Calcium Signaling Regulated by Cellular Membrane Systems and Calcium Homeostasis Perturbed in Alzheimer’s Disease
 Dong-Xu Huang1
Dong-Xu Huang1  Xin Yu1 Wen-Jun Yu1 Xin-Min Zhang2 Chang Liu3 Hong-Ping Liu3 Yue Sun4
Xin Yu1 Wen-Jun Yu1 Xin-Min Zhang2 Chang Liu3 Hong-Ping Liu3 Yue Sun4  Zi-Ping Jiang1*
Zi-Ping Jiang1*- 1Department of Hand and Foot Surgery, The First Hospital of Jilin University, Changchun, China
- 2Department of Anesthesiology, The First Hospital of Jilin University, Changchun, China
- 3Department of Neurology, The First Hospital of Jilin University, Changchun, China
- 4Deparment of The First Operating Room, The First Hospital of Jilin University, Changchun, China
Although anything that changes spatiotemporally could be a signal, cells, particularly neurons, precisely manipulate calcium ion (Ca2+) to transmit information. Ca2+ homeostasis is indispensable for neuronal functions and survival. The cytosolic Ca2+ concentration ([Ca2+]CYT) is regulated by channels, pumps, and exchangers on cellular membrane systems. Under physiological conditions, both endoplasmic reticulum (ER) and mitochondria function as intracellular Ca2+ buffers. Furthermore, efficient and effective Ca2+ flux is observed at the ER-mitochondria membrane contact site (ERMCS), an intracellular membrane juxtaposition, where Ca2+ is released from the ER followed by mitochondrial Ca2+ uptake in sequence. Hence, the ER intraluminal Ca2+ concentration ([Ca2+]ER), the mitochondrial matrix Ca2+ concentration ([Ca2+]MT), and the [Ca2+]CYT are related to each other. Ca2+ signaling dysregulation and Ca2+ dyshomeostasis are associated with Alzheimer’s disease (AD), an irreversible neurodegenerative disease. The present review summarizes the cellular and molecular mechanism underlying Ca2+ signaling regulation and Ca2+ homeostasis maintenance at ER and mitochondria levels, focusing on AD. Integrating the amyloid hypothesis and the calcium hypothesis of AD may further our understanding of pathogenesis in neurodegeneration, provide therapeutic targets for chronic neurodegenerative disease in the central nervous system.
Introduction
The intraneuronal calcium ion (Ca2+) homeostasis is indispensable for neuronal functions and survival, even death (Miller, 1991; Berridge, 1998). Mainly, Ca2+ functions as a second messenger: the spatiotemporal change of the cytosolic Ca2+ concentration ([Ca2+]CYT), also known as the Ca2+ signal, is one of the ways that cells convey various information either intracellularly or intercellularly (Berridge et al., 1998). Additionally, Ca2+ acts as a carrier of positive electrical current, which enters into the cytosol and depolarizes the transmembrane potential (Byrne et al., 2014).
At the molecular level, the [Ca2+]CYT is regulated by channels, ATPase pumps, and ion exchangers on cellular membrane systems (the plasma membrane and intracellular membranes), as well as Ca2+-binding proteins in the cytosol (Byrne et al., 2014). At the subcellular level, at least two organelles, endoplasmic reticulum (ER) and mitochondria, have participated in the regulation of [Ca2+]CYT either respectively or interactively (Martonosi, 1984; Miller, 1991; Spät et al., 2008). Structurally, the ER extends into every inner domain in neurons, and mitochondria tend to localize in intraneuronal compartments that consume massive ATPs, such as synapses (Sheng and Cai, 2012; Wu et al., 2017). Functionally, both the ER and mitochondria act as internal Ca2+ sources and sinks; namely, both organelles possess the role of buffering the [Ca2+]CYT (Miller, 1991; Berridge, 1998; Spät et al., 2008). Collectively, both the endoplasmic reticulum intraluminal Ca2+ concentration ([Ca2+]ER) and the mitochondrial matrix Ca2+ concentration ([Ca2+]MT) fluctuate simultaneously with [Ca2+]CYT (Figure 1). Moreover, efficient and effective Ca2+ flux is observed at the ER-mitochondria contact site (ERMCS), where the two organelles are intimately apposed (Wu et al., 2018). Briefly, Ca2+ is released from the ER lumen followed by mitochondrial Ca2+ uptake into the mitochondrial matrix through the outer and inner mitochondrial membranes in sequence (Rizzuto et al., 2012).
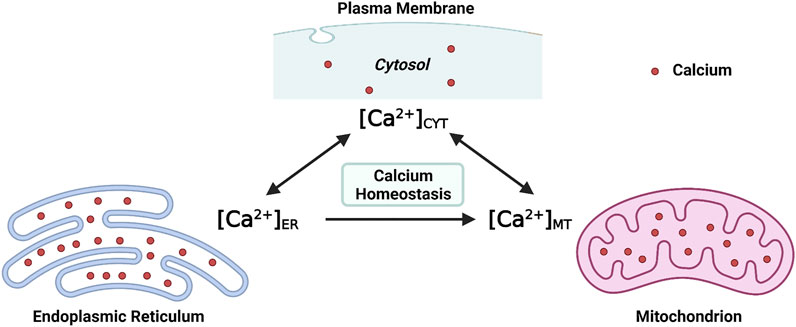
FIGURE 1. Maintaining the intraneuronal calcium homeostasis: the endoplasmic reticulum intraluminal Ca2+ concentration ([Ca2+]ER) and the mitochondrial matrix Ca2+ concentration ([Ca2+]MT) fluctuate with the cytosolic Ca2+ concentration ([Ca2+]CYT) (Created with BioRender.com).
Maintaining the physiological level of [Ca2+]CYT, [Ca2+]ER, and [Ca2+]MT is essential for intraneuronal Ca2+ homeostasis. When the neuronal Ca2+ signaling is dysregulated, neurons will undergo excitotoxicity or apoptosis (Lipton and Rosenberg, 1994; Berridge et al., 1999). The intraneuronal Ca2+ dyshomeostasis contributes to neurodegenerative diseases such as Alzheimer’s disease (AD), an irreversible chronic neurodegenerative disease without effective treatment (Pchitskaya et al., 2018). The underlying cellular and molecular mechanisms which regulate Ca2+ signaling and maintain intracellular Ca2+ homeostasis, particularly by the ER and mitochondria, are summarized in the present review, focusing on AD.
Endoplasmic Reticulam is the Calcium Source Inside the Neuron
Subcellular Structures Formed by ER in the Neuron
The ER extends into every portion of the neuron to form an elaborate network, also considered as “a neuron within a neuron” (Berridge, 1998; Wu et al., 2017). The ER membrane, which connects with the nuclear envelope, also connects with the plasma membrane to form various types of specialized regions named the subsurface cisternae (located in the soma and initial dendrites, similar to the triadic junction in myocytes), the cisternae organelle (multilayered subsurface cisternae situated in the initial segment of the axon), the hypolemmal cisternae (located in the axon), and the spine apparatus (located in the dendritic spine) (Berridge, 1998).
Two Primary ER Ca2+ Channels: InsP3R and RyR
Types and Distribution of ER Ca2+ Channels
As in other cell types, neuronal ER also contains the inositol 1,4,5-triphosphate receptor (InsP3R) and the ryanodine receptor (RyR), sharing similar characteristics (Galione et al., 1993; Striggow and Ehrlich, 1996). Structurally, InsP3Rs are homo- or hetero-tetrameric assemblies that own three isoforms, namely type 1 (InsP3R1), type 2 (InsP3R2), type 3 (InsP3R3) (Taylor, 1998; Spät et al., 2008). Similarly, RyRs are tetrameric proteins that possess three subtypes: RyR1, the skeletal muscle type; RyR2, the cardiac muscle type; RyR3, the brain type (Querfurth et al., 1997; Spät et al., 2008). Functionally, InsP3Rs and RyRs are chemically-gated Ca2+ channels that evoke the regenerative Ca2+ wave from the ER lumen to the cytosol, also known as the Ca2+-induced Ca2+ release (CICR) (Martonosi, 1984; Berridge, 1998; Spät et al., 2008). Seemingly, InsP3Rs and RyRs have evolved from the same ancestor owing to the similarities (Berridge, 1997).
Spatially, InsP3Rs and RyRs share similar but not identical distributions in neurons (Berridge, 1998). From the subcellular perspective, InsP3Rs spread widely within the neuron, while RyRs localize predominantly in the soma (Walton et al., 1991; Kuwajima et al., 1992; Takei et al., 1992). Concerning mouse hippocampal neurons, both RyRs and InsP3Rs coexist densely within the soma; but are distributed heterogeneously within dendrites: RyRs are restricted to the proximal region of dendrites, InsP3Rs are found in the whole region of dendrites (Seymour-Laurent and Barish, 1995). Intriguingly, inspecting dendrites of chicken cerebellum Purkinje cells, there are only InsP3Rs and no RyRs within the dendritic spine, but there are both InsP3Rs and RyRs within the dendritic shaft (Walton et al., 1991). From the anatomical perspective, the cardiac muscle type RyR2, which conducts the Ca2+-elicitated Ca2+ release, is detected throughout the brain; nevertheless, the skeletal muscle type RyR1, which performs the depolarization-evoked Ca2+ release, is seen exclusively in the cerebellum; the brain type RyR3 is distributed within the hippocampus, cortex, and corpus striatum (Kuwajima et al., 1992; Querfurth et al., 1997).
Elementary and Global Ca2+ Signals from ER
Neuronal Ca2+ signal initiates with increasing of [Ca2+]CYT, which is followed by decreasing of [Ca2+]CYT to the resting level (Miller, 1991). Although various types of Ca2+ signals are named in different ways, it is less important to focus on the terminology but essential for identifying their characteristics (Berridge et al., 1999).
The elementary Ca2+ signals originating from ER Ca2+ channel own hierarchical characteristics (Bootman et al., 1997; Berridge et al., 1999). At the fundamental level, the “blip” from InsP3R and the “quark” from RyR are analogous, both of which are evoked from a single channel (Bootman et al., 1997). At the intermediate level, the “puff” from InsP3Rs and the “spark” from RyRs are similar, both of which are liberated from clusters of channels (Bootman et al., 1997). These elementary Ca2+ signals are characterized by a quick rise period followed by a slow recovery period (Berridge, 1997). The underlying mechanism is that the opening of the channel leads to a plume of Ca2+ releasing from ER lumen; after the channel’s closing, the released Ca2+ plume dissipates slowly by diffusion (Berridge, 1997).
