Disturb mitochondrial associated proteostasis: Neurodegeneration and imperfect ageing
 Yuvraj Anandrao Jagtap1† Prashant Kumar1† Sumit Kinger1 Ankur Rakesh Dubey1 Akash Choudhary1 Ravi Kumar Gutti2
Yuvraj Anandrao Jagtap1† Prashant Kumar1† Sumit Kinger1 Ankur Rakesh Dubey1 Akash Choudhary1 Ravi Kumar Gutti2  Sarika Singh3
Sarika Singh3  Hem Chandra Jha4
Hem Chandra Jha4  Krishna Mohan Poluri5
Krishna Mohan Poluri5  Amit Mishra1*
Amit Mishra1*- 1Cellular and Molecular Neurobiology Unit, Indian Institute of Technology Jodhpur, Jodhpur, Rajasthan, India
- 2Department of Biochemistry, School of Life Sciences, University of Hyderabad, Hyderabad, India
- 3Division of Neuroscience and Ageing Biology, Division of Toxicology and Experimental Medicine, CSIR-Central Drug Research Institute, Lucknow, India
- 4Infection Bioengineering Group, Department of Biosciences and Biomedical Engineering, Indian Institute of Technology Indore, Indore, Simrol, India
- 5Department of Biotechnology, Indian Institute of Technology Roorkee, Centre for Nanotechnology, Indian Institute of Technology Roorkee, Roorkee, Uttarakhand, India
The disturbance in mitochondrial functions and homeostasis are the major features of neuron degenerative conditions, like Parkinson’s disease, Amyotrophic Lateral Sclerosis, and Alzheimer’s disease, along with protein misfolding. The aberrantly folded proteins are known to link with impaired mitochondrial pathways, further contributing to disease pathogenesis. Despite their central significance, the implications of mitochondrial homeostasis disruption on other organelles and cellular processes remain insufficiently explored. Here, we have reviewed the dysfunction in mitochondrial physiology, under neuron degenerating conditions. The disease misfolded proteins impact quality control mechanisms of mitochondria, such as fission, fusion, mitophagy, and proteasomal clearance, to the detriment of neuron. The adversely affected mitochondrial functional roles, like oxidative phosphorylation, calcium homeostasis, and biomolecule synthesis as well as its axes and contacts with endoplasmic reticulum and lysosomes are also discussed. Mitochondria sense and respond to multiple cytotoxic stress to make cell adapt and survive, though chronic dysfunction leads to cell death. Mitochondria and their proteins can be candidates for biomarkers and therapeutic targets. Investigation of internetworking between mitochondria and neurodegeneration proteins can enhance our holistic understanding of such conditions and help in designing more targeted therapies.
1 Introduction
The cellular system continuously performs multiple biochemical reactions to maintain its homeostasis essential for sustaining life. These biochemical reactions need energy generated by the organelle mitochondria, also recognized as “cellular powerhouse.” In 1898, Carl Benda coined the term mitochondria for the intracellular structures noted by Richard Altman (1890) that are ubiquitously found in eukaryotes (Ernster and Schatz, 1981). Altman showed the involvement of these structures in cellular metabolism and suggested its existence to be symbiotic. Lynn Margulis further laid down the basis for the endosymbiotic theory, where she proposed the hypothesis; that engulfment of prokaryotes (capable of aerobic metabolism) by heterotrophic anaerobe for survival in the oxygen-rich environment (Sagan, 1967). Although numerous reports support this theory, several other theories were also suggested for origin of mitochondria (Gray, 2012; Martin et al., 2015; Dunn, 2017). The mitochondria are essential for cellular energy and perform numerous other functions equally vital for normal cell functioning. The altered functioning of mitochondria is reported in several pathological conditions, including metabolic, neurodevelopmental, neurodegeneration, and cancer (Vyas et al., 2016; Srivastava, 2017; Ortiz-Gonzalez, 2021). Here in this review, we have focused on how mitochondrial damage and dysfunction are linked with multiple neurodegenerative diseases. Considering mitochondria’s complex and essential functions in diverse cellular pathways and signaling, maintaining its health and dynamics is critical. Several quality control pathways are engaged in mitochondrial homeostasis and its proper functioning during stress conditions. Various neurodegenerative disease-causing proteins are observed to alter this homeostasis by interfering with the mitochondrial pathways affecting their normal functioning (Briston and Hicks, 2018). Furthermore, how these mitochondrial deformities lead to other cellular defects is also reviewed. Identifying the multiple mitochondria-associated disease targets and pathways can help in detailed understanding of disease pathology and developing more specified therapeutic options.
1.1 Structural and functional importance of mitochondria
The advancement in electron microscopy has enhanced our structural understanding of mitochondria. The mitochondria exist with outer mitochondrial membrane (OMM) isolating it from cytoplasm. Inner mitochondrial membrane (IMM) folded in cristae-like structures provides a site for complexes of electron transport chain (ETC.) (ATP synthesis). The space between these two membranes, i.e., intermembrane space (IMS), contains cytochrome c (Cyt C), essential for induction of the cellular death pathway via apoptosis (Wang and Youle, 2009). Mitochondria perform numerous functions, like regulation of cytosolic calcium levels and formation of biomolecules (cholesterol, steroid, and heme), and generate reactive oxygen species (ROS) (Lathrop and Timko, 1993; Gunter et al., 1994; Miller, 2013) (Figure 1).
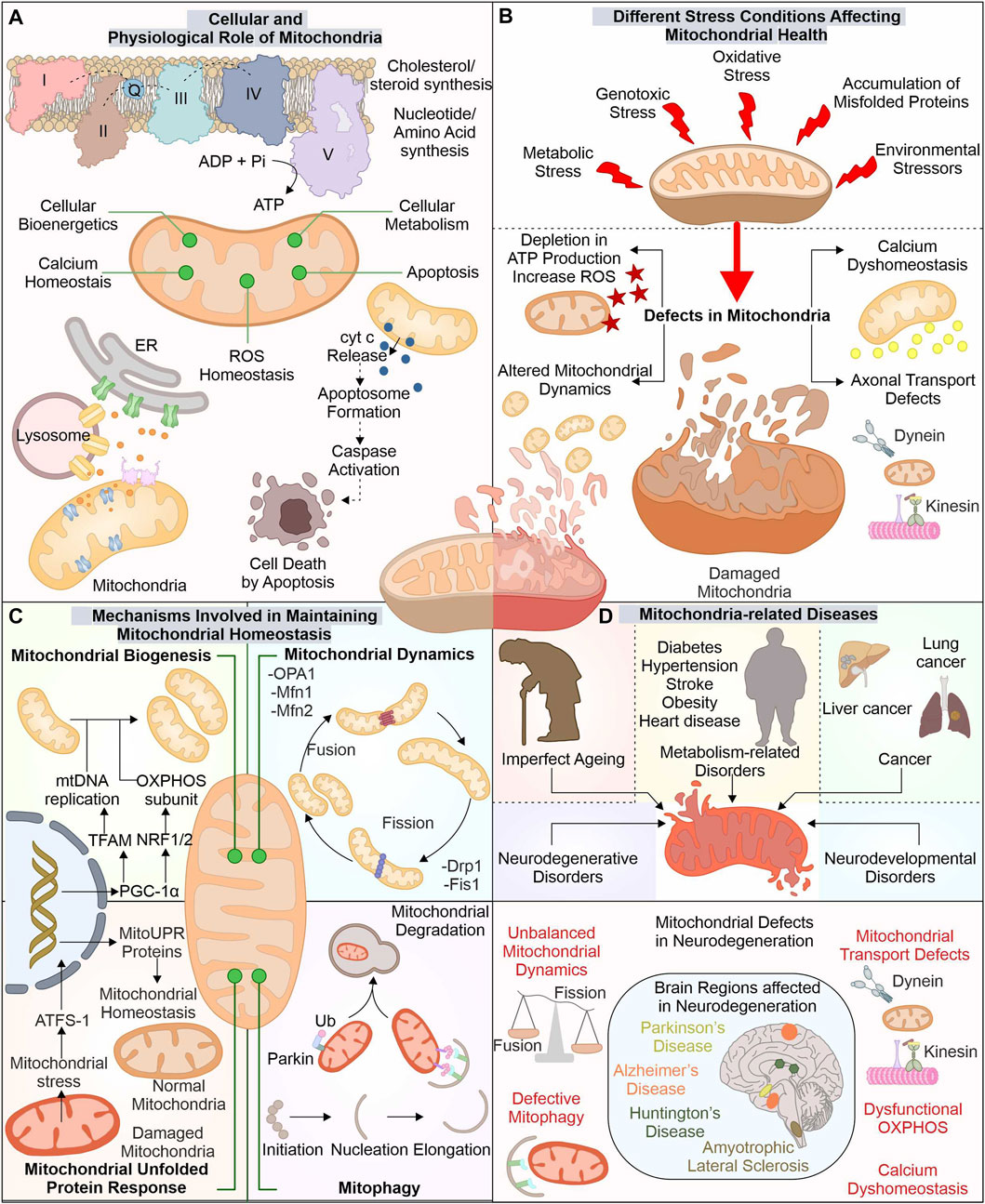
FIGURE 1. Mitochondria: The key player in cellular function and its implication in diverse diseases. Besides producing ATP for numerous cellular pathways, mitochondria also regulate ROS and calcium levels, synthesize several biomolecules, and control cell death via the apoptosis pathway (A). Different stresses affect these mitochondrial functions leading to mitochondrial dysfunction (B). Mitochondria possess several pathways to avoid such damage by controlling the synthesis of new mitochondria, morphology, and eliminating any dysfunctional mitochondria (C). The altered function of these mitostasis mechanisms is observed in several disease conditions, including neuronal degenerative disorders such as ALS, AD, PD, and HD (D).
The OMM tethers with different organelles with the help of numerous membrane-localized proteins. Mitochondria interact with endoplasmic reticulum (ER) membrane via mitochondrial-associated ER membrane (MAM) proteins; its altered association is reported in diseases like neurodegeneration (Lee and Min, 2018). The IMM is divided into two separate components, i.e., inner boundary membrane (IBM), present close to OMM, and the cristae membrane (CM), having larger section folded into the mitochondrial matrix, with cristae junctions connecting these two parts (Frey et al., 2002). The protein composition in these separate membranes differs as CM has a high number of enzymes involved in OXPHOS (Vogel et al., 2006). The different cellular stress factors can damage mitochondrial health leading to its altered functions with depletion in ATP production and induced ROS levels generating excessive oxidative damage. These stress factors can include exposure to ultraviolet (UV) light, excessive glucose levels, genotoxic stress, and H2O2 production, which affect the morphology of mitochondria (Tondera et al., 2009; Trudeau et al., 2011; Iqbal and Hood, 2014).
1.2 Mechanisms involved in maintaining mitochondrial homeostasis
The cell has developed different mechanisms and pathways to survive and adapt during stressful conditions, including mitochondrial quality control mechanisms. Which modulate the mitochondrial functions and their number as per the cellular requirements. The biogenesis of new mitochondria is does not occur through a de novo pathway, but is divided from the existing one. The formation of new mitochondria needs replication of mitochondria DNA (mtDNA) and expression of multiple mitochondrial proteins. Nearly 1,136 genes are known till now with expressing proteins of mitochondrial origin. The mitochondrial protein synthesis is controlled by nuclear genome (∼99%) and mtDNA (13 proteins) (Scarpulla, 2008; Rath et al., 2021). The biogenesis of mitochondria is a highly controlled mechanism requiring concerted expression of mtDNA and nucleus DNA. PGC-1 acts as a master controller for biogenesis of mitochondria by modulating the transcription factors (TF) required for mtDNA copying and its gene expression (Wu et al., 1999). The PGC-1 levels are controlled by different cellular signals, such as cellular energy and metabolism, where AMPK and SIRT1 act as modulators of PGC-1 activity (Scarpulla, 2011). The targets of PGC-1 include ERRα, NRF1, and NRF2, which modulate mitochondrial biogenesis gene expression. NRF1/2 controls mitochondrial respiratory chain protein synthesis, membrane protein import complexes, and other TFs (TFAM, TFB1M, and TFB2M) (Bruni et al., 2010; Satoh et al., 2013; Yang et al., 2014). TFAM maintains number of mtDNA and is crucial in biogenesis as its inactivity alters mtDNA copies and OXPHOS (Larsson et al., 1998). The depletion of PGC-1α activity is reported in aging and associated disorders (Sahin and DePinho, 2012; Picca et al., 2013).
Furthermore, fission and fusion pathways regulate mitochondrial dynamics under changing metabolic requirements of cells. As during metabolic and environmental stress, the damaged mitochondria are fused with healthy ones to reduce the damage. Moreover, through the mitochondrial fission mechanism, the damaged mitochondrial part is removed (Youle and van der Bliek, 2012; van der Bliek et al., 2013). Drp1 (Dnm1 in yeast) protein is involved in fission pathway (Bleazard et al., 1999; Smirnova et al., 2001). Fis1 protein aids in the recruitment and proper distribution of Drp1 at the site of mitochondrial division. Further, Drp1, through its GTPase activity, helps in membrane constriction and, finally, division of organelle (Mozdy et al., 2000; Ingerman et al., 2005). The OMM and IMM fusion is controlled through separate set of proteins. Mfn1 and Mfn2 recruited at the OMM interact with adjoining mitochondria-localized mitofusion proteins. Further, their GTPase activity helps in the fusion between these two separate mitochondrial membranes (Santel and Fuller, 2001). The IMM fusion is controlled by another GTPase protein, OPA1, localized at the IMM protruding into IMS (Olichon et al., 2002). OMA1, an IMM localized protease, can decrease the OPA1 activity and prevent the fusion of damaged mitochondria with decreased membrane potential (ΔΨm) and ATP levels (Head et al., 2009; Youle and van der Bliek, 2012).
Mitochondrial chaperones and proteases are also involved in maintaining mitochondrial health by either refolding or removing unfolded proteins and mediating their degradation. Numerous such chaperones are distributed in mitochondria performing specified functions. At OMM, Hsp90, together with TOM, assists in the import of mitochondrial proteins. Further, HtrA2 (at IMS) helps translocate proteins from outer to inner mitochondrial membrane. At IMM, chaperones such as COX17p control the folding of, ETC., complex proteins. Finally, at the mitochondrial matrix, different chaperones and co-chaperones (Hsp60-Hsp10, mtHsp70-HSC20, DNAJA3, ERAL1) regulate the maintenance of matrix proteins, mtDNA, and mitoribosome (Castro et al., 2018). Furthermore, proteases like ClpP and LonP (Matrix localized), m-AAA and i-AAA proteases (IMM), and HtrA2 (IMS), target degradation of misfolded and damaged proteins. Furthermore, these proteases also regulate fusion and fission by modulating proteins involved in these pathways (de Sagarra et al., 1999; Leonhard et al., 2000; Bota and Davies, 2002; Martins et al., 2004). The OMM localized unfolded proteins are removed through assistance from cytosolic proteasome system (Livnat-Levanon and Glickman, 2011).
However, with the increased accumulation of unfolded proteins and oxidative stress, mitochondria activate retrograde signaling, also called mitochondrial unfolded protein response (UPRmt) (Haynes et al., 2007; Kohler et al., 2015; Oliveira and Hood, 2018). The ectopic expression of non-native ornithine transcarbamylase in COS-7 cells causes its accumulation in mitochondria (Zhao et al., 2002). This mitochondrial accumulation of misfolded protein was found to induce different genes, including chaperones (Hsp60, mtDnaJ, Hsp10), proteases (YMEL1 and ClpP) along with complex I subunit and membrane-localized Tim17A import protein (Zhao et al., 2002; Aldridge et al., 2007). HAF-1 represses the import of ATFS-1 (ATF5 in humans) transcription factor in defective mitochondria leading to its translocation to the nucleus (Fiorese et al., 2016; Shpilka and Haynes, 2018). ATFS-1 with DVE-1 and UBL-5 remodulate chromatin structure, inducing mitochondrial related gene transcripts production such as chaperones (Hsp60, Dnj-10) and antioxidants gene transcription factor (Skn-1) (Nargund et al., 2012; Shpilka and Haynes, 2018). Mitophagy is another quality control pathway where selective removal of altered mitochondria occurs. PINK1 and E3 ubiquitin ligase Parkin are crucial components of mitophagy. The proteolytic cleavage inhibition of PINK1 in mitochondria with reduced membrane potential causes its stabilization on the mitochondrial surface (Matsuda et al., 2010; Narendra et al., 2010; Fallaize et al., 2015). Furthermore, the stabilized PINK1 phosphorylates and recruits Parkin to mitochondria, inducing its ubiquitination activity (Kim et al., 2008; Narendra et al., 2008; Matsuda et al., 2010; Sha et al., 2010). The PINK1-recruited Parkin initiates the ubiquitination of multiple proteins localized to OMM, targeting its degradation by ubiquitin-proteasome system (UPS). The clearance of multiple such OMM localized proteins (Mfn1, Mfn2) via UPS is an essential checkpoint for mitophagy (Chan et al., 2011).
1.3 Mitochondrial impairments in complex disorders: Focus on neurodegeneration
The cellular importance of mitochondria is well acknowledged; therefore, it is not surprising that any damage or dysfunction in this organelle leads to disease conditions (McInnes, 2013; Hsu et al., 2016; Monzio Compagnoni et al., 2020). The mitochondria-related diseases commonly occur because of mutations (spontaneous/inherited) in nuclear and mtDNA encoding proteins for mitochondrial functions such as OXPHOS complexes, mitochondrial quality control proteins, and enzymes involved in mtDNA preservation and expression of genes (Gorman et al., 2016). Mutations in mtDNA are reported in diseases such as Leigh syndrome, LHON, and MERRF (Gorman et al., 2015; Gorman et al., 2016). Further multiple metabolic disorders such as diabetes, hypertension, stroke, obesity, and heart disease were also reported with mitochondrial impairment (Bhatti et al., 2017). Earlier the Warburg effect observed in cancer cells was thought to be linked with mitochondrial dysfunction; however, later studies confirm the importance of mitochondrial energetics in survival and growth of cancer (Wallace, 2012). The augmented activity of HIF1α and FOS–JUN inducing cancer cell proliferation was identified to be associated with increased mitochondrial ROS levels. Moreover, mitochondrial calcium dyshomeostasis leads to cytosolic accumulation of calcium, further inducing the metastatic potential of cancer cells by activating NF-κB (Wallace, 2012).
