 Eleanor Raffan1 Liam A. Hurst2 Saeed Al Turki2,3 Gillian Carpenter4 Carol Scott2 Allan Daly2 Alison Coffey2 Sanjeev Bhaskar2 Eleanor Howard2 Naz Khan5 Helen Kingston5 Aarno Palotie2 David B. Savage1 Mark O’Driscoll4 Claire Smith6 Stephen O’Rahilly1 Inês Barroso1,2* Robert K. Semple1*
Eleanor Raffan1 Liam A. Hurst2 Saeed Al Turki2,3 Gillian Carpenter4 Carol Scott2 Allan Daly2 Alison Coffey2 Sanjeev Bhaskar2 Eleanor Howard2 Naz Khan5 Helen Kingston5 Aarno Palotie2 David B. Savage1 Mark O’Driscoll4 Claire Smith6 Stephen O’Rahilly1 Inês Barroso1,2* Robert K. Semple1*
- 1 Institute of Metabolic Science, University of Cambridge Metabolic Research Laboratories, Cambridge, UK
- 2 The Wellcome Trust Sanger Institute, Hinxton, UK
- 3 Department of Pathology, National Guard Health Affairs, Riyadh, Saudi Arabia
- 4 Human DNA Damage Response Disorders Group, Genome Damage and Stability Centre, University of Sussex, Brighton, UK
- 5 Genetic Medicine, Manchester Academic Health Science Centre, University of Manchester, Manchester, UK
- 6 East Lancashire Hospitals NHS Trust, Royal Blackburn Hospital, Lancashire, UK
Genetic diagnosis of inherited metabolic disease is conventionally achieved through syndrome recognition and targeted gene sequencing, but many patients receive no specific diagnosis. Next-generation sequencing allied to capture of expressed sequences from genomic DNA now offers a powerful new diagnostic approach. Barriers to routine diagnostic use include cost, and the complexity of interpreting results arising from simultaneous identification of large numbers of variants. We applied exome-wide sequencing to an individual, 16-year-old daughter of consanguineous parents with a novel syndrome of short stature, severe insulin resistance, ptosis, and microcephaly. Pulldown of expressed sequences from genomic DNA followed by massively parallel sequencing was undertaken. Single nucleotide variants were called using SAMtools prior to filtering based on sequence quality and existence in control genomes and exomes. Of 485 genetic variants predicted to alter protein sequence and absent from control data, 24 were homozygous in the patient. One mutation – the p.Arg732X mutation in the WRN gene – has previously been reported in Werner’s syndrome (WS). On re-evaluation of the patient several early features of WS were detected including loss of fat from the extremities and frontal hair thinning. Lymphoblastoid cells from the proband exhibited a defective decatenation checkpoint, consistent with loss of WRN activity. We have thus diagnosed WS some 15 years earlier than average, permitting aggressive prophylactic therapy and screening for WS complications, illustrating the potential of exome-wide sequencing to achieve early diagnosis and change management of rare autosomal recessive disease, even in individual patients of consanguineous parentage with apparently novel syndromes.
Introduction
Molecular diagnosis of novel Mendelian disorders has traditionally relied upon genetic linkage studies in one or more pedigrees with multiple affected members. Wider assessment of the penetrance and phenotypic spectrum associated with the causal genetic defect(s) thus identified then permits targeted genetic testing in a clinical diagnostic setting. However despite the major advances achieved using this paradigm, it is estimated that fewer than half of the allelic variants responsible for Mendelian disease have been identified to date (Ng et al., 2010a). One explanation for this lies in the limitations of positional cloning approaches, which may be thwarted by availability of only very small numbers of affected patients, sometimes exacerbated by locus heterogeneity in phenotypically similar disease. A further barrier arises in the clinical application of genetic knowledge, which may be confounded when patients are affected by an atypical syndrome, sometimes because of co-inheritance of more than one pathogenic mutation, or where early clinical signs of a syndrome are non-specific. As a result definitive diagnosis may be prevented or seriously delayed.
The rapidly developing technology of exome-wide sequencing has enormous potential to enhance the efficiency of molecular genetic diagnosis of inherited disease by circumventing barriers both to making novel associations between genes and disease, and to applying this research-derived knowledge to clinical diagnostics. This offers the opportunity for greatly improved prospective management of known clinical complications. Recent improvements in efficiency of exome capture combined with next-generation sequencing mean that sequencing of increasing numbers of exomes is being reported. Initial reports studied healthy individuals (Ng et al., 2008, 2009; Hedges et al., 2009) or those affected by Mendelian disorders with a known genetic basis (Ng et al., 2009) as proof of the concept that exome-wide sequencing can uncover pathogenic allelic variants. Subsequently exome sequencing (and to a lesser extent whole genome sequencing; Lupski et al., 2010; Rios et al., 2010) has been applied to identifying novel causes of Mendelian disease, using sequencing of unrelated individuals with highly similar phenotypes (Gilissen et al., 2010; Hoischen et al., 2010; Lalonde et al., 2010; Musunuru et al., 2010; Ng et al., 2010a,b), by studying multiple affected and unaffected individuals from one family (Bilgüvar et al., 2010; Krawitz et al., 2010) or by focusing on variants found in areas of interest from gene mapping (Bilgüvar et al., 2010; Sirmaci et al., 2010; Walsh et al., 2010).
A small number of pathogenic mutations have been identified after sequencing the exome of just one patient with no other affected family members; in these cases the variants have occurred in genes previously implicated in Mendelian disease (Choi et al., 2009; Bonnefond et al., 2010; Rios et al., 2010). However, the large number of rare sequence variants harbored by every exome poses a major challenge to the routine diagnostic use of the technology for individual patients. Nevertheless such evaluation of a single patient with no classically identifiable Mendelian syndrome is likely to be the most common application of exome-wide sequencing in clinical practice. This report now expands upon early experience by describing the diagnostic use of exome-wide sequencing in a patient with a novel clinical syndrome including severe insulin resistance, leading to molecular diagnosis of Werner’s syndrome some 10–20 years in advance of the usual time of diagnosis.
