 Evgenia Globa1*
Evgenia Globa1* Natalia Zelinska1Yulia Shcherbak2Joelle Bignon-Topalovic3
Natalia Zelinska1Yulia Shcherbak2Joelle Bignon-Topalovic3 Anu Bashamboo3
Anu Bashamboo3 Ken MсElreavey3
Ken MсElreavey3- 1Ukrainian Scientific and Practical Center of Endocrine Surgery, Transplantation of Endocrine Organs and Tissues of the Ministry of Health of Ukraine, Kyiv, Ukraine
- 2National Children’s Specialized Hospital OHMATDYT of the Ministry of Health of Ukraine, Kyiv, Ukraine
- 3Human Developmental Genetics, Institute Pasteur, Paris, France
Background: The clinical profile and genetics of individuals with Disorders/Differences of Sex Development (DSD) has not been reported in Ukraine.
Materials and Methods: We established the Ukrainian DSD Register and identified 682 DSD patients. This cohort includes, 357 patients (52.3% [303 patients with Turner syndrome)] with sex chromosome DSD, 119 (17.5%) with 46,XY DSD and 206 (30.2%) with 46,XX DSD. Patients with sex chromosome DSD and congenital adrenal hyperplasia (CAH, n=185) were excluded from further studies. Fluorescence in situ hybridization (FISH) was performed for eight 46,XX boys. 79 patients underwent Whole Exome Sequencing (WES).
Results: The majority of patients with 46,XY and 46,XX DSD (n=140), were raised as female (56.3% and 61.9% respectively). WES (n=79) identified pathogenic (P) or likely pathogenic (LP) variants in 43% of the cohort. P/LP variants were identified in the androgen receptor (AR) and NR5A1 genes (20.2%). Variants in other DSD genes including AMHR2, HSD17B3, MYRF, ANOS1, FGFR11, WT1, DHX37, SRD5A1, GATA4, TBCE, CACNA1A and GLI2 were identified in 22.8% of cases. 83.3% of all P/LP variants are novel. 35.3% of patients with a genetic diagnosis had an atypical clinical presentation. A known pathogenic variant in WDR11, which was reported to cause congenital hypogonadotropic hypogonadism (CHH), was identified in individuals with primary hypogonadism.
Conclusions: WES is a powerful tool to identify novel causal variants in patients with DSD, including a significant minority that have an atypical clinical presentation. Our data suggest that heterozygous variants in the WDR11 gene are unlikely to cause of CHH.
Highlights
•What is already known on this topic? DSD is a group of rare conditions that are defined by a discordance of the chromosomal, gonadal or phenotypic features. The genetic cause is known in a minority of patients and obtaining a genetic etiology is challenging due to a variable clinical presentation. A small number of studies have applied WES to large cohorts of DSD.
•What does this study add? This study describes the main clinical features and genetic findings in a large cohort of DSD patients from Ukraine. The most common genetic causes of DSD were variants in the AR and NR5A1 genes. A significant number (35.3%) of patients with a genetic diagnosis had an atypical clinical presentation. A variant in WDR11, previously reported to cause CHH, was identified in individuals with primary hypogonadism suggesting that heterozygous variants in this gene may not always cause CHH.
Introduction
Disorders of Sex Development (DSD) is a group of rare conditions that are defined by a discordance of the chromosomal, gonadal or phenotypic features of the internal and/or external genitalia (1). The development of the gonads is a complex process governed by a combination of genetic networks and hormonal signaling. Given the complexity of gonad formation and differentiation, comprehensive genetic testing is recognized as a key element in the investigation of patients with DSD (2). On the basis of the underlying etiology, DSD can be further divided into several subclasses, such as primary disorders of gonadal development, hormone secretion or hormone action and syndromic conditions (1).
The most common cause of 46,XY DSD is the disruption of sex hormone synthesis or anomalies of their receptors, such as variants in the androgen receptor (AR). Approximately 30-45% of patients with partial androgen insensitivity syndrome (PAIS) carry pathogenic variants in the AR (3, 4). In contrast, more than 80% of 46,XY DSD raised as females, have pathogenic variants in the AR gene (4). Anomalies of testis-determination result in gonadal dysgenesis and are usually caused by the pathogenic variants in the SRY, NR5A1 and DHX37 genes (15%, 10-13% and 10-12% respectively) (4–6). Pathogenic variants in the MAP3K1, MAMLD1, HSD17B3, SRD5A2 and DAX1 genes are relatively infrequent (5-10%) (7, 8). The most common presentation of 46,XX DSD is congenital adrenal hyperplasia (CAH) that is caused by pathogenic variants in the CYP21A2 gene (9). Translocation of the testis-determining gene SRY from the Y chromosome to the X chromosome is responsible for up to 90% of cases of 46,XX testicular or ovotesticular DSD (10).
Defining the genetic causes of DSD using a gene by gene approach can identify the molecular etiology in up to 64% of cases (11, 12). Targeted next generation sequencing (tNGS) gene panels can result in a diagnostic yield of 43% for 46,XY DSD (13). Excluding patients with CAH, pathogenic variants in the AR, NR5A1, SRD5A2, ZFPM2, HSD17B3 and DHH genes are the most frequent causes of 46,XY DSD (13). However, since the underlying genetic etiology of DSD can vary depending on geography and ancestry, the diagnostic yield may differ from one region to another. Whole Exome Sequencing (WES) can overcome these problems by theoretically sequencing all of the genes in the human genome. It also allows data to be reanalyzed as new genetic causes are identified. Currently, more than 60 genes that cause DSD are known (5, 14). A genetic diagnosis allows further knowledge-based clinical management as well as genetic counseling on variant transmission, fertility and the risk of malignancy. Despite the power of WES, the interpretation and reporting of genetic variants is challenging and secondary findings that are not related to the primary condition may arise. Since a small number of studies have used WES on large DSD cohorts, the aim of this study was to determine the main clinical features and the genetic findings of a large cohort of DSD patients from Ukraine.
Materials and Methods
Patients
The Ukrainian DSD Registry was created in 2014 to include children diagnosed with DSD, identified by regional Ukrainian pediatric endocrinologists, gynecologists, urologists and contains 682 patients. Chromosomal DSD was found in 357 patients (52.3%, of which 303 patients had Turner syndrome (84.8%)). In the 46,XX DSD group (n=206, 30.2%) CAH was diagnosed in 185 patients (89.8%). We also identified 140 cases (20.5%) with 46,XY (n=119) or 46,XX DSD (n=21). Of these DNA samples were obtained from 79 patients (56.4%) from 75 unrelated families for the further genetic testing using WES.
In this cohort study the diagnosis of DSD was done based on clinical evaluation, laboratory and imaging examination in according to the Consensus Statement on Management of Intersex Disorders (1). Before WES all patients underwent routine clinical examination, hormonal tests and instrumental diagnostics (including assessment of bone age, ultrasound (US) and/or MRI if necessary). For patients who undergone gonadectomy, additional data included - age at gonadectomy, indication for this procedure and histology result. All patients had a karyotype determined by standard methods, and for eight 46,XX boys, molecular cytogenetic studies (FISH) was done to determine a Y-X translocation of the SRY locus.
The main inclusion criteria included the following - ambiguous external genitalia (female genitalia with an enlarged clitoris, posterior labial fusion, or an inguinal/labial mass and/or inguinal hernia or male genitalia with bilateral undescended testes, micropenis, isolated perineal hypospadias, or mild hypospadias with undescended testis), delayed or incomplete puberty, virilization with typical female external genitalia, primary amenorrhea, breast development in a typical male, a discordance between the genital appearance and the karyotype and family history of DSD (1). Patients with specific chromosomal DSD anomalies (e.g., Turner syndrome, Klinefelter syndrome etc.) and those with CAH were excluded from further study.