The exposure to different stress conditions and impaired quality control pathways further aggravates the mitochondrial damage altering its normal functions. The neuronal cells (affected in neurodegeneration), with their complex cellular structure and postmitotic nature, depend on mitochondrial energy for numerous pathways and are the majorly affected cells due to their altered function. Mitochondrial dynamics, morphology, and function changes are reported in amyotrophic lateral sclerosis (ALS) disease models (Magrane et al., 2014; Stribl et al., 2014). Furthermore, Huntington’s disease was observed to have mitochondrial defects, altered OXPHOS, and impaired transport of mitochondrial proteins (Kim et al., 2010; Yano et al., 2014). Alzheimer’s disease patients were also reported with altered mitochondrial morphology, defective biogenesis, and reduced levels of respiratory complexes involved in, ETC (Maurer et al., 2000; Manczak et al., 2004; Baloyannis, 2006; Sheng et al., 2012). The mitochondrial quality control defects are also apparent in Parkinson’s disease (PD) patients having PINK1, DJ-1, and Parkin gene mutations (Kitada et al., 1998; Bonifati et al., 2003; Valente et al., 2004). Also, mitochondrial energy defects with deficient capacity of complex I are reported in PD (Gatt et al., 2016).
2 Deciphering the link between amyotrophic lateral sclerosis and mitochondrial impairment
ALS is identified with motor neuronal deterioration in brain and spinal cord region that regulate muscle activity. ALS is also recognized by the name “Charcot’s disease” for the contribution made by Jean-Martin Charcot to understanding ALS pathology (Goetz, 2000). ALS patients can be characterized based on symptoms such as limb onset ALS, where muscle cramps, weakness, or stiffness in the arm/leg are observed. In contrast, patients with bulbar onset ALS have swallowing and speaking difficulties, which are associated with atrophy of the tongue (Goetz, 2000; Hardiman et al., 2017). During pathogenesis of ALS, the initial muscle damage further causes paralysis of other muscles in the late stage of disease, resulting in death often due to respiratory insufficiency (Corcia et al., 2008). Numerous ALS (familial and sporadic) disease-causing mutations are identified in different genes (Renton et al., 2014; Weishaupt et al., 2016). ALS’s most frequently mutated genes include C9orf72, SOD1, TDP-43, and FUS (Figure 2). The gain or loss of function due to these mutations alters their cellular role. Furthermore, these pathogenic variants also affect cellular organelles such as mitochondria. The altered mitochondrial bioenergetics in the presence of ALS-associated pathogenic proteins also disturbs other cellular pathways generating a cytotoxic environment. Additionally, the inability of quality control pathways to maintain homeostasis by alleviating or restricting the damage caused by defective mitochondria results in cell death.
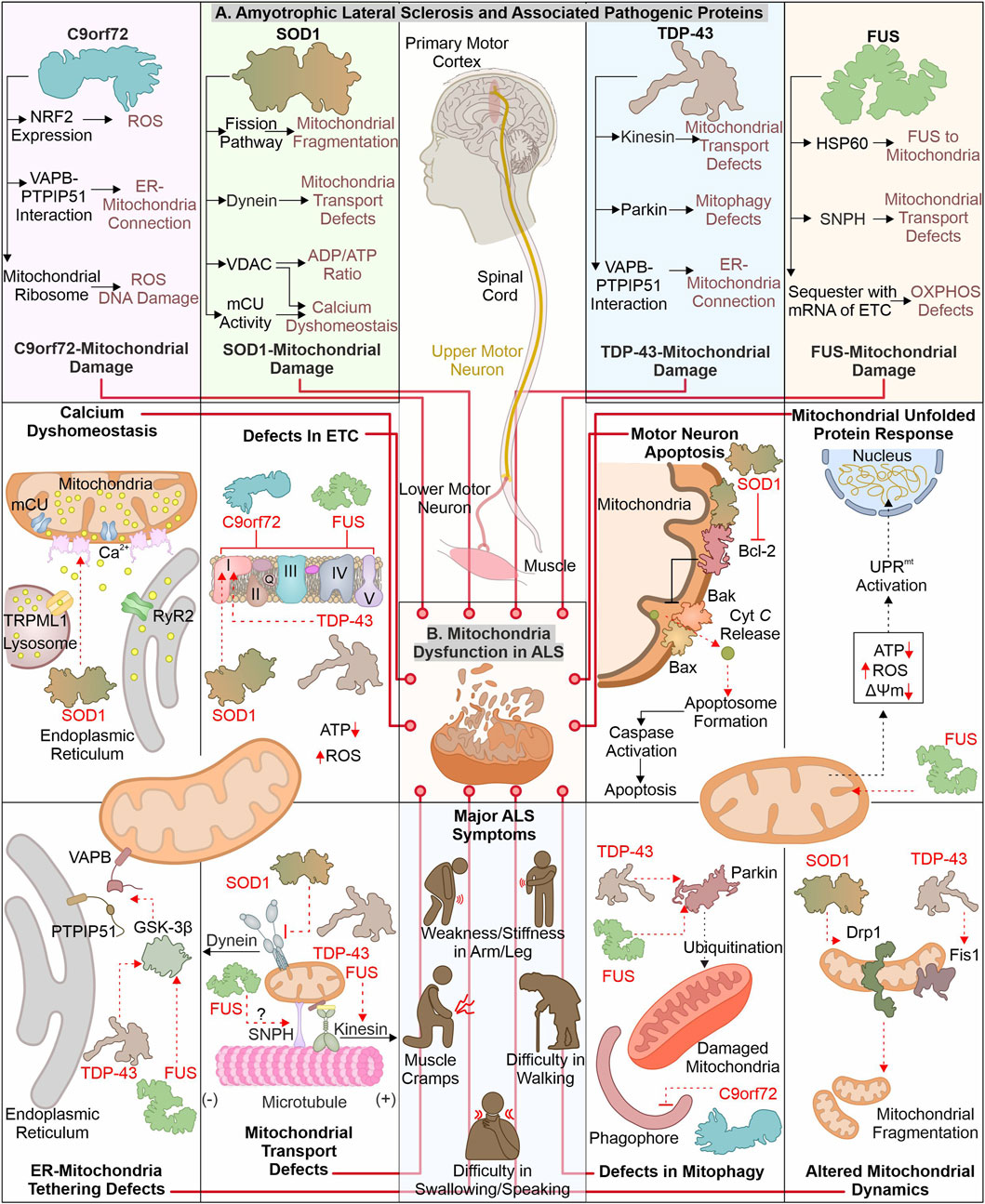
FIGURE 2. ALS-linked mutant proteins contribute to mitochondrial impairment. ALS is a complex neuronal degenerative condition where motor neurons (brain and spinal cord) are affected. Various signs, like muscle cramps, stiffness in the arm, and swallowing and speaking difficulties, characterize ALS patients. Different disease-causing proteins are identified to interact and affect mitochondrial proteins causing mitochondrial damage at several levels (A). The interaction of ALS-linked pathogenic proteins (TDP-43, SOD1, C9orf72, and FUS) with mitochondria and associated proteins causes its dysfunction, leading to calcium dyshomeostasis, altered OXPHOS, mitochondrial dynamics, and mitophagy (B).
2.1 How are mitochondrial defects associated with the loss or gain of functions in C9orf72?
The mutations in the C9orf72 are considered as the frequently occurring genetic insult in ALS/FTD disease process. Mutations in C9orf72 ALS/FTD patients having a hexanucleotide (GGGGCC) repeats expansion (HRE) positioned at first intron of gene were reported in 2011 (DeJesus-Hernandez et al., 2011; Renton et al., 2011). The gene encoding C9orf72 is present on chromosome 9p21.2 (human) (Boxer et al., 2011). Two isoforms of C9orf72 are reported in humans, i.e., C9-S (exon 2–5, 222 amino acids) and C9-L (exon 2–11, 481 amino acids). The C9-L isoform is principally present in the brain; however, its decreased levels are reported in ALS (Frick et al., 2018). 2–30 HRE are considered normal, whereas patients with C9orf72 ALS are reported to have nearly thousands of repeats (Beck et al., 2013; Benussi et al., 2014; Hubers et al., 2014). The involvement of C9orf72 in ALS pathology remains debated as gain as well as loss of function are described to cause ALS. Multiple studies report that decreased C9orf72 at mRNA and protein levels are linked with disease pathology (Gijselinck et al., 2012; Waite et al., 2014; Saberi et al., 2018). Moreover, the higher levels of C9orf72 were found to have advantageous effects (van Blitterswijk et al., 2015). Conversely, a gain of function (at RNA or protein level) is also reported in ALS pathology. The RNA transcribed from disease-linked C9orf72 tends to form aggregates in nuclear foci (Donnelly et al., 2013; Sareen et al., 2013). Moreover, the production of different dipeptide repeats (DPRs) from expanded mRNA is also involved in ALS pathomechanism (Mori et al., 2013). Various such DPRs, including poly- (GP), (PA), (GR), (GA), and (PR), are reported in the cytoplasm of neurons (Ash et al., 2013).
The pathogenic variants of C9orf72 were found to cause mitochondrial defects where the mouse model expressing poly-(GR) form shows neuronal loss and behavioral changes. Furthermore, poly-(GR) induces ATP5A1 (subunit of complex V) proteasomal degradation reducing its levels and compromising mitochondrial function. The reduction in poly-(GR) or increased expression of ATP5A1 was found to have protective effects by reducing the neurotoxicity caused due to poly-(GR) (Choi et al., 2019). The RNAseq analysis of ALS patient tissue samples shows a decreased expression of mtDNA-encoded ETC. complexes, causing failure of mitochondrial bioenergetics. Moreover, defects in the axonal mitochondrial movement are also reported in patient-derived iPSCs (Mehta et al., 2021). The interaction of poly(GR)80 with mitochondrial ribosomal proteins is also reported to cause mitochondrial dysfunction. Further, the increased oxidative stress in such conditions induces DNA damage observed in the C9orf72 neurons (Lopez-Gonzalez et al., 2016). Onesto et al. report an increase in ROS levels in cells expressing mutated C9orf72 altering mitochondrial activity and might be the mechanism behind the cell death observed in ALS (Onesto et al., 2016). Moreover, the expression of redox genes was also observed to be impaired in ALS, where expression of the master regulator of redox genes, i.e., NRF2, is also reported to decrease in cells expressing DPRs (Jimenez-Villegas et al., 2022). On the other hand, the loss of C9orf72 functions is also linked with mitochondrial abnormalities, where C9orf72 controls assembly of complex-I, maintaining OXPHOS. The AIFM1/CHCHD4 mediates the import of C9orf72 to IMS from the cytosol, where C9orf72 inhibits the degradation of TIMMDC1, a chaperone involved in complex I formation (Wang et al., 2021). The C9orf72 DPRs are also shown to disrupt calcium homeostasis in neurons by affecting the ER-mitochondria association. Where C9orf72 mediated, GSK-3β activation disrupts ER-mitochondria (VAPB-PTPIP51) linkage (Gomez-Suaga et al., 2022). The involvement of C9orf72 in autophagy and lysosomal pathway is recently reported; further its depletion may result in autophagy defects observed in ALS pathology (Sullivan et al., 2016).
2.2 How mitochondrial dysfunction caused by mutant SOD1 contributes to neuronal death
Cu/Zn superoxide dismutase 1 (SOD1) was initially recognized by erythocuprein, which is essential in removing superoxide radicals generated during oxygen utilization in cellular systems (McCord and Fridovich, 1969). The SOD1 gene is positioned on chromosome 21q22.11 (human), having five exons encoding 154 extended amino acid chains forming monomers of the homodimeric SOD1 enzyme. Each monomer has a binding site for Cu+ and Zn2+, which aids in the stabilization and catalytic activity of SOD1 (Siddique et al., 1991; Wei et al., 2017). SOD1 is predominantly present in the different regions of motor neurons, including dendrites, axons, and cell bodies (Pardo et al., 1995). Furthermore, it principally exists in cytosolic and mitochondrial regions where copper chaperone CCS helps import SOD1 to IMS of mitochondria (Field et al., 2003). SOD1 converts superoxide radicals generated in mitochondria (during OXPHOS) into H2O2 and molecular oxygen (Zelko et al., 2002). Finally, glutathione peroxidase transforms hydrogen peroxide to H2O, reducing the damage caused by superoxide radicals. Numerous mutations are identified in SOD1 that alter its metal binding properties and catalytic activity. The presence of such multiple mutations is also confirmed in fALS (Rosen, 1993; Hayward et al., 2002). The pathogenic variants of SOD1 affect mitochondrial functions and result in cellular death by apoptosis (Pasinelli et al., 2004).
The build-up of disease-causing SOD1 in mitochondria (spinal cord) is reported to affect its normal function (Liu et al., 2004). Further, mutated SOD1 interacts with mitochondria-associated Bcl2 protein, where this interaction causes structural changes in Bcl2 protein. The altered structure of Bcl2 (exposed BH3 domain) changes the mitochondrial structure, and ΔΨm results in Cyt C release, activating neuronal death by apoptosis (Pedrini et al., 2010). Furthermore, mutated SOD1 interaction with VDAC1 protein (at OMM) is also identified in mitochondria (spinal cord). This interaction alters the conductance of VDAC1, resulting in an increased cytosolic ADP/ATP ratio and a decrease in membrane potential (Israelson et al., 2010). The VDAC1 also provides a docking site for the hexokinase I (HK1), and this interaction helps to regulate ROS production and apoptosis (da-Silva et al., 2004). The depleted HK1 expression in the mouse spine compared to the brain is reported and may be a prime reason for selective damage of spinal cord mitochondria (Israelson et al., 2010). Recently Magri et al. identified SOD1 mutant and HK1 vying for VDAC1 and treatment of the N-terminal of HK1 in NSC34 cells protected mitochondria by inhibiting SOD1 (G93A) and VDAC1 interaction (Magri et al., 2016; Magri et al., 2021). The disturbed homeostasis of intracellular calcium is a key hallmark of ALS models, and mitochondria are also involved in calcium homeostasis. A study executed on the mutant SOD1 (G93A) expressing mice shows a decline in mitochondrial calcium uptake due to altered activity of mCU even though their expression appears normal (Damiano et al., 2006; Fuchs et al., 2013). The mutant SOD1 also modifies the mitochondrial dynamics, where it is found to induce levels of Drp1 and reduces the levels of OPA1, resulting in increased mitochondrial fragmentation (Raimondi et al., 2006; Ferri et al., 2010). In continuation, mitofusion activators in SOD1 (G93A) mice delay the ALS pathology by reducing mitochondria-related abnormalities (Dang et al., 2022). Furthermore, the mutated SOD1 interactions with the dynein protein is found to affect the retrograde transport of mitochondria (Zhang et al., 2007).
2.3 Disturbance of mitochondrial homeostasis: TDP-43 involvement
TDP-43 (43 kDa) is key disease-causing protein initially noted in ubiquitin-positive inclusions in different segments of CNS (neocortex, hippocampus, and spine) of ALS patients (Arai et al., 2006; Neumann et al., 2006). Multiple disease-linked TDP-43 mutants involvement in ALS pathogenicity are reported. TDP-43 gene is situated on chromosome 1p36.22 (human) (Sreedharan et al., 2008). TDP-43 shows structural similarity with heterogenous ribonucleoproteins (hnRNPs). It is principally found in nuclei and is involved in RNA processing (Buratti and Baralle, 2008; Freibaum et al., 2010). Conversely, cytoplasmic build-up of mutant TDP-43 is observed, affecting RNA processing of multiple proteins involved in neurodevelopment (Polymenidou et al., 2011; Tollervey et al., 2011). Both loss-and gain-of-function is evident in the TDP-43 associated pathomechanism (Sephton et al., 2010; Wils et al., 2010). TDP-43 is inherently inclined to form aggregates, where its C-terminal is crucial for spontaneous aggregate formation. Moreover, numerous mutations in this domain are identified, causing aggregate formation and inducing the associated pathogenicity (Johnson et al., 2009).
The disease-linked TDP-43 expressed in cells causes mitochondrial defects leading to cellular death. The TDP-43 depletes activity of complex I, resulting in ΔΨm loss (Lu et al., 2012). Interestingly, induced UCP2 levels were also observed in TDP-43 transfected cells showing a response against cellular injury. Recently, study has suggested that TDP-43 (full-length) may cause mitochondrial damage compared to truncated forms of TDP-43. TDP-43 interaction with mitochondrial mRNAs synthesizing complex I subunit (ND3/6), causing the impairment in its assembly and activity. The defective assembly of complex I further generate oxidative stress, inducing the formation of oligomers from truncated TDP-43 (Salvatori et al., 2018). Another study reports interacting partners of TDP-43, including VDAC1, PHB2, and Mfn2. TDP-43 with these interactors may modulate the autophagic clearance of mitochondria and its dynamics; however, the mechanistic details for this modulation are not entirely known (Davis et al., 2018). The increased levels of Fis1 and induced mitochondrial fragmentation are also reported in fibroblasts expressing mutant TDP-43 (Onesto et al., 2016).
The expression of TDP-43 (either truncated or full-length) was found to activate mitophagy with increased LC3 and decreased p62 levels. Furthermore, TDP-43 carboxy-terminal selectively reduces the VDAC1 levels (Hong et al., 2012). FUS and TDP-43 regulate the Parkin levels, and reduction in TDP-43 and FUS cause a decrease in Parkin levels (Lagier-Tourenne et al., 2012). ALS-associated disturbed ER-mitochondria connections also involve TDP-43, where activation of GSK-3β in TDP-43 dependent manner disrupts VAPB–PTPIP51 interaction. These disrupted connections also generate calcium disbalance leading to increased cytosolic calcium, further affecting other cellular processes, including axonal mitochondrial transport (Stoica et al., 2014). The anterograde mitochondrial movement requires kinesin-1, which interacts with mitochondria via Miro1 protein. Miro1 is a calcium-sensitive protein, and excess calcium impairs this transport pathway (Saxton and Hollenbeck, 2012). Moreover, TDP-43 and FUS control kinesin expression (Xiao et al., 2011; Colombrita et al., 2012). Following this, the altered mobility of mitochondria is observed in ALS disease models like Drosophila and patient-derived spinal motor neurons (Baldwin et al., 2016; Kreiter et al., 2018).
2.4 Altered function of FUS protein causes ALS-linked mitochondrial defects
FUS (RNA-interacting protein) is another ALS pathology protein, and mutation in this protein forms cytosol-localized aggregates in motor neurons affected in ALS (King et al., 2012). Initially, FUS has been identified as an oncogene causing liposarcoma and is alternatively termed as translocated in liposarcoma (TLS) (Crozat et al., 1993). FUS is one of FET family members, comprising other proteins, EWS and TAF15 (Tan and Manley, 2009). The gene encoding the FUS protein is situated in chromosome 16p11.2 (human), comprising 15 exons (Deng et al., 2014). The different domains in FUS protein include the SYGQ-rich region, RGG, RRM, ZnF domain, and nuclear localization and export signal sequence (Wang et al., 2015a). FUS is predominantly located at the nucleus, with its prominent role in DNA damage repair, gene expression, RNA processing, a regulator of cell division, and also implicated in stress response (Sama et al., 2014). The initial recognition of FUS as an oncoprotein led the research toward finding its involvement in cancer. In 2009, numerous mutations in FUS were reported to be linked with fALS. These mutations cause its mislocalization to the cytoplasm and forming inclusions responsible for the degeneration of motor neurons (Kwiatkowski et al., 2009; Vance et al., 2009). The NLS in the FUS protein is essential for its translocation to cellular nuclei by interacting with Karyopherinβ2, and mutation in this sequence results in its cytoplasmic retention (Bosco et al., 2010; Zhang and Chook, 2012).
