 Yuan-Ming Zhang
Yuan-Ming Zhang Zhenyu Jia2
Zhenyu Jia2 Jim M. Dunwell
Jim M. Dunwell- 1Crop Information Center, College of Plant Science and Technology, Huazhong Agricultural University, Wuhan, China
- 2Department of Botany and Plant Sciences, University of California, Riverside, Riverside, CA, United States
- 3School of Agriculture, Policy and Development, University of Reading, Reading, United Kingdom
Editorial on the Research Topic
The Applications of New Multi-Locus GWAS Methodologies in the Genetic Dissection of Complex Traits
Since the establishment of the mixed linear model (MLM) method for genome-wide association studies (GWAS) by Zhang et al. (2005) and Yu et al. (2006), a series of new MLM-based methods have been proposed (Feng et al., 2016). These methods have been widely used in genetic dissection of complex and omics-related traits (Figure 1), especially in conjunction with the development of advanced genomic sequencing technologies. However, most existing methods are based on single marker association in genome-wide scans with population structure and polygenic background controls. To control false positive rate, Bonferroni correction for multiple tests is frequently adopted. This stringent correction results in the exclusion of important loci, especially for large experimental error inherent in field experiments of crop genetics. To address this issue, multi-locus GWAS methodologies have been recommended, i.e., mrMLM (Wang et al., 2016), ISIS EM-BLASSO (Tamba et al., 2017), pLARmEB (Zhang et al., 2017), FASTmrEMMA (Wen et al., 2018a), pKWmEB (Ren et al., 2018), and FASTmrMLM (Zhang and Tamba, 2018). Here we summarize their advantages and potential limitations for using these methods (Table 1).
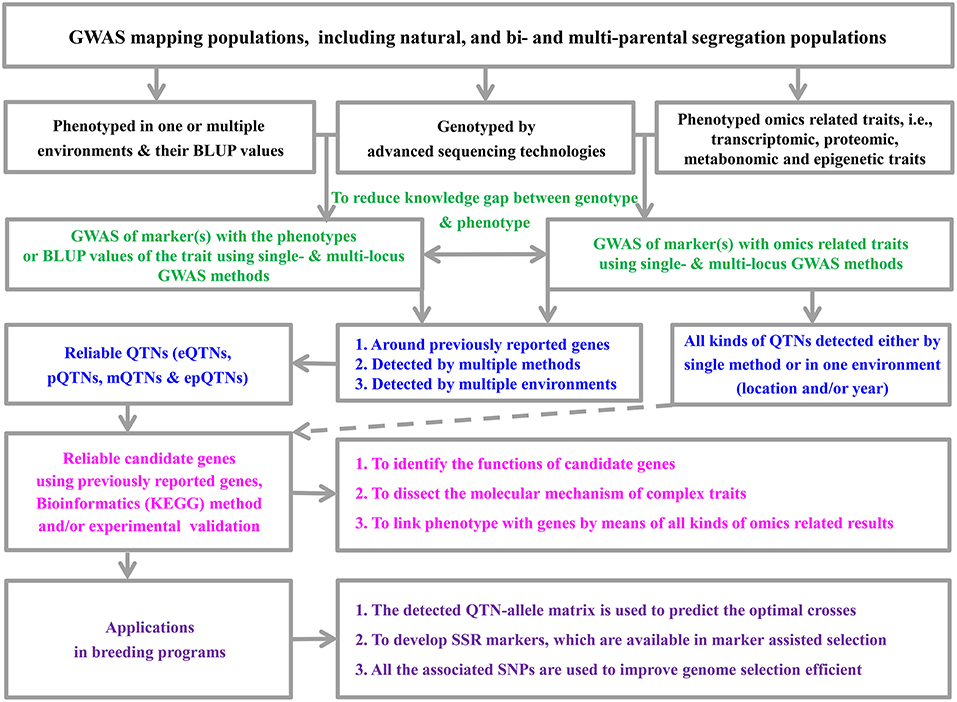
Figure 1. The pipeline framework of genome-wide association studies and their application.