These elementary Ca2+ signals construct the global Ca2+ signals, such as waves (at the subcellular level) and oscillations or spikes (at the whole-cell level) (Bootman et al., 1997; Berridge et al., 1999). Ca2+ waves propagate by regional Ca2+ diffusions and neighbor Ca2+ regenerations, based on the CICR, a positive feedback mechanism (Bootman et al., 1997). Furthermore, CICR is regulated by the positive and negative feedback influence of Ca2+ on the InsP3R or RyRs, which are discussed later (Berridge, 1997). Under high, intermediate, low positive feedback CICR, the Ca2+ waves, respectively, are continuous, saltatory, and abortive (Bootman et al., 1997).
Regulation of InsP3R Ca2+ Channel
The Ca2+-release activity from the opened InsP3R, at least, is regulated by the InsP3, [Ca2+]CYT, and [Ca2+]ER. Under a modest concentration of InsP3, the opening of InsP3R is biphasically regulated by cytosolic Ca2+: the low [Ca2+]CYT (<1 μM) can activate InsP3R; in contrast, the high [Ca2+]CYT (>1–10 μM) can inhibit the channel (Bootman and Lipp, 1999). Under the circumstance mentioned above, the original graph describing the probability of the InsP3R opening against the [Ca2+]CYT level reveals a bell-shaped curve (Bootman and Lipp, 1999). The ascending portion of the bell-shaped curve yields the positive feedback effect of the [Ca2+]CYT on the InsP3R opening, which allows the localized elementary Ca2+ signal to spread regeneratively as Ca2+ waves (Berridge, 1997; Sun et al., 1998). The descending portion of the bell-shaped curve represents the negative feedback dependence of the InsP3R opening on the [Ca2+]CYT, which terminates the elementary Ca2+ signal (Berridge, 1997; Sun et al., 1998).
Constructively, Adkins and Taylor suggest that InsP3 acts as a molecular switch that converts the InsP3R from a condition under which only an inhibitory Ca2+-binding site is feasible to one under which only a stimulatory Ca2+-binding site is viable (Adkins and Taylor, 1999). Sequentially, two steps are required for opening the InsP3R: initially, it becomes a liganded InsP3R by binding with InsP3; subsequently, it becomes an active InsP3R by binding with Ca2+ at the stimulatory Ca2+-binding site (Adkins and Taylor, 1999).
Nevertheless, the bell-shaped dependence of the InsP3R opening on the [Ca2+]CYT is not always expected. If the high [Ca2+]CYT (100 μM) is applied secondary to the maximal concentration of InsP3 (10 μM), the cytosolic Ca2+ fails to inhibit the Ca2+ release from the liganded InsP3R; in turn, if the high [Ca2+]CYT (100 μM) is given before the InsP3 (10 μM), the cytosolic Ca2+ can entirely inhibit the Ca2+ release from the unliganded InsP3R (Adkins and Taylor, 1999). Moreover, the liganded InsP3R owns a limited time window beyond which it undergoes intrinsic inactivation, and then the cytosolic Ca2+ cannot activate the InsP3R (Bootman and Lipp, 1999). Notably, although the opening of InsP3R requires binding with both InsP3 and Ca2+, it might not necessarily need the cytosolic Ca2+ (Bootman and Lipp, 1999). When [Ca2+]ER is low, the opening of InsP3R requires both InsP3 and cytosolic Ca2+; however, when [Ca2+]ER is high, there is no requirement for cytosolic Ca2+, it is enough for InsP3 itself to open the InsP3R (Missiaen et al., 1994).
Collectively, at the high InsP3 level and the low [Ca2+]ER level, the high [Ca2+]CYT cannot inhibit InsP3R because most InsP3Rs are liganded (Adkins and Taylor, 1999). At the low InsP3 level and the high [Ca2+]ER level, the low [Ca2+]CYT cannot activate InsP3R due to InsP3 alone can open the InsP3R (Missiaen et al., 1994).
Regulation of RyR Ca2+ Channel
The RyR is opened and releases Ca2+ into the cytosol by Ca2+ binding with the high-affinity stimulatory site; the Ca2+ is released until the local [Ca2+]CYT rises to the point where the low-affinity inhibitory site is bound, resulting in the RyR closing, which is the mechanism of CICR mediated by RyR (Payne et al., 2013). RyR1 and RyR2 are studied extensively in skeletal myocyte and cardiac myocyte, respectively. Dihydropyridine receptor (DHPR)-coupled RyR1 is opened upon depolarization of the plasma membrane and then is closed upon repolarization; subsequently, the surrounding uncoupled RyR1 is regeneratively opened under the CICR mechanism (Berridge, 1997). RyR2 is opened by the brief cytosolic Ca2+ pulse from DHPR, which is activated upon depolarization of the plasma membrane; approximately four RyR2s together evoke the Ca2+ quark, then these quarks turn to sparks, finally to waves (Berridge, 1997). Additionally, the activation of RyR is also regulated by the [Ca2+]ER level (Györke and Györke, 1998). Similar to InsP3R, when [Ca2+]ER is overloaded, the Ca2+-release activity of RyR is also significantly potentiated (Cheng et al., 1996).
Mitochondria are Calcium Buffers Inside the Neuron
Mitochondria-Linked Cytosolic Ca2+ Buffering
In addition to synthesizing adenosine triphosphate (ATP), another primary function of mitochondria is buffering intracellular Ca2+ (Miller, 1991). Neuronal mitochondria segregate Ca2+ under both physiological and pathological conditions (Miller, 1991). The Ca2+ buffering ability of mitochondria may lead to the accumulation of abundant Ca2+ in a particular domain in neurons (Rizzuto et al., 2012). Mitochondria may function as the last line against the exaggerated [Ca2+]CYT, which may be fatal for cells when other intracellular Ca2+-regulating mechanisms are exhausted (Martonosi, 1984). It is considered that the majority of mitochondria are generated in the soma, and the dysfunctional mitochondria return to the soma for degradation (Sheng and Cai, 2012).
Mitochondria usually cluster in neuronal domains with high demand for ATP, such as presynaptic and postsynaptic terminals (Tang and Zucker, 1997). In neurons, mitochondria located in proximal to Ca2+ channels, such as NMDAR on the postsynaptic density, can accumulate the cytosolic Ca2+ and prevent the propagation of Ca2+ waves, a global Ca2+ signal (Rizzuto et al., 2012). In the post-tetanic potentiation, mitochondria in the presynaptic terminal regulate the [Ca2+]CYT by buffering extra intraneuronal Ca2+: during tetanic stimulation, mitochondria take up Ca2+; after tetanic stimulation, mitochondria release Ca2+ into the cytosol, maintaining the [Ca2+]CYT at a relatively high level (Tang and Zucker, 1997).
Mitochondria-Located Ca2+ Machinery
Logically, the entrance of Ca2+ into the mitochondrial matrix requires passing through two intracellular membranes: the outer mitochondrial membrane (OMM) and the inner mitochondrial membrane (IMM). The OMM is permeable to ions attributed to the massive expression of voltage-dependent anion channels (VDAC) (Rizzuto et al., 2012). The notion that the expression level of VDACs seems to be the bottleneck of mitochondrial Ca2+ uptake is supported by the demonstration that over-expression of VDACs potentiates [Ca2+]MT; in contrast, down-regulation of VDACs attenuates [Ca2+]MT (Madesh and Hajnóczky, 2001; Rapizzi et al., 2002). Among three isoforms of VDACs (VDAC1, VDAC2, VDAC3), the VDAC1 isoform selectively interacted with InsP3R3 to transmit Ca2+ signal into the mitochondrial matrix that associates with apoptosis (De Stefani et al., 2012). Consistently, in the Chinese hamster ovary cell models that express all three isoforms of InsP3Rs, the InsP3R3 preferentially conducts Ca2+ signal into the mitochondria to induce apoptosis (Mendes et al., 2005).
The mitochondrial calcium uniporter (MCU) on the IMM can rapidly accumulate Ca2+ into the mitochondria matrix across the electrochemical gradient (Gunter and Gunter, 1994). MCU selectively binds Ca2+ with extremely high affinity (KD ≤ 2 nM) (Kirichok et al., 2004). MCU contains two transmembrane domains and significantly potentiates mitochondrial Ca2+ uptake after over-expression (De Stefani et al., 2011). Acidic residues, a binding site for ruthenium red and its analogs (the most potent inhibitors of MCU), reside in the highly conserved motif between the two transmembrane domains and are essential for the entire activity of MCU (Baughman et al., 2011). The mitochondrial calcium uptake 1 (MICU1) protein interacts directly with MCU to regulate the rapid Ca2+ uptake of mitochondria (Perocchi et al., 2010).
Calcium Cross-Talk Through Endoplasmic Reticulam-Mitochondria Contact Site
The ER has distributed the entire intracellular space from the nucleus to the plasma membrane intertwining all organelles, including mitochondria (Giorgi et al., 2009; Lebiedzinska et al., 2009; Wu et al., 2018). The ER network in which mitochondria are embedded exists in all compartments of neurons (Wu et al., 2017; Wu et al., 2018). The endoplasmic reticulum-mitochondria contact site (ERMCS) is abundant in every neuronal domain from the soma to dendrites and the axon (Wu et al., 2017). The distance between the two membranes in ERMCS is less than 200 nm (Rizzuto et al., 1998). Since the early 1960s, several different contact sites between the opposing membranes have been identified, such as the plasma membrane-ER contact site and the plasma membrane-mitochondria contact site (Lebiedzinska et al., 2009).
Mitochondrial Ca2+ uptake can occur at the ERMCS, where a high concentration of Ca2+ transports from the ER lumen into the mitochondrial matrix (Rizzuto et al., 2012). Briefly, Ca2+ releases from ER membrane via the InsP3R, followed by mitochondrial Ca2+ uptake by the VDAC on OMM, subsequently by the MCU on the IMM (Figure 2) (Rizzuto et al., 2012). Mitochondrial Ca2+ uptake can regulate the activity of InsP3R by decreasing the [Ca2+]CYT nearby ER membrane (Rizzuto et al., 2012). During Ca2+ absorbing by mitochondria, the [Ca2+]CYT near the InsP3R mouth is not high enough to block the channel; hence the InsP3R sustain opening, and the Ca2+ release from ER is prolonged (Boitier et al., 1999; Hajnóczky et al., 1999; Rizzuto et al., 2012). The increased ERMCS may induce the mitochondrial Ca2+ overload following Ca2+ release from the ER; conversely, the decreased ERMCS may impair Ca2+-dependent mitochondrial metabolism (Csordás et al., 2006; Lebiedzinska et al., 2009). As mentioned before, InsP3R3-VDAC1 interaction seems to play a major role in Ca2+ fluxion in ERMCS (Mendes et al., 2005; De Stefani et al., 2012). Collectively, [Ca2+]ER, [Ca2+]CYT, and [Ca2+]MT are simultaneously regulated by ERMCS.