The cytosolic accumulation of FUS also affects mitochondrial health, causing bioenergetic disbalance in a cell. The involvement of mitochondrial chaperone HSP60 in mitochondrial targeting of mutated FUS is reported by (Deng et al. 2015). Furthermore, the mutant FUS also binds with mitochondrial chaperones (Hsp70 and Hsp90), involved in transport of mitochondrial proteins from cytosol to mitochondria (Wang et al., 2015b). Mitochondria-localized FUS protein inhibits the ATP synthase formation by interacting with its catalytic subunit ATP5B. The FUS-dependent altered mitochondrial function activates UPRmt, and its extended activation results in neuronal death (Deng et al., 2018). Furthermore, the sequestration of mutant FUS protein with multiple mRNAs encoding, ETC. proteins is also reported. The aggregation of mRNA inhibits their translation, resulting in decreased levels of respiratory chain proteins affecting mitochondrial bioenergetics (Tsai et al., 2020). Nakaya and Maragkakis’s report also signify the defects in mitochondrial morphology in the presence of mutant FUS (R495X), which alters the expression of mitochondrial genes (Nakaya and Maragkakis, 2018). Mutant FUS may also be involved in the ER-mitochondria communication defects as it is identified to activate GSK-3β responsible for the VAPB–PTPIP51 defective interaction (Stoica et al., 2016). ALS-associated axonal cargo transport defects may involve FUS protein as it is recently recognized to interact with Syntaphilin (involved in mitochondrial tethering) and a mutated form of FUS alters this interaction causing mitochondrial abnormalities observed in ALS (Salam et al., 2021).
3 Unravelling the Alzheimer's disease associated mitochondrial impairments
Alzheimer’s disease (AD) is complex neurological disorder generating atrophy of brain due to neuronal cells death resulting in dementia, majorly occurring at a later stage of life. The characteristic of AD is the intracytoplasmic neurofibrillary tangles (NFTs) made-up of proteins known as tau and extracellular senile plaques consisting of amyloid beta (Aβ). These pathogenic aggregates cause cellular defects, including mitochondrial damage, resulting in apoptosis-mediated neuronal death (Kumar et al., 2018). The toxic forms of tau and Aβ are extensively reported to adversely impact the neuronal cells of AD patients by impairing mitochondrial energy generation and antioxidation capacity (Figure 3).
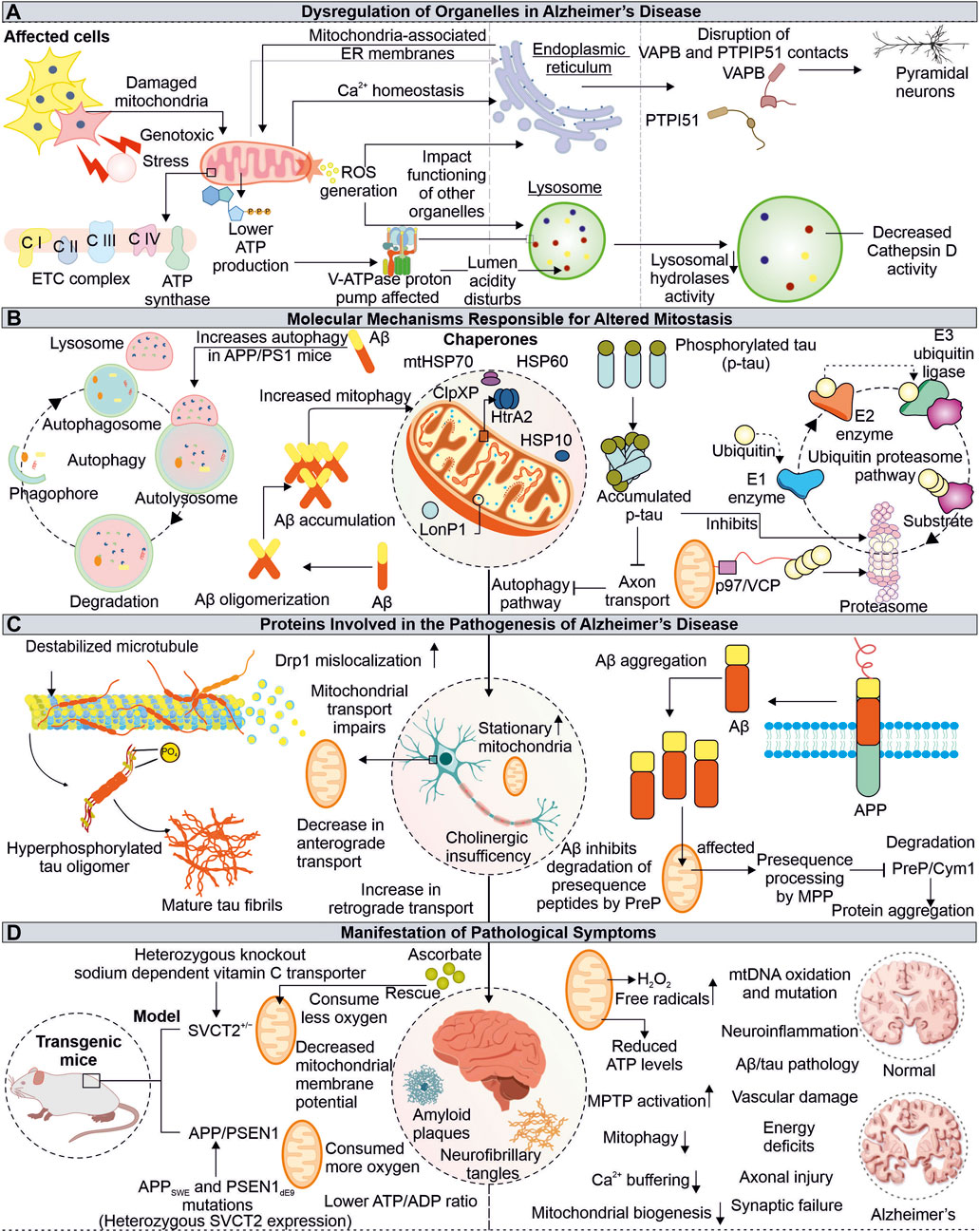
FIGURE 3. Impaired functions of Mitochondria in Alzheimer’s disease. Mitochondrial dysfunction due to pathogenic proteins from AD depletes the cellular energy, further affecting other organelles such as ER and lysosome (A). The pathogenic Aβ and tau proteins are also identified to impair cellular quality control pathways, including autophagy and proteasome (B). The tau and Aβ alter the normal mitochondrial functions and axonal transport (C). The AD-associated physiological changes are linked with mitochondrial damage, where the misfolded Aβ and tau affect mitochondrial function at different levels (D).
3.1 Defective clearance of Aβ at mitochondria may affect import of mitochondrial proteins
In amyloidogenesis, step-wise splicing of β-amyloid precursor protein (APP) by β- and γ-secretases produce 37–43 amino acid-containing peptide known as Aβ (Haass et al., 2012). These Aβ peptides are amphipathic, forming amyloid fibers observed in AD patient brain sections (Soreghan et al., 1994; Hayden and Teplow, 2013). The animal models subjected to human Aβ42 oligomers have been shown to affect the number of synapses along with memory formation defects (Selkoe and Hardy, 2016). Furthermore, the binding of Aβ oligomers with metal ions like copper and iron can lead to a generation of ROS. The ROS further damages other cellular elements like lipids leading to neurotoxicity and cell death (Cheignon et al., 2018). The intracellular processing of APP at the site of mitochondria and ER contacts (MAM) leads to the production and internalization of Aβ to the mitochondria, where involvement of translocase of the outer membrane (TOM) is reported (Hansson Petersen et al., 2008; Area-Gomez et al., 2009). The Aβ targeted at mitochondria is degraded by hPreP, limiting its mitochondrial damage (Falkevall et al., 2006). However, during increased ROS levels, the inactivation of hPreP generates an Aβ build-up in mitochondria resulting in AD-associated death of neurons. Furthermore, reduced hPreP levels were reported in AD patient brains compared to control; thus, clearance of Aβ via hPreP can be explored as a therapeutic option (Alikhani et al., 2011; Teixeira et al., 2012).
Additionally, the occurrence of Aβ inhibits PreP resulting in accumulation of the presequence peptides usually processed by mitochondrial processing peptidase (MPP). The accumulation of presequence peptides inhibits MPP activity via a feedback mechanism, impairing mitochondrial presequence proteins' transport and resulting in mitochondrial impairments (Mossmann et al., 2014). The interaction of mitochondria located Aβ and Abeta-binding alcohol dehydrogenase (ABAD) alters ABAD function, leading to ROS generation, further causing mitochondrial dysfunction and death of neurons (Lustbader et al., 2004; Chen and Yan, 2007). Different inhibitors of ABAD-Aβ interaction are being tested for their possibility of reducing Aβ-linked neurotoxicity, and further development in this area can provide us with a viable therapeutic choice (Morsy and Trippier, 2019). Another approach for Alzheimer’s treatment is the clearance of Aβ from AD patient’s brains. Multiple drugs have been tried to achieve this objective with minimal success in clinical trials (Huang et al., 2020; Karran and De Strooper, 2022). Moreover, antibodies-based therapeutics for targeted clearance of Aβ were also explored, however recently FDA-approved aducanumab use for AD treatment remains controversial (Sevigny et al., 2016; Tampi et al., 2021). This further raise questions about correlation of Aβ pathology in AD-associated cognitive impairments, whether it is the cause or a feature of AD.
3.2 Tau associated mitochondrial defects and role of antioxidants in mitigating oxidative stress
Tau proteins are known to be associated with a microtubule, and AD-associated altered tau protein was also reported to cause mitochondrial defects. The hyperphosphorylated tau protein are identified in AD pathomechanism (Iqbal et al., 2010). The expression of tau protein was found to affect the cellular transport of multiple organelles, including mitochondria (Ebneth et al., 1998). Furthermore, the anterograde transport of mitochondria is affected in tau-overexpressed cells (Shahpasand et al., 2012). The mitochondrial dynamics defects, such as elongated mitochondria, are observed in human tau-expressed neuronal cells from Drosophila and mouse models. The impaired mitochondrial localization of Drp1 results in defective mitochondrial fission leading to elongation in mitochondria (DuBoff et al., 2012). Moreover, the aberrant interaction of Drp1 with hyperphosphorylated tau and Aβ leads to increased mitochondrial fission and fragmentation, damaging neuronal health (Manczak and Reddy, 2012). The combined toxic effects of Aβ and truncated tau (cleaved at Asp421) on mitochondria are observed in primary cortical neurons where the number of stationary mitochondria was increased along with loss of mitochondrial potential (Quintanilla et al., 2012).
The neuronal damage due to high oxidative stress in presence of AD-associated pathogenic proteins is another major phenomenon observed in AD patients (Uttara et al., 2009) (Lin and Beal, 2006). Further, the impaired antioxidant defense mechanism aggravates this neuronal damage, where numerous mitochondrial mechanisms, including its biogenesis, axonal transport, and dynamics, are also affected. The treatment of the mitochondria-specific antioxidant SS31 alleviates the above-mentioned effects (Calkins et al., 2011). The cellular system and mitochondria possess multiple antioxidant enzymes and ROS scavengers to regulate ROS levels (Mailloux, 2018). The decreased ROS scavengers, such as ascorbate (Vitamin C), accelerate the disease progression in transgenic APP/PSEN1 mice due to mitochondrial dysfunction. The altered levels of ascorbate can be used as a biological marker for the early detection of mitochondrial defects linked to disorders. Moreover, their decreased levels can be improved by dietary supplementation in elderly peoples prone to disease (Dixit et al., 2017).
4 How does mitochondrial dysfunction in AD lead to lysosomal and endoplasmic reticulum defects?
Mitochondrial communication with other cellular organelles is essential for maintaining homeostasis. The AD-associated mitochondrial impairments not only disturb cellular bioenergetics but also affects the functioning of other organelles, like lysosomes and ER. Although these organelles are compartmentalized and function independently in the cellular environment, they depend on mitochondria for energy. Different ER and lysosome defects associated with mitochondrial damage are reported in AD, which underlies the complex pathomechanism of neurodegeneration.
4.1 Alzheimer’s disease-related mitochondrial abnormalities and their impact on lysosome functions
The lysosomes are membrane-bound organelles engaged in degrading intracellular (autophagic pathway) and extracellular (endocytic pathway) components (Saftig and Klumperman, 2009). The acidic pH (4.5–5) of lysosome lumen drives its hydrolytic enzymes to carry out this activity. The vacuolar (H+) ATPase (or V-ATPase) drives proton into the lumen of lysosome using ATP and is primarily responsible for maintaining lysosome pH gradients (Mindell, 2012). The lysosomal acidification is impacted by V-ATPase dysfunction, which inhibits substrate clearance and causes a variety of illnesses, including neurodegenerative diseases (Song et al., 2020). Mitochondrial impairment causes a shortage of ATP, which can impact the V-ATPase activity; thus, lysosomal lumen pH maintenance is disturbed in disease conditions (Stepien et al., 2020). The initial belief is that the lysosome and mitochondria functions were independent but recently changed, with numerous studies showing their interdependency. Moreover, either lysosome or mitochondrial impaired function may affect others' functions, generating complex pathogenic conditions such as neuronal degeneration (Audano et al., 2018). The cortex and hippocampus tissue sections from APP/PS1 mice model show an accumulation of lysosomal marker lysosome-associated membrane protein 1 (LAMP1) along with enlarged lysosome structures around the aggregates of Aβ. This also represents impaired lysosomal degradation capacity. Further, the positive modulation of lysosomal enzyme cathepsin B (CatB) by PADK shows improved clearance of Aβ (Hwang et al., 2019). The decreased activity of CatB upon V-ATPase inhibition is also reported (Kubisch et al., 2014). This highlights the importance of V-ATPase in CatB-mediated clearance of Aβ and the connection of V-ATPase impairments due to mitochondria dysfunction observed in AD. Cathepsin D (CatD) is also reported to involve in clearing Aβ and tau, and its inhibition by Aβ42 is observed in the early onset of AD and can be utilized as a potential biomarker (Chai et al., 2019; Suire et al., 2020).
4.2 Interrupted endoplasmic reticulum-mitochondria communication in pathophysiology of Alzheimer’s disease
Mitochondria contacts with ER via MAMs are essential in multiple physiological functions of cells, including Ca2+ homeostasis, lipid synthesis, mitochondrial dynamics regulation, apoptosis, inflammation, and autophagy. The impaired crosstalk between these crucial organelles can be seen in multiple diseases, including neurodegeneration (Missiroli et al., 2018; Barazzuol et al., 2021). The enhanced interaction between Bcl-xL and IP3R3 at MAM provides cellular protection during stress conditions, which is achieved by improved mitochondria-ER communication, calcium homeostasis, and cellular bioenergetics (Williams et al., 2016). Numerous crucial neural processes are modulated through the mitochondria and ER cross-talk, where multiple tethering proteins from both organelles crucially regulate this communication. One such connection is formed by the interaction of the surface proteins such as VAPB-PTPIP51 and is reported to be disrupted in degenerating neuronal conditions, like ALS, Parkinson, and FTD. Recently, the importance of these tethering proteins in autophagy regulation is reported, where loss of VAPB-PTPIP51 found to affect formation of autophagosome (Gomez-Suaga et al., 2017; Gómez-Suaga et al., 2019). Moreover, in post-mortem AD brain tissue analysis, the disrupted association between VAPB-PTPIP51 is observed in pyramidal neurons (temporal cortex) at the late stages of AD (Lau et al., 2020).
5 Pathogenic tau and Aβ affects cellular quality control pathways
The UPS and autophagy are crucial and essential cellular quality control pathways for maintaining homeostasis by clearance of misfolded and non-required proteins (Yang and Klionsky, 2010; Pohl and Dikic, 2019). These pathways also preserve the quality of cellular organelles, such as mitochondria. UPS is reported to modulate several mitochondrial pathways like its dynamics, mitophagy, and biogenesis (Ross et al., 2015; Bragoszewski et al., 2017). Moreover, the significance of cytosolic UPS in degrading OMM-localized proteins is also reported. Cdc48/VCP/p97 recruited at OMM assists in translocating them from mitochondria to cytosol (Fang et al., 2015). The inhibition of proteasome activity by Aβ is known further to accumulate Aβ and tau in aging-related disorders like AD (Oddo, 2008; Tseng et al., 2008). The impaired proteasome activity may damage mitochondrial health by altering its different quality control mechanisms, which further need to be studied and can be utilized as target AD-related therapeutics. The pathological interaction of p-tau and Aβ with Drp1 protein and Aβ-PINK1/Parkin affects the removal of damaged mitochondria from neurons. Furthermore, the age-linked accumulation of p-tau and Aβ causes depletion of multiple autophagy proteins (Reddy and Oliver, 2019). However, another report also suggests the induction of autophagy and mitophagy in cellular models treated with Aβ but not at sufficient levels to reduce Aβ. Moreover, reduced mitochondrial biogenesis and its increased fragmentation and mitophagy are observed in early pathogenic events in APP/PS1 mice (de la Cueva et al., 2022).
6 How do mitochondrial impairments and energy defects contribute to Parkinson's disease pathology?
Parkinson’s disease (PD) is often linked to imperfect aging and neuronal degeneration, with increasing occurrence globally. The disease symptoms include various motor function defects, frequently initiated with tremors, further progressing to loss of balance, muscle stiffness, and difficulty in coordination (Pringsheim et al., 2014; Kalia and Lang, 2015; Dorsey, 2018). Dopaminergic neurons exist in substantia nigra pars compacta (SNpc) are primarily affected in PD patients. Lewy bodies comprising aggregates of α-synuclein protein are identified in brain sections of PD patients (Figure 4). The continuous research on understanding the disease pathomechanism has improved our knowledge, and the involvement of multiple affected processes has been identified, such as increased oxidative stress, proteostasis defects, calcium dyshomeostasis, impaired axonal transport, and mitochondrial damage (Poewe et al., 2017). Research further highlights the mitochondrial impairments observed in PD patients with various mitochondria-associated proteins involved in PD pathology (Abou-Sleiman et al., 2006; Park et al., 2018).