The clinical presentation of 46,XY DSD (n=71) patients who underwent WES included (i) DSD of undefined origin (n=22, including 16 patients with severe ambiguous genitalia (AG) at birth) with a broad spectrum of phenotypes for which the underlying cause was unknown; (ii) a suspected disorder in androgen synthesis or action (DASA) [n=18, including 2 patients with Persistent Müllerian Duct Syndrome (PMDS)]; (iii) confirmed or probable gonadal dysgenesis (GD) (n=14); (iv) testicular regression syndrome (TRS) (n=12); (v) Kallmann syndrome (n=4) and (vi) a patient with 46,XY ovotesticular DSD (n=1). The 46,XX patients (n=8) who underwent WES, consisted of five girls with a primary hypogonadism (PH) without virilization, whilst two had signs of androgen excess (Prader 3-4) and one phenotypic male who presented with testicular DSD.
Genetic Testing
Exome sequencing of genomic DNA was performed for 79 patients. Enrichment for WES was generated with Agilent SureSelect Human All Exon V4, followed by paired-end sequencing on the Illumina HiSeq2000 platform with TruSeq v3 chemistry. Data analysis was performed from the sequencing platform using manufacturer’s proprietary software. All reads were aligned against the human reference genome (NCBI, GRCh37/hg19 or GRCh38/hg38) via Burrows-Wheeler aligner. Single-nucleotide variants and small insertions and deletions (InDel) were selected with GATK version 1.6. Picard version 1.62 (http://broadinstitute.github.io/picard/) and SAMtools version 0.1.18 were used to mark duplicate reads and to process the BAM files manipulations, respectively. For each case, single-nucleotide polymorphism (SNP) and indel variants were annotated to dbSNP 138 identifiers using the Genome Analysis Toolkit (GATK) Unified Genotyper. The SNP Effect Predictor bioinformatics tools on the Ensembl website (http://www.ensembl.org/homosapiens/userdata/uploadvariations), gnomAD (https://gnomad.broadinstitute.org/) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) were used to annotated the novel variants, followed by manual screening of all variants by using the Human Gene Mutation Database Professional Biobase (http://www.biobaseinternational.com/product/hgmd/). Potentially pathogenic variants were confirmed by Sanger sequencing.
Results
The Ukrainian DSD Registry has 682 patients, consisting of 357 (52.3%) individuals with chromosomal DSD, 119 (17.5%) individuals with 46,XY DSD and 206 patients with 46,XX DSD (30.2%), (Figure 1A). Molecular genetic studies (WES) were performed in a selected group of patients (n=79) from 75 unrelated families with 46,XY and 46,XX DSD.
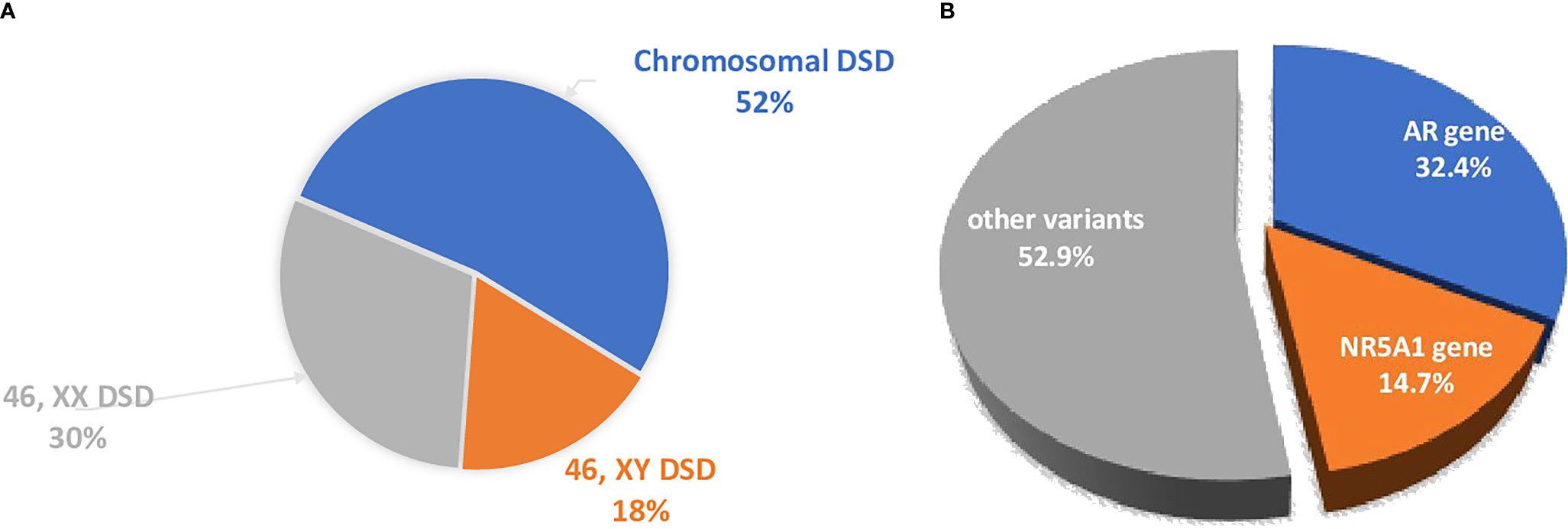
Figure 1 (A) The distribution of the different forms of DSD in the Ukrainian DSD registry. 46,XX DSD cohort included 89.8% CAH patients. (B) The distribution of the main genetic causes of DSD were analyzed by WES. The dominant causes are due to variants in the AR and NR5A1.
All patients included in the study, underwent follow-up examinations and have been fully analyzed. The majority of affected individuals included in the WES study, were raised as female (56.3% 46,XY DSD and 61.9% 46,XX DSD). P/LP variants were identified in 34/79 patients (43%), however two patients had P variants that could either not explain all of the phenotype, or the phenotypic expression was atypical for the gene (Table 1). Of the thirty-four patients carrying P/LP variants, twenty-eight patients (35.4%) carried variants that have not been reported in association with the pathology, nor have they been reported in any public variant database [variants of unknown significance (VUS)].
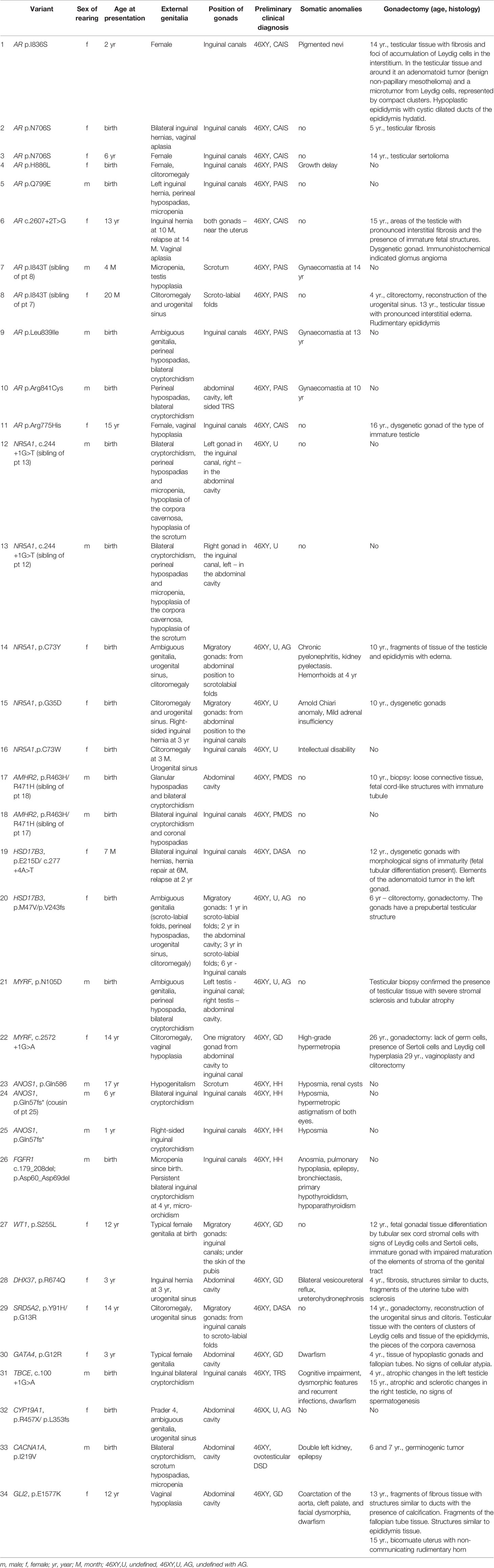
Table 1 Clinical details of the individuals with Pathogenic or Likely Pathogenic variants causing DSD identified by WES.
