Advantages and Limitations of Gene Therapy and Gene Editing for Friedreich’s Ataxia
 Anusha Sivakumar
Anusha Sivakumar Stephanie Cherqui
Stephanie Cherqui- Division of Genetics, Department of Pediatrics, University of California, San Diego, San Diego, CA, United States
Friedreich’s ataxia (FRDA) is an inherited, multisystemic disorder predominantly caused by GAA hyper expansion in intron 1 of frataxin (FXN) gene. This expansion mutation transcriptionally represses FXN, a mitochondrial protein that is required for iron metabolism and mitochondrial homeostasis, leading to neurodegerative and cardiac dysfunction. Current therapeutic options for FRDA are focused on improving mitochondrial function and increasing frataxin expression through pharmacological interventions but are not effective in delaying or preventing the neurodegeneration in clinical trials. Recent research on in vivo and ex vivo gene therapy methods in FRDA animal and cell models showcase its promise as a one-time therapy for FRDA. In this review, we provide an overview on the current and emerging prospects of gene therapy for FRDA, with specific focus on advantages of CRISPR/Cas9-mediated gene editing of FXN as a viable option to restore endogenous frataxin expression. We also assess the potential of ex vivo gene editing in hematopoietic stem and progenitor cells as a potential autologous transplantation therapeutic option and discuss its advantages in tackling FRDA-specific safety aspects for clinical translation.
Introduction
Friedreich’s ataxia (FRDA) is the most common inherited human ataxia with an incidence of 1 in 50,000 individuals. FRDA is an autosomal recessive disorder resulting from deficiency of the mitochondrial protein, frataxin (FXN) (Campuzano et al., 1996; Delatycki et al., 2000). The most common mutation is the expansion of GAA trinucleotide repeats in the first intron of the gene leading to decreased FXN transcription due to heterochromatin formation and/or epigenetic modification (Gottesfeld, 2019). The protein level in patients ranges between 5 and 35% of the levels in healthy individuals (Gottesfeld et al., 2013). The age of onset and severity of the symptoms correlate with the number of repeats, which can vary between 66 and 1,700 (Dürr et al., 1996; Epplen et al., 1997). Point mutations have also been described in rare cases, but always at the heterozygote state along with a GAA expansion mutation (Campuzano et al., 1996; Li et al., 2013). Although the exact function of FXN is still unclear, it is predicted to assist in the biogenesis of mitochondrial iron-sulfur clusters (Tsai and Barondeau, 2010; González-Cabo and Palau, 2013). Thus, frataxin deficiency results in altered cellular iron metabolism, increased mitochondrial iron load, decreased mitochondrial energy production and biogenesis as well as increased oxidative stress.
FRDA is a progressively lethal multi-systemic disease. The primary pathological cause of the neuropathy is the progressive loss of large sensory neurons in the dorsal root ganglia (DRG) affecting the peripheral and central nervous systems (CNS) (Pandolfo, 2009; Koeppen and Mazurkiewicz, 2013). This progressive neurodegeneration leads to loss of motor skills and muscle degeneration, and ultimately the inability to walk, within 10–15 years of onset. Heart abnormalities cause premature death in 60–80% of the affected individuals and is usually the primary cause of death (Weidemann et al., 2013; Perdomini et al., 2014).
Currently, there are no effective treatment for FRDA. Clinical trials with antioxidants (idebenone and coenzyme Q10), iron chelators (deferipone) and epigenetic modulators (RG2833, nicotinamide) failed to prove efficacy in patients (Arpa et al., 2014; Libri et al., 2014; Soragni et al., 2014) but omaveloxolone, an NRF2 activator, improved neurological functions in Phase 2 clinical trial (Lynch et al., 2021). Gene therapy for FRDA is also under active investigation and studies of additive gene therapy using viral vectors carrying FXN cDNA have reported promising outcomes in vitro and in vivo. An adeno-associated virus (AAV)-based gene therapy product, LX2006, targeting FRDA cardiomyopathy, was recently approved for Phase 1/2 clinical trial by the United States Food & Drug Administration (FDA) following promising preclinical studies (Salami et al., 2020). An alternative approach for gene therapy is gene editing, which allows manipulating eukaryotic genomes using target-specific engineered nucleases. Gene editing has the key advantage of correcting the defective gene in situ, keeping their internal regulation system intact. Several gene editing nucleases like the zinc finger nucleases (ZFN), transcription activator-like effector (TALE) nucleases (TALENS), and CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats-associated nuclease Cas9) exist and are continuously evolving with new generation variants (Gaj et al., 2013). CRISPR/Cas9 is gaining more traction in recent years due to its specificity, efficiency in editing, and simplicity in design.
Post-mitotic cells like neurons and cardiomyocytes, the primary cell types affected by FRDA, pose significant challenges for gene therapy methods and in multisystemic disorders, systemic expression of the protein is critical for rescue of disease phenotype. In such cases, ex vivo gene therapy using hematopoietic stem and progenitor cells (HSPC) are versatile, safe and efficient delivery vehicles, and have been widely used in regenerative and cell replacement therapy including neurodegenerative disorders (Porter et al., 2011; DiGiusto et al., 2013; Drakopoulou et al., 2013; Zhang et al., 2013; Eichler et al., 2017a; Massaro et al., 2021). These cells can migrate to and differentiate into tissue-specific macrophages for delivering organelles such as mitochondria and lysosomes, proteins, ions and microRNAs (Naphade et al., 2015; Dupont et al., 2018). Thus, a single infusion of gene-corrected stem cells residing in the bone marrow niche will become a reservoir of healthy cells for lifespan of the patient. This is a potentially viable approach for FRDA for ensuring sustained systemic delivery of frataxin to the injured organs. In this review, we report on the studies and progress being made in in vivo gene therapy, gene editing and ex vivo gene therapy for FRDA with a special focus on the potential of ex vivo gene editing as a new therapeutic avenue for FRDA, and discuss its advantages in tackling FRDA-specific safety aspects for clinical translation.
Additive Gene Therapy for Friedreich’s Ataxia
Because FRDA is a monogenic disease caused by reduction of FXN expression, gene addition using viral vectors represent a promising strategy. Lentiviruses (LV), herpes simplex virus type 1 (HSV-1) and AAVs have been used as delivery vehicles of FXN for in vivo and in vitro studies of FRDA. LV expressing human FXN (hFXN) under CMV (human immediate-early cytomegalovirus) promoter and HSV-1 carrying full genomic DNA of FXN, with its endogenous promoter, enhancer elements and introns, have shown therapeutic benefits in human fibroblasts derived from FRDA patients, resulting in the partial or complete restoration of the normal cellular phenotype in response to oxidative stress (Fleming et al., 2005; Gomez-Sebastian et al., 2007). Similarly, HSV-1 vector carrying hFXN cDNA, injected into the brainstem of a conditional neuronal Fxn-knockout mice, having 20–60% reduced FXN expression in olivary neurons, led to the recovery of the motor coordination of the treated mice by restoring FXN expression to that of physiological levels (Lim et al., 2007). The promise of AAV9 serotype carrying human SMN1 in improving survival and motor functions of wheelchair-bound children with spinal muscular atrophy (SMA) (Mendell et al., 2017) spurred translation of several explorative research to clinical trials for neurodegenerative diseases with AAVs. These are single-stranded DNA viruses suitable for CNS associated diseases as they can infect both dividing and non-dividing cells with selective serotypes like the AAV9 and AAVrh10 capable of crossing the blood brain barrier (BBB), the major hurdle in gene therapy for neurodegenerative diseases (Kantor et al., 2014; Zhang et al., 2011). Both AAV9 and AAVrh10 have seen applications for FRDA, either carrying the FXN transgene or transcription activators that induce endogenous FXN expression (Table 1). Trembley and colleagues used AAV9 containing TALE gene fused with a transcription activator domain (TAD) to target the proximal promoter of FXN gene and induced its expression in vitro and in vivo. A series of studies with TALEs and TADs led to the identification of TALE-VP64, a tetrameric repeat of VP16 protein of Herpes simplex virus that acts as a strong transcription activator upon binding to the promoter sequence. Under the control of CAG promoter, TALE-VP64 enhanced FXN gene expression by 1.6–1.9-fold and mature frataxin protein expression by 1.4-fold in FRDA patient-derived fibroblasts (Tremblay et al., 2012). The same TALE-VP64, when delivered intraperitonially in the YG8R mouse model of FRDA using AAV9 vector, increased hFXN mRNA and protein expression levels in the liver, heart, and skeletal muscle (Chapdelaine et al., 2016). YG8R mice are the original transgenic humanized animal model of FRDA generated by Dr. Pook that expresses two human FXN transgenes, containing 82 and 190 GAA repeats, in a murine frataxin null background (fxn−/−FXN+) (Al-Mahdawi et al., 2006). However, despite injection of high viral copy numbers, AAV9-carrying TALE-VP64 expression in the brain was low and did not improve frataxin expression. The therapeutic effect of AAV9 vector containing hFXN transgene was also tested in the MCK-Cre and NSE-Cre mouse models of FRDA. MCK-Cre mice is a cardiac and striated muscle conditional Fxn-knockout (KO) mouse model with Cre transgene being expressed under the Muscle Creatine Kinase promoter, and the NSE-Cre is conditional Fxn-KO in neurons using the Neuron-Specific Enolase promoter, with partial KO in other organs (Puccio et al., 2001). Both these models are considered acute models of FRDA because they display early and severe onset of symptoms with a short-life expectancy of < 40 days caused by complete absence of Fxn in the targeted tissues. Intraperitoneal injection of AAV9 vector containing hFXN transgene expressed under CB (CMV enhancer/chicken beta-actin) promoter increased the life span of NSE-Cre mice by 3-fold and MCK-Cre by 2.7 fold, improved locomotion in NSE-Cre mice, and reduced cardiac hypertrophy and improved the overall cardiac function in MCK-Cre mice (Gérard et al., 2014). AAVrh10, a serotype of AAV isolated from rhesus monkey, is shown to have similar tropism to the CNS than AAV9 (Tanguy et al., 2015). When AAVrh10 containing hFXN expressed under CAG promoter was intraperitonially injected into MCK-Cre mice, it increased frataxin expression in the heart, leading to preservation of the hemodynamic parameters and cardiac output, and also complete reversal of the cardiomyopathy after disease onset (Perdomini et al., 2014). Indeed, cardiomyocytes with severe energy failure and ultrastructure disorganization could be rescued and remodeled by this gene therapy approach. Similarly, Salami et al. (2020) also demonstrate the therapeutic potential of AAVrh10-hFXN for the cardiac phenotype including improved cardiac ejection fraction and myocardial fractional shortening using a cardiac-specific partial Fxn knockout model, the αMyhc mice. These mice have 51% reduced expression of frataxin in the heart and associated cardiac pathology, as the Cre expression is driven by a cardiac specific αMyhc promoter.