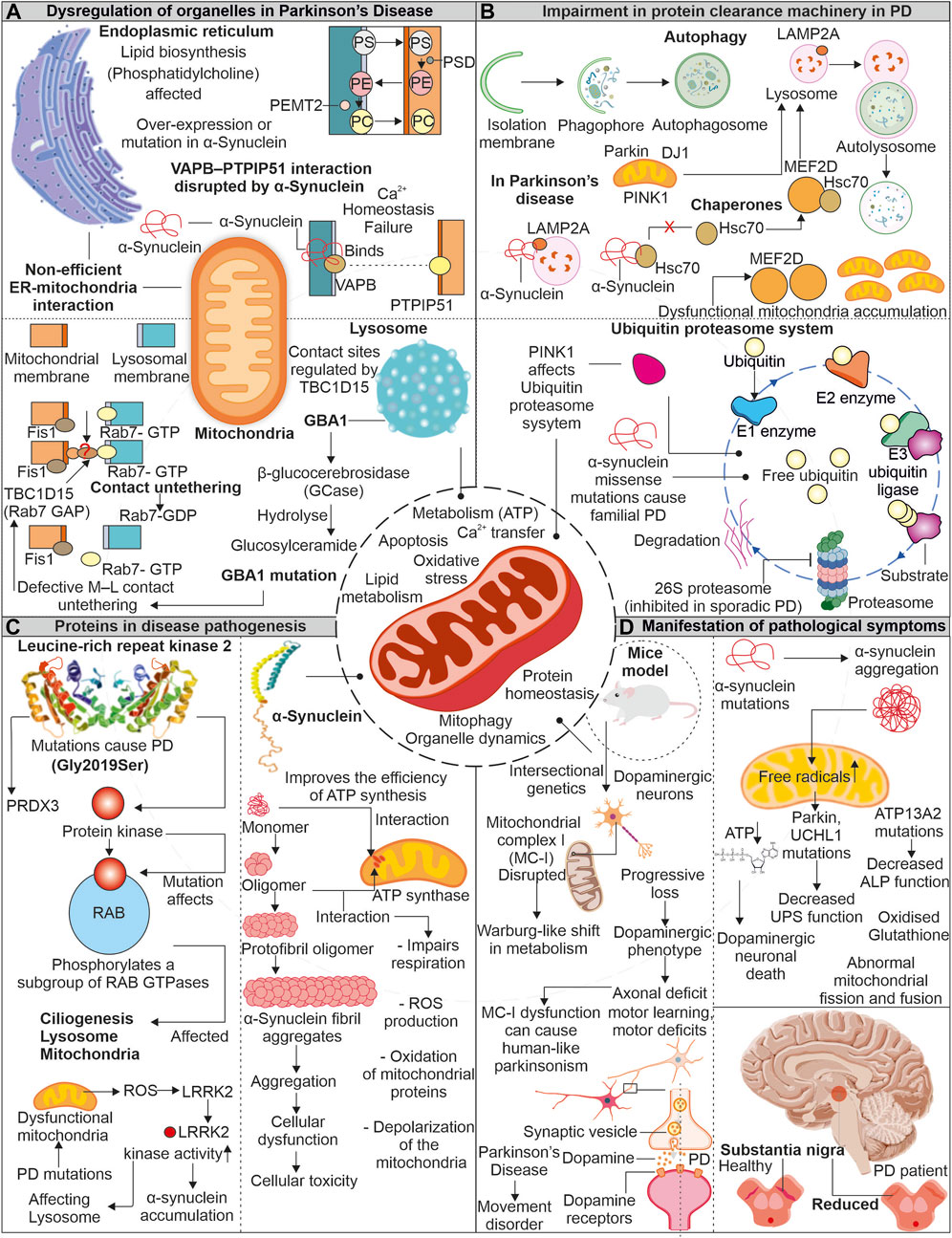
FIGURE 4. Mitochondrial impairment in Parkinson’s disease: Unraveling the connection. In the pathogenesis of the disease, mitochondria show dysfunctionality which affects other organelles functioning. Enzymes of lysosome like GBA1 is impaired due to mutation in PD, which affects the association of lysosome with mitochondria. Similarly, α-synuclein protein interferes with ER and mitochondrial association (A). The impaired proteins involved in disease pathogenesis interact with autophagy and the UPS and interfere with the clearance of dysfunctional mitochondria (B). Two proteins LRRK2 and α-synuclein, are reportedly intricate in disease pathogenesis. These proteins impair the functionality of mitochondria and increase oxidative stress (C). Due to dysfunctional mitochondria, the dopamine-producing neurons are affected and later affect the brain region (D).
6.1 Mutant proteins are implicated in the mitochondrial damage observed in Parkinson’s disease
The α-synuclein is a neuronal protein and acts as a chaperone promoting the assembly of SNARE-complex present in pre-synapse (Burre et al., 2010). The α-synuclein is known to affect dopaminergic neuronal mitochondria. Devi et al. reported increased α-synuclein accumulation, but not its lower levels in mitochondria generate bioenergetic defects by impairing the activity of complex I (Devi et al., 2008). Whereas recently, the unfolded monomeric form of α-synuclein is reported to interact and induce ATP synthase function. The increased levels of α-synuclein at pre-synapse are also suggested to positively modulate ATP synthase providing higher energy required for synaptic transmission mechanisms (Ludtmann et al., 2016). Moreover, the same group also showed the negative effect of oligomeric α-synuclein on ATP synthase, impairing mitochondrial respiration. Further, the depolarization of mitochondria in increased ROS levels causes opening of mitochondrial permeability transition pore (mPTP) (Ludtmann et al., 2018). Furthermore, the selectively impaired complex I activity is found to induce parkinsonism, where the altered release of dopamine is suggested as the prime mechanism for motor dysfunction (Gonzalez-Rodriguez et al., 2021). Intracellular oligomerization of α-synuclein is reported to occur at the membrane surface, where the mitochondrial lipid cardiolipin plays a prominent role in the oligomer formation. The hiPSC derived from α-synuclein (A53T) PD patients shows sequestration of cardiolipin in aggregates of lipid-protein complexes. Further, these aggregates were shown to cause mitochondrial defects leading to neuronal cell death (Choi et al., 2022). Moreover, the oligomers and fibrillar forms of α-synuclein can independently generate ROS in occurrence of metal ions (Deas et al., 2016). α-synuclein’s toxic oligomeric forms can harm cellular systems in various ways, such as damaging the cytoskeleton, hindering protein clearance, disrupting membranes, and causing mitochondrial depolarization (Roberts and Brown, 2015).
Leucine-rich repeat kinase 2 (LRRK2) is another protein from PD pathology, and multiple disease-causing mutations are identified in the LRRK2 gene. From several identified mutations, the G2019S mutation (kinase domain) is frequently observed in familial and sporadic PD (Gandhi et al., 2009). Furthermore, LRRK2 (G2019S) mutation was found to affect mitochondrial H2O2 homeostasis by altering the activity of mitochondria-localized peroxiredoxin-3 (PRDX3), a major H2O2 scavenger (Angeles et al., 2014). The increased mitochondrial fragmentation was observed in cells expressing LRRK2 (G2019S), which induces Drp1 phosphorylation at T595, increasing its activity (Su and Qi, 2013). Elevated oxidative stress in PD-affected neurons is a well-known fact. Further, this stress is reported to enhance LRRK2 kinase function, similar to LRRK2 (G2019S) (Rocha et al., 2022). The increased kinase LRRK2 activity is further found to induce phosphorylation of α-synuclein, leading to its enhanced accumulation observed in PD (Qing et al., 2009). Moreover, both proteins' co-localization is also reported in PD brain tissue sections and Lewy body inclusions (Guerreiro et al., 2013).
7 Parkinson's disease-associated mitochondrial defects contribute to the impairment of endoplasmic reticulum and lysosomal functions
Mitochondria continuously communicate with other cell organelles and are vital for numerous pathways functioning. Mitochondria interact with ER to regulate cytoplasmic calcium levels and lipid synthesis. Moreover, it is also essential for fission, fusion, and UPRmt. The disturbance in MAM contact sites is reported in numerous neurodegenerative disorders, including PD. Further, lysosomes and mitochondria are also associated via multiple membrane proteins regulating metabolite transfer and organelle dynamics. The disruption in these tethering proteins is also reported in PD (Wilson and Metzakopian, 2021; Cisneros et al., 2022).
7.1 Disrupted mitochondria- endoplasmic reticulum contacts in Parkinson’s disease
The mutated form of α-synuclein is identified to disturb the VAPB- PTPIP51 (ER-Mitochondria) association, further generating calcium dyshomeostasis and mitochondrial bioenergetic imbalances. The dopaminergic neurons with α-synuclein mutations also confirm the disturbed connection between VAPB-PTPIP51. However, this disrupted connection was not linked with GSK-3β altered activity and suggested that the binding of mutant α-synuclein with VAPB results in disrupted ER-mitochondrial association observed in PD (Paillusson et al., 2017). The ER-mitochondrial association is also essential for transporting multiple lipids required in mitochondrial functioning (Flis and Daum, 2013). Mitochondria and ER contact sites are necessary for synthesizing specific lipids like phosphatidylcholine (PC), where multiple intermediate steps require contact between these organelles (Vance, 1990; Flis and Daum, 2013). The phosphatidylserine (PS) generated in the presence of PS synthase 1/2 is further transported to mitochondria for transforming into phosphatidylethanolamine (PE), where PSD (a decarboxylase) role is necessary. The importance of PE at mitochondria for its morphology and function is reported. Again this PE from mitochondria can be transferred back into ER, where PEMT2 can convert it back to PC (Steenbergen et al., 2005; Joshi et al., 2012). Furthermore, PD models with the loss of Parkin/PINK1 functions show defects in sleep patterns and circadian cycles. Although the mitochondrial deformities were not predominantly observed in such conditions, the increased ER-mitochondrial associations were evident. This induced ER-mitochondrial association leads to the depletion of ER levels of phosphatidylserine, generating defects in the synthesis of neuropeptide-containing vesicles. Moreover, phosphatidylserine supplementation in disease models mitigates defective sleep patterns with improved synthesis of neuropeptide-containing vesicles (Valadas et al., 2018).
7.2 How do lysosomal abnormalities are associated with impaired mitochondria in Parkinson’s disease?
The mitochondrial contacts with lysosomes allow the organelles to communicate while maintaining cellular homeostasis. Various mitochondria-localized proteins control these organelle connections. The RAB7 protein from the lysosome assists in tethering with mitochondria, where Fis1 recruited TBC1D15 disrupts these connections by converting RAB7 from GTP (active) to GDP (inactive) form (Wong et al., 2018; Wong et al., 2019). The lysosomal functions are essential for removing the misfolded proteins, and its impaired activity is observed in PD. This lysosomal dysfunction is linked with impaired mitochondria. The increase in oxidized dopamine due to mitochondria-linked oxidative stress led to a decrease in lysosomal glucocerebrosidase activity, which further accumulates α-synuclein (Burbulla et al., 2017). The lysosomal and autophagic defects are observed in PD patient-derived neurons with a mutation in lysosomal enzyme β-glucocerebrosidase. Further, these neurons also represent calcium dyshomeostasis with increased cytosol calcium levels (Schondorf et al., 2014). Restoring β-glucocerebrosidase enzyme activity improves the lysosome-linked clearance of α-synuclein (Mazzulli et al., 2016). The mitochondrial respiration and axonal transport are also affected in PD patient-derived dopaminergic neurons having the β-glucocerebrosidase mutation. The observed mitochondrial defects were linked with altered mitochondria-lysosome contacts, where prolonged tethering due to decreased activity of TBC1D15 is identified (Kim et al., 2021). The altered activity of lysosome-linked ATP13A2 affects lysosomes at multiple levels; further, it impairs autophagy, resulting in α-synuclein aggregation (Zhang et al., 2022).
8 Autophagy and proteasomal defects associated with Parkinson disease
Chaperone-mediated autophagy (CMA), a selective type of autophagy, aids in removal of soluble cytosolic protein having a KFERQ-like pentapeptide sequence. Cytosolic Hsc70 and co-chaperones such as the HSC70-HSP90 organizing protein (HOP), HSP40, and CHIP identify this sequence on target proteins, further facilitating their translocation across the lysosomal membrane via LAMP2A receptor and resulting in protein breakdown (Kaushik and Cuervo, 2018). The native α-synuclein acts as a substrate for CMA-dependent degradation (Vogiatzi et al., 2008). Moreover, the mutant α-synuclein (A53T and A30P) was observed to inhibit the CMA by interacting with LAMP2A, further affecting the degradation of other CMA substrates and contributing to increased cellular stress (Cuervo et al., 2004). The transcriptional controller MEF2 was shown to have a crucial role in neuronal survival (Mao et al., 1999). Their suppression causes the death of neurons while increasing their activity guards against harmful cellular stress (Tang et al., 2005; Smith et al., 2006). The suppressed CMA in the presence of α-synuclein affects the interaction of MEF2D with Hsc70, resulting in its accumulation and loss of function, causing neuronal death (Yang et al., 2009). The neuronal accumulation of multiple PD-associated misfolded proteins positive for ubiquitin also suggests the impairment of another quality control pathway, i.e., UPS. Furthermore, PD-linked Lewy bodies were reported positive for 20S proteasome and α-synuclein protein. Moreover, the impaired proteolytic activity due to α-synuclein oligomers is also shown (Lindersson et al., 2004). PINK1 mutants from PD pathology generates mitochondrial bioenergetic defects, which further affect other cellular energy-dependent processes including UPS-mediated degradation (Liu et al., 2009).
9 Mitochondria: A promising therapeutic target for neurodegenerative disorders
The decline in mitochondrial function during aging and associated disorders like neurodegeneration has received much attention, and there are extensive efforts underway to develop pharmacological treatments that can restore the potential and integrity of these crucial organelles. A large number of pharmacological modulators, both natural and synthetic, are being studied for their ability to reduce mitochondrial stress by targeting different pathways, including mitochondrial OXPHOS, ROS homeostasis, and metabolic processes. Furthermore, several other pathways, such as mitochondrial biogenesis, dynamics, and degradation, are also considered in developing therapeutics against mitochondria-associated disorders.
The depleted energy production by mitochondria due to various cellular stresses such as increased ROS levels and calcium dyshomeostasis, significantly affects energy-intensive cells like neurons. Scientists have explored various molecules that could potentially improve the functioning of the, ETC. For example, riboflavin and idebenone enhance the transfer of electrons, while others, such as thiamine and dichloroacetate, increase the availability of, ETC., substrates (Bernsen et al., 1993; Barshop et al., 2004; Haefeli et al., 2011; Mermigkis et al., 2013). The promising effects of idebenone are noted in multiple studies on individuals with Alzheimer’s disease (Senin et al., 1992). In addition, researchers have studied various natural compounds for their ability to regulate mitochondrial oxidative stress. For example, saponins derived from Panax japonicus and Panax notoginseng show neuroprotective effects by reducing mitochondrial damage through the induction of antioxidant responses (Zhou et al., 2018; Wan et al., 2020). Besides this, several mitochondrial antioxidants, including MitoQ, Mitotempo, and Mito apocynin, protects mitochondria from oxidative damage (Hu and Li, 2016; Brenza et al., 2017; Young and Franklin, 2019).
Another approach to address energy deficiency involves increasing the number of mitochondria in cells by targeting transcription factors participating in formation of new mitochondria, such as PGC-1α. Pioglitazone, a type of thiazolidinedione, has been shown to have protective effects in several neurological diseases (Masciopinto et al., 2012; Morato et al., 2013). Pioglitazone induces mitochondrial biogenesis and mtDNA copy number by targeting transcription factors such as PGC-1α (Bogacka et al., 2005). Furthermore, various studies show the protective effects of directly or indirectly activating mitochondrial biogenesis with compounds such as bezafibrate, resveratrol, and AICAR (Komen and Thorburn, 2014). It is evident that mitochondrial dynamics are altered in various neurodegenerative diseases, and different pharmacological interventions that can modulate the proteins involved in this process are investigated. Echinacoside (ECH) treatment shows neuroprotective effects by inducing mitochondrial fusion via increased transcription of Mfn2 (Zeng et al., 2021). Treatment with liquiritigenin, a flavonoid, has been found to induce mitochondrial fusion and protects against Aβ cytotoxicity (Liu et al., 2011; Jo et al., 2016).
Additionally, inhibiting the mitochondrial fission pathway has been found to have therapeutic benefits. For example, the use of mitochondrial division inhibitor-1 (Mdivi-1) decreases mitochondrial fragmentation (inhibit Drp1) in cells treated with Aβ, as well as maintains ΔΨm and prevents Cyto C release (Cassidy-Stone et al., 2008; Xie et al., 2014). Various modulators of mitophagy that have shown beneficial effects by removing damaged or altered mitochondria are identified. Urolithin A induces autophagic removal of altered mitochondria and extended lifespan in C. elegans (Ryu et al., 2016). Spermidine treatment was found to induce both the mitophagy as well as biogenesis of mitochondria in aged mice heart cells (Messerer et al., 2023). Metformin, a drug used in treating T2D, has been found to enhance mitochondrial function by restoring, ETC., proteins and promoting mitophagy (de Maranon et al., 2022).
10 Conclusion and future outlook
Mitochondria are crucial cell organelles well-recognized for their involvement in ATP production, and they also take part in numerous cellular signaling and pathways. Primarily, these include homeostasis of calcium, synthesis of biomolecules, and regulation of cell death by apoptosis. The impairment in such mitochondrial functions is detected in numerous pathological conditions, including metabolic, neurodevelopmental, neurodegeneration, and cancer. Moreover, mitochondrial dysfunction at early stages of neurodegeneration is prominently observed in ALS, AD, and PD. The neurons affected in neurodegenerative disease are significantly dependent on mitochondria for numerous functions, for instance, neurogenesis, neuronal development, neurotransmission, and neuronal repair pathways. Different neurodegenerative disease-causing proteins are identified to affect the mitochondria resulting in bioenergetic defects and increased ROS production. The ALS-linked C9orf72, FUS, SOD1, TDP-43, and PD-linked α-synuclein oligomers were found to affect mitochondrial, ETC., protein function by either influencing their activity, expression of subunits or directly impairing their assembly. The OXPHOS-impaired mitochondria act as source of ROS, inducing oxidative stress in neurons. Moreover, the inadequate antioxidant response further worsens the damage. The age-linked depletion in ROS homeostasis and other quality control pathways further aggravates the mitochondria damage leading to cell death by apoptosis.