Materials and Methods
Exome-Wide Sequencing
The proband and her parents gave full informed consent for the use of exome-wide sequencing, in line with procedures approved by the NHS Cambridgeshire three research ethics committee, and in accordance with the principles of Helsinki. Specifically, consent was given to seek genetic defects that were believed to explain the proband’s clinical disease, and to inform the proband and her parents of these, but not to inform her of any incidentally detected genetic variation, whether or not possibly conferring risk of other, unrelated disease.
Proband DNA was screened for pathogenic copy number variants with data from Affymetrix SNP 6.0 ultra high density chip analyzed using Genotyping Console (TM) software (Affymetrix, Santa Clara, USA). For exome sequencing, 20 μg of DNA were sheared to 100–400 bp using a Covaris S2 following manufacturer’s protocols and using the following settings: Duty Cycle, 20%; Intensity, 5.0; Cycles/burst, 200; Duration, 90; Mode, Freq Sweeping. The sheared sample was quantified on a Bioanalyzer (Agilent, Santa Clara, USA). Fifteen micrograms of sheared DNA were end-repaired, A-tailed, and Illumina sequencing adapters ligated to the resulting fragments using the Illumina Paired-End DNA Sample Prep kit and protocol with the slight modification that the gel size selection step was replaced with SPRI bead purification (Beckman Coulter, Brea, USA). Five micrograms of the library were hybridized to a NimbleGen Sequence Capture 2.1 M Human Exome Array programmed to capture the NCBI consensus coding sequence (Pruitt et al., 2009; Roche NimbleGen, Madison, USA) following the manufacturer’s protocols with the modification that no pre-hybridization PCR was performed. The captured sample was washed and eluted in 50 μl of PCR-Grade water following manufacturer’s protocols. The eluted sample was added to a mix containing 2 mM MgCl2, 0.2 mM dNTPs, 0.5 μM PE.1, 0.5 μM PE.2, and 3 units of Platinum® Pfx DNA Polymerase (Invitrogen, Carlsbad, USA), and aliquoted into three individual wells of a plate and amplified using the following conditions: 94°C for 5 min followed by 20 cycles of 94°C for 15 s, 58°C for 30 s, 72°C for 30 s, and a final extension of 72°C for 5 min. PCR products were purified using SPRI beads prior to sequencing. Captured libraries were sequenced on the Illumina Genome Analyzer II platform (Illumina, Little Chesterford, UK) as 54-bp paired-end reads.
Data Analysis
MAQ (Li et al., 2008) software package v0.6.5 was used to map sequence reads to the human genome reference sequence (NCBI build 36). Duplicate fragments were removed using MAQ (v0.6.5) rmdup command. Alignments were converted from native MAQ map to SAM format using SAMtools software (Li and Durbin, 2009). Base qualities were recalibrated using The Genome Analysis Toolkit (GATK) early access v02Oct2009 (McKenna et al., 2010). Single nucleotide variants (SNVs) were called using SAMtools v0.1.7 (Li and Durbin, 2009). SNVs were filtered to include only those with a minimum SNP quality of 25, minimum genotype quality of 20, a minimum depth of 4× and maximum depth of 1200×. SNVs were reported in VCF (variant calling file)1 format v3.3.
Because of the rarity of the patient’s syndrome we assumed that the causative mutation would not be present in control individuals. Perl scripts were used to filter SNVs against the NCBI dbSNP database Build ID 1292, then against SNVs in 179 individuals included in the 1000 Genomes Project pilot 1 data release (April 2009, 100328; Durbin et al., 2010)3 and exome-wide sequencing data from 236 control individuals exomes. Variants were annotated against Ensembl version 54 and were defined as coding if they were non-synonymous, resulted in loss or gain of a stop codon, or occurred within essential splice sites. Since we hypothesized that the patient would have inherited a common ancestral allele from her consanguineous parents, Perl scripts were written to exclude SNVs which were not flanked by similarly homozygous SNPs, with the likely practical effect of excluding variants falsely called as homozygous in areas of low sequence depth.
Confirmatory Sanger Sequencing
Variants which remained after filtering were confirmed by Sanger sequencing in the proband and both parents. PCR primers were designed using Primer3, and PCR products were purified using Agencourt® CleanSEQ® reagents (Agencourt Bioscience, Beverly, MA, USA) and sequenced in both directions using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, PerkinElmer, Foster City, CA, USA) and an ABI 3730 DNA sequencer (Applied Biosystems). Sequence analysis was performed using Sequencher software (Gene Codes, Ann Arbor, MI, USA).
Cell Culture
Epstein Barr virus-transformed lymphoblastoid cells (LBLs) were generated as described previously (Pressman and Rotter, 1991), and maintained in RPMI with 15% fetal calf serum, 2 mmol/l l-glutamine, 100 U/l penicillin, and 100 μg/ml streptomycin in 5% ambient CO2.
Western Blotting
Whole cell extracts were resolved on 6% SDS-polyacrylamide gels followed by semi-dry transfer onto a PVDF membrane. Primary antibodies used were anti-ATR (H-300) and anti-MCM2 (N-19), both from Santa Cruz.
Decatenation Checkpoint Assay
Exponentially growing LBLs were treated with the topoisomerase inhibitor ICRF-183 (150 μM) for 4 h in the presence of colcimed (0.2 μg/ml), pelleted, swollen in 75 mM KCl for 10 min, fixed in Carnoy’s solution (3:1 methanol:acetic acid) for 10 min prior to staining with DAPI. Cells were then immobilized on poly-l-lysine coated slides by cytocentrifugation. The mitotic index and levels of pseudo-mitosis were determined using a Zeiss Axioplan microscope.
Assessment of Cellular G2-M Checkpoint
For ATR-dependent G2-M arrest cells were untreated or irradiated with 5 J/m2 UV-C (UV) and immediately seeded into complete medium, incubated for 2 h, before being cytospun onto poly-l-lysine coated slides, and stained with DAPI and anti-phosphoHistone H3 as previously described (Alderton et al., 2006). Cells from a patient with a previously described homozygous hypomorphic ATR mutation were used as positive controls (O’Driscoll et al., 2003). For the ataxia telangiectasia mutated (ATM)-dependent G2-M arrest cells were either untreated or irradiated with 2 Gy ionizing radiation (IR) and processed as above.
