 Xiang Song
Xiang Song Zhiyong Yu
Zhiyong Yu- Department of Breast Cancer Center, Shandong Cancer Hospital and Institute, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, China
Gefitinib resistance in triple negative breast cancer (TNBC) is a growing important concern. Glutathione peroxidase 4 (GPX4) is a main regulator of ferroptosis, which is pivotal for TNBC cell growth. We investigated GPX4-mediated ferroptosis in gefitinib sensitivity in TNBC. Gefitinib resistant TNBC cells MDA-MB-231/Gef and HS578T/Gef were constructed and treated with lentivirus sh-GPX4 and ferroptosis inhibitor ferrostatin-1. GPX4 expression, cell viability and apoptosis were detected. Malondialdehyde (MDA), glutathione (GSH), reactive oxygen species (ROS) levels were evaluated. The levels of ferroptosis-related proteins were detected. Subcutaneous tumor model was established in nude mice, and gefitinib was intraperitoneally injected to evaluate tumor growth, apoptosis, and Ki-67 expression. GPX4 was increased in gefitinib-resistant cells. After silencing GPX4, the inhibition rate of cell viability was increased, the limitation of colony formation ability was reduced, apoptosis rate was increased, and the sensitivity of cells to gefitinib was improved. After silencing GPX4, MDA and ROS production were increased, while GSH was decreased. Silencing GPX4 promoted ferroptosis. Inhibition of GPX4 promoted gefitinib sensitivity by promoting cell ferroptosis. In vivo experiments also revealed that inhibition of GPX4 enhanced the anticancer effect of gefitinib through promoting ferroptosis. Overall, inhibition of GPX4 stimulated ferroptosis and enhanced TNBC cell sensitivity to gefitinib.
Introduction
Breast cancer (BC) is the most common female tumor and leading cause of death among women (1). Triple-negative breast cancer (TNBC) accounts for about 20% of BC cases (2). TNBC is a distinct subtype of BC characterized by aggressive phenotype, high recurrence rates, high visceral metastasis rate, and worse outcomes (3–5). Molecular heterogeneity is prominent in the TNBC subtype, and is reflected by the obvious prognostic and patient’s sensitivity to chemotherapy treatment, which is the only available systemic therapy currently (1). Although TNBC patients benefit from standard chemotherapy, they still face a high recurrence rate and a high possibility of drug resistance, leaving TNBC a major challenge in the clinic (4, 6). Clinical trial data show that gefitinib is well tolerated in patients with a wide range of tumor types (7). Gefitinib, an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, has shown both anti-proliferative and anti-tumoral activity in BC (8, 9). However, MDAMB-231 cells are resistant to clinically relevant doses of geftinib (10). Therefore, an understanding of the cellular targets of gefitinib will allow the discovery of biomarkers for predicting outcomes and provide information for overcoming gefitinib resistance in TNBC.
Targeting EGFR kinase activity by gefitinib insufficiently inhibits TNBC cell proliferation (11). Therefore, some synergistic interactions with other genes or drugs are paramount. Ferroptosis is a distinct type of cell death from the traditional apoptosis and necrosis, which is an iron-dependent cell death initiated and progressed by lipid peroxide accumulation and reactive oxygen species (ROS) production (12, 13). Ferroptosis is implicated in various diseases, including neurodegenerative diseases and cancers (14). Additionally, siramesine, a lysosome disrupting agent, and lapatinib, an EGFR inhibitor, elicit ferroptosis in a synergistic manner in BC cell lines by altering iron regulation (15). TNBC cells are reportedly sensitive to erastin-induced ferroptosis (16).
Furthermore, ferroptosis is specifically activated by missing glutathione peroxidase 4 (GPX4) activities (17). GPX4, an antioxidant defense enzyme is functional to repair oxidative damage to lipids and is a leading inhibitor of ferroptosis (18). Ferroptosis is featured with GSH depletion, disrupted GPX4 redox defense, inflammation, and detrimental lipid ROS formation (19, 20). However, whether GPX4-mediated ferroptosis is promising for gefitinib resistance in TNBC has not been reported. Therefore, we determined the role of GPX4-mediated ferroptosis in the mechanism of gefitinib resistance in TNBC cells, which could provide a new theoretical basis for the protective effects of GPX4 and contribute to developing effective drugs for TNBC in the future.
Methods
Treatment of Gef-Resistant Cells
Human TNBC cell lines MDA-MB-231 (ATCC® CRM-HTB-26™) and HS578T (ATCC® HTB-126™) (ATCC, VA, USA) were cultured in Dulbecco’s modified eagle’s medium (DMEM, 30-2002, ATCC) at 37℃ with 10% fetal bovine serum (FBS, 16000044, GIBCO, NY, USA). In order to construct Gef-resistant cells, MDA-MB-231 and HS578T cells were cultured in the medium with increasing concentration of gefitinib for 11 months [the highest concentration of gefitinib was 3 μM (21)]. Finally, the Gef-resistant cells MDA-MB-231/Gef and HS578T/Gef were stored in the medium containing 3 μM gefitinib (22). Gefitinib was purchased from MedChemExpress (HY-50895, Shanghai, China).
The Gef-resistant cells were assigned into sh-NC group (treated with lentivirus small hairpin RNA-negative control), sh-GPX4 group (treated with lentivirus sh-GPX4), sh-NC + DMSO group (treated with sh-NC + dimethyl sulphoxide), sh-GPX4 + DMSO (treated with lentivirus sh-GPX4 + DMSO), and sh-GPX4 + Ferrostatin-1 group (treated with lentivirus sh-GPX4 and 1 μM Ferrostatin-1) (23). Ferrostatin-1 is an inhibitor of ferroptosis and purchased from MedChemExpress (HY-100579). The titer of lentivirus was 1× 108 Tu/ml.
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was extracted using RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and reverse transcribed into cDNA using the reverse transcription kit (RR047A, Takara, Japan). RT-qPCR was performed using SYBR® Premix Ex Taq™ II (Perfect Real Time) kit (DRR081, Takara, Japan) and real-time fluorescent qPCR instrument (ABI 7500, ABI, Foster City, CA, USA). The amplification procedures were pre-denaturation at 95°C for 30 s, and 40 cycles of 95°C 5 s, and 60°C 34 s. Each sample is provided with three duplicated wells. The primers were synthesized by Sangon Biotech (Shanghai) Co., Ltd. (Shanghai, China) (Table 1). Using GAPDH as an internal parameter, the relative expression of GPX4 was calculated by 2-ΔΔCT method (24).

Table 1 Primer sequences for RT-qPCR.