Mitochondria possess different quality control mechanisms to circumvent such situations, including chaperones and proteases. UPRmt activated upon increase in mitochondrial stress communicates with the nucleus for induced expression of mitostasis proteins. The dynamics and autophagy of mitochondria also regulate its health by either diluting the mitochondrial damage (fusion) or degrading dysfunctional mitochondria (fission and mitophagy). The pathogenic misfolded proteins from neurodegenerative diseases are known to interfere with these mitostasis pathways. The AD (Aβ, tau), ALS (SOD1), and PD (LRRK2) pathogenic proteins interact and alter fission proteins inducing aberrant mitochondrial fragmentation. These misfolded proteins can disturb mitochondrial contact with ER and lysosomes, resulting in impairment of calcium homeostasis and autophagy. The increased cytosolic calcium due to dysfunctional ER-mitochondria connection is also linked with defective mitochondrial transport. The negative impact of disease-misfolded proteins on cellular protein quality control (PQC) machinery hampers mitochondrial quality control, intensifying the damage to multiple mitochondria-dependent cellular processes. Despite the existence of many sophisticated methods for understanding the pathogenesis of disorders, no effective treatments are currently available for mitochondrial-associated diseases. Therapeutic approaches for mitochondrial diseases are presently limited to enhancing electron transfer in OXPHOS, using antioxidants, and stimulating mitochondrial biogenesis, among others. Furthermore, mitochondrial gene therapy, mitochondrial protein replacement therapy, mitochondrial transplantation, and nanoparticle drug delivery can be potential ways to advance mitochondrial disease therapeutics.
Developing therapeutic agents and strategies for neurodegeneration is a challenging task, given the insufficient understanding of complexity of their initiation and pathogenesis. Mitochondria can serve as ideal candidates for the connecting link between multiple complex degenerating disorders. Understanding role of mitochondria, mechanism of their dyshomeostasis, and its effect on cellular health and survival can provide us insight into the basic mechanistic aspects of neurodegeneration. The investigation of mitochondrial dysfunction in network with other associated organelles in neurodegeneration allows us to target mitochondria specifically or in combination with other organelles. The current therapeutic landscape includes many targets for neuronal degeneration, such as cholinergic, glutamatergic, and dopaminergic systems, but the clinical results are less than satisfactory. The multiple mitochondrial molecules and pathways discussed in this review can be investigated for their diagnostic and therapeutic potential and advancing the understanding of neurodegeneration.
Author contributions
YJ, PK, SK, AD, and AC under the concept and performed the figures formation and also prepare the first draft in line with the framework of manuscript. RG, SS, HJ, and KP analysed the prepared framework and perform critical observations of the manuscript and helped inline the manuscript finalization. AM overall contributed concept design, analysed the entire content of manuscript and draft finalization of manuscript. All authors discussed the manuscript and contributed to the final manuscript.
Funding
AM received research grant EMR/2016/000716 from Science and Engineering Research Board (SERB), Department of Science & Technology, Government of India and from BRNS grant to (AM) 54/14/16/2020-BRNS-Government of India.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abou-Sleiman, P. M., Muqit, M. M., and Wood, N. W. J. N. R. N. (2006). Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat. Rev. Neurosci. 7 (3), 207–219. doi:10.1038/nrn1868
Aldridge, J. E., Horibe, T., and Hoogenraad, N. J. (2007). Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One 2 (9), e874. doi:10.1371/journal.pone.0000874
Alikhani, N., Guo, L., Yan, S., Du, H., Pinho, C. M., Chen, J. X., et al. (2011). Decreased proteolytic activity of the mitochondrial amyloid-beta degrading enzyme, PreP peptidasome, in Alzheimer's disease brain mitochondria. J. Alzheimers Dis. 27 (1), 75–87. doi:10.3233/JAD-2011-101716
Angeles, D. C., Ho, P., Chua, L. L., Wang, C., Yap, Y. W., Ng, C., et al. (2014). Thiol peroxidases ameliorate LRRK2 mutant-induced mitochondrial and dopaminergic neuronal degeneration in Drosophila. Hum. Mol. Genet. 23 (12), 3157–3165. doi:10.1093/hmg/ddu026
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351 (3), 602–611. doi:10.1016/j.bbrc.2006.10.093
Area-Gomez, E., de Groof, A. J. C., Boldogh, I., Bird, T. D., Gibson, G. E., Koehler, C. M., et al. (2009). Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 175 (5), 1810–1816. doi:10.2353/ajpath.2009.090219
Ash, P. E., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., Dejesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77 (4), 639–646. doi:10.1016/j.neuron.2013.02.004
Audano, M., Schneider, A., and Mitro, N. (2018). Mitochondria, lysosomes, and dysfunction: Their meaning in neurodegeneration. J. Neurochem. 147 (3), 291–309. doi:10.1111/jnc.14471
Baldwin, K. R., Godena, V. K., Hewitt, V. L., and Whitworth, A. J. (2016). Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum. Mol. Genet. 25 (12), 2378–2392. doi:10.1093/hmg/ddw105
Baloyannis, S. J. (2006). Mitochondrial alterations in Alzheimer's disease. J. Alzheimers Dis. 9 (2), 119–126. doi:10.3233/jad-2006-9204
Barazzuol, L., Giamogante, F., and Calì, T. J. C. C. (2021). Mitochondria associated membranes (MAMs): Architecture and physiopathological role. Cell Calcium 94, 102343. doi:10.1016/j.ceca.2020.102343
Barshop, B. A., Naviaux, R. K., McGowan, K. A., Levine, F., Nyhan, W. L., Loupis-Geller, A., et al. (2004). Chronic treatment of mitochondrial disease patients with dichloroacetate. Mol. Genet. Metab. 83 (1-2), 138–149. doi:10.1016/j.ymgme.2004.06.009
Beck, J., Poulter, M., Hensman, D., Rohrer, J. D., Mahoney, C. J., Adamson, G., et al. (2013). Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am. J. Hum. Genet. 92 (3), 345–353. doi:10.1016/j.ajhg.2013.01.011
Benussi, L., Rossi, G., Glionna, M., Tonoli, E., Piccoli, E., Fostinelli, S., et al. (2014). C9ORF72 hexanucleotide repeat number in frontotemporal lobar degeneration: A genotype-phenotype correlation study. J. Alzheimers Dis. 38 (4), 799–808. doi:10.3233/JAD-131028
Bernsen, P. L., Gabreels, F. J., Ruitenbeek, W., and Hamburger, H. L. (1993). Treatment of complex I deficiency with riboflavin. J. Neurol. Sci. 118 (2), 181–187. doi:10.1016/0022-510x(93)90108-b
Bhatti, J. S., Bhatti, G. K., and Reddy, P. H. (2017). Mitochondrial dysfunction and oxidative stress in metabolic disorders - a step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 1863 (5), 1066–1077. doi:10.1016/j.bbadis.2016.11.010
Bleazard, W., McCaffery, J. M., King, E. J., Bale, S., Mozdy, A., Tieu, Q., et al. (1999). The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1 (5), 298–304. doi:10.1038/13014
Bogacka, I., Xie, H., Bray, G. A., and Smith, S. R. (2005). Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes 54 (5), 1392–1399. doi:10.2337/diabetes.54.5.1392
Bonifati, V., Rizzu, P., van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E., et al. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299 (5604), 256–259. doi:10.1126/science.1077209
Bosco, D. A., Lemay, N., Ko, H. K., Zhou, H., Burke, C., Kwiatkowski, T. J., et al. (2010). Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 19 (21), 4160–4175. doi:10.1093/hmg/ddq335
Bota, D. A., and Davies, K. J. (2002). Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat. Cell Biol. 4 (9), 674–680. doi:10.1038/ncb836
Boxer, A. L., Mackenzie, I. R., Boeve, B. F., Baker, M., Seeley, W. W., Crook, R., et al. (2011). Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J. Neurol. Neurosurg. Psychiatry 82 (2), 196–203. doi:10.1136/jnnp.2009.204081
Bragoszewski, P., Turek, M., and Chacinska, A. (2017). Control of mitochondrial biogenesis and function by the ubiquitin-proteasome system. Open Biol. 7, 170007. doi:10.1098/rsob.170007
Brenza, T. M., Ghaisas, S., Ramirez, J. E. V., Harischandra, D., Anantharam, V., Kalyanaraman, B., et al. (2017). Neuronal protection against oxidative insult by polyanhydride nanoparticle-based mitochondria-targeted antioxidant therapy. Nanomedicine 13 (3), 809–820. doi:10.1016/j.nano.2016.10.004
Briston, T., and Hicks, A. R. (2018). Mitochondrial dysfunction and neurodegenerative proteinopathies: Mechanisms and prospects for therapeutic intervention. Biochem. Soc. Trans. 46 (4), 829–842. doi:10.1042/BST20180025
Bruni, F., Polosa, P. L., Gadaleta, M. N., Cantatore, P., and Roberti, M. (2010). Nuclear respiratory factor 2 induces the expression of many but not all human proteins acting in mitochondrial DNA transcription and replication. J. Biol. Chem. 285 (6), 3939–3948. doi:10.1074/jbc.M109.044305
Buratti, E., and Baralle, F. E. (2008). Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front. Biosci. 13, 867–878. doi:10.2741/2727
Burbulla, L. F., Song, P., Mazzulli, J. R., Zampese, E., Wong, Y. C., Jeon, S., et al. (2017). Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science 357 (6357), 1255–1261. doi:10.1126/science.aam9080
Burre, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Sudhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329 (5999), 1663–1667. doi:10.1126/science.1195227
Calkins, M. J., Manczak, M., Mao, P., Shirendeb, U., and Reddy, P. H. (2011). Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum. Mol. Genet. 20 (23), 4515–4529. doi:10.1093/hmg/ddr381
Cassidy-Stone, A., Chipuk, J. E., Ingerman, E., Song, C., Yoo, C., Kuwana, T., et al. (2008). Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14 (2), 193–204. doi:10.1016/j.devcel.2007.11.019
Castro, J. P., Wardelmann, K., Grune, T., and Kleinridders, A. (2018). Mitochondrial chaperones in the brain: Safeguarding brain health and metabolism? Front. Endocrinol. (Lausanne) 9, 196. doi:10.3389/fendo.2018.00196
Chai, Y. L., Chong, J. R., Weng, J., Howlett, D., Halsey, A., Lee, J. H., et al. (2019). Lysosomal cathepsin D is upregulated in Alzheimer's disease neocortex and may be a marker for neurofibrillary degeneration. Brain Pathol. 29 (1), 63–74. doi:10.1111/bpa.12631
Chan, N. C., Salazar, A. M., Pham, A. H., Sweredoski, M. J., Kolawa, N. J., Graham, R. L. J., et al. (2011). Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20 (9), 1726–1737. doi:10.1093/hmg/ddr048
Cheignon, C., ToMas, M., Bonnefont-Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2018). Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol. 14, 450–464. doi:10.1016/j.redox.2017.10.014
Chen, J. X., and Yan, S. D. (2007). Amyloid-beta-induced mitochondrial dysfunction. J. Alzheimers Dis. 12 (2), 177–184. doi:10.3233/jad-2007-12208
Choi, M. L., Chappard, A., Singh, B. P., Maclachlan, C., Rodrigues, M., Fedotova, E. I., et al. (2022). Pathological structural conversion of α-synuclein at the mitochondria induces neuronal toxicity. Nat. Neurosci. 25 (9), 1134–1148. doi:10.1038/s41593-022-01140-3
Choi, S. Y., Lopez-Gonzalez, R., Krishnan, G., Phillips, H. L., Li, A. N., Seeley, W. W., et al. (2019). C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat. Neurosci. 22 (6), 851–862. doi:10.1038/s41593-019-0397-0
Cisneros, J., Belton, T. B., and Shum, G. C. (2022). Mitochondria-lysosome contact site dynamics and misregulation in neurodegenerative diseases. Trends Neurosci. 45. doi:10.1016/j.tins.2022.01.005
Colombrita, C., Onesto, E., Megiorni, F., Pizzuti, A., Baralle, F. E., Buratti, E., et al. (2012). TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J. Biol. Chem. 287 (19), 15635–15647. doi:10.1074/jbc.M111.333450
Corcia, P., Pradat, P. F., Salachas, F., Bruneteau, G., Forestier, N. l., Seilhean, D., et al. (2008). Causes of death in a post-mortem series of ALS patients. Amyotroph. Lateral Scler. 9 (1), 59–62. doi:10.1080/17482960701656940
Crozat, A., MaNdahl, N., and Ron, D. (1993). Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 363 (6430), 640–644. doi:10.1038/363640a0
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T., and Sulzer, D. (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305 (5688), 1292–1295. doi:10.1126/science.1101738
da-Silva, W. S., Gomez-Puyou, A., de Gomez-Puyou, M. T., Moreno-Sanchez, R., De Felice, F. G., de Meis, L., et al. (2004). Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 279 (38), 39846–39855. doi:10.1074/jbc.M403835200
Damiano, M., Starkov, A. A., Petri, S., Kipiani, K., Kiaei, M., Mattiazzi, M., et al. (2006). Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J. Neurochem. 96 (5), 1349–1361. doi:10.1111/j.1471-4159.2006.03619.x
Dang, X., Zhang, L., Franco, A., and Dorn, G. W. (2022). Activating mitofusins interrupts mitochondrial degeneration and delays disease progression in SOD1 mutant amyotrophic lateral sclerosis. Hum. Mol. Genet. 2022, ddac287. doi:10.1093/hmg/ddac287
Davis, S. A., Itaman, S., Khalid-Janney, C. M., Sherard, J. A., Dowell, J. A., Cairns, N. J., et al. (2018). TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 678, 8–15. doi:10.1016/j.neulet.2018.04.053
de la Cueva, M., Antequera, D., Ordonez-Gutierrez, L., Wandosell, F., Camins, A., Carro, E., et al. (2022). Amyloid-β impairs mitochondrial dynamics and autophagy in Alzheimer’s disease experimental models. Sci. Rep. 12 (1), 10092. doi:10.1038/s41598-022-13683-3
de Maranon, A. M., Diaz-Pozo, P., Canet, F., Diaz-Morales, N., Abad-Jimenez, Z., Lopez-Domenech, S., et al. (2022). Metformin modulates mitochondrial function and mitophagy in peripheral blood mononuclear cells from type 2 diabetic patients. Redox Biol. 53, 102342. doi:10.1016/j.redox.2022.102342
de Sagarra, M. R., Mayo, I., Marco, S., Rodriguez-Vilarino, S., Oliva, J., Carrascosa, J. L., et al. (1999). Mitochondrial localization and oligomeric structure of HClpP, the human homologue of E. coli ClpP. J. Mol. Biol. 292 (4), 819–825. doi:10.1006/jmbi.1999.3121
Deas, E., Cremades, N., Angelova, P. R., Ludtmann, M. H. R., Yao, Z., Chen, S., et al. (2016). Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson's disease. Antioxid. Redox Signal 24 (7), 376–391. doi:10.1089/ars.2015.6343
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72 (2), 245–256. doi:10.1016/j.neuron.2011.09.011
Deng, H., Gao, K., and Jankovic, J. (2014). The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 10 (6), 337–348. doi:10.1038/nrneurol.2014.78
Deng, J., Wang, P., Chen, X., Cheng, H., Liu, J., Fushimi, K., et al. (2018). FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. U. S. A. 115 (41), E9678–E9686. doi:10.1073/pnas.1806655115
Deng, J., Yang, M., Chen, Y., Chen, X., Liu, J., Sun, S., et al. (2015). FUS interacts with HSP60 to promote mitochondrial damage. PLoS Genet. 11 (9), e1005357. doi:10.1371/journal.pgen.1005357
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283 (14), 9089–9100. doi:10.1074/jbc.M710012200
Dixit, S., Fessel, J. P., and Harrison, F. E. (2017). Mitochondrial dysfunction in the APP/PSEN1 mouse model of Alzheimer's disease and a novel protective role for ascorbate. Free Radic. Biol. Med. 112, 515–523. doi:10.1016/j.freeradbiomed.2017.08.021
Donnelly, C. J., Zhang, P. W., Pham, J. T., Haeusler, A. R., Heusler, A. R., Mistry, N. A., et al. (2013). RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80 (2), 415–428. doi:10.1016/j.neuron.2013.10.015
Dorsey, E. R. (2018). Global, regional, and national burden of Parkinson's disease, 1990–2016: A systematic analysis for the global burden of disease study 2016. Lancet Neurol. 17 (11), 939–953.