Results
Case History
The proband was born at term to parents of Pakistani origin who were first cousins. Her birth weight was 2.94 kg (10th centile) and head circumference 32 cm (3rd centile). Bilateral ptosis was noted at birth. There was mild motor developmental delay with hypertonia of the upper limbs, but development was otherwise normal. At 6 years old acanthosis nigricans was noted (Figure 1A), and at 8 years old both height and weight were progressing along the 50th centile, while head circumference was well below the 3rd centile (Figure 1B). There was mild hirsutism and signs of early puberty (Tanner B2P1). Oral glucose tolerance testing showed impaired glucose tolerance. At 10 years old insulin resistant diabetes was diagnosed with a fasting blood glucose of 8.3 mmol/l [normal range (NR) 3.9–5.5], concomitant plasma insulin of >990 pmol/l (NR 0–60) and HbA1c of 6.8% (NR 4.0–6.5). Serum alanine aminotransferase was elevated at 96 IU/l (NR 7–56), triglyceride was elevated at 4.2 mmol/l (NR < 2.2), and high density lipoprotein (HDL)-cholesterol was low at 0.5 mmol/l (NR > 0.9). Sex hormone binding globulin was low at 11.4 nmol/l (NR 25–110). As all these features are characteristic of insulin resistance with hepatic steatosis, metformin, and insulin were introduced. At this stage no further pubertal progression was apparent, but marked acanthosis nigricans persisted.
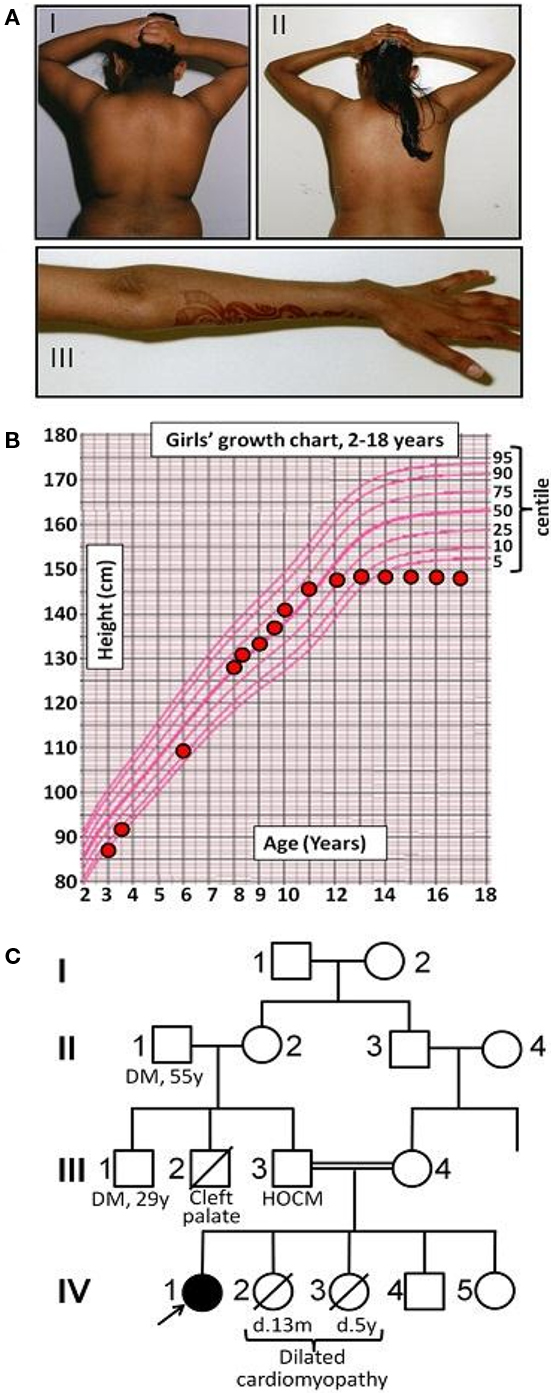
Figure 1. Early clinical features of Werner’s syndrome in a 16-year-old female patient from a consanguineous family. (A) Clinical images of proband at (I) 7 years old and (II, III) 16 years old, showing truncal obesity and acanthosis nigricans in the neck and flexures at both ages, and marked loss of distal subcutaneous tissue from the arms by 16 years. (B) Growth chart showing failure of pubertal growth spurt, typical of Werner’s syndrome. (C) Pedigree diagram (HOCM, hypertrophic obstructive cardiomyopathy; DM, type II diabetes mellitus).
Extensive biochemical and genetic testing was undertaken in an unsuccessful attempt to find a unifying genetic diagnosis. Normal investigations included a karyotype, blood mitochondrial DNA screen (for MELAS, MERRF, LHON, and NARP mutations and deletions), muscle mitochondrial DNA screen, respiratory chain analysis of frozen muscle homogenate, CSF and blood lactate, serial echocardiograms, transferrin immunoelectrophoresis, serum creatine kinase, flash electroretinogram, flash visual evoked potentials, urine amino and organic acids, sialic acid and oligosaccharides, and TORCH screen. Muscle biopsy showed only sparse focal lipid accumulation within muscle fibers. Computed tomography of the brain showed scattered foci of intracranial calcification in the right parietal region, in the right basal ganglia, and in the left lateral ventricle.
Abnormal liver function tests persisted, but near normoglycemia was restored, allowing cessation of insulin and maintenance treatment with metformin. (Table 1) Menarche occurred at 11 years and 1 month, but there was no discernible pubertal growth spurt, with linear growth ceasing at 12 years and a final height between the 0.4th and 2nd centile, and weight between the 2nd and 9th centile (Figure 1B). On re-evaluation at 16 years old she remained hirsute and oligomenorrhoeic with marked acanthosis nigricans (Figure 1A), microcephaly, and bilateral ptosis. Echocardiographic appearances were consistently normal.