Western Blot Analysis
Total protein was extracted using strong radio-immunoprecipitation assay lysis buffer (Boster Biological Technology Co., Ltd, Wuhan, Hubei, China), and the protein concentration was detected using a bicinchoninic acid protein assay kit (Boster). The protein was separated via 10% electrophoresis, transferred to polyvinylidene fluoride membranes, and blocked with 5% bovine serum albumin for 2 h to block the nonspecific binding. Afterward, the membranes underwent an overnight incubation with diluted primary antibodies GPX4 (ab125066, 1:2,000, Abcam, MA, USA), Ki-67 (ab16667, 1:1,000, Abcam), proliferating cell nuclear antigen (PCNA) (ab92552, 1:5,000, Abcam), Cleaved caspase-3 (ab2302, 1:1,000, Abcam), Cleaved caspase-9 (ab2324, 1:1,000, Abcam), acyl-CoA synthetase 4 (ACSL4) (ab155282, 1:10,000, Abcam), cyclooxygenase-2 (PTGS2) (ab15191, 1:1,000, Abcam), nicotinamide-adenine dinucleotide phosphate (NADPH) oxidase 1 (NOX1) (ab55831, 1:1,000, Abcam), ferritin heavy chain 1 (FTH1) (ab75972, 1:2,000, Abcam), and rabbit polyclonal β-actin (ab8227, 1:2,000, Abcam) at 4°C. Following washings, the membranes were incubated for 1 h with horseradish peroxidase (HRP)-labeled goat anti-rabbit secondary antibody immunoglobulin G (ab205718, 1:2,000, Abcam). The protein was developed using an enhanced chemiluminescence working solution (Millipore, Billerica, MA, USA). Image Pro Plus 6.0 (media cybernetics, USA) was used to quantify the gray level of each band, and β-actin was used as an internal reference. Each experiment was repeated three times (25).
Cell Counting Kit-8 (CCK-8) Assay
After 48 h of transfection, the cells were seeded in 96-well plates at 0.4×104 cells/well. Then the cells were treated with different doses of gefitinib (0, 2, 4, 6, 8, and 10 μM) for 48 h. CCK-8 assay kit (HY-K0301, MedChemExpress) was used to detect cell viability at 450 nm. The average inhibition rate of cell activity at each concentration was calculated (22, 23).
Colony Formation Assay
After 48 h of transfection, 1,000 cells were seeded in 24-well plates. As previously described (26), the colonies were observed by crystal violet staining after 2 weeks of culture.
Flow Cytometry
Annexin V FITC/PI double staining was utilized for apoptosis detection. After 48 h of transfection, cells were adjusted to 1×106 cells/well. Cells were fixed overnight with 70% precooled ethanol solution at 4°C, and then 100 μl cell suspension (no less than 106 cells/ml) was resuspended in 200 μl binding buffer. Then, the cells were stained with 10 μl Annexin V-FITC and 5 μl PI for 15 min in the dark. After the addition of 300 μl binding buffer, apoptosis at 488 nm was measured by flow cytometry, with 2×104 cells each time (27).
Determination of Malondialdehyde (MDA) and Glutathione (GSH)
MDA level and GSH level were measured using the Lipid Peroxidation Assay Kit (ab118970, Abcam) and the Glutathione Assay Kit (CS0260, Sigma, St. Louis, MO, USA), respectively.
Assessment of ROS
After 48 hours of transfection, the cells were seeded in 6-well plates. After 8 h, the cells were stained with 20 μM 2’,7’-dichlorofluorescein diacetates (Beyotime Biotechnology, Shanghai, China) in the dark at 37°C for 30 min. The ROS level in the cells was observed by a fluorescence microscope (28).
JC-1 Mitoscreen Assay
At 24 h after transfection, mitochondrial membrane potential (MMP) assay kit with JC-1 (C2006, Beyotime) was used to detect the MMP. The cells were imaged with a fluorescence microscope (28).
Determination of Intracellular Iron Concentration
According to the instructions of the manufacturer of the iron ion kit (MAK025, Sigma), the cells were added into the buffer for iron determination. After mixing, the cells were centrifuged at 4°C at 13,000 g for 10 min to obtain the supernatant. At 25°C, 50 μl supernatant and 50 μl buffer were incubated in 96-well microplate for 30 min. Then the mixture was incubated with 200 μl reagent mixture in the dark at 25°C for 30 min, and then the absorbance value at 593 nm was measured to calculate the iron ion level.
Xenograft Tumors in Nude Mice
Six-week-old female BALB nude (nu/nu) mice were purchased from Hunan SJA laboratory animal Co., Ltd. (Changsha, China; n = 40) and kept in a specific pathological free environment. In order to induce tumor in vivo, BALB/C nude mice were randomly allocated into four groups: sh-NC, sh-NC + Gef, sh-GPX4, and sh-GPX4 + Gef, with 10 mice in each group. Stable transfected cell lines were constructed by lentivirus infection. About 2×106 cells were suspended in 200 μl phosphate-buffered saline (PBS). These cells were injected subcutaneously into the right hind leg of nude mice. Seven days after subcutaneous injection, gefitinib (10 mg/kg, once every other day) was intraperitoneally injected into sh-NC + Gef group and sh-GPX4 + Gef group for 2 weeks. On the 28th day of subcutaneous injection, all tumors were collected, and the following tests were carried out.
TUNEL Staining
The tumor tissue was fixed with 4% paraformaldehyde, dehydrated and cleared, and then paraffin embedded. The tumor tissue was sliced into 5 μm sections for staining. After dewaxing, the tissue sections were treated with conventional histological methods. To block peroxidase in tissues, the samples were placed in ethanol solution containing 3% hydrogen peroxide for 15 min. After washing, the samples were incubated with protease K for 20 min. After washing, the slides were incubated with the reaction solution of TUNEL staining kit. Then samples were incubated with 2,4-diaminobutyric acid (DAB) solution for 15 min, washed with PBS, and stained with hematoxylin. Finally, cells with brown nuclei were assessed as TUNEL-positive cells (29, 30).
Immunohistochemistry
The parraffined sections were deparaffinized and hydrated, and the endogenous peroxidase activity was quenched by incubation in 0.3% H2O2 at 37°C for 30 min. After washing with PBS, the tissue sections were boiled in 10 mmol/L citrate buffer (pH 6.0) at 100°C for 30 min. Once cooled to room temperature, sections were sealed with 5% normal goat serum at 37°C for 1 h, followed by an overnight incubation with Ki-67 (ab16667, 1:200, Abcam) at 4°C. After washing three times in PBS, the tissue sections were incubated with the secondary antibody IgG (ab205718; 1:2,000) at 37°C for 1 h. After washing in PBS for three times, the tissue sections were then incubated with HRP-bound streptomyces avidin (1:1,000) at 37°C for 45 min. After that, the sections were stained with freshly prepared DAB. All tissue sections were counterstained with hematoxylin. Finally, the stained sections were analyzed under Olympus BX51 microscope (31).
Statistical Analysis
SPSS 21.0 (IBM Corp., Armonk, NY, USA) was applied for data analysis. The measurement data are expressed in the form of mean ± standard deviation. Firstly, the normal distribution and variance homogeneity tests were conducted. If the data conformed to the normal distribution and homogeneity of variance, then the t test was used for comparison analysis between two groups. One-way or two-way analysis of variance (ANOVA) was used for comparison analysis among multiple groups, followed by Tukey’s multiple comparisons test. If the data did not conform to the normal distribution or homogeneity of variance, the rank sum test was carried out. The p <0.05 meant statistically significant.