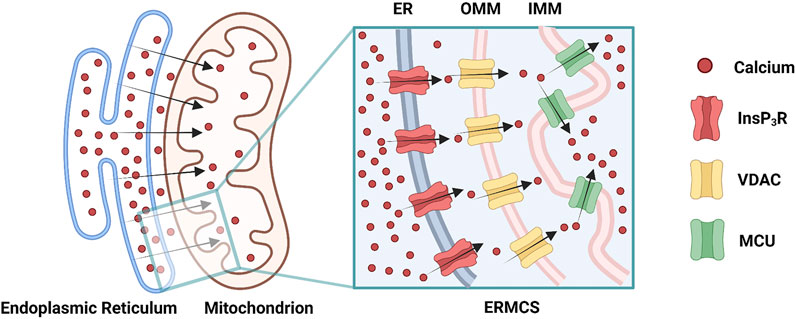
FIGURE 2. Ca2+ transmits through the endoplasmic reticulam-mitochondria contact site (ERMCS): Ca2+ releases from the endoplasmic reticulam (ER) membrane via the inositol 1,4,5-triphosphate receptor (InsP3R), followed by mitochondrial Ca2+ uptake by the voltage-dependent anion channel (VDAC) on the outer mitochondrial membrane (OMM), subsequently by the mitochondrial calcium uniporter (MCU) on the inner mitochondrial membrane (IMM) (Created with BioRender.com).
Alzheimer’s Disease: Irreversible Neurodegeneration Without Effective Therapies
Characteristics of Alzheimer’s Disease
Alzheimer’s disease (AD), first described in 1907 (Alzheimer et al., 1995), is a type of chronic neurodegenerative disease growing in number, which has brought physical sufferings, psychological stresses, and economic burden to individuals, families, and society (Alzheimer’sAssociation, 2020). Regrettably, there are no available medications for slowing, ceasing, or reversing the neuronal pathological progression that causes neurodegenerative symptoms and makes AD fatal (Alzheimer’sAssociation, 2020). Merely five drugs improving symptoms of AD have been approved by the Food and Drug Administration (FDA): three cholinesterase inhibitors (galantamine, rivastigmine, donepezil); one NMDAR blocker (memantine); one concomitant agent (memantine and donepezil) (Kumar et al., 2015; Atri, 2019; Alzheimer’sAssociation, 2020). Additionally, tacrine, a cholinesterase inhibitor approved once by FDA, is discontinued in the United States due to severe side effects, such as liver damage (Kumar et al., 2015; Alzheimer’sAssociation, 2016). Until 2021, 126 agents are in clinical trials for AD in the United States, and most investigational new drugs target modification of AD (Cummings et al., 2021). Recently, the repurposing and repositioning of conventional drugs is considered an alternative strategy for cancer therapy (Heckman-Stoddard et al., 2017; Huang et al., 2021). The same strategy could facilitate the identification of novel therapy for AD (Ballard et al., 2020).
Pathologically, the senile plaques (also known as β-amyloid plaques or neuritic plaques) and the neurofibrillary tangles (NFT) (also known as tau tangles or dystrophic neurites), observed inside and outside neurons, respectively, are two of several neuropathological features related to AD (Selkoe and Hardy, 2016; Alzheimer’sAssociation, 2020).
Based on the age of morbidity, Alzheimer’s disease is divided into two subtypes: the early-onset AD (EOAD), ranging from 30 years to 60 or 65 years; the late-onset AD (LOAD), defined with an onset age later than 60 or 65 years (Bekris et al., 2010). At the inheritance level, EOAD is characterized by the hereditary form, also known as the familial AD (FAD); by contrast, LOAD is typically termed as the sporadic AD (SAD) (Selkoe and Hardy, 2016; Kozlov et al., 2017).
Genetics of Alzheimer’s Disease
Mutations in the amyloid precursor protein (APP), presenilin-1 (PSEN1), and presenilin-2 (PSEN2) genes are genetically associated with FAD (Bekris et al., 2010). The APP gene resides on chromosome 21 (Selkoe, 1994). Indeed, individuals with Down syndrome (DS) have an increased risk of developing AD owing to trisomy 21 (Alzheimer’sAssociation, 2020). The PSEN1 gene, residing in chromosome 14, encodes the presenilin-1 protein of 467 amino acids which contains nine transmembrane domains; the PSEN2 gene, residing in chromosome 1, encodes the presenilin-2 protein of 448 amino acids topologically 67% identical to the presenilin-1 protein (Levy-Lahad et al., 1995; Sherrington et al., 1995; Cook et al., 1996; Leissring et al., 1999a; Laudon et al., 2005; Bekris et al., 2010). Mutations in the APP gene account for less than 5% of all FAD cases, mutations in the PSEN1 gene are responsible for approximately 70% of early-onset FAD (Van Broeckhoven, 1995). Consequently, mutations in the PSEN1 gene are the most common cause of presenile FAD; by contrast, mutations in the PSEN2 gene are a rare cause (Bekris et al., 2010). Mutations in the apolipoprotein E (APOE) gene, residing in chromosome 19, fulfill a significant role in SAD (Bertram and Tanzi, 2004; Bekris et al., 2010). Less than one hundred families with mutations in the APP gene, as well as several hundred families with mutations in the PSEN1 gene and the PSEN2 gene have been reported worldwide, hence the FAD cases would occur in less than 1% of all AD cases (Bekris et al., 2010; Castellani and Smith, 2011). More than 90% of individuals with AD would suffer the sporadic type of this disease (Bekris et al., 2010).
Integrating Amyloid Hypothesis and Calcium Hypothesis of Alzheimer's Disease
Following the “amyloid hypothesis” of AD, initiated by the study of Glenner and Wong in 1984, the accumulation of the amyloid-β (Aβ) peptide is the predominant force of AD-related pathogenesis, including plaques, tangles, synapse loss, and neuronal death (Glenner and Wong, 1984; Tanzi and Bertram, 2005). Although there are still several controversies (Castellani and Smith, 2011; Kozlov et al., 2017), the amyloid hypothesis, supported by many preclinical and clinical studies, has become the primary model of AD pathogenesis and has provided potential therapeutic targets for AD treatments (Selkoe and Hardy, 2016).
The “calcium hypothesis” of AD, which regards the persistent intraneuronal Ca2+ dyshomeostasis as one of the early causes of AD, is first proposed by Khachaturian based on limited direct evidence in the 1980s (Khachaturian, 1994; LaFerla, 2002). Growing lines of evidence have emerged to support the calcium hypothesis (Mattson et al., 2000). Ca2+ regulates a series of neuronal functions, such as neurotransmitter release and synaptic plasticity; in turn, neurons own precise mechanisms to sustain the Ca2+ homeostasis (LaFerla, 2002). For the intraneuronal Ca2+ dyshomeostasis to trigger the AD pathology, the Ca2+ signal perturbation must be an initial phenotype of AD, and the Ca2+ signaling dysregulation can affect the Aβ accumulation and the tau protein hyperphosphorylation (LaFerla, 2002). Although the former is still controversial (LaFerla, 2002), the latter is well accepted by viable evidence (Mattson, 1990; Mattson et al., 1993).
The relationship between the amyloid hypothesis and other potential hypotheses of AD may not conflict with one theory against another (Selkoe and Hardy, 2016). Moreover, integrating the amyloid hypothesis (Hardy and Selkoe, 2002; Bekris et al., 2010) and the calcium hypothesis (LaFerla, 2002) may further the understanding of Alzheimer’s disease pathogenesis. The calcium hypothesis remains compelling, and targeting selective calcium pathways would be a competitive therapeutic approach for AD (LaFerla, 2002).
Amyloid-B Peptide is Associated With Calcium Dyshomeostasis in Alzheimer's Disease
Aβ Forms Ca2+-Permeable Channel
Aβ peptides form Ca2+-permeable channels (also known as Aβ channels) on the plasma membrane and disrupt Ca2+ homeostasis by rapidly elevating intracellular Ca2+ concentration, leading to neuronal death in AD (Figure 3) (Arispe et al., 1993; Arispe et al., 1994b). The physical and chemical characteristics of Aβ peptides enable the formation of the β-sheet and subsequent aggregation into dimers and, even, large oligomers, which form β-barrel structures for the cation-selective permeability, particularly for Ca2+ (Figure 3) (Kagan et al., 2002). The nanomole (nM)-level concentrations of Aβ42 can form Ca2+-permeable channels, which elevate [Ca2+]CYT levels and rapidly elicit the degeneration of cultured endothelial cells (Bhatia et al., 2000). When incorporating Aβ40 into the artificial bilayer membrane, Ca2+ permeates through the opened Aβ channels (Arispe et al., 1993). The Ca2+ influxes through these channels would prevail due to the most significant electrochemical gradient between extracellular Ca2+ concentration and [Ca2+]CYT (Arispe et al., 1993; Arispe et al., 1994a). For a neuron with a single Aβ channel in opening state, the corresponding Ca2+ influx would increase the [Ca2+]CYT level at a rate of 5 μmol per second (5 μM/s), exhausting the neuronal Ca2+ buffering capacity rapidly, subsequently leading to the neurotoxicity (Arispe et al., 1993; Arispe et al., 1994a).
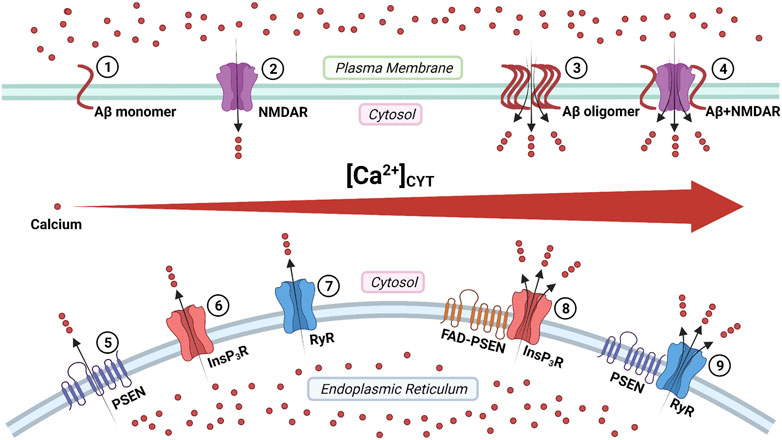
FIGURE 3. The regulation of calcium signaling by cellular membrane systems: ①③ amyloid-β (Aβ) peptide monomers aggregate into the oligomer which forms Ca2+-permeable channel; ②④ the Aβ accumulation promotes the persistent Ca2+ signal through the N-methyl-D-aspartate receptor (NMDAR); ⑤ the presenilin (PSEN) holoprotein functions as the endoplasmic reticulum (ER) passive Ca2+ leak channel; ⑥⑧ the enhanced Ca2+ signal by the familial Alzheimer's disease- causing mutant presenilin (FAD-PSEN) is the inositol 1,4,5-triphosphate receptor (InsP3R) dependent; ⑦⑨ the interaction between the PSEN and the ryanodine receptor (RyR) regulates the Ca2+ signal (Created with BioRender.com).