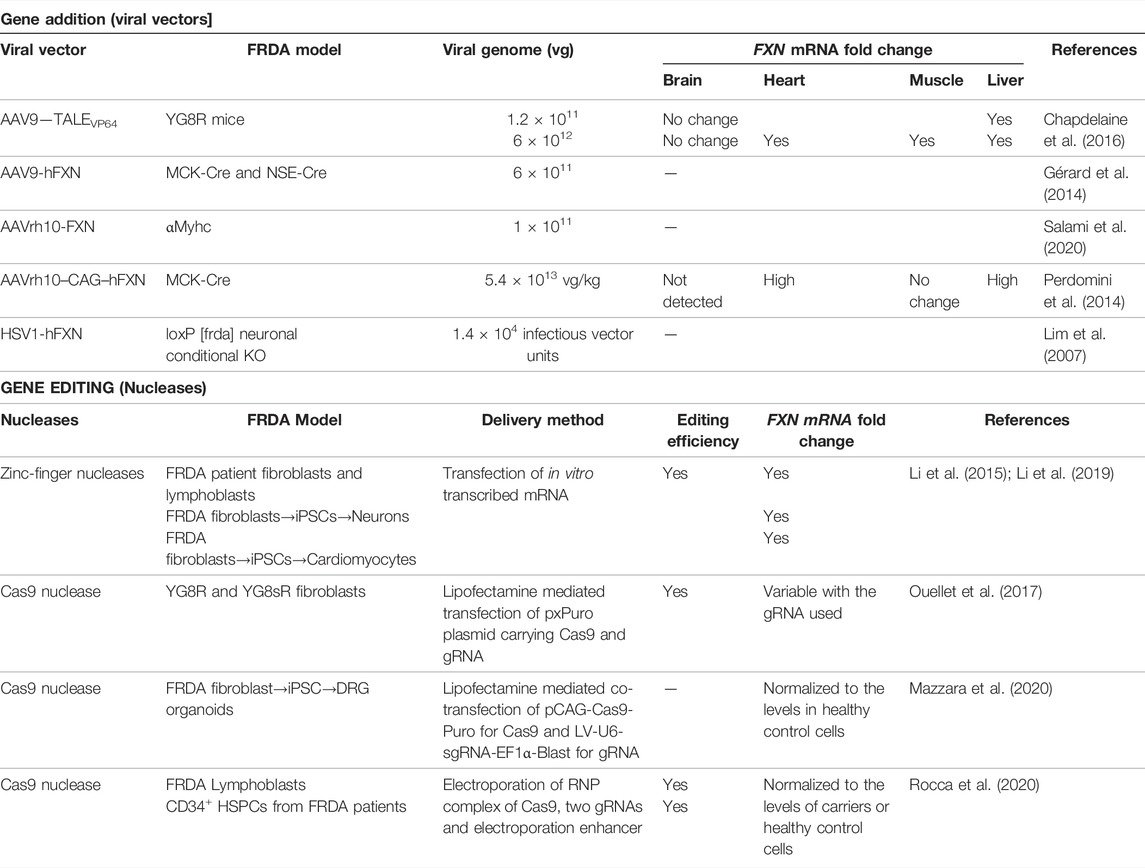
TABLE 1. Feasibility of gene therapy methods in increasing FXN expression, in vitro and in vivo.
These preclinical studies demonstrate the potential of viral vectors as delivery vehicles for frataxin transgene, leading to disease phenotype rescue, either cardiac or neurological. However, both the clinical complications must be treated in FRDA as the patients lose their motor function with progressively worsening cardiac functions. With gene additive therapy, it is also critical to ensure that frataxin expression is tightly controlled. Indeed, frataxin is expressed at a relatively low level even in healthy individuals (Campuzano et al., 1996; Campuzano et al., 1997), and while some studies showed that overexpression of frataxin was not harmful and even had positive effects (Ristow et al., 2000; Shoichet et al., 2002; Miranda et al., 2004; Schulz et al., 2010), others showed that this was deleterious (Navarro et al., 2011; Wang et al., 2014). Transgenic flies overexpressing human frataxin (hFXN) had reduced viability with neurologic and muscular defects (Navarro et al., 2011), and overexpression of frataxin homologue in a yeast model enhanced oxidative stress and iron accumulation (Wang et al., 2014). Further, AAVrh10-mediated frataxin overexpression in the MCK-Cre mice heart promoted mitochondrial ultrastructural damages, impaired respiratory complex functions, and caused myocardial fibrosis (Belbellaa et al., 2020). The study also showed overexpression of frataxin even with lower dose of AAVrh10-hFXN still led to cardiotoxicity. More recently, a Phase 1 clinical study on recombinant fusion protein delivering functional frataxin to mitochondria (CTI 1601) was put on hold as the study investigators reported mortality of non-human primates in toxicological studies in the high dose cohort (Vyas et al., 2012; LarimarTherapeutics, 2022).
In addition to the toxicity associated with frataxin overexpression, in vivo gene therapy using viral vectors inherently poses potential safety and logistic concerns: 1) localized delivery by direct viral injection to affected sites poses challenges in accessing sites such as heart, brain, and DRG, and leads only to tissue-specific rescue; 2) systemic AAV delivery remains difficult in humans due to the high levels of vector necessary, leading to vector synthesis and safety concerns including potential immune reaction. This is particularly concerning in light of the recent reports on severe adverse events and deaths in AAV-based gene therapy clinical trials. Three participants in the high dose cohort of the X-linked Myotubular Myopathy clinical trial developed liver dysfunction and sepsis, leading to their death (Morales et al., 2020; AstellasPharma, 2021). More recently, an additional participant belonging to the low dose cohort developed hepatic abnormalities and was reported dead and investigation into the cause is underway. The Phase 1b clinical trial for Duchenne muscular dystrophy clinical trial is on hold due to the death of a subject (Bryson, 2021), and severe adverse events such as low platelet counts and kidney injury were also reported on another trial (SolidBiosciences, 2019). Similarly, in the SMA clinical trial, thrombotic microangiopathy were seen in 3 infants that may be due to an immune reaction to the therapy (Chand et al., 2021). Therefore, while gene addition mediated by viral vectors holds promising therapeutic potential, it is essential to address the toxicity associated with sustained systemic frataxin expression and high dose AAV vector for future clinical application of this strategy.
Gene Editing for Friedreich’s Ataxia
Genome editing has been in the development for several decades but the advent of CRISPR/Cas9 and its ease-of-use have rendered preclinical studies and clinical translation for multisystemic diseases much more accessible. All three engineered nucleases, ZFN, TALEN, and CRISPR/Cas9 induce double strand DNA breaks (DSBs) in targeted DNA that are then repaired by the cell’s innate DNA repair mechanisms, the homology-directed repair (HDR) or non-homologous end joining (NHEJ) (Li et al., 2020). While NHEJ is error-prone and occur during any cell cycle phase, HDR is more efficient in repairing and preferentially occurs during S or G2 phase, using sister chromatid as the template. In FRDA, because the GAA expansion mutation is located in the intron 1 of the FXN gene, removal of the repeats can be done by creating DSBs in the 5′- and 3′- sites flanking the repeat region without disturbing the coding sequence and be maintained under its endogenous promoter/enhancers. In addition, mutation carriers do not display the disease phenotype, thus correcting one allele should lead to cellular phenotype correction (Coppola et al., 2011). Genome editing approaches for reactivating endogenous FXN have been studied for FRDA, both in vitro and in vivo (Table 1) and its significant advantages are schematized in Figure 1.