Results
Gefitinib Induced GPX4 Expression in Gef-Resistant TNBC Cells
TNBC cell lines MDA-MB-231 and HS578T were selected to construct Gef-resistant strains. The levels of GPX4 were detected by RT-qPCR and Western blot. The results showed that the expression of GPX4 in the resistant strains was higher than that in the sensitive strains (Figures 1A, B) (p < 0.05). This indicated that gefitinib can induce the expression of GPX4 in TNBC cell lines MDA-MB-231 and HS578T.
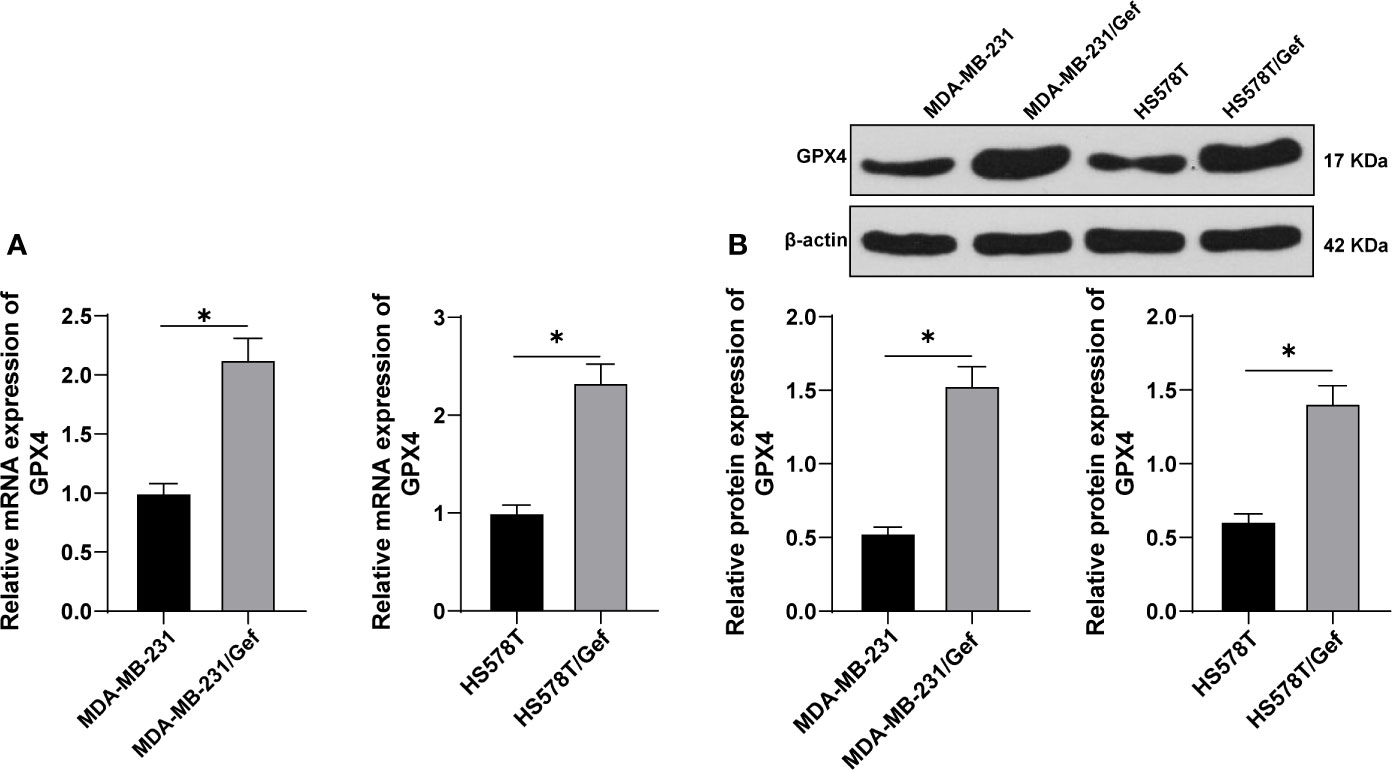
Figure 1 Gefitinib induced GPX4 expression in Gef-resistant TNBC cells. (A, B), The levels of GPX4 were detected by RT-qPCR and Western blot. The cell experiment was repeated 3 times. The data were expressed as mean ± standard deviation and analyzed by the t test. *p < 0.05.
GPX4 Inhibition Increased Gefitinib Sensibility
To detect the relationship between GPX4 and gefitinib sensitivity, we silenced GPX4 in gefitinib resistant cell lines MDA-MB-231 and HS578T. RT-qPCR showed that GPX4 expression in sh-GPX4 group was lower than that in sh-NC group (Figure 2A). CCK-8 and colony formation assay were used to detect the proliferation of gefitinib resistant cell lines after silencing GPX4. With the increase of gefitinib concentration, the inhibition rate of cell activity increased significantly. Compared with sh-NC group, sh-GPX4 group significantly increased the cell activity inhibition rate (Figure 2B), and decreased cell colony formation (Figure 2C). Then we measured the apoptosis rate by flow cytometry (Figure 2D), and utilized Western blot to measure proliferation-related proteins Ki-67 and PCNA and apoptosis-related proteins Cleaved caspase-3 and Cleaved caspase-9 (Figure 2E) (all p < 0.05). Compared with sh-NC group, the apoptosis rate of sh-GPX4 group was clearly increased, the levels of Ki-67 and PCNA were significantly decreased, and the levels of Cleaved caspase-3 and Cleaved caspase-9 was increased in sh-GPX4 group. These results suggested that inhibition of GPX4 can increase the sensitivity of TNBC cell lines to gefitinib.
