Corrigendum: Analysis of Pathogenic Pseudoexons Reveals Novel Mechanisms Driving Cryptic Splicing
 Niall P. Keegan
Niall P. Keegan Steve D. Wilton
Steve D. Wilton Sue Fletcher
Sue Fletcher- 1Centre for Molecular Medicine and Innovative Therapeutics, Health Futures Institute, Murdoch University, Perth, WA, Australia
- 2Centre for Neuromuscular and Neurological Disorders, Perron Institute for Neurological and Translational Science, The University of Western Australia, Perth, WA, Australia
Understanding pre-mRNA splicing is crucial to accurately diagnosing and treating genetic diseases. However, mutations that alter splicing can exert highly diverse effects. Of all the known types of splicing mutations, perhaps the rarest and most difficult to predict are those that activate pseudoexons, sometimes also called cryptic exons. Unlike other splicing mutations that either destroy or redirect existing splice events, pseudoexon mutations appear to create entirely new exons within introns. Since exon definition in vertebrates requires coordinated arrangements of numerous RNA motifs, one might expect that pseudoexons would only arise when rearrangements of intronic DNA create novel exons by chance. Surprisingly, although such mutations do occur, a far more common cause of pseudoexons is deep-intronic single nucleotide variants, raising the question of why these latent exon-like tracts near the mutation sites have not already been purged from the genome by the evolutionary advantage of more efficient splicing. Possible answers may lie in deep intronic splicing processes such as recursive splicing or poison exon splicing. Because these processes utilize intronic motifs that benignly engage with the spliceosome, the regions involved may be more susceptible to exonization than other intronic regions would be. We speculated that a comprehensive study of reported pseudoexons might detect alignments with known deep intronic splice sites and could also permit the characterisation of novel pseudoexon categories. In this report, we present and analyse a catalogue of over 400 published pseudoexon splice events. In addition to confirming prior observations of the most common pseudoexon mutation types, the size of this catalogue also enabled us to suggest new categories for some of the rarer types of pseudoexon mutation. By comparing our catalogue against published datasets of non-canonical splice events, we also found that 15.7% of pseudoexons exhibit some splicing activity at one or both of their splice sites in non-mutant cells. Importantly, this included seven examples of experimentally confirmed recursive splice sites, confirming for the first time a long-suspected link between these two splicing phenomena. These findings have the potential to improve the fidelity of genetic diagnostics and reveal new targets for splice-modulating therapies.
1 Introduction
According to the current release of Ensembl (104.38), the 3.1 gigabases of the human genome are estimated to contain over 44,000 genes, just over 20,000 of which encode proteins (Howe et al., 2021). Based on statistics provided by Piovesan et al. (2019), protein-coding genes account for 41% of the total human genome, although exons, the transcribed segments that are retained in the mature mRNA, comprise only 4.65% of this fraction, or 1.91% of the total genome.
During and after transcription pre-mRNAs undergo splicing, a process whereby introns are excised from the pre-mRNA molecule and the ends of the flanking exons are ligated together by a multi-molecular assembly called the spliceosome. This splicing process needs to be both consistent and accurate, since an error of even a single nucleotide could render an entire transcript functionless or cause it to encode a toxic product. However, this does not mean that splicing must be perfect. Occasional splicing errors are inevitable even in healthy cells (Alexieva et al., 2021), and these aberrant transcripts are generally well-managed by error-detecting systems such as nonsense-mediated decay (NMD) (Hug et al., 2016)—but never without some energy cost to the cell.
There is, therefore, an ancient and relentless evolutionary pressure on all eukaryotes to splice pre-mRNAs as efficiently as possible. This raises the question of why evolution allows introns to persist in the first place. Much effort has been devoted to investigating this question, and there appears to be no single answer (Chorev and Carmel, 2012; Jo and Choi, 2015). Stated briefly, introns contain numerous regulatory elements that enable alternative splicing and fine control of gene expression, and the presence of these elements may have indirectly modulated the efficiency of natural selection for eukaryotic life on Earth. Recent research also reveals a regulatory role for conserved exon-like sequence elements within some introns, such as poison exons, decoy exons and recursive splice sites (Conboy, 2021). However, this does not imply that every nucleotide of every intron is of equal importance as a nucleotide of coding sequence. Pathogenic mutations are discovered within known coding regions at a rate of at least 25% (Sawyer et al., 2016), much higher than the exonic proportion of genes (4.65%), indicating that mutations in introns are generally better tolerated.
Pathogenic mutations within introns are frequently found to affect splicing. Splicing mutations account for about 9% of all identified pathogenic mutations, though this figure also includes exonic mutations with splicing impact (Stenson et al., 2017). Most pathogenic splicing mutations weaken the definition of conserved branch point, acceptor site or donor site motifs of canonical exons, resulting in skipping of whole or partial exons, or inclusion of partial or whole introns (Abramowicz and Gos 2018). In some cases, however, a mutation will cause part of an intron to be erroneously spliced into mature transcripts as if it were an exon. These inclusions, when they occur, are called pseudoexons (PEs) or cryptic exons.
Mutations that cause pathogenic PEs in germline cells could accurately be described as ‘rare but ubiquitous.’ They are rare in that they appear to account for very few unique splicing events, yet they are also ubiquitous in the sense that they can potentially arise in any gene with at least one intron, and do not exhibit any noticeable bias towards particular genes or cell types. Consequently, any new insights into how and why pathogenic PEs arise may have implications for numerous genetic diseases.
Surprisingly, there have been very few focused studies on the general characteristics of pseudoexons. Královičová and Vořechovský (2007) examined the exonic splice enhancer and silencer (ESE and ESS) content of pathogenic PEs and found that they were intermediate between introns and canonical exons. A 2010 study by Vořechovský investigated the correlation of PEs with transposable elements and found that MIRs (Mammalian-wide interspersed repeats) and antisense Alu elements were statistically overrepresented in pseudoexon sequences, which they attributed to exon-like characteristics that these elements naturally possess. Dhir and Buratti (2010) directly examined the mutations that activate PEs and proposed that they could be divided into five distinct categories: splice site or branch point creation, ESE gain or ESS loss, internal deletion, intragenic inversion, and loss of a flanking canonical splice site. Subsequent observations by Romano et al. (2013) of PEs in cancer-associated genes implicitly supported Dhir and Buratti’s categories, although Romano et al., additionally noted the possibility of PEs arising through other, as-yet uncharacterised mechanisms. More recently, Vaz-Drago et al. (2017) collated additional examples of PEs fitting Dhir and Buratti’s first two categories and observed a distribution of PE sizes similar to that of internal canonical exons. Lastly, our own review of pseudoexons in the DMD gene (Keegan 2020) examined the coincidence of PEs with reported recursive splicing, and suggested mutation-driven exonization of recursive splice sites as an explanatory mechanism for some PEs.
Although this handful of reviews provided many useful insights into the mechanisms of PE pathogenesis, all of them (including our own) were limited by their small sample sizes. For the older reviews in particular, this impediment was largely attributable to the scarcity of pseudoexon reports available at the time they were written, although a lack of clarity in how some primary reports presented their pseudoexon data may also have contributed. In recent years, the rate of reports of new pseudoexons has continued to accelerate as new technologies make RNA analysis faster, cheaper and more accurate. Therefore, it is timely to undertake a new and comprehensive analysis of pseudoexons and their instigating mutations.
In this report, we present a catalogue of 413 germline pseudoexon variants, which were as many as we could find through a thorough search of the literature. To our knowledge, this is the first time a PE dataset of this size has been assembled. Our analysis of this data discovered that 15.7% of PEs exhibit splicing activity at one or both of their splice sites in non-mutant cells, suggesting that many reported PEs might be more accurately reclassified as mutant variants of intronic splice regulating elements within introns, such as poison exons or decoy exons. Importantly, these shared intronic splice sites include seven empirically-verified recursive splice sites, confirming for the first time a long-suspected link between these two splicing phenomena.
Additionally, while our examination of the mutations that cause PEs largely supported the observations of Dhir and Buratti (2010), the expanded sample size of our catalogue allowed us to suggest refinements and additions to their original five categories.
2 Materials and Methods
Although PEs have been observed in a highly diverse range of genes and cell types, they remain a relatively rare splicing phenomenon, and it was therefore unavoidable that any analysis of their characteristics would require some degree of compromise between specificity and sample size. In this section we will outline the criteria we adopted for determining what data to include in our analyses, what data to exclude, and why.
2.1 Working Definition of “Pseudoexon”
Our intention for this report was not to analyse all forms of cryptic splicing, but specifically those instances where splicing of non-canonical exons in a gene increased as the result of pathogenic mutations in that gene.
In a previous report on DMD gene PEs (Keegan, 2020) we suggested the following definition for PEs:
“[A pseudoexon is] any continuous tract of a transcribed gene that: 1) does not overlap, adjoin or duplicate any sense-strand sequence of that gene’s canonical exons; 2) bears an acceptor splice site motif at its 5′ end and a donor splice site motif at its 3′ end; and, 3) via both these motifs, is spliced into a measurable proportion of the mature transcripts of that gene in at least one proband.”
We adopted a streamlined version of that definition for this report:
A germline pseudoexon is any continuous tract of a transcribed gene that: 1) does not overlap, adjoin, or duplicate any sense-strand sequence of a canonical exon; and 2) is spliced into mature transcripts of that gene in non-cancer cells of at least one proband 3) partly or wholly due to mutation in that gene.
This modified definition allowed for the inclusion of PEs spliced via non-canonical motifs and PEs spliced in first-exon or last-exon orientations. It also enforces the exclusion of canonical exons introduced into other genes via gene fusions, which could technically have been classed as PEs under the previous phrasing.
2.1.1 Exclusion of Cancer Pseudoexons
While many reports have detailed the correlation of PEs and various forms of cancer, we chose to limit our analysis to only those PEs that arose from germline mutations. This was because cancer cells often exhibit idiosyncratic changes in splice factor expression, and a general relaxation of splicing stringency, in comparison to non-cancer cells (Zhang et al., 2021). As such, we judged it would not be valid to make like-for-like comparisons between PEs in cancers and those in germline cells. However, we did include PEs arising from germline mutations in cancer-associated genes, such as NF1 and ATM, as in these cases carcinogenesis appeared to be a result of PE inclusion rather than the cause of it.
2.1.2 Exclusion of Pseudoexons in Non-Humans
Although PEs have been observed in other animal species (e.g., Smith et al., 2007; Gómez-Grau et al., 2017), such observations are even rarer than they are in humans, a disparity that probably stems from the lower level of interest in mutation analysis in non-human species. We determined that the inclusion of PEs from non-human species would only offer a modest increase to our study’s sample size at the cost of greatly generalising its conclusions, and therefore limited its scope to human PEs only.
2.1.3 Inclusion of Pseudoexons Identified via Transfected Minigene/Midigene Constructs
Wherever possible, it is ideal for investigations of splicing mutations to use RNA from patient cells that natively express the gene of interest. Unfortunately, this is not a practical option for many genes. For example, the Stargardt disease gene ABCA4 is primarily expressed in kidney and retinal cells, and it is rare that a biopsy of either of these internal tissues can be justified. Instead, many researchers utilise HEK293T cells (immortalized human embryonic kidney cells), which they transform with minigene constructs of the ABCA4 region of interest to model the effects of the mutation. In other cases, transformed COS cells (immortalised simian kidney fibroblast-like cells) or transformed HeLa cells (immortalised cervical cancer cell line) are used for similar purposes.
We elected to include in our dataset most of the PEs identified via minigene constructs, provided that the minigene constructs contained, at minimum, the entire intron surrounding the PE and both the flanking canonical exons. This minimised the chance of including a PE produced by construct-specific changes to its proximal sequence elements instead of the patient’s mutation. For the same reason, we also decided that if the effects of a splicing mutation were observed in both modified and unmodified cells, only the observations from the unmodified cells would be considered.
2.1.4 Inclusion of ‘Terminal-Pseudoexons’
Virtually all of the reports surveyed in this analysis described internal PEs, i.e., PEs that were spliced into a gene transcript somewhere between its canonical first and last exons. However, there were some reports that appeared to detail “terminal-pseudoexons” (tPEs) arising from mutations that caused non-canonical sequence inclusions at the 3′ ends of largely canonical transcripts. We determined that these putative tPEs would only be classed as such, and included in our catalogue, if they could meet three criteria:
(1) They possessed a novel acceptor site.
(2) They did not overlap or adjoin any sense-oriented canonical exon sequence.
(3) They possessed a functional polyadenylation site.
This third criteria is perhaps the most important, as it distinguishes “true” tPEs from more common events such as incomplete splicing and/or partial intron inclusion, which typically result in rapid nonsense-mediated decay of the affected transcript. We therefore only included tPEs if the supporting RNA analysis directly confirmed a de novo polyadenylation site, either via 3′ RACE or through whole transcript sequencing.
2.2 Quality and Method of RNA Sequencing
The primary sources collated in this report span nearly 40 years of genetics research. As such, the RNA sequencing methods used by these sources run the full gamut of technologies, from S1 nuclease mapping to Nanopore. With few exceptions, we were agnostic towards the RNA sequencing technology used, provided that the PE sequence and splice sites could be mapped to the genomic reference sequence with a high degree of certainty. The level of detail provided in most reports made this a straightforward process, especially for those that had included Sanger sequence traces of the PE splice site junctions or Varnomen-format descriptions matched to specific reference sequences. In other cases, it was necessary to deduce PE sequences from precise but indirect details, such as the stated length of the PE relative to its instigating mutation.
In some reports, the stated boundaries of one or more PEs appeared to be exceptions to the U2-type GY-AG splicing that predominates in the dataset and did not fit the established motifs of U12-type splice sites either (Turunen et al., 2013). Given the rarity of such non-canonical exon boundaries in the human transcriptome (Parada et al., 2014), we judged that it was appropriate to exercise additional scrutiny, and we only included non-canonically spliced PEs if their supporting sequence data was unequivocal.
In cases where the published detail of a PE report was insufficient to determine the PE sequence, we contacted the corresponding authors with requests for further detail and have cited those that graciously responded as “Pers Comms” where appropriate.
2.3 Quantity of Pseudoexon Inclusion in Mature Transcripts
Our dataset does not incorporate quantitative data for the frequency of inclusion of each PE. While it could be argued that this weakens the analyses in some respects—since PEs with 10% inclusion are treated identically to PEs with 100% inclusion—we reasoned that avoiding quantitative analysis would be the “lesser of two evils.” Collectively, this study’s source reports show enormous variation in the genes studied, the types of cells used, and the methods of RNA analysis, with many producing sequence data that was non-quantitative or at best semi-quantitative. Accurate and objective standardisation of these data would have been all but impossible and risked generating misleading conclusions.