Table 1. Comparison of single- and multi-locus GWAS methodologies.
Multi-locus Genome-wide Association Studies for Complex Traits
Comparison of GWAS Methodologies
Our methodological papers have showed their advantages in terms of quantitative trait nucleotide (QTN) detection power and QTN effect estimation accuracy over existing methods (Wang et al., 2016; Tamba et al., 2017; Zhang et al., 2017; Ren et al., 2018; Wen et al., 2018a). This conclusion has been echoed in a number of other applied studies in this Research Topic. For example, Ma et al. and Zhang et al. indicated that mrMLM, FASTmrEMMA, pLARmEB, and ISIS EM-BLASSO outperform the R package GAPIT, with ISIS EM-BLASSO being the most powerful multi-locus approach. Xu et al. compared one single-locus method (GEMMA) and three multi-locus methods (FASTmrEMMA, FarmCPU, and LASSO) in the genetic dissection of starch pasting properties in maize. As a result, FASTmrEMMA detected the most QTNs (29), followed by FarmCPU (19) and LASSO (12), and GEMMA detected the least QTNs (7). In the genetic dissection of salt tolerance traits in rice, Cui et al. compared all the six multi-locus approaches and identified the most co-detected QTNs from ISIS EM-BLASSO. Peng et al. used our six multi-locus GWAS methods to analyze 20 free amino acid levels in kernels of bread wheat (Triticum aestivum L.) and found the reliability and complementarity of these methods. In the detection of small-effect QTNs for fiber-quality related traits in the early-maturity varieties of upland cotton, Su et al. claimed that the multi-locus GWAS methods are more powerful and robust than the MLM method in TASSEL v5.0. Hou et al. demonstrated that 20 QTNs were associated with drought stress response using mrMLM, while three QTNs were associated with resistance to Verticillium wilt using EMMAX. Although the above studies have shown the advantages of multi-locus GWAS methods over single-locus GWAS methods, Chang et al., He et al., Li et al., and Xu et al. recommended the combination of single-locus methods and/or multi-locus methods to improve the detection power and robustness of GWAS, and Cui et al. recommended adding a bin analysis to the models or developing a hybrid method that merges the results from different methods. Our previous results in the analysis of real and simulated dataset support the above recommendations.
In addition, Liu et al. adopted four multi-locus GWAS algorithms (mrMLM, FASTmrEMMA, ISIS EM-BLASSO, and pLARmEB) to dissect the genetic foundation for fiber quality and yield component traits in RILs. As a result, a significant number of QTNs were found to coincide with the physical regions of the confidence intervals of reported QTLs, demonstrating the effectiveness and feasibility of multi-locus GWAS methods in RILs.
The Critical P-Value or LOD Score for Significant QTN
In single-locus GWAS, one key concern is the high false positive rate (FPR). To reduce FPR, Bonferroni correction is frequently applied in the single-locus methods, including EMMAX (Kang et al., 2010), GEMMA (Zhou and Stephens, 2012), ECMLM (Li et al., 2014), and MLM (Yu et al., 2006). In human genetics, the genome-wide significance P-value threshold of 5 × 10−8 has become a standard for common-variant GWAS (Barsh et al., 2012; Fadista et al., 2016; Chang et al., 2018). However, this correction or the critical P-value in human genetics is too stringent to detect certain associated loci for complex traits in crop genetics. To address this issue, a modified Bonferroni correction has been proposed; in other words, the number of markers (m) in the correction formulas is replaced by the effective number of markers (me) (Wang et al., 2016; Guan et al.). In real data analysis in crop genetics, some subjective and less stringent P-value thresholds for significant level are frequently applied owing to large experimental error, i.e., 1/m (m is the number of markers) (Li et al.; Xu et al.), 10−5 (Misra et al.), and 10−4 (Chang et al.). To balance high QTN detection power and low false positive rate, Xu et al. replaced Bonferroni correction by a less stringent criterion (1/m) for GEMMA, and a satisfactory result was achieved in their Monte Carlo simulation studies.