46,ХY DSD Cohort
The main genetic causes of DSD in the XY cohort that underwent WES are pathogenic variants in the AR and NR5A1 genes (16/79, 20.2%; Tables 1, 2 and Figure 1B).
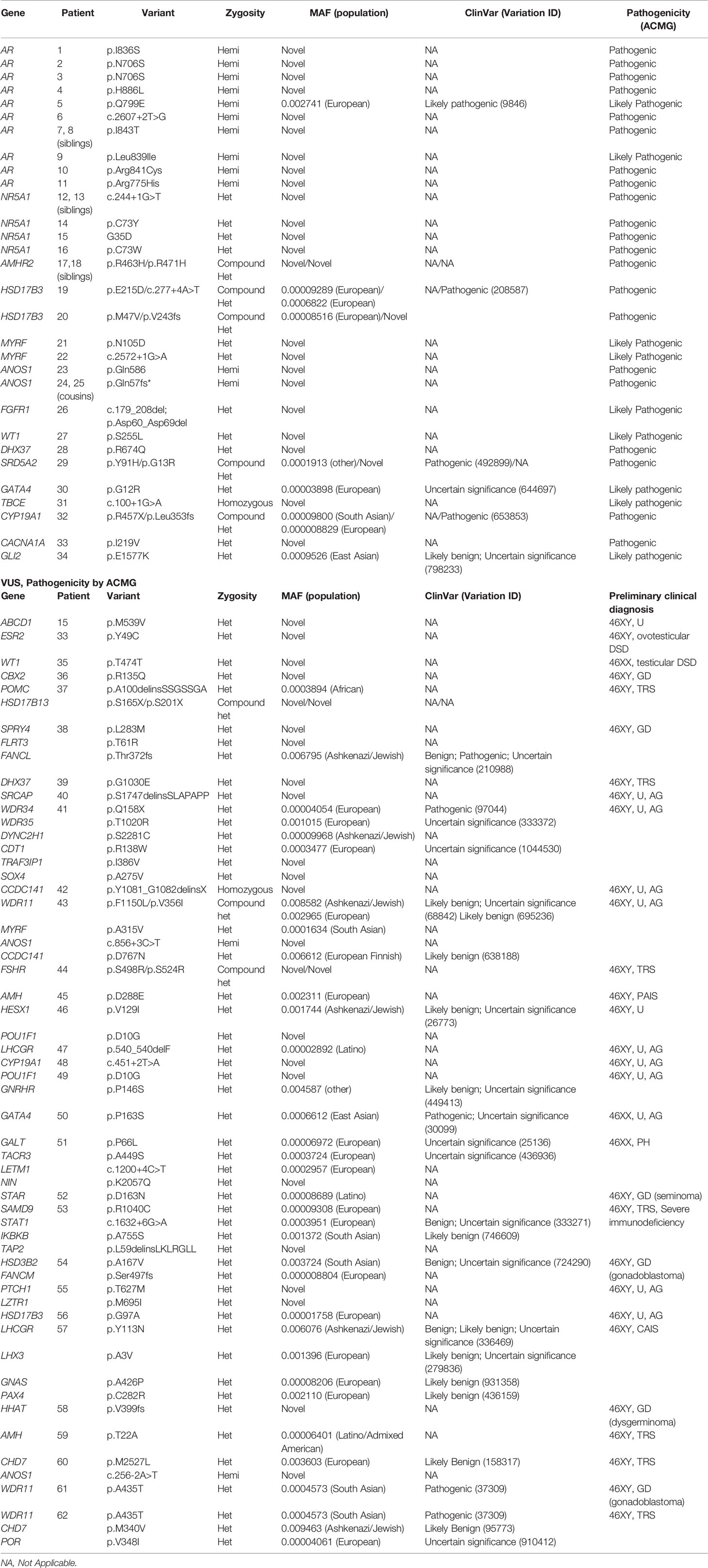
Table 2 Genetic causes of DSD in patients from the Ukraine DSD Register.
Eleven patients from 10 families carried hemizygous pathogenic variants in the AR gene. Of the nine AR variants associated with XY DSD, eight have not been reported to cause either PAIS or CAIS and they are absent from public SNP databases (Table 2). Eight of the variants are missense and one is an essential splice donor site. Of the eleven patients, five were classified as CAIS and seven registered as female. Two girls were defined as PAIS due to clitoromegaly. Six females had a gonadectomy between 6 and 16 years old, and of these, one girl was diagnosed with a sertolioma at 14 years (Table 1). Four of the eleven patients were registered as male with a PAIS phenotype. Cases 7 and 8 are siblings (AR p.I843T variant), where one was raised as a boy and the other as a girl. The former presented with pubertal delay and micropenia. One patient with AR p.Leu839Ile had atypical genitalia at birth and was registered as female, but child’s gender was reassigned to male at 11 months.
The second most common genetic cause of DSD in the XY cohort are variants in the NR5A1 gene (Tables 1, 2 and Figure 1B). Five individuals from 4 families carried heterozygous pathogenic variants in the NR5A1 gene. Two were raised as boys and three as girls. All variants are novel and consists of three missense variants, all located within the zinc-finger DNA-binding domain (G35D, p.C73W, p.C73Y; Table 1). The other splice site variant (c.244+1G>T) was carried by twin boys born after IVF treatment. At birth they presented with bilateral cryptorchidism, perineal hypospadias and micropenia. Family history indicated that the father and grandfather had a similar DSD phenotype at birth and underwent surgery of the external genitalia and cryptorhidism correction. The NR5A1 variant p.G35D was carried by a girl who presented with clitoromegaly and urogenital sinus at birth. Before karyotyping she was diagnosed with CAH and glucocorticoids were prescribed, but were stopped at the age of 4 years. A follow up examination after receiving of the WES result at 9 years revealed signs of moderate primary hypocorticism (ACTH 122 pg/ml (normal range 6-55), cortisol daily urine 42.4 μg/day (normal range 58-403). Two girls who carried the NR5A1 p.C73Y and p.C73W variants presented at birth with clitoromegaly and the gonads were located in the inguinal canals.
Two siblings (cases 17 and 18) with compound heterozygous variant in the AMHR2 gene (p.R463H/p.R471H) were reported previously (15).
