Myocardial disturbances of intermediary metabolism in Barth syndrome
 Amanda A. Greenwell
Amanda A. Greenwell Seyed Amirhossein Tabatabaei Dakhili
Seyed Amirhossein Tabatabaei Dakhili John R. Ussher
John R. Ussher- 1Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, AB, Canada
- 2Women and Children's Health Research Institute, University of Alberta, Edmonton, AB, Canada
Barth Syndrome (BTHS) is a rare X-linked mitochondrial disorder due to mutations in the gene TAFAZZIN, which leads to immature cardiolipin (CL) remodeling and is characterized by the development of cardiomyopathy. The immature CL remodeling in BTHS results in electron transport chain respiratory defects and destabilization of supercomplexes, thereby impairing ATP production. Thus, BTHS-related cardiomyopathy appears to share metabolic characteristics of the failing heart being an “engine out of fuel.” As CL associates with numerous mitochondrial enzymes involved in ATP production, BTHS is also characterized by several defects in intermediary energy metabolism. Herein we will describe the primary disturbances in intermediary energy metabolism relating to the heart's major fuel sources, fatty acids, carbohydrates, ketones, and amino acids. In addition, we will interrogate whether these disturbances represent potential metabolic targets for alleviating BTHS-related cardiomyopathy.
Introduction
Cardiomyopathy is the most common clinical feature and primary driver of disease outcomes in Barth syndrome (BTHS; Online Mendelian Inheritance in Man [OMIM] 302060), a rare X-linked mitochondrial disorder first described by Dr. Peter Barth and colleagues in 1983 (1, 2). Despite the broad variability in clinical presentation, which can include features such as neutropenia, skeletal myopathy, exercise intolerance, 3-methylglutaconic aciduria, and pre-pubertal growth retardation, cardiomyopathy manifests in approximately 90% of males with BTHS (3, 4). However, the severity and specific cardiomyopathic phenotype can vary both between individuals and throughout disease progression, with no definitive genotype-phenotype correlations having yet been identified. Dilated cardiomyopathy is most common, however other forms including restrictive (5), hypertrophic, and hypertrophic-dilated cardiomyopathy have also been reported (2, 6).
BTHS is caused by mutations in the TAFAZZIN gene, formerly notated as TAZ, on chromosome Xq28, which encodes for the protein tafazzin, a phospholipid transacylase responsible for cardiolipin (CL) remodeling (3, 7). CL is a unique, dimeric, tetra-acyl phospholipid found almost exclusively in the inner mitochondrial membrane (IMM) with essential roles in the maintenance of mitochondrial morphology, and in fundamental mitochondrial functions including fission-fusion dynamics, mitophagy, apoptosis, and energy metabolism (8–10). Impaired cardiac ATP production is a principal contributor to the development and progression of heart failure (11, 12), which appears to also be present in BTHS. Indeed, the use of 31P magnetic resonance spectroscopy to measure the phosphocreatine to ATP ratio (PCr/ATP), an index of cardiac high-energy phosphate metabolism and energy status, was reduced in young adults (18–36 years of age) and children/adolescents with BTHS compared to healthy participants (13, 14). Nonetheless, the molecular mechanisms linking TAFAZZIN deficiency to perturbed myocardial energetics are presently not well-characterized. Importantly, evidence that impairments in oxidative metabolism are substrate specific in BTHS may indicate a primary defect in upstream intermediary metabolism pathways (15–17). Accordingly, the focus of this mini-review will be to characterize the alterations in intermediary metabolism that accompany TAFAZZIN deficiency, and to discuss the potential of targeting these pathways as a therapeutic approach to mitigate the development and progression of BTHS-related cardiomyopathy (Figure 1).