Theoretically, correction for multiple tests is unnecessary in multi-locus GWAS because all the potential genes or loci for complex traits are fitted to a single linear model and their effects are estimated and tested simultaneously. For example, 0.05 was chosen as the P-value threshold in QTN detection of Khan et al. (2018). Although relaxing the stringency of significance level in multi-locus GWAS can identify more hits, confidence in these hits will drop significantly. Thus, Segura et al. (2012) and Liu et al. (2016) imposed Bonferroni correction on QTN detection in their multi-locus GWAS methods. Our results indicated that Bonferroni correction in multi-locus GWAS of (Segura et al., 2012) and Liu et al. (2016) may be too stringent, while the cutoff of 0.05 in multi-locus GWAS of Khan et al. (2018) may be too relaxed due to the fact that a significance level of 0.05 can result in a high false positive rate. Lü et al. simply used LOD score ≥ 5 as a threshold for QTN detection in their multi-locus GWAS. Based on our studies, we proposed using LOD = 3.0 (or P = 0.0002) as a cutoff in multi-locus GWAS to balance the high power and low false positive rate for QTN detection.
Heritability Missing in GWAS
Heritability missing is a common issue in GWAS (Maher, 2008). Human geneticists ascribe heritability missing to a few reasons, including rare alleles, gene-by-gene and gene-by-environment interactions, and miniature genetic effects of DNA variants that can hardly reach the level of genome-wide significance (Eichler et al., 2010). In our opinion, the stringent threshold in genome-wide detection is also a factor, because certain QTNs cannot meet the significant level if such P-value cutoff is applied. This viewpoint is supported by the simulation results of Xu et al.
In most GWAS methodologies, the genotypes of a SNP, for example, QQ, Qq, and qq, are conventionally coded as 2, 1, and 0, respectively. Thus, the estimated QTN effect is actually the average effect of allelic substitution, being a + (q − p)d. Let a + (q − p)d = 0, then d = a/(p − q). Where p takes different values, such as p = 0.1, 0.3, 0.5, 0.7, and 0.9, so d = −1.25a, −2.5 a, ∞, 2.5 a, and 1.25 a, respectively, indicating the difficulty in the detection of QTNs with over-dominance. This may be another reason for the heritability missing.
New methodologies have been proposed to handle heritability missing, for example, GCTA (Yang et al., 2011) and GREML-LDMS (Yang et al., 2015). In this Research Topic, we suggest that part of the missing heritability may be regained by using multi-locus GWAS methods, since more QTNs can be detected and overall estimated heritability will be increased.
How to Determine Reliable QTNs and Mine Reliable Candidate Genes?
How to Determine Reliable QTNs?
Firstly, when several multi-locus methods are used to analyze a same dataset, the QTNs identified by multiple approaches are usually reliable. For example, all the 31 genomic regions associated with four photosynthesis related traits were detected by at least three multi-locus methods in Lü et al., five QTNs associated with forage quality-related traits were detected by at least two methods in Li et al., and all the common QTNs either between single-locus methods and multi-locus methods, or across several multi-locus methods were declared in Misra et al. Secondly, the QTNs near previously reported trait-associated genes should be reliable. For example, the QTNs around genes GRMZM2G163761, GRMZM2G412611, and GRMZM2G066749 likely contribute to the callus regenerative capacity (Ma et al.), the QTNs around genes GRMZM2G032628 (ae1) and GRMZM2G392988 may be associated with starch biosynthesis (Xu et al.), and the QTNs around genes Gh_D102255 and Gh_A13G0187 perhaps participate in cellular activities for fiber elongation (Liu et al.). Finally, the QTNs identified across various environments (locations and/or years) are also reliable, i.e., Liu et al. identified 57 QTNs that were associated with cotton fiber quality and yield components in at least two environments; Hu et al. repeatedly detected 39 QTNs clusters to be associated with 14 agronomic traits in 122 barley doubled haploid lines in multiple environments; Zhang et al. repeatedly detected 22 common QTNs to be associated with protein content in 144 soybean four-way recombinant inbred lines in 20 environments.