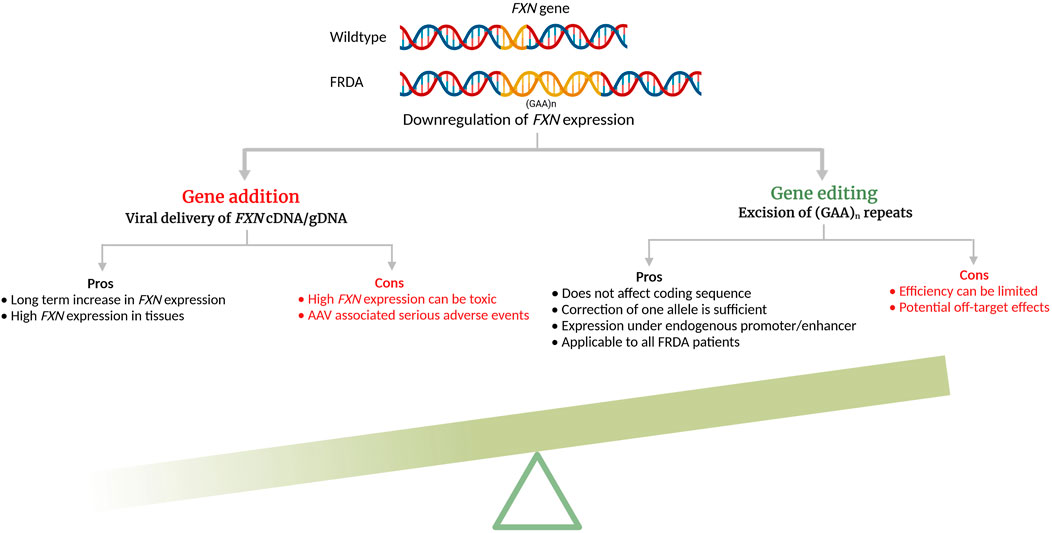
FIGURE 1. Pros and cons of additive gene therapy and gene editing for Friedreich’s ataxia (cDNA—complimentary DNA; gDNA—genomic DNA). The schematic was created with BioRender.com.
Nucleofection of ZFN mRNA flanking the GAA repeats mutation in the intron 1 of FXN led to an editing efficiency of 2.3% and 6.7% in single clones derived from FRDA lymphoblasts and fibroblasts, respectively, and increased its mRNA expression by ∼2.5–4.5 fold in both the lines (Li et al., 2015). When the edited fibroblasts were reprogrammed into induced pluripotent stem cell (iPSC) -derived neurons, they retained allele correction, and exhibited improved mitochondrial and cellular functions (Li et al., 2015). Similarly, when differentiated into iPSC-derived cardiomyocytes, FXN mRNA expression was increased ∼3-fold and genes associated with cardiac hypertrophy development were alleviated (Li et al., 2019). While clinical trials with ZFNs are still undertaken for mucopolisaccharidosis II, HIV/AIDS, transfusion dependent β-thalassemia and others (Tebas et al., 2014; First in vivo human genome, 2018; Walters et al., 2021), the design complexity and gene editing efficacy level have limited their widespread application.
The CRISPR/Cas9 system has supplanted ZFNs in terms of higher efficiency, minimal off-target effects, and a simplified three-component system, offering a modular way of editing the genome. CRISPR/Cas9 allows robust RNA-guided genome modifications in multiple eukaryotic systems (Cho et al., 2013; Cong et al., 2013; Mali et al., 2013) using a target specific CRISPR RNA (crRNA) and a universal trans-activating CRISPR RNA (tracrRNA) that form the guideRNA (gRNA) for specific cleavage by Cas9 (Karvelis et al., 2013). The tracrRNA and crRNA can be chemically fused to form a single guide RNA (sgRNA) that reduces the CRISPR/Cas9 technology to a two-component system. Delivery of AAV vector carrying both Cas9, under CMV promoter, and gRNA, under U6 Pol III promoter, in murine fibroblasts isolated from YG8R and YG8sR mice, led to editing efficiency ranging from 21.6 to 50% depending on the gRNA (Ouellet et al., 2017). YG8sR mice are derived from YG8R and carry a single hFXN transgene with 190 GAA repeats (Anjomani Virmouni et al., 2015). Similarly, LV mediated delivery of Cas9, under CAG promoter, and gRNA, under U6 promoter, were used by Mazzara et al. to show that excision of complete intron 1 as opposed to the GAA expansion only, overcame the epigenetic repression and improved in vitro survival of DRG organoids (DRGO) differentiated from FRDA patient-derived iPSC clones (Mazzara et al., 2020). These DRGOs also demonstrated improved mitochondrial biogenesis and function with enhanced axonal spreading. Taken together, these in vitro studies showed that gene editing to remove the GAA hyper expansion is an efficient strategy for FRDA leading to increased FXN mRNA and protein expression, and cellular phenotype improvement, and CRISPR/Cas9 is particularly attractive for this purpose. However, despite its higher efficiency, its use in vivo is still limited due to potential safety concern associated with vector-mediated delivery of Cas9 that would result in long-term expression of this protein, increasing the risks for off-target activity (Pattanayak et al., 2013; Merkle et al., 2015). Indeed, off-target effects are highly reliant on the specificity of the gRNAs to the target sequence and the duration of the editing system within the cells and are extensively reviewed elsewhere (Listgarten et al., 2018; Atkins et al., 2021; Vicente et al., 2021). In contrast, the use of ex vivo gene editing would have the critical benefits of having the Cas9 delivered transiently in cells that can be characterized for gene editing efficiency, potential off-target effects and other safety features, prior to transplant.
Ex Vivo Gene Editing for Friedreich’s Ataxia
Ex vivo gene therapy using hematopoietic stem and progenitor cell (HSPC) has been on the rise for treating inherited, immunological, metabolic, and neurodegenerative disorders (Massaro et al., 2021; DiGiusto et al., 2013; Drakopoulou et al., 2013; Eichler et al., 2017a; Porter et al., 2011; Zhang et al., 2013; Tucci et al., 2021). Ex vivo HSPC gene therapy has potential key advantages: 1) it avoids immune reaction during an autologous transplantation procedure (Drysdale et al., 2020), 2) it may treat all the complications by a single infusion of hematopoietic stem cells (Epah and Schäfer, 2021); 3) ex vivo gene modification of the patients’ cells will occur in a controlled environment allowing cell characterization prior to transplantation (Soni and Kohn, 2019); 4) it potentially provides once-in-a-lifetime intervention as engrafted, gene modified HSPCs will constitute a long-term reservoir of repopulating healthy cells in the bone marrow; stable transgene expression of over 15 years is reported for severe combined immunodeficiency caused by adenosine deaminase deficiency (ADA-SCID) (Cicalese et al., 2016); 5) HSPC-derived monocyte/macrophages can cross the BBB and engraft long-term in the CNS as microglia-like cells in the context of neurodegenerative disorders and after myeloablative conditioning (Capotondo et al., 2012; Peterson et al., 2019). and, 6) HSPC-derived macrophages and microglia can deliver lacking protein/enzyme to the disease cells in the injured tissues (Tan et al., 2019). Clinical trials using gene-modified autologous CD34+ HSPCs are being undertaken for genetic diseases such as X-SCID, ADA-SCID, Wiskott-Aldrich syndrome, metachromatic leukodystrophy, X-linked cerebral adrenoleukodystrophy, and mucopolysaccharidosis type I (Gentner et al., 2021; Mamcarz et al., 2019; De Ravin et al., 2016; Kohn et al., 2021; Magnani et al., 2022; Ma et al., 2021; Ferrua and Aiuti, 2017; Morris et al., 2017; Biffi et al., 2013; Eichler et al., 2017b; Fumagalli et al., 2022). Currently, our lab is conducting a phase 1/2 clinical trial for cystinosis (ClinicalTrials.gov Identifier: NCT03897361), a multisystemic lysosomal storage disorder, characterized by accumulation of cystine in all tissues and due to mutations or deletions in CTNS gene, encoding a lysosomal cystine transporter (Cherqui, 2021). Single infusion of ex vivo gene-corrected HSPCs using a self-inactivated lentiviral vector carrying CTNS cDNA in the mouse model of cystinosis led to long-term preservation of the kidney (Yeagy et al., 2011), eye (Rocca et al., 2015) and thyroid (Gaide Chevronnay et al., 2016) functions. The mechanism underlying this therapeutic effect involves the tissue engraftment of HSPCs and differentiation into macrophages, which provide “healthy lysosomes” carrying the functional cystinosin to the diseased cells via extension of tunneling nanotubes (TNTs) (Naphade et al., 2015). Because mitochondria can also be transferred through TNTs (Domhan et al., 2011; Vallabhaneni et al., 2012), we tested the impact of HSPC transplantation on FRDA using the YG8R murine model (Rocca et al., 2017). A single systemic transplantation of wildtype bone marrow HSPCs in YG8R mice prevented the neurological complications and muscle weakness in the treated mice, with functional, histological and biochemical properties comparable to WT mice as opposed to non-treated YG8R mice or treated with YG8R HSPCs. Cardiac iron deposits were also prevented in old YG8R mice. Abundant HSPCs engrafted into affected tissues and differentiated into microglia in brain and spinal cord, and macrophages in DRG, heart and muscle, and led to frataxin transfer to the diseased neurons and myocytes. Another study (Kemp et al., 2018) subsequently reported similar results where transplantation of wild type bone marrow (BM) cells to YG8R FRDA mice improved motor coordination, rescued neurobehavioral deficits and resulted in engraftment of bone marrow-derived macrophages/microglia in DRG, spinal cord and cerebellum. Altogether, these data represent the proof of concept that the different complications associated with FRDA could be treated by a HSPC transplantation. However, gene addition in HSPCs would potentially lead to the same toxicity issue associated with FXN overexpression and thus, gene editing represents a better option.