Furthermore, two girls (cases 19, 20) presented with compound heterozygous variants in the HSD17B3 gene. One presented at 7 months due to the presence of bilateral inguinal hernias and surgery was performed. At 10 years old US of the pelvic organs and inguinal canals revealed the absence of the uterus and the location of the gonads in the inguinal canals. She carried rare HSD17B3 variants (p.E215D/c.277+4A>T). A second girl with rare and novel HSD17B3 variants (p.M47V/p.V243fs) presented at birth had ambiguous genitalia (scroto-labial folds, perineal hypospadias, urogenital sinus) and the location of the gonads in the scroto-labial folds.
Two individuals carried LP and novel MYRF variants. A boy (case 21, p.N105D) presented at birth with ambiguous genitalia and was registered as a female. The child’s gender was reassigned to male after the discovery of a 46,XY karyotype at 1 month. There were no other somatic phenotypic anomalies. A girl (case 22) carries a heterozygous MYRF loss-of-function (LOF) variant (c.2572+1G>A). She presented at 14 years of age with primary amenorrhea, lack of secondary sexual characteristics and high-grade hypermetropia. She also had PH, hirsutism and vaginal hypoplasia. US of the pelvic organs indicated a hypoplastic uterus and right migratory gonad (from the abdominal cavity to the inguinal canal). The left gonad was absent. She underwent a gonadectomy and vaginoplasty with clitorectomy at the age of 26 and 29 years old respectively.
Four boys were diagnosed with hypo-/anosmic hypogonadotropic hypogonadism (HH). In three of the boys two novel hemizygous P variants were identified in the ANOS1 gene (Table 2). Case 23, carrying an ANOS1 c.1756C>T (p.Gln586) variant, presented with hypogenitalism (micro-orchidism, testis volume 0.65 ml) and hyposmia at 17 years old. Serum inhibin B and AMH levels were normal. Standard short HCG test (three injections for three consecutive days of 1500/m2 IU of HCG) was negative, however long HCG test was positive (doubled total testosterone level on day 11th), but this did not result either in further testosterone increasing nor testis enlargement based on the US data and clinical examination 19 days after the test. Considering the absence of a history of cryptorchidism and the normal inhibin B and AMH levels, the child underwent a subsequent treatment with Follitropin alfa, which led to the enlargement of the testicular volume by 77% and the penile, but testosterone levels did not increase. The MRI of the brain confirmed a hypoplasia of the olfactory bulbs. Two cousins with hyposmia (cases 24-25) carried an ANOS1 c.171_181del (p.Gln57fs*) variant inherited from their mothers, and bilateral inguinal cryptorchidism developed after 6 years of age in a proband, but in his cousin right-sided inguinal cryptorchidism and micropenia presented at 1 year of age and he had orchidopexy at 5 and 9 years without an effect. Both siblings did not receive treatment until 14 and 15 years respectively. The GnRH stimulation test confirmed HH and a standard short HCG test was positive in both patients. Serum inhibin B and AMH levels were normal. Treatment with HCG at 14 and 15 years of age lead to the descent of the testes into the scrotum and increased testosterone levels after 10 weeks of treatment in both patients. Case 26 with an FGFR1 (c.179_208del; p.Asp60_Asp69del) pathogenic variant presented at 2 years with micropenia that was first observed at birth. A persistent bilateral inguinal cryptorchidism appeared at 4 years. Serum inhibin B and AMH levels were low. The GnRH stimulation test confirmed HH and a standard short HCG test was negative. The child also has pulmonary hypoplasia and bronchiectasis. Subsequent treatment with HCG was ineffective and he underwent an orchidopexy at 9 years. His mother and grandmother reported the presence of anosmia. At the age of 9 years new clinical features (primary hypoparathyroidism, hypothyroidism and epilepsy) were noted.
P/LP variants in the genes WT1, DHX37, SRD5A1, GATA4, TBCE, CACNA1A and GLI2 were identified in single individuals (Tables 1, 2). A 46,XY girl with normal renal function carried a heterozygous WT1 p.S255L variant that was inherited from her healthy mother. She presented at 12 years with primary amenorrhea and the absence of secondary sexual characteristics. US of the inguinal canals revealed the presence of gonads and she underwent gonadectomy. The DHX37 variant was reported previously (5). Case 29, who carries compound heterozygous variants in the SRD5A2 gene, presented at 14 years with primary amenorrhea, clitoromegaly and absence of secondary sexual characteristics. US of the inguinal canals revealed the presence of gonads with subsequent gonadectomy. Within the cohort a single individual carried a LP, rare missense variant in the GATA4 gene (p.G12R) that was inherited from her healthy mother. The girl presented at the age of 3 years with significant growth retardation (> - 2SD) and her karyotype revealed a minimal mosaicism (93.3% of nuclei with locus Yq12) (46, XY. nuc ish Xp11.1- q.11.1) (DXZ1 *1), Yq12 (DYZ1-) [25]/Xp11.1-q.11.1 (DXZ1*1), Yq12 (DYZ1 * 1) [375]). Further examination revealed PH, and US confirmed the presence of the uterus and gonads in the abdominal cavity. Gonadectomy was done at the age of 4 years due to a significant growth retardation (> - 2SD) and the need to initiate treatment with growth hormone. Ultrasound of the heart (EchoCG) was normal in both the mother and child carrying the GATA4 variant. Case 31, a 46,XY boy, was born with SGA and inguinal bilateral cryptorchidism. He carries a homozygous splice site variant (c.100+1G>A) in the TBCE gene. Biallelic variants of TBCE are associated with Hypoparathyroidism-Retardation-Dysmorphism Syndrome and Kenny-Caffey Syndrome (OMIM 604934). He underwent two stages of surgery (at 3 and 4 years), after which however, the right testis remained in the inguinal canal and the left testis was removed because of its atrophy. He also had cognitive impairment, dysmorphic features and recurrent infections. At the age of 14 years, further examination showed a PH and dwarfism (height -2.8 SD) with the normal clonidine test, but low IGF-1 level (>- 2SD). Final gonadectomy was done at the age of 15 years.
Two of the patients in the cohort carried variants that contributed to their phenotype, but the association of these variants with DSD is unclear. Case 33, is a 46,XY boy who presented at birth with bilateral cryptorchidism, scrotum hypospadias, micropenia and a double left kidney. He underwent gonadectomy in 2 stages because of ovotesticular DSD at 6 and 7 years. Histopathology indicated a germinogenic tumor and the child had subsequent chemotherapy. At 13 years the boy developed epilepsy. He carries a heterozygous CACNA1A p.I219V variant. Variants in the CACNA1A gene are associated with epilepsy (16), but to our knowledge variants in this gene are not associated with DSD. The second individual, case 34, is a female who presented with short stature, PH (FSH 111.1 mIU/ml, LH 24.2 mIU/ml), absence of the uterus and gonads and vaginal hypoplasia at the age of 12 years. The child also presented with congenital coarctation of the aorta, cleft palate, and facial dysmorphia. She carries a GLI2 p.E1577K variant. Variants in this gene are associated with autosomal dominant holoprosencephaly 9 and Cooler-Jones syndrome. These syndromes are characterized by hypopituitarism with dwarfism, developmental delay, polydactyly and facial dysmorphia, and this can explain most of the phenotype of the girl (17). However, the DSD associated with GLI2 variants are considered secondary to the pituitary anomalies and they are not associated with PH. Gonadectomy was done at the age of 13 years. After the gonadectomy and initiation of the replacement therapy with growth hormone therapy, she was subsequently found to have an additional structure near the uterus with a fluid component from a pelvic ultrasound, performed as part of this study. Subsequent surgery confirmed the presence of a detached rudimentary uterine horn.