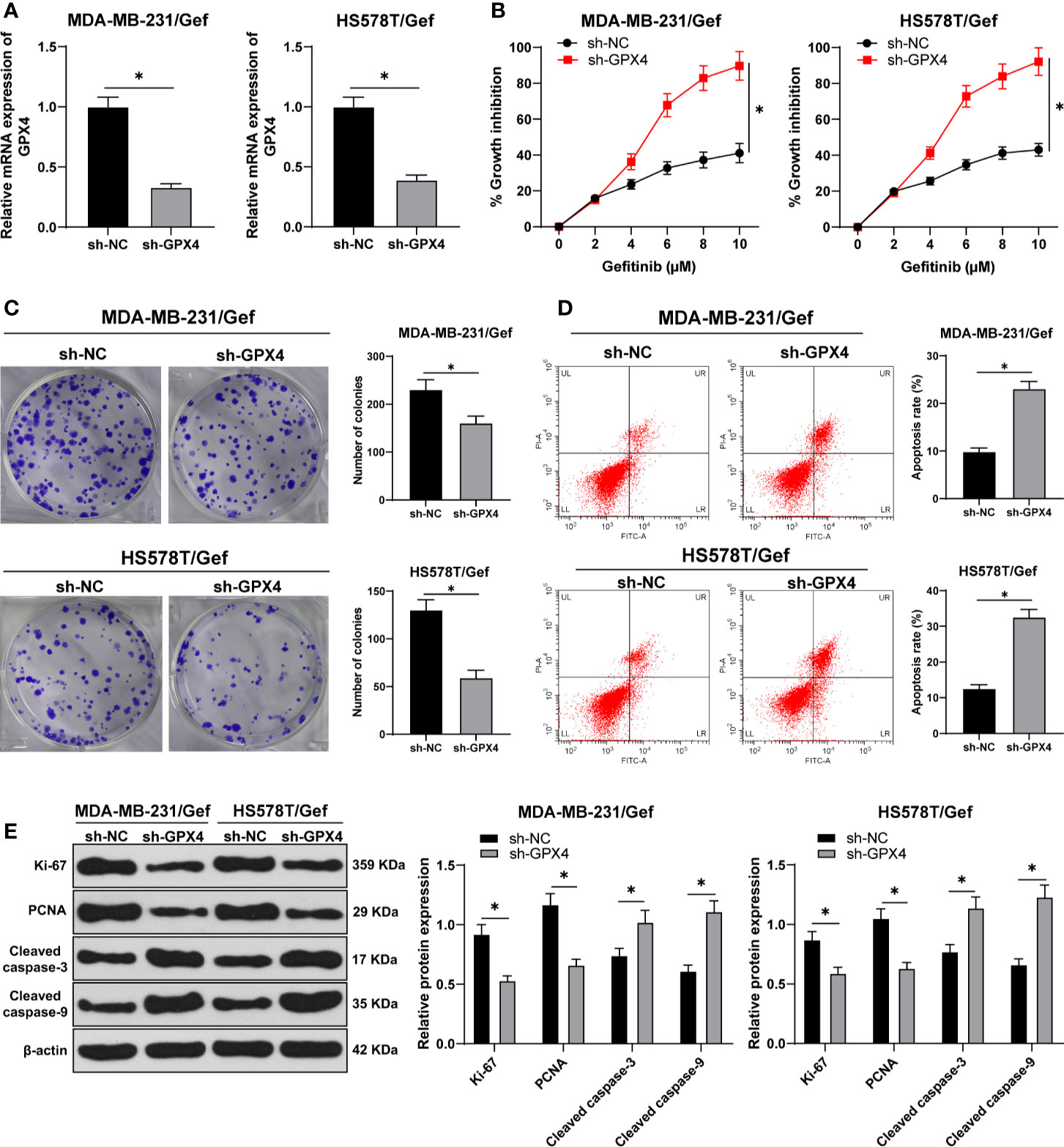
Figure 2 Inhibition of GPX4 can increase the sensitivity of TNBC cell lines to gefitinib. (A) The expression of GPX4 was detected by RT-qPCR; (B) CCK-8 was used to detect the inhibition rate of cell viability after different concentrations of gefitinib (0, 2, 4, 6, 8, 10 μm); (C) Colony formation assay was used to detect the ability of cell clone formation; (D) Flow cytometry was used to detect the apoptosis; (E) Western blot was used to detect the expression of Ki-67, PCNA, Cleaved caspase-3, and Cleaved caspase-9. The cell experiment was repeated 3 times. The data were expressed as mean ± standard deviation and analyzed by the t test (panels A/C/D) and two-way ANOVA (panels B/E), followed by Tukey’s multiple comparisons test. *p < 0.05.
GPX4 Negatively Regulated Ferroptosis in Gef-Resistant Cells
Ferroptosis is very important for the survival of TNBC cells; ferroptosis is an iron dependent non apoptotic cell death, which is closely related to GPX4 (28). We first found that compared with sh-NC group, the MDA level was significantly increased after GPX4 silencing (Figure 3A), and GSH level was decreased (Figure 3B). Furthermore, we observed that ROS production increased significantly after GPX4 silencing (Figure 3C) and MMP reduced (Figure 3D). After that, we detected the intracellular iron concentration. Compared with sh-NC group, the intracellular iron concentration was significantly increased after GPx4 silencing (Figure 3E). We detected the levels of ferroptosis-related proteins ACSL4, PTGS2, NOX1, and FTH1 by Western blot. Compared with sh-NC group, the levels of ACSL4, PTGS2, and Nox1 were notably upregulated, while FTH1 levels were significantly downregulated in the sh-GPX4 group (Figures 3F, G) (all p < 0.05). The above results showed the same trend in MDA-MB-231/Gef and HS578T/Gef cells, indicating that inhibition of GPX4 can promote ferroptosis in gefitinib resistant cells.
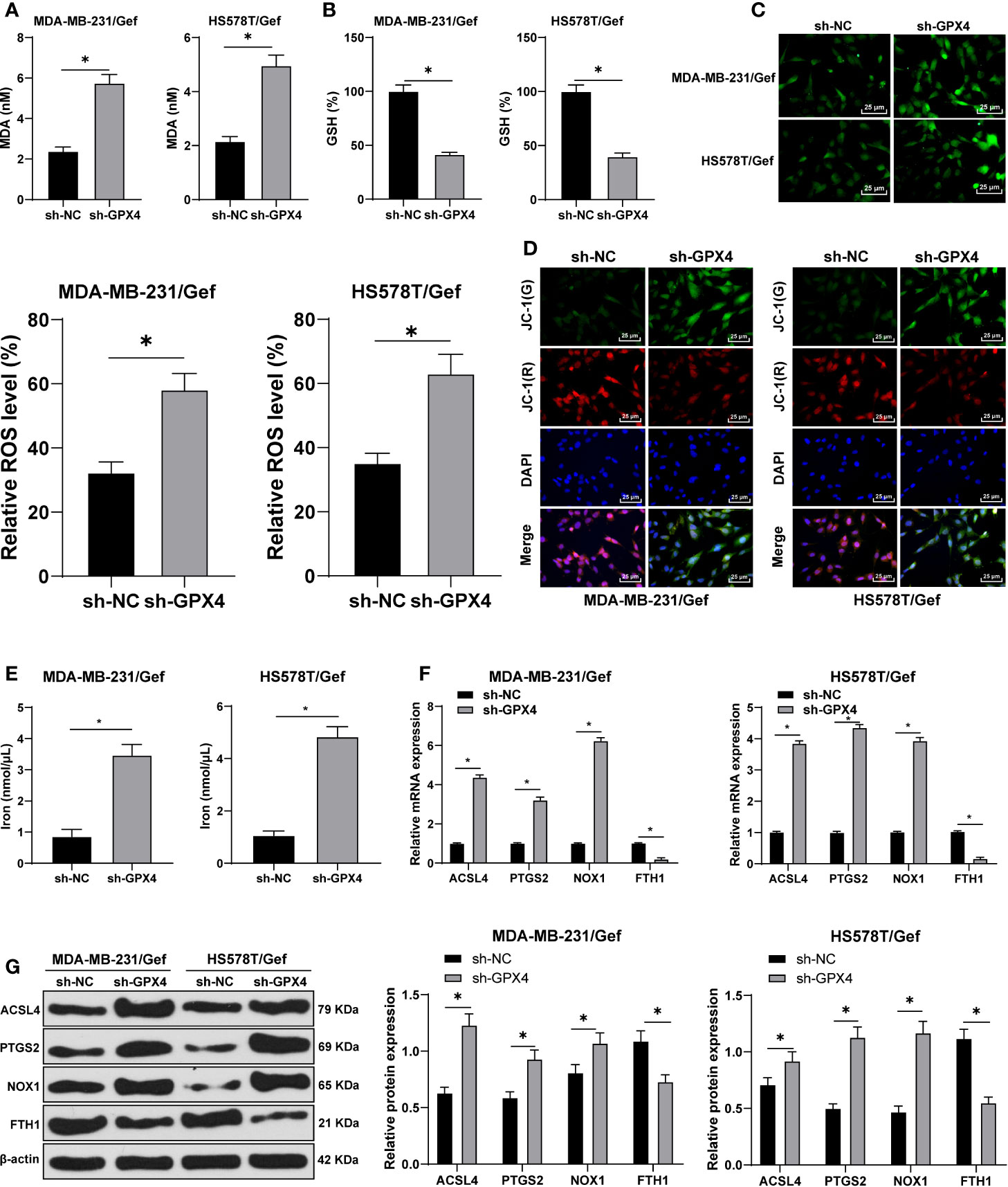
Figure 3 Inhibition of GPX4 can promote ferroptosis in gefitinib resistant cells. (A) MDA level in MDA-MB-231 and HS578T cells detected by a kit; (B) GSH level in MDA-MB-231 and HS578T cells detected by a kit; (C) ROS production was detected by fluorescence microscope (left) and fluorescence quantitative analysis (right), 400 ×, scale bar = 25 μM; (D) mitochondrial membrane potential test, 200 ×, scale bar = 50 μm; (E) the intracellular iron concentration was detected by iron ion kit; (F, G) RT-qPCR and Western blot detected the levels of ferroptosis-related proteins ACSl4, PTGS2, NOX1, and FTH1. The cell experiment was repeated 3 times. The data were expressed as mean ± standard deviation and analyzed by the t test (panels A/B/C) and two-way ANOVA (E) followed by Tukey’s multiple comparisons test. *p < 0.05.