How to Mine Reliable Candidate Genes?
All known genes in the regions around reliable QTNs potentially contribute to the traits of interest. However, only a subset of them may be reliable candidate genes which are worthy of further investigation. We can use homolog (previously reported genes) in other species, e.g., Arabidopsis thaliana, to mine reliable candidate genes in these regions. For example, WOX2 in Arabidopsis has been reported to increase the rate of resistant seedlings from transformed immature embryos in maize and, therefore, the homologous gene GRMZM2G108933 might play an important role in controlling maize callus regeneration (Ma et al.). Bioinformatics approaches, such as the KEGG pathway analytic tool, may be used for mining reliable candidate genes and relevant gene networks. For example, two genes (LOC_Os01g45760 and LOC_Os10g04860) are found to be involved in auxin biosynthesis in rice using KEGG (Cui et al.). Experimental validations are often needed to confirm the associations between these candidate genes and the traits of interest. For instance, RNA-seq analysis and qRT-PCR experiments verified that four genes (RD2, HAT22, PIP2, and PP2C) are associated with drought tolerance in cotton (Hou et al.); genomic DNA sequencing showed that two candidate genes BnaA08g08280D and BnaC03g60080D are different between the high- and low-oleic acid lines (Guan et al.). The combined use of GWAS and experimental validation has great potential for detection of new genes and their biological functions. For example, a new gene GRMZM2G065083 was found by Xu et al. to play a critical role in starch biosynthesis in maize by being involved in the gluconeogenesis process, hexose biosynthetic and metabolic process, and glucose-6-phosphate isomerase activity, providing insights into the molecular mechanism underlying the pasting properties of maize starch.
Important genes may be missed if we only select consensus QTNs identified by more than one methodology or in more than one experiment/environment. In practice, we found that some QTNs detected by only one multi-locus method or one environment may lead to important discoveries. These QTNs may be used to mine candidate genes through network analysis using bioinformatics analysis and/or experimental validation.
How to Make Use of the GWAS Results?
The main product of GWAS includes the detected QTNs and the candidate genes nearby. Three approaches are available for applying these results to breeding programs. Firstly, one can organize the detected QTN-allele matrix as the population genetic constitution to facilitate the selection of optimal crosses. For example, the top 10 optimal crosses were predicted according to their 95th percentile weighted average values (Khan et al., 2018). Secondly, we can develop SSR markers around the reliable QTNs and utilize them in marker assisted selection of crops (Li et al., 2018). Thirdly, all the SNPs that are significantly associated with the trait of interest can be used for improving genome selection (He et al.; He et al., 2019).
Figure 1 summarizes how to design a GWAS to identify QTNs and mine candidate genes, of which the biological functions may be further investigated or validated at a molecular level.
Future Perspectives
It is becoming common to use multiple statistical methods to detect major quantitative trait loci (QTLs) in the linkage analyses of complex traits. Thus, we recommend using a few GWAS methods, especially several multi-locus GWAS methods which do not need correction for multiple comparisons, to investigate complex traits. However, not all QTNs can be identified by all these methods, posing difficulties for using these GWAS results. This may be ascribed to the fact that the various GWAS models are based upon different genetic or statistical assumptions. Possible solutions have been provided in this editorial to compare the results from various GWAS models and screen for candidate QTNs or genes, facilitating the subsequent validation or application.
Interaction at different omics levels, including QTL-by-environment and QTL-by-QTL interactions, can be detected with various software programs in linkage analysis. Nevertheless, methods and software programs with comparable function are quite limited in GWAS, especially for the studies of quantitative traits in natural populations where large numbers of genomic markers are analyzed. The number of variables in GWAS models will increase sharply if interactions are considered, challenging both computational efficiency and detection power. Multicollinearity among highly saturated and linked markers is another issue in GWAS, which impairs the efficiency and accuracy of the current statistical methods. Innovative strategies are needed to distill many thousands of variables by removing the redundant genomic markers such that the computational burden and impact from multicollinearity can be reduced and the studies of interactions made more feasible.