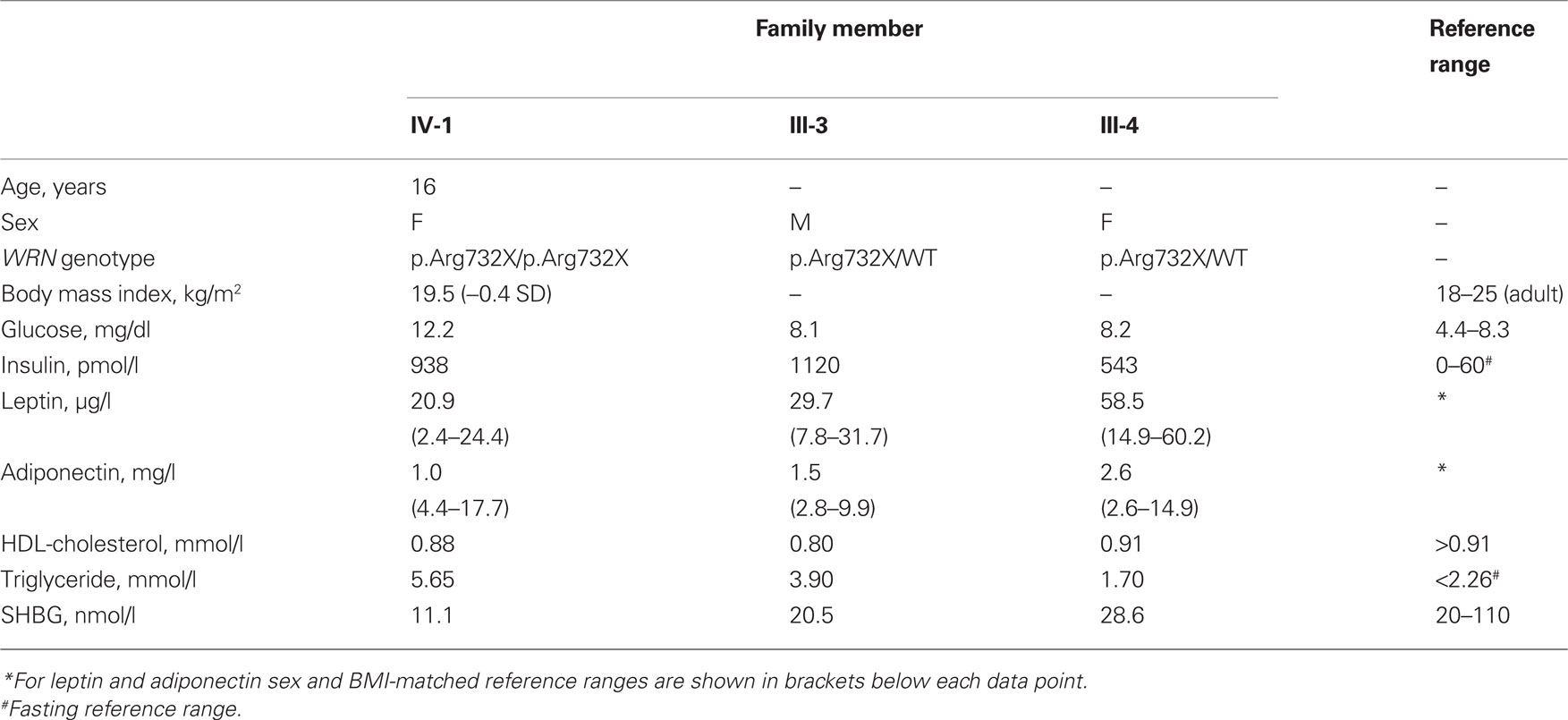
Table 1. Biochemical profile of proband and parents (postprandial).
Two of the proband’s younger sisters died in early childhood from dilated cardiomyopathy, while her father had hypertrophic obstructive cardiomyopathy diagnosed on clinical grounds. One paternal uncle had cleft palate and motor developmental delay, while another paternal uncle and grandfather had type 2 diabetes mellitus (Figure 1C).
At this stage further genetic evaluation was undertaken on a research basis. The proband was the only affected child of first cousins, and thus had a predicted coefficient of inbreeding of 0.0625. This means that 6.25% of her genome was likely to be identical by descent. Allied to the lack of obvious candidate genes to explain her novel syndrome, this meant that neither targeted sequencing of genes falling within homozygous regions, nor undertaking conventional autozygosity mapping were likely to reveal the pathogenic mutation. For this reason exome-wide sequencing of genomic DNA from the proband was undertaken.
Exome-Wide Sequencing
Overall 140,768,194 sequencing reads (281,536,388 paired end reads) were generated, of which 61.87% were uniquely mapped to the targeted exome with mean coverage depth of 155.35. The initial list of 22,344 variants was refined to a final 24 rare homozygous variants predicted to affect protein sequence, and lying in regions homozygous by descent (Figure 2).

Figure 2. Data analysis algorithm for whole exome sequencing data and location of pathogenic WRN mutation. Data analysis algorithm used for filtering all single nucleotide variants (SNVs) identified using exome-wide sequencing, with numbers of variants left at each filtering step.
Validation and Parental Genotyping
Sanger sequencing confirmed 21 of the 24 to be homozygous in the proband. Of those, 17 were heterozygous in both parents, 2 were homozygous in one parent and heterozygous in the other, and 2 were homozygous in both parents. Three SNVs called homozygous on exome-wide sequencing were found to be either heterozygous (2) or homozygous (1) for the reference allele by Sanger sequencing in the proband. Of the 21 confirmed homozygous SNVs, only the p.Arg732X (c.2982C > T) nonsense mutation in the WRN gene (GenBank NM_000553.4; NP_000544.2), described in at least three unrelated patients with Werner syndrome (MIM #277700), had previously been implicated in Mendelian disease. Both parents were heterozygous for the mutation. None of the other 20 genes harboring homozygous mutations had previously been associated with any aspect of the proband’s clinical presentation, and nor was any plausible mechanistic basis for any such association apparent. Moreover, on re-evaluation of the patient in the light of this result, frontal hair thinning and loss of subcutaneous tissue from the extremities (Figure 1A) were noted, both consistent with evolving Werner Syndrome. Importantly, because the informed consent given by the proband and her family for exome-wide sequencing permitted disclosure to the proband or third parties only of variants believed by the investigators to account for her clinical disease, these other variants are not listed in this report.
The WRN gene encodes a RecQ-like 3′-5′ DNA helicase with additional 5′-3′ exonuclease activity, and in common with nearly all pathogenic WRN mutations, the p.Arg732X mutation truncates the protein before the critical helicase domain (Figure 3A). As the cellular consequences of this mutation have not previously been described, and in view of the atypical presentation, further cellular evidence for loss of function of the WRN helicase was sought.