GPX4 Inhibition Increased Gefitinib Sensibility by Promoting Ferroptosis
To further confirm that the regulation of gefitinib sensitivity by GPX4 is achieved by affecting ferroptosis, we silenced GPX4 in TNBC cell line MDA-MB-231 and used ferrostatin-1 to inhibit ferroptosis. GPX4 levels were detected by RT-qPCR and Western blot. The results showed that GPX4 levels were effectively reduced after silencing GPX4, but partially restored by ferrostatin-1 (Figures 4A, B). After silencing GPX4, MDA level increased significantly (Figure 4C), GSH level decreased (Figure 4D), and ROS production increased (Figure 4E) (all p < 0.05). However, inhibition of ferroptosis by ferrostatin-1 partially reversed the effect of sh-GPX4. These results further indicated that GPX4 can negatively regulate ferroptosis.
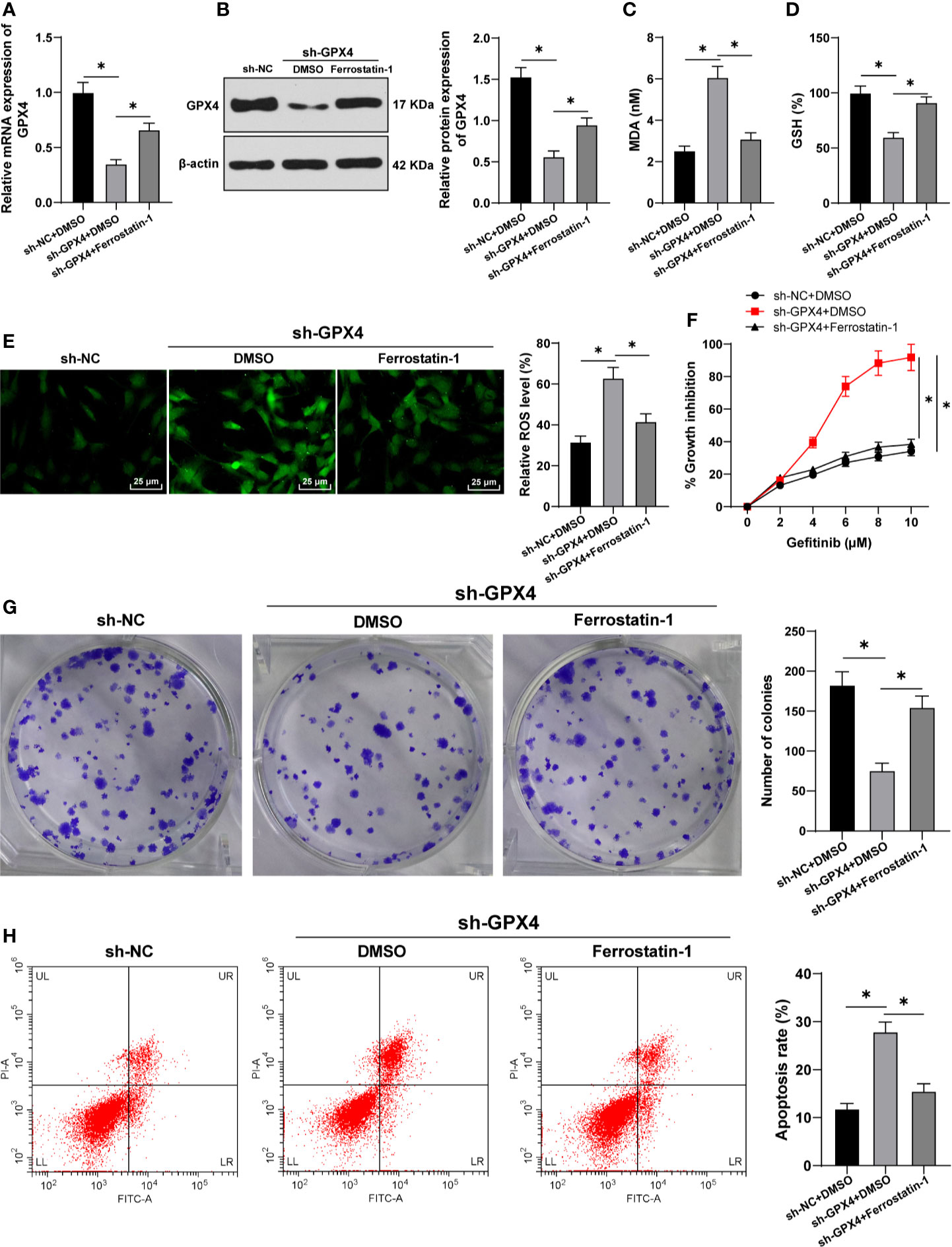
Figure 4 GPX4 inhibition increased gefitinib sensibility by promoting ferroptosis. While silencing GPX4 in TNBC cell line MDA-MB-231, ferrostatin-1 was used to inhibit ferroptosis. (A, B) The levels of GPX4 were detected by RT-qPCR and Western blot; (C) MDA level in MDA-MB-231 and HS578T cells detected by a kit; (D) GSH level in MDA-MB-231 and HS578T cells detected by a kit; (E) ROS production was detected by fluorescence microscope (left) and fluorescence quantitative analysis (right), 400 ×, scale bar = 25 μM; (F) CCK-8 was used to detect the inhibition rate of cell viability after different concentrations of gefitinib (0, 2, 4, 6, 8, 10 μm); (G) Colony formation assay was used to detect the ability of cell clone formation; (H) Flow cytometry was used to detect the apoptosis. The cell experiment was repeated 3 times. The data were expressed as mean ± standard deviation and analyzed by one-way ANOVA (A–E, G–H) and two-way ANOVA (F) followed by Tukey’s multiple comparisons test. *p < 0.05.