2.4 Search Criteria
Literature search was performed using Google Scholar and the Murdoch University academic research portal FindIt. Individual searches of the following terms were conducted through both portals: “pseudoexon,” “pseudo exon,” “cryptic exon,” and “deep intronic.” The first three of these terms comprise the most common descriptors for PEs, while the fourth term served as a “safety net” to return any publications of deep intronic splicing mutations that may have reported on PEs using unexpected terminology.
In addition to scrutinising each paper for useful data, we also performed searches within each paper for the key terms mentioned above and investigated any references that were cited against these mentions. This allowed us to discover additional PE reports that, for various reasons, had escaped capture by our direct searches.
Similarly, we are indebted to the authors of several previous reviews of PEs, whose works led us to additional primary sources that had eluded discovery through the above methods (Královičová and Vořechovský, 2007; Dhir and Buratti 2010; Vořechovský 2010; Romano et al., 2013; Vaz-Drago et al., 2017). We have also incorporated data for DMD PEs that was originally collated for a previous report (Keegan 2020).
2.5 Construction of Pseudoexon Catalogue
2.5.1 Transcribed Pseudoexon Features
The rarity of PEs, coupled with broad variability in how they were reported, made it unfeasible to automate their annotation. Therefore, all annotation of PE data was performed manually by the authors. This approach was further justified post hoc by discoveries of minor inconsistencies in numerous PE reports (e.g., stated splice sites or lengths that differed from those shown in the published figures), errors that would have escaped detection by any automated process and led to inclusion of inaccurate data.
For each PE, annotated data fields included the name of the affected gene using current nomenclature, as listed on the Genecards Human Gene Database (Stelzer et al., 2016); the sequence of the PE; the sequences of the flanking exons to which it was spliced; the cell or tissue type(s) in which its splicing was observed; the Varnomen cDNA code for the instigating mutation(s) if present and known (den Dunnen et al., 2016); additional notes if relevant; and citations for the primary sources of the data.
While transcript reference sequences are included for the encompassing gene of each PE, in many cases a specific refseq ID was not explicitly declared in the source. The listed reference sequences instead correspond to the lowest-numbered transcript variant (usually TV1) that matched the splicing patterns observed and have been included to allow the use of cDNA-type Varnomen mutation codes, which are more human-readable than genomic codes. Chromosomal coordinates and all other genomic features refer to the most recent human genome assembly, GRCh38.p13.
2.5.2 Intron Numbering
Historically, unique identifiers have been employed when referring to the introns of certain genes, such as NF1 and CLRN. For consistency, we have ignored these in favour of simple 1-to-n numbering for all transcript variants, but caution the reader that this may create the appearance of discrepancies when referring to some cited reports.
2.5.3 Assignment of Unique Pseudoexon IDs
Each pseudoexon was assigned a unique ID according to the name of its gene, the number of the encompassing intron, and an alphanumeric identifier according to its similarity to other pseudoexons in that intron. For example, two pseudoexons with completely distinct sequence were reported in the 17th intron of the ATM gene and were IDed as ATM-17-1 and ATM-17-2, while the two PEs reported in ATM intron 27 were IDed as ATM-27-1a and ATM-27-1b due to sharing an acceptor site.
2.5.4 Citations
While descriptions of most PEs were limited to a single report, some were reported multiple times by different research groups and to varying levels of detail. An extreme example of this is CFTR-7-1, a prevalent disease allele for cystic fibrosis that has been studied extensively. In the interest of clarity, we limited our citations for each PE to its earliest known report, including later reports only if they substantially added to the characterisation or were already cited for unique observations of other PEs.
2.5.5 Derived Features
Maximum entropy (MaxEnt) splice site scores were calculated via the Burge Laboratory’s MaxEntScan web-tool (Yeo and Burge, 2004). Sizes and distances were directly calculated using spreadsheet formulae.
3 Pseudoexon Mutation Characteristics
3.1 Pseudoexon Splice Motif Mutations
3.1.1 Pseudoexon Donor Motif Mutations
Donor splice site mutations are the most frequently observed cause of PE pathogenesis, comprising 210 of the 359 catalogued distinct mutations. Of these 210, 202 are single nucleotide variants (SNVs), and the frequency of mutation for each nucleotide position relative to the donor site (Figure 1, right) approximates the degree of nucleotide conservation observed for that position in canonical sites (Ma et al., 2015). This may be a logical consequence of how the spliceosome binds to candidate donor sites: because the effect of nucleotide identity on spliceosome binding varies greatly across the motif, changes at the most essential positions will have the greatest effect and the best chance of “breaking through” the silencing mechanisms that would otherwise prevent detectable levels of splicing. This is corroborated by our observation that no PE in our dataset was instigated by an SNV at the donor site −3 position, despite this nucleotide falling inside the donor site motif. Because the −3 position is not highly conserved, any change in this nucleotide is unlikely to cause a noticeably pathogenic increase in PE inclusion.
FIGURE 1
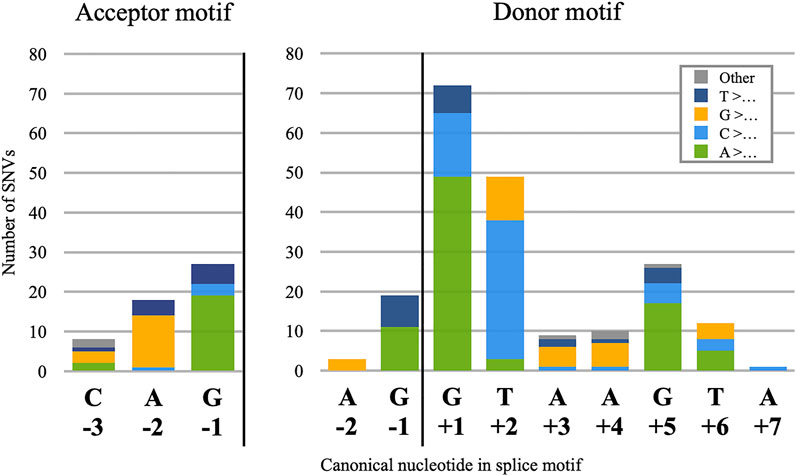
FIGURE 1. Positional frequencies of 255 pseudoexon splice-motif SNVs. Stacked columns are categorised by the identity of the reference nucleotides, which in 249 of the 255 cases were mutated to the most frequently observed nucleotide at that position of the motifs, as shown on the X-axis. Exceptions (6) are categorised as “Other.”
However, we did note a single PE with a C>A SNV 7 nt 3′ of the donor splice site (MMUT-11-1b). Although the donor site motif is traditionally considered to end at the 6th 3′ nucleotide, a comprehensive analysis of human splice sites (Ma et al., 2015) reveals a substantial bias towards purines at the +7 position (59% A/G). As this MMUT SNV is a pyrimidine-to-purine transition, the simplest explanation of it is as a donor site mutation.
3.1.2 Pseudoexon Acceptor Motif Mutations
Acceptor splice-site mutations account for 53 of the catalogued PE mutations, and all but one of those (DMD-30-1) were SNVs. Despite the greater size of the acceptor motif compared to the donor, pathogenic acceptor site SNVs were limited almost entirely to positions −2 and −1, with only an additional eight at −3 (Figure 1, left). As with the distribution of donor site mutations, this appears to be a result of low conservation at positions +1 to +3 and the low impact of individual nucleotides in the −20 to −4 range, with pathogenic mutations in this latter region apparently impacting branch point definition more than they did acceptor site definition. We also noted a single case of an SNV at the PE acceptor +3 position, (ABRAXAS1-5-1) but chose to analyse this as an internal mutation since its effect on the acceptor splice score was negligible.
Our observations of position frequency in PE donor and acceptor splice motif SNVs generally accorded with those of Krawczak et al. (2007), who examined a much larger set of gain-of-function splice mutations; and with those of Sakaguchi and Suyama (2021), who examined a smaller dataset consisting entirely of novel PEs. We also noted that the number of transition SNVs—purine-to-purine or pyrimidine-to-pyrimidine nucleotide changes—was approximately double that of transversion mutations, at 165 and 92, respectively. This accords with prior observations of how often each mutation type is generally observed in the human genome (Jiang and Zhao 2006).
3.2 Pseudoexon Internal Mutations
We catalogued 37 examples of PEs caused by sequence changes between the PE splice site motifs. In five of these examples (COL4A5-37-1, DMD-11-2, DMD-34-1, DMD-48-1 and GNAS-AS1-4-1—see Supplementary Table S1) the mutation was a >10 kb deletion that brought a latent acceptor-donor motif pair into conjunction. In these cases, we assumed that sheer distance between the splice sites was the chief silencing element that had been lost and did not analyse these further. Of the remaining 32 cases, there was one PE (GLA-4-1) caused by a 113 nt insertion, three PEs caused by 2–4 nt deletions, and 28 PEs caused by SNVs (Supplementary Table S2).
The positions and predicted effects of the 31 unique mutations showed much greater variability than the mutations affecting PE splice motifs. Although we could not distinguish any obvious patterns in their locations within the PEs, we noted that the primary reports consistently described these mutations as gains of ESE motifs and/or losses of ESS motifs within the PEs. Most of these assessments were made via an assortment of RNA motif analysis utilities, some of which are no longer available. We therefore standardised our re-analysis to a single utility, HExoSplice, which was designed specifically for analysing this type of mutation (Ke et al., 2011; Tubeuf et al., 2020). Because HExoSplice only calculates scores for SNVs, we derived scores for the deletion and insertion mutations manually by subtracting the total score for the wild-type exon from the total score of the mutant. Impressively, HExoSplice correctly predicted the directionality of 29 out of the 31 mutations with a net increase in the score (∆Hx). There were no obvious similarities between the two SNVs with negative ∆Hx scores (ABCA4-30-1c and DMD-32-1b). We suggest that in both these cases, the mutations may have altered binding of splicing factors specific to those genes or cells, as this kind of specific effect cannot be accurately predicted by a generalised tool such as HExoSplice.
3.3 Pseudoexon Branch Point Mutations
Just 14 of the 359 PE mutations were ultimately classified as altering pseudoexon branch point definition, and these mutations showed considerable variation in their nature and location relative to the PE acceptor site (Figure 2). Historically, branch point mutations have been poorly understood and have proven more challenging to predict than other splicing mutations (Canson et al., 2020). Despite this variability we observed that the mutations we had catalogued were remarkably consistent in their effects on branch point characteristics (Table 1).
FIGURE 2
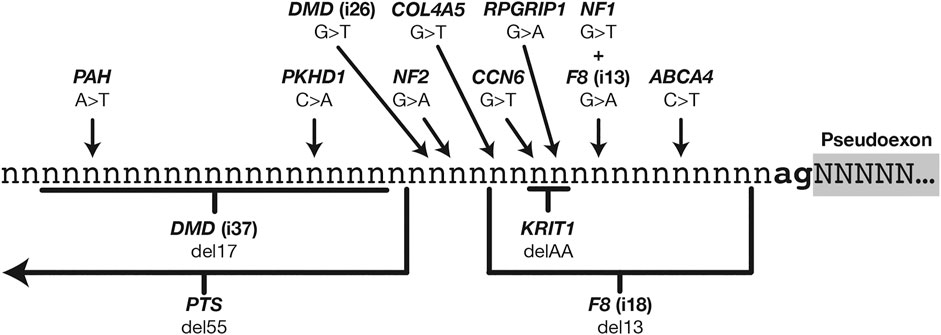
FIGURE 2. Relative locations of 14 mutations that instigate pseudoexons via enhancement or creation of branch point motifs.
TABLE 1
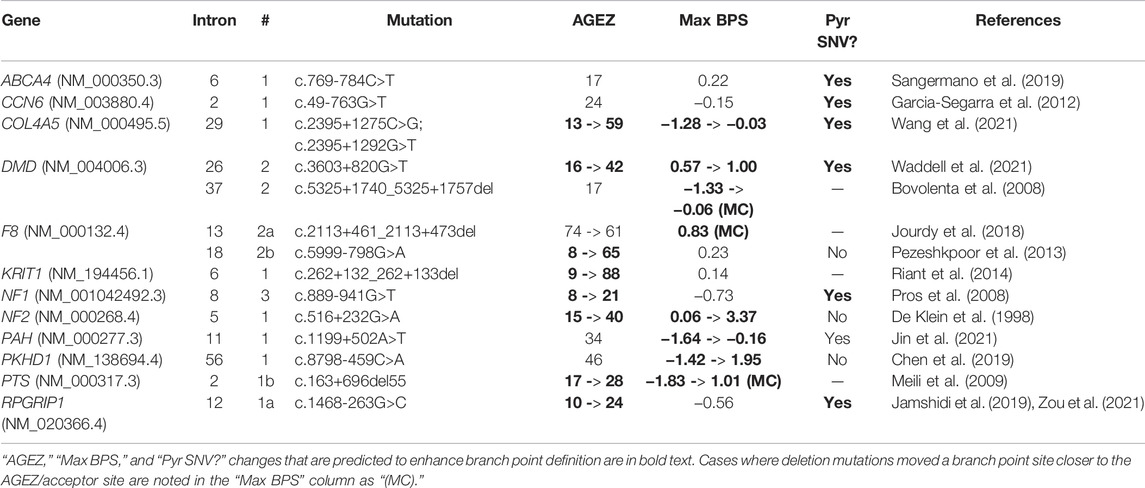
TABLE 1. Pseudoexon mutations that enhance pseudoexon branch point motifs.
Branch point definition requires two elements: a functional branch point site (the motif for which is weakly conserved in U2 introns) and a continuous AG-exclusion zone (AGEZ) connecting the branch point to a downstream acceptor splice site. In 12 out of the 14 cases shown here, the effect of the mutation was to “close the circuit” between an acceptor site and a branch point, through some combination of improving an in-range branch site motif (as predicted by SVM-BPfinder with “AGEZ only” selected—see Corvelo et al., 2010), increasing the size of the AGEZ, or moving an existing branch point closer to an AGEZ (Gooding et al., 2006).