46,ХХ DSD Cohort
A lower genetic diagnostic yield of WES was found for 46,XX DSD compared with 46,XY DSD cohort (12.5% and 46.5% accordingly), (Figures 2A, B). In the former group 61.9% (n=13) were raised as girls and 38.1% (n=8) were raised as boys. Three of the eight boys carried the SRY gene, which explains the phenotype. Two of the three 46,XX SRY+ boys had cryptorchidism and two had severe somatic anomalies (severe myopathy in one and cognitive deficiency and anorectal atresia in another). Of the patients with 46,XX DSD with female presentation (n=13), six patients had a PH without virilization, whilst others had signs of androgen excess (Prader 3-5). WES performed for one boy and seven girls (n=8) and identified rare biallelic and pathogenic CYP19A1 variant (p.R457X/p.L353fs) in one child (case 32) who presented with ambiguous genitalia and was registered as a male. At the age of 8 months, following the discovery of a 46,XX SRY- negative karyotype the child’s gender was reassigned to female. At one year of age, she was diagnosed with fluorocolpos, with a spontaneous regression. Repeated hormonal investigations at the age of 5 years old indicated PH (FSH 22 mIU/ml). At 10 years the child underwent plastic surgery for the urogenital sinus. At this time the girl had elevated FSH level (34.4 mIU/ml).
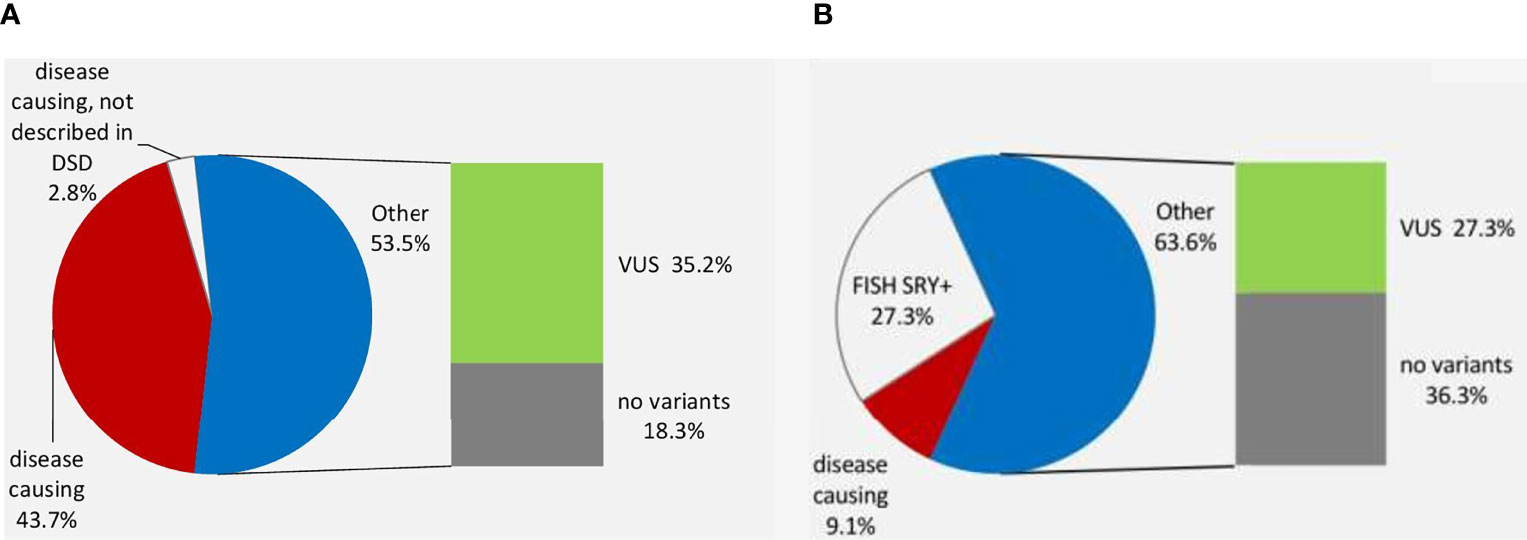
Figure 2 The genetic diagnosis of 46, XY DSD (A) and 46,XX DSD (B).
VUS variants were found in 28/79 patients (35.4%) and their contribution to DSD is currently unknown, however two patients with P variants (case 15 and 33) also carry VUS and potentially can have a digenic disease (Table 2 and Figures 2A, B).
Discussion
A single study reported a diagnostic yield of 35% following WES on a large DSD cohort (18). Other studies, on smaller patient numbers using either tNGS or WES approaches, have reported a diagnostic yield of 23%-69.5% (11, 13, 19–26). The difference in yield is attributed to the makeup of patients’ cohorts included in each study. The highest diagnostic rate (64%) was found by Laino et all (11), presumably due to the inclusion of patients with CAH. In all studies a higher diagnostic yield was found for 46,XY DSD patients compared to 46,XX (13, 20, 27, 28). In our study we found that the diagnostic yield after WES was higher than previously reported (18) for either P/LP variants and VUS (43% vs 35% and 35.4% vs 15% accordingly). Pathogenic variants in the AR gene (n=11) and the NR5A1 gene (n=5) were the most common cause (47%) of DSD among patients with a genetic diagnosis (Table 1 and Figure 1B). 83.3% of all P/LP variants were novel.
Two cases with variants in DHX37 contributed to the discovery of this gene as a new cause of DSD (5), making this approach especially promising for DSD patients that do not have a genetic etiology. The majority of patients in the cohort are unexplained, but 35.4% carry one or more VUS that require careful ongoing evaluation in order to establish causality. For example, a 46,XX DSD male with a de novo synonymous variant in WT1 p.T474T (case 34), is considered a good candidate for DSD based on previous reports (29), but requires further studies to establish pathogenicity.
An important finding in this study are variants involving heterozygous WDR11 and DSD. Heterozygous variants in WDR11 are proposed as a cause of autosomal dominant CHH (30, 31), including two variants p.A435T and p.F1150L (30). Here, we identified two patients carrying the heterozygous p.A435T variant (cases 60 and 61), who presented with severe PH (46,XY female with GD and a gonadoblastoma and a 46,XY male with TRS). Both variants were inherited from their healthy mothers. A recent study identified biallelic LOF WDR11 variants in association with a complex familial phenotype of microcephaly and intellectual disability (32). There were no reproductive phenotype reported in all affected individuals as well as carriers (32). Together with our data, this suggests that heterozygous WDR11 variants are unlikely to cause CHH and questions their contribution to any reproductive phenotype. However further studies are needed to formally establish the role of WDR11 in human disorders.
For the DSD patients in whom WES identified P/LP variants (n=34), the most common variants among males (n=15) were AR variants (n=4), HH with FGFR1 and ANOS1 (n=4, including two affected cousins), an NR5A1 variant in twin boys (n=2) and AMHR2 variants in siblings (n=2). In patients who were registered as females (n=19), the most common etiology of DSD (36.8%) were AR gene variants (n=7). This differs from published data (7, 8), and may be due to the exclusion of patients with classical CAH from our study. According to the international Consensus (1), registration of female sex is recommended for 46,XX patients with CAH, or CAIS and 46,XY patients with LH receptor deficiency. Registration of male sex is recommended for 5α-reductase deficiency, since 60% of them later self-identify as males, and for 17β-HSD3 deficiency, because more than 50% of patients later self-identify as male. In our cohort all patients with 46,XY DSD and pathogenic HSD17B3 and SRD5A2 variants were raised as females. It is generally believed that in the process of the patient’ sex registration the potential quality of sexual life is a key factor, as available evidence suggests that genital anatomy is less influenced than other important factors associated with interpersonal relationships (33).