Zhang et al. (2018) showed that the explained heritability increases with sample size in GWAS, and also estimated that the required sample size may range from a few hundred thousand to multiple millions to account for most of the heritability. The samples used in crop genetics, however, is often small, therefore, increasing sample size in crop GWAS has a great potential in future research.
With the rapid advances in various technologies, other types of omic data, including transcriptomic, proteomic, metabolomic and epigenetic data, have been recently exploited in crop research (Peng et al.; Wen et al., 2018b). These multi-omic variables may be treated as additional traits in GWAS, which promises to reduce knowledge gap between genotype and phenotype and will eventually benefit selective breeding. For example, omic-traits (at various layers) that are mapped to the same genomic locations with agronomic traits will provide multi-dimensional insights of genetic architectures and the underlying biological pathways. We believe multi-locus GWAS methodologies will become useful and popular tools for analysis of omics big datasets and help understand the mysterious world of genetics.
Author Contributions
Y-MZ, ZJ, and JMD contributed to manuscript writing, provided important interpretations, and revised the work. All the authors checked and confirmed the final version of the manuscript.
Funding
The work was supported by the National Natural Science Foundation of China (31571268, 31871242, and U1602261), and Huazhong Agricultural University Scientific and Technological Self-innovation Foundation (2014RC020).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Chengsong Zhu at Southwestern Medical Center, University of Texas and Dr. Jia Wen at Department of Bioinformatics and Genomics, University of North Carolina at Charlotte for their comments and suggestions.
References
Barsh, G. S., Copenhaver, G. P., Gibson, G., and Williams, S. M. (2012). Guidelines for genome-wide association studies. PLoS Genet. 8:e1002812. doi: 10.1371/journal.pgen.1002812
Chang, M., He, L., and Cai, L. (2018). An overview of genome-wide association studies. Methods Mol. Biol. 1754, 97–108. doi: 10.1007/978-1-4939-7717-8_6
Eichler, E. E., Flint, J., Gibson, G., Kong, A., Leal, S. M., Moore, J. H., et al. (2010). Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet. 11, 446–450. doi: 10.1038/nrg2809
Fadista, J., Manning, A. K., Florez, J. C., and Groop, L. (2016). The (in)famous GWAS P-value threshold revisited and updated for low-frequency variants. Eur J. Hum. Genet. 24, 1202–1205. doi: 10.1038/ejhg.2015.269
Feng, J. Y., Wen, Y. J., Zhang, J., and Zhang, Y. M. (2016). Advances on methodologies for genome-wide association studies in plants. Acta Agron. Sin. 42, 945–956. doi: 10.3724/SP.J.1006.2016.00945
He, L., Xiao, J., Rashid, K. Y., Jia, G., Li, P., Yao, Z., et al. (2019). Evaluation of genomic prediction for pasmo resistance in flax. Int. J. Mol. Sci. 20:E359. doi: 10.3390/ijms20020359
Kang, H. M., Sul, J. H., Service, S. K., Zaitlen, N. A., Kong, S. Y., Freimer, N. B., et al. (2010). Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354. doi: 10.1038/ng.548
Khan, M. A., Tong, F., Wang, W., He, J., Zhao, T., and Gai, J. (2018). Analysis of QTL-allele system conferring drought tolerance at seedling stage in a nested association mapping population of soybean [Glycine max (L.) Merr.] using a novel GWAS procedure. Planta 248, 947–962. doi: 10.1007/s00425-018-2952-4
Li, C. X., Xu, W. G., Guo, R., Zhang, J. Z., Qi, X. L., Hu, L., et al. (2018). Molecular marker assisted breeding and genome composition analysis of Zhengmai 7698, an elite winter wheat cultivar. Sci. Rep. 8:322. doi: 10.1038/s41598-017-18726-8