Then, we detected the cell activity by CCK-8 and colony formation assays. The inhibition rate of cell activity was clearly increased after silencing GPX4, while the effect of sh-GPX4 was partially reversed by ferrostatin-1 (Figures 4F, G). In addition, flow cytometry showed that the apoptosis rate was increased after silencing GPX4, and ferrostatin-1 partially reversed the promoting effect of sh-GPX4 on apoptosis (Figure 4H) (all p < 0.05). All these results suggested that inhibition of GPX4 promoted gefitinib sensitivity by promoting cell ferroptosis.
GPX4 Inhibition Increased the Anti-Tumor Effect of Gefitinib by Promoting Ferroptosis
After constructing the subcutaneous tumor model in nude mice, sh-GPX4 treatment significantly reduced the growth rate of tumor in nude mice, and further increased the inhibitory effect of gefitinib on tumor (Figure 5A). Gefitinib promoted GPX4 expression, while sh-GPX4 treatment effectively inhibited GPX4 expression (Figure 5B). After treatment with gefitinib alone or silencing GPX4 alone, MDA was significantly increased, while GSH was decreased. Silencing GPX4 could further enhance the regulation of gefitinib on MDA and GSH (Figures 5C, D). Finally, TUNEL staining and immunohistochemistry were used to detect the apoptosis and Ki-67 expression. The results showed that gefitinib alone or silencing GPX4 alone notably increased the apoptosis rate and decreased Ki-67 expression. After gefitinib treatment and silencing GPX4, the apoptosis rate was further increased, and Ki-67 expression was further decreased (Figures 5E, F). All in all, inhibition of GPX4 enhanced the anticancer effect of gefitinib in vivo by promoting cell ferroptosis.
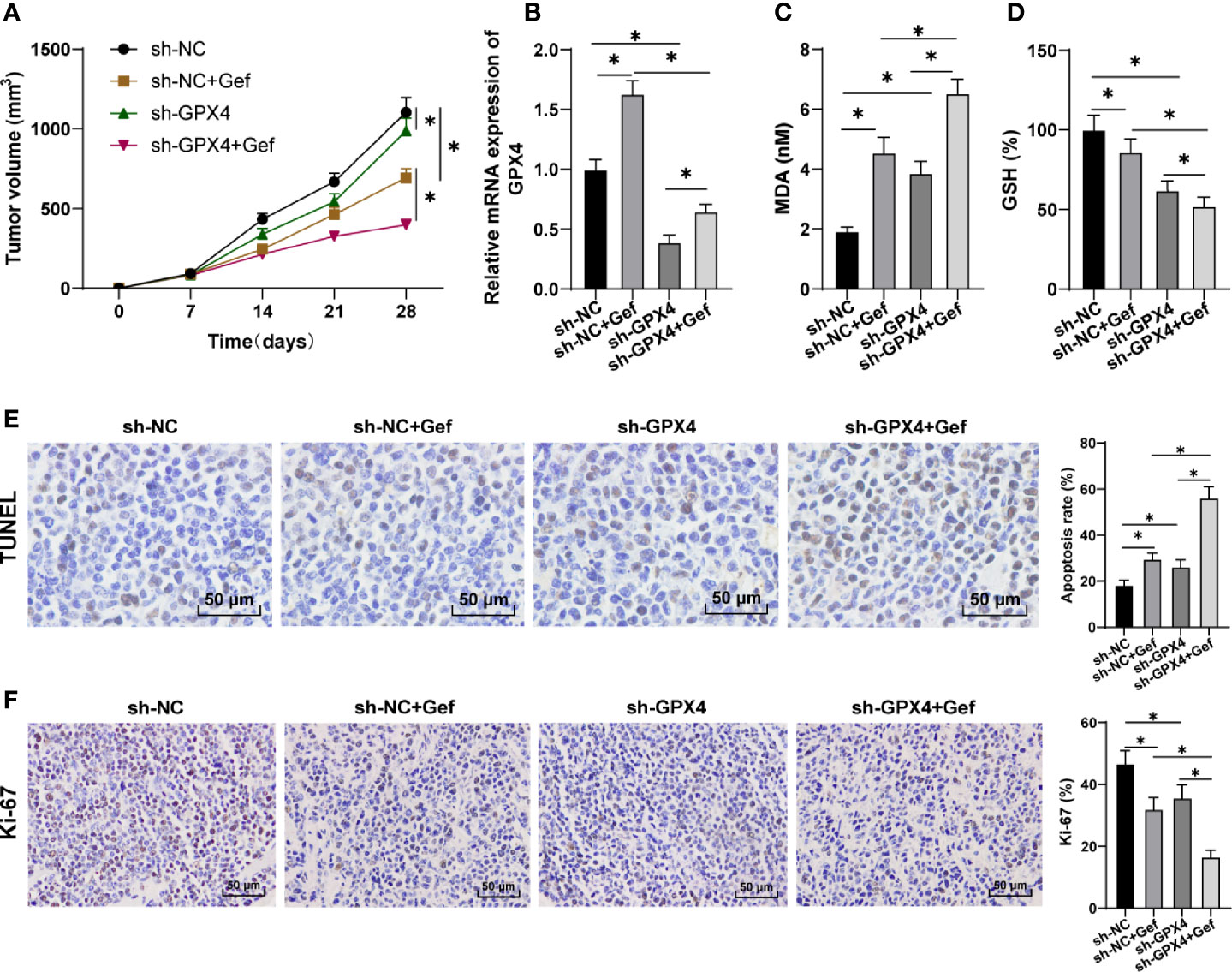
Figure 5 GPX4 inhibition increased the anti-tumor effect of gefitinib by promoting ferroptosis. (A) Tumor growth curve; (B) GPX4 expression was detected by RT-qPCR; (C) MDA level was detected by a kit; (D) GSH level was detected by a kit; (E) TUNEL staining was used to detect apoptosis, 400 ×, scale bar = 25 μm; (F) Immunohistochemistry was used to detect the expression of Ki-67, 400 ×, scale bar = 25 μm; n = 10. The data were expressed as mean ± standard deviation and analyzed by one-way ANOVA (B–F) and two-way ANOVA (A) followed by Tukey’s multiple comparisons test. *p < 0.05.
Discussion
Inhibition of ferroptosis is potential to facilitate sorafenib (an EGFR inhibitor) resistance to cancer cells (23, 32). Previous study has identified a potential therapeutic benefit of blocking EGFR with gefitinib in basal-like subtypes of TNBC in vitro (21). Besides, GPX4 is a promising target for killing therapy-resistant cancer cells via ferroptosis (33). However, the role of GPX4-mediated ferroptosis in gefitinib sensitivity in TNBC cells is largely unknown. Herein, this study focused on the possible mechanism of GPX4 in gefitinib sensitivity in TNBC cells by monitoring ferroptosis.