Two exceptions to this rule were ABCA4-6-1 and CCN6-2-1, both instigated by SNVs that modestly increased their acceptor site scores (6.63 -> 7.12 and 3.12 -> 5.43, respectively) but did not affect their AGEZ size or branch point scores. Although we could defensibly have classified these as acceptor-motif mutations, we noted that every other acceptor-motif mutation fell within three nucleotides 5′ of the PE (Figure 1, left) and would therefore directly affect binding of the U2AF35 spliceosome component (Voith von Voithenberg et al., 2016). We reasoned that the positioning of the ABCA4-6-1 and CCN6-2-1 mutations within their respective polypyrimidine tracts suggested at least some interaction with other spliceosome components. A third mutation similar to these (c.639+861C>T) was reported in GLA (Filoni et al., 2008), although the affected exon in this case was subsequently classified as a canonical exon of transcript variant NR_164783 (Supplementary Table S3). A causative mechanism was not empirically confirmed for any of these three mutations. However, a fourth mutation, similar in type but opposite in effect, was reported by van der Wal et al. (2017). This mutation (c.-32-13T > G) occurred 13 nt 5′ of exon 2 in the gene GAA and caused skipping of all or part of exon 2. In this case, Van der Wal et al., were able to empirically demonstrate that the splicing disruption arose from a loss of U2AF65 binding at the mutation site.
The U2AF65 protein is a U2 spliceosome component that binds pyrimidine tracts 5′ of exon acceptor sites, interacting with the U2 snRNP to facilitate branch point recognition (Valcárcel et al., 1996). It has multiple known pyrimidine-rich binding motifs (Paz et al., 2014; Drewe-Boss et al., 2018). Since the ABCA4-6-1 and CCN6-2-1 mutations are both SNVs that create new thymine nucleotides, we theorised that the true pathogenic effect of these mutations is creation or enhancement of a U2AF65 binding site. Unfortunately, we could not test this prediction as there is at present no in silico tool for predicting U2AF65 binding that incorporates all known motifs. We therefore added “new pyrimidine nucleotide” as a crude predictor of U2AF65 binding.
We also noted that these three mutations are predicted by Splice Aid 2 (Piva et al., 2012) to create new PTBP1 (hnRNP I) binding sites. Although PTBP1 plays a complex role in splicing (Lou et al., 1999; Han et al., 2014) it is generally observed to silence nearby exons. However, in some cases PTBP1 has been shown to indirectly enhance 3′ exon inclusion by antagonising splicing repressors (Paradis et al., 2007) or preventing erroneous binding of U2AF65 (Sutandy et al., 2018), so we cannot rule out a possible mechanistic role for PTBP1 in these three mutations. There is clearly a need for an accurate and comprehensive in silico tool that can predict the effects of all types of branch point mutations, as they remain something of a blind spot in splice mutation research.
3.4 Distal Mutations
All the pathogenic mutations discussed thus far have entailed some form of direct improvement to the exon-like characteristics of the PE, be it the splice sites, branch point or local enhancer/silencer balance. However, we have also catalogued 35 cases of PEs being instigated by intragenic mutations outside the span of the PE’s branch point and donor site motif, sometimes multiple canonical exons and tens of kilobases away. When viewed individually, the aetiology of these PEs may appear baffling, but examining them en masse reveals many important common features.
3.4.1 Decreased Definition in Adjacent Canonical Exons
In their 2010 report, Dhir and Buratti proposed that loss of a canonical splice site “facing” a pseudoexon—i.e., a 5′ donor site or a 3′ acceptor site–may be a general mechanism of pseudoexon pathogenesis. Impressively, although their prediction was based on just five supporting examples (BRCA2-20-1a, CFTR-3-1, IDS-3-1a, IDS-3-1d and MMUT-11-1d) we found that it was generally supported by the characteristics of 13 of the additional mutations we collated (Supplementary Figure S1), albeit with refinements to the original terms of their category.
The ten examples shown in Supplementary Figure S1A most closely fit the terms of Dhir and Buratti’s original category, as all entailed loss or weakening of a 5′ donor or 3′ acceptor site. This includes one mutation that appeared to weaken a 3′ branch point (GAA-1-1), thereby indirectly weakening definition of the acceptor site, and one case (DGKE-5-1) where a mutation created a cryptic upstream donor site but left the original intact, indirectly weakening its definition via competitive effects. In addition, we also observed three examples of mutations that induced skipping of a whole exon (Supplementary Figure S1B)—including an alternative transcript variant caused by the same GAA-1-1 branch point mutation shown in Supplementary Figure S1A—and three examples of mutations that caused a net loss of enhancers within a flanking canonical exon (Supplementary Figure S1C), as indicated by their negative ∆Hx scores. We were also interested to note that in every case where a PE was spliced to an alternative flanking splice site—whether through cryptic splice site activation or whole exon skipping - the new site always had a lower MaxEnt score than the unmutated original.
However, this pattern is further complicated by three cases of PEs instigated by mutations in “next-but-one” neighbour exons that were not directly spliced to the PE (Supplementary Figure S1D). These cases may be a consequence of the exons near these PEs having tightly linked splicing fates, e.g., a loss of definition in MMUT exon 4 exerts a similar effect as a hypothetical loss of definition in exon 5 would. An analogous example of this kind of splicing effect can be seen in cases where a mutation within a single canonical exon caused skipping of both that exon and adjacent, unmutated exons (Fisher et al., 1993; Suzuki et al., 2016). We also note that there is substantial evidence of exons in many genes being consistently spliced out of transcription order (Takahara et al., 2002; Attanasio et al., 2003; Kim et al., 2017) and speculate that this, too, may contribute to the aetiology of some highly distal pseudoexon mutations. In general, exon-like tracts in late-spliced introns could be more vulnerable to the knock-on effects of splicing changes elsewhere in the maturing transcript, while those in early-spliced introns remain insulated by sheer distance.
We also observed that one of the three mutations shown in Supplementary Figure S1D (DMD-3-1a) was an intra-exon mutation with a positive ∆Hx score, indicating an increase in the definition strength of that exon. Although this may represent a valid counterexample to the prevailing trend of pathogenically decreased exon definition, we tentatively dismissed it as another of the inevitable minority of incorrect predictions made by the HExoSplice algorithm and note that one of the two other such examples we observed also occurred in the DMD gene (DMD-32-1b—see Supplementary Table S2).
Considered in aggregate, these observations suggest the common mechanism underlying these PEs is a comparative weakening of definition in the successfully spliced neighbouring exons; and vice versa, that the definition strength of exons flanking an intron can be an important mechanism for silencing latent PEs within. This hypothesis is supported by prior observations of stronger splice motifs in exons flanking large introns (Farlow et al., 2012), since larger introns could generally be assumed to have a higher chance of containing at least one latent PE. The infrequency with which these types of PEs arise, compared to PEs generated by direct enhancement of their donor or acceptor splice sites, may be a result of general selective pressure against splice-competent elements within intron tracts. We also speculate that pseudoexons of the kind seen in Supplementary Figure S1C may be more common than suspected and could account for the pathogenic nature of some synonymous variants (Shi et al., 2019), perhaps having escaped detection in prior experiments due to nonsense-mediated decay (NMD) of the affected transcripts.
3.4.2 Novel Pseudoexon Mutation Categories
We observed 17 examples of PE mutations that bore no resemblance to Dhir and Buratti’s five categories, some of which bore sufficient similarities to each other to justify new categories (Supplementary Figure S2).
Proximity to a Directly Mutated Pseudoexon in the Same Intron
The first of these categories comprises four cases where PEs were apparently instigated by the activation of a second PE in the same intron (Supplementary Figure S2A). In all four cases the mutation in the ‘primary’ PE created or enhanced a donor site, and all occurred in introns of similar sizes, ranging from 1,212 nt (MYBPC3 intron 20) to 2,582 nt (MYBPC3 intron 12). It is also notable that in each of these cases, the primary PE introduces a flanking splice site as strong or stronger than that of the nearby canonical exon. For example, the primary PE in F8-12-2a introduces a downstream acceptor site stronger (MaxEnt = 8.80) than that of exon 19 (MaxEnt = 6.91). This would appear to contradict the pattern of PEs arising from weakening of flanking exon definition. However, it may be that in these cases the intron subdivision caused by splicing of the primary PEs leads to a disruption of splicing co-ordination (Drexler et al., 2020), and a consequent loss of silencing of the secondary PE that exceeds the expected gain of silencing from the primary PEs’ strong flanking splice sites. An assay of intron splicing order in cells carrying these mutations, or other similar mutations, may shed much light on the processes involved.
Loss of Upstream Polyadenylation Motifs
There was a single case of a PE arising from an intragenic region due to an upstream deletion (EPCAM-7-1, Supplementary Figure S2B). While we would otherwise hesitate to define an entire category by a single exemplar, in this case the causative mechanism is straightforward enough to justify it: Genomic deletion of the latter two exons of EPCAM, which necessarily entailed deletion of that gene’s polyadenylation signals, permitted transcription to continue through the intergenic region and into the 3′ gene MSH2, which shares EPCAM’s sense-strand orientation. By chance, this novel “intergenic intron” contained a 111 nt tract that was sufficiently exon-like to be spliced to the neighbouring canonical internal exons, namely EPCAM exon 7 and MSH2 exon 2 (Ligtenberg et al., 2009).
Although this is the only example of this type of PE that we catalogued, similar PEs could occur in other cases where a genomic polyadenylation site deletion is followed by a 3′ gene with the same strand orientation and at least one intron, and where the splice sites involved are in sufficient proximity.
Change to Proximal Intronic Splice Motifs
We catalogued six cases where PEs were instigated by mutations within the same intron but beyond the PE splice motifs and branch points (Supplementary Figure S2C). Three of these mutations were 3′ of the PE (FBOX38-9-1, MFGE8-6-1, and NPHP3-3-1) and three were 5′ of the PE (DMD-56-1, NR2E3-7-2, and RPGR-9-1). Here we must clarify that although DMD-56-1 bore deletions both 5′ and 3′ of the PE, the reporting authors empirically demonstrated that only the 5′ deletion caused PE inclusion (Khelifi et al., 2011).
The MFGE8-6 and NPHP3-3 mutations were both SNVs similar distances from the PE donor sites (43 and 50 nt, respectively) that created new predicted FUS (hnRNP P2) binding motifs (Piva et al., 2012). Studies in mouse cells (Ishigaki et al., 2012) have shown that FUS binding along flanking introns can regulate alternative exon splicing in neuronal cells, so it is possible that a perturbation of normal FUS binding is responsible for these PEs escaping silencing.
A similarly positioned SNV in FBXO38-9-1 destroyed a predicted binding site for hnRNP K (Piva et al., 2012). This is consistent with a recent report demonstrating that hnRNP K depletion can lead to a widespread increase in cryptic exon inclusion, and that at least some of these cryptic exons are ordinarily silenced by hnRNP K binding within 100 nt of the 3′ intron (Bampton et al., 2021).
Unfortunately, there are few such similarities to connect the three PEs with 5´ distal mutations. The first, DMD-56-1, is caused by a 592nt deletion ending 26 nt 5´ of the PE acceptor site. The authors experimentally excluded modified branch point definition as a causative factor for this PE, but despite thorough experimentation with minigene assays they could not positively identify which components of the deleted region were responsible for the PE’s inclusion or what their mode of action was. The second PE with a distal 5´ mutation, NR2E3-7-2, was instigated by an SNV 581 nt upstream that altered multiple splice factor binding sites, making it difficult to predict which, if any, are mechanistically responsible. Incidentally, this was the same mutation that created an acceptor motif AG dinucleotide in NR2E3-7-1, though unlike the examples discussed in the first category of Novel Pseudoexon Mutation Categories, these two PEs exhibit mutually exclusive splicing.
In the third PE with a distal 5′ mutation, RPGR-9-1, a TTAAA motif is created 53 nt from the acceptor site. This motif is predicted to bind KHDRBS1 (Sam68) and/or KHDRBS3 (SLM-2), two splicing factors with high homology and similar effects on pre-mRNA splicing (Danilenko et al., 2017). In particular, KHDRBS1 has been shown to aid in the splicing of introns bearing Alu retrotransposon sequences (Pagliarini et al., 2020). Two such Alu elements occur within RPGR intron 9 (Supplementary Figure S3). Although the true pathology of this mutation is yet to be empirically determined, it may be that a disruption to KHDRBS1-mediated splicing is responsible for the RPGR-9-1 pathogenesis.
Unknown Mechanisms
In six cases, the connection between the identified mutation and the PE was unclear (Supplementary Figure S2D) and these cases bore no similarities to other catalogued examples. However, we note that DYSF-51-1b is an identical sequence inclusion to DYSF-51-1a (which arose from a PE donor site mutation), and has been observed at low levels in cells from healthy donors (Gonorazky et al., 2019); and similarly, that DMD-3-1a is also instigated by a 1 nt deletion in exon 5 (Supplementary Figure S1D).
3.5 Summary of Pseudoexon Mutation Analysis
Our pseudoexon catalogue, which is to date the most comprehensive ever assembled, confirms that PEs are most frequently instigated by direct mutation of their local splicing motifs; that the most frequently mutated components are the PE donor and acceptor splice motifs; and that the predominant type of instigating mutation is single nucleotide substitution. These findings support previously published observations of smaller pseudoexon datasets, which we gratefully acknowledge as secondary data sources for this catalogue. We add to this several novel classifications for rarer types of PE-instigating mutation, the most well-supported of these being mutations that weaken definition of adjacent canonical exons.
4 Latent Factors Contributing to Pseudoexon Splicing
Considering the complexity and stringency of vertebrate exon definition, in conjunction with the observation that single-nucleotide substitutions are the most frequent cause of PE pathogenesis, we are forced to question why these exon-like intron tracts exist in the first place. Even if the reference allele of a given PE is ultimately excluded from mature transcripts by the lack of one crucial splice motif, the presence of all the other exonic motifs might still encourage abortive “false start” activity by the spliceosome, wasting energy and unnecessarily prolonging mRNA maturation.
It may be that the latent elements of some PEs are mildly deleterious in this way but persist in the genome simply as another of evolution’s myriad compromises and works-in-progress. However, we must also examine the alternative explanation that these latent elements persist due to their spliceosome interactions being benign or even beneficial, and consider the various forms these interactions may take.
4.1 Canonical Exon Splice Variants
The earliest reported PE to meet the criteria of this catalogue (Dobkin et al., 1983) predates the completion of the first rough draft of the human genome project by nearly 18 years (Lander et al., 2001), and the years that intervened and followed these milestones have seen numerous revisions to the official coding sequences and transcript variants of thousands of genes. An inevitable side-effect of this progress is that many splicing phenomena initially reported as PEs have subsequently been reclassified as either canonical exons or mutant splice variants thereof. In the course of assembling and curating this catalogue, we separately collated 35 such examples (Supplementary Table S3). These examples could not be included in any of our PE analyses since there is no meaningful distinction between them and other canonical exon splice mutations. However, they serve as a useful reminder of the difficulty in distinguishing PEs from mutant variants of as-yet-unannotated canonical exons, especially if the canonical exons are expressed at low frequencies or in unexamined cell types (Ray et al., 2020). We expect that progress in transcriptomics will eventually necessitate similar reclassification for at least some of the PEs in this catalogue.