Aβ Activates NMDAR
The N-methyl-D-aspartate receptor (NMDAR) is named by its specific agonist, N-methyl-D-aspartate (NMDA), which does not occur naturally. NMDARs belong to one ionotropic family of glutamate receptors located on the plasma membrane. NMDARs can integrate two extracellular chemical stimuli (glycine and glutamate) and one membrane electrical stimulus (the depolarization of the plasma membrane) into the Ca2+ signal (Lipton and Rosenberg, 1994; Furukawa et al., 2005). Structurally, NMDARs constitute three families of subunits: glycine-binding NR1, which owns eight isoforms; glutamate-binding NR2, including NR2A, NR2B, NR2C, and NR2D; glycine-binding NR3, including NR3A and NR3B (Cull-Candy and Leszkiewicz, 2004; Furukawa et al., 2005). Functional NMDARs are tetrameric assemblies composed of two copies of NR1/NR2 heterodimers, sometimes NR1/NR3 heterodimers (Chen and Wyllie, 2006). Moreover, identical or diverse NR2 subunits form di-heteromeric assemblies (such as NR1-NR1-NR2A-NR2A, NR1-NR1-NR2B-NR2B) or tri-heteromeric assemblies (such as NR1-NR1-NR2A-NR2B, NR1-NR1-NR2B-NR2D) (Cull-Candy and Leszkiewicz, 2004; Köhr, 2006). Additionally, massive excitatory and inhibitory neurons encode at least two types of NR2 subunits to give rise to di-heteromeric or tri-heteromeric NMDARs in the same neuron (Köhr, 2006). Speculatively, at least 80 kinds of NMDAR subtypes may exist in the central nervous system (Cull-Candy and Leszkiewicz, 2004).
The overstimulation of NMDARs generates massive Ca2+ influxes that overexcite neurons, finally leading to neuronal death (a pathological condition also known as excitotoxicity) (Lipton and Rosenberg, 1994; Lynch and Guttmann, 2002). The Aβ accumulation promotes the persistent Ca2+ influx through NMDARs, leading to neuronal excitotoxicity at the early stage AD (Figure 3) (Parameshwaran et al., 2008). Furthermore, the monomeric and oligomeric Aβ42 elevate the [Ca2+]CYT level by activating the NR2B subunit of NMDARs in cultured cortical neurons (Ferreira et al., 2012). In turn, prolonged activation of extrasynaptic NMDARs, not synaptic NMDARs, promotes the production of Aβ in cultured cortical neurons (Lesné et al., 2005; Bordji et al., 2010). It reveals a positive feedback interaction between Aβ and NMDAR.
NMDAR-Related Mitochondrial Ca2+ Uptake
Notably, compared with non-NMDARs or voltage-gated Ca2+ channels, NMDAR-related mitochondrial Ca2+ uptake is faster and tighter (Peng and Greenamyre, 1998). When neuronal [Ca2+]CYT is elevated by NMDARs, the cytosol Ca2+ is segregated by the mitochondrial Ca2+ uptake; meanwhile, the mitochondrial Ca2+ transient persistently depolarizes the mitochondrial membrane potential (ΔΨ), causing the opening of the permeability transition pore (PTP) and further depolarising the ΔΨ, which parallels with the level of neuronal death (Schinder et al., 1996). Furthermore, under the circumstance in which the [Ca2+]CYT elevated vastly, mitochondria divert their function from ATP synthesis to Ca2+ accumulation (Lipton and Rosenberg, 1994). Additionally, the lack of ATP synthesis affects Na+-K+-ATPase activity and results in plasm membrane depolarization, which alleviates the Mg2+ block of NMDARs and further activates NMDARs (Greene and Greenamyre, 1996). Mitochondrial Ca2+ uptake regulates NMDAR activity under a positive feedback mechanism.
Considering the fundamental role of NMDARs in normal synaptic functions, a complete antagonism of NMDARs generates the majority of side effects, such as severe memory impairment (Hardingham and Bading, 2010; Mota et al., 2014). Coincidentally, extrasynaptic NMDARs have been largely associated with neuronal excitotoxicity (Hardingham and Bading, 2010), and extrasynaptic NMDARs mainly contain NR2B subunits (Petralia, 2012). Thus, the selective blockage of extrasynaptic NR2B subunits may be a potential strategy to prevent synaptic dysfunction in AD (Mota et al., 2014).
Presenilins are Related to Calcium Dyshomeostasis in Alzheimer's Disease
PSENs regulate Ca2+ signaling, and FAD-causing mutant PSENs perturb Ca2+ homeostasis (Leissring et al., 2000; LaFerla, 2002). Spatially, both PSEN1 and PSEN2 are mainly found on the ER membrane (Kovacs et al., 1996) and are widely expressed throughout the central nervous system (Cribbs et al., 1996). A series of FAD-causing mutant PSENs disrupt Ca2+ signaling (LaFerla, 2002). PSEN1-deficient neurons also reveal an increased [Ca2+]CYT level after exposure to H2O2 (Nakajima et al., 2001). Indeed, PSENs do not contain any Ca2+-binding motif, so presenilins may interact with several Ca2+-binding proteins to regulate Ca2+ signaling (LaFerla, 2002).
Cleaved Presenilins on the Plasma Membrane Possess γ-secretase Activity
The well-known function of PSENs is to provide the catalytic component of the γ-secretase complex, a membrane-embedded protease for several integral membrane proteins (De Strooper et al., 1998; Wolfe et al., 1999). PSEN has nine transmembrane domains (TMD) (Laudon et al., 2005). During maturation, PSEN is cleaved into a 30 KDa amino-terminal fragment (NTF) and a 20 KDa carboxy-terminal fragment (CTF) within a cytosol sizeable hydrophilic loop between TMD-6 and TMD-7 by endoproteolysis (Wolfe et al., 1999). Immature (or un-cleaved) presenilin holoproteins are localized on the ER membrane (Annaert et al., 1999). The endoproteolytic cleavage of PSEN holoproteins occurs on the ER membrane (Tandon and Fraser, 2002; Honarnejad and Herms, 2012). The cleaved PSEN (a heterodimer of NTF and CTF), together with anterior pharynx-defective 1 (APH-1), presenilin enhancer 2 (PEN-2), and nicastrin (all are ER transmembrane proteins), form the γ-secretase complex (De Strooper, 2003; Cheung et al., 2010; Honarnejad and Herms, 2012). The γ-secretase complex forms on the ER membrane and subsequently traffics to the Golgi apparatus, finally housed on the plasma membrane to generate Aβ peptide from APP (De Strooper et al., 2012; Honarnejad and Herms, 2012).
Presenilin Holoproteins on the ER Membrane Function as Ca2+-Leaking Channels
Under the two suppositions that the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) acts with 100% efficiency and the [Ca2+]CYT level is 0.1 μM, the calculated upper limit value of the [Ca2+]ER is 2,400 μM (Tu et al., 2006). In contrast, by directing measurement, the estimated [Ca2+]ER level range is from 100 to 500 μM (Hofer, 1999). The leakiness of Ca2+ from the ER lumen to cytosol may explain the [Ca2+]ER level difference mentioned above (Tu et al., 2006).
Tu and colleagues initially proposed the “presenilin calcium leak channel hypothesis”, in which the un-cleaved PSEN holoprotein functions as an ER passive Ca2+ leak channel independently from its γ-secretase activity, based on their sophisticated experiments with PSEN1/PSEN2 double knockout mouse embryonic fibroblasts (DKO-MEFs) (Figure 3) (Tu et al., 2006). The perturbed intracellular Ca2+ signaling in DKO-MEFs manifests as the potentiated amplitude of bradykinin-induced Ca2+ response, the exaggerated content of ionomycin-sensitive Ca2+ pool, and the reduced rate of thapsigargin-induced Ca2+ leak, compared with the wild-type control (Tu et al., 2006). Subsequently, in their rescue experiments, the expression of PSEN1WT and PSEN2WT successfully rescue Ca2+ signaling abnormalities, but PSEN1M146V and PSEN2N141I do not (Tu et al., 2006). Similarly, in planar lipid bilayers (BLM), the PSEN1WT and PSEN2WT can form a low-conductance divalent-cation-permeable channel, but PSEN1M146V and PSEN2N141I can not (Tu et al., 2006).
Quantitatively, the directly-measured [Ca2+]ER level in DKO-MEFs (190 μM) is approximately 2-fold higher than it is in wild-type control (87 μM); moreover, it is calculated that PSENs account for 80% of the ER endogenous Ca2+-leaking ability (Tu et al., 2006). Additionally, PSEN1D257A, a mutation of catalytic aspartate indispensable for γ-secretase activity, forms a channel in BLM and alleviates all Ca2+ signaling perturbation in DKO-MEFs; specifically, PSEN1ΔE9 is a gain-of-function mutation that leads to Ca2+ over-leak from ER (Tu et al., 2006), likely contributing to elevated [Ca2+]CYT and depleted [Ca2+]ER (Bezprozvanny and Mattson, 2008). The presenilin calcium leak channel hypothesis is supported by Bandara and colleagues who investigated the role of PSEN2 in regulating [Ca2+]ER using a fluorescence resonance energy transfer (FRET) probe (Bandara et al., 2013). The knockdown of PSEN2 significantly increases the [Ca2+]ER level, and the overexpression of PSEN2 decreases the [Ca2+]ER level (Bandara et al., 2013).
Adversely, Kasri and colleagues showed opposite conclusions: the increased Ca2+ leak from ER and the decreased [Ca2+]ER level in the same DKO-MEFs model (Kasri et al., 2006). The presenilin calcium leak channel hypothesis is under suspicion by directly measuring ER Ca2+ dynamics (Shilling et al., 2012).
FAD-Causing Mutant Presenilins Increase the Probability of InsP3R Opening
In 1994, Ito and colleagues first demonstrated that the InsP3-mediated Ca2+ liberation was potentiated in the skin fibroblast from AD patients (later known to harbor the PSEN1A246Q mutation, a FAD-causing mutation) (Ito et al., 1994; LaFerla, 2002). In 1999, Leissring and colleagues found that the InsP3-mediated Ca2+ liberation was enhanced in the Xenopus oocytes model, expressing PSEN1M146V, PSEN2N141I, and PSEN2M239V, all of which are FAD-causing mutations (Leissring et al., 1999a; Leissring et al., 1999b). The underlying mechanism is that FAD-causing mutant PSENs (PSEN1M146L, PSEN1L166P, PSEN1A246E, PSEN1G384A, PSEN2N141I) significantly elevate the probability of InsP3R opening compared with wild-type control (Cheung et al., 2008; Cheung et al., 2010). Interestingly, γ-secretase-eliminated mutant PSENs (PSEN1D257A, PSEN1D385A) also considerably enhance the InsP3R opening, which indicates that the γ-secretase activity of PSEN is not required for its influence on InsP3R opening (Cheung et al., 2010). Suppression of InsP3R1 expression genetically by 50% can normalize the enhanced InsP3R-mediated Ca2+ signaling associated with FAD-causing mutant PSENs (PSEN1M146V) and profoundly decreases both Aβ accumulation and tau protein hyperphosphorylation in cortical and hippocampal neurons of transgenic mice (Shilling et al., 2014). These lines of evidence support that the enhanced intraneuronal Ca2+ signaling by FAD-causing mutant PSENs is InsP3R dependent, and targeting the InsP3 signaling pathway could be a potential therapeutic strategy for FAD (Figure 3) (Shilling et al., 2014).