DuBoff, B., Gotz, J., and Feany, M. B. (2012). Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 75 (4), 618–632. doi:10.1016/j.neuron.2012.06.026
Dunn, C. D. (2017). Some liked it hot: A hypothesis regarding establishment of the proto-mitochondrial endosymbiont during eukaryogenesis. J. Mol. Evol. 85 (3-4), 99–106. doi:10.1007/s00239-017-9809-5
Ebneth, A., Stamer, K., Illenberger, S., Trinczek, B., and MandElkow, E. (1998). Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer's disease. J. Cell Biol. 143 (3), 777–794. doi:10.1083/jcb.143.3.777
Ernster, L., and Schatz, G. (1981). Mitochondria: A historical review. J. Cell Biol. 91 (3), 227s–255s. doi:10.1083/jcb.91.3.227s
Falkevall, A., Alikhani, N., Bhushan, S., Pavlov, P. F., Busch, K., Johnson, K. A., et al. (2006). Degradation of the amyloid beta-protein by the novel mitochondrial peptidasome. Prep. J. Biol. Chem. 281 (39), 29096–29104. doi:10.1074/jbc.M602532200
Fallaize, D., Chin, L. S., and Li, L. (2015). Differential submitochondrial localization of PINK1 as a molecular switch for mediating distinct mitochondrial signaling pathways. Cell Signal 27 (12), 2543–2554. doi:10.1016/j.cellsig.2015.09.020
Fang, L., Hemion, C., Pinho Ferreira Bento, A. C., Bippes, C. C., Flammer, J., and Neutzner, A. (2015). Mitochondrial function in neuronal cells depends on p97/VCP/Cdc48-mediated quality control. Front. Cell Neurosci. 9, 16. doi:10.3389/fncel.2015.00016
Ferri, A., Fiorenzo, P., Nencini, M., Cozzolino, M., Pesaresi, M. G., Valle, C., et al. (2010). Glutaredoxin 2 prevents aggregation of mutant SOD1 in mitochondria and abolishes its toxicity. Hum. Mol. Genet. 19 (22), 4529–4542. doi:10.1093/hmg/ddq383
Field, L. S., Furukawa, Y., O'Halloran, T. V., and Culotta, V. C. (2003). Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J. Biol. Chem. 278 (30), 28052–28059. doi:10.1074/jbc.M304296200
Fiorese, C. J., Schulz, A. M., Lin, Y. F., Rosin, N., Pellegrino, M. W., and Haynes, C. M. (2016). The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 26 (15), 2037–2043. doi:10.1016/j.cub.2016.06.002
Flis, V. V., and Daum, G. J. (2013). Lipid transport between the endoplasmic reticulum and mitochondria. Cold Spring Harb. Perspect. Biol. 5 (6), a013235. doi:10.1101/cshperspect.a013235
Freibaum, B. D., Chitta, R. K., High, A. A., and Taylor, J. P. (2010). Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 9 (2), 1104–1120. doi:10.1021/pr901076y
Frey, T. G., Renken, C. W., and Perkins, G. A. (2002). Insight into mitochondrial structure and function from electron tomography. Biochim. Biophys. Acta 1555 (1-3), 196–203. doi:10.1016/s0005-2728(02)00278-5
Frick, P., Sellier, C., Mackenzie, I. R. A., Cheng, C. Y., Tahraoui-Bories, J., Martinat, C., et al. (2018). Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol. Commun. 6 (1), 72. doi:10.1186/s40478-018-0579-0
Fuchs, A., Kutterer, S., Muhling, T., Duda, J., Schutz, B., Liss, B., et al. (2013). Selective mitochondrial Ca2+ uptake deficit in disease endstage vulnerable motoneurons of the SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Physiol. 591 (10), 2723–2745. doi:10.1113/jphysiol.2012.247981
Gandhi, P. N., Chen, S. G., and Wilson-Delfosse, A. L. (2009). Leucine-rich repeat kinase 2 (LRRK2): A key player in the pathogenesis of Parkinson's disease. J. Neurosci. Res. 87 (6), 1283–1295. doi:10.1002/jnr.21949
Gatt, A. P., Duncan, O. F., Attems, J., Francis, P. T., Ballard, C. G., and Bateman, J. M. (2016). Dementia in Parkinson's disease is associated with enhanced mitochondrial complex I deficiency. Mov. Disord. 31 (3), 352–359. doi:10.1002/mds.26513
Gijselinck, I., Van Langenhove, T., van der Zee, J., Sleegers, K., Philtjens, S., Kleinberger, G., et al. (2012). A C9orf72 promoter repeat expansion in a flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: A gene identification study. Lancet Neurol. 11 (1), 54–65. doi:10.1016/S1474-4422(11)70261-7
Goetz, C. G. (2000). Amyotrophic lateral sclerosis: Early contributions of jean-martin Charcot. Muscle Nerve 23 (3), 336–343. doi:10.1002/(sici)1097-4598(200003)23:3<336::aid-mus4>3.0.co;2-l
Gomez-Suaga, P., Morotz, G. M., Markovinovic, A., Martin-Guerrero, S. M., Preza, E., Arias, N., et al. (2022). Disruption of ER-mitochondria tethering and signalling in C9orf72-associated amyotrophic lateral sclerosis and frontotemporal dementia. Aging Cell 21 (2), e13549. doi:10.1111/acel.13549
Gomez-Suaga, P., Paillusson, S., Stoica, R., Noble, W., Hanger, D. P., and Miller, C. C. J. (2017). The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr. Biol. 27 (3), 371–385. doi:10.1016/j.cub.2016.12.038
Gómez-Suaga, P., Perez-Nievas, B. G., Glennon, E. B., Lau, D. H. W., Paillusson, S., Morotz, G. M., et al. (2019). The VAPB-PTPIP51 endoplasmic reticulum-mitochondria tethering proteins are present in neuronal synapses and regulate synaptic activity. Acta Neuropathol. Commun. 7 (1), 35–13. doi:10.1186/s40478-019-0688-4
Gonzalez-Rodriguez, P., Zampese, E., Stout, K. A., Guzman, J. N., Ilijic, E., Yang, B., et al. (2021). Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 599 (7886), 650–656. doi:10.1038/s41586-021-04059-0
Gorman, G. S., Chinnery, P. F., DiMauro, S., Hirano, M., Koga, Y., McFarland, R., et al. (2016). Mitochondrial diseases. Nat. Rev. Dis. Prim. 2, 16080. doi:10.1038/nrdp.2016.80
Gorman, G. S., Schaefer, A. M., Ng, Y., Gomez, N., Blakely, E. L., Alston, C. L., et al. (2015). Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 77 (5), 753–759. doi:10.1002/ana.24362
Gray, M. W. (2012). Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 4 (9), a011403. doi:10.1101/cshperspect.a011403
Guerreiro, P. S., Huang, Y., Gysbers, A., Cheng, D., Gai, W. P., Outeiro, T. F., et al. (2013). LRRK2 interactions with alpha-synuclein in Parkinson's disease brains and in cell models. J. Mol. Med. Berl. 91 (4), 513–522. doi:10.1007/s00109-012-0984-y
Gunter, T. E., Gunter, K. K., Sheu, S. S., and Gavin, C. E. (1994). Mitochondrial calcium transport: Physiological and pathological relevance. Am. J. Physiol. 267 (2), C313–C339. doi:10.1152/ajpcell.1994.267.2.C313
Haass, C., Kaether, C., Thinakaran, G., and Sisodia, S. (2012). Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2 (5), a006270. doi:10.1101/cshperspect.a006270
Haefeli, R. H., Erb, M., Gemperli, A. C., Robay, D., Courdier Fruh, I., Anklin, C., et al. (2011). NQO1-dependent redox cycling of idebenone: Effects on cellular redox potential and energy levels. PLoS One 6 (3), e17963. doi:10.1371/journal.pone.0017963
Hansson Petersen, C. A., Alikhani, N., Behbahani, H., Wiehager, B., Pavlov, P. F., Alafuzoff, I., et al. (2008). The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. U. S. A. 105 (35), 13145–13150. doi:10.1073/pnas.0806192105
Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E. M., Logroscino, G., Robberecht, W., et al. (2017). Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 3, 17071. doi:10.1038/nrdp.2017.71
Hayden, E. Y., and Teplow, D. B. (2013). Amyloid beta-protein oligomers and Alzheimer's disease. Alzheimers Res. Ther. 5 (6), 60. doi:10.1186/alzrt226
Haynes, C. M., Petrova, K., Benedetti, C., Yang, Y., and Ron, D. (2007). ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 13 (4), 467–480. doi:10.1016/j.devcel.2007.07.016
Hayward, L. J., Rodriguez, J. A., Kim, J. W., Tiwari, A., Goto, J. J., Cabelli, D. E., et al. (2002). Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 277 (18), 15923–15931. doi:10.1074/jbc.M112087200
Head, B., Griparic, L., Amiri, M., Gandre-Babbe, S., and van der Bliek, A. M. (2009). Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 187 (7), 959–966. doi:10.1083/jcb.200906083
Hong, K., Li, Y., Duan, W., Guo, Y., Jiang, H., Li, W., et al. (2012). Full-length TDP-43 and its C-terminal fragments activate mitophagy in NSC34 cell line. Neurosci. Lett. 530 (2), 144–149. doi:10.1016/j.neulet.2012.10.003
Hsu, C. C., Tseng, L. M., and Lee, H. C. (2016). Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. (Maywood) 241 (12), 1281–1295. doi:10.1177/1535370216641787
Hu, H., and Li, M. (2016). Mitochondria-targeted antioxidant mitotempo protects mitochondrial function against amyloid beta toxicity in primary cultured mouse neurons. Biochem. Biophys. Res. Commun. 478 (1), 174–180. doi:10.1016/j.bbrc.2016.07.071
Huang, L. K., Chao, S. P., and Hu, C. J. (2020). Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 27 (1), 18. doi:10.1186/s12929-019-0609-7
Hubers, A., Marroquin, N., Schmoll, B., Vielhaber, S., Just, M., Mayer, B., et al. (2014). Polymerase chain reaction and Southern blot-based analysis of the C9orf72 hexanucleotide repeat in different motor neuron diseases. Neurobiol. Aging 35 (5), 1214–e6. doi:10.1016/j.neurobiolaging.2013.11.034
Hwang, J., Estick, C. M., Ikonne, U. S., Butler, D., Pait, M. C., Elliott, L. H., et al. (2019). The role of lysosomes in a broad disease-modifying approach evaluated across transgenic mouse models of Alzheimer's disease and Parkinson's disease and models of mild cognitive impairment. Int. J. Mol. Sci. 20 (18), 4432. doi:10.3390/ijms20184432
Ingerman, E., Perkins, E. M., Marino, M., Mears, J. A., McCaffery, J. M., Hinshaw, J. E., et al. (2005). Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 170 (7), 1021–1027. doi:10.1083/jcb.200506078
Iqbal, K., Liu, F., Gong, C. X., and Grundke-Iqbal, I. (2010). Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 7 (8), 656–664. doi:10.2174/156720510793611592
Iqbal, S., and Hood, D. A. (2014). Oxidative stress-induced mitochondrial fragmentation and movement in skeletal muscle myoblasts. Am. J. Physiol. Cell Physiol. 306 (12), C1176–C1183. doi:10.1152/ajpcell.00017.2014
Israelson, A., Arbel, N., Da Cruz, S., Ilieva, H., Yamanaka, K., Shoshan-Barmatz, V., et al. (2010). Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 67 (4), 575–587. doi:10.1016/j.neuron.2010.07.019
Jimenez-Villegas, J., Kirby, J., Mata, A., Cadenas, S., Turner, M. R., Malaspina, A., et al. (2022). Dipeptide repeat pathology in C9orf72-ALS is associated with redox, mitochondrial and NRF2 pathway imbalance. Antioxidants (Basel) 11 (10), 1897. doi:10.3390/antiox11101897
Jo, D. S., Shin, D. W., Park, S. J., Bae, J. E., Kim, J. B., Park, N. Y., et al. (2016). Attenuation of Aβ toxicity by promotion of mitochondrial fusion in neuroblastoma cells by liquiritigenin. Arch. Pharm. Res. 39 (8), 1137–1143. doi:10.1007/s12272-016-0780-2
Johnson, B. S., Snead, D., Lee, J. J., McCaffery, J. M., Shorter, J., and Gitler, A. D. (2009). TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 284 (30), 20329–20339. doi:10.1074/jbc.M109.010264
Joshi, A. S., Thompson, M. N., Fei, N., Huttemann, M., and Greenberg, M. L. (2012). Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J. Biol. Chem. 287 (21), 17589–17597. doi:10.1074/jbc.M111.330167
Kalia, L. V., and Lang, A. E. J. T. L. (2015). Parkinson's disease. Lancet 386 (9996), 896–912. doi:10.1016/S0140-6736(14)61393-3
Karran, E., and De Strooper, B. (2022). The amyloid hypothesis in alzheimer disease: New insights from new therapeutics. Nat. Rev. Drug Discov. 21 (4), 306–318. doi:10.1038/s41573-022-00391-w
Kaushik, S., and Cuervo, A. M. (2018). The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 19 (6), 365–381. doi:10.1038/s41580-018-0001-6
Kim, J., Moody, J. P., Edgerly, C. K., Bordiuk, O. L., Cormier, K., Smith, K., et al. (2010). Mitochondrial loss, dysfunction and altered dynamics in Huntington's disease. Hum. Mol. Genet. 19 (20), 3919–3935. doi:10.1093/hmg/ddq306
Kim, S., Wong, Y. C., Gao, F., and Krainc, D. (2021). Dysregulation of mitochondria-lysosome contacts by GBA1 dysfunction in dopaminergic neuronal models of Parkinson’s disease. Nat. Commun. 12 (1), 1807–1814. doi:10.1038/s41467-021-22113-3
Kim, Y., Park, J., Kim, S., Song, S., Kwon, S. K., Lee, S. H., et al. (2008). PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem. Biophys. Res. Commun. 377 (3), 975–980. doi:10.1016/j.bbrc.2008.10.104
King, O. D., Gitler, A. D., and Shorter, J. (2012). The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 1462, 61–80. doi:10.1016/j.brainres.2012.01.016
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., MinoShima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392 (6676), 605–608. doi:10.1038/33416
Kohler, F., Muller-Rischart, A. K., Conradt, B., and Rolland, S. G. (2015). The loss of LRPPRC function induces the mitochondrial unfolded protein response. Aging (Albany NY) 7 (9), 701–717. doi:10.18632/aging.100812
Komen, J. C., and Thorburn, D. R. (2014). Turn up the power - pharmacological activation of mitochondrial biogenesis in mouse models. Br. J. Pharmacol. 171 (8), 1818–1836. doi:10.1111/bph.12413
Kreiter, N., Pal, A., Lojewski, X., Corcia, P., Naujock, M., Reinhardt, P., et al. (2018). Age-dependent neurodegeneration and organelle transport deficiencies in mutant TDP43 patient-derived neurons are independent of TDP43 aggregation. Neurobiol. Dis. 115, 167–181. doi:10.1016/j.nbd.2018.03.010
Kubisch, R., Frohlich, T., Arnold, G. J., Schreiner, L., von Schwarzenberg, K., Roidl, A., et al. (2014). V-ATPase inhibition by archazolid leads to lysosomal dysfunction resulting in impaired cathepsin B activation in vivo. Int. J. Cancer 134 (10), 2478–2488. doi:10.1002/ijc.28562
Kwiatkowski, T. J., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323 (5918), 1205–1208. doi:10.1126/science.1166066
Lagier-Tourenne, C., Polymenidou, M., Hutt, K. R., Vu, A. Q., Baughn, M., Huelga, S. C., et al. (2012). Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 15 (11), 1488–1497. doi:10.1038/nn.3230
Larsson, N. G., Wang, J., WilHelmsson, H., Oldfors, A., Rustin, P., Lewandoski, M., et al. (1998). Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 18 (3), 231–236. doi:10.1038/ng0398-231
Lathrop, J. T., and Timko, M. P. (1993). Regulation by heme of mitochondrial protein transport through a conserved amino acid motif. Science 259 (5094), 522–525. doi:10.1126/science.8424176
Lau, D. H. W., Paillusson, S., Hartopp, N., Rupawala, H., Morotz, G. M., Gomez-Suaga, P., et al. (2020). Disruption of endoplasmic reticulum-mitochondria tethering proteins in post-mortem Alzheimer's disease brain. Neurobiol. Dis. 143, 105020. doi:10.1016/j.nbd.2020.105020
Lee, S., and Min, K. T. (2018). The interface between ER and mitochondria: Molecular compositions and functions. Mol. Cells 41 (12), 1000–1007. doi:10.14348/molcells.2018.0438
Leonhard, K., Guiard, B., Pellecchia, G., TzAgoloff, A., Neupert, W., and Langer, T. (2000). Membrane protein degradation by AAA proteases in mitochondria: Extraction of substrates from either membrane surface. Mol. Cell 5 (4), 629–638. doi:10.1016/s1097-2765(00)80242-7
Lin, M. T., and Beal, M. F. J. N. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443 (7113), 787–795. doi:10.1038/nature05292
Lindersson, E., Beedholm, R., Hojrup, P., Moos, T., Gai, W., Hendil, K. B., et al. (2004). Proteasomal inhibition by alpha-synuclein filaments and oligomers. J. Biol. Chem. 279 (13), 12924–12934. doi:10.1074/jbc.M306390200
Liu, J., Lillo, C., Jonsson, P. A., Vande Velde, C., Ward, C. M., Miller, T. M., et al. (2004). Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 43 (1), 5–17. doi:10.1016/j.neuron.2004.06.016
Liu, R. T., Tang, J. T., Zou, L. B., Fu, J. Y., and Lu, Q. J. (2011). Liquiritigenin attenuates the learning and memory deficits in an amyloid protein precursor transgenic mouse model and the underlying mechanisms. Eur. J. Pharmacol. 669 (1-3), 76–83. doi:10.1016/j.ejphar.2011.07.051
Liu, W., Vives-Bauza, C., Acin-Perez-, R., Yamamoto, A., Tan, Y., Li, Y., et al. (2009). PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and alpha-synuclein aggregation in cell culture models of Parkinson's disease. PLoS One 4 (2), e4597. doi:10.1371/journal.pone.0004597
Livnat-Levanon, N., and Glickman, M. H. (2011). Ubiquitin-proteasome system and mitochondria - reciprocity. Biochim. Biophys. Acta 1809 (2), 80–87. doi:10.1016/j.bbagrm.2010.07.005
Lopez-Gonzalez, R., Lu, Y., Gendron, T. F., Karydas, A., Tran, H., Yang, D., et al. (2016). Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 92 (2), 383–391. doi:10.1016/j.neuron.2016.09.015
Lu, J., Duan, W., Guo, Y., Jiang, H., Li, Z., Huang, J., et al. (2012). Mitochondrial dysfunction in human TDP-43 transfected NSC34 cell lines and the protective effect of dimethoxy curcumin. Brain Res. Bull. 89 (5-6), 185–190. doi:10.1016/j.brainresbull.2012.09.005
Ludtmann, M. H., Angelova, P. R., Ninkina, N. N., Gandhi, S., Buchman, V. L., and Abramov, A. Y. (2016). Monomeric alpha-synuclein exerts a physiological role on brain ATP synthase. J. Neurosci. 36 (41), 10510–10521. doi:10.1523/JNEUROSCI.1659-16.2016
Ludtmann, M. H. R., Angelova, P. R., Horrocks, M. H., Choi, M. L., Rodrigues, M., Baev, A. Y., et al. (2018). α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson's disease. Nat. Commun. 9 (1), 2293. doi:10.1038/s41467-018-04422-2