4.2 Novel or Unannotated Canonical Exons
Having excluded from of our catalogue those PEs that coincided with known canonical exons, we attempted to annotate additional examples of PEs that might undergo this reclassification in future. Our criteria for inclusion were 1) the PE must show evidence of splicing in normal cells for at least one of its splice sites, either in the original report, in non-cancer cell spliced expressed sequence tags (ESTs) from the UCSC Genome Browser’s “Spliced ESTs” track (Kent et al., 2002) or in paired-end RNAseq data (Sibley et al., 2015); and 2) inclusion of the PE in the mature transcript must be predicted not to trigger NMD.
A total of six PEs met these criteria (Supplementary Table S4). In all six cases, NMD avoidance was predicted due to preservation of the open reading frame and absence of any novel stop codons. We also allowed for cases where a transcript variant with a premature stop codon may have escaped NMD due the stop codon being introduced less than 55 nt from the final splice junction of the transcript (Zhang et al., 1998) but did not find any examples that met this criterion.
4.3 Poison Exons and Decoy Exons
In recent years the term “poison exons” has been steadily gaining prominence in literature related to cryptic splicing phenomena. Carvill and Mefford (2020) characterised poison exons as conserved, alternatively spliced exons containing one or more premature termination codons that are spliced into unneeded transcripts to prevent their translation and target them for nonsense-mediated decay. “Decoy” exons behave similarly but are characterised by their additional capacity to non-productively interact with adjacent canonical splice sites, thereby promoting whole intron retention (Conboy, 2021).
There is a clear overlap between the definitions of poison/decoy exons and PEs, although the phenomena are not identical. Both describe non-canonical exon inclusions that generally impair the translation of full-length, functional protein from the affected transcript; but while PEs arise due to intragenic mutations and are often deleterious to the health of the patient, poison exons are a normal component of splicing that may contribute to fine-control of gene expression and are presumably beneficial, or at the very least benign.
Given the similarities between PEs and poison exons, and the relative novelty of the latter term, the intriguing possibility emerges that some of the splicing phenomena historically reported as PEs might be better re-classified as poison exons, or splice variants thereof.
Having already determined the concurring splice site reads between our PE catalogue, and ESTs and RNAseq data (see Section 4.1) we separately tabulated all those examples where evidence supported their splicing in normal cells, but which did not preserve the transcript open reading frame (Supplementary Table S5).
A possible reason for the high number of candidate poison exons seen in NF1 and DMD is the exceptional size and high intron count of these genes. These features unavoidably entail a long transcription and maturation time, which must be reconciled with the fact that the quantity of any encoded protein that the cell needs can change dramatically in a matter of seconds. The more poison exons a transcript contains, the more possible time-points there are for interrupting the reading frame and preventing an unneeded transcript from reaching functional maturation.
Of the 413 catalogued PEs, for 65 (15.7%) we found evidence of splicing of at least one splice site in normal cells. This is a remarkably high concordance when one considers that, for the most part, splicing of putative PEs in normal cells is not something that has been systematically investigated; as such, what supporting evidence there is exists largely by chance. As RNAseq becomes more commonplace and is applied with greater sensitivity and read depth to a broader range of cell types, it may emerge that many more PEs—perhaps even a majority—originated as benign rare exons or functional exon-like intronic sites.
4.4 Recursive Splice Sites
In a previous report focused on PEs in the DMD gene (Keegan 2020), we examined the possibility that some PEs may arise from the errant splicing of predicted recursive splice sites (RSSes). Here we sought to examine this possibility as it applies to our total set of PEs. This task was complicated by the fact that there is as yet no consensus on the precise definition of RSSes and how best to experimentally verify their presence. For example, the criteria employed by Zhang et al. (2018) required that a putative RS-exon should bear an agGT tetranucleotide at the acceptor site and that the nucleotides around the acceptor site should be highly conserved, while the approach of Wan et al. (2021) was agnostic to sequence conservation.
We searched the splice site coordinates of our PE dataset against five published datasets of recursive splice sets (Kelly et al., 2015; Sibley et al., 2015; Blázquez et al., 2018; Zhang et al., 2018; Wan et al., 2021). We did not find any matches in Kelly et al. (2015) or in the filtered results of Sibley et al. (2015), but we did discover seven matches in the filtered results of the other three reports—five in Wan et al. (2021) and one in each of Blázquez et al. (2018) and Zhang et al. (2018) (Table 2). To our knowledge, this is the first conclusive evidence supporting our earlier hypothesis that pathogenic PEs can arise from mutations near recursive splice sites (Keegan 2020). Additionally, we were interested to note that six of the seven recursive splice sites were also spliced as components of putative poison exons in normal cells (Supplementary Table S5), with COL4A5-6-1 being the exception. This may indicate that these sites serve a dual purpose in splicing regulation, though this remains to be confirmed through functional studies.
TABLE 2
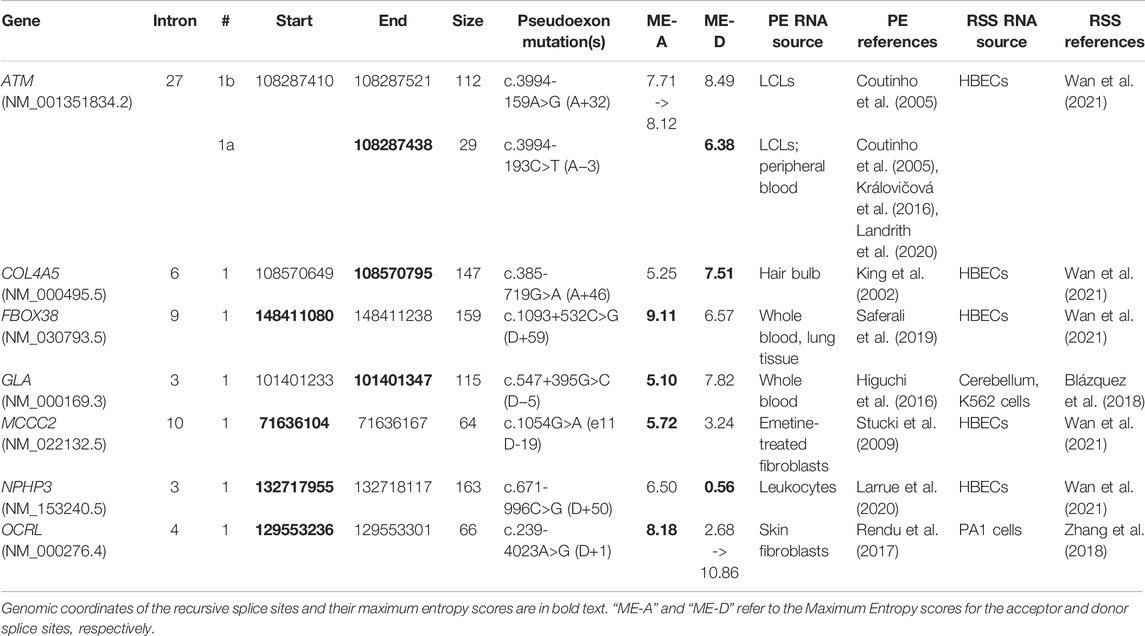
TABLE 2. Pseudoexons associated with seven confirmed intronic recursive splice sites.
5 Unique Cases and Additional Observations
5.1 No Known Pseudoexons are Processed by the U12 Spliceosome
The minor spliceosome, or U12 spliceosome, processes just 0.37% of all human introns (Olthof et al., 2019). Type-U12 introns can most easily be recognised by their highly conserved donor-site (UTATCCT) and branch point (CCTTUAY) motifs, and their tolerance for AT-AC terminal dinucleotides—although the latter feature is not present in all U12 introns and GT-AC, AT-AG or GT-AG terminal dinucleotide pairs are also observed (Turunen et al., 2013).
A search of the donor sites of all catalogued PEs and their 5′ spliced exons discovered a single example of a UTATCCT donor site motif, at the donor site of LHCGR-6-1b. Although this donor site scores low as a U2 splice site (MaxEnt = 0.48), there was no type-U12 CCTTUAY branch-point motif near the acceptor site of the 3′ exon 7, and the canonical 5′ exon 6 did not have a type-U12 donor site. This indicates that the termini of LHCGR intron 6 have evolved to be removed via the predominant mode of U2 splicing. It therefore appears that this PE is spliced via the U2-spliceosome and not the U12. Therefore, we concluded that no U12-spliced PEs are reported in this dataset, although we did note that STK11-1-1 occurs in a U2 intron that is 5′ adjacent to a known U12 intron (Hastings et al., 2005).
While a type-U12 PE may yet be reported, it is unsurprising that none have been discovered thus far. The great majority of reported PEs have been observed as singletons that are directly spliced to canonical upstream and downstream exons in the mature transcript. This means that each of the two PE splicing reactions involves one neighbouring canonical splice motif that has evolved for optimal interaction with a particular spliceosome. From this we can infer that the mode of a PE’s splicing will largely be determined by the splicing mode of its encompassing intron, i.e., a U2-spliced PE cannot arise within a U12 intron or vice versa. A similar hypothesis was suggested by Qu et al. (2017) in their analysis of U12 splice mutations. Because only 0.37% of human introns are type-U12 (Olthof et al., 2019) the genomic range within which a U12 PE could plausibly arise is vanishingly small. However, exceptions may occur if a mutation that prevents proper recognition of the splice motifs of a U12 intron results in cryptic U2 splicing. Madan et al. (2015) observed such cryptic U2 splicing of a U12 intron of WDR41, though this was the result of knockdown of the splice factor ZRSR2, rather than mutations in WDR41 itself.
5.2 Pseudoexons With Non-AG Acceptor Sites Occur Rarely but Unpredictably
We catalogued four examples of PE variants with non-AG acceptor site dinucleotides. Three of these (RB1-14-1b, RB1-14-1c, and RB1-14-1d) arose from the 5′ junction of a single LINE-1 retrotransposon insertion in RB1 intron 14 (Rodríguez-Martín et al., 2016). These three variants share a common U2-type donor site, but each have unique non-canonical acceptor sites that were confirmed through Sanger sequencing. This LINE-1 insertion also induced an additional PE variant with a canonical acceptor site (RB1-14-1a).
The fourth non-canonical PE (NF1-39-1a) was observed in NF1 as the result of a donor-site-creating SNV. Like the RB1 PEs, NF1-39-1a bears a canonical donor site and a non-canonical acceptor site and shares its donor site with a wholly canonical variant, NF1-39-1b.
A report by Parada et al. (2014), examined common features of 184 non-canonical splice sites, and the authors observed therein that the terminal dinucleotides of most non-canonical splice sites differ from the canonical AG or GY pairs by only a single nucleotide. This holds true for the RB1 non-canonical PEs, which have CG, AT and AT respectively as their acceptor-site terminal dinucleotides, and for NF1-39-1a, which has a TG dinucleotide. We speculate that this one-nucleotide rule is observed because varying only a single nucleotide minimises the amount of resistance that must be overcome to “persuade” the spliceosome to cleave at a non-AG/GY dinucleotide.
Unfortunately, there are few other established hallmarks for human non-canonical exons that these PEs can be compared against. Burset et al. (2000) suggested that non-canonical splice sites may parasitically exploit the presence of nearby canonical splice motifs to recruit the spliceosome, an hypothesis supported by the alternative canonical acceptor sites observed in RB1-14-1a and NF1-39-1b. However, even if this “parasite” model accounts for spliceosome recruitment, it still begs the question of why the non-canonical splice sites are used at all when workable canonical sites are available. Similarly, although Parada et al. (2014) detected a higher density of ESEs and intronic splice enhancers around non-canonical sites, it is not valid to apply their statistical analysis to just four additional sites. Deriving a complete explanation for why these two mutations in RB1 and NF1 created PEs with non-canonical splice sites, when so many other similar mutations in these and other genes did not, therefore remains as a challenge for future researchers.
5.3 Terminal Pseudoexons are Both Rare and Difficult to Detect Without Third-Generation Sequencing Technologies
We catalogued two examples of terminal pseudoexons (tPEs), each arising from unique mutations in ARHGEF9 (Figure 3A) and F8 (Figure 3B). Although it is difficult to generalise from just two observations, there are obvious similarities between these cases that are worth noting. Both the ARGHEF9 and F8 genes are carried on the q arm of the X-chromosome, albeit at opposite ends (Figure 3C), and in both cases the instigating mutations entail large sequence rearrangements that moved the canonical 3′ end of the gene out of splicing range of the upstream exons. In the case of ARHGEF-6-2, this mutation is a balanced crossover with chromosome 18, while the F8 gene of the second patient bears a 3.8 Mb insertion of chromosome X intergenic sequence. Notably, the region inserted into F8 in F8-25-1 encompasses ARHGEF9 along with 11 other protein-coding genes (not shown). Interestingly, although the ARHGEF9 mutation was originally described as creating two tPEs—one 5′ of the breakpoint in the normal intron 6 sequence, and one in the translocated chromosome 18 sequence—we found that the first of these terminal exons shares its polyadenylation site with ARHGEF-IT1, a noncoding and largely uncharacterised two-exon transcript nested within ARHGEF intron 6. Because it shares sequence with a canonical exon, this mutant terminal exon therefore does not meet the criteria for classification as a tPE.
FIGURE 3
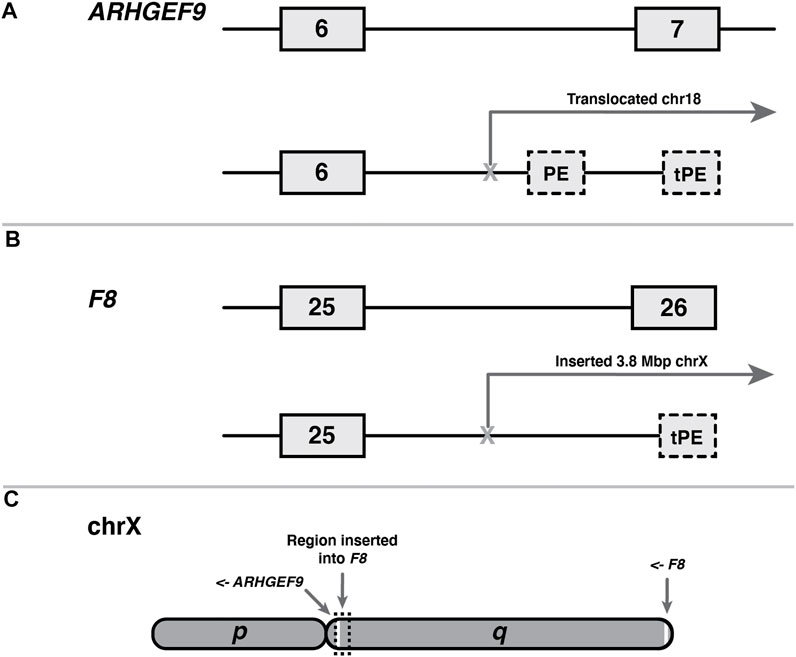
FIGURE 3. Shared features of two terminal pseudoexons (tPEs). (A) ARHGEF9 tPE (ARHGEF9-6-2) and internal PE (ARHGEF9-6-1) arising from within a translocated region of chromosome 18. (B) F8 tPE (F8-25-1) arising within intragenic sequence of a transposed 3.8 Mb tract of the X chromosome. (C) Relative locations, to scale, of the affected genes on the X chromosome q-arm, and genomic origin of transposition in patient B. Arrows indicate reading directions of each gene.