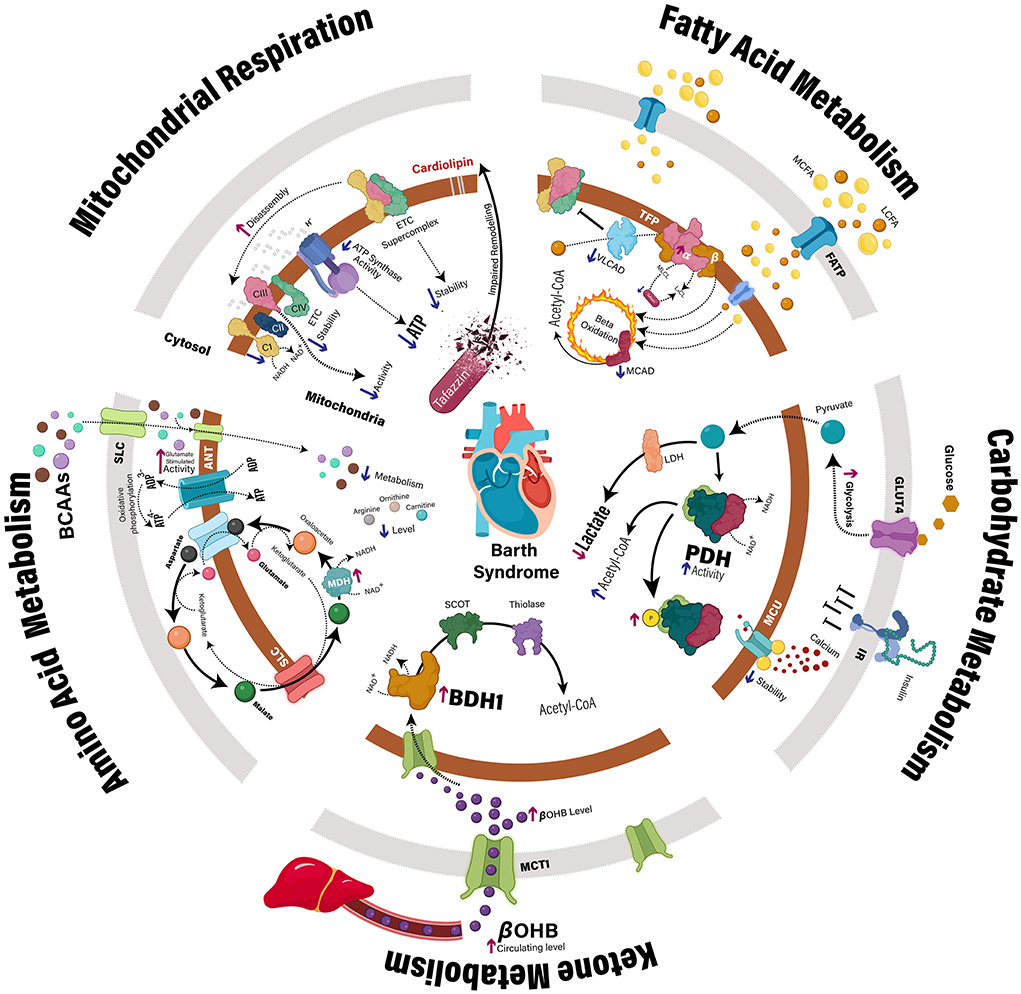
Figure 1. Myocardial metabolic disturbances in Barth syndrome. Clinical and preclinical studies have identified multiple mechanisms that may contribute to impaired cardiac ATP production in the context of TAFAZZIN deficiency. In addition to direct impairment of the mitochondrial electron transport chain, specific defects have also been identified relating to the intermediary metabolism of fatty acids, carbohydrates, ketones, and amino acids. ANT, adenine nucleotide translocase; BCAA, branched-chain amino acid, BDH1, β-hydroxybutyrate dehydrogenase; βOHB, β-hydroxybutyrate; CI-IV, complex 1–4; GLUT4, glucose transporter type 4; IR, insulin receptor; LCFA, long-chain fatty acids; LDH, lactate dehydrogenase; MCAD, medium-chain acyl-CoA dehydrogenase; MCFA, medium-chain fatty acids; MCU, mitochondrial calcium uniporter; MDH, malate dehydrogenase; P, phosphate group; PDH, pyruvate dehydrogenase; SCOT, succinyl-CoA:3-ketoacid CoA transferase; SLC, solute carrier; TFP, mitochondrial trifunctional protein; VLCAD, very long-chain acyl-CoA dehydrogenase.
Mitochondrial respiratory abnormalities in Barth syndrome
Notably, TAFAZZIN deficiency is associated with an impairment of mitochondrial respiratory capacity and ATP production, which to date, has largely been attributed to destabilization of the IMM electron transport chain (ETC) (18). Normal production of ATP in healthy mitochondria involves the oxidation of fatty acids, glucose, ketones, and amino acids to produce acetyl-CoA, which enters the Krebs cycle resulting in the generation of reducing equivalents (i.e., NADH and FADH2) by its 4 dehydrogenase enzymes. As these reducing equivalents deliver electrons to complex I (NADH) and II (FADH2) of the ETC, an electrochemical gradient across the IMM is created as electrons are passed along the ETC through a series of redox reactions, which drives the production of ATP by ATP synthase (complex V) (Figure 1).
In a Tafazzin deficient mouse model that relies on cre-mediated deletion of the Tafazzin gene containing loxP sites flanking exons 5–10, label-free quantitative proteomics revealed reductions in the relative abundances of all ETC complexes (complexes I-V) (19). Moreover, liquid chromatography (LC)-mass spectrometry (MS)/MS analysis of the cardiac proteome demonstrated a reduction in complex 1 (following normalization to citrate synthase) in left ventricular (LV) tissue from individuals with BTHS compared to non-failing control heart samples (20). Of interest, CL has been shown to be essential for the assembly of ETC supercomplexes, and supercomplexes were shown to be destabilized in hearts of Tafazzin knockdown (TazKD) mice secondary to doxycycline-mediated induction of a short-hairpin RNA against Tafazzin (21). Analysis of cardiac mitochondria isolated from cardiac-specific Tafazzin deficient mice prior to the development of cardiomyopathy, also revealed structural remodeling of the respiratory chain with a shift from high molecular weight supercomplexes in the ETC toward lower molecular weight forms such as heterooligomers and individual complexes (22). Furthermore, complex III activity is decreased in cardiac mitochondria isolated from TazKD mice (15, 23, 24), whereas complex V (F1F0-ATP synthase) activity is decreased in both TazKD mice and inducible pluripotent stem cell-derived cardiomyocytes from BTHS patients (15, 25). Although the detrimental consequences of TAFAZZIN deficiency on the structural integrity and optimal functioning of the ETC are well-defined, several studies have identified specific derangements in upstream intermediary energy metabolism pathways that we will herein interrogate (15–17). Such observations illustrate that mechanisms beyond respiratory chain dysfunction may contribute to the cardiac energy deficit in BTHS-related cardiomyopathy, while representing possible metabolic targets for intervention.