Li, M., Liu, X., Bradbury, P., Yu, J., Zhang, Y. M., Todhunter, R. J., et al. (2014). Enrichment of statistical power for genome-wide association studies. BMC Biol. 12:73. doi: 10.1186/s12915-014-0073-5
Liu, X., Huang, M., Fan, B., Buckler, E. S., and Zhang, Z. (2016). Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 12:e1005767. doi: 10.1371/journal.pgen.1005767
Maher, B. (2008). Personal genomes: the case of the missing heritability. Nature 456, 18–21. doi: 10.1038/456018a
Ren, W. L., Wen, Y. J., Dunwell, J. M., and Zhang, Y. M. (2018). pKWmEB: integration of Kruskal-Wallis test with empirical bayes under polygenic background control for multi-locus genome-wide association study. Heredity 120, 208–218. doi: 10.1038/s41437-017-0007-4
Segura, V., Vilhjálmsson, B. J., Platt, A., Korte, A., Seren, Ü., Long, Q., et al. (2012). An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 44, 825–830. doi: 10.1038/ng.2314
Tamba, C. L., Ni, Y. L., and Zhang, Y. M. (2017). Iterative sure independence screening EM-Bayesian LASSO algorithm for multi-locus genome-wide association studies. PLoS Comput. Biol. 13:e1005357. doi: 10.1371/journal.pcbi.1005357
Wang, S. B., Feng, J. Y., Ren, W. L., Huang, B., Zhou, L., Wen, Y. J., et al. (2016). Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 6:19444. doi: 10.1038/srep19444
Wen, Y. J., Zhang, H., Ni, Y. L., Huang, B., Zhang, J., Feng, J. Y., et al. (2018a). Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 19, 700–712. doi: 10.1093/bib/bbw145
Wen, Y. J., Zhang, Y. W., Zhang, J., Feng, J. Y., Dunwell, J. M., and Zhang, Y. M. (2018b). An efficient multi-locus mixed model framework for the detection of small and linked QTLs in F2. Brief. Bioinform. doi: 10.1093/bib/bby058. [Epub ahead of print].
Yang, J., Bakshi, A., Zhu, Z., Hemani, G., Vinkhuyzen, A. A., Lee, S. H., et al. (2015). Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat. Genet. 47, 1114–1120. doi: 10.1038/ng.3390
Yang, J., Lee, S. H., Goddard, M. E., and Visscher, P. M. (2011). GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. doi: 10.1016/j.ajhg.2010.11.011
Yu, J., Pressoir, G., Briggs, W. H., Bi, I. V., Yamasaki, M., Doebley, J. F., et al. (2006). A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208. doi: 10.1038/ng1702
Zhang, J., Feng, J. Y., Ni, Y. L., Wen, Y. J., Niu, Y., Tamba, C. L., et al. (2017). pLARmEB: integration of least angle regression with empirical bayes for multi-locus genome-wide association studies. Heredity 118, 517–524. doi: 10.1038/hdy.2017.8
Zhang, Y., Qi, G., Park, J. H., and Chatterjee, N. (2018). Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat. Genet. 50, 1318–1326. doi: 10.1038/s41588-018-0193-x
Zhang, Y. M., Mao, Y., Xie, C., Smith, H., Luo, L., and Xu, S. (2005). Mapping quantitative trait loci using naturally occurring genetic variance among commercial inbred lines of maize (Zea mays L.). Genetics 169, 2267–2275. doi: 10.1534/genetics.104.033217
Zhang, Y. M., and Tamba, C. L. (2018). A fast mrMLM algorithm for multi-locus genome-wide association studies. bioRxiv [Preprint]. doi: 10.1101/341784
Keywords: genome-wide association study, mixed linear model, multi-locus model, mrMLM, omics big dataset
Citation: Zhang Y-M, Jia Z and Dunwell JM (2019) Editorial: The Applications of New Multi-Locus GWAS Methodologies in the Genetic Dissection of Complex Traits. Front. Plant Sci. 10:100. doi: 10.3389/fpls.2019.00100
Received: 21 December 2018; Accepted: 22 January 2019;
Published: 11 February 2019.
Edited and reviewed by: Luigi Cattivelli, Council for Agricultural and Economics Research, Italy
Copyright © 2019 Zhang, Jia and Dunwell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuan-Ming Zhang, soyzhang@mail.hzau.edu.cn