The fact that tPEs are so rare in comparison to internal PEs is surprising when one considers that the defining hallmarks of a last exon are comparably well-defined to those of an internal exon, and therefore they should be expected to arise from random mutation at roughly the same frequency. The requirement for a functional acceptor site is similar in both exon types, and the requirements for polyadenylation site definition (Kaida, 2016) do not appear very much stricter than those for donor site definition. Furthermore, last exons usually contain a stop codon, a requirement that most PEs meet by default, and last exons also exhibit a much broader range of sizes than internal exons (Movassat et al., 2019).
We suggest that there are at least three compounding causes for the low discovery rate of tPEs. The first is that the laboratory techniques required to confirm a tPE make them considerably more difficult to discover than internal PEs. Many internal PEs were detected serendipitously when researchers noticed unusually large products from their RT-PCRs of flanking canonical exons, but this method of discovery is only possible if the RT-PCR primer target sites are present on both sides of the mRNA insertion, and this is not the case for tPEs as they are not spliced to any 3′ exon. Any RT-PCR of the canonical exons flanking a tPCR would produce either low abundance products of the expected size (if some level of normal splicing is still present) or no products at all. Even if the researcher eventually discovers the acceptor splice site of the tPE, their subsequent failure to detect an active donor site may lead them to conclude that the effect of the mutation is partial intron inclusion and arrest of splicing.
A third possible contributing factor is that terminal exon definition may be a stricter process than it appears. At the very least, the similarity of the two mutations described here suggests that the absence of competition from downstream canonical exons is a contributing factor, which is something that can only occur after large-scale sequence rearrangements such as these. Conversely, the effect of an EPCAM terminal exon deletion mutation (Supplementary Figure S2B) was to induce a fusion transcript with MSH2 but no novel polyadenylation site, as in this case a latent intergenic pseudoexon combined with the chromosomal proximity of MSH2 provided viable splicing partners.
Regardless of the true frequency of tPEs, it is worth noting that the aforementioned barriers to their detection do not apply to third generation sequencing technologies like Nanopore, which are largely agnostic in their detection of polyadenylated transcripts. As the uptake of Nanopore and other third-generation RNA sequencing technologies continues to increase, there may be a corresponding increase in the discovery rate of tPEs.
6 Conclusion
Pathogenic pseudoexons primarily arise from mutations that directly enhance their donor or acceptor site motifs. However, other types of instigating mutation are also observed less frequently, but with consistent features, many of which are characterised for the first time in this report. In rare cases, the splicing pathology of a PE was highly idiosyncratic and could not be properly categorised due to a lack of similar supporting examples. These findings advance our understanding of how mutations give rise to pathogenic pseudoexons, but also highlight that our understanding is still far from complete.
We also discovered seven examples of pseudoexons that coincide with recently confirmed recursive splice sites, conclusively demonstrating that functional exon-like intron elements can be converted to pseudoexons when favourable mutations arise nearby. Although it remains to be determined how many pseudoexons arise in this way, we found that 15.7% of pseudoexons showed evidence of splicing at one or both of their splice sites in cells from healthy donors, a figure that is likely to increase further as the fidelity and quantity of RNAseq data continues to improve.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
NK conceived of the project. All authors contributed to project design and developed its rationale. NK and SF defined scope and criteria for data inclusion and exclusion and decided methods of analysis. NK performed research, catalogue curation and data analysis and wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
Funding was provided by the Australian Commonwealth Government Research Training Program Scholarship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank the Australian Commonwealth Government Research Training Program and Murdoch University for the provision of the PhD stipend for NK, and funding and computing resources, and their colleagues at the Centre for Molecular Medicine and Innovative Therapeutics for advice and constructive criticism.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.806946/full#supplementary-material
Supplementary Figure 1 | Pseudoexons instigated by weakened definition of nearby canonical exons. Pseudoexons are shown as medium grey dash-line boxes, with flanking canonical exons in light grey and intervening introns as solid black lines. Sizes are not to scale. Dash-lines elsewhere indicate altered splicing of canonical exons. Roman numerals within pseudoexons (I–IV) indicate identical sequence inclusions (see also Supplementary Figure S2). Selected relevant features for each mutation are indicated where appropriate, with full details and references provided in Supplementary Table S1. Numbers above exon ends indicate acceptor and donor Maximum Entropy splice site scores, with “NF” indicating a site that is non-functional in the reference (NF -> score) or mutant (score -> NF) allele due to lacking an essential AG-GY dinucleotide. Changes to exon HExoSplice scores are shown above the affected exons as “∆Hx,” other motif changes are indicated with vertical lines and labelled as “± name-of-motif.” Vertical brackets indicate common features of enclosed splice events. Note that GAA-1-1 is shown twice due to the splicing effects of its mutation fitting both the (A,B) sub-categories.
Supplementary Figure 2 | Pseudoexons instigated by novel or unknown mechanisms. Pseudoexons are shown as medium grey dash-line boxes, with flanking canonical exons in light grey and intervening introns as solid black lines. Sizes are not to scale. Dash-lines elsewhere indicate altered splicing of canonical exons. Roman numeral “IV” indicates an identical sequence inclusion to one shown in Supplementary Figure S1. Selected relevant features for each mutation are indicated where appropriate, with full details and references provided in Supplementary Table S1. Numbers above exon ends indicate acceptor and donor Maximum Entropy splice site scores, with “NF” indicating a site that is non-functional in the reference (NF -> score) or mutant (score -> NF) allele due to lacking an essential AG-GY dinucleotide. “+D” indicates one or more gain-of-donor-motif mutations. Other motif changes are indicated with vertical lines and labelled as “± name-of-motif.” Vertical brackets indicate common features of enclosed splice events. *Variant-induced increase in splicing of FBXO38-9-1 was low (from 8 to 13% of FBXO38 transcripts) but statistically significant (Saferali et al., 2019).
Supplementary Figure 3 | A G>A substitution within RPGR intron 9 creates a binding motif for KHDRSB1 (Sam68), potentially altering the splicing of an adjacent pseudoexon (RPGR-9-1) that originates within an AluSx element. Image captured from UCSC Genome Browser, GRCh38/hg38. Location of TTAAA motif indicated with a thick vertical red line, and additional annotations are written in red text. Scale at top shows chromosome X coordinates. “GENCODE V36” track shows aligned transcripts, with gene names on the left, solid bars indicating exons and horizontal lines indicating introns. Arrows (< and >) on introns indicate transcript reading direction relative to chromosome + strand. “Repeating Elements” track shows alignments of SINE or LINE elements as solid rectangles, with darker shading indicating higher homology to the reference sequence for that element.
References
Agrawal, A., Hamvas, A., Cole, F. S., Wambach, J. A., Wegner, D., Coghill, C., et al. (2012). An Intronic ABCA3 Mutation that Is Responsible for Respiratory Disease. Pediatr. Res. 71 (6), 633–637. doi:10.1038/pr.2012.21
Ajeawung, N. F., Nguyen, T. T. M., Lu, L., Kucharski, T. J., Rousseau, J., Molidperee, S., et al. (2019). Mutations in ANAPC1, Encoding a Scaffold Subunit of the Anaphase-Promoting Complex, Cause Rothmund-Thomson Syndrome Type 1. Am. J. Hum. Genet. 105 (3), 625–630. doi:10.1016/j.ajhg.2019.06.011
Akawi, N., Al-Gazali, L., and Ali, B. (2012). Clinical and Molecular Analysis of UAE Fibrochondrogenesis Patients Expands the Phenotype and Reveals Two COL11A1 Homozygous Null Mutations. Clin. Genet. 82 (2), 147–156. doi:10.1111/j.1399-0004.2011.01734.x
Albert, S., Garanto, A., Sangermano, R., Khan, M., Bax, N. M., Hoyng, C. B., et al. (2018). Identification and Rescue of Splice Defects Caused by Two Neighboring Deep-Intronic ABCA4 Mutations Underlying Stargardt Disease. Am. J. Hum. Genet. 102 (4), 517–527. doi:10.1016/j.ajhg.2018.02.008
Alexieva, D., Long, Y., Sarkar, R., Dhayan, H., Bruet, E., Winston, R., et al. (2020). Background Splicing and Genetic Disease. Research Square [Preprint]. doi:10.21203/rs.3.rs-92665/v1
Alves, S., Mangas, M., Prata, M. J., Ribeiro, G., Lopes, L., Ribeiro, H., et al. (2006). Molecular Characterization of Portuguese Patients with Mucopolysaccharidosis Type II Shows Evidence that the IDS Gene Is Prone to Splicing Mutations. J. Inherit. Metab. Dis. 29 (6), 743–754. doi:10.1007/s10545-006-0403-z
Aminoff, M., Carter, J. E., Chadwick, R. B., Johnson, C., Gräsbeck, R., Abdelaal, M. A., et al. (1999). Mutations in CUBN, Encoding the Intrinsic Factor-Vitamin B12 Receptor, Cubilin, Cause Hereditary Megaloblastic Anaemia 1. Nat. Genet. 21 (3), 309–313. doi:10.1038/6831
Anczuków, O., Buisson, M., Léoné, M., Coutanson, C., Lasset, C., Calender, A., et al. (2012). BRCA2 Deep Intronic Mutation Causing Activation of a Cryptic Exon: Opening toward a New Preventive Therapeutic Strategy. Clin. Cancer Res. 18 (18), 4903–4909. doi:10.1158/1078-0432.CCR-12-1100
Abramowicz, A., and Gos, M. (2018). Splicing Mutations in Human Genetic Disorders: Examples, Detection, and Confirmation. J. Appl. Genet. 59 (3), 253–268. doi:10.1007/s13353-018-0444-7
Ars, E., Kruyer, H., Morell, M., Pros, E., Serra, E., Ravella, A., et al. (2003). Recurrent Mutations in the NF1 Gene Are Common Among Neurofibromatosis Type 1 Patients. J. Med. Genet. 40 (6), e82. doi:10.1136/jmg.40.6.e82
Ars, E., Serra, E., de la Luna, S., Estivill, X., and Lázaro, C. (2000). Cold Shock Induces the Insertion of a Cryptic Exon in the Neurofibromatosis Type 1 (NF1) mRNA. Nucleic Acids Res. 28 (6), 1307–1312. doi:10.1093/nar/28.6.1307
Attanasio, C., David, A., and Neerman-Arbez, M. (2003). Outcome of Donor Splice Site Mutations Accounting for Congenital Afibrinogenemia Reflects Order of Intron Removal in the Fibrinogen Alpha Gene (FGA). Blood 101 (5), 1851–1856. doi:10.1182/blood-2002-03-0853
Bach, J. E., Müller, C. R., and Rost, S. (2016). Mini-gene Assays Confirm the Splicing Effect of Deep Intronic Variants in the Factor VIII Gene. Thromb. Haemost. 115 (1), 222–224. doi:10.1160/TH15-05-0399
Backers, L., Parton, B., De Bruyne, M., Tavernier, S. J., Van Den Bogaert, K., Lambrecht, B. N., et al. (2021). Missing Heritability in Bloom Syndrome: First Report of a Deep Intronic Variant Leading to Pseudo‐exon Activation in the BLM Gene. Clin. Genet. 99 (2), 292–297. doi:10.1111/cge.13859
Bagnall, R. D., Ingles, J., Dinger, M. E., Cowley, M. J., Ross, S. B., Minoche, A. E., et al. (2018). Whole Genome Sequencing Improves Outcomes of Genetic Testing in Patients with Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 72 (4), 419–429. doi:10.1016/j.jacc.2018.04.078
Bagnall, R. D., Waseem, N. H., Green, P. M., Colvin, B., Lee, C., and Giannelli, F. (1999). Creation of a Novel Donor Splice Site in Intron 1 of the Factor VIII Gene Leads to Activation of a 191 Bp Cryptic Exon in Two Haemophilia A Patients. Br. J. Haematol. 107 (4), 766–771. doi:10.1046/j.1365-2141.1999.01767.x