Perturbations in myocardial fatty acid metabolism in Barth syndrome
Fatty acids are the predominant fuel source for the heart and an impaired capacity to effectively utilize fatty acids can contribute to an energy deficit and subsequent cardiac dysfunction (12, 26, 27), which appears to be present in BTHS. Myocardial fatty acid extraction and uptake following positron emission tomography (PET) imaging with [1–11C]palmitate were significantly reduced in young adults with BTHS compared to healthy, age-matched controls, though myocardial fatty acid β-oxidation remained similar (14). Furthermore, cardiac mitochondria isolated from TazKD mice at 2 months or 4–6 months of age, and from 2-month-old mice with a cardiac-specific Tafazzin deficiency demonstrated a significant repression of state 3 (ADP-stimulated) respiration rates supported by palmitoylcarnitine and malate (15, 16, 22). A decrease in palmitoylcarnitine supported state 3 respiration was also observed in permeabilized cardiac fibers with intact mitochondrial matrices from 4–6-month-old TazKD mice with preserved cardiac function, as determined by high-resolution respirometry (16). Protein expression of fatty acid binding protein and acyl-CoA synthetase, which mediate intracellular fatty acid transport and fatty acid esterification to CoA, respectively, were similar in LV tissue from male patients with BTHS and non-failing hearts (20). Furthermore, similar enzymatic activities of carnitine palmitoyltransferase I and 2 (CPT-1 and CPT-2) were observed in LV tissue homogenates from male patients with BTHS-related cardiomyopathy compared to non-failing controls (20). This suggests that BTHS-related defects in myocardial fatty acid metabolism may be downstream of mitochondrial fatty acid uptake. Supporting this premise, respiratory capacity of cardiac mitochondria from TazKD mice was decreased compared to wild-type (WT) littermates using palmitoylcarnitine, which bypasses CPT-1, whereas respiration with palmitoyl CoA plus carnitine was not reduced, providing further evidence against a specific defect in mitochondrial fatty acid uptake (16).
Alternatively, impaired tafazzin-mediated CL remodeling in BTHS may result in a selective block in long-chain fatty acid β-oxidation. Protein levels of very long-chain acyl CoA dehydrogenase (VLCAD) were decreased in LV tissue from male BTHS subjects compared to age-matched, non-failing male subjects and male subjects diagnosed with idiopathic dilated cardiomyopathy, as assessed by both immunoblotting and LC-MS/MS based proteomic profiling (20). Likewise, VLCAD protein expression was also decreased in cardiac mitochondria from 4–6-month-old TazKD mice compared to their WT littermates, as determined via LC-MS/MS based proteomic profiling (16). This is consistent with evidence of disrupted interactions between VLCAD and ETC supercomplexes in isolated cardiac mitochondria from 3-month-old TazKD mice (21). Of interest, protein expression of the alpha subunit of the trifunctional protein complex (TFPα) of fatty acid β-oxidation is elevated in heart tissue from BTHS subjects (20). As TFPα also possesses monolysocardiolipin (MLCL) acyltransferase activity, further investigation is required to determine whether the accumulation of MLCL in BTHS may impair the β-oxidation function of TFPα by shifting toward MLCL reacylation. Further evidence for a general defect in the enzymatic machinery of fatty acid β-oxidation in BTHS is also seen by the decline in medium-chain acyl-CoA dehydrogenase protein expression and a reduction in octanoylcarnitine-supported respiration in cardiac mitochondria from TazKD mice (16).
While the above described studies allude to impaired myocardial fatty acid β-oxidation in BTHS, there are limitations with assessing intermediary metabolism in isolated mitochondria where key cellular regulators (i.e., malonyl CoA) may be removed, and measures of protein expression do not necessarily reflect flux (28). Indeed, palmitate oxidation rates measured using [9, 10-3H]palmitate were elevated in the isolated working hearts of 8–10-week-old TazKD mice compared to their WT littermates (17). The reported increase in palmitate oxidation was not associated with enhanced gene or protein expression of β-oxidation enzymes and was surprisingly associated with an increase in myocardial triacylglycerol content, as well as a reduction in mRNA expression of the fatty acid transporter, cluster of differentiation 36. Reasons that may explain the elevated palmitate oxidation in the isolated working heart are unclear, though substrate oxidation rates in the working heart operate at a much higher workload than that of isolated mitochondria, and these perfusions were performed in the presence of insulin.
Despite these discrepancies in myocardial fatty acid β-oxidation in BTHS, current dogma in the field posits that stimulating myocardial fatty acid β-oxidation may be a novel approach to improve cardiac function in heart failure via increasing ATP production to support contraction (12). Supporting such a strategy as a therapeutic approach in BTHS, the pan-peroxisome proliferator activated receptor (PPAR) agonist, bezafibrate, is currently being investigated in the CARDIOlipin MANipulation (CARDIOMAN) trial (29). PPAR agonists, in particular PPARα, promote fatty acid β-oxidation via transcriptional upregulation of genes encoding for enzymes involved in fatty acid β-oxidation (30). Intriguingly, treatment of 3-month-old TazKD mice with bezafibrate for 4-months prevented the development of dilated cardiomyopathy and systolic dysfunction, which was associated with increased expression of genes involved in multiple energy metabolism pathways (31). Although the results of the CARDIOMAN trial are not yet published, a summary of the initial results of this single center, double-blinded, randomized, placebo-controlled crossover study of 11 participants with BTHS who were administered bezafibrate treatment for 15-weeks, was released by the Barth Syndrome Foundation. Assessment of cardiac function by ultrasound echocardiography revealed a significant improvement in heart strain at rest but not during peak exercise, and a trend toward improved heart chamber size. However, given that neither traditional cardiac parameters nor quality of life was changed significantly, these findings of cardiac improvement may not be clinically relevant (https://www.barthsyndrome.org/research/clinicaltrials/cardioman.html, accessed June 03, 2022). Taken together, these bezafibrate-mediated cardiac improvements in preclinical and clinical studies are encouraging and suggest that stimulating myocardial fatty acid β-oxidation may have utility in BTHS. Nonetheless, it has been suggested by others that systemic activation of PPARα with fibrates actually decreases myocardial fatty acid β-oxidation due to the stimulation of hepatic fatty acid β-oxidation and subsequent reduction in circulating lipids (32). Hence, further studies are still required to delineate whether the specific enhancement of cardiac fatty acid β-oxidation is indeed effective in alleviating BTHS-related cardiomyopathy.