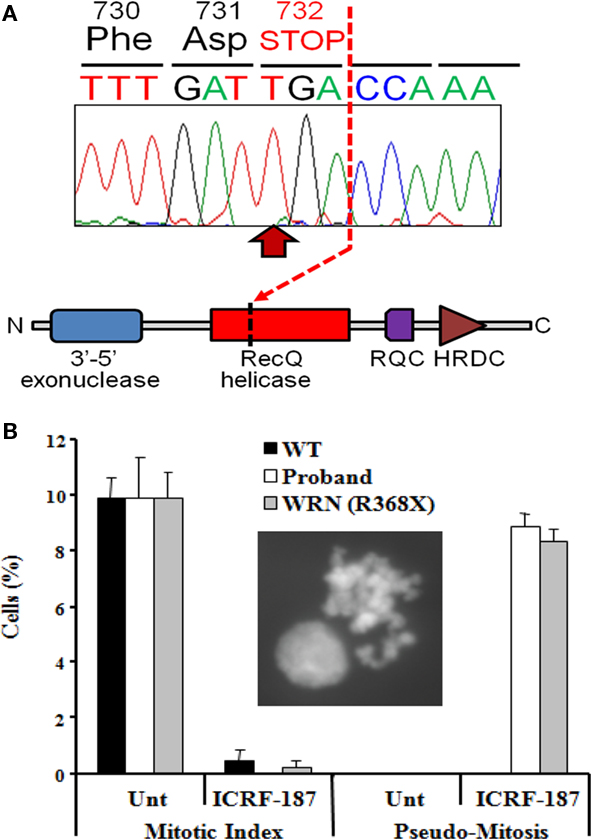
Figure 3. Identification of a loss-of-function mutation in the WRN helicase (A). Homozygous p.Arg732X mutation in the WRN gene found in the proband, leading to truncation of the RecQ helicase domain and loss of both RecQ C-terminal (RQC) and helicase RNAseD C-terminal (HRDC) domains which mediate interaction with DNA and proteins by the WRN protein. (B) Impairment of the DNA decatenation checkpoint in lymphoblastoid cells from the proband. The inset shows a representative pseudo-mitosis. WT, wild type; Unt, untreated; WRN (R368X), cells from a Werner’s syndrome patient homozygous for the p.Arg368X pathogenic variant.
Decatenation Checkpoint Assay
As a consequence of DNA replication, the DNA strands of sister chromatids become intimately entangled, or catenated. To prevent chromosomal mis-segregation or DNA breakage during the subsequent anaphase it is essential that such catenations are resolved (Damelin and Bestor, 2007), a process that is thought to be critically dependent on the activity of topoisomerase II (Luo et al., 2009). Delay in decatenation, for example through chemical inhibition of Topo II using the bisdioxopiperazine family of catalytic inhibitors, activates the decatenation checkpoint (DCC), and prevents entry into mitosis (Damelin and Bestor, 2007). The WRN DNA helicase has previously been implicated as an important component of the DNA DCC mechanism (Franchitto et al., 2003), with WRN deficient cells failing to show cell cycle arrest in response to topoisomerase inhibition. We thus sought to assess integrity of the DCC in LBLs from the proband as an indirect index of loss of function of the WRN helicase. Treatment of LBLs from both a wild type control and the proband with the topoisomerase inhibitor ICRF-187 induced strong G2-arrest, as determined by a dramatic decrease in mitotic index (Figure 3B). However only cells from the proband exhibited a defective DCC, indicated by increased levels of pseudo-mitosis following this treatment (Figure 3B). These findings are strongly supportive of loss of the cellular function of the WRN protein, and are in keeping with a clinical diagnosis of Werner syndrome.
ATR Immunoblotting and G2-M Checkpoint Function
Both the proband and her mother were also heterozygous for a late essential splice donor site mutation in the Ataxia telangiestasia and Rad3-related (ATR) gene (IVS43 + 2T > C). A homozygous hypomorphic mutation of ATR, which encodes a key DNA damage-sensing kinase, has previously been reported to cause Seckel syndrome, which includes severe microcephaly and growth retardation (O’Driscoll et al., 2003). ATR signaling responds to single-stranded regions of DNA generated at stalled replication forks and ATM-dependent signaling is activated in response to DNA double strand breaks. Detection of DNA breaks leads to G2-M checkpoint arrest. Since the relative microcephaly of the proband was not adequately explained by Werner syndrome, ATR expression and function were assessed in LBLs from the proband. No difference in either the size or expression of the ATR protein was apparent compared to a wild type control (Figure 4A). Furthermore, LBLs from the proband exhibited a proficient DNA double strand break-induced ATM-dependent G2-M arrest following ionizing radiation (Figure 4B).
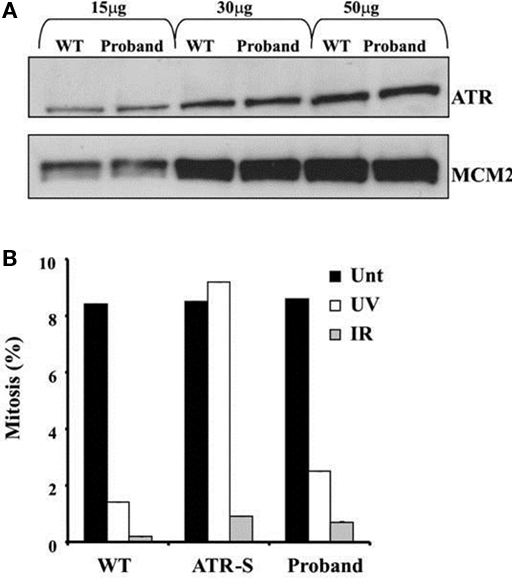
Figure 4. (A) ATR protein expression in EBV-transformed lymphoblastoid cells from the proband compared to a wildtype control (WT). MCM2 is shown as a loading control. (B) Preserved G2-M cell cycle checkpoint in response to either ultraviolet light (UV) or ionizing radiation (IR), assessed by the rate of mitosis compared to untreated cells (Unt). Cells from a patient with biallelic hypomorphic mutation of ATR (ATR-S) are shown as a positive control for ATR dysfunction.