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., et al. (2004). ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 304 (5669), 448–452. doi:10.1126/science.1091230
Magrane, J., Cortez, C., Gan, W. B., and Manfredi, G. (2014). Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 23 (6), 1413–1424. doi:10.1093/hmg/ddt528
Magri, A., Belfiore, R., Reina, S., Tomasello, M. F., Di Rosa, M. C., Guarino, F., et al. (2016). Hexokinase I N-terminal based peptide prevents the VDAC1-SOD1 G93A interaction and re-establishes ALS cell viability. Sci. Rep. 6, 34802. doi:10.1038/srep34802
Magri, A., Risiglione, P., Caccamo, A., Formicola, B., Tomasello, M. F., Arrigoni, C., et al. (2021). Small hexokinase 1 peptide against toxic SOD1 G93A mitochondrial accumulation in ALS rescues the ATP-related respiration. Biomedicines 9 (8), 948. doi:10.3390/biomedicines9080948
Mailloux, R. J. (2018). Mitochondrial antioxidants and the maintenance of cellular hydrogen peroxide levels. longevity 2018, 7857251. doi:10.1155/2018/7857251
Manczak, M., Park, B. S., Jung, Y., and Reddy, P. H. (2004). Differential expression of oxidative phosphorylation genes in patients with Alzheimer's disease: Implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 5 (2), 147–162. doi:10.1385/NMM:5:2:147
Manczak, M., and Reddy, P. H. (2012). Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer's disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 21 (11), 2538–2547. doi:10.1093/hmg/dds072
Mao, Z., Bonni, A., Xia, F., Nadal-Vicens, M., and Greenberg, M. E. (1999). Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 286 (5440), 785–790. doi:10.1126/science.286.5440.785
Martin, W. F., Garg, S., and Zimorski, V. (2015). Endosymbiotic theories for eukaryote origin. Philos. Trans. R. Soc. Lond B Biol. Sci. 370 (1678), 20140330. doi:10.1098/rstb.2014.0330
Martins, L. M., Morrison, A., Klupsch, K., Fedele, V., Moisoi, N., Teismann, P., et al. (2004). Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol. Cell Biol. 24 (22), 9848–9862. doi:10.1128/MCB.24.22.9848-9862.2004
Masciopinto, F., Di Pietro, N., Corona, C., BoMba, M., Pipino, C., Curcio, M., et al. (2012). Effects of long-term treatment with pioglitazone on cognition and glucose metabolism of PS1-KI, 3xTg-AD, and wild-type mice. Cell Death Dis. 3 (12), e448. doi:10.1038/cddis.2012.189
Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C. A., et al. (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189 (2), 211–221. doi:10.1083/jcb.200910140
Maurer, I., Zierz, S., and Moller, H. J. (2000). A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 21 (3), 455–462. doi:10.1016/s0197-4580(00)00112-3
Mazzulli, J. R., Zunke, F., Tsunemi, T., Toker, N. J., Jeon, S., Burbulla, L. F., et al. (2016). Activation of beta-glucocerebrosidase reduces pathological alpha-synuclein and restores lysosomal function in Parkinson's patient midbrain neurons. J. Neurosci. 36 (29), 7693–7706. doi:10.1523/JNEUROSCI.0628-16.2016
McCord, J. M., and Fridovich, I. (1969). Superoxide dismutase. J. Biol. Chem. 244 (22), 6049–6055. doi:10.1016/s0021-9258(18)63504-5
McInnes, J. (2013). Mitochondrial-associated metabolic disorders: Foundations, pathologies and recent progress. Nutr. Metab. (Lond) 10 (1), 63. doi:10.1186/1743-7075-10-63
Mehta, A. R., Gregory, J. M., Dando, O., Carter, R. N., Burr, K., Nanda, J., et al. (2021). Mitochondrial bioenergetic deficits in C9orf72 amyotrophic lateral sclerosis motor neurons cause dysfunctional axonal homeostasis. Acta Neuropathol. 141 (2), 257–279. doi:10.1007/s00401-020-02252-5
Mermigkis, C., Bouloukaki, I., Mastorodemos, V., Plaitakis, A., Alogdianakis, V., Siafakas, N., et al. (2013). Medical treatment with thiamine, coenzyme Q, vitamins E and C, and carnitine improved obstructive sleep apnea in an adult case of Leigh disease. Sleep. Breath. 17 (4), 1129–1135. doi:10.1007/s11325-013-0816-5
Messerer, J., Wrede, C., Schipke, J., Brandenberger, C., Abdellatif, M., Eisenberg, T., et al. (2023). Spermidine supplementation influences mitochondrial number and morphology in the heart of aged mice. J. Anat. 242 (1), 91–101. doi:10.1111/joa.13618
Miller, W. L. (2013). Steroid hormone synthesis in mitochondria. Mol. Cell Endocrinol. 379 (1-2), 62–73. doi:10.1016/j.mce.2013.04.014
Mindell, J. A. (2012). Lysosomal acidification mechanisms. Annu. Rev. Physiol. 74, 69–86. doi:10.1146/annurev-physiol-012110-142317
Missiroli, S., Patergnani, S., and Caroccia, N. (2018). Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 9 (3), 1–14. doi:10.1038/s41419-017-0027-2
Monzio Compagnoni, G., Di Fonzo, A., Corti, S., Comi, G. P., Bresolin, N., and Masliah, E. (2020). The role of mitochondria in neurodegenerative diseases: The lesson from Alzheimer's disease and Parkinson's disease. Mol. Neurobiol. 57 (7), 2959–2980. doi:10.1007/s12035-020-01926-1
Morato, L., Galino, J., Ruiz, M., Calingasan, N. Y., Starkov, A. A., Dumont, M., et al. (2013). Pioglitazone halts axonal degeneration in a mouse model of X-linked adrenoleukodystrophy. Brain 136 (8), 2432–2443. doi:10.1093/brain/awt143
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339 (6125), 1335–1338. doi:10.1126/science.1232927
Morsy, A., and Trippier, P. C. (2019). Amyloid-binding alcohol dehydrogenase (ABAD) inhibitors for the treatment of Alzheimer's disease. J. Med. Chem. 62 (9), 4252–4264. doi:10.1021/acs.jmedchem.8b01530
Mossmann, D., Vogtle, F. N., Taskin, A. A., Teixeira, P. F., Ring, J., Burkhart, J. M., et al. (2014). Amyloid-beta peptide induces mitochondrial dysfunction by inhibition of preprotein maturation. Cell Metab. 20 (4), 662–669. doi:10.1016/j.cmet.2014.07.024
Mozdy, A. D., McCaffery, J. M., and Shaw, J. M. (2000). Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J. Cell Biol. 151 (2), 367–380. doi:10.1083/jcb.151.2.367
Nakaya, T., and Maragkakis, M. (2018). Amyotrophic Lateral Sclerosis associated FUS mutation shortens mitochondria and induces neurotoxicity. Sci. Rep. 8 (1), 15575. doi:10.1038/s41598-018-33964-0
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., et al. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8 (1), e1000298. doi:10.1371/journal.pbio.1000298
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183 (5), 795–803. doi:10.1083/jcb.200809125
Nargund, A. M., Pellegrino, M. W., Fiorese, C. J., Baker, B. M., and Haynes, C. M. (2012). Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337 (6094), 587–590. doi:10.1126/science.1223560
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314 (5796), 130–133. doi:10.1126/science.1134108
Oddo, S. J. (2008). The ubiquitin proteasome system in Alzheimer's disease. J. Cell. Mol. Med. 12 (2), 363–373. doi:10.1111/j.1582-4934.2008.00276.x
Olichon, A., Emorine, L. J., Descoins, E., Pelloquin, L., Brichese, L., Gas, N., et al. (2002). The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 523 (1-3), 171–176. doi:10.1016/s0014-5793(02)02985-x
Oliveira, A. N., and Hood, D. A. (2018). Effect of Tim23 knockdown in vivo on mitochondrial protein import and retrograde signaling to the UPR(mt) in muscle. Am. J. Physiol. Cell Physiol. 315 (4), C516–C526. doi:10.1152/ajpcell.00275.2017
Onesto, E., Colombrita, C., Gumina, V., Borghi, M. O., Dusi, S., Doretti, A., et al. (2016). Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 4 (1), 47. doi:10.1186/s40478-016-0316-5
Ortiz-Gonzalez, X. R. (2021). Mitochondrial dysfunction: A common denominator in neurodevelopmental disorders? Dev. Neurosci. 43 (3-4), 222–229. doi:10.1159/000517870
Paillusson, S., Gomez-Suaga, P., Stoica, R., Little, D., Gissen, P., Devine, M. J., et al. (2017). α-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol. 134 (1), 129–149. doi:10.1007/s00401-017-1704-z
Pardo, C. A., Xu, Z., Borchelt, D. R., Price, D. L., Sisodia, S. S., and Cleveland, D. W. (1995). Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc. Natl. Acad. Sci. U. S. A. 92 (4), 954–958. doi:10.1073/pnas.92.4.954
Park, J.-S., Davis, R. L., and Sue, C. M. (2018). Mitochondrial dysfunction in Parkinson’s disease: New mechanistic insights and therapeutic perspectives. Curr. Neurol. Neurosci. Rep. 18 (5), 21–11. doi:10.1007/s11910-018-0829-3
Pasinelli, P., Belford, M. E., Lennon, N., Bacskai, B. J., Hyman, B. T., Trotti, D., et al. (2004). Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 43 (1), 19–30. doi:10.1016/j.neuron.2004.06.021
Pedrini, S., Sau, D., Guareschi, S., Bogush, M., Brown, R. H., Naniche, N., et al. (2010). ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum. Mol. Genet. 19 (15), 2974–2986. doi:10.1093/hmg/ddq202
Picca, A., Pesce, V., Fracasso, F., Joseph, A. M., Leeuwenburgh, C., and Lezza, A. M. S. (2013). Aging and calorie restriction oppositely affect mitochondrial biogenesis through TFAM binding at both origins of mitochondrial DNA replication in rat liver. PLoS One 8 (9), e74644. doi:10.1371/journal.pone.0074644
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Prim. 3, 17013. doi:10.1038/nrdp.2017.13
Pohl, C., and Dikic, I. (2019). Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 366 (6467), 818–822. doi:10.1126/science.aax3769
Polymenidou, M., Lagier-Tourenne, C., Hutt, K. R., Huelga, S. C., Moran, J., Liang, T. Y., et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14 (4), 459–468. doi:10.1038/nn.2779
Pringsheim, T., Jette, N., Frolkis, A., and Steeves, T. D. L. (2014). The prevalence of Parkinson's disease: A systematic review and meta-analysis. Mov. Disord. 29 (13), 1583–1590. doi:10.1002/mds.25945
Qing, H., Wong, W., McGeer, E. G., and McGeer, P. L. (2009). Lrrk2 phosphorylates alpha synuclein at serine 129: Parkinson disease implications. Biochem. Biophys. Res. Commun. 387 (1), 149–152. doi:10.1016/j.bbrc.2009.06.142
Quintanilla, R. A., Dolan, P. J., Jin, Y. N., and Johnson, G. V. W. (2012). Truncated tau and Aβ cooperatively impair mitochondria in primary neurons. Neurobiol. Aging 33 (3), 619 e25–e35. doi:10.1016/j.neurobiolaging.2011.02.007
Raimondi, A., Mangolini, A., Rizzardini, M., Tartari, S., Massari, S., Bendotti, C., et al. (2006). Cell culture models to investigate the selective vulnerability of motoneuronal mitochondria to familial ALS-linked G93ASOD1. Eur. J. Neurosci. 24 (2), 387–399. doi:10.1111/j.1460-9568.2006.04922.x
Rath, S., Sharma, R., Gupta, R., Ast, T., Chan, C., Durham, T. J., et al. (2021). MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49 (D1), D1541–D1547. doi:10.1093/nar/gkaa1011
Reddy, P. H., and Oliver, D. M. (2019). Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer's disease. Cells 8 (5), 488. doi:10.3390/cells8050488
Renton, A. E., Chio, A., and Traynor, B. J. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17 (1), 17–23. doi:10.1038/nn.3584
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72 (2), 257–268. doi:10.1016/j.neuron.2011.09.010
Roberts, H. L., and Brown, D. R. (2015). Seeking a mechanism for the toxicity of oligomeric alpha-synuclein. Biomolecules 5 (2), 282–305. doi:10.3390/biom5020282
Rocha, E. M., Keeney, M. T., Di Maio, R., De Miranda, B. R., and Greenamyre, J. T. (2022). LRRK2 and idiopathic Parkinson's disease. Trends Neurosci. 45 (3), 224–236. doi:10.1016/j.tins.2021.12.002
Rosen, D. R. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362 (6415), 362–62. doi:10.1038/364362c0
Ross, J. M., Olson, L., and Coppotelli, G. (2015). Mitochondrial and ubiquitin proteasome system dysfunction in ageing and disease: Two sides of the same coin? Int. J. Mol. Sci. 16 (8), 19458–19476. doi:10.3390/ijms160819458
Ryu, D., Mouchiroud, L., Andreux, P. A., Katsyuba, E., Moullan, N., Nicolet-Dit-Felix, A. A., et al. (2016). Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 22 (8), 879–888. doi:10.1038/nm.4132
Saberi, S., Stauffer, J. E., Jiang, J., Garcia, S. D., Taylor, A. E., Schulte, D., et al. (2018). Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. 135 (3), 459–474. doi:10.1007/s00401-017-1793-8
Saftig, P., and Klumperman, J. (2009). Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 10 (9), 623–635. doi:10.1038/nrm2745
Sagan, L. (1967). On the origin of mitosing cells. J. Theor. Biol. 14 (3), 255–274. doi:10.1016/0022-5193(67)90079-3
Sahin, E., and DePinho, R. A. (2012). Axis of ageing: Telomeres, p53 and mitochondria. Nat. Rev. Mol. Cell Biol. 13 (6), 397–404. doi:10.1038/nrm3352
Salam, S., Tacconelli, S., Smith, B. N., Mitchell, J. C., Glennon, E., Nikolaou, N., et al. (2021). Identification of a novel interaction of FUS and syntaphilin may explain synaptic and mitochondrial abnormalities caused by ALS mutations. Sci. Rep. 11 (1), 13613. doi:10.1038/s41598-021-93189-6
Salvatori, I., Ferri, A., Scaricamazza, S., Giovannelli, I., Serrano, A., Rossi, S., et al. (2018). Differential toxicity of TAR DNA-binding protein 43 isoforms depends on their submitochondrial localization in neuronal cells. J. Neurochem. 146 (5), 585–597. doi:10.1111/jnc.14465
Sama, R. R., Ward, C. L., and Bosco, D. A. (2014). Functions of FUS/TLS from DNA repair to stress response: Implications for ALS. ASN Neuro 6 (4), 1759091414544472. doi:10.1177/1759091414544472
Santel, A., and Fuller, M. T. (2001). Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 114 (5), 867–874. doi:10.1242/jcs.114.5.867
Sareen, D., O'Rourke, J. G., Meera, P., Muhammad, A. K. M. G., Grant, S., Simpkinson, M., et al. (2013). Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5 (208), 208ra149. doi:10.1126/scitranslmed.3007529
Satoh, J., Kawana, N., and Yamamoto, Y. (2013). Pathway analysis of ChIP-seq-based NRF1 target genes suggests a logical hypothesis of their involvement in the pathogenesis of neurodegenerative diseases. Gene Regul. Syst. Bio 7, 139–152. doi:10.4137/GRSB.S13204
Saxton, W. M., and Hollenbeck, P. J. (2012). The axonal transport of mitochondria. J. Cell Sci. 125 (9), 2095–2104. doi:10.1242/jcs.053850
Scarpulla, R. C. (2011). Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813 (7), 1269–1278. doi:10.1016/j.bbamcr.2010.09.019
Scarpulla, R. C. (2008). Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 88 (2), 611–638. doi:10.1152/physrev.00025.2007
Schondorf, D. C., Aureli, M., McAllister, F. E., Hindley, C. J., Mayer, F., Schmid, B., et al. (2014). iPSC-derived neurons from GBA1-associated Parkinson's disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 5, 4028. doi:10.1038/ncomms5028
Selkoe, D. J., and Hardy, J. J. E. (2016). The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 8 (6), 595–608. doi:10.15252/emmm.201606210
Senin, U., Parnetti, L., Barbagallo-Sangiorgi, G., Bartorelli, L., Bocola, V., Capurso, A., et al. (1992). Idebenone in senile dementia of alzheimer type: A multicentre study. Arch. Gerontol. Geriatr. 15 (3), 249–260. doi:10.1016/0167-4943(92)90060-h
Sephton, C. F., Good, S. K., Atkin, S., Dewey, C. M., Mayer, P., Herz, J., et al. (2010). TDP-43 is a developmentally regulated protein essential for early embryonic development. J. Biol. Chem. 285 (9), 6826–6834. doi:10.1074/jbc.M109.061846
Sevigny, J., Chiao, P., Bussiere, T., Weinreb, P. H., Williams, L., Maier, M., et al. (2016). The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature 537 (7618), 50–56. doi:10.1038/nature19323
Sha, D., Chin, L. S., and Li, L. (2010). Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-kappaB signaling. Hum. Mol. Genet. 19 (2), 352–363. doi:10.1093/hmg/ddp501
Shahpasand, K., Uemura, I., Saito, T., Asano, T., Hata, K., Shibata, K., et al. (2012). Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer's disease. J. Neurosci. 32 (7), 2430–2441. doi:10.1523/JNEUROSCI.5927-11.2012
Sheng, B., Wang, X., Su, B., Lee, H. g., Casadesus, G., Perry, G., et al. (2012). Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer's disease. J. Neurochem. 120 (3), 419–429. doi:10.1111/j.1471-4159.2011.07581.x
Shpilka, T., and Haynes, C. M. (2018). The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 19 (2), 109–120. doi:10.1038/nrm.2017.110
Siddique, T., Figlewicz, D. A., Pericak-Vance, M. A., Haines, J. L., Rouleau, G., Jeffers, A. J., et al. (1991). Linkage of a gene causing familial amyotrophic lateral sclerosis to chromosome 21 and evidence of genetic-locus heterogeneity. N. Engl. J. Med. 324 (20), 1381–1384. doi:10.1056/NEJM199105163242001
Smirnova, E., Griparic, L., Shurland, D. L., and van der Bliek, A. M. (2001). Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12 (8), 2245–2256. doi:10.1091/mbc.12.8.2245
Smith, P. D., Mount, M. P., Shree, R., Callaghan, S., Slack, R. S., Anisman, H., et al. (2006). Calpain-regulated p35/cdk5 plays a central role in dopaminergic neuron death through modulation of the transcription factor myocyte enhancer factor 2. J. Neurosci. 26 (2), 440–447. doi:10.1523/JNEUROSCI.2875-05.2006
Song, Q., Meng, B., Xu, H., and Mao, Z. (2020). The emerging roles of vacuolar-type ATPase-dependent Lysosomal acidification in neurodegenerative diseases. Transl. Neurodegener. 9 (1), 17–14. doi:10.1186/s40035-020-00196-0