Perturbations in myocardial carbohydrate metabolism in Barth syndrome
Numerous forms of cardiovascular disease (e.g., ischemic heart disease, heart failure, diabetic cardiomyopathy) are characterized by an impairment in myocardial glucose oxidation, often due to defects in pyruvate dehydrogenase (PDH), the rate-limiting enzyme of glucose oxidation (33–35). This metabolic perturbation also appears to be present in BTHS-related cardiomyopathy, as circulating lactate levels are elevated in individuals with BTHS (36), consistent with uncoupled glucose metabolism as glycolytically-derived pyruvate is shunted to lactate vs. being oxidized via PDH. Further evidence for a glucose oxidation defect in BTHS has been reported in several studies utilizing the TazKD mouse model and Tafazzin deficient cell lines. Utilizing a XF24 Seahorse bioanalyzer, neonatal cardiac myocytes isolated from TazKD mice demonstrated an ~40% decrease in oxygen consumption coincident with an increase in extracellular acidification rates, suggestive of uncoupled glucose metabolism due to elevated glycolysis and decreased glucose oxidation (23). Furthermore, ADP-stimulated respiration in isolated cardiac subsarcolemmal and intermyofibrillar mitochondria, as well as permeabilized cardiac muscle fibers, was significantly decreased with pyruvate and malate as substrates in 4–6-month-old TazKD mice vs. their WT littermates (16). Conversely, no impairment in ADP-stimulated respiration with pyruvate as a substrate was observed in isolated cardiac mitochondria from 2-month-old TazKD mice vs. their WT littermates (15). In addition, C2C12 mouse myoblasts subjected to CRISPR/Cas9-mediated knockout of Tafazzin demonstrated decreased incorporation of [U-13C]glucose into acetyl-CoA (37). This reduction was associated with increased inhibitory phosphorylation of PDH at serine 293, resulting in an approximate 50% decrease in enzymatic activity that could be rescued by incubation with exogenous CL. Deficiency of mature CL may also blunt Ca2+-dependent PDH dephosphorylation by reducing the abundance and stability of the mitochondrial calcium uptake protein 1, the primary regulator of the mitochondrial calcium uniporter (MCU), thereby impairing mitochondrial Ca2+ uptake and PDH activation (38). A cardiac-specific downregulation of the MCU pore-forming MCUa subunit has also been observed in 10-week-old TazKD mice, which abrogates Ca2+ uptake in isolated cardiac mitochondria (39) and could further explain impairments in PDH activity in BTHS. This impairment in normal Ca2+ handling was associated with increased myofilament Ca2+ sensitivity and decreased cross-bridge cycling velocity, thereby contributing to diastolic dysfunction in TazKD mice and consistent with observations that a cardiac-specific deficiency of PDH promotes diastolic dysfunction (40).
BTHS-related impairments in myocardial glucose oxidation using [U-14C]glucose have also been observed in aerobically perfused isolated working hearts from 8–10-week-old TazKD mice, which was also associated with impaired PDH activity compared to their WT littermates (17). However, this reduction in myocardial PDH activity was independent of alterations in phosphorylation or acetylation. Of interest, the insulin-stimulated enhancement of glucose oxidation rates was also blunted in the TazKD isolated working heart suggestive of cardiac insulin resistance, which coincided with a reduction in the gene expression of glucose transporter 4. Evidence of increased and decreased circulating glucose and insulin levels, respectively, and of reduced pancreatic islet insulin secretion in TazKD mice provide further support for the paradigm of impaired insulin action in the context of Tafazzin deficiency (17, 41). PET imaging studies with [1-11C]glucose in individuals with BTHS (age 18–36 years) contrast the majority of findings in preclinical models of BTHS, as myocardial glucose extraction fraction, uptake and utilization were significantly elevated compared to healthy age-matched controls (14). However, given that myocardial glucose oxidation rates were not directly assessed in these subjects, it is plausible that the elevated myocardial glycolytic rates observed in BTHS may explain the observed increase in myocardial glucose utilization.
As the stimulation of glucose oxidation in the heart has been shown to improve cardiac function in ischemic heart disease, heart failure, and diabetic cardiomyopathy (33, 35, 42–44), this may be an exciting metabolic approach to alleviate BTHS-related cardiomyopathy. Surprisingly, treatment of 6-week-old TazKD mice for 6-weeks with dichloroacetate (DCA; 70 mM in the drinking water), a pyruvate analog that inhibits PDH kinase to prevent inhibitory phosphorylation of PDH, did not alleviate their hypertrophic cardiomyopathy phenotype despite an enhancement of PDH activity (45). Reasons that DCA may have been devoid of benefit in TazKD mice may be due to the possibility that despite PDH activity and glucose oxidation being elevated, the immature CL remodeling and destabilized ETC supercomplexes would still be present, resulting in no net improvement in myocardial ATP production. Despite the failure of DCA to alleviate hypertrophic cardiomyopathy in TazKD mice, further investigation is needed to determine whether the pharmacological optimization of glucose oxidation may still have clinical utility in BTHS. On the contrary, it is also possible that any metabolic intervention to improve myocardial oxidative metabolism in BTHS will fail unless the CL-induced defects in ETC function are also resolved.