Autologous transplantation of CRISPR/Cas9–mediated gene edited CD34+ HPSCs has seen its first success in the rare diseases, β-thalassemia and sickle cell disease (SCD) (Frangoul et al., 2021a). For both diseases, targeted disruption in the specific transcription factor binding site on BCL11A erythroid enhancer leading to downregulation of BCL11A in CD34+ HSPCs, reactivated the production of fetal hemoglobin in adult-stage erythroid cells. These cells showed gene editing ranging from 9.5 to 87.0% (Wu et al., 2019). After a year, the patients had high level of fetal globin, and were transfusion-free for β-thalassemia, and with no episode of vaso-occlusion for SCD. A similar approach was undertaken by our group to remove the GAA repeats in FRDA patients’ CD34+ HSPCs using RNP mediated delivery of Cas9 protein pre-complexed with gRNA (Rocca et al., 2020). RNP complexes are well tolerated by CD34+ HSPCs [Table 2, (Cromer et al., 2018)] and addresses the clinical concerns with cloning Cas9 and gRNAs on vectors and associated viral genotoxicity. In addition, it was previously shown that transfected Cas9/RNP complex cleave chromosomal DNA almost immediately after delivery and are degraded rapidly in cells, reducing off-target effects, and increases gene editing efficiency in mouse and human HSPCs: ∼60% and ∼75%, respectively, with good cell viability: ∼80% and ∼69%, respectively (Gundry et al., 2016; Lattanzi et al., 2019). RNP complexes are also widely used in preclinical studies of CD34+ HSPC mediated therapy, some of which are listed in Table 2.

TABLE 2. Ribonucleoprotein (RNP) mediated delivery of CRISPR/Cas9 in patient derived CD34+ HSPCs.
Gene editing protocol for FRDA was first optimized in FRDA patients’ lymphoblasts and healthy HSPCs using two separate gRNA surrounding the GAA expansion region and editing efficiency ranged from 39.8 to 61.9% and 32.6–49.8%, respectively (Rocca et al., 2020). Directed removal of FXN GAA expansion was then tested in CD34+ HSPCs from FRDA patients, and we demonstrated gene editing of 12.1–55.9% with a mean of 29.66%, across multiple patients, and with excision of >1,000 repeats. Increased FXN expression was observed in the edited cells with significant correlation between the proportion of gene editing and level of FXN expression. Furthermore, xenotransplantation of genome edited CD34+ cells into non-obese diabetic (NOD) severe combined immunodeficiency (SCID) Il2rg−/− (NSG) mice demonstrated that our editing approach did not alter the in vivo hematopoietic repopulation and differentiation capacity. Altogether, these data represent the manufacturing feasibility of a gene editing strategy for CD34+ cells from FRDA patients and is schematized in Figure 2. Our previous in vivo study in YG8R mice demonstrated that 30% of wildtype donor cell engraftment was sufficient to fully rescue the FRDA phenotype, and we obtained up to 55.9% of FXN of gene editing efficiency in patients’ CD34+ HSPCs, suggesting that this approach could reach therapeutic threshold for FRDA.
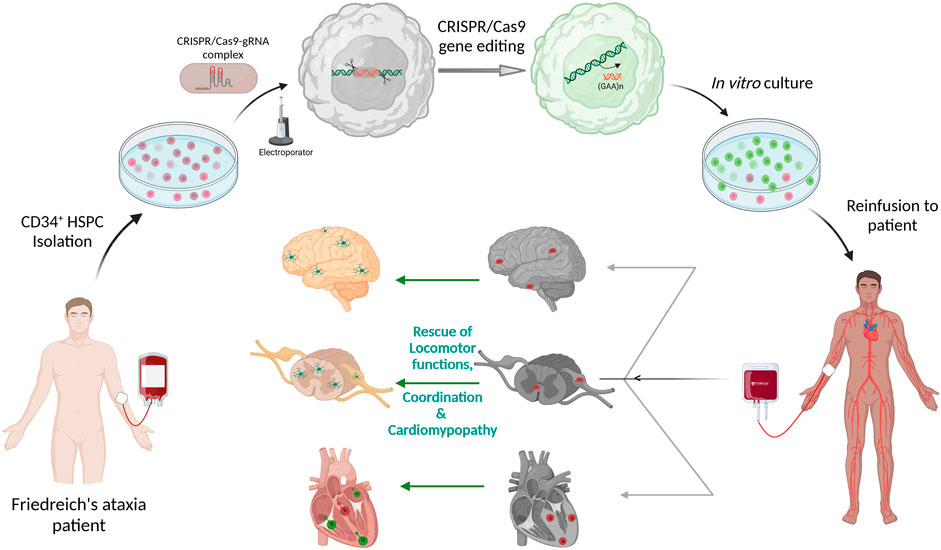
FIGURE 2. Schematic representation of Autologous transplantation of CRISPR/Cas9 gene-edited CD34+ Hematopoietic Stem and Progenitor Cells (HSPCs) for Friedreich’s ataxia. CD34+ HSPCs isolated from peripheral blood of Friedreich’s ataxia patient will be electroporated with pre-complexed CRISPR/Cas9-guideRNA ribonucleoprotein complex for excision of the (GAA)n repeat sequences in intron 1 of FXN. These gene-edited HSPCs will be put back in culture and then be reinfused to the same patient. The gene-corrected HSPCs are expected to engraft into the disease tissues such as the heart, brain, spinal cord and dorsal root ganglion to deliver frataxin to the neurons and myocytes. The schematic was created with BioRender.com.
A potential issue associated with the clinical translation of CRISPR/Cas9 edited CD34+ HSPC is the transient overexpression of p53 due to CRISPR/Cas9-induced DSBs and its downstream negative cell cycle regulator, CDKN1A/p21, leading to cell cycle arrest for damage response proteins to repair (Schiroli et al., 2019). This results in gene edited CD34+ cell proliferation delay and thus potentially decrease editing efficiency after transplantation. Despite this set back, CRISPR/Cas9 mediated gene editing has demonstrated encouraging results in current clinical trials (Frangoul et al., 2021b) and has spurred research on understanding and reducing the impact of transient p53 expression in the context of gene editing-induced DSBs. Indeed, knockdown or knockout of p53 or inhibiting its activity with small molecules partially restores cell cycle progression, and also improve gene editing (Ihry et al., 2018; Riesenberg and Maricic, 2018; Geisinger and Stearns, 2020) but long-term inhibition of p53 can increase selection of cells tolerant towards DNA damage and risk tumorigenesis, a specific concern in cells aimed for clinical translation. This had led to exploring transient silencing of p53 or reversible inhibition of the protein. As such, transfection of mRNA encoding dominant negative mutant of p53, GSE56, in human cord blood HSPCs improved cell proliferation after Cas9-mediated DSB induction, and increased HDR efficiency by 50% without affecting the self-renewal capacity of HSPCs in vivo (Ferrari et al., 2020). Thus, ensuring a delicate balance between efficiency of gene editing and controlling the DNA damage-repair mechanism, while maintaining the stem cell potency of CD34+ HSPC, is necessary to ensure a safe and efficacious ex vivo gene editing method for FRDA and other diseases.
Conclusion
Gene therapy and genome editing have transformed the treatment options for genetic disorders. Each approach comes with their own limitations and assessing the risk:benefit ratio for therapeutic intervention is highly disease-specific. Gene editing to remove the GAA expansion in the intron 1 of FXN for FRDA presents critical advantages, considering the toxicity of overexpression of the protein. Because of the systemic character of the disease, gene editing of patients’ autologous HSPCs appear like an attractive option as intelligent and widespread delivery vehicles to obtain a stable, sustained and regulated expression of frataxin in all the appropriate tissues. It also brings new perspectives to regenerative medicine, showing its applicability for multi-compartment disorders involving deficient mitochondrial function and addressing the pressing and systemic unmet medical need.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by the National Institutes of Health (NIH) R01-NS108965, and Friedreich’s ataxia Research Alliance (FARA) and FARA-Australia.
Conflict of Interest
SC is co-inventor on a patent entitled “Methods of treating lysosomal disorders” (#20378-101530) and “Methods of treating mitochondrial disorders” (#20378-101911), and is a cofounder, shareholder and a member of both the Scientific Board and board of directors of Papillon Therapeutics Inc. SC serves as a consultant for AVROBIO, Inc. and receives compensation for these services. SC also serves as a member of the Scientific Review Board and Board of Trustees of the Cystinosis Research Foundation. The terms of this arrangement have been reviewed and approved by the University of California San Diego in accordance with its conflict-of-interest policies.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Reference
Al-Mahdawi, S., Pinto, R. M., Varshney, D., Lawrence, L., Lowrie, M. B., Hughes, S., et al. (2006). GAA Repeat Expansion Mutation Mouse Models of Friedreich Ataxia Exhibit Oxidative Stress Leading to Progressive Neuronal and Cardiac Pathology. Genomics 88 (5), 580–590. doi:10.1016/j.ygeno.2006.06.015
Anjomani Virmouni, S., Ezzatizadeh, V., Sandi, C., Sandi, M., Al-Mahdawi, S., Chutake, Y., et al. (2015). A Novel GAA-Repeat-Expansion-Based Mouse Model of Friedreich's Ataxia. Dis. Model Mech. 8 (3), 225–235. doi:10.1242/dmm.018952
Arpa, J., Sanz-Gallego, I., Rodríguez-de-Rivera, F. J., Domínguez-Melcón, F. J., Prefasi, D., Oliva-Navarro, J., et al. (2014). Triple Therapy with Deferiprone, Idebenone and Riboflavin in Friedreich's Ataxia - Open-Label Trial. Acta Neurol. Scand. 129 (1), 32–40. doi:10.1111/ane.12141
AstellasPharma, Astellas Provides Update on ASPIRO Clinical Trial of AT132 in Patients with X-Linked Myotubular Myopathy. 2021.