Discussion
Genetic loss of function of the WRN helicase in humans produces Werner syndrome, a progeria syndrome in which features of premature aging including cataracts, scleroderma, atherosclerosis, and malignancies begin to be become clinically manifest in the 3rd decade of life. Insulin resistance and diabetes are also well recognized features of the condition, and have been reported at a young age (Epstein et al., 1966). Although clinical diagnostic criteria for Werner’s syndrome have been suggested (Nakura et al., 1994; Goto, 1997; The International Registry of Werner’s syndrome)4, these are only applicable to the later stages of the disease once frank progeria has become apparent.
We describe the case of a 16-year girl in whom no unifying diagnosis had been made despite intensive and expensive investigation in the 10 years since she first presented with clinical features of severe insulin resistance. Although she still does not fulfill clinical diagnostic criteria for Werner syndrome, the high penetrance of mutations in the WRN gene and the defective DCC seen in her LBLs, allied to the frontal hair thinning, loss of subcutaneous tissue from the extremities, and lack of pubertal growth spurt, has permitted a confident diagnosis of Werner syndrome to be made.
Although IR is a characteristic feature of Werner syndrome, both the proband’s parents were also markedly insulin resistant, as evidenced by hyperinsulinemia and suppressed adiponectin and SHBG (Table 1). Her father, who was also obese, had striking acanthosis nigricans, and both parents had dyslipidemia and hyperinsulinemia. No metabolic evaluation of people with heterozygous WRN mutations have been reported, so it is possible that the mutation carried by both parents does account for their tendency to severe IR, however given the high background prevalence of IR and type 2 diabetes in South Asian populations, this may instead by caused by other mutations, acting either independently of or synergistically with the WRN mutation.
The early clinical features of Werner syndrome, such as failure of the pubertal growth spurt, are relatively non-specific, and because clinical diagnostic criteria rely on complications that usually arise only in the 3rd decade onward, the average age at diagnosis of Werner syndrome is in the late 30s. In this case a further confounding factor was the co-existence of microcephaly and bilateral ptosis, neither of which have been reported in Werner syndrome. It is most likely that these features are attributable to co-inherited recessive disease, leading to superimposition of more than one genetic syndrome, a phenomenon that is likely to be common in families with high degrees of consanguinity.
Although inspection of the list of rare homozygous variants found in the proband did not reveal any previously described genetic defects associated with microcephaly and ptosis, both the proband and her mother were heterozygous for a late essential splice donor site mutation in the ATR gene (IVS43 + 2T > C). As loss of ATR function in humans has been reported to cause Seckel syndrome, which includes severe microcephaly (O’Driscoll et al., 2003), and as both WRN and ATR have been reported to function in series in some DNA damage checkpoints (Otterlei et al., 2006; Damelin and Bestor, 2007), we speculated that co-inheritance of WRN loss of function may result in mild loss of ATR function being clinically expressed in the form of microcephaly. However the normal electrophoretic appearance of ATR protein in cells from the proband, and preservation of the G2-M DNA damage checkpoint argues against significant loss of ATR function, though a subtle effect cannot be ruled out.
Early diagnosis of Werner syndrome in this case is likely to have considerable clinical benefits. It will enable (1) Early and concerted use of established primary preventative therapies in the proband, such as statins for cardiovascular disease and skin care to reduce the risk of ulceration, (2) Prospective screening and intervention for major complications of Werner’s syndrome including cancers, atherosclerosis and cataracts, and (3) Consideration of more experimental therapies aimed at slowing the disease process. An example of this is high dose vitamin C therapy, recently shown to retard the premature aging process in a rodent model of Werner syndrome (Massip et al., 2010). In addition, molecular confirmation of the diagnosis enables carrier testing for other members of the family to be offered, with antenatal diagnosis available for couples at risk of having affected children.
In this case, a very stringent filter was used to remove variants occurring in any of the 415 available control genomes and exomes, yielding a small list of candidate homozygous mutations, among which a highly likely pathogenic mutation was found. However caution must be exercised in the use of such filtering in the setting of autosomal recessive disease, where carriers may plausibly exist in the control population. In some cases a less stringent filter removing only variants found above a certain allele frequency in controls may be more useful. Where to set this filter will rely in large part on the rarity of the syndrome being studied, and where no plausible pathogenic mutations are revealed, a progressive relaxation of the filtering stringency may be tried.
This report illustrates the decisive diagnostic potential of exome-wide sequencing in the early diagnosis of rare genetic disease in individual patients before the phenotype is fully evolved, especially where corroborated by a reliable cellular assay. This approach is likely to have particular utility where clinical recognition of genetic syndromes is confounded by the superimposition of unrelated clinical features due to co-inherited mutations, as may be seen particularly with high degree parental consanguinity. Further efforts to define the correct place for whole exome genetic sequencing in the diagnostic workup of such patients is warranted.
Conflict of Interest Statement
Inês Barroso and her spouse own stock in the companies GlaxoSmithKline (GSK) and Incyte (INCY).
Acknowledgments
We are grateful to John Burton, Mike Quail, Hazel Arbury, and Matthew Humphries for technical assistance in exome pulldown, library preparation, and sequencing. Stephen O’Rahilly, Inês Barroso, Robert K. Semple, and Eleanor Raffan are supported by the Wellcome Trust (grant numbers 078986/Z/06/Z, 077016/Z/05/Z, 080952/Z/06/Z, and 087678/Z/08/Z respectively) and the Medical Research Council Centre for Obesity and Related Disorders and the United Kingdom National Institute for Health Research Cambridge Biomedical Research Centre. MOD is a Cancer Research UK Senior Fellow (grant C24110/A8300).
Footnotes
- ^http://www.1000genomes.org/wiki/doku.php?id = 1000_genomes:analysis:vcf3.3
- ^ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606/database/organism_data/b129/
- ^ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/pilot_data/release/2010_03/pilot1/README_SRP000031.2010_03_snps
- ^http://www.pathology.washington.edu/research/werner/registry/registry.html
References
Alderton, G. K., Galbiati, L., Griffith, E., Surinya, K. H., Neitzel, H., Jackson, A. P., Jeggo, P. A., and O’Driscoll, M. (2006). Regulation of mitotic entry by microcephalin and its overlap with ATR signalling. Nat. Cell Biol. 8, 725–733.