Perturbations in myocardial ketone metabolism in Barth syndrome
Observations that the failing heart increases its reliance on ketones as an oxidative fuel source (26, 46) has led to increased investigation of potential perturbations in myocardial ketone metabolism during the pathology of cardiovascular disease. Whether a similar shift in myocardial substrate utilization occurs in BTHS remains enigmatic, though metabolomics studies in 23 individuals with BTHS demonstrated a 1.8-fold increase in circulating β-hydroxybutyrate (βOHB) levels in comparison to 15 age-matched individuals not known to have an inborn error of metabolism (47). An increase in myocardial βOHB content was also observed in LV samples from BTHS subjects compared to age-matched non-failing control heart samples (20). Furthermore, an ~4-fold increase in myocardial protein expression of the ketone oxidation enzyme, βOHB dehydrogenase 1 (BDH1), was observed in 10-week-old TazKD mice vs. their WT littermates. Interestingly, this increased BDH1 expression did not translate into an increase in βOHB oxidation rates assessed during isolated working perfusions. As myocardial ketone oxidation rates are highly dependent on ketone delivery to the heart (48), it is plausible that identical perfusate βOHB concentrations (0.8 mM) was a limiting factor in the aforementioned study, and that ketone oxidation rates are elevated in vivo in TazKD mice.
It has been proposed that the salutary actions of sodium-glucose cotransporter-2 inhibitors in heart failure with reduced or preserved ejection fraction in the presence or absence of diabetes may be due in part to increasing circulating ketone levels and subsequent myocardial ketone oxidation (49–51). Accordingly, interventions that optimize cardiac ketone oxidation may represent a potential target to alleviate BTHS-related cardiomyopathy. Moreover, such a strategy may prove to be a more viable metabolic target, due to the enzymes of ketone oxidation not being as reliant on interactions with CL for their activity, as they are primarily localized to the mitochondrial matrix (52, 53).
Perturbations in myocardial amino acid metabolism in Barth syndrome
Although amino acids contribute minimally to cardiac ATP production under normal physiological conditions (54), combined glutamate and malate-supported state 3 respiration was upregulated by 45–68% in cardiac mitochondria isolated from TazKD mice 4–6 months of age compared to their WT littermates (16). Because TAFAZZIN deficiency may reduce CoA availability, it was postulated that the increased reliance on glutamate metabolism manifests since glutamate can be oxidized through the malate-aspartate shuttle, which generates NADH through CoA-independent reactions. The upregulated glutamate oxidation in cardiac mitochondria from TazKD mice was associated with increased protein expression of malate dehydrogenase, a key enzyme of the malate-aspartate shuttle (55). Likewise, a 25% increase in glutamate-stimulated state 3 respiration was also reported in isolated cardiac mitochondria from 2-month-old TazKD mice (15). This particular study also reported a 6-fold increase in glutamate-stimulated activity of the CL-regulated adenine nucleotide translocase in TazKD mouse cardiac mitochondria, thus suggesting that aberrant CL remodeling may influence substrate selectivity of the adenine nucleotide translocase and overall ETC flux.
Circulating branched-chain amino acids (BCAAs) are positively associated with cardiovascular disease (26, 56), and there appears to be both transcriptional and proteomic downregulation of pathways involved in BCAA metabolism in hearts from TazKD mice at 2–3 months of age (15, 21). A decrease in cardiac BCAA metabolism, depending on where the restriction is, may lead to increased levels of myocardial BCAAs and their correspondent keto acids, the latter of which are proposed to explain how BCAA metabolism promotes cardiac insulin resistance and cardiac hypertrophy (57). Alternatively, it has been suggested that amino acid utilization is enhanced in individuals with BTHS vs. activity matched healthy control subjects, as they exhibited decreased serum levels of amino acids involved in Krebs Cycle anaplerosis, including arginine, ornithine and citrulline (58). Nonetheless, an increased reliance on amino acid metabolism to support energetic demands could potentially result in increased proteolysis of skeletal and cardiac muscle, which may contribute to the decreased lean mass and cardiac dysfunction characteristic of the BTHS phenotype (3). Supporting this perspective, a higher whole-body leucine rate of appearance per kg of fat-free mass, a measure of proteolysis, demonstrated a trend to being associated with worsened LV function as determined by a lower LV global strain in adolescents and young adults with BTHS (58). While there appears to be several disturbances in myocardial amino acid metabolism present in BTHS, further investigation will be required to determine whether such disturbances are a viable metabolic target to alleviate BTHS-related cardiomyopathy.
Summary, conclusions & future directions
The development of multiple murine models of BTHS has greatly advanced our understanding of the metabolic perturbations present in BTHS, where it is clear that not only defects in ETC respiratory function, but also intermediary metabolism characterize this rare genetic disorder. As optimizing cardiac intermediary energy metabolism is being extensively pursued as a pharmacological strategy to improve cardiac function in heart failure, such an approach may also have clinical utility for managing BTHS-related cardiomyopathy. Unfortunately, the majority of studies that have tried to specifically optimize cardiac intermediary metabolism have yet to yield any major success. It does remain possible that any intervention that stimulates cardiac intermediary metabolism in BTHS in the absence of correcting the ETC respiratory defects, would not succeed since augmented oxidation of fuel (i.e., glucose) would not translate into increased ATP production to support cardiac function. As such, in order for metabolic therapy to yield benefit in BTHS it may need to be coupled with interventions that stabilize ETC respiratory function. Indeed, elamipretide is a cell-permeable, aromatic-cationic mitochondria-targeting tetrapeptide that localizes to the IMM, where it selectively associates with CL (59, 60) and is currently being tested in phase 2 studies for the treatment of BTHS (5). Although improvements in primary and secondary outcome measures were not observed in subjects with BTHS after 12 weeks during part 1 of the study, during the 36 week extension of part 2, exercise capacity, patient fatigue, 6-min walk test distances, and cardiac stroke volumes demonstrated improvement. Taken together, it will be imperative for future studies to assess whether elamipretide in combination with a metabolic therapy like DCA, which has been safely used in humans, may be a more effective approach to alleviate BTHS-related cardiomyopathy.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This study was supported by a Project Grant from the Canadian Institutes of Health Research (CIHR) to JRU. AAG was supported by a Vanier Canada Graduate Scholarship from the CIHR. JRU is a Tier 2 Canada Research Chair (Pharmacotherapy of Energy Metabolism in Obesity).