Atkins, A., Chung, C.-H., Allen, A. G., Dampier, W., Gurrola, T. E., Sariyer, I. K., et al. (2021). Off-Target Analysis in Gene Editing and Applications for Clinical Translation of CRISPR/Cas9 in HIV-1 Therapy. Front. Genome Ed. 3, 673022. doi:10.3389/fgeed.2021.673022
Belbellaa, B., Reutenauer, L., Messaddeq, N., Monassier, L., and Puccio, H. (2020). High Levels of Frataxin Overexpression Lead to Mitochondrial and Cardiac Toxicity in Mouse Models. Mol. Ther. - Methods & Clin. Dev. 19, 120–138. doi:10.1016/j.omtm.2020.08.018
Biffi, A., Montini, E., Lorioli, L., Cesani, M., Fumagalli, F., Plati, T., et al. (2013). Lentiviral Hematopoietic Stem Cell Gene Therapy Benefits Metachromatic Leukodystrophy. Science 341 (6148), 1233158. doi:10.1126/science.1233158
Campuzano, V., Montermini, L., Lutz, Y., Cova, L., Hindelang, C., Jiralerspong, S., et al. (1997). Frataxin Is Reduced in Friedreich Ataxia Patients and Is Associated with Mitochondrial Membranes. Hum. Mol. Genet. 6 (11), 1771–1780. doi:10.1093/hmg/6.11.1771
Campuzano, V., Montermini, L., Moltò, M. D., Pianese, L., Cossée, M., Cavalcanti, F., et al. (1996). Friedreich's Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet Repeat Expansion. Science 271 (5254), 1423–1427. doi:10.1126/science.271.5254.1423
Capotondo, A., Milazzo, R., Politi, L. S., Quattrini, A., Palini, A., Plati, T., et al. (2012). Brain Conditioning Is Instrumental for Successful Microglia Reconstitution Following Hematopoietic Stem Cell Transplantation. Proc. Natl. Acad. Sci. U.S.A. 109 (37), 15018–15023. doi:10.1073/pnas.1205858109
Chand, D. H., Zaidman, C., Arya, K., Millner, R., Farrar, M. A., Mackie, F. E., et al. (2021). Thrombotic Microangiopathy Following Onasemnogene Abeparvovec for Spinal Muscular Atrophy: A Case Series. J. Pediatr. 231, 265–268. doi:10.1016/j.jpeds.2020.11.054
Chapdelaine, P., Gérard, C., Sanchez, N., Cherif, K., Rousseau, J., Ouellet, D. L., et al. (2016). Development of an AAV9 Coding for a 3XFLAG-TALEfrat#8-VP64 Able to Increase In Vivo the Human Frataxin in YG8R Mice. Gene Ther. 23 (7), 606–614. doi:10.1038/gt.2016.36
Cherqui, S. (2021). Hematopoietic Stem Cell Gene Therapy for Cystinosis: From Bench-To-Bedside. Cells 10 (12). doi:10.3390/cells10123273
Cho, S. W., Kim, S., Kim, J. M., and Kim, J.-S. (2013). Targeted Genome Engineering in Human Cells with the Cas9 RNA-Guided Endonuclease. Nat. Biotechnol. 31 (3), 230–232. doi:10.1038/nbt.2507
Cicalese, M. P., Ferrua, F., Castagnaro, L., Pajno, R., Barzaghi, F., Giannelli, S., et al. (2016). Update on the Safety and Efficacy of Retroviral Gene Therapy for Immunodeficiency Due to Adenosine Deaminase Deficiency. Blood 128 (1), 45–54. doi:10.1182/blood-2016-01-688226
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 339 (6121), 819–823. doi:10.1126/science.1231143
Coppola, G., Burnett, R., Perlman, S., Versano, R., Gao, F., Plasterer, H., et al. (2011). A Gene Expression Phenotype in Lymphocytes from Friedreich Ataxia Patients. Ann. Neurol. 70 (5), 790–804. doi:10.1002/ana.22526
Cromer, M. K., Vaidyanathan, S., Ryan, D. E., Curry, B., Lucas, A. B., Camarena, J., et al. (2018). Global Transcriptional Response to CRISPR/Cas9-AAV6-Based Genome Editing in CD34+ Hematopoietic Stem and Progenitor Cells. Mol. Ther. 26 (10), 2431–2442. doi:10.1016/j.ymthe.2018.06.002
De Ravin, S. S., Li, L., Wu, X., Choi, U., Allen, C., Koontz, S., et al. (2017). CRISPR-Cas9 Gene Repair of Hematopoietic Stem Cells from Patients with X-Linked Chronic Granulomatous Disease. Sci. Transl. Med. 9 (372). doi:10.1126/scitranslmed.aah3480
De Ravin, S. S., Wu, X., Moir, S., Anaya-O'Brien, S., Kwatemaa, N., Littel, P., et al. (2016). Erratum for the Research Article: "Lentiviral Hematopoietic Stem Cell Gene Therapy for X-Linked Severe Combined Immunodeficiency. Sci. Transl. Med. 8 (335), 341er5. doi:10.1126/scitranslmed.aag1383
Delatycki, M. B., Williamson, R., and Forrest, S. M. (2000). Friedreich Ataxia: an Overview. J. Med. Genet. 37 (1), 1–8. doi:10.1136/jmg.37.1.1
DeWitt, M. A., Magis, W., Bray, N. L., Wang, T., Berman, J. R., Urbinati, F., et al. Efficient Correction of the Sickle Mutation in Human Hematopoietic Stem Cells Using a Cas9 Ribonucleoprotein Complex. bioRxiv, 2016: p. 036236.
DiGiusto, D., Stan, R., Krishnan, A., Li, H., Rossi, J., and Zaia, J. (2013). Development of Hematopoietic Stem Cell Based Gene Therapy for HIV-1 Infection: Considerations for Proof of Concept Studies and Translation to Standard Medical Practice. Viruses 5 (11), 2898–2919. doi:10.3390/v5112898
Domhan, S., Ma, L., Tai, A., Anaya, Z., Beheshti, A., Zeier, M., et al. (2011). Intercellular Communication by Exchange of Cytoplasmic Material via Tunneling Nano-Tube like Structures in Primary Human Renal Epithelial Cells. PLoS One 6 (6), e21283. doi:10.1371/journal.pone.0021283
Drakopoulou, E., Papanikolaou, E., Georgomanoli, M., and Anagnou, N. (2013). Towards More Successful Gene Therapy Clinical Trials for β-Thalassemia. Cmm 13 (8), 1314–1330. doi:10.2174/15665240113139990064
Drysdale, C. M., Tisdale, J. F., and Uchida, N. (2020). Immunoresponse to Gene-Modified Hematopoietic Stem Cells. Mol. Ther. - Methods & Clin. Dev. 16, 42–49. doi:10.1016/j.omtm.2019.10.010
Dupont, M., Souriant, S., Lugo-Villarino, G., Maridonneau-Parini, I., and Vérollet, C. (2018). Tunneling Nanotubes: Intimate Communication between Myeloid Cells. Front. Immunol. 9, 43. doi:10.3389/fimmu.2018.00043
Dürr, A., Cossee, M., Agid, Y., Campuzano, V., Mignard, C., Penet, C., et al. (1996). Clinical and Genetic Abnormalities in Patients with Friedreich's Ataxia. N. Engl. J. Med. 335 (16), 1169–1175. doi:10.1056/nejm199610173351601
Eichler, F., Duncan, C., Musolino, P. L., Orchard, P. J., De Oliveira, S., Thrasher, A. J., et al. (2017). Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 377 (17), 1630–1638. doi:10.1056/NEJMoa1700554
Eichler, F., Duncan, C., Musolino, P. L., Orchard, P. J., De Oliveira, S., Thrasher, A. J., et al. (2017). Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 377 (17), 1630–1638. doi:10.1056/nejmoa1700554
Epah, J., and Schäfer, R. (2021). Implications of Hematopoietic Stem Cells Heterogeneity for Gene Therapies. Gene Ther. 28 (9), 528–541. doi:10.1038/s41434-021-00229-x
Epplen, C., Epplen, J. r. T., Frank, G., Miterski, B., Santos, E. J. M., and Schöls, L. (1997). Differential Stability of the (GAA) N Tract in the Friedreich Ataxia (STM7) Gene. Hum. Genet. 99 (6), 834–836. doi:10.1007/s004390050458
Ferrari, S., Jacob, A., Beretta, S., Unali, G., Albano, L., Vavassori, V., et al. (2020). Efficient Gene Editing of Human Long-Term Hematopoietic Stem Cells Validated by Clonal Tracking. Nat. Biotechnol. 38 (11), 1298–1308. doi:10.1038/s41587-020-0551-y
Ferrua, F., and Aiuti, A. (2017). Twenty-Five Years of Gene Therapy for ADA-SCID: FromBubble Babiesto an Approved Drug. Hum. Gene Ther. 28 (11), 972–981. doi:10.1089/hum.2017.175