Cytosolic Amino-Terminal Fragment of Presenilins Regulates RyR-Mediated Ca2+ Release
Payne and colleagues identified a novel mechanism under which the interaction between the cytosolic amino-terminal fragment of presenilin (PSEN-NTFCYT) and RyR regulates the Ca2+ signal from ER (Figure 3) (Payne et al., 2013). Physiological normal Ca2+ concentration (10 nM < [Ca2+]CYT < 1 μM) and pathological high Ca2+ concentration ([Ca2+]CYT > 10 μM) are required for the cytosolic amino-terminal fragment residues 1–82 of presenilin-1 (PSEN1-NTFCYT1-82) and the cytosolic amino-terminal fragment residues 1–87 of presenilin-2 (PSEN2-NTFCYT1-87) to bind RyR, respectively (Hayrapetyan et al., 2008; Rybalchenko et al., 2008; Payne et al., 2013). After PSEN1-NTFCYT1-82 binding RyR at normal [Ca2+]CYT, the single RyR opening probability and mean currents are potentiated, causing an increased rate of Ca2+ release (Figure 3) (Rybalchenko et al., 2008; Payne et al., 2013). Hence, the whole-neuron net Ca2+ release from ER is reduced due to the inhibitory Ca2+ concentration being reached in a shorter time (Rybalchenko et al., 2008; Payne et al., 2013). After PSEN2-NTFCYT1-87 binding RyR at high [Ca2+]CYT, the low-affinity inhibitory Ca2+-binding site is blocked, resulting in more elevated [Ca2+]CYT is required to close the RyR, which represent a potential feedforward mechanism of Ca2+ dysregulation (Hayrapetyan et al., 2008; Payne et al., 2013).
Discussion
For receiving information about the changing environment, cells evolved the ability to signal (Clapham, 2007). Even though the precise definition of the signal is still controversial, it is recently stated that anything that changes could be a signal (Chakravorty, 2018). Ca2+ is elegantly manipulated by cells, particularly neurons, as a second messenger (Clapham, 2007). The unequal distribution of ions inside and outside neurons, such as K+, Na+, and Cl−, keeps the cellular function by generating the resting membrane potential and holds the neuronal volume by maintaining the osmotic balance (Byrne et al., 2014). It is widely known that the large gradient between extracellular and intracellular Ca2+ concentration levels is the most significant among particles with electrical charges. Cells possess numerous molecular machinery to regulate the Ca2+ distribution spatially and temporally; simultaneously, numbers of biochemical reactions are controlled by intracellular Ca2+. Therefore, the Ca2+ signal can transmit various information throughout the cells, and neurons are no exception (Berridge et al., 2000).
The generation and termination of the Ca2+ signal are featured as increasing [Ca2+]CYT and decreasing [Ca2+]CYT, respectively (Miller, 1991). Multiple Ca2+ channels exist in the various compartment of neurons to perform separate functions (Berridge et al., 2000). The [Ca2+]CYT is changed by extracellular stimuli through directly activating the gated Ca2+ channels on the plasma membrane or indirectly triggering the Ca2+-release channels on intracellular Ca2+ stores (Takei et al., 1992). In turn, Ca2+, released from ER, can alter transmembrane potential and regulate the excitability of neurons (Berridge, 1998). Spatiotemporally different Ca2+ signals modulate a series of neuronal functions, such as neurotransmitter release, post-tetanic potentiation, long-term potentiation (LTP), and long-term depression (LTD) (Purves et al., 2018). For example, large and fast Ca2+ signals evoke LTP, and small and slow Ca2+ signals trigger LTD (Purves et al., 2018). For neurons under physiological conditions, [Ca2+]CYT, [Ca2+]ER, and [Ca2+]MT are at a subtle equilibrium level. Both ER and mitochondria can shape the [Ca2+]CYT. In addition, the Ca2+ in the ER lumen can transmit into the mitochondrial matrix through ERMCS (Wu et al., 2018). Collectively, maintaining the Ca2+ homeostasis is vital for neurons.
Dysregulation in Ca2+ signaling has been reported in neurodegenerative diseases, such as AD, Parkinson’s disease (PD), and Huntington’s disease (HD) (Bezprozvanny and Mattson, 2008; Sheng and Cai, 2012; Pchitskaya et al., 2018). The [Ca2+]ER is overfilled in AD, whereas depleted in PD and HD (Pchitskaya et al., 2018). In Caenorhabditis elegans, mutations in the SEL-12 (the PSEN ortholog) can elevate the [Ca2+]MT level, and reducing the Ca2+ signal from ER to mitochondria normalizes the [Ca2+]MT level and the mitochondrial function (Sarasija et al., 2018). In neurons, mitochondria dysfunction is recognized as a final pathway in neurodegeneration (Friedman et al., 2010; Rizzuto et al., 2012). Area-Gomez and colleagues observed that PSENs are abundant in ERMCS (Area-Gomez et al., 2009), later the same research team demonstrated that mutations in PSEN1, PSEN2, and APP can upregulate the function of ERMCS (Area-Gomez et al., 2012). Moreover, variations in ERMCS likely influence the cellular Ca2+ homeostasis (Area-Gomez et al., 2012).
The present review summarizes the intracellular Ca2+ signaling regulated by molecular machinery on cellular membrane systems and the Ca2+ dyshomeostasis linked to Aβ and presenilins. Connecting the amyloid hypothesis with the calcium hypothesis may further the understanding of Alzheimer’s disease pathogenesis. At ER and mitochondria levels, understanding the regulation of cellular Ca2+ signaling and the mechanism underlying neuronal Ca2+ dyshomeostasis in AD may provide therapeutic targets for chronic neuronal degeneration disease in the central nervous system.
Author Contributions
Z-PJ conceived the project. D-XH, XY, W-JY, X-MZ, CL, H-PL, and YS searched and prepared references. D-XH wrote the manuscript and designed figures.
Funding
This work was financially supported by the Natural Science Foundation of Jilin Province (20210101310JC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Thanks to Dr. Huan-Fa Yi for critical comments and invaluable advice on this manuscript and the Central Laboratory of the First Hospital of Jilin University-the Eastern Division for support. Thanks to the Department of Pathology of the First Hospital of Jilin University-the Eastern Division for supporting this work.
References
Adkins, C. E., and Taylor, C. W. (1999). Lateral Inhibition of Inositol 1,4,5-trisphosphate Receptors by Cytosolic Ca2+. Curr. Biol. 9 (19), 1115–1118. doi:10.1016/s0960-9822(99)80481-3
Alzheimer’sAssociation (2016). 2016 Alzheimer's Disease Facts and Figures. Alzheimer's Demen. 12 (4), 459–509. doi:10.1016/j.jalz.2016.03.001
Alzheimer’sAssociation (2020). 2020 Alzheimer's Disease Facts and Figures. Alzheimers Dement 16 (3), 391–460. doi:10.1002/alz.12068
Annaert, W. G., Levesque, L., Craessaerts, K., Dierinck, I., Snellings, G., Westaway, D., et al. (1999). Presenilin 1 Controls γ-Secretase Processing of Amyloid Precursor Protein in Pre-golgi Compartments of Hippocampal Neurons. J. Cel Biol. 147 (2), 277–294. doi:10.1083/jcb.147.2.277
Area-Gomez, E., de Groof, A. J. C., Boldogh, I., Bird, T. D., Gibson, G. E., Koehler, C. M., et al. (2009). Presenilins Are Enriched in Endoplasmic Reticulum Membranes Associated with Mitochondria. Am. J. Pathol. 175 (5), 1810–1816. doi:10.2353/ajpath.2009.090219
Area-Gomez, E., Del Carmen Lara Castillo, M., Tambini, M. D., Guardia-Laguarta, C., de Groof, A. J. C., Madra, M., et al. (2012). Upregulated Function of Mitochondria-Associated ER Membranes in Alzheimer Disease. Embo j 31 (21), 4106–4123. doi:10.1038/emboj.2012.202
Arispe, N., Pollard, H. B., and Rojas, E. (1994b). Beta-Amyloid Ca2+-Channel Hypothesis for Neuronal Death in Alzheimer Disease. Mol. Cel Biochem. 140 (2), 119–125. doi:10.1007/bf00926750
Arispe, N., Pollard, H. B., and Rojas, E. (1994a). The Ability of Amyloid β-Protein [AβP (1-40)] to Form Ca2+ Channels Provides a Mechanism for Neuronal Death in Alzheimer's Disease. Ann. N. Y Acad. Sci. 747, 256–266. doi:10.1111/j.1749-6632.1994.tb44414.x
Arispe, N., Rojas, E., and Pollard, H. B. (1993). Alzheimer Disease Amyloid Beta Protein Forms Calcium Channels in Bilayer Membranes: Blockade by Tromethamine and Aluminum. Proc. Natl. Acad. Sci. 90 (2), 567–571. doi:10.1073/pnas.90.2.567
Atri, A. (2019). Current and Future Treatments in Alzheimer's Disease. Semin. Neurol. 39 (2), 227–240. doi:10.1055/s-0039-1678581
Ballard, C., Aarsland, D., Cummings, J., O’Brien, J., Mills, R., Molinuevo, J. L., et al. (2020). Drug Repositioning and Repurposing for Alzheimer Disease. Nat. Rev. Neurol. 16 (12), 661–673. doi:10.1038/s41582-020-0397-4
Bandara, S., Malmersjö, S., and Meyer, T. (2013). Regulators of Calcium Homeostasis Identified by Inference of Kinetic Model Parameters from Live Single Cells Perturbed by siRNA. Sci. Signal. 6 (283), ra56. doi:10.1126/scisignal.2003649
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative Genomics Identifies MCU as an Essential Component of the Mitochondrial Calcium Uniporter. Nature 476 (7360), 341–345. doi:10.1038/nature10234
Bekris, L. M., Yu, C.-E., Bird, T. D., and Tsuang, D. W. (2010). Review Article: Genetics of Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 23 (4), 213–227. doi:10.1177/0891988710383571
Berridge, M. J., Bootman, M. D., and Lipp, P. (1998). Calcium - a Life and Death Signal. Nature 395 (6703), 645–648. doi:10.1038/27094
Berridge, M. J. (1997). Elementary and Global Aspects of Calcium Signalling. J. Physiol. 499 (Pt 2Pt 2), 291–306. doi:10.1113/jphysiol.1997.sp021927
Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The Versatility and Universality of Calcium Signalling. Nat. Rev. Mol. Cel Biol. 1 (1), 11–21. doi:10.1038/35036035
Berridge, M. J. (1998). Neuronal Calcium Signaling. Neuron 21 (1), 13–26. doi:10.1016/s0896-6273(00)80510-3
Berridge, M., Lipp, P., and Bootman, M. (1999). Calcium Signalling. Curr. Biol. 9 (5), R157–R159. doi:10.1016/s0960-9822(99)80101-8
Bertram, L., and Tanzi, R. E. (2004). Alzheimer's Disease: One Disorder, Too many Genes? Hum. Mol. Genet. 13 Spec No 1, 135R–141R. doi:10.1093/hmg/ddh077
Bezprozvanny, I., and Mattson, M. P. (2008). Neuronal Calcium Mishandling and the Pathogenesis of Alzheimer's Disease. Trends Neurosciences 31 (9), 454–463. doi:10.1016/j.tins.2008.06.005
Bhatia, R., Lin, H., and Lal, R. (2000). Fresh and Globular Amyloid β Protein (1-42) Induces Rapid Cellular Degeneration: Evidence for AβP Channel‐mediated Cellular Toxicity. FASEB j. 14 (9), 1233–1243. doi:10.1096/fasebj.14.9.1233
Boitier, E., Rea, R., and Duchen, M. R. (1999). Mitochondria Exert a Negative Feedback on the Propagation of Intracellular Ca2+ Waves in Rat Cortical Astrocytes. J. Cel Biol. 145 (4), 795–808. doi:10.1083/jcb.145.4.795
Bootman, M. D., and Lipp, P. (1999). Calcium Signalling: Ringing Changes to the 'bell-shaped Curve'. Curr. Biol. 9 (23), R876–R878. doi:10.1016/s0960-9822(00)80072-x
Bootman, M., Niggli, E., Berridge, M., and Lipp, P. (1997). Imaging the Hierarchical Ca2+ Signalling System in HeLa Cells. J. Physiol. 499 (Pt 2), 307–314. doi:10.1113/jphysiol.1997.sp021928
Bordji, K., Becerril-Ortega, J., Nicole, O., and Buisson, A. (2010). Activation of Extrasynaptic, but Not Synaptic, NMDA Receptors Modifies Amyloid Precursor Protein Expression Pattern and Increases Amyloid- Production. J. Neurosci. 30 (47), 15927–15942. doi:10.1523/jneurosci.3021-10.2010
Broeckhoven, C. V. (1995). Presenilins and Alzheimer Disease. Nat. Genet. 11 (3), 230–232. doi:10.1038/ng1195-230
Byrne, J. H., Heidelberger, R., and Neal Waxham, M. (2014). From Molecules to Networks: An Introduction to Cellular and Molecular Neuroscience. United Kingdom, United States: Academic Press.