Acknowledgments
The figure in this review was created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Barth PG, Scholte HR, Berden JA, Van der Klei-Van Moorsel JM, Luyt-Houwen IE, Van 't Veer-Korthof ET, et al. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci. (1983) 62:327–35. doi: 10.1016/0022-510X(83)90209-5
2. Taylor C, Rao ES, Pierre G, Chronopoulou E, Hornby B, Heyman A, et al. Clinical presentation and natural history of Barth syndrome: an overview. J Inherit Metab Dis. (2022) 45:7–16. doi: 10.1002/jimd.12422
3. Jefferies JL. Barth syndrome. Am J Med Genet C Semin Med Genet. (2013) 163C:198–205 doi: 10.1002/ajmg.c.31372
4. Kang SL, Forsey J, Dudley D, Steward CG, Tsai-Goodman B. Clinical characteristics and outcomes of cardiomyopathy in Barth syndrome: the UK experience. Pediatr Cardiol. (2016) 37:167–17. doi: 10.1007/s00246-015-1260-z
5. Reid Thompson W, Hornby B, Manuel R, Bradley E, Laux J, Carr J, et al. A phase 2/3 randomized clinical trial followed by an open-label extension to evaluate the effectiveness of elamipretide in Barth syndrome, a genetic disorder of mitochondrial cardiolipin metabolism. Genet Med. (2021) 23:471–8. doi: 10.1038/s41436-020-01006-8
6. Rigaud C, Lebre AS, Touraine R, Beaupain B, Ottolenghi C, Chabli A, et al. Natural history of Barth syndrome: a national cohort study of 22 patients. Orphanet J Rare Dis. (2013) 8:70. doi: 10.1186/1750-1172-8-70
7. Garlid AO, Schaffer CT, Kim J, Bhatt H, Guevara-Gonzalez V, Ping P, et al. TAZ encodes tafazzin, a transacylase essential for cardiolipin formation and central to the etiology of Barth syndrome. Gene. (2020) 726:144148. doi: 10.1016/j.gene.2019.144148
8. Mcmillin JB, Dowhan W. Cardiolipin and apoptosis. Biochim Biophys Acta. (2002) 1585:97–107. doi: 10.1016/s1388-1981(02)00329-3
9. Gaspard GJ, Mcmaster CR. Cardiolipin metabolism and its causal role in the etiology of the inherited cardiomyopathy Barth syndrome. Chem Phys Lipids. (2015) 193:1–10. doi: 10.1016/j.chemphyslip.2015.09.005
10. Shen Z, Li Y, Gasparski AN, Abeliovich H, Greenberg ML. Cardiolipin regulates mitophagy through the protein kinase C pathway. J Biol Chem. (2017) 292:2916–23. doi: 10.1074/jbc.M116.753574
11. Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. (2007) 356:1140–51. doi: 10.1056/NEJMra063052
12. Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac Energy Metabolism in Heart Failure. Circ Res. (2021) 128:1487–513. doi: 10.1161/CIRCRESAHA.121.318241
13. Bashir A, Bohnert KL, Reeds DN, Peterson LR, Bittel AJ, de Las Fuentes L. Impaired cardiac and skeletal muscle bioenergetics in children, adolescents, and young adults with Barth syndrome. Physiol Rep. (2017) 5:e13130. doi: 10.14814/phy2.13130
14. Cade WT, Laforest R, Bohnert KL, Reeds DN, Bittel AJ, de Las Fuentes L, et al. Myocardial glucose and fatty acid metabolism is altered and associated with lower cardiac function in young adults with Barth syndrome. J Nucl Cardiol. (2021) 28:1649–59. doi: 10.1007/s12350-019-01933-3
15. Kiebish MA, Yang K, Liu X, Mancuso DJ, Guan S, Zhao Z, et al. Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J Lipid Res. (2013) 54:1312–25. doi: 10.1194/jlr.M034728
16. Le CH, Benage LG, Specht KS, Puma Li, Mulligan LC, Heuberger CMGC, et al. Tafazzin deficiency impairs CoA-dependent oxidative metabolism in cardiac mitochondria. J Biol Chem. (2020) 295:12485–97. doi: 10.1074/jbc.RA119.011229
17. Greenwell AA, Gopal K, Altamimi TR, Saed CT, Wang F, Tabatabaei Dakhili SA, et al. Barth syndrome-related cardiomyopathy is associated with a reduction in myocardial glucose oxidation. Am J Physiol Heart Circ Physiol. (2021) 320:H2255–69. doi: 10.1152/ajpheart.00873.2020
18. Dudek J, Maack C. Mechano-energetic aspects of Barth syndrome. J Inherit Metab Dis. (2022) 45:82–98. doi: 10.1002/jimd.12427
19. Xu Y, Erdjument-Bromage H, Phoon CKL, Neubert TA, Ren M, Schlame M, et al. Cardiolipin remodeling enables protein crowding in the inner mitochondrial membrane. EMBO J. (2021) 40:e108428. doi: 10.15252/embj.2021108428
20. Chatfield KC, Sparagna GC, Specht KS, Whitcomb LA, Omar AK, Miyamoto SD, et al. Long-chain fatty acid oxidation and respiratory complex I deficiencies distinguish Barth Syndrome from idiopathic pediatric cardiomyopathy. J Inherit Metab Dis. (2022) 45:111–24. doi: 10.1002/jimd.12459
21. Huang Y, Powers C, Madala SK, Greis KD, Haffey WD, Towbin JA, et al. Cardiac metabolic pathways affected in the mouse model of Barth syndrome. PLoS ONE. (2015) 10:e0128561. doi: 10.1371/journal.pone.0128561