GPX4 is positively related to chemoresistance of anticancer drug lapatinib and IC50 of lapatinib (an inhibitor of EGFR) (34). In this study, we found that GPX4 expression in the resistant TNBC cell strains was higher than that in the sensitive strains. This indicated that gefitinib can induce the expression of GPX4 in TNBC cell lines MDA-MB-231 and HS578T. As reported, inhibition of tumor propellant GPX4 enhances anticancer effect of chemotherapeutic drugs (34). Then we silenced GPX4 expression in MDA-MB-231/Gef and HS578T/Gef cells to test its effect on gefitinib sensitivity. With the increase of gefitinib concentrations, the inhibition rate of cell activity was increased, and cell colony formation was decreased significantly. The apoptosis rate of sh-GPX4-treated cells was clearly increased, the levels of Ki-67 and PCNA were decreased, and the levels of Cleaved caspase-3 and Cleaved caspase-9 were increased. Similarly, in resistant cells overexpressing GPX4, GPX4 inhibitor overcomes resistance to erlotinib, and knockdown of GPX4 inhibits the migration of resistant cells (35). Decreased protein level of GPX4 is accompanied with increased levels of cleaved caspase 3 and iron concentrations (36). These results suggested that inhibition of GPX4 can increase the sensitivity of TNBC cell lines to gefitinib.
Increasing evidences have indicated that promoting ferroptosis is a promising approach to attenuate drug resistance in cancer chemotherapy (37, 38). Given the therapeutic promise for inducing ferroptosis in drug-resistant cancers, a potent GPX4 inhibitor is paramount (39, 40). Inhibition of GPX4 leads to the induction of ferroptosis (41), especially in drug-resistant tumors (42, 43). After GPX4 silencing, our data revealed that MDA level was notably increased, and GSH level was decreased, ROS production was increased significantly and MMP was reduced. Ferroptosis is very important for the survival of TNBC cells and is closely related to GPX4 (28). Lipid ROS increases after suppression of GPX4 (32). Elimination of ROS sensitizes the BC cells to gefitinib (44). GSH is the most decreased cellular metabolite during ferroptosis, and GSH depletion causes loss of cellular antioxidant and inhibition of GPXs (13, 45). Ferroptosis induces GSH depletion, disrupted GPX4 function and ROS production (14, 19, 46). Moreover, the levels of ACSL4, PTGS2 and NOX1 were notably upregulated, while FTH1 was downregulated in the sh-GPX4 group. ACSL4 is preferentially expressed in TNBC cell lines and cells with GPX4-ACSL4 double knockout show marked resistance to ferroptosis (47). ACSL4 inhibition is a viable therapeutic approach to preventing ferroptosis-related diseases (48). The above results showed the same trends in MDA-MB-231/Gef and HS578T/Gef cells, indicating that inhibition of GPX4 can promote ferroptosis in gefitinib resistant cells.
To further confirm that the regulation of gefitinib sensitivity by GPX4 is achieved by affecting ferroptosis, we silenced GPX4 in TNBC cell line MDA-MB-231 and used ferrostatin-1 to inhibit ferroptosis. GPX4 expression was effectively reduced after silencing GPX4, but partially restored by ferrostatin-1. Consistently, ferrostatin-1 distinctly raises the GPX4 and FTH1 expression and blocks the PTGS2 and ACSL4 level (49). After silencing GPX4, MDA level was increased, GSH level was decreased, and ROS production was increased, and apoptosis rate was elevated. However, inhibition of ferroptosis by ferrostatin-1 partially reversed the effect of sh-GPX4. The enhancement of ROS generation in H1650 and H1975 Gef-resistant cells leads to impairment of tumor growth and induction of cell apoptosis (50). In lung cancer cell lines, cell proliferation is decreased with GPX4 knockdown and this inhibition could be reversed by ferrostatin-1, the specific inhibitor of ferroptosis (14, 51). These results further indicated that GPX4 can negatively regulate ferroptosis, and ferrostatin-1 partially reversed the effect of sh-GPX4. Furthermore, sh-GPX4 treatment reduced the growth rate of tumor in nude mice, and further strengthened the inhibitory effect of gefitinib on tumors. All in all, inhibition of GPX4 enhanced the anticancer effect of gefitinib in vivo by promoting cell ferroptosis.
In summary, this study simply revealed that GPX4 knockdown can enhance the sensitivity of TNBC cells to gefitinib by promoting cell ferroptosis, but the regulatory mechanism of GPX4 expression in gefitinib resistant cells has not been studied in depth. Due to the current funding and cycle limitations, we cannot study more inhibitors for GPX4 ablation in this paper. But we will apply more inhibitors for sh-GPX4 lines to ferroptosis research in the future, in order to provide more information on the mechanism of inhibiting lipid peroxidation. In the future, we will study the regulation mechanism of GPX4 expression in gefitinib resistant TNBC cells from the perspective of transcriptional regulation.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics Statement
This study was reviewed and approved by the ethics committee of Shandong Cancer Hospital and Institute.
Author Contributions
Conceptualization: XS and XW. Validation, research, resources, data reviewing, and writing: ZL. Review and editing: ZY. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gregorio AC, Lacerda M, Figueiredo P, Simoes S, Dias S, Moreira JN. Therapeutic Implications of the Molecular and Immune Landscape of Triple-Negative Breast Cancer. Pathol Oncol Res 24:701–16.
2. Costa RLB, Han HS, Gradishar WJ. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: a review. Breast Cancer Res Treat 169:397–406.
3. Khosravi-Shahi P, Cabezon-Gutierrez L, Aparicio Salcedo MI. State of art of advanced triple negative breast cancer. Breast J 25:967–70.
4. Sun X, Wang M, Wang M, Yu X, Guo J, Sun T, et al. Metabolic Reprogramming in Triple-Negative Breast Cancer. Front Oncol 10:428.
5. Tang Q, Ouyang H, He D, Yu C, Tang G. MicroRNA-based potential diagnostic, prognostic and therapeutic applications in triple-negative breast cancer. Artif Cells Nanomed Biotechnol 47:2800–9.
6. Yang X, Zhao L, Pei J, Wang Z, Zhang J, Wang B. CELF6 modulates triple-negative breast cancer progression by regulating the stability of FBP1 mRNA. Breast Cancer Res Treat 183:71–82.
7. Von Pawel J. Gefitinib (Iressa, ZD1839): a novel targeted approach for the treatment of solid tumors. Bull Cancer 91:E70–6.
8. Bernsdorf M, Ingvar C, Jorgensen L, Tuxen MK, Jakobsen EH, Saetersdal A, et al. Effect of adding gefitinib to neoadjuvant chemotherapy in estrogen receptor negative early breast cancer in a randomized phase II trial. Breast Cancer Res Treat 126:463–70.
9. Girgert R, Emons G, Grundker C. 17beta-estradiol-induced growth of triple-negative breast cancer cells is prevented by the reduction of GPER expression after treatment with gefitinib. Oncol Rep 37:1212–8.