Bampton, A., Gatt, A., Humphrey, J., Cappelli, S., Bhattacharya, D., Foti, S., et al. (2021). HnRNP K Mislocalisation Is a Novel Protein Pathology of Frontotemporal Lobar Degeneration and Ageing and Leads to Cryptic Splicing. Acta Neuropathol. 142 (4), 609–627. doi:10.1007/s00401-021-02340-0
Baressi, R., Di Blasi, C., Negri, T., Brugnoni, R., Vitali, A., Felisari, G., et al. (2000). Disruption of Heart Sarcoglycan Complex and Severe Cardiomyopathy Caused by Beta Sarcoglycan Mutations. J. Med. Genet. 37 (2), 102–107. doi:10.1136/jmg.37.2.102
Baskin, B., Gibson, W. T., and Ray, P. N. (2011). Duchenne Muscular Dystrophy Caused by a Complex Rearrangement between Intron 43 of the DMD Gene and Chromosome 4. Neuromuscul. Disord. 21 (3), 178–182. doi:10.1016/j.nmd.2010.11.008
Bauwens, M., Garanto, A., Sangermano, R., Naessens, S., Weisschuh, N., De Zaeytijd, J., et al. (2019). ABCA4-associated Disease as a Model for Missing Heritability in Autosomal Recessive Disorders: Novel Noncoding Splice, Cis-Regulatory, Structural, and Recurrent Hypomorphic Variants. Genet. Med. 21 (8), 1761–1771. doi:10.1038/s41436-018-0420-y
Baux, D., Vaché, C., Blanchet, C., Willems, M., Baudoin, C., Moclyn, M., et al. (2017). Combined Genetic Approaches Yield a 48% Diagnostic Rate in a Large Cohort of French Hearing-Impaired Patients. Sci. Rep. 7 (1), 16783. doi:10.1038/s41598-017-16846-9
Bergougnoux, A., Délétang, K., Pommier, A., Varilh, J., Houriez, F., Altieri, J. P., et al. (2019). Functional Characterization and Phenotypic Spectrum of Three Recurrent Disease-Causing Deep Intronic Variants of the CFTR Gene. J. Cystic Fibrosis 18 (4), 468–475. doi:10.1016/j.jcf.2018.10.012
Bergsma, A. J., in ‘t Groen, S. L., Verheijen, F. W., van der Ploeg, A. T., and Pijnappel, W. P. (2016). In 't Groen, S.L., Verheijen, F.W., van der Ploeg, A.T., and Pijnappel, WFrom Cryptic Toward Canonical Pre-mRNA Splicing in Pompe Disease: a Pipeline for the Development of Antisense Oligonucleotides. Mol. Ther. - Nucleic Acids 5 (9), e361. doi:10.1038/mtna.2016.75
Béroud, C., Carrié, A., Beldjord, C., Deburgrave, N., Llense, S., Carelle, N., et al. (2004). Dystrophinopathy Caused by Mid-intronic Substitutions Activating Cryptic Exons in the DMD Gene. Neuromuscul. Disord. 14 (1), 10–18. doi:10.1016/s0960-8966(03)00169-x
Bhola, Z., Smith, M. J., Byers, H. J., Miles, E. K., Evans, D. G., and Newman, W. G. (2014). Intronic Splicing Mutations in PTCH1 Cause Gorlin Syndrome. Fam. Cancer 13 (3), 477–480. doi:10.1007/s10689-014-9712-9
Blázquez, L., Azpitarte, M., Sáenz, A., Goicoechea, M., Otaegui, D., Ferrer, X., et al. (2008). Characterization of Novel CAPN3 Isoforms in white Blood Cells: an Alternative Approach for Limb-Girdle Muscular Dystrophy 2A Diagnosis. Neurogenetics 9 (3), 173–182. doi:10.1007/s10048-008-0129-1
Blázquez, L., Emmett, W., Faraway, R., Pineda, J. M. B., Bajew, S., Gohr, A., et al. (2018). Exon Junction Complex Shapes the Transcriptome by Repressing Recursive Splicing. Mol. Cell 72 (3), 496–509. doi:10.1016/j.molcel.2018.09.033
Bodle, E. E., Zhu, W., Velez-Bartolomei, F., Tesi-Rocha, A., Liu, P., and Bernstein, J. A. (2021). Combined Genome Sequencing and RNA Analysis Reveals and Characterizes a Deep Intronic Variant in IGHMBP2 in a Patient with Spinal Muscular Atrophy with Respiratory Distress Type 1. Pediatr. Neurol. 114, 16–20. doi:10.1016/j.pediatrneurol.2020.09.011
Boisson, B., Honda, Y., Ajiro, M., Bustamante, J., Bendavid, M., Gennery, A. R., et al. (2019). Rescue of Recurrent Deep Intronic Mutation Underlying Cell Type-dependent Quantitative NEMO Deficiency. J. Clin. Invest. 129 (2), 583–597. doi:10.1172/JCI124011
Bolduc, V., Foley, A. R., Solomon-Degefa, H., Sarathy, A., Donkervoort, S., Hu, Y., et al. (2019). A Recurrent COL6A1 Pseudoexon Insertion Causes Muscular Dystrophy and Is Effectively Targeted by Splice-Correction Therapies. JCI Insight 4 (6), 1. doi:10.1172/jci.insight.124403
Bonifert, T., Karle, K. N., Tonagel, F., Batra, M., Wilhelm, C., Theurer, Y., et al. (2014). Pure and Syndromic Optic Atrophy Explained by Deep Intronic OPA1 Mutations and an Intralocus Modifier. Brain 137 (Pt 8), 2164–2177. doi:10.1093/brain/awu165
Bonini, J., Varilh, J., Raynal, C., Thèze, C., Beyne, E., Audrezet, M.-P., et al. (2015). Small-scale High-Throughput Sequencing-Based Identification of New Therapeutic Tools in Cystic Fibrosis. Genet. Med. 17 (10), 796–806. doi:10.1038/gim.2014.194
Bovolenta, M., Neri, M., Fini, S., Fabris, M., Trabanelli, C., Venturoli, A., et al. (2008). A Novel Custom High Density-Comparative Genomic Hybridization Array Detects Common Rearrangements as Well as Deep Intronic Mutations in Dystrophinopathies. BMC Genomics 9, 572. doi:10.1186/1471-2164-9-572
Brasil, S., Viecelli, H. M., Meili, D., Rassi, A., Desviat, L. R., Pérez, B., et al. (2011). Pseudoexon Exclusion by Antisense Therapy in 6-Pyruvoyl-Tetrahydropterin Synthase Deficiency. Hum. Mutat. 32 (9), 1019–1027. doi:10.1002/humu.21529
Braun, T. A., Mullins, R. F., Wagner, A. H., Andorf, J. L., Johnston, R. M., Bakall, B. B., et al. (2013). Non-exomic and Synonymous Variants in ABCA4 Are an Important Cause of Stargardt Disease. Hum. Mol. Genet. 22 (25), 5136–5145. doi:10.1093/hmg/ddt367
Brinckmann, A., Mischung, C., Bässmann, I., Kühnisch, J., Schuelke, M., Tinschert, S., et al. (2007). Detection of novelNF1 Mutations and Rapid Mutation Prescreening with Pyrosequencing. Electrophoresis 28 (23), 4295–4301. doi:10.1002/elps.200700118
Broeders, M., Smits, K., Goynuk, B., Oussoren, E., van den Hout, H. J. M. P., Bergsma, A. J., et al. (2020). A Generic Assay to Detect Aberrant ARSB Splicing and mRNA Degradation for the Molecular Diagnosis of MPS VI. Mol. Ther. - Methods Clin. Dev. 19, 174–185. doi:10.1016/j.omtm.2020.09.004
Bryen, S. J., Oates, E. C., Evesson, F. J., Lu, J. K., Waddell, L. B., Joshi, H., et al. (2021). Pathogenic Deep Intronic MTM1 Variant Activates a Pseudo-exon Encoding a Nonsense Codon Resulting in Severe X-Linked Myotubular Myopathy. Eur. J. Hum. Genet. 29 (1), 61–66. doi:10.1038/s41431-020-00715-7
Burset, M., Seledtsov, I. A., and Solovyev, V. V. (2000). Analysis of Canonical and Non-canonical Splice Sites in Mammalian Genomes. Nucleic Acids Res. 28 (21), 4364–4375. doi:10.1093/nar/28.21.4364
Bustamante, J., Aksu, G., Vogt, G., de Beaucoudrey, L., Genel, F., Chapgier, A., et al. (2007). BCG-osis and Tuberculosis in a Child with Chronic Granulomatous Disease. J. Allergy Clin. Immunol. 120 (1), 32–38. doi:10.1016/j.jaci.2007.04.034
Caciotti, A., Tonin, R., Mort, M., Cooper, D. N., Gasperini, S., Rigoldi, M., et al. (2018). Mis-splicing of the GALNS Gene Resulting from Deep Intronic Mutations as a Cause of Morquio a Disease. BMC Med. Genet. 19 (1), 183. doi:10.1186/s12881-018-0694-6
Cagliani, R., Sironi, M., Ciafaloni, E., Bardoni, A., Fortunato, F., Prelle, A., et al. (2004). An Intragenic Deletion/inversion Event in the DMD Gene Determines a Novel Exon Creation and Results in a BMD Phenotype. Hum. Genet. 115 (1), 13–18. doi:10.1007/s00439-004-1118-6
Campeau, E., Dupuis, L., Leclerc, D., and Gravel, R. A. (1999). Detection of a Normally Rare Transcript in Propionic Acidemia Patients with mRNA Destabilizing Mutations in the PCCA Gene. Hum. Mol. Genet. 8 (1), 107–113. doi:10.1093/hmg/8.1.107
Canson, D., Glubb, D., and Spurdle, A. B. (2020). Variant Effect on Splicing Regulatory Elements, Branchpoint Usage, and Pseudoexonization: Strategies to Enhance Bioinformatic Prediction Using Hereditary Cancer Genes as Exemplars. Hum. Mutat. 41 (10), 1705–1721. doi:10.1002/humu.24074
Caparrós-Martín, J. A., De Luca, A., Cartault, F., Aglan, M., Temtamy, S., Otaify, G. A., et al. (2015). Specific Variants in WDR35 Cause a Distinctive Form of Ellis-van Creveld Syndrome by Disrupting the Recruitment of the EvC Complex and SMO into the Cilium. Hum. Mol. Genet. 24 (14), 4126–4137. doi:10.1093/hmg/ddv152
Carmody, D., Park, S.-Y., Ye, H., Perrone, M. E., Alkorta-Aranburu, G., Highland, H. M., et al. (2015). Continued Lessons from theINSgene: an Intronic Mutation Causing Diabetes through a Novel Mechanism. J. Med. Genet. 52 (9), 612–616. doi:10.1136/jmedgenet-2015-103220
Carvill, G. L., and Mefford, H. C. (2020). Poison Exons in Neurodevelopment and Disease. Curr Opin Genet Dev 65, 98–102. doi:10.1016/j.gde.2020.05.030
Cassini, T. A., Duncan, L., Rives, L. C., Newman, J. H., Phillips, J. A., Koziura, M. E., et al. (2019). Whole Genome Sequencing Reveals Novel IGHMBP2 Variant Leading to Unique Cryptic Splice-Site and Charcot-Marie-Tooth Phenotype with Early Onset Symptoms. Mol. Genet. Genomic Med. 7 (6), e00676. doi:10.1002/mgg3.676
Castaman, G., Giacomelli, S. H., Mancuso, M. E., D'Andrea, G., Santacroce, R., Sanna, S., et al. (2011). Deep Intronic Variations May Cause Mild Hemophilia A. J. Thromb. Haemost. 9 (8), 1541–1548. doi:10.1111/j.1538-7836.2011.04408.x
Castellanos, E., Rosas, I., Rosas, I., Solanes, A., Bielsa, I., Lázaro, C., et al. (2013). In Vitro antisense Therapeutics for a Deep Intronic Mutation Causing Neurofibromatosis Type 2. Eur. J. Hum. Genet. 21 (7), 769–773. doi:10.1038/ejhg.2012.261
Castoldi, E., Duckers, C., Radu, C., Spiezia, L., Rossetto, V., Tagariello, G., et al. (2011). Homozygous F5 Deep-Intronic Splicing Mutation Resulting in Severe Factor V Deficiency and Undetectable Thrombin Generation in Platelet-Rich Plasma. J. Thromb. Haemost. 9 (5), 959–968. doi:10.1111/j.1538-7836.2011.04237.x
Catania, A., Ardissone, A., Verrigni, D., Legati, A., Newman, J. H., Phillips, J. A., Koziura, M. E., et al. (2019). Whole Genome Sequencing Reveals Novel IGHMBP2 Variant Leading to Unique Cryptic Splice-Site and Charcot-Marie-Tooth Phenotype with Early Onset Symptoms. Mol. Genet. Genomic Med. 7 (6), e00676. doi:10.1002/mgg3.676
Cavalieri, S., Pozzi, E., Gatti, R. A., and Brusco, A. (2013). Deep-intronic ATM Mutation Detected by Genomic Resequencing and Corrected In Vitro by Antisense Morpholino Oligonucleotide (AMO). Eur. J. Hum. Genet. 21 (7), 774–778. doi:10.1038/ejhg.2012.266
Cavestro, C., Panteghini, C., Reale, C., Nasca, A., Fenu, S., Salsano, E., et al. (2021). Novel Deep Intronic Mutation in PLA2G6 Causing Early-Onset Parkinson's Disease with Brain Iron Accumulation through Pseudo-exon Activation. Neurogenetics 22 (4), 347–351. doi:10.1007/s10048-021-00667-0
Chang, C. Y., Perng, C. L., Cheng, S. N., Hu, S. H., Wu, T. Y., Lin, S. Y., et al. (2019). Deep Intronic Variant c.5999‐277G>A of F8 Gene May Be a Hot Spot Mutation for Mild Hemophilia A Patients without Mutation in Exonic DNA. Eur. J. Haematol. 103 (1), 47–55. doi:10.1111/ejh.13242
Chatron, N., Schluth‐Bolard, C., Frétigny, M., Labalme, A., Vilchez, G., Castet, S. M., et al. (2019). Severe Hemophilia A Caused by an Unbalanced Chromosomal Rearrangement Identified Using Nanopore Sequencing. J. Thromb. Haemost. 17 (7), 1097–1103. doi:10.1111/jth.14460
Chen, J., Ma, N., Zhao, X., Li, W., Zhang, Q., Yuan, S., et al. (2019). A Rare Deep Intronic Mutation of PKHD1 Gene, c.8798-459 C > A, Causes Autosomal Recessive Polycystic Kidney Disease by Pseudoexon Activation. J. Hum. Genet. 64 (3), 207–214. doi:10.1038/s10038-018-0550-8
Chen, X., Truong, T.-T. N., Weaver, J., Bove, B. A., Cattie, K., Armstrong, B. A., et al. (2006). Intronic Alterations inBRCA1andBRCA2: Effect on mRNA Splicing Fidelity and Expression. Hum. Mutat. 27 (5), 427–435. doi:10.1002/humu.20319
Cheng, T. C., Orkin, S. H., Antonarakis, S. E., Potter, M. J., Sexton, J. P., Markham, A. F., et al. (1984). Beta-Thalassemia in Chinese: Use of In Vivo RNA Analysis and Oligonucleotide Hybridization in Systematic Characterization of Molecular Defects. Proc. Natl. Acad. Sci. 81 (9), 2821–2825. doi:10.1073/pnas.81.9.2821
Chillón, M., Dörk, T., Casals, T., Giménez, J., Fonknechten, N., Will, K., et al. (1995). A Novel Donor Splice Site in Intron 11 of the CFTR Gene, Created by Mutation 1811+1.6kbA-->G, Produces a New Exon: High Frequency in Spanish Cystic Fibrosis Chromosomes and Association with Severe Phenotype. Am. J. Hum. Genet. 56 (3), 623–629.