First In Vivo Human Genome Editing Trial. Nat. Biotechnol., 2018. 36(1): p. 5.doi:10.1038/nbt0118-5b
Fleming, J., Spinoulas, A., Zheng, M., Cunningham, S. C., Ginn, S. L., McQuilty, R. C., et al. (2005). Partial Correction of Sensitivity to Oxidant Stress in Friedreich Ataxia Patient Fibroblasts by Frataxin-Encoding Adeno-Associated Virus and Lentivirus Vectors. Hum. Gene Ther. 16 (8), 947–956. doi:10.1089/hum.2005.16.947
Frangoul, H., Altshuler, D., Domenica Cappellini, M., Chen, Y-S., Domm, J., and Eustace, B. K. (2021). CRISPR-Cas9 Gene Editing for Sickle Cell Disease and Beta-Thalassemia. N. Engl. J. Med. 384 (23), e91. doi:10.1056/nejmoa2031054
Frangoul, H., Altshuler, D., Cappellini, M. D., Chen, Y.-S., Domm, J., Eustace, B. K., et al. (2021). CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 384 (3), 252–260. doi:10.1056/nejmoa2031054
Fumagalli, F., Calbi, V., Natali Sora, M. G., Sessa, M., Baldoli, C., Rancoita, P. M. V., et al. (2022). Lentiviral Haematopoietic Stem-Cell Gene Therapy for Early-Onset Metachromatic Leukodystrophy: Long-Term Results from a Non-randomised, Open-Label, Phase 1/2 Trial and Expanded Access. Lancet 399 (10322), 372–383. doi:10.1016/s0140-6736(21)02017-1
Gaide Chevronnay, H. P., Janssens, V., Van Der Smissen, P., Rocca, C. J., Liao, X. H., Refetoff, S., et al. (2016). Hematopoietic Stem Cells Transplantation Can Normalize Thyroid Function in a Cystinosis Mouse Model. Endocrinology 157 (4), 1363–1371. doi:10.1210/en.2015-1762
Gaj, T., Gersbach, C. A., and Barbas, C. F. (2013). ZFN, TALEN, and CRISPR/Cas-based Methods for Genome Engineering. Trends Biotechnol. 31 (7), 397–405. doi:10.1016/j.tibtech.2013.04.004
Geisinger, J. M., and Stearns, T. (2020). CRISPR/Cas9 Treatment Causes Extended TP53-dependent Cell Cycle Arrest in Human Cells. Nucleic Acids Res. 48 (16), 9067–9081. doi:10.1093/nar/gkaa603
Gentner, B., Tucci, F., Galimberti, S., Fumagalli, F., De Pellegrin, M., Silvani, P., et al. (2021). Hematopoietic Stem- and Progenitor-Cell Gene Therapy for Hurler Syndrome. N. Engl. J. Med. 385 (21), 1929–1940. doi:10.1056/NEJMoa2106596
Gérard, C., Xiao, X., Filali, M., Coulombe, Z., Arsenault, M., Couet, J., et al. (2014). An AAV9 Coding for Frataxin Clearly Improved the Symptoms and Prolonged the Life of Friedreich Ataxia Mouse Models. Mol. Ther. - Methods & Clin. Dev. 1, 14044. doi:10.1038/mtm.2014.44
Gomez-Ospina, N., Scharenberg, S. G., Mostrel, N., Bak, R. O., Mantri, S., Quadros, R. M., et al. (2019). Human Genome-Edited Hematopoietic Stem Cells Phenotypically Correct Mucopolysaccharidosis Type I. Nat. Commun. 10 (1), 4045. doi:10.1038/s41467-019-11962-8
Gomez-Sebastian, S., Gimenez-Cassina, A., Diaz-Nido, J., Lim, F., and Wade-Martins, R. (2007). Infectious Delivery and Expression of a 135 Kb Human FRDA Genomic DNA Locus Complements Friedreich's Ataxia Deficiency in Human Cells. Mol. Ther. 15 (2), 248–254. doi:10.1038/sj.mt.6300021
González-Cabo, P., and Palau, F. (2013). Mitochondrial Pathophysiology in Friedreich's Ataxia. J. Neurochem. 126 (Suppl. 1), 53–64. doi:10.1111/jnc.12303
Gottesfeld, J. M. (2019). Molecular Mechanisms and Therapeutics for the GAA·TTC Expansion Disease Friedreich Ataxia. Neurotherapeutics 16 (4), 1032–1049. doi:10.1007/s13311-019-00764-x
Gottesfeld, J. M., Rusche, J. R., and Pandolfo, M. (2013). Increasing Frataxin Gene Expression with Histone Deacetylase Inhibitors as a Therapeutic Approach for Friedreich's Ataxia. J. Neurochem. 126 (Suppl. 1), 147–154. doi:10.1111/jnc.12302
Gundry, M. C., Brunetti, L., Lin, A., Mayle, A. E., Kitano, A., Wagner, D., et al. (2016). Highly Efficient Genome Editing of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. Cell Rep. 17 (5), 1453–1461. doi:10.1016/j.celrep.2016.09.092
Ihry, R. J., Worringer, K. A., Salick, M. R., Frias, E., Ho, D., Theriault, K., et al. (2018). p53 Inhibits CRISPR-Cas9 Engineering in Human Pluripotent Stem Cells. Nat. Med. 24 (7), 939–946. doi:10.1038/s41591-018-0050-6
Kantor, B., Bailey, R. M., Wimberly, K., Kalburgi, S. N., and Gray, S. J. (2014). Methods for Gene Transfer to the Central Nervous System. Adv. Genet. 87, 125–197. doi:10.1016/b978-0-12-800149-3.00003-2
Karvelis, T., Gasiunas, G., Miksys, A., Barrangou, R., Horvath, P., and Siksnys, V. (2013). crRNA and tracrRNA Guide Cas9-Mediated DNA Interference inStreptococcus Thermophilus. RNA Biol. 10 (5), 841–851. doi:10.4161/rna.24203
Kemp, K. C., Hares, K., Redondo, J., Cook, A. J., Haynes, H. R., Burton, B. R., et al. (2018). Bone Marrow Transplantation Stimulates Neural Repair in Friedreich's Ataxia Mice. Ann. Neurol. 83 (4), 779–793. doi:10.1002/ana.25207
Koeppen, A. H., and Mazurkiewicz, J. E. (2013). Friedreich Ataxia: Neuropathology Revised. J. Neuropathol. Exp. Neurol. 72 (2), 78–90. doi:10.1097/nen.0b013e31827e5762
Kohn, D. B., Booth, C., Shaw, K. L., Xu-Bayford, J., Garabedian, E., Trevisan, V., et al. (2021). Autologous Ex Vivo Lentiviral Gene Therapy for Adenosine Deaminase Deficiency. N. Engl. J. Med. 384 (21), 2002–2013. doi:10.1056/NEJMoa2027675
Kuo, C. Y., Long, J. D., Campo-Fernandez, B., de Oliveira, S., Cooper, A. R., Romero, Z., et al. (2018). Site-Specific Gene Editing of Human Hematopoietic Stem Cells for X-Linked Hyper-IgM Syndrome. Cell Rep. 23 (9), 2606–2616. doi:10.1016/j.celrep.2018.04.103
Lattanzi, A., Meneghini, V., Pavani, G., Amor, F., Ramadier, S., Felix, T., et al. (2019). Optimization of CRISPR/Cas9 Delivery to Human Hematopoietic Stem and Progenitor Cells for Therapeutic Genomic Rearrangements. Mol. Ther. 27 (1), 137–150. doi:10.1016/j.ymthe.2018.10.008
Li, H., Gakh, O., Smith, D. Y., Ranatunga, W. K., and Isaya, G. (2013). Missense Mutations Linked to Friedreich Ataxia Have Different but Synergistic Effects on Mitochondrial Frataxin Isoforms. J. Biol. Chem. 288 (6), 4116–4127. doi:10.1074/jbc.m112.435263
Li, H., Yang, Y., Hong, W., Huang, M., Wu, M., and Zhao, X. (2020). Applications of Genome Editing Technology in the Targeted Therapy of Human Diseases: Mechanisms, Advances and Prospects. Sig Transduct. Target Ther. 5 (1), 1. doi:10.1038/s41392-019-0089-y
Li, J., Rozwadowska, N., Clark, A., Fil, D., Napierala, J. S., and Napierala, M. (2019). Excision of the Expanded GAA Repeats Corrects Cardiomyopathy Phenotypes of iPSC-Derived Friedreich's Ataxia Cardiomyocytes. Stem Cell Res. 40, 101529. doi:10.1016/j.scr.2019.101529
Li, Y., Polak, U., Bhalla, A. D., Rozwadowska, N., Butler, J. S., Lynch, D. R., et al. (2015). Excision of Expanded GAA Repeats Alleviates the Molecular Phenotype of Friedreich's Ataxia. Mol. Ther. 23 (6), 1055–1065. doi:10.1038/mt.2015.41
Libri, V., Yandim, C., Athanasopoulos, S., Loyse, N., Natisvili, T., Law, P. P., et al. (2014). Epigenetic and Neurological Effects and Safety of High-Dose Nicotinamide in Patients with Friedreich's Ataxia: an Exploratory, Open-Label, Dose-Escalation Study. Lancet 384 (9942), 504–513. doi:10.1016/s0140-6736(14)60382-2