10. McGovern UB, Francis RE, Peck B, Guest SK, Wang J, Myatt SS, et al. Gefitinib (Iressa) represses FOXM1 expression via FOXO3a in breast cancer. Mol Cancer Ther 8:582–91.
11. McLaughlin RP, He J, van der Noord VE, Redel J, Foekens JA, Martens JWM, et al. A kinase inhibitor screen identifies a dual cdc7/CDK9 inhibitor to sensitise triple-negative breast cancer to EGFR-targeted therapy. Breast Cancer Res 21:77.
13. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156:317–31.
14. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–72.
15. Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med 133:144–52.
16. Yu H, Guo P, Xie X, Wang Y, Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med 21:648–57.
17. Maiorino M, Conrad M, Ursini F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid Redox Signal 29:61–74.
18. Chen L, Hambright WS, Na R, Ran Q. Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. J Biol Chem 290:28097–106.
19. Jelinek A, Heyder L, Daude M, Plessner M, Krippner S, Grosse R, et al. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic Biol Med 117:45–57.
20. Li X, Wang TX, Huang X, Li Y, Sun T, Zang S, et al. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int 40:1378–94.
21. Yi YW, Hong W, Kang HJ, Kim HJ, Zhao W, Wang A, et al. Inhibition of the PI3K/AKT pathway potentiates cytotoxicity of EGFR kinase inhibitors in triple-negative breast cancer cells. J Cell Mol Med 17:648–56.
22. Ge P, Cao L, Chen X, Jing R, Yue W. miR-762 activation confers acquired resistance to gefitinib in non-small cell lung cancer. BMC Cancer 19:1203.
23. Sun X, Niu X, Chen R, He W, Chen D, Kang R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 64:488–500.
24. Guerriero E, Capone F, Accardo M, Sorice A, Costantini M, Colonna G, et al. GPX4 and GPX7 over-expression in human hepatocellular carcinoma tissues. Eur J Histochem 59:2540.
25. Wu Q, Yi X. Down-regulation of Long Noncoding RNA MALAT1 Protects Hippocampal Neurons Against Excessive Autophagy and Apoptosis via the PI3K/Akt Signaling Pathway in Rats with Epilepsy. J Mol Neurosci 65:234–45.
26. Wang P, Yu J, Zhang L. The nuclear function of p53 is required for PUMA-mediated apoptosis induced by DNA damage. Proc Natl Acad Sci U S A 104:4054–9.
27. Cai H, Yang X, Gao Y, Xu Z, Yu B, Xu T, et al. Exosomal MicroRNA-9-3p Secreted from BMSCs Downregulates ESM1 to Suppress the Development of Bladder Cancer. Mol Ther Nucleic Acids 18:787–800.
28. Yu M, Gai C, Li Z, Ding D, Zheng J, Zhang W, et al. Targeted exosome-encapsulated erastin induced ferroptosis in triple negative breast cancer cells. Cancer Sci 110:3173–82.
29. Fani M, Mohammadipour A, Ebrahimzadeh-Bideskan A. The effect of crocin on testicular tissue and sperm parameters of mice offspring from mothers exposed to atrazine during pregnancy and lactation periods: An experimental study. Int J Reprod BioMed (Yazd) 16:519–28.
30. Repasy T, Martinez N, Lee J, West K, Li W, Kornfeld H. Bacillary replication and macrophage necrosis are determinants of neutrophil recruitment in tuberculosis. Microbes Infect 17:564–74.
31. Zeng Q, Wu Z, Duan H, Jiang X, Tu T, Lu D, et al. Impaired tumor angiogenesis and VEGF-induced pathway in endothelial CD146 knockout mice. Protein Cell 5:445–56.
32. Poursaitidis I, Wang X, Crighton T, Labuschagne C, Mason D, Cramer SL, et al. Oncogene-Selective Sensitivity to Synchronous Cell Death following Modulation of the Amino Acid Nutrient Cystine. Cell Rep 18:2547–56.
33. Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ, et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol 16:497–506.
34. Zhang X, Sui S, Wang L, Li H, Zhang L, Xu S, et al. Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J Cell Physiol 235:3425–37.
35. Ma CS, Lv QM, Zhang KR, Tang YB, Zhang YF, Shen Y, et al. NRF2-GPX4/SOD2 axis imparts resistance to EGFR-tyrosine kinase inhibitors in non-small-cell lung cancer cells. Acta Pharmacol Sin (2020). doi: 10.1038/s41401-020-0443-1
36. Wang C, Yuan W, Hu A, Lin J, Xia Z, Yang CF, et al. Dexmedetomidine alleviated sepsisinduced myocardial ferroptosis and septic heart injury. Mol Med Rep 22:175–84.
37. Liu Q, Wang K. The induction of ferroptosis by impairing STAT3/Nrf2/GPx4 signaling enhances the sensitivity of osteosarcoma cells to cisplatin. Cell Biol Int 43:1245–56.
38. Wenz C, Faust D, Linz B, Turmann C, Nikolova T, Dietrich C. Cell-cell contacts protect against t-BuOOH-induced cellular damage and ferroptosis in vitro. Arch Toxicol 93:1265–79.
39. Roh JL, Kim EH, Jang HJ, Park JY, Shin D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett 381:96–103.
40. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547:453–7.
41. Forcina GC, Dixon SJ. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 19:e1800311.
42. Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol 14:507–15.
43. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551:247–50.
44. Giordano CR, Mueller KL, Terlecky LJ, Krentz KA, Bollig-Fischer A, Terlecky SR, et al. A targeted enzyme approach to sensitization of tyrosine kinase inhibitor-resistant breast cancer cells. Exp Cell Res 318:2014–21.
47. Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr Top Microbiol Immunol 403:143–70.
48. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13:91–8.
49. Chen Y, Zhang P, Chen W, Chen G. Ferroptosis mediated DSS-induced ulcerative colitis associated with Nrf2/HO-1 signaling pathway. Immunol Lett 225:9–15.
50. Li X, Fan XX, Jiang ZB, Loo WT, Yao XJ, Leung EL, et al. Shikonin inhibits gefitinib-resistant non-small cell lung cancer by inhibiting TrxR and activating the EGFR proteasomal degradation pathway. Pharmacol Res 115:45–55.
Keywords: triple negative breast cancer, gefitinib, GPX4, ferroptosis, sensitivity
Citation: Song X, Wang X, Liu Z and Yu Z (2020) Role of GPX4-Mediated Ferroptosis in the Sensitivity of Triple Negative Breast Cancer Cells to Gefitinib. Front. Oncol. 10:597434. doi: 10.3389/fonc.2020.597434
Received: 21 August 2020; Accepted: 26 October 2020;
Published: 23 December 2020.
Edited by:
Anindita Chakrabarty, Shiv Nadar University, IndiaReviewed by:
Tasaduq Wani, University of Oxford, United KingdomEkta Khattar, SVKM’s Narsee Monjee Institute of Management Studies – NMIMS, India
Copyright © 2020 Song, Wang, Liu and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiyong Yu, zyyu@sdfmu.edu.cn