Chiu, Y.-H., Chang, Y.-C., Chang, Y.-H., Niu, D.-M., Yang, Y.-L., Ye, J., et al. (2012). Mutation Spectrum of and Founder Effects Affecting the PTS Gene in East Asian Populations. J. Hum. Genet. 57 (2), 145–152. doi:10.1038/jhg.2011.146
Chmel, N., Danescu, S., Gruler, A., Kiritsi, D., Bruckner-Tuderman, L., Kreuter, A., et al. (2015). A Deep-Intronic FERMT1 Mutation Causes Kindler Syndrome: An Explanation for Genetically Unsolved Cases. J. Invest. Dermatol. 135 (11), 2876–2879. doi:10.1038/jid.2015.227
Chorev, M., and Carmel, L. (2012). The Function of Introns. Front. Gene 3, 55. doi:10.3389/fgene.2012.00055
Chorin, O., Yachelevich, N., Mohamed, K., Moscatelli, I., Pappas, J., Henriksen, K., et al. (2020). Transcriptome Sequencing Identifies a Noncoding, Deep Intronic Variant in CLCN7 Causing Autosomal Recessive Osteopetrosis. Mol. Genet. Genomic Med. 8 (10), e1405. doi:10.1002/mgg3.1405
Christie, P. T., Harding, B., Nesbit, M. A., Whyte, M. P., and Thakker, R. V. (2001). X-linked Hypophosphatemia Attributable to Pseudoexons of the PHEX Gene. J. Clin. Endocrinol. Metab. 86 (8), 3840–3844. doi:10.1210/jcem.86.8.7730
Chung, W. Y., Cho, M. H., Gu, Y.-R., Leem, S.-H., and Cheong, H. I. (2012). Medullary Sponge Kidney Detected in the Pediatric Age. J. Korean Soc. Pediatr. Nephrol. 16 (2), 1. doi:10.3339/jkspn.2012.16.2.109
Clemens, D. J., Tester, D. J., Marty, I., and Ackerman, M. J. (2020). Phenotype-guided Whole Genome Analysis in a Patient with Genetically Elusive Long QT Syndrome Yields a Novel TRDN-Encoded Triadin Pathogenetic Substrate for Triadin Knockout Syndrome and Reveals a Novel Primate-specific Cardiac TRDN Transcript. Heart Rhythm 17 (6), 1017–1024. doi:10.1016/j.hrthm.2020.01.012
Clendenning, M., Buchanan, D. D., Walsh, M. D., Nagler, B., Rosty, C., Thompson, B., et al. (2011). Mutation Deep within an Intron of MSH2 Causes Lynch Syndrome. Fam. Cancer 10 (2), 297–301. doi:10.1007/s10689-011-9427-0
Conboy, J. G. (2021). Unannotated Splicing Regulatory Elements in Deep Intron Space. Wiley Interdiscip. Rev. RNA 12 (5), e1656. doi:10.1002/wrna.1656
Corrigan, A., Arenas, M., Escuredo, E., Fairbanks, L., and Marinaki, A. (2011). HPRT Deficiency: Identification of Twenty-Four Novel Variants Including an Unusual Deep Intronic Mutation. Nucleosides, Nucleotides and Nucleic Acids 30 (12), 1260–1265. doi:10.1080/15257770.2011.590172
Corvelo, A., Hallegger, M., Smith, C. W. J., and Eyras, E. (2010). Genome-wide Association between branch point Properties and Alternative Splicing. Plos Comput. Biol. 6 (11), e1001016. doi:10.1371/journal.pcbi.1001016
Costantino, L., Claut, L., Paracchini, V., Coviello, D. A., Colombo, C., Porcaro, L., et al. (2010). A Novel Donor Splice Site Characterized by CFTR mRNA Analysis Induces a New Pseudo-exon in CF Patients. J. Cystic Fibrosis 9 (6), 411–418. doi:10.1016/j.jcf.2010.08.009
Cottrell, E., Maharaj, A., Williams, J., Chatterjee, S., Cirillo, G., Miraglia Del Giudice, E., et al. (2021). Growth Hormone Receptor (GHR) 6Ω Pseudoexon Activation: a Novel Cause of Severe Growth Hormone Insensitivity. J. Clin. Endocrinol. Metab. 107 (1), 401–416. doi:10.1210/clinem/dgab550
Coutinho, G., Xie, J., Du, L., Brusco, A., Krainer, A. R., and Gatti, R. A. (2005). Functional Significance of a Deep Intronic Mutation in the ATM Gene and Evidence for an Alternative Exon 28a. Hum. Mutat. 25 (2), 118–124. doi:10.1002/humu.20170
Coutinho, M. F., da Silva Santos, L., Lacerda, L., Quental, S., Wibrand, F., Lund, A. M., et al. (2012). Alu-Alu Recombination Underlying the First Large Genomic Deletion in GlcNAc-Phosphotransferase Alpha/Beta (GNPTAB) Gene in a MLII Alpha/Beta Patient. JIMD Rep. 4, 117–124. doi:10.1007/8904_2011_83
Covello, G., Ibrahim, G. H., Bacchi, N., Casarosa, S., and Denti, M. A. (2021). Exon Skipping via Chimeric Antisense U1 snRNAs to Correct Retinitis Pigmentosa GTPase-Regulator (RPGR) Splice Defect. bioRxiv [preprint]. doi:10.1101/2021.06.26.449721 (Accessed September 09, 2021)
Cummings, B. B., Marshall, J. L., Tukiainen, T., Lek, M., Donkervoort, S., Foley, A. R., et al. (2017). Improving Genetic Diagnosis in Mendelian Disease with Transcriptome Sequencing. Sci. Transl Med. 9 (386), 1. doi:10.1126/scitranslmed.aal5209
Danilenko, M., Dalgliesh, C., Pagliarini, V., Naro, C., Ehrmann, I., Feracci, M., et al. (2017). Binding Site Density Enables Paralog-specific Activity of SLM2 and Sam68 Proteins in Neurexin2 AS4 Splicing Control. Nucleic Acids Res. 45 (7), 4120–4130. doi:10.1093/nar/gkw1277
Davis, R. L., Homer, V. M., George, P. M., and Brennan, S. O. (2009). A Deep Intronic Mutation inFGBcreates a Consensus Exonic Splicing Enhancer Motif that Results in Afibrinogenemia Caused by Aberrant mRNA Splicing, Which Can Be Corrected In Vitro with Antisense Oligonucleotide Treatment. Hum. Mutat. 30 (2), 221–227. doi:10.1002/humu.20839
de Boer, M., van Leeuwen, K., Geissler, J., Weemaes, C. M., van den Berg, T. K., Kuijpers, T. W., et al. (2014). Primary Immunodeficiency Caused by an Exonized Retroposed Gene Copy Inserted in theCYBBGene. Hum. Mutat. 35 (4), 486–496. doi:10.1002/humu.22519
De Gasperi, R., Gama Sosa, M. A., Sartorato, E. L., Battistini, S., MacFarlane, H., Gusella, J. F., et al. (1996). Molecular Heterogeneity of Late-Onset Forms of Globoid-Cell Leukodystrophy. Am. J. Hum. Genet. 59 (6), 1233–1242.
De Klein, A., Riegman, P. H., Bijlsma, E. K., Heldoorn, A., Muijtjens, M., den Bakker, M. A., et al. (1998). A G-->A Transition Creates a branch point Sequence and Activation of a Cryptic Exon, Resulting in the Hereditary Disorder Neurofibromatosis 2. Hum. Mol. Genet. 7 (3), 393–398. doi:10.1093/hmg/7.3.393
Dear, A., Daly, J., Brennan, S. O., Tuckfield, A., and George, P. M. (2006). An Intronic Mutation within FGB (IVS1+2076 Ag) Is Associated with Afibrinogenemia and Recurrent Transient Ischemic Attacks. J. Thromb. Haemost. 4 (2), 471–472. doi:10.1111/j.1538-7836.2006.01722.x
Deburgrave, N., Daoud, F., Llense, S., Barbot, J. C., Récan, D., Peccate, C., et al. (2007). Protein- and mRNA-Based Phenotype-Genotype Correlations in DMD/BMD with point Mutations and Molecular Basis for BMD with Nonsense and Frameshift Mutations in the DMD Gene. Hum. Mutat. 28 (2), 183–195. doi:10.1002/humu.20422
Dehainault, C., Michaux, D., Pagès-Berhouet, S., Caux-Moncoutier, V., Doz, F., Desjardins, L., et al. (2007). A Deep Intronic Mutation in the RB1 Gene Leads to Intronic Sequence Exonisation. Eur. J. Hum. Genet. 15 (4), 473–477. doi:10.1038/sj.ejhg.5201787
den Dunnen, J. T., Dalgleish, R., Maglott, D. R., Hart, R. K., Greenblatt, M. S., McGowan-Jordan, J., et al. (2016). HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 37 (6), 564–569. doi:10.1002/humu.22981
den Hollander, A. I., Koenekoop, R. K., Yzer, S., Lopez, I., Arends, M. L., Voesenek, K. E. J., et al. (2006). Mutations in the CEP290 (NPHP6) Gene Are a Frequent Cause of Leber Congenital Amaurosis. Am. J. Hum. Genet. 79 (3), 556–561. doi:10.1086/507318
Dericquebourg, A., Jourdy, Y., Fretigny, M., Lienhart, A., Claeyssens, S., Ternisien, C., et al. (2020). Identification of New F8 Deep Intronic Variations in Patients with Haemophilia A. Haemophilia 26 (5), 847–854. doi:10.1111/hae.14134
Dhir, A., and Buratti, E. (2010). Alternative Splicing: Role of Pseudoexons in Human Disease and Potential Therapeutic Strategies. FEBS J. 277 (4), 841–855. doi:10.1111/j.1742-4658.2009.07520.x
Di Scipio, M., Tavares, E., Deshmukh, S., Audo, I., Green-Sanderson, K., Zubak, Y., et al. (2020). Phenotype Driven Analysis of Whole Genome Sequencing Identifies Deep Intronic Variants that Cause Retinal Dystrophies by Aberrant Exonization. Invest. Ophthalmol. Vis. Sci. 61 (10), 36. doi:10.1167/iovs.61.10.36
Dobkin, C., and Bank, A. (1985). Reversibility of IVS 2 Missplicing in a Mutant Human Beta-Globin Gene. J. Biol. Chem. 260 (30), 16332–16337. doi:10.1016/s0021-9258(17)36241-5
Dobkin, C., Pergolizzi, R. G., Bahre, P., and Bank, A. (1983). Abnormal Splice in a Mutant Human Beta-Globin Gene Not at the Site of a Mutation. Proc. Natl. Acad. Sci. 80 (5), 1184–1188. doi:10.1073/pnas.80.5.1184
Dominov, J. A., Uyan, Ö., McKenna‐Yasek, D., Nallamilli, B. R. R., Kergourlay, V., Bartoli, M., et al. (2019). Correction of Pseudoexon Splicing Caused by a Novel Intronic Dysferlin Mutation. Ann. Clin. Transl Neurol. 6 (4), 642–654. doi:10.1002/acn3.738
Dominov, J. A., Uyan, Ö., Sapp, P. C., McKenna‐Yasek, D., Nallamilli, B. R. R., Hegde, M., et al. (2014). A Novel Dysferlin Mutant Pseudoexon Bypassed with Antisense Oligonucleotides. Ann. Clin. Transl Neurol. 1 (9), 703–720. doi:10.1002/acn3.96
Drewe-Boss, P., Wessels, H.-H., and Ohler, U. (2018). omniCLIP: Probabilistic Identification of Protein-RNA Interactions from CLIP-Seq Data. Genome Biol. 19 (1), 183. doi:10.1186/s13059-018-1521-2
Drexler, H. L., Choquet, K., and Churchman, L. S. (2020). Splicing Kinetics and Coordination Revealed by Direct Nascent RNA Sequencing through Nanopores. Mol. Cell 77 (5), 985–998. e988. doi:10.1016/j.molcel.2019.11.017
Dwi Pramono, Z. A., Takeshima, Y., Surono, A., Ishida, T., and Matsuo, M. (2000). A Novel Cryptic Exon in Intron 2 of the Human Dystrophin Gene Evolved from an Intron by Acquiring Consensus Sequences for Splicing at Different Stages of Anthropoid Evolution. Biochem. Biophysical Res. Commun. 267 (1), 321–328. doi:10.1006/bbrc.1999.1962
Ellingford, J. M., Thomas, H. B., Rowlands, C., Arno, G., Beaman, G., Gomes-Silva, B., et al. (2019). Functional and In-Silico Interrogation of Rare Genomic Variants Impacting RNA Splicing for the Diagnosis of Genomic Disorders. BioRxiv. doi:10.1101/781088 (Accessed June 09, 2021)
Engel, K., Nuoffer, J.-M., Mühlhausen, C., Klaus, V., Largiadèr, C. R., Tsiakas, K., et al. (2008). Analysis of mRNA Transcripts Improves the success Rate of Molecular Genetic Testing in OTC Deficiency. Mol. Genet. Metab. 94 (3), 292–297. doi:10.1016/j.ymgme.2008.03.009
Evans, D. G., Bowers, N., Burkitt-Wright, E., Miles, E., Garg, S., Scott-Kitching, V., et al. (2016). Comprehensive RNA Analysis of the NF1 Gene in Classically Affected NF1 Affected Individuals Meeting NIH Criteria Has High Sensitivity and Mutation Negative Testing Is Reassuring in Isolated Cases with Pigmentary Features Only. EBioMedicine 7, 212–220. doi:10.1016/j.ebiom.2016.04.005
Faà, V., Incani, F., Meloni, A., Corda, D., Masala, M., Baffico, A. M., et al. (2009). Characterization of a Disease-Associated Mutation Affecting a Putative Splicing Regulatory Element in Intron 6b of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Gene. J. Biol. Chem. 284 (44), 30024–30031. doi:10.1074/jbc.M109.032623
Fadaie, Z., Khan, M., Del Pozo‐Valero, M., Cornelis, S. S., Ayuso, C., Cremers, F. P. M., et al. (2019). Identification of Splice Defects Due to Noncanonical Splice Site or Deep‐intronic Variants in ABCA4. Hum. Mutat. 40 (12), 2365–2376. doi:10.1002/humu.23890
Fadaie, Z., Whelan, L., Ben-Yosef, T., Dockery, A., Corradi, Z., Gilissen, C., et al. (2021). Whole genome sequencing and in vitro splice assays reveal genetic causes for inherited retinal diseases. NPJ Genom Med 6 (1), 97. doi:10.1038/s41525-021-00261-1
Falzarano, M. S., Grilli, A., Zia, S., Fang, M., Rossi, R., Gualandi, F., et al. (2022). RNA-seq in DMD Urinary Stem Cells Recognized Muscle-Related Transcription Signatures and Addressed the Identification of Atypical Mutations by Whole-Genome Sequencing. Hum. Genet. Genomics Adv. 3 (1), 100054. doi:10.1016/j.xhgg.2021.100054
Fanin, M., Torella, A., Savarese, M., Nigro, V., and Angelini, C. (2015). GYG1gene Mutations in a Family with Polyglucosan Body Myopathy. Neurol. Genet. 1 (3), e21. doi:10.1212/NXG.0000000000000021
Farlow, A., Dolezal, M., Hua, L., and Schlötterer, C. (2012). The Genomic Signature of Splicing-Coupled Selection Differs between Long and Short Introns. Mol. Biol. Evol. 29 (1), 21–24. doi:10.1093/molbev/msr201
Ferlini, A., Galié, N., Merlini, L., Sewry, C., Branzi, A., and Muntoni, F. (1998). A Novel Alu-like Element Rearranged in the Dystrophin Gene Causes a Splicing Mutation in a Family with X-Linked Dilated Cardiomyopathy. Am. J. Hum. Genet. 63 (2), 436–446. doi:10.1086/301952
Ferlini, A., and Muntoni, F. (1998). The 5′ Region of Intron 11 of the Dystrophin Gene Contains Target Sequences for Mobile Elements and Three Overlapping ORFs. Biochem. Biophysical Res. Commun. 242 (2), 401–406. doi:10.1006/bbrc.1997.7976
Fernández-Rodríguez, J., Castellsagué, J., Benito, L., Benavente, Y., Capellá, G., Blanco, I., et al. (2011). A Mild Neurofibromatosis Type 1 Phenotype Produced by the Combination of the Benign Nature of a Leaky NF1-Splice Mutation and the Presence of a Complex Mosaicism. Hum. Mutat. 32 (7), 705–709. doi:10.1002/humu.21500
Ferraresi, P., Balestra, D., Guittard, C., Buthiau, D., Pan-Petesh, B., Maestri, I., et al. (2020). Next-generation Sequencing and Recombinant Expression Characterized Aberrant Splicing Mechanisms and provided Correction Strategies in Factor VII Deficiency. Haematologica 105 (3), 829–837. doi:10.3324/haematol.2019.217539
Filoni, C., Caciotti, A., Carraresi, L., Donati, M. A., Mignani, R., Parini, R., et al. (2008). Unbalanced GLA mRNAs Ratio Quantified by Real-Time PCR in Fabry Patients' Fibroblasts Results in Fabry Disease. Eur. J. Hum. Genet. 16 (11), 1311–1317. doi:10.1038/ejhg.2008.109
Fisher, C. W., Fisher, C. R., Chuang, J. L., Lau, K. S., Chuang, D. T., and Cox, R. P. (1993). Occurrence of a 2-bp (AT) Deletion Allele and a Nonsense (G-To-T) Mutant Allele at the E2 (DBT) Locus of Six Patients with maple Syrup Urine Disease: Multiple-Exon Skipping as a Secondary Effect of the Mutations. Am. J. Hum. Genet. 52 (2), 414–424.