Lim, F., Palomo, G. M., Mauritz, C., Giménez-Cassina, A., Illana, B., Wandosell, F., et al. (2007). Functional Recovery in a Friedreich's Ataxia Mouse Model by Frataxin Gene Transfer Using an HSV-1 Amplicon Vector. Mol. Ther. 15 (6), 1072–1078. doi:10.1038/sj.mt.6300143
Listgarten, J., Weinstein, M., Kleinstiver, B. P., Sousa, A. A., Joung, J. K., Crawford, J., et al. (2018). Prediction of Off-Target Activities for the End-To-End Design of CRISPR Guide RNAs. Nat. Biomed. Eng. 2 (1), 38–47. doi:10.1038/s41551-017-0178-6
Lynch, D. R., Chin, M. P., Delatycki, M. B., Subramony, S. H., Corti, M., Hoyle, J. C., et al. (2021). Safety and Efficacy of Omaveloxolone in Friedreich Ataxia ( MOXIe Study). Ann. Neurology 89 (2), 212–225. doi:10.1002/ana.25934
Ma, C. Y., Li, C., Zhou, X., Zhang, Z., Jiang, H., Liu, H., et al. (2021). Management of Adrenoleukodystrophy: From Pre-clinical Studies to the Development of New Therapies. Biomed. Pharmacother. 143, 112214. doi:10.1016/j.biopha.2021.112214
Magnani, A., Semeraro, M., Adam, F., Booth, C., Dupré, L., Morris, E. C., et al. (2022). Long-term Safety and Efficacy of Lentiviral Hematopoietic Stem/progenitor Cell Gene Therapy for Wiskott-Aldrich Syndrome. Nat. Med. 28 (1), 71–80. doi:10.1038/s41591-021-01641-x
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., et al. (2013). RNA-guided Human Genome Engineering via Cas9. Science 339 (6121), 823–826. doi:10.1126/science.1232033
Mamcarz, E., Zhou, S., Lockey, T., Abdelsamed, H., Cross, S. J., Kang, G., et al. (2019). Lentiviral Gene Therapy Combined with Low-Dose Busulfan in Infants with SCID-X1. N. Engl. J. Med. 380 (16), 1525–1534. doi:10.1056/nejmoa1815408
Massaro, G., Geard, A. F., Liu, W., Coombe-Tennant, O., Waddington, S. N., Baruteau, J., et al. (2021). Gene Therapy for Lysosomal Storage Disorders: Ongoing Studies and Clinical Development. Biomolecules 11 (4). doi:10.3390/biom11040611
Mazzara, P. G., Muggeo, S., Luoni, M., Massimino, L., Zaghi, M., Valverde, P. T.-T., et al. (2020). Frataxin Gene Editing Rescues Friedreich's Ataxia Pathology in Dorsal Root Ganglia Organoid-Derived Sensory Neurons. Nat. Commun. 11 (1), 4178. doi:10.1038/s41467-020-17954-3
Mendell, J. R., Al-Zaidy, S., Shell, R., Arnold, W. D., Rodino-Klapac, L. R., Prior, T. W., et al. (2017). Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 377 (18), 1713–1722. doi:10.1056/nejmoa1706198
Merkle, F. T., Neuhausser, W. M., Santos, D., Valen, E., Gagnon, J. A., Maas, K., et al. (2015). Efficient CRISPR-Cas9-Mediated Generation of Knockin Human Pluripotent Stem Cells Lacking Undesired Mutations at the Targeted Locus. Cell Rep. 11 (6), 875–883. doi:10.1016/j.celrep.2015.04.007
Miranda, C. J., Santos, M. M., Ohshima, K., Tessaro, M., Sequeiros, J., and Pandolfo, M. (2004). Frataxin Overexpressing Mice. FEBS Lett. 572 (1-3), 281–288. doi:10.1016/j.febslet.2004.07.022
Morales, L., Gambhir, Y., Bennett, J., and Stedman, H. H. (2020). Broader Implications of Progressive Liver Dysfunction and Lethal Sepsis in Two Boys Following Systemic High-Dose AAV. Mol. Ther. 28 (8), 1753–1755. doi:10.1016/j.ymthe.2020.07.009
Morris, E. C., Fox, T., Chakraverty, R., Tendeiro, R., Snell, K., Rivat, C., et al. (2017). Gene Therapy for Wiskott-Aldrich Syndrome in a Severely Affected Adult. Blood 130 (11), 1327–1335. doi:10.1182/blood-2017-04-777136
Naphade, S., Sharma, J., Gaide Chevronnay, H. P., Shook, M. A., Yeagy, B. A., Rocca, C. J., et al. (2015). Brief Reports: Lysosomal Cross-Correction by Hematopoietic Stem Cell-Derived Macrophages via Tunneling Nanotubes. Stem Cells 33 (1), 301–309. doi:10.1002/stem.1835
Navarro, J. A., Llorens, J. V., Soriano, S., Botella, J. A., Schneuwly, S., Martínez-Sebastián, M. J., et al. (2011). Overexpression of Human and Fly Frataxins in Drosophila Provokes Deleterious Effects at Biochemical, Physiological and Developmental Levels. PLoS One 6 (7), e21017. doi:10.1371/journal.pone.0021017
Ouellet, D. L., Cherif, K., Rousseau, J., and Tremblay, J. P. (2017). Deletion of the GAA Repeats from the Human Frataxin Gene Using the CRISPR-Cas9 System in YG8R-Derived Cells and Mouse Models of Friedreich Ataxia. Gene Ther. 24 (5), 265–274. doi:10.1038/gt.2016.89
Pandolfo, M. (2009). Friedreich Ataxia: the Clinical Picture. J. Neurol. 256 Suppl 1 (Suppl. 1), 3–8. doi:10.1007/s00415-009-1002-3
Park, S. H., Lee, C. M., Dever, D. P., Davis, T. H., Camarena, J., Srifa, W., et al. (2019). Highly Efficient Editing of the β-globin Gene in Patient-Derived Hematopoietic Stem and Progenitor Cells to Treat Sickle Cell Disease. Nucleic Acids Res. 47 (15), 7955–7972. doi:10.1093/nar/gkz475
Pattanayak, V., Lin, S., Guilinger, J. P., Ma, E., Doudna, J. A., and Liu, D. R. (2013). High-throughput Profiling of Off-Target DNA Cleavage Reveals RNA-Programmed Cas9 Nuclease Specificity. Nat. Biotechnol. 31 (9), 839–843. doi:10.1038/nbt.2673
Perdomini, M., Belbellaa, B., Monassier, L., Reutenauer, L., Messaddeq, N., Cartier, N., et al. (2014). Prevention and Reversal of Severe Mitochondrial Cardiomyopathy by Gene Therapy in a Mouse Model of Friedreich's Ataxia. Nat. Med. 20 (5), 542–547. doi:10.1038/nm.3510
Peterson, C. W., Adair, J. E., Wohlfahrt, M. E., Deleage, C., Radtke, S., Rust, B., et al. (2019). Autologous, Gene-Modified Hematopoietic Stem and Progenitor Cells Repopulate the Central Nervous System with Distinct Clonal Variants. Stem Cell Rep. 13 (1), 91–104. doi:10.1016/j.stemcr.2019.05.016
Porter, D. L., Levine, B. L., Kalos, M., Bagg, A., and June, C. H. (2011). Chimeric Antigen Receptor-Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 365 (8), 725–733. doi:10.1056/nejmoa1103849
Puccio, H., Simon, D., Cossée, M., Criqui-Filipe, P., Tiziano, F., Melki, J., et al. (2001). Mouse Models for Friedreich Ataxia Exhibit Cardiomyopathy, Sensory Nerve Defect and Fe-S Enzyme Deficiency Followed by Intramitochondrial Iron Deposits. Nat. Genet. 27 (2), 181–186. doi:10.1038/84818
Rai, R., Romito, M., Rivers, E., Turchiano, G., Blattner, G., Vetharoy, W., et al. (2020). Targeted Gene Correction of Human Hematopoietic Stem Cells for the Treatment of Wiskott - Aldrich Syndrome. Nat. Commun. 11 (1), 4034. doi:10.1038/s41467-020-17626-2
Riesenberg, S., and Maricic, T. (2018). Targeting Repair Pathways with Small Molecules Increases Precise Genome Editing in Pluripotent Stem Cells. Nat. Commun. 9 (1), 2164. doi:10.1038/s41467-018-04609-7
Ristow, M., Pfister, M. F., Yee, A. J., Schubert, M., Michael, L., Zhang, C.-Y., et al. (2000). Frataxin Activates Mitochondrial Energy Conversion and Oxidative Phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 97 (22), 12239–12243. doi:10.1073/pnas.220403797
Rocca, C. J., Goodman, S. M., Dulin, J. N., Haquang, J. H., Gertsman, I., Blondelle, J., et al. (2017). Transplantation of Wild-type Mouse Hematopoietic Stem and Progenitor Cells Ameliorates Deficits in a Mouse Model of Friedreich's Ataxia. Sci. Transl. Med. 9 (413). doi:10.1126/scitranslmed.aaj2347