Castellani, R. J., and Smith, M. A. (2011). Compounding Artefacts with Uncertainty, and an Amyloid cascade Hypothesis that Is 'too Big to Fail'. J. Pathol. 224 (2), 147–152. doi:10.1002/path.2885
Chakravorty, P. (2018). What Is a Signal? [Lecture Notes]. IEEE Signal. Process. Mag. 35 (5), 175–177. doi:10.1109/MSP.2018.2832195
Chen, P. E., and Wyllie, D. J. A. (2006). Pharmacological Insights Obtained from Structure-Function Studies of Ionotropic Glutamate Receptors. Br. J. Pharmacol. 147 (8), 839–853. doi:10.1038/sj.bjp.0706689
Cheng, H., Lederer, M. R., Lederer, W. J., and Cannell, M. B. (1996). Calcium sparks and [Ca2+]i Waves in Cardiac Myocytes. Am. J. Physiology-Cell Physiol. 270 (1 Pt 1), C148–C159. doi:10.1152/ajpcell.1996.270.1.C148
Cheung, K.-H., Mei, L., Mak, D.-O. D., Hayashi, I., Iwatsubo, T., Kang, D. E., et al. (2010). Gain-of-Function Enhancement of IP 3 Receptor Modal Gating by Familial Alzheimer's Disease-Linked Presenilin Mutants in Human Cells and Mouse Neurons. Sci. Signal. 3 (114), ra22. doi:10.1126/scisignal.2000818
Cheung, K.-H., Shineman, D., Müller, M., Cárdenas, C., Mei, L., Yang, J., et al. (2008). Mechanism of Ca2+ Disruption in Alzheimer's Disease by Presenilin Regulation of InsP3 Receptor Channel Gating. Neuron 58 (6), 871–883. doi:10.1016/j.neuron.2008.04.015
Cook, D. G., Sung, J. C., Golde, T. E., Felsenstein, K. M., Wojczyk, B. S., Tanzi, R. E., et al. (1996). Expression and Analysis of Presenilin 1 in a Human Neuronal System: Localization in Cell Bodies and Dendrites. Proc. Natl. Acad. Sci. 93 (17), 9223–9228. doi:10.1073/pnas.93.17.9223
Cribbs, D. H., Chen, L. S., Bende, S. M., and LaFerla, F. M. (1996). Widespread Neuronal Expression of the Presenilin-1 Early-Onset Alzheimer's Disease Gene in the Murine Brain. Am. J. Pathol. 148 (6), 1797–1806.
Csordás, G., Renken, C., Várnai, P., Walter, L., Weaver, D., Buttle, K. F., et al. (2006). Structural and Functional Features and Significance of the Physical Linkage between ER and Mitochondria. J. Cel Biol. 174 (7), 915–921. doi:10.1083/jcb.200604016
Cull-Candy, S. G., and Leszkiewicz, D. N. (2004). Role of Distinct NMDA Receptor Subtypes at central Synapses. Sci. STKE 2004 (255), re16. doi:10.1126/stke.2552004re16
Cummings, J., Lee, G., Zhong, K., Fonseca, J., and Taghva, K. (2021). Alzheimer's Disease Drug Development Pipeline: 2021. Alzheimer's Demen. Translational Res. Clin. Interventions 7 (1), e12179. doi:10.1002/trc2.12179
De Stefani, D., Bononi, A., Romagnoli, A., Messina, A., De Pinto, V., Pinton, P., et al. (2012). VDAC1 Selectively Transfers Apoptotic Ca2+ Signals to Mitochondria. Cell Death Differ 19 (2), 267–273. doi:10.1038/cdd.2011.92
De Stefani, D., Raffaello, A., Teardo, E., Szabò, I., and Rizzuto, R. (2011). A Forty-Kilodalton Protein of the Inner Membrane Is the Mitochondrial Calcium Uniporter. Nature 476 (7360), 336–340. doi:10.1038/nature10230
De Strooper, B. (2003). Aph-1, Pen-2, and Nicastrin with Presenilin Generate an Active γ-Secretase Complex. Neuron 38 (1), 9–12. doi:10.1016/s0896-6273(03)00205-8
De Strooper, B., Iwatsubo, T., and Wolfe, M. S. (2012). Presenilins and -Secretase: Structure, Function, and Role in Alzheimer Disease. Cold Spring Harbor Perspect. Med. 2 (1), a006304. doi:10.1101/cshperspect.a006304
De Strooper, B., Saftig, P., Craessaerts, K., Vanderstichele, H., Guhde, G., Annaert, W., et al. (1998). Deficiency of Presenilin-1 Inhibits the normal Cleavage of Amyloid Precursor Protein. Nature 391 (6665), 387–390. doi:10.1038/34910
Epstein, F. H., Lipton, S. A., and Rosenberg, P. A. (1994). Excitatory Amino Acids as a Final Common Pathway for Neurologic Disorders. N. Engl. J. Med. 330 (9), 613–622. doi:10.1056/nejm199403033300907
Ferreira, I. L., Bajouco, L. M., Mota, S. I., Auberson, Y. P., Oliveira, C. R., and Rego, A. C. (2012). Amyloid Beta Peptide 1-42 Disturbs Intracellular Calcium Homeostasis through Activation of GluN2B-Containing N-Methyl-D-Aspartate Receptors in Cortical Cultures. Cell Calcium 51 (2), 95–106. doi:10.1016/j.ceca.2011.11.008
Friedman, J. R., Webster, B. M., Mastronarde, D. N., Verhey, K. J., and Voeltz, G. K. (2010). ER Sliding Dynamics and ER-Mitochondrial Contacts Occur on Acetylated Microtubules. J. Cel Biol. 190 (3), 363–375. doi:10.1083/jcb.200911024
Furukawa, H., Singh, S. K., Mancusso, R., and Gouaux, E. (2005). Subunit Arrangement and Function in NMDA Receptors. Nature 438 (7065), 185–192. doi:10.1038/nature04089
Galione, A., McDougall, A., Busa, W. B., Willmott, N., Gillot, I., and Whitaker, M. (1993). Redundant Mechanisms of Calcium-Induced Calcium Release Underlying Calcium Waves during Fertilization of Sea Urchin Eggs. Science 261 (5119), 348–352. doi:10.1126/science.8392748
Giorgi, C., De Stefani, D., Bononi, A., Rizzuto, R., and Pinton, P. (2009). Structural and Functional Link between the Mitochondrial Network and the Endoplasmic Reticulum. Int. J. Biochem. Cel Biol. 41 (10), 1817–1827. doi:10.1016/j.biocel.2009.04.010
Glenner, G. G., and Wong, C. W. (1984). Alzheimer's Disease: Initial Report of the Purification and Characterization of a Novel Cerebrovascular Amyloid Protein. Biochem. Biophysical Res. Commun. 120 (3), 885–890. doi:10.1016/s0006-291x(84)80190-4
Greene, J. G., and Greenamyre, J. T. (1996). Manipulation of Membrane Potential Modulates Malonate-Induced Striatal Excitotoxicity In Vivo. J. Neurochem. 66 (2), 637–643. doi:10.1046/j.1471-4159.1996.66020637.x
Gunter, K. K., and Gunter, T. E. (1994). Transport of Calcium by Mitochondria. J. Bioenerg. Biomembr 26 (5), 471–485. doi:10.1007/bf00762732
Györke, I., and Györke, S. (1998). Regulation of the Cardiac Ryanodine Receptor Channel by Luminal Ca2+ Involves Luminal Ca2+ Sensing Sites. Biophys. J. 75 (6), 2801–2810. doi:10.1016/s0006-3495(98)77723-9
Hajnóczky, G., Hager, R., and Thomas, A. P. (1999). Mitochondria Suppress Local Feedback Activation of Inositol 1,4,5-Trisphosphate Receptors by Ca2+. J. Biol. Chem. 274 (20), 14157–14162. doi:10.1074/jbc.274.20.14157
Hardingham, G. E., and Bading, H. (2010). Synaptic versus Extrasynaptic NMDA Receptor Signalling: Implications for Neurodegenerative Disorders. Nat. Rev. Neurosci. 11 (10), 682–696. doi:10.1038/nrn2911
Hardy, J., and Selkoe, D. J. (2002). The Amyloid Hypothesis of Alzheimer's Disease: Progress and Problems on the Road to Therapeutics. Science 297 (5580), 353–356. doi:10.1126/science.1072994
Hayrapetyan, V., Rybalchenko, V., Rybalchenko, N., and Koulen, P. (2008). The N-Terminus of Presenilin-2 Increases Single Channel Activity of Brain Ryanodine Receptors through Direct Protein-Protein Interaction. Cell Calcium 44 (5), 507–518. doi:10.1016/j.ceca.2008.03.004
Heckman-Stoddard, B. M., DeCensi, A., Sahasrabuddhe, V. V., and Ford, L. G. (2017). Repurposing Metformin for the Prevention of Cancer and Cancer Recurrence. Diabetologia 60 (9), 1639–1647. doi:10.1007/s00125-017-4372-6
Hofer, A. M. (1999). Measurement of Free [Ca2+] Changes in Agonist-Sensitive Internal Stores Using Compartmentalized Fluorescent Indicators. Methods Mol. Biol. 114, 249–266. doi:10.1385/1-59259-250-3:249
Honarnejad, K., and Herms, J. (2012). Presenilins: Role in Calcium Homeostasis. Int. J. Biochem. Cel Biol. 44 (11), 1983–1986. doi:10.1016/j.biocel.2012.07.019
Huang, Y., Zhou, Z., Zhang, J., Hao, Z., He, Y., Wu, Z., et al. (2021). lncRNA MALAT1 Participates in Metformin Inhibiting the Proliferation of Breast Cancer Cell. J. Cel Mol Med 25 (15), 7135–7145. doi:10.1111/jcmm.16742