Fitzgerald, J., Feist, C., Dietz, P., Moore, S., and Basel, D. (2020). A Deep Intronic Variant Activates a Pseudoexon in the MTM1 Gene in a Family with X-Linked Myotubular Myopathy. Mol. Syndromol 11 (5-6), 264–270. doi:10.1159/000510286
Flanagan, S. E., Xie, W., Caswell, R., Damhuis, A., Vianey-Saban, C., Akcay, T., et al. (2013). Next-generation Sequencing Reveals Deep Intronic Cryptic ABCC8 and HADH Splicing Founder Mutations Causing Hyperinsulinism by Pseudoexon Activation. Am. J. Hum. Genet. 92 (1), 131–136. doi:10.1016/j.ajhg.2012.11.017
Friedrich, K., Lee, L., Leistritz, D. F., Nürnberg, G., Saha, B., Hisama, F. M., et al. (2010). WRN Mutations in Werner Syndrome Patients: Genomic Rearrangements, Unusual Intronic Mutations and Ethnic-specific Alterations. Hum. Genet. 128 (1), 103–111. doi:10.1007/s00439-010-0832-5
Frio, T. R., McGee, T. L., Wade, N. M., Iseli, C., Beckmann, J. S., Berson, E. L., et al. (2009). A Single-Base Substitution within an Intronic Repetitive Element Causes Dominant Retinitis Pigmentosa with Reduced Penetrance. Hum. Mutat. 30 (9), 1340–1347. doi:10.1002/humu.21071
Garcia Segarra, N., Mittaz, L., Campos-Xavier, A. B., Bartels, C. F., Tuysuz, B., Alanay, Y., et al. (2012). The Diagnostic challenge of Progressive Pseudorheumatoid Dysplasia (PPRD): a Review of Clinical Features, Radiographic Features, and WISP3 Mutations in 63 Affected Individuals. Am. J. Med. Genet. 160C (3), 217–229. doi:10.1002/ajmg.c.31333
Garea, J. S. (2006). Identification and Characterisation of CFTR Mutations at Transcript Level and Association with Disease Severity in Cystic Fibrosis PhD Dissertation. Bern, Switzerland: Universität Bern.
Geng, X., Liu, Y., Ren, X., Guan, Y., Wang, Y., Mao, B., et al. (2018). Novel NTRK1 Mutations in Chinese Patients with Congenital Insensitivity to Pain with Anhidrosis. Mol. Pain 14, 1744806918781140. doi:10.1177/1744806918781140
Gillis, E., Kempers, M., Salemink, S., Timmermans, J., Cheriex, E. C., Bekkers, S. C. A. M., et al. (2014). AnFBN1Deep Intronic Mutation in a Familial Case of Marfan Syndrome: An Explanation for Genetically Unsolved Cases? Hum. Mutat. 35 (5), 571–574. doi:10.1002/humu.22540
Gómez-Grau, M., Albaigès, J., Casas, J., Auladell, C., Dierssen, M., Vilageliu, L., et al. (2017). New Murine Niemann-Pick Type C Models Bearing a Pseudoexon-Generating Mutation Recapitulate the Main Neurobehavioural and Molecular Features of the Disease. Sci. Rep. 7, 41931. doi:10.1038/srep41931
Gonçalves, A., Oliveira, J., Coelho, T., Taipa, R., Melo-Pires, M., Sousa, M., et al. (2017). Exonization of an Intronic LINE-1 Element Causing Becker Muscular Dystrophy as a Novel Mutational Mechanism in Dystrophin Gene. Basel: Genes, 8. doi:10.3390/genes8100253
Gonorazky, H. D., Naumenko, S., Ramani, A. K., Nelakuditi, V., Mashouri, P., Wang, P., et al. (2019). Expanding the Boundaries of RNA Sequencing as a Diagnostic Tool for Rare Mendelian Disease. Am. J. Hum. Genet. 104 (3), 466–483. doi:10.1016/j.ajhg.2019.01.012
Gonorazky, H., Liang, M., Cummings, B., Lek, M., Micallef, J., Hawkins, C., et al. (2016). RNA Seq Analysis for the Diagnosis of Muscular Dystrophy. Ann. Clin. Transl Neurol. 3 (1), 55–60. doi:10.1002/acn3.267
Gooding, C., Clark, F., Wollerton, M. C., Grellscheid, S.-N., Groom, H., and Smith, C. W. (2006). A Class of Human Exons with Predicted Distant branch Points Revealed by Analysis of AG Dinucleotide Exclusion Zones. Genome Biol. 7 (1), R1. doi:10.1186/gb-2006-7-1-r1
Goossens, R., van den Boogaard, M. L., Lemmers, R. J. L. F., Balog, J., van der Vliet, P. J., Willemsen, I. M., et al. (2019). Intronic SMCHD1 Variants in FSHD: Testing the Potential for CRISPR-Cas9 Genome Editing. J. Med. Genet. 56 (12), 828–837. doi:10.1136/jmedgenet-2019-106402
Greer, K., Mizzi, K., Rice, E., Kuster, L., Barrero, R. A., Bellgard, M. I., et al. (2015). Pseudoexon Activation Increases Phenotype Severity in a Becker Muscular Dystrophy Patient. Mol. Genet. Genomic Med. 3 (4), 320–326. doi:10.1002/mgg3.144
Grodecká, L., Kováčová, T., Kramárek, M., Seneca, S., Stouffs, K., De Laet, C., et al. (2017). Detailed Molecular Characterization of a Novel IDS Exonic Mutation Associated with Multiple Pseudoexon Activation. J. Mol. Med. 95 (3), 299–309. doi:10.1007/s00109-016-1484-2
Gualandi, F., Rimessi, P., Trabanelli, C., Spitali, P., Neri, M., Patarnello, T., et al. (2006). Intronic Breakpoint Definition and Transcription Analysis in DMD/BMD Patients with Deletion/duplication at the 5′ Mutation Hot Spot of the Dystrophin Gene. Gene 370, 26–33. doi:10.1016/j.gene.2005.11.002
Guo, D. C., Gupta, P., Tran-Fadulu, V., Guidry, T. V., Leduc, M. S., Schaefer, F. V., et al. (2008). An FBN1 Pseudoexon Mutation in a Patient with Marfan Syndrome: Confirmation of Cryptic Mutations Leading to Disease. J. Hum. Genet. 53 (11-12), 1007–1011. doi:10.1007/s10038-008-0334-7
Guo, L., Bertola, D. R., Takanohashi, A., Saito, A., Segawa, Y., Yokota, T., et al. (2019). Bi-allelic CSF1R Mutations Cause Skeletal Dysplasia of Dysosteosclerosis-Pyle Disease Spectrum and Degenerative Encephalopathy with Brain Malformation. Am. J. Hum. Genet. 104 (5), 925–935. doi:10.1016/j.ajhg.2019.03.004
Gurvich, O. L., Tuohy, T. M., Howard, M. T., Finkel, R. S., Medne, L., Anderson, C. B., et al. (2008). DMDpseudoexon Mutations: Splicing Efficiency, Phenotype, and Potential Therapy. Ann. Neurol. 63 (1), 81–89. doi:10.1002/ana.21290
Hamanaka, K., Miyatake, S., Koshimizu, E., Tsurusaki, Y., Mitsuhashi, S., Iwama, K., et al. (2019). RNA Sequencing Solved the Most Common but Unrecognized NEB Pathogenic Variant in Japanese Nemaline Myopathy. Genet. Med. 21 (7), 1629–1638. doi:10.1038/s41436-018-0360-6
Han, A., Stoilov, P., Linares, A. J., Zhou, Y., Fu, X.-D., and Black, D. L. (2014). De Novo prediction of PTBP1 Binding and Splicing Targets Reveals Unexpected Features of its RNA Recognition and Function. Plos Comput. Biol. 10 (1), e1003442. doi:10.1371/journal.pcbi.1003442
Hastings, M. L., Resta, N., Traum, D., Stella, A., Guanti, G., and Krainer, A. R. (2005). An LKB1 AT-AC Intron Mutation Causes Peutz-Jeghers Syndrome via Splicing at Noncanonical Cryptic Splice Sites. Nat. Struct. Mol. Biol. 12 (1), 54–59. doi:10.1038/nsmb873
Helman, G., Compton, A. G., Hock, D. H., Walkiewicz, M., Brett, G. R., Pais, L., et al. (2021). Multiomic Analysis Elucidates Complex I Deficiency Caused by a Deep Intronic Variant in NDUFB10. Hum. Mutat. 42 (1), 19–24. doi:10.1002/humu.24135
Highsmith, W. E., Burch, L. H., Zhou, Z., Olsen, J. C., Boat, T. E., Spock, A., et al. (1994). A Novel Mutation in the Cystic Fibrosis Gene in Patients with Pulmonary Disease but normal Sweat Chloride Concentrations. N. Engl. J. Med. 331 (15), 974–980. doi:10.1056/NEJM199410133311503
Higuchi, T., Kobayashi, M., Ogata, J., Kaneshiro, E., Shimada, Y., Kobayashi, H., et al. (2016). Identification of Cryptic Novel α-Galactosidase A Gene Mutations: Abnormal mRNA Splicing and Large Deletions. JIMD Rep. 30, 63–72. doi:10.1007/8904_2015_475
Hilgert, N., Topsakal, V., van Dinther, J., Offeciers, E., Van de Heyning, P., and Van Camp, G. (2008). A Splice-Site Mutation and Overexpression of MYO6 Cause a Similar Phenotype in Two Families with Autosomal Dominant Hearing Loss. Eur. J. Hum. Genet. 16 (5), 593–602. doi:10.1038/sj.ejhg.5202000
Hiraide, T., Nakashima, M., Ikeda, T., Tanaka, D., Osaka, H., and Saitsu, H. (2020). Identification of a Deep Intronic POLR3A Variant Causing Inclusion of a Pseudoexon Derived from an Alu Element in Pol III-Related Leukodystrophy. J. Hum. Genet. 65 (10), 921–925. doi:10.1038/s10038-020-0786-y
Holliday, M., Singer, E. S., Ross, S. B., Lim, S., Lal, S., Ingles, J., et al. (2021). Transcriptome Sequencing of Patients with Hypertrophic Cardiomyopathy Reveals Novel Splice-Altering Variants in MYBPC3. Circ. Genom Precis Med. 14 (2), e003202. doi:10.1161/CIRCGEN.120.003202
Homolova, K., Zavadakova, P., Doktor, T. K., Schroeder, L. D., Kozich, V., and Andresen, B. S. (2010). The Deep Intronic c.903+469T>C Mutation in the MTRR Gene Creates an SF2/ASF Binding Exonic Splicing Enhancer, Which Leads to Pseudoexon Activation and Causes the cblE Type of Homocystinuria. Hum. Mutat. 31 (4), 437–444. doi:10.1002/humu.21206
Horinouchi, T., Nozu, K., Yamamura, T., Minamikawa, S., Omori, T., Nakanishi, K., et al. (2018). Detection of Splicing Abnormalities and Genotype-Phenotype Correlation in X-Linked Alport Syndrome. Jasn 29 (8), 2244–2254. doi:10.1681/ASN.2018030228
Howe, K. L., Achuthan, P., Allen, J., Allen, J., Alvarez-Jarreta, J., Amode, M. R., et al. (2021). Ensembl 2021. Nucleic Acids Res. 49 (D1), D884–D891. doi:10.1093/nar/gkaa942
Hug, N., Longman, D., and Cáceres, J. F. (2016). Mechanism and Regulation of the Nonsense-Mediated Decay Pathway. Nucleic Acids Res. 44 (4), 1483–1495. doi:10.1093/nar/gkw010
Huizing, M., Anikster, Y., Fitzpatrick, D. L., Jeong, A