Rocca, C. J., Kreymerman, A., Ur, S. N., Frizzi, K. E., Naphade, S., Lau, A., et al. (2015). Treatment of Inherited Eye Defects by Systemic Hematopoietic Stem Cell Transplantation. Invest. Ophthalmol. Vis. Sci. 56 (12), 7214–7223. doi:10.1167/iovs.15-17107
Rocca, C. J., Rainaldi, J. N., Sharma, J., Shi, Y., Haquang, J. H., Luebeck, J., et al. (2020). CRISPR-Cas9 Gene Editing of Hematopoietic Stem Cells from Patients with Friedreich's Ataxia. Mol. Ther. - Methods & Clin. Dev. 17, 1026–1036. doi:10.1016/j.omtm.2020.04.018
Román-Rodríguez, F. J., Ugalde, L., Álvarez, L., Díez, B., Ramírez, M. J., Risueño, C., et al. (2019). NHEJ-mediated Repair of CRISPR-Cas9-Induced DNA Breaks Efficiently Corrects Mutations in HSPCs from Patients with Fanconi Anemia. Cell Stem Cell 25 (5), 607–621.e7. doi:10.1016/j.stem.2019.08.016
Salami, C. O., Jackson, K., Jose, C., Alyass, L., Cisse, G. I., De, B. P., et al. (2020). Stress-Induced Mouse Model of the Cardiac Manifestations of Friedreich's Ataxia Corrected by AAV-Mediated Gene Therapy. Hum. Gene Ther. 31 (15-16), 819–827. doi:10.1089/hum.2019.363
Schiroli, G., Conti, A., Ferrari, S., della Volpe, L., Jacob, A., Albano, L., et al. (2019). Precise Gene Editing Preserves Hematopoietic Stem Cell Function Following Transient P53-Mediated DNA Damage Response. Cell Stem Cell 24 (4), 551–565.e8. doi:10.1016/j.stem.2019.02.019
Schulz, T. J., Westermann, D., Isken, F., Voigt, A., Laube, B., Thierbach, R., et al. (2010). Activation of Mitochondrial Energy Metabolism Protects against Cardiac Failure. Aging 2 (11), 843–853. doi:10.18632/aging.100234
Shoichet, S. A., Bäumer, A. T., Stamenkovic, D., Sauer, H., Pfeiffer, A. F. H., Ronald Kahn, C., et al. (2002). Frataxin Promotes Antioxidant Defense in a Thiol-dependent Manner Resulting in Diminished Malignant Transformation In Vitro. Hum. Mol. Genet. 11 (7), 815–821. doi:10.1093/hmg/11.7.815
Soni, S., and Kohn, D. B. (2019). Chemistry, Manufacturing and Controls for Gene Modified Hematopoietic Stem Cells. Cytotherapy 21 (3), 358–366. doi:10.1016/j.jcyt.2018.12.001
Soragni, E., Miao, W., Iudicello, M., Jacoby, D., De Mercanti, S., Clerico, M., et al. (2014). Epigenetic Therapy for Friedreich Ataxia. Ann. Neurol. 76 (4), 489–508. doi:10.1002/ana.24260
Tan, E. Y., Boelens, J. J., Jones, S. A., and Wynn, R. F. (2019). Hematopoietic Stem Cell Transplantation in Inborn Errors of Metabolism. Front. Pediatr. 7, 433. doi:10.3389/fped.2019.00433
Tanguy, Y., Biferi, M. G., Besse, A., Astord, S., Cohen-Tannoudji, M., Marais, T., et al. (2015). Systemic AAVrh10 Provides Higher Transgene Expression Than AAV9 in the Brain and the Spinal Cord of Neonatal Mice. Front. Mol. Neurosci. 8, 36. doi:10.3389/fnmol.2015.00036
Tebas, P., Stein, D., Tang, W. W., Frank, I., Wang, S. Q., Lee, G., et al. (2014). Gene Editing ofCCR5in Autologous CD4 T Cells of Persons Infected with HIV. N. Engl. J. Med. 370 (10), 901–910. doi:10.1056/nejmoa1300662
Tran, N. T., Graf, R., Wulf-Goldenberg, A., Stecklum, M., Strauß, G., Kühn, R., et al. (2020). CRISPR-Cas9-Mediated ELANE Mutation Correction in Hematopoietic Stem and Progenitor Cells to Treat Severe Congenital Neutropenia. Mol. Ther. 28 (12), 2621–2634. doi:10.1016/j.ymthe.2020.08.004
Tremblay, J. P., Chapdelaine, P., Coulombe, Z., and Rousseau, J. (2012). Transcription Activator-like Effector Proteins Induce the Expression of the Frataxin Gene. Hum. Gene Ther. 23 (8), 883–890. doi:10.1089/hum.2012.034
Tsai, C.-L., and Barondeau, D. P. (2010). Human Frataxin Is an Allosteric Switch that Activates the Fe−S Cluster Biosynthetic Complex. Biochemistry 49 (43), 9132–9139. doi:10.1021/bi1013062
Tucci, F., Scaramuzza, S., Aiuti, A., and Mortellaro, A. (2021). Update on Clinical Ex Vivo Hematopoietic Stem Cell Gene Therapy for Inherited Monogenic Diseases. Mol. Ther. 29 (2), 489–504. doi:10.1016/j.ymthe.2020.11.020
Vallabhaneni, K. C., Haller, H., and Dumler, I. (2012). Vascular Smooth Muscle Cells Initiate Proliferation of Mesenchymal Stem Cells by Mitochondrial Transfer via Tunneling Nanotubes. Stem Cells Dev. 21 (17), 3104–3113. doi:10.1089/scd.2011.0691
Vicente, M. M., Chaves-Ferreira, M., Jorge, J. M. P., Proença, J. T., and Barreto, V. M. (2021). The Off-Targets of Clustered Regularly Interspaced Short Palindromic Repeats Gene Editing. Front. Cell Dev. Biol. 9, 718466. doi:10.3389/fcell.2021.718466
Vyas, P. M., Tomamichel, W. J., Pride, P. M., Babbey, C. M., Wang, Q., Mercier, J., et al. (2012). A TAT-Frataxin Fusion Protein Increases Lifespan and Cardiac Function in a Conditional Friedreich's Ataxia Mouse Model. Hum. Mol. Genet. 21 (6), 1230–1247. doi:10.1093/hmg/ddr554
Walters, M. C., Smith, A. R., Schiller, G. J., Esrick, E. B., Williams, D. A., Gogoleva, T., et al. (2021). Updated Results of a Phase 1/2 Clinical Study of Zinc Finger Nuclease-Mediated Editing of BCL11A in Autologous Hematopoietic Stem Cells for Transfusion-dependent Beta Thalassemia. Blood 138 (Suppl. 1), 3974. doi:10.1182/blood-2021-147907
Wang, Y., Wang, Y., Marcus, S., and Busenlehner, L. S. (2014). The Role of Frataxin in Fission Yeast Iron Metabolism: Implications for Friedreich's Ataxia. Biochimica Biophysica Acta (BBA) - General Subj. 1840 (10), 3022–3033. doi:10.1016/j.bbagen.2014.06.017
Weidemann, F., Störk, S., Liu, D., Hu, K., Herrmann, S., Ertl, G., et al. (2013). Cardiomyopathy of Friedreich Ataxia. J. Neurochem. 126 (Suppl. 1), 88–93. doi:10.1111/jnc.12217
Wu, Y., Zeng, J., Roscoe, B. P., Liu, P., Yao, Q., Lazzarotto, C. R., et al. (2019). Highly Efficient Therapeutic Gene Editing of Human Hematopoietic Stem Cells. Nat. Med. 25 (5), 776–783. doi:10.1038/s41591-019-0401-y
Yeagy, B. A., Harrison, F., Gubler, M.-C., Koziol, J. A., Salomon, D. R., and Cherqui, S. (2011). Kidney Preservation by Bone Marrow Cell Transplantation in Hereditary Nephropathy. Kidney Int. 79 (11), 1198–1206. doi:10.1038/ki.2010.537
Zhang, H., Yang, B., Mu, X., Ahmed, S. S., Su, Q., He, R., et al. (2011). Several rAAV Vectors Efficiently Cross the Blood-Brain Barrier and Transduce Neurons and Astrocytes in the Neonatal Mouse Central Nervous System. Mol. Ther. 19 (8), 1440–1448. doi:10.1038/mt.2011.98
Keywords: Friedreich’s ataxia, CRISPR/Cas9 gene editing, AAV, gene therapy, hematopoietic stem and progenitor cells, gene editing
Citation: Sivakumar A and Cherqui S (2022) Advantages and Limitations of Gene Therapy and Gene Editing for Friedreich’s Ataxia. Front. Genome Ed. 4:903139. doi: 10.3389/fgeed.2022.903139
Received: 23 March 2022; Accepted: 21 April 2022;
Published: 17 May 2022.
Edited by:
Guilherme Baldo, Federal University of Rio Grande do Sul, BrazilCopyright © 2022 Sivakumar and Cherqui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephanie Cherqui, scherqui@ucsd.edu