Ito, E., Oka, K., Etcheberrigaray, R., Nelson, T. J., McPhie, D. L., Tofel-Grehl, B., et al. (1994). Internal Ca2+ Mobilization Is Altered in Fibroblasts from Patients with Alzheimer Disease. Proc. Natl. Acad. Sci. 91 (2), 534–538. doi:10.1073/pnas.91.2.534
Kagan, B. L., Hirakura, Y., Azimov, R., Azimova, R., and Lin, M.-C. (2002). The Channel Hypothesis of Alzheimer's Disease: Current Status. Peptides 23 (7), 1311–1315. doi:10.1016/s0196-9781(02)00067-0
Kasri, N. N., Kocks, S. L., Verbert, L., Hébert, S. S., Callewaert, G., Parys, J. B., et al. (2006). Up-regulation of Inositol 1,4,5-trisphosphate Receptor Type 1 Is Responsible for a Decreased Endoplasmic-Reticulum Ca2+ Content in Presenilin Double Knock-Out Cells. Cell Calcium 40 (1), 41–51. doi:10.1016/j.ceca.2006.03.005
Khachaturian, Z. S. (1994). Calcium Hypothesis of Alzheimer's Disease and Brain Aginga. Ann. N. Y Acad. Sci. 747, 1–11. doi:10.1111/j.1749-6632.1994.tb44398.x
Kirichok, Y., Krapivinsky, G., and Clapham, D. E. (2004). The Mitochondrial Calcium Uniporter Is a Highly Selective Ion Channel. Nature 427 (6972), 360–364. doi:10.1038/nature02246
Köhr, G. (2006). NMDA Receptor Function: Subunit Composition versus Spatial Distribution. Cell Tissue Res. 326 (2), 439–446. doi:10.1007/s00441-006-0273-6
Kovacs, D. M., Fausett, H. J., Page, K. J., Kim, T.-W., Moir, R. D., Merriam, D. E., et al. (1996). Alzheimer-associated Presenilins 1 and 2 : Neuronal Expression in Brain and Localization to Intracellular Membranes in Mammalian Cells. Nat. Med. 2 (2), 224–229. doi:10.1038/nm0296-224
Kozlov, S., Afonin, A., Evsyukov, I., and Bondarenko, A. (2017). Alzheimer's Disease: as it Was in the Beginning. Rev. Neurosci. 28 (8), 825–843. doi:10.1515/revneuro-2017-0006
Kumar, A., Singh, A., and Ekavali, (2015). A Review on Alzheimer's Disease Pathophysiology and its Management: an Update. Pharmacol. Rep. 67 (2), 195–203. doi:10.1016/j.pharep.2014.09.004
Kuwajima, G., Futatsugi, A., Niinobe, M., Nakanishi, S., and Mikoshiba, K. (1992). Two Types of Ryanodine Receptors in Mouse Brain: Skeletal Muscle Type Exclusively in Purkinje Cells and Cardiac Muscle Type in Various Neurons. Neuron 9 (6), 1133–1142. doi:10.1016/0896-6273(92)90071-k
LaFerla, F. M. (2002). Calcium Dyshomeostasis and Intracellular Signalling in Alzheimer's Disease. Nat. Rev. Neurosci. 3 (11), 862–872. doi:10.1038/nrn960
Laudon, H., Hansson, E. M., Melén, K., Bergman, A., Farmery, M. R., Winblad, B., et al. (2005). A Nine-Transmembrane Domain Topology for Presenilin 1. J. Biol. Chem. 280 (42), 35352–35360. doi:10.1074/jbc.M507217200
Lebiedzinska, M., Szabadkai, G., Jones, A. W. E., Duszynski, J., and Wieckowski, M. R. (2009). Interactions between the Endoplasmic Reticulum, Mitochondria, Plasma Membrane and Other Subcellular Organelles. Int. J. Biochem. Cel Biol. 41 (10), 1805–1816. doi:10.1016/j.biocel.2009.02.017
Leissring, M. A., Akbari, Y., Fanger, C. M., Cahalan, M. D., Mattson, M. P., and LaFerla, F. M. (2000). Capacitative Calcium Entry Deficits and Elevated Luminal Calcium Content in Mutant Presenilin-1 Knockin Mice. J. Cel Biol. 149 (4), 793–798. doi:10.1083/jcb.149.4.793
Leissring, M. A., Parker, I., and LaFerla, F. M. (1999a). Presenilin-2 Mutations Modulate Amplitude and Kinetics of Inositol 1,4,5-Trisphosphate-Mediated Calcium Signals. J. Biol. Chem. 274 (46), 32535–32538. doi:10.1074/jbc.274.46.32535
Leissring, M. A., Paul, B. A., Parker, I., Cotman, C. W., and LaFerla, F. M. (1999b). Alzheimer's Presenilin-1 Mutation Potentiates Inositol 1,4,5-Trisphosphate-Mediated Calcium Signaling in Xenopus. J. Neurochem. 72 (3), 1061–1068. doi:10.1046/j.1471-4159.1999.0721061.x
Lesne, S., Ali, C., Gabriel, C., Croci, N., MacKenzie, E. T., Glabe, C. G., et al. (2005). NMDA Receptor Activation Inhibits -Secretase and Promotes Neuronal Amyloid- Production. J. Neurosci. 25 (41), 9367–9377. doi:10.1523/jneurosci.0849-05.2005
Levy-Lahad, E., Wijsman, E. M., Nemens, E., Anderson, L., Goddard, K. A. B., Weber, J. L., et al. (1995). A Familial Alzheimer's Disease Locus on Chromosome 1. Science 269 (5226), 970–973. doi:10.1126/science.7638621
Lynch, D. R., and Guttmann, R. P. (2002). Excitotoxicity: Perspectives Based onN-Methyl-D-Aspartate Receptor Subtypes. J. Pharmacol. Exp. Ther. 300 (3), 717–723. doi:10.1124/jpet.300.3.717
Madesh, M., and Hajnóczky, G. (2001). VDAC-dependent Permeabilization of the Outer Mitochondrial Membrane by Superoxide Induces Rapid and Massive Cytochrome C Release. J. Cel Biol. 155 (6), 1003–1016. doi:10.1083/jcb.200105057
Martonosi, A. N. (1984). Mechanisms of Ca2+ Release from Sarcoplasmic Reticulum of Skeletal Muscle. Physiol. Rev. 64 (4), 1240–1320. doi:10.1152/physrev.1984.64.4.1240
Mattson, M. P. (1990). Antigenic Changes Similar to Those Seen in Neurofibrillary Tangles Are Elicited by Glutamate and Ca2+ Influx in Cultured Hippocampal Neurons. Neuron 4 (1), 105–117. doi:10.1016/0896-6273(90)90447-n
Mattson, M. P., LaFerla, F. M., Chan, S. L., Leissring, M. A., Shepel, P. N., and Geiger, J. D. (2000). Calcium Signaling in the ER: its Role in Neuronal Plasticity and Neurodegenerative Disorders. Trends Neurosciences 23 (5), 222–229. doi:10.1016/s0166-2236(00)01548-4
Mattson, M. P., Lovell, M. A., Ehmann, W. D., and Markesbery, W. R. (1993). Comparison of the Effects of Elevated Intracellular Aluminum and Calcium Levels on Neuronal Survival and Tau Immunoreactivity. Brain Res. 602 (1), 21–31. doi:10.1016/0006-8993(93)90236-g
Mendes, C. C. P., Gomes, D. A., Thompson, M., Souto, N. C., Goes, T. S., Goes, A. M., et al. (2005). The Type III Inositol 1,4,5-trisphosphate Receptor Preferentially Transmits Apoptotic Ca2+ Signals into Mitochondria. J. Biol. Chem. 280 (49), 40892–40900. doi:10.1074/jbc.M506623200
Miller, R. (1991). The Control of Neuronal Ca2+ Homeostasis. Prog. Neurobiol. 37 (3), 255–285. doi:10.1016/0301-0082(91)90028-y
Missiaen, L., De Smedt, H., Parys, J. B., and Casteels, R. (1994). Co-activation of Inositol Trisphosphate-Induced Ca2+ Release by Cytosolic Ca2+ Is Loading-dependent. J. Biol. Chem. 269 (10), 7238–7242. doi:10.1016/s0021-9258(17)37273-3
Mota, S. I., Ferreira, I. L., and Rego, A. C. (2014). Dysfunctional Synapse in Alzheimer's Disease - A Focus on NMDA Receptors. Neuropharmacology 76 A, 16–26. doi:10.1016/j.neuropharm.2013.08.013
Nakajima, M., Miura, M., Aosaki, T., and Shirasawa, T. (2001). Deficiency of Presenilin-1 Increases Calcium-dependent Vulnerability of Neurons to Oxidative Stress In Vitro. J. Neurochem. 78 (4), 807–814. doi:10.1046/j.1471-4159.2001.00478.x
Parameshwaran, K., Dhanasekaran, M., and Suppiramaniam, V. (2008). Amyloid Beta Peptides and Glutamatergic Synaptic Dysregulation. Exp. Neurol. 210 (1), 7–13. doi:10.1016/j.expneurol.2007.10.008
Payne, A. J., Gerdes, B. C., Naumchuk, Y., McCalley, A. E., Kaja, S., and Koulen, P. (2013). Presenilins Regulate the Cellular Activity of Ryanodine Receptors Differentially through Isotype-specific N-Terminal Cysteines. Exp. Neurol. 250, 143–150. doi:10.1016/j.expneurol.2013.09.001
Pchitskaya, E., Popugaeva, E., and Bezprozvanny, I. (2018). Calcium Signaling and Molecular Mechanisms Underlying Neurodegenerative Diseases. Cell Calcium 70, 87–94. doi:10.1016/j.ceca.2017.06.008
Peng, T. I., and Greenamyre, J. T. (1998). Privileged Access to Mitochondria of Calcium Influx through N-Methyl-D-Aspartate Receptors. Mol. Pharmacol. 53 (6), 974–980.