22. Zhu S, Chen Z, Zhu M, Shen Y, Leon LJ, Chi L, et al. Cardiolipin remodeling defects impair mitochondrial architecture and function in a murine model of Barth Syndrome cardiomyopathy. Circ Heart Fail. (2021) 14:e008289. doi: 10.1161/CIRCHEARTFAILURE.121.008289
23. Powers C, Huang Y, Strauss A, Khuchua Z. Diminished exercise capacity and mitochondrial bc1 complex deficiency in tafazzin-knockdown mice. Front Physiol. (2013) 4:74. doi: 10.3389/fphys.2013.00074
24. Dudek J, Cheng IF, Chowdhury A, Wozny K, Balleininger M, Reinhold R, et al. Cardiac-specific succinate dehydrogenase deficiency in Barth syndrome. EMBO Mol Med. (2016) 8:139–54. doi: 10.15252/emmm.201505644
25. Wang G, Mccain ML, Yang L, He A, Pasqualini FS, Agarwal A, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. (2014) 20:616–23. doi: 10.1038/nm.3545
26. Ussher JR, Elmariah S, Gerszten RE, Dyck JR. The emerging role of metabolomics in the diagnosis and prognosis of cardiovascular disease. J Am Coll Cardiol. (2016) 68:2850–70. doi: 10.1016/j.jacc.2016.09.972
27. Murashige D, Jang C, Neinast M, Edwards JJ, Cowan A, Hyman MC, et al. Comprehensive quantification of fuel use by the failing and non-failing human heart. Science. (2020) 370:364–8. doi: 10.1126/science.abc8861
28. Taegtmeyer H, Young ME, Lopaschuk GD, Abel ED, Brunengraber H, Darley-Usmar V, et al. Assessing cardiac metabolism: a scientific statement from the american heart association. Circ Res. (2016) 118:1659–701. doi: 10.1161/RES.0000000000000097
29. Dabner L, Pieles GE, Steward CG, Hamilton-Shield JP, Ness AR, Rogers CA, et al. Treatment of Barth Syndrome by Cardiolipin Manipulation (CARDIOMAN) with bezafibrate: protocol for a randomized placebo-controlled pilot trial conducted in the nationally commissioned Barth syndrome service. JMIR Res Protoc. (2021) 10:e22533. doi: 10.2196/22533
30. Khuchua Z, Glukhov AI, Strauss AW, Javadov S. Elucidating the beneficial role of PPAR Agonists in Cardiac Diseases. Int J Mol Sci. (2018) 19:3464. doi: 10.3390/ijms19113464
31. Schafer C, Moore V, Dasgupta N, Javadov S, James JF, Glukhov AI, et al. The effects of PPAR stimulation on cardiac metabolic pathways in Barth syndrome mice. Front Pharmacol. (2018) 9:318. doi: 10.3389/fphar.2018.00318
32. Aasum E, Khalid AM, Gudbrandsen OA, How OJ, Berge RK, Larsen TS, et al. Fenofibrate modulates cardiac and hepatic metabolism and increases ischemic tolerance in diet-induced obese mice. J Mol Cell Cardiol. (2008) 44:201–9. doi: 10.1016/j.yjmcc.2007.08.020
33. Ussher JR, Wang W, Gandhi M, Keung W, Samokhvalov V, Oka T, et al. Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc Res. (2012) 94:359–69. doi: 10.1093/cvr/cvs129
34. Masoud WG, Ussher JR, Wang W, Jaswal JS, Wagg CS, Dyck JR, et al. Failing mouse hearts utilize energy inefficiently and benefit from improved coupling of glycolysis and glucose oxidation. Cardiovasc Res. (2014) 101:30–8. doi: 10.1093/cvr/cvt216
35. Gopal K, Al Batran R, Altamimi TR, Greenwell AA, Saed CT, Tabatabaei Dakhili SA, et al. FoxO1 inhibition alleviates type 2 diabetes-related diastolic dysfunction by increasing myocardial pyruvate dehydrogenase activity. Cell Rep. (2021) 35:108935. doi: 10.1016/j.celrep.2021.108935
36. Ferri L, Donati MA, Funghini S, Malvagia S, Catarzi S, Lugli L, et al. New clinical and molecular insights on Barth syndrome. Orphanet J Rare Dis. (2013) 8:27. doi: 10.1186/1750-1172-8-27
37. Li Y, Lou W, Raja V, Denis S, Yu W, Schmidtke MW, et al. Cardiolipin-induced activation of pyruvate dehydrogenase links mitochondrial lipid biosynthesis to TCA cycle function. J Biol Chem. (2019) 294:11568–78. doi: 10.1074/jbc.RA119.009037
38. Ghosh S, Zulkifli M, Joshi A, Venkatesan M, Cristel A, Vishnu N, et al. MCU-complex-mediated mitochondrial calcium signaling is impaired in Barth syndrome. Hum Mol Genet. (2022) 31:376–85. doi: 10.1093/hmg/ddab254
39. Bertero E, Nickel A, Kohlhaas M, Hohl M, Sequeira V, Brune C, et al. Loss of mitochondrial Ca(2+) uniporter limits inotropic reserve and provides trigger and substrate for arrhythmias in Barth syndrome cardiomyopathy. Circulation. (2021) 144:1694–713. doi: 10.1161/CIRCULATIONAHA.121.053755
40. Gopal K, Almutairi M, Al Batran R, Eaton F, Gandhi M, Ussher JR, et al. Cardiac-Specific deletion of pyruvate dehydrogenase impairs glucose oxidation rates and induces diastolic dysfunction. Front Cardiovasc Med. (2018) 5:17. doi: 10.3389/fcvm.2018.00017
41. Cole LK, Agarwal P, Doucette CA, Fonseca M, Xiang B, Sparagna GC, et al. Tafazzin deficiency reduces basal insulin secretion and mitochondrial function in pancreatic islets from male mice. Endocrinology. (2021) 162:102 doi: 10.1210/endocr/bqab102
42. Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail. (2010) 3:420–30. doi: 10.1161/CIRCHEARTFAILURE.109.888479