Bilgüvar, K., Oztürk, A. K., Louvi, A., Kwan, K. Y., Choi, M., Tatli, B., Yalnizog?lu, D., Tüysüz, B., Cag?layan, A. O., Gökben, S., Kaymakçalan, H., Barak, T., Bakirciog?lu, M., Yasuno, K., Ho, W., Sanders, S., Zhu, Y., Yilmaz, S., Dinçer, A., Johnson, M. H., Bronen, R. A., Koçer, N., Per, H., Mane, S., Pamir, M. N., Yalçinkaya, C., Kumandaş, S., Topçu, M., Ozmen, M., Sestan, N., Lifton, R. P., State, M. W., and Günel, M. (2010). Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467, 207–210.
Bonnefond, A., Durand, E., Sand, O., Graeve, F. D., Gallina, S., Busiah, K., Lobbens, S., Simon, A., Bellanné-Chantelot, C., Létourneau, L., Scharfmann, R., Delplanque, J., Sladek, R., Polak, M., Vaxillaire, M., and Froguel, P. (2010). Molecular diagnosis of neonatal diabetes mellitus using next-generation sequencing of the whole exome. PLoS ONE 5, e13630. doi: 10.1371/journal.pone.0013630
Choi, M., Scholl, U. I., Ji, W., Liu, T., Tikhonova, I. R., Zumbo, P., Nayir, A., Bakkalog?lu, A. i., Ozen, S., Sanjad, S., Nelson-Williams, C., Farhi, A., Mane, S., and Lifton, R. P. (2009). Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. U.S.A. 106, 19096–19101.
Durbin, R. M., Abecasis, G. R., Altshuler, D. L., Auton, A., Brooks, L. D., Gibbs, R. A., Hurles, M. E., and McVean, G. A. (2010). A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073.
Epstein, C. J., Martin, G. M., Schultz, A. L., and Motulsky, A. G. (1966). Werner’s syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine 45, 177–221.
Franchitto, A., Oshima, J., and Pichierri, P. (2003). The G2-phase decatenation checkpoint is defective in Werner syndrome cells. Cancer Res. 63, 3289–3295.
Gilissen, C., Arts, H. H., Hoischen, A., Spruijt, L., Mans, D. A., Arts, P., van Lier, B., Steehouwer, M., van Reeuwijk, J., Kant, S. G., Roepman, R., Knoers, N. V. A. M., Veltman, J. A., and Brunner, H. G. (2010). Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am. J. Hum. Genet. 87, 418–423.
Goto, M. (1997). Hierarchical deterioration of body systems in Werner’s syndrome: implications for normal ageing. Mech. Ageing Dev. 98, 239–254.
Hedges, D. J., Burges, D., Powell, E., Almonte, C., Huang, J., Young, S., Boese, B., Schmidt, M., Pericak-Vance, M. A., Martin, E., Zhang, X., Harkins, T. T., and Zuchner, S. (2009). Exome sequencing of a multigenerational human pedigree. PLoS ONE 4, e8232. doi: 10.1371/journal.pone.0008232
Hoischen, A., van Bon, B. W. M., Gilissen, C., Arts, P., van Lier, B., Steehouwer, M., de Vries, P., de Reuver, R., Wieskamp, N., Mortier, G., Devriendt, K., Amorim, M. Z., Revencu, N., Kidd, A., Barbosa, M., Turner, A., Smith, J., Oley, C., Henderson, A., Hayes, I. M., Thompson, E. M., Brunner, H. G., de Vries, B. B. A., and Veltman, J. A. (2010). De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat. Genet. 42, 483–485.
Krawitz, P. M., Schweiger, M. R., Rödelsperger, C., Marcelis, C., Kölsch, U., Meisel, C., Stephani, F., Kinoshita, T., Murakami, Y., Bauer, S., Isau, M., Fischer, A., Dahl, A., Kerick, M., Hecht, J., Köhler, S., Jäger, M., Grünhagen, J., de Condor, B. J., Doelken, S., Brunner, H. G., Meinecke, P., Passarge, E., Thompson, M. D., Cole, D. E., Horn, D., Roscioli, T., Mundlos, S., and Robinson, P. N. (2010). Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat. Genet. 40, 827–829.
Lalonde, E., Albrecht, S., Ha, K. C., Jacob, K., Bolduc, N., Polychronakos, C., Dechelotte, P., Majewski, J., and Jabado, N. (2010). Unexpected allelic heterogeneity and spectrum of mutations in Fowler syndrome revealed by next-generation exome sequencing. Hum. Mutat. 31, 918–923.
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760.
Li, H., Ruan, J., and Durbin, R. (2008). Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18, 1851–1858.
Luo, K., Yuan, J., Chen, J., and Lou, Z. (2009). Topoisomerase IIalpha controls the decatenation checkpoint. Nat. Cell Biol. 11, 204–210.
Lupski, J. R., Reid, J. G., Gonzaga-Jauregui, C., Deiros, D. R., Chen, D. C. Y., Nazareth, L., Bainbridge, M., Dinh, H., Jing, C., Wheeler, D. A., McGuire, A. L., Zhang, F., Stankiewicz, P., Halperin, J. J., Yang, C., Gehman, C., Guo, D., Irikat, R. K., Tom, W., Fantin, N. J., Muzny, D. M., and Gibbs, R. A. (2010). Whole-genome sequencing in a patient with Charcot–Marie–Tooth neuropathy. N. Engl. J. Med. 362, 1181–1191.
Massip, L., Garand, C., Paquet, E. R., Cogger, V. C., O’Reilly, J. N., Tworek, L., Hatherell, A., Taylor, C. G., Thorin, E., Zahradka, P., Le Couteur, D. G., and Lebel, M. (2010). Vitamin C restores healthy aging in a mouse model for Werner syndrome. FASEB J. 24, 158–172.
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly, M., and Depristo, M. A. (2010). The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303.
Musunuru, K., Pirruccello, J. P., Do, R., Peloso, G. M., Guiducci, C., Sougnez, C., Garimella, K. V., Fisher, S., Abreu, J., Barry, A. J., Fennell, T., Banks, E., Ambrogio, L., Cibulskis, K., Kernytsky, A., Gonzalez, E., Rudzicz, N., Engert, J. C., Depristo, M. A., Daly, M. J., Cohen, J. C., Hobbs, H. H., Altshuler, D., Schonfeld, G., Gabriel, S. B., Yue, P. and Kathiresan, S. (2010) Exome Sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N. Engl. J. Med. 363, 2220–2227.