Perocchi, F., Gohil, V. M., Girgis, H. S., Bao, X. R., McCombs, J. E., Palmer, A. E., et al. (2010). MICU1 Encodes a Mitochondrial EF Hand Protein Required for Ca2+ Uptake. Nature 467 (7313), 291–296. doi:10.1038/nature09358
Petralia, R. S. (20122012). Distribution of Extrasynaptic NMDA Receptors on Neurons. Scientific World J. 2012, 1–11. doi:10.1100/2012/267120
Purves, D., Augustine, G. J., Fitzpatrick, D., Hall, W. C., LaMantia, A.-S., Mooney, R. D., et al. (2018). Neuroscience. Sunderland, Massachusetts: Oxford University Press.
Querfurth, H. W., Jiang, J., Geiger, J. D., and Selkoe, D. J. (1997). Caffeine Stimulates Amyloid β-Peptide Release from β-Amyloid Precursor Protein-Transfected HEK293 Cells. J. Neurochem. 69 (4), 1580–1591. doi:10.1046/j.1471-4159.1997.69041580.x
Rapizzi, E., Pinton, P., Szabadkai, G., Wieckowski, M. R., Vandecasteele, G., Baird, G., et al. (2002). Recombinant Expression of the Voltage-dependent Anion Channel Enhances the Transfer of Ca2+ Microdomains to Mitochondria. J. Cel Biol. 159 (4), 613–624. doi:10.1083/jcb.200205091
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as Sensors and Regulators of Calcium Signalling. Nat. Rev. Mol. Cel Biol. 13 (9), 566–578. doi:10.1038/nrm3412
Rizzuto, R., Pinton, P., Carrington, W., Fay, F. S., Fogarty, K. E., Lifshitz, L. M., et al. (1998). Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca 2+ Responses. Science 280 (5370), 1763–1766. doi:10.1126/science.280.5370.1763
Rybalchenko, V., Hwang, S.-Y., Rybalchenko, N., and Koulen, P. (2008). The Cytosolic N-Terminus of Presenilin-1 Potentiates Mouse Ryanodine Receptor Single Channel Activity. Int. J. Biochem. Cel Biol. 40 (1), 84–97. doi:10.1016/j.biocel.2007.06.023
Sarasija, S., Laboy, J. T., Ashkavand, Z., Bonner, J., Tang, Y., and Norman, K. R. (2018). Presenilin Mutations Deregulate Mitochondrial Ca2+ Homeostasis and Metabolic Activity Causing Neurodegeneration in Caenorhabditis elegans. Elife 7. doi:10.7554/eLife.33052
Schinder, A. F., Olson, E. C., Spitzer, N. C., and Montal, M. (1996). Mitochondrial Dysfunction Is a Primary Event in Glutamate Neurotoxicity. J. Neurosci. 16 (19), 6125–6133. doi:10.1523/jneurosci.16-19-06125.1996
Selkoe, D. J., and Hardy, J. (2016). The Amyloid Hypothesis of Alzheimer's Disease at 25 Years. EMBO Mol. Med. 8 (6), 595–608. doi:10.15252/emmm.201606210
Selkoe, D. J. (1994). Normal and Abnormal Biology of the Beta-Amyloid Precursor Protein. Annu. Rev. Neurosci. 17, 489–517. doi:10.1146/annurev.ne.17.030194.002421
Seymour-Laurent, K., and Barish, M. (1995). Inositol 1,4,5-trisphosphate and Ryanodine Receptor Distributions and Patterns of Acetylcholine- and Caffeine-Induced Calcium Release in Cultured Mouse Hippocampal Neurons. J. Neurosci. 15 (4), 2592–2608. doi:10.1523/jneurosci.15-04-02592.1995
Sheng, Z.-H., and Cai, Q. (2012). Mitochondrial Transport in Neurons: Impact on Synaptic Homeostasis and Neurodegeneration. Nat. Rev. Neurosci. 13 (2), 77–93. doi:10.1038/nrn3156
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., et al. (1995). Cloning of a Gene Bearing Missense Mutations in Early-Onset Familial Alzheimer's Disease. Nature 375 (6534), 754–760. doi:10.1038/375754a0
Shilling, D., Mak, D.-O. D., Kang, D. E., and Foskett, J. K. (2012). Lack of Evidence for Presenilins as Endoplasmic Reticulum Ca2+ Leak Channels. J. Biol. Chem. 287 (14), 10933–10944. doi:10.1074/jbc.M111.300491
Shilling, D., Muller, M., Takano, H., Daniel Mak, D.-O., Abel, T., Coulter, D. A., et al. (2014). Suppression of InsP3 Receptor-Mediated Ca2+ Signaling Alleviates Mutant Presenilin-Linked Familial Alzheimer's Disease Pathogenesis. J. Neurosci. 34 (20), 6910–6923. doi:10.1523/jneurosci.5441-13.2014
Spat, A., Szanda, G., Csordas, G., and Hajnoczky, G. (2008). High- and Low-calcium-dependent Mechanisms of Mitochondrial Calcium Signalling. Cell Calcium 44 (1), 51–63. doi:10.1016/j.ceca.2007.11.015
Stelzmann, R. A., Norman Schnitzlein, H., Reed Murtagh, F., and Murtagh, F. R. (1995). An English translation of alzheimer's 1907 paper, "ber eine eigenartige erkankung der hirnrinde". Clin. Anat. 8 (6), 429–431. doi:10.1002/ca.980080612
Striggow, F., and Ehrlich, B. E. (1996). Ligand-gated Calcium Channels inside and Out. Curr. Opin. Cel Biol. 8 (4), 490–495. doi:10.1016/s0955-0674(96)80025-1
Sun, X.-P., Callamaras, N., Marchant, J. S., and Parker, I. (1998). A Continuum of InsP3-Mediated Elementary Ca2+signalling Events inXenopusoocytes. J. Physiol. 509 (Pt 1), 67–80. doi:10.1111/j.1469-7793.1998.067bo.x
Takei, K., Stukenbrok, H., Metcalf, A., Mignery, G., Sudhof, T., Volpe, P., et al. (1992). Ca2+ Stores in Purkinje Neurons: Endoplasmic Reticulum Subcompartments Demonstrated by the Heterogeneous Distribution of the InsP3 Receptor, Ca(2+)-ATPase, and Calsequestrin. J. Neurosci. 12 (2), 489–505. doi:10.1523/jneurosci.12-02-00489.1992
Tandon, A., and Fraser, P. (2002). The Presenilins. Genome Biol. 3 (11), reviews3014.1. doi:10.1186/gb-2002-3-11-reviews3014
Tang, Y.-g., and Zucker, R. S. (1997). Mitochondrial Involvement in post-tetanic Potentiation of Synaptic Transmission. Neuron 18 (3), 483–491. doi:10.1016/s0896-6273(00)81248-9
Tanzi, R. E., and Bertram, L. (2005). Twenty Years of the Alzheimer's Disease Amyloid Hypothesis: a Genetic Perspective. Cell 120 (4), 545–555. doi:10.1016/j.cell.2005.02.008
Taylor, C. W. (1998). Inositol Trisphosphate Receptors: Ca2+-Modulated Intracellular Ca2+ Channels. Biochim. Biophys. Acta 1436 (1-2), 19–33. doi:10.1016/s0005-2760(98)00122-2
Tu, H., Nelson, O., Bezprozvanny, A., Wang, Z., Lee, S.-F., Hao, Y.-H., et al. (2006). Presenilins Form ER Ca2+ Leak Channels, a Function Disrupted by Familial Alzheimer's Disease-Linked Mutations. Cell 126 (5), 981–993. doi:10.1016/j.cell.2006.06.059
Walton, P. D., Airey, J. A., Sutko, J. L., Beck, C. F., Mignery, G. A., Südhof, T. C., et al. (1991). Ryanodine and Inositol Trisphosphate Receptors Coexist in Avian Cerebellar Purkinje Neurons. J. Cel Biol. 113 (5), 1145–1157. doi:10.1083/jcb.113.5.1145
Wolfe, M. S., Xia, W., Ostaszewski, B. L., Diehl, T. S., Kimberly, W. T., and Selkoe, D. J. (1999). Two Transmembrane Aspartates in Presenilin-1 Required for Presenilin Endoproteolysis and γ-secretase Activity. Nature 398 (6727), 513–517. doi:10.1038/19077
Wu, H., Carvalho, P., and Voeltz, G. K. (2018). Here, There, and Everywhere: The Importance of ER Membrane Contact Sites. Science 361 (6401). doi:10.1126/science.aan5835
Keywords: calcium signaling, calcium homeostasis, endoplasmic reticulum, mitochondria, membrane contact site, Alzheimer’s disease
Citation: Huang D-X, Yu X, Yu W-J, Zhang X-M, Liu C, Liu H-P, Sun Y and Jiang Z-P (2022) Calcium Signaling Regulated by Cellular Membrane Systems and Calcium Homeostasis Perturbed in Alzheimer’s Disease. Front. Cell Dev. Biol. 10:834962. doi: 10.3389/fcell.2022.834962
Received: 14 December 2021; Accepted: 31 January 2022;
Published: 25 February 2022.
Edited by:
Yongye Huang, Northeastern University, ChinaReviewed by:
Reza Raeisossadati, Universidade Federal do ABC, BrazilKenneth Norman, Albany Medical College, United States
Linlin Xu, Shandong University, China
Copyright © 2022 Huang, Yu, Yu, Zhang, Liu, Liu, Sun and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zi-Ping Jiang, waterjzp@jlu.edu.cn