The WES diagnostic rate differed between the DSD subcategories. This was highest in patients with a clinical suspicion of Kallmann syndrome (n=100%) and with DASA (83.3%), which corresponds to previous findings (13). In contrast, the diagnostic yield was lower in GD patients (35.7%) as well as in the DSD group of undefined etiology with AG (U, AG) (22.2%), and was lowest in the TRS cohort (8.3%). VUS were the most frequent in the U, AG and TRS patients (35.7% and 25% respectively). No patient with 46,XX PH (n=5) carried with P/LP variants. The number of 46,XX DSD patients was lower than 46,XY in the cohort since CAH patients were excluded. The diagnostic yield was higher for 46,XY compared to 46,XX DSD groups (46.5% and 12.5% respectively), which is similar to that reported elsewhere (13, 20, 27, 28). Of the eight 46,XX DSD patients who underwent WES, only a single case with CYP19A1 was confirmed.
A significant number (12/34) of patients with a genetic diagnosis had atypical clinical presentations. An example is the presence of hypospadias in both siblings with PMDS (cases 17-18), where the uterus was not solidly attached to the testis in one of patients (15). This raises the question of how many cases with a simple bilateral inguinal cryptorchidism due to AMH/AMHR2 variants may be missed. GATA4 and WT1 variants were observed in the absence of neither heart or kidney anomalies respectively, inherited from their unaffected mothers. This may be due to incomplete penetrance, or oligogenic mechanisms (34). The MYRF LOF variant reported here in a child with gonadal dysgenesis and nanophthalmos is, to our knowledge, the first case with this combination of phenotypes. Other examples of atypical presentation include a heterozygous LOF TBCE variant in a syndromic form of 46,XY DSD. Pathogenic variants in TBCE are associated with neurodevelopmental syndromes, hypoparathyroidism-retardation-dysmorphism, Kenny-Caffey syndrome (35, 36) and with hypopituitarism (including GH insufficiency, hypocortisolemia and CHH) (37). However, probable PH was also described in association with extreme growth failure, dysmorphic features and hypoparathyroidism (38). In our case hypoparathyroidism was absent in the presence of PH (FSH 85.9 mIU/ml). Variants in the CYP19A1 gene cause aromatase deficiency and are considered a rare recessive disease with about 40 cases of aromatase deficiency reported (39). We report biallelic variants in CYP19A1 with an atypical presentation consisting of virilization in the child (development of clitoromegaly and the formation of the urogenital sinus), an absence of ovarian cysts, and FSH and LH levels that increased after 5 years of age. The individual harboring a GLI2 variant had a bicornuate uterus with non-communicating rudimentary horn, which has not been previously described in 46,XY DSD patients to our knowledge. An atypical clinical presentation was found in two boys with HH. Pathogenic variants in ANOS1 and FGFR1 cause Kallmann’s syndrome (40). In our cohort, four patients (cases 24-25) had a classical clinical picture of Kallmann’s syndrome, but a boy (case 23) with the ANOS1 variant p.Gln586 had increased serum testosterone levels but did not have enlargement of testis after the long HCG test with normal inhibin B and AMH levels. The prepubertal testicular volume (<4 ml), low serum inhibin B concentration and a history of cryptorchidism are described as negative predictors of stimulating treatment response (41) as we observed in case 26, but in case 23 both treatment modalities (Follitropin alfa and HCG) were ineffective where only micro-orchidism was present. This suggests that a small testicular volume by itself can be considered as unfavorable predictor, which differs from published data (42) and ‘primary’ hypogonadism in GnRH non-responders with ANOS1 variants should not be excluded (43). However, two cousins with an ANOS1 LOF variant and with a history of either untreated cryptorchidism, small testicular volume (<4 ml) but normal AMH and inhibin B levels had a good response to HCG treatment. A boy (case 26) with HH who carried an FGFR1 variant had novel extragenital features, including pulmonary hypoplasia, bronchiectasis, primary hypothyroidism, hypoparathyroidism and epilepsy. Activating variants in FGFR1 were reported in patients with osteoglophonic dysplasia and hypophosphatemia, moreover in one family all family members with FGFR1 p.Y372C died due to affected respiratory function (44). Further studies of the possible impact of FGFR1 on pulmonary and other extragenital diseases in patients with HH are needed. Also, two patients with CACNA1 and GLI2 variants presented with PH that has not been previously reported. Overall, 10/32 of patients with P/LP variants in genes known to cause DSD, had atypical clinical presentations.
Almost all DSD patients in our cohort had non-specific clinical signs (e.g., ambiguous genitalia, clitoromegaly, urogenital sinus, hypospadias, inguinal hernias, cryptorchidism, etc.), with non-specific changes in hormonal levels. WES is recommended for these patients, since predicting which gene may be involved is challenging. Determining the etiology is important to assess the risk of gonadal malignancy, since these patients are at an increased risk (1, 33). In our cohort nine 46,XY DSD individuals had a malignant gonadal tumor. Of these only two patients carried P variants (AR and CACNA1A; 22.2%). Overall, of the nine 46,XY patients, eight were female (with unambiguously female phenotype, i.e. with severe undervirilization) with the exception of three patients who presented with clitoris enlargement at different age (at the earliest at 4 years) and one with AR gene variant with location of gonads in the inguinal canals. All tumors were seminomatous and chemotherapy was required for 3/9 patients. In 8/9 patients the gonads were located in the abdominal cavity (7 females/1 male). Although the risk of malignancy is considered low in ovotestis and CAIS and before the pubertal age (1, 33), two of these patients had malignant tumors at an early age. In the entire cohort, three patients had gonadal malignant tumors before 10 years of age. This data supports previous studies where a combination of severe undervirilization and location of the gonads in the abdominal cavity are the risk factors for gonadal tumors (45, 46). However, their early age of onset in 33.3% of patients in our study is also rare finding.
Conclusions
The most common genetic causes of DSD in this study are P/LP variants in the AR and NR5A1 genes (20.2% of entire WES cohort and 47% among patients with a genetic diagnosis). Remarkably, almost 84% of all P/LP variants have not been reported elsewhere and a significant number of patients (35.3%) carrying these variants had atypical clinical presentations. This indicates that WES is the approach of choice to obtain a genetic diagnosis in these conditions, which can be difficult to define based on the clinical presentation and hormonal data. Our data also question the contribution of WDR11 variants to CHH.
Data Availability Statement
The data presented in the study are deposited in the ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) repository, accession numbers (VCV000492789, VCV000974911, VCV000658124, VCV001202584, VCV001202585, VCV001202591.1, VCV001205840.1, VCV001202603.1, VCV000935575, VCV001202587, VCV001202586, VCV001202588, VCV001202589, VCV000929443, VCV000981464, VCV000631595.5, VCV001199399.1 and VCV001199405.1).
Ethics Statement
The studies involving human participants were reviewed and approved by Local ethical committee of Ukrainian Scientific and Practical Center of Endocrine Surgery, Transplantation of Endocrine Organs and Tissues of the Ministry of Health of Ukraine, (№ 34, 26.12.2016). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
EG performed a clinical investigation of patients at the initial stage and follow-up, was responsible for conception and design of the study, data acquisition, preparation of the manuscript, finding relevant references, and final approval of the manuscript. NZ performed a clinical investigation of patients at the initial stage and follow-up; designed the analyses; reviewed and edited the manuscript. YS performed a clinical investigation of 15 patients at the initial stage. JB-T, AB, and KM performed and interpreted genetic testing; conceptualized and designed the study; and critically reviewed and revised the manuscript. KM is the guarantor and approved the final manuscript as submitted.