43. Le Page LM, Rider OJ, Lewis AJ, Ball V, Clarke K, Johansson E, et al. Increasing Pyruvate dehydrogenase flux as a treatment for diabetic cardiomyopathy: a combined 13C hyperpolarized magnetic resonance and echocardiography study. Diabetes. (2015) 64:2735–43. doi: 10.2337/db14-1560
44. Almutairi M, Gopal K, Greenwell AA, Young A, Gill R, Aburasayn H, et al. The GLP-1 receptor agonist liraglutide increases myocardial glucose oxidation rates via indirect mechanisms and mitigates experimental diabetic cardiomyopathy. Can J Cardiol. (2021) 37:140–50. doi: 10.1016/j.cjca.2020.02.098
45. Greenwell AA, Saed CT, Gopal K, Tabatabaei Dakhili SA, Kruger J, Eaton F, et al. Inhibition of pyruvate dehydrogenase kinase does not improve cardiac abnormalities in a mouse model of human Barth syndrome. Circulation. (2021) 144:A11800. doi: 10.1161/circ.144.suppl_1.11800
46. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, et al. The failing heart relies on ketone bodies as a fuel. Circulation. (2016) 133:698–705. doi: 10.1161/CIRCULATIONAHA.115.017355
47. Sandlers Y, Mercier K, Pathmasiri W, Carlson J, Mcritchie S, Sumner S, et al. Metabolomics reveals new mechanisms for pathogenesis in Barth syndrome and introduces novel roles for cardiolipin in cellular function. PLoS ONE. (2016) 11:e0151802. doi: 10.1371/journal.pone.0151802
48. Ho KL, Karwi QG, Wagg C, Zhang L, Vo K, Altamimi T, et al. Ketones can become the major fuel source for the heart but do not increase cardiac efficiency. Cardiovasc Res. (2021) 117:1178–87. doi: 10.1093/cvr/cvaa143
49. Verma S, Rawat S, Ho KL, Wagg CS, Zhang L, Teoh H, et al. Empagliflozin increases cardiac energy production in diabetes: novel translational insights into the heart failure benefits of SGLT2 Inhibitors. JACC Basic Transl Sci. (2018) 3:575–87. doi: 10.1016/j.jacbts.2018.07.006
50. Santos-Gallego CG, Requena-Ibanez JA, San Antonio R, Ishikawa K, Watanabe S, Picatoste B, et al. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J Am Coll Cardiol. (2019) 73:1931–44. doi: 10.1016/j.jacc.2019.01.056
51. Lopaschuk GD, Verma S. Mechanisms of cardiovascular benefits of sodium glucose Co-transporter 2 (SGLT2) inhibitors: a state-of-the-art review. JACC Basic Transl Sci. (2020) 5:632–44. doi: 10.1016/j.jacbts.2020.02.004
52. Robinson AM, Williamson DH. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev. (1980) 60:143–87. doi: 10.1152/physrev.1980.60.1.143
53. Sumegi B, Srere PA. Binding of the enzymes of fatty acid beta-oxidation and some related enzymes to pig heart inner mitochondrial membrane. J Biol Chem. (1984) 259:8748–52. doi: 10.1016/S0021-9258(17)47216-4
54. Karwi QG, Lopaschuk GD. Branched-Chain amino acid metabolism in the failing heart. Cardiovasc Drugs Ther. (2022). doi: 10.1007/s10557-022-07320-4
55. Yang L, Shi Q, Ho DJ, Starkov AA, Wille EJ, Xu H, et al. Mice deficient in dihydrolipoyl succinyl transferase show increased vulnerability to mitochondrial toxins. Neurobiol Dis. (2009) 36:320–30. doi: 10.1016/j.nbd.2009.07.023
56. Shah SH, Kraus WE, Newgard CB. Metabolomic profiling for the identification of novel biomarkers and mechanisms related to common cardiovascular diseases: form and function. Circulation. (2012) 126:1110–20. doi: 10.1161/CIRCULATIONAHA.111.060368
57. Walejko JM, Christopher BA, Crown SB, Zhang GF, Pickar-Oliver A, Yoneshiro T, et al. Branched-chain alpha-ketoacids are preferentially reaminated and activate protein synthesis in the heart. Nat Commun. (2021) 12:1680. doi: 10.1038/s41467-021-21962-2
58. Cade WT, Spencer CT, Reeds DN, Waggoner AD, O'Connor R, Maisenbacher M, et al. Substrate metabolism during basal and hyperinsulinemic conditions in adolescents and young-adults with Barth syndrome. J Inherit Metab Dis. (2013) 36:91–101 doi: 10.1007/s10545-012-9486-x
59. Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. (2014) 171:2029–50. doi: 10.1111/bph.12461
Keywords: Barth syndrome (BTHS), cardiomyopathy, cardiac energetics, glucose oxidation, fatty acid oxidation, ketone oxidation
Citation: Greenwell AA, Tabatabaei Dakhili SA and Ussher JR (2022) Myocardial disturbances of intermediary metabolism in Barth syndrome. Front. Cardiovasc. Med. 9:981972. doi: 10.3389/fcvm.2022.981972
Received: 29 June 2022; Accepted: 25 July 2022;
Published: 10 August 2022.
Edited by:
Diederik Wouter Dimitri Kuster, Amsterdam University Medical Center, NetherlandsReviewed by:
Vasco Sequeira, University Hospital Würzburg, GermanyCopyright © 2022 Greenwell, Tabatabaei Dakhili and Ussher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John R. Ussher, jussher@ualberta.ca