1097/00005072-199908000-00001
Wimo, A., Winblad, B., and Jönsson, L. (2010). The worldwide societal costs of dementia: estimates for 2009. Alzheimer’s Dement. 6, 98–103. doi: 10.1016/j.jalz.2010.01.010
Wines-Samuelson, M., Schulte, E. C., Smith, M. J., Aoki, C., Liu, X., Kelleher, R. J., et al. (2010). Characterization of age-dependent and progressive cortical neuronal degeneration in Presenilin conditional mutant mice. PLoS One 5:e10195. doi: 10.1371/journal.pone.0010195
Wolfe, M. S. (2006). The γ-secretase complex: membrane-embedded proteolytic ensemble. Biochemistry 45, 7931–7939. doi: 10.1021/bi060799c
Wolfe, M. S., and Kopan, R. (2004). Intramembrane proteolysis: theme and variations. Science 305, 1119–1123. doi: 10.1126/science.1096187
Wolfe, M. S., Xia, W., Ostaszewski, B. L., Diehl, T. S., Kimberly, W. T., and Selkoe, D. J. (1999). Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 398, 513–517. doi: 10.1038/19077
Xiong, C., Jasielec, M. S., Weng, H., Fagan, A. M., Benzinger, T. L. S., Head, D., et al. (2016). Longitudinal relationships among biomarkers for Alzheimer disease in the adult children study. Neurology 86, 1499–1506. doi: 10.1212/WNL.0000000000002593
Yan, R., Blenkowski, M. J., Shuck, M. E., Miao, H., Tory, M. C., Pauley, A. M., et al. (1999). Membrane-anchored aspartyl protease with Alzheimer’s disease β- secretase activity. Nature 402, 533–537. doi: 10.1038/990107
Yang, G., Zhou, R., Zhou, Q., Guo, X., Yan, C., Ke, M., et al. (2019). Structural basis of Notch recognition by human γ-secretase. Nature 565, 192–197. doi: 10.1038/s41586-018-0813-8
Young, A. L., Oxtoby, N. P., Daga, P., Cash, D. M., Fox, N. C., Ourselin, S., et al. (2014). A data-driven model of biomarker changes in sporadic Alzheimer’s disease. Brain 137, 2564–2577. doi: 10.1093/brain/awu176
Zhao, J., Xiao, Y., Liu, X., Kim, S., Wu, X., Barros, M., et al. (2020). Substrate interaction inhibits γ-secretase production of amyloid-β peptides. Chem. Commun. 56, 2578–2581. doi: 10.1039/c9cc09170j
Keywords: Alzheimer’s disease, amyloid-β, β-secretase inhibitor, γ-secretase inhibitors, γ-secretase modulator, clinical trial
Citation: Zhao J, Liu X, Xia W, Zhang Y and Wang C (2020) Targeting Amyloidogenic Processing of APP in Alzheimer’s Disease. Front. Mol. Neurosci. 13:137. doi: 10.3389/fnmol.2020.00137
Received: 04 May 2020; Accepted: 08 July 2020;
Published: 04 August 2020.
Edited by:
Maria Rosário Almeida, Universidade do Porto, PortugalReviewed by:
Homira Behbahani, Karolinska Institutet (KI), SwedenHong Qing, Beijing Institute of Technology, China
Copyright © 2020 Zhao, Liu, Xia, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunyu Wang, wangc5@rpi.edu