Soreghan, B., Kosmoski, J., and Glabe, C. (1994). Surfactant properties of Alzheimer's A beta peptides and the mechanism of amyloid aggregation. J. Biol. Chem. 269 (46), 28551–28554. doi:10.1016/s0021-9258(19)61939-3
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319 (5870), 1668–1672. doi:10.1126/science.1154584
Srivastava, S. (2017). The mitochondrial basis of aging and age-related disorders. Genes (Basel) 8 (12), 398. doi:10.3390/genes8120398
Steenbergen, R., Nanowski, T. S., Beigneux, A., Kulinski, A., Young, S. G., and Vance, J. E. (2005). Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J. Biol. Chem. 280 (48), 40032–40040. doi:10.1074/jbc.M506510200
Stepien, K. M., Roncaroli, F., Turton, N., Hendriksz, C. J., Roberts, M., Heaton, R. A., et al. (2020). Mechanisms of mitochondrial dysfunction in lysosomal storage disorders: A review. J. Clin. Med. 9 (8), 2596. doi:10.3390/jcm9082596
Stoica, R., De Vos, K. J., Paillusson, S., Mueller, S., Sancho, R. M., Lau, K. F., et al. (2014). ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5, 3996. doi:10.1038/ncomms4996
Stoica, R., Paillusson, S., Gomez-Suaga, P., Mitchell, J. C., Lau, D. H., Gray, E. H., et al. (2016). ALS/FTD-associated FUS activates GSK-3β to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep. 17 (9), 1326–1342. doi:10.15252/embr.201541726
Stribl, C., Samara, A., Trumbach, D., Peis, R., Neumann, M., Fuchs, H., et al. (2014). Mitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43. J. Biol. Chem. 289 (15), 10769–10784. doi:10.1074/jbc.M113.515940
Su, Y. C., and Qi, X. (2013). Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum. Mol. Genet. 22 (22), 4545–4561. doi:10.1093/hmg/ddt301
Suire, C. N., Abdul-Hay, S. O., Sahara, T., Kang, D., Brizuela, M. K., Saftig, P., et al. (2020). Cathepsin D regulates cerebral Aβ42/40 ratios via differential degradation of Aβ42 and Aβ40. Alzheimers Res. Ther. 12 (1), 80–13. doi:10.1186/s13195-020-00649-8
Sullivan, P. M., Zhou, X., Robins, A. M., Paushter, D. H., Kim, D., Smolka, M. B., et al. (2016). The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 4 (1), 51. doi:10.1186/s40478-016-0324-5
Tampi, R. R., Forester, B. P., and Agronin, M. (2021). Aducanumab: Evidence from clinical trial data and controversies. Drugs Context 10, 1–9. doi:10.7573/dic.2021-7-3
Tan, A. Y., and Manley, J. L. (2009). The TET family of proteins: Functions and roles in disease. J. Mol. Cell Biol. 1 (2), 82–92. doi:10.1093/jmcb/mjp025
Tang, X., Wang, X., Gong, X., Tong, M., Park, D., Xia, Z., et al. (2005). Cyclin-dependent kinase 5 mediates neurotoxin-induced degradation of the transcription factor myocyte enhancer factor 2. J. Neurosci. 25 (19), 4823–4834. doi:10.1523/JNEUROSCI.1331-05.2005
Teixeira, P. F., Pinho, C. M., Branca, R. M., Lehtio, J., Levine, R. L., and Glaser, E. (2012). In vitro oxidative inactivation of human presequence protease (hPreP). Free Radic. Biol. Med. 53 (11), 2188–2195. doi:10.1016/j.freeradbiomed.2012.09.039
Tollervey, J. R., Curk, T., Rogelj, B., Briese, M., Cereda, M., Kayikci, M., et al. (2011). Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14 (4), 452–458. doi:10.1038/nn.2778
Tondera, D., Grandemange, S., Jourdain, A., Karbowski, M., Mattenberger, Y., Herzig, S., et al. (2009). SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 28 (11), 1589–1600. doi:10.1038/emboj.2009.89
Trudeau, K., Molina, A. J., and Roy, S. (2011). High glucose induces mitochondrial morphology and metabolic changes in retinal pericytes. Invest Ophthalmol. Vis. Sci. 52 (12), 8657–8664. doi:10.1167/iovs.11-7934
Tsai, Y. L., Coady, T. H., Lu, L., Zheng, D., Alland, I., Tian, B., et al. (2020). ALS/FTD-associated protein FUS induces mitochondrial dysfunction by preferentially sequestering respiratory chain complex mRNAs. Genes Dev. 34 (11-12), 785–805. doi:10.1101/gad.335836.119
Tseng, B. P., Green, K. N., Chan, J. L., Blurton-Jones, M., and LaFerla, F. M. (2008). Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol. Aging 29 (11), 1607–1618. doi:10.1016/j.neurobiolaging.2007.04.014
Uttara, B., Singh, A. V., Zamboni, P., and Mahajan, R. T. (2009). Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 7 (1), 65–74. doi:10.2174/157015909787602823
Valadas, J. S., Esposito, G., Vandekerkhove, D., Miskiewicz, K., Deaulmerie, L., Raitano, S., et al. (2018). ER lipid defects in neuropeptidergic neurons impair sleep patterns in Parkinson's disease. Neuron 98 (6), 1155–1169. doi:10.1016/j.neuron.2018.05.022
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M. K., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304 (5674), 1158–1160. doi:10.1126/science.1096284
van Blitterswijk, M., Gendron, T. F., Baker, M. C., DeJesus-Hernandez, M., Finch, N. A., Brown, P. H., et al. (2015). Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol. 130 (6), 863–876. doi:10.1007/s00401-015-1480-6
van der Bliek, A. M., Shen, Q., and Kawajiri, S. (2013). Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 5 (6), a011072. doi:10.1101/cshperspect.a011072
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323 (5918), 1208–1211. doi:10.1126/science.1165942
Vance, J. E. J. (1990). Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265 (13), 7248–7256. doi:10.1016/s0021-9258(19)39106-9
Vogel, F., Bornhovd, C., Neupert, W., and Reichert, A. S. (2006). Dynamic subcompartmentalization of the mitochondrial inner membrane. J. Cell Biol. 175 (2), 237–247. doi:10.1083/jcb.200605138
Vogiatzi, T., Xilouri, M., Vekrellis, K., and Stefanis, L. (2008). Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 283 (35), 23542–23556. doi:10.1074/jbc.M801992200
Vyas, S., Zaganjor, E., and Haigis, M. C. (2016). Mitochondria and cancer. Cell 166 (3), 555–566. doi:10.1016/j.cell.2016.07.002
Waite, A. J., Baumer, D., East, S., Neal, J., Morris, H. R., Ansorge, O., et al. (2014). Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging 35 (7), 1779 e5–e1779. doi:10.1016/j.neurobiolaging.2014.01.016
Wallace, D. C. (2012). Mitochondria and cancer. Nat. Rev. Cancer 12 (10), 685–698. doi:10.1038/nrc3365
Wan, J. Z., Wang, R., Zhou, Z. Y., Deng, L. L., Zhang, C. C., Liu, C. Q., et al. (2020). Saponins of Panax japonicus confer neuroprotection against brain aging through mitochondrial related oxidative stress and autophagy in rats. Curr. Pharm. Biotechnol. 21 (8), 667–680. doi:10.2174/1389201021666191216114815
Wang, C., and Youle, R. J. (2009). The role of mitochondria in apoptosis*. Annu. Rev. Genet. 43, 95–118. doi:10.1146/annurev-genet-102108-134850
Wang, T., Jiang, X., Chen, G., and Xu, J. (2015). Interaction of amyotrophic lateral sclerosis/frontotemporal lobar degeneration-associated fused-in-sarcoma with proteins involved in metabolic and protein degradation pathways. Neurobiol. Aging 36 (1), 527–535. doi:10.1016/j.neurobiolaging.2014.07.044
Wang, T., Liu, H., Itoh, K., Oh, S., Zhao, L., Murata, D., et al. (2021). C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 33 (3), 531–546. doi:10.1016/j.cmet.2021.01.005
Wang, X., Schwartz, J. C., and Cech, T. R. (2015). Nucleic acid-binding specificity of human FUS protein. Nucleic Acids Res. 43 (15), 7535–7543. doi:10.1093/nar/gkv679
Wei, Q., Zhou, Q., Chen, Y., Ou, R., Cao, B., Xu, Y., et al. (2017). Analysis of SOD1 mutations in a Chinese population with amyotrophic lateral sclerosis: A case-control study and literature review. Sci. Rep. 7, 44606. doi:10.1038/srep44606
Weishaupt, J. H., Hyman, T., and Dikic, I. (2016). Common molecular pathways in amyotrophic lateral sclerosis and frontotemporal dementia. Trends Mol. Med. 22 (9), 769–783. doi:10.1016/j.molmed.2016.07.005
Williams, A., Hayashi, T., Wolozny, D., Yin, B., Su, T. C., Betenbaugh, M. J., et al. (2016). The non-apoptotic action of bcl-xL: Regulating Ca2+ signaling and bioenergetics at the ER-mitochondrion interface. J. Bioenerg. Biomembr. 48 (3), 211–225. doi:10.1007/s10863-016-9664-x
Wils, H., Kleinberger, G., Janssens, J., Pereson, S., Joris, G., Cuijt, I., et al. (2010). TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. U. S. A. 107 (8), 3858–3863. doi:10.1073/pnas.0912417107
Wilson, E. L., and Metzakopian, E. (2021). ER-Mitochondria contact sites in neurodegeneration: Genetic screening approaches to investigate novel disease mechanisms. Cell Death Differ. 28 (6), 1804–1821. doi:10.1038/s41418-020-00705-8
Wong, Y. C., Kim, S., Peng, W., and Krainc, D. (2019). Regulation and function of mitochondria–lysosome membrane contact sites in cellular homeostasis. Trends Cell Biol. 29 (6), 500–513. doi:10.1016/j.tcb.2019.02.004
Wong, Y. C., Ysselstein, D., and Krainc, D. J. N. (2018). Mitochondria–lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554 (7692), 382–386. doi:10.1038/nature25486
Wu, Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., et al. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98 (1), 115–124. doi:10.1016/S0092-8674(00)80611-X
Xiao, S., Sanelli, T., Dib, S., Sheps, D., Findlater, J., Bilbao, J., et al. (2011). RNA targets of TDP-43 identified by UV-CLIP are deregulated in ALS. Mol. Cell Neurosci. 47 (3), 167–180. doi:10.1016/j.mcn.2011.02.013
Xie, N., Wang, C., Lian, Y., Wu, C., Zhang, H., and Zhang, Q. (2014). Inhibition of mitochondrial fission attenuates Aβ-induced microglia apoptosis. Neuroscience 256, 36–42. doi:10.1016/j.neuroscience.2013.10.011
Yang, Q., She, H., Gearing, M., Colla, E., Lee, M., Shacka, J. J., et al. (2009). Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 323 (5910), 124–127. doi:10.1126/science.1166088
Yang, Z. F., Drumea, K., Mott, S., Wang, J., and Rosmarin, A. G. (2014). GABP transcription factor (nuclear respiratory factor 2) is required for mitochondrial biogenesis. Mol. Cell Biol. 34 (17), 3194–3201. doi:10.1128/MCB.00492-12
Yang, Z., and Klionsky, D. J. (2010). Eaten alive: A history of macroautophagy. Nat. Cell Biol. 12 (9), 814–822. doi:10.1038/ncb0910-814
Yano, H., Baranov, S. V., Baranova, O. V., Kim, J., Pan, Y., Yablonska, S., et al. (2014). Inhibition of mitochondrial protein import by mutant huntingtin. Nat. Neurosci. 17 (6), 822–831. doi:10.1038/nn.3721
Youle, R. J., and van der Bliek, A. M. (2012). Mitochondrial fission, fusion, and stress. Science 337 (6098), 1062–1065. doi:10.1126/science.1219855
Young, M. L., and Franklin, J. L. (2019). The mitochondria-targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg-AD mice. Mol. Cell Neurosci. 101, 103409. doi:10.1016/j.mcn.2019.103409
Zelko, I. N., Mariani, T. J., and Folz, R. J. (2002). Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 33 (3), 337–349. doi:10.1016/s0891-5849(02)00905-x
Zeng, K. W., Wang, J. K., Wang, L. C., Guo, Q., Liu, T. T., Wang, F. J., et al. (2021). Small molecule induces mitochondrial fusion for neuroprotection via targeting CK2 without affecting its conventional kinase activity. Signal Transduct. Target Ther. 6 (1), 71. doi:10.1038/s41392-020-00447-6
Zhang, F., Strom, A. L., Fukada, K., Lee, S., Hayward, L. J., and Zhu, H. (2007). Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex. J. Biol. Chem. 282 (22), 16691–16699. doi:10.1074/jbc.M609743200
Zhang, F., Wu, Z., Long, F., Tan, J., Gong, N., Li, X., et al. (2022). The roles of ATP13A2 gene mutations leading to abnormal aggregation of alpha-synuclein in Parkinson's disease. Front. Cell Neurosci. 16, 927682. doi:10.3389/fncel.2022.927682
Zhang, Z. C., and Chook, Y. M. (2012). Structural and energetic basis of ALS-causing mutations in the atypical proline-tyrosine nuclear localization signal of the Fused in Sarcoma protein (FUS). Proc. Natl. Acad. Sci. U. S. A. 109 (30), 12017–12021. doi:10.1073/pnas.1207247109
Zhao, Q., Wang, J., Levichkin, I. V., Stasinopoulos, S., Ryan, M. T., and Hoogenraad, N. J. (2002). A mitochondrial specific stress response in mammalian cells. EMBO J. 21 (17), 4411–4419. doi:10.1093/emboj/cdf445
Zhou, Z., Wang, J., Song, Y., He, Y., Zhang, C., Liu, C., et al. (2018). Panax notoginseng saponins attenuate cardiomyocyte apoptosis through mitochondrial pathway in natural aging rats. Phytother. Res. 32 (2), 243–250. doi:10.1002/ptr.5961
Glossary
AD Alzheimer’s disease
AICAR 5-Aminoimidazole-4-carboxamide ribonucleotide
AIFM1 apoptosis inducing factor mitochondrial 1
APP Amyloid precursor protein
ATP5A1 ATP synthase F1 subunit alpha
Aβ Amyloid beta
Bcl2 B-cell leukemia/lymphoma two protein
C9orf72 chromosome nine open reading frame 72
CatB Cathepsin B
CatD Cathepsin D
CCS copper chaperone for superoxide dismutase
CHCHD4 Coiled-coil-helix-coiled-coil-helix domain-containing protein 4
Clp Caseinolytic proteins
ClpP Caseinolytic protease P
CM Cristae membrane
CMA Chaperone-mediated autophagy
CNS Central nervous system
COX17p Cytochrome c oxidase copper chaperone
Cyt C Cytochrome C
DNAJA3DnaJ homolog subfamily A member 3 mitochondrial
Dnm1 Dynamin-related protein DNM1
DPRs Dipeptide repeats
Drp1 Dynamin-related protein 1
ER Endoplasmic reticulum
ERAL1 ERA-like protein 1
ERRα Estrogen-related receptor α
ETC Electron transport chain
EWS Ewing Sarcoma
fALS familial ALS
Fis1 Mitochondrial fission one protein
FTD Frontotemporal dementia
fTLD Frontotemporal lobar degeneration
FUS Fused in sarcoma
GSK3β Glycogen synthase kinase-3 beta
HIF1α Hypoxia inducible factor 1 subunit alpha
HK1 hexokinase I
hnRNPs Heterogenous ribonucleoproteins
HOP HSC70-HSP90 organizing protein
hPreP Human presequence protease
HRE hexanucleotide (GGGGCC) repeats expansion
HSC20 Heat shock cognate protein 20
HtrA2 High temperature requirement protein A2
i-AAA Intermembrane space AAA
IBM Inner boundary membrane
IMM Inner mitochondrial membrane
IMS Intermembrane space
IP3R3 Type 3 inositol 1,4,5-tris-phosphate receptors
iPSCs Induced pluripotent stem cells
LAMP1 lysosome-associated membrane protein 1
LC3 Light chain 3
LHON Leber hereditary optic neuropathy
LonP Lon protease
MAM mitochondrial-associated ER membrane
MBR Metal-binding region
MCU mitochondrial calcium uniporter
MERRF Myoclonic epilepsy with ragged red fibers
Mfn1 Mitofusin-1
Mfn2 mitofusin two
MICU1 Mitochondrial calcium uptake one
mtDNA mitochondria DNA
ND3 NADH-ubiquinone oxidoreductase chain 3
NFTs Neurofibrillary tangles
NF-κB Nuclear factor kappa B
NHK1 HK1-N-terminal based peptide
NLS Nuclear localization signal
NRF1 Nuclear respiratory factor 1
NRF2 Nuclear factor erythroid 2-Related Factor 2
OMA1 Overlapping With The M-AAA Protease 1 Homolog
OMM Outer mitochondrial membrane
OPA1 optic atrophy protein 1
OPTN Optineurin
OXPHOS oxidative phosphorylation
PADK Z-Phe-Ala-diazomethylketone
PC Phosphatidylcholine
PD Parkinson’s disease
PE Phosphatidylethanolamine
PEMT2 Phosphatidylethanolamine N-methyltransferase
PFN1 Profilin one
PGC-1α Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha
PHB2 Prohibitin-2
PINK1 PTEN-induced putative kinase 1
poly (GA) Glycine-Alanine
poly(GP) Glycine-Proline
poly(GR) Glycine-Arginine
poly(PA) Proline-Alanine
poly (PR) Proline-Arginine
PS Phosphatidylserine
PSD Phosphatidylserine decarboxylase
PTPIP51 Protein tyrosine phosphatase interacting protein-51
RGG Arg–Gly–Gly-rich motif
ROS Reactive oxygen species
RRM RNA-recognition motif
SNpc substantia nigra pars compacta
SOD1 Cu/Zn superoxide dismutase one
SQSTM1 Sequestosome one
SYGQ-rich region (serine tyrosine glycine glutamine)-rich region
T2D type 2 diabetes
TAF15 TATA-box binding protein associated factor 15
TBC1D15 TBC1 domain family member 15
TDP-43 TAR DNA-binding protein 43
TFAM transcription factor A Mitochondrial
TFB1M Transcription Factor B1 Mitochondrial
TFB2M Transcription factor B2 Mitochondrial
TIMMDC1 Translocase of inner mitochondrial membrane domain containing one
TLS Translocated in liposarcoma
TOM Translocase of the outer membrane
UBQLN2 Ubiquilin two
UCP2 Uncoupling protein two
UPRmt Mitochondrial unfolded protein response
UV Ultraviolet
VAPB Vesicle-associated membrane-protein-associated protein B
V-ATPase Vacuolar (H+) ATPase
VCP Valosin-containing protein
VDAC1 Voltage-dependent anion-selective channel protein 1
WTL Wild-type like
ZnF Zinc-finger motif
Keywords: neurodegeneration, mitostasis, autophagy, proteasome, oxidative stress, mitochondria
Citation: Jagtap YA, Kumar P, Kinger S, Dubey AR, Choudhary A, Gutti RK, Singh S, Jha HC, Poluri KM and Mishra A (2023) Disturb mitochondrial associated proteostasis: Neurodegeneration and imperfect ageing. Front. Cell Dev. Biol. 11:1146564. doi: 10.3389/fcell.2023.1146564
Received: 17 January 2023; Accepted: 27 February 2023;
Published: 10 March 2023.
Edited by:
Subodh Kumar, Texas Tech University Health Sciences Center, United StatesReviewed by:
Jasdeep Singh, University of Denver, United StatesVikash Dubey, Indian Institute of Technology (BHU), India
Copyright © 2023 Jagtap, Kumar, Kinger, Dubey, Choudhary, Gutti, Singh, Jha, Poluri and Mishra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amit Mishra, amit@iitj.ac.in
†These authors have contributed equally to this work