Funding
This work is funded in part by a research grant from the European Society of Pediatric Endocrinology, and by the Agence Nationale de la Recherche (ANR), ANR-10-LABX-73 REVIVE, ANR-17-CE14-0038-01, ANR-19-CE14-0022 and ANR-19-CE14-0012 and by the Ministry of Health of Ukraine (0117U003036).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank all Ukrainian regional pediatric endocrinologists and gynecologists for referring patients: Pogadaeva N, Enhovatova V, Horoshaya O, Shevchenko I, Behytova T, Malashonok V, Siryk N, Slepyan E, Ivanenko L, Bachinska I, Gavrilova I, Kolesnik N, and cytogeneticists Kulbalaeva S, Kurakova V.
References
1. Lee PA, Houk P, Ahmed FS, Hughes IA. Consensus Statement on Management of Intersex Disorders. Pediatrics (2006) 118(2):488–500. doi: 10.1542/peds.2006-0738
2. Kyriakou A, Lucas-Herald A, McGowan R, Tobias E, Ahmed FS. Disorders of Sex Development: Advances in Genetic Diagnosis and Challenges in Management. Adv Genomics Genet (2015) 5:165–77. doi: 10.2147/AGG.S53226
3. Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. The Androgen Receptor Gene Mutations Database (ARDB): 2012 Update. Hum Mutat (2012) 33:887–94. doi: 10.1002/humu.22046
4. Ahmed SF, Bashamboo A, Lucas-Herald A, McElreavey K. Understanding the Genetic Aetiology in Patients With XY DSD. Br Med Bull (2013) 106:67–89. doi: 10.1093/bmb/ldt008
5. McElreavey K, Jorgensen A, Eozenou C, Merel T, Bignon-Topalovic J, Tan DS, et al. Pathogenic Variants in the DEAH-Box RNA Helicase DHX37 are a Frequent Cause of 46,XY Gonadal Dysgenesis and 46,XY Testicular Regression Syndrome. Genet Med (2020) 22(1):150–9. doi: 10.1038/s41436-019-0606-y
6. Barseghyan H, Délot E, Vilain E. New Genomic Technologies: An Aid for Diagnosis of Disorders of Sex Development. Hormone Metab Res (2015) 47(05):312–20. doi: 10.1055/s-0035-1548831
7. Kalfa N, Fukami M, Philibert P, Audran F, Pienkowski C, Weill J, et al. Screening of MAMLD1 Mutations in 70 Children With 46,XY DSD: Identification and Functional Analysis of Two New Mutations. PloS One (2012) 7(3):e32505. doi: 10.1371/journal.pone.0032505
8. Ahmed SF, Cheng A, Dovey L, Hawkins JR, Martin H, Rowland J, et al. Phenotypic Features, Androgen Receptor Binding, and Mutational Analysis in 278 Clinical Cases Reported as Androgen Insensitivity Syndrome. J Clin Endocrinol Metab (2000) 85(2):658–65. doi: 10.1210/jc.85.2.658
9. White PC, Speiser PW. Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. Endocr Rev (2000) 21(3):245–91. doi: 10.1210/edrv.21.3.0398
10. Wu QY, Li N, Li WW, Li TF, Zhang C, Cui YX, et al. Clinical, Molecular and Cytogenetic Analysis of 46,XX Testicular Disorder of Sex Development With SRY-Positive. BMC Urol (2014) 14:1–5. doi: 10.1186/1471-2490-14-70
11. Laino L, Majore S, Preziosi N, Grammatico B, De Bernardo C, Scommegna S, et al. Disorders of Sex Development: A Genetic Study of Patients in a Multidisciplinary Clinic. Endocr Connect (2014) 3(4):180–92. doi: 10.1530/EC-14-0085
12. Baxter RM, Vilain E. Translational Genetics for Diagnosis of Human Disorders of Sex Development. Annu Rev Genom Hum Genet (2013) 14:371–92. doi: 10.1146/annurev-genom-091212-153417
13. Eggers S, Sadedin S, van den Bergen JA, Robevska G, Ohnesorg T, Hewitt J, et al. Disorders of Sex Development: Insights From Targeted Gene Sequencing of a Large International Patient Cohort. Genome Biol (2016) 17:243. doi: 10.1186/s13059-016-1105-y
14. Audi L, Ahmed SF, Krone N, Cools M, McElreavey K, Holterhus PM, et al. The EU COST Action. GENETICS IN ENDOCRINOLOGY: Approaches to Molecular Genetic Diagnosis in the Management of Differences/Disorders of Sex Development (DSD): Position Paper of EU COST Action BM 1303 ‘Dsdnet’. Eur J Endocrinol (2018) 179(4):R197–206. doi: 10.1530/EJE-18-0256
15. Globa E, Zelinska N, Siryk N, Bashamboo A, McElreavey K. Atypical Clinical Presentation of Persistent Müllerian Duct Syndrome in Siblings. Sex Dev (2020) 14(1-6):27–32. doi: 10.1159/000512844
16. Damaj L, Lupien-Meilleur A, Lortie A, Riou É, Ospina LH, Gagnon L, et al. CACNA1A Haploinsufficiency Causes Cognitive Impairment, Autism and Epileptic Encephalopathy With Mild Cerebellar Symptoms. Eur J Hum Genet (2015) 23(11):1505–12. doi: 10.1038/ejhg.2015.21
17. Bertolacini CDP, Ribeiro-Bicudo LA, Petrin A, Richieri-Costa A, Murray JC. Clinical Findings in Patients With GLI2 Mutations–Phenotypic Variability. Clin Genet (2012) 81:70–5. doi: 10.1111/j.1399-0004.2010.01606.x
18. Baxter RM, Arboleda VA, Lee H, Barseghyan H, Adam MP, Fechner PY, et al. Exome Sequencing for the Diagnosis of 46,XY Disorders of Sex Development. J Clin Endocrinol Metab (2015) 100(2):E333–44. doi: 10.1210/jc.2014-2605
19. Délot EC, Papp JC, Workgroup DTG, Sandberg DE, Vilain E. Genetics of Disorders of Sex Development: The DSD-TRN Experience. Endocrinol Metab Clinics North America (2017) 46(2):519–37. doi: 10.1016/j.ecl.2017.01.015
20. Xu Y, Wang Y, Li N, Yao R, Li G, Li J, et al. New Insights From Unbiased Panel and Whole-Exome Sequencing in a Large Chinese Cohort With Disorders of Sex Development. Eur J Endocrinol (2019) 181(3):311–23. doi: 10.1530/EJE-19-0111
21. Dong Y, Yi Y, Yao H, Yang Z, Hu H, Liu J, et al. Targeted Next-Generation Sequencing Identification of Mutations in Patients With Disorders of Sex Development. BMC Med Genet (2016) 17:23. doi: 10.1186/s12881-016-0286-2
22. Fan Y, Zhang X, Wang L, Wang R, Huang Z, Sun Y, et al. Diagnostic Application of Targeted Next-Generation Sequencing of 80 Genes Associated With Disorders of Sexual Development. Sci Rep (2017) 7:44536. doi: 10.1038/srep44536
23. Kim JH, Kang E, Heo SH, Kim GH, Jang JH, Cho EH, et al. Diagnostic Yield of Targeted Gene Panel Sequencing to Identify the Genetic Etiology of Disorders of Sex Development. Mol Cell Endocrinol (2017) 444:19–25. doi: 10.1016/j.mce.2017.01.037
24. Özen S, Onay H, Atik T, Solmaz AE, Özkınay F, Gökşen D, et al. Rapid Molecular Genetic Diagnosis With Next-Generation Sequencing in 46,XY Disorders of Sex Development Cases: Efficiency and Cost Assessment. Hormone Res Paediatrics (2017) 87(2):81–7. doi: 10.1159/000452995