Nakura, J., Wijsman, E. M., Miki, T., Kamino, K., Yu, C. E., Oshima, J., Fukuchi, K., Weber, J. L., Piussan, C., Melaragno, M. I., Epstein, C. J., Scappaticci, S., Fraccaro, M., Matusmura, T., Murano, S., Yoshida, S., Fujiwara, Y., Saida, T., Ogihara, T., Martin, G. M., and Schellenberg, G. D. (1994). Homozygosity mapping of the Werner syndrome locus (WRN). Genomics 23, 600–608.
Ng, P. C., Levy, S., Huang, J., Stockwell, T. B., Walenz, B. P., Li, K., Axelrod, N., Busam, D. A., Strausberg, R. L., and Venter, J. C. (2008). Genetic variation in an individual human exome. PLoS Genet. 4, e1000160. doi: 10.1371/journal.pgen.1000160
Ng, S. B., Bigham, A. W., Buckingham, K. J., Hannibal, M. C., McMillin, M. J., Gildersleeve, H. I., Beck, A. E., Tabor, H. K., Cooper, G. M., Mefford, H. C., Lee, C., Turner, E. H., Smith, J. D., Rieder, M. J., Yoshiura, K., Matsumoto, N., Ohta, T., Niikawa, N., Nickerson, D. A., Bamshad, M. J., and Shendure, J. (2010a). Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 42, 790–793.
Ng, S. B., Buckingham, K. J., Lee, C., Bigham, A. W., Tabor, H. K., Dent, K. M., Huff, C. D., Shannon, P. T., Jabs, E. W., Nickerson, D. A., Shendure, J., and Bamshad, M. J. (2010b). Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 42, 30–35.
Ng, S. B., Turner, E. H., Robertson, P. D., Flygare, S. D., Bigham, A. W., Lee, C., Shaffer, T., Wong, M., Bhattacharjee, A., Eichler, E. E., Bamshad, M., Nickerson, D. A., and Shendure, J. (2009). Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461, 272–276.
O’Driscoll, M., Ruiz-Perez, V. L., Woods, C. G., Jeggo, P. A., and Goodship, J. A. (2003). A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 33, 497–501.
Otterlei, M., Bruheim, P., Ahn, B., Bussen, W., Karmakar, P., Baynton, K., and Bohr, V. A. (2006). Werner syndrome protein participates in a complex with RAD51, RAD54, RAD54B and ATR in response to ICL-induced replication arrest. J. Cell. Sci. 119, 5137–5146.
Pressman, S., and Rotter, J. I. (1991). Epstein-Barr virus transformation of cryopreserved lymphocytes: prolonged experience with technique. Am. J. Hum. Genet. 49, 467.
Pruitt, K. D., Harrow, J., Harte, R. A., Wallin, C., Diekhans, M., Maglott, D. R., Searle, S., Farrell, C. M., Loveland, J. E., Ruef, B. J., Hart, E., Suner, M. M., Landrum, M. J., Aken, B., Ayling, S., Baertsch, R., Fernandez-Banet, J., Cherry, J. L., Curwen, V., Dicuccio, M., Kellis, M., Lee, J., Lin, M. F., Schuster, M., Shkeda, A., Amid, C., Brown, G., Dukhanina, O., Frankish, A., Hart, J., Maidak, B. L., Mudge, J., Murphy, M. R., Murphy, T., Rajan, J., Rajput, B., Riddick, L. D., Snow, C., Steward, C., Webb, D., Weber, J. A., Wilming, L., Wu, W., Birney, E., Haussler, D., Hubbard, T., Ostell, J., Durbin, R., and Lipman, D. (2009). The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 19, 1316–1323.
Rios, J., Stein, E., Shendure, J., Hobbs, H. H., and Cohen, J. C. (2010). Identification by whole-genome resequencing of gene defect responsible for severe hypercholesterolemia. Hum. Mol. Genet. 19, 4313–4318.
Sirmaci, A., Walsh, T., Akay, H., Spiliopoulos, M., Sakalar, Y. l. r. m. B., Hasanefendiog?lu-Bayrak, A., Duman, D., Farooq, A., King, M.-C., and Tekin, M. (2010). MASP1 mutations in patients with facial, umbilical, coccygeal, and auditory findings of Carnevale, Malpuech, OSA, and Michels Syndromes. Am. J. Hum. Genet. 87, 679–686.
Walsh, T., Shahin, H., Elkan-Miller, T., Lee, M. K., Thornton, A. M., Roeb, W., Rayyan, A. A., Loulus, S., Avraham, K. B., King, M.-C., and Kanaan, M. (2010). Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am. J. Hum. Genet. 87, 90–94.
Keywords: Werner’s syndrome, whole exome sequencing, insulin resistance, diabetes, WRN
Citation: Raffan E, Hurst LA, Al Turki S, Carpenter G, Scott C, Daly A, Coffey A, Bhaskar S, Howard E, Khan N, Kingston H, Palotie A, Savage DB, O’Driscoll M, Smith C, O’Rahilly S, Barroso I and Semple RK (2011) Early diagnosis of Werner’s syndrome using exome-wide sequencing in a single, atypical patient. Front. Endocrin. 2:8. doi: 10.3389/fendo.2011.00008
Received: 26 January 2011;
Paper pending published: 12 February 2011;
Accepted: 18 March 2011;
Published online: 29 March 2011.
Edited by:
Sian Ellard, University of Exeter, UKReviewed by:
Michael Weedon, University of Exeter, UKCharles Shaw-Smith, Peninsula Medical School, UK
Copyright: © 2011 Raffan, Hurst, Al Turki, Carpenter, Scott, Daly, Coffey, Bhaskar, Howard, Khan, Kingston, Palotie, Savage, O’Driscoll, Smith, O’Rahilly, Barroso and Semple . This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Robert Semple, University of Cambridge Metabolic Research Laboratories, Institute of Metabolic Science, Addenbrooke’s Hospital, Box 289, Cambridge CB2 0QQ, UK. e-mail: rks16@cam.ac.uk;
Inês Barroso, The Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SA, UK. e-mail: ib1@sanger.ac.uk