25. Wang H, Zhang L, Wang N, Zhu H, Han B, Sun F, et al. Next-Generation Sequencing Reveals Genetic Landscape in 46, XY Disorders of Sexual Development Patients With Variable Phenotypes. Hum Genet (2018) 137(3):265–27. doi: 10.1007/s00439-018-1879-y
26. Hughes LA, McKay Bounford K, Webb E, Dasani P, Clokie S, Chandran H, et al. Next Generation Sequencing (NGS) to Improve the Diagnosis and Management of Patients With Disorders of Sex Development (DSD). Endocr Connect (2019) 8(2):100–10. doi: 10.1530/EC-18-0376
27. Buonocore F, Achermann JC. Human Sex Development: Targeted Technologies to Improve Diagnosis. Genome Biol (2016) 17:257. doi: 10.1186/s13059-016-1128-4
28. Hiort O. The Differential Role of Androgens in Early Human Sex Development. BMC Med (2013) 11:152. doi: 10.1186/1741-7015-11-152
29. Eozenou C, Gonen N, Touzon MS, Jorgensen A, Yatsenko SA, Fusee L, et al. Testis Formation in XX Individuals Resulting From Novel Pathogenic Variants in Wilms' Tumor 1 (WT1) Gene. Proc Natl Acad Sci USA (2020) 117(24):13680–8. doi: 10.1073/pnas.1921676117
30. Kim HG, Ahn JW, Kurth I, Ullmann R, Kim HT, Kulharya A, et al. WDR11 a WD Protein That Interacts With Transcription Factor EMX1, is Mutated in Idiopathic Hypogonadotropic Hypogonadism and Kallmann Syndrome. Am J Hum Genet (2010) 87(4):465–79. doi: 10.1016/j.ajhg.2010.08.018
31. Kim YJ, Osborn DP, Lee JY, Araki M, Araki K, Mohun T, et al. WDR11-Mediated Hedgehog Signalling Defects Underlie a New Ciliopathy Related to Kallmann Syndrome. EMBO Rep (2018) 19(2):269–89. doi: 10.15252/embr.201744632
32. Haag N, Tan E-C, Begemann M, Buschmann L, Kraft F, Holschbach P, et al. Biallelic Loss-of-Function Variants in WDR11 are Associated With Microcephaly and Intellectual Disability. Eur J Hum Genet (2021) 29:1663–8. doi: 10.1038/s41431-021-00943-5
33. Lee PA, Nordenstrom A, Houk CP, Ahmed SF, Auchus R, Baratz A, et al. Global Disorders of Sex Development Update Since 2006: Perceptions, Approach and Care. Horm Res Paediatr (2016) 85(3):158–80. doi: 10.1159/000442975
34. Martinez de LaPiscina I, de Mingo C, Riedl S, Rodriguez A, Pandey AV, Fernández-Cancio M, et al. GATA4 Variants in Individuals With a 46,XY Disorder of Sex Development (DSD) May or May Not Be Associated With Cardiac Defects Depending on Second Hits in Other DSD Genes. Front Endocrinol (Lausanne) (2018) 9:142. doi: 10.3389/fendo.2018.00142
35. Antonella S, Gilbert B, Teresa R. TBCE Mutations Cause Early-Onset Progressive Encephalopathy With Distal Spinal Muscular Atrophy. Am J Hum Genet (2016) 99(4):974–83. doi: 10.1016/j.ajhg.2016.08.006
36. Parvari R, Hershkovitz E, Grossman N, Gorodischer R, Loeys B, Zecic A, et al. HRD/Autosomal Recessive Kenny-Caffey Syndrome Consortium Mutation of TBCE Causes Hypoparathyroidism-Retardation-Dysmorphism and Autosomal Recessive Kenny-Caffey Syndrome. Nat Genet (2002) 32:448–52. doi: 10.1038/ng1012
37. Raja P, Dan K, Martin P, Al-Khawari M, Hindmarsh Peter C, Dattani Mehul T. Mutation in the TBCE Gene Is Associated With Hypoparathyroidism-Retardation-Dysmorphism Syndrome Featuring Pituitary Hormone Deficiencies and Hypoplasia of the Anterior Pituitary and the Corpus Callosum. J Clin Endocrinol Metab (2009) 94(8):2686–91. doi: 10.1210/jc.2008-2788
38. Soliman AT, Darwish A, AlSalmi I, Asfour M. Defective Growth Hormone Secretion and Hypogonadism in the New Syndrome of Congenital Hypoparathyroidism, Growth Failure and Dysmorphic Features. Indian J Pediatr (1996) 63(5):679–82. doi: 10.1007/BF02730821
39. Fatma D, Serdar C. A Novel Homozygous CYP19A1 Gene Mutation: Aromatase Deficiency Mimicking Congenital Adrenal Hyperplasia in an Infant Without Obvious Maternal Virilisation. J Clin Res Pediatr Endocrinol (2019) 11(2):196–201. doi: 10.4274/jcrpe.galenos.2018.2018.0140
40. del Castillo I, Cohen-Salmon M, Blanchard S, Lutfalla G, Petit C. Structure of the X-Linked Kallmann Syndrome Gene and its Homologous Pseudogene on the Y Chromosome. Nat Genet (1992) 2(4):305–10. doi: 10.1038/ng1292-305
41. Pitteloud N, Hayes FJ, Dwyer A, Boepple PA, Lee H, Crowley WF Jr. Predictors of Outcome of Long-Term GnRH Therapy in Men With Idiopathic Hypogonadotropic Hypogonadism. J Clin Endocrinol Metab (2002) 87:4128–36. doi: 10.1210/jc.2002-020518
42. Dwyer AA, Sykiotis GP, Hayes FJ, Boepple PA, Lee H, Loughlin KR, et al. Trial of Recombinant Follicle-Stimulating Hormone Pretreatment for GnRH-Induced Fertility in Patients With Congenital Hypogonadotropic Hypogonadism. J Clin Endocrinol Metab (2013) 98:E1790–1795. doi: 10.1210/jc.2013-2518
43. Lee J, Kim Y, Ataliotis P, Kim H-G, Kim D-W, Bennett DC, et al. Loss of Kallmann Syndrome-Associated Gene WDR11 Disrupts Primordial Germ Cell Development by Affecting Canonical and Non-Canonical Hedgehog Signalling. doi: 10.1101/2020.09.06.284927
44. White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, et al. Mutations That Cause Osteoglophonic Dysplasia Define Novel Roles for FGFR1 in Bone Elongation. Am J Hum Genet (2005) 76(2):361–7. doi: 10.1086/427956
45. Cools M, Wolffenbuttel KP, Drop SLS, Oosterhuis JW, Looijenga LHJ. Gonadal Development and Tumor Formation at the Crossroads of Male and Female Sex Determination. Sex Dev (2011) 5:167–80. doi: 10.1159/000329477
Keywords: 46,XY and 46,XX disorders of sex development, genes, karyotype, phenotype, whole exome sequencing (WES)
Citation: Globa E, Zelinska N, Shcherbak Y, Bignon-Topalovic J, Bashamboo A and MсElreavey K (2022) Disorders of Sex Development in a Large Ukrainian Cohort: Clinical Diversity and Genetic Findings. Front. Endocrinol. 13:810782. doi: 10.3389/fendo.2022.810782
Received: 07 November 2021; Accepted: 31 January 2022;
Published: 21 March 2022.
Edited by:
Ani Amelia Zainuddin, National University of Malaysia, MalaysiaReviewed by:
Claire Bouvattier, Université Paris-Saclay, FranceSonia R. Grover, Royal Children’s Hospital, Australia
Copyright © 2022 Globa, Zelinska, Shcherbak, Bignon-Topalovic, Bashamboo and MсElreavey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Evgenia Globa, ie.globa@i.ua; orcid.org/0000-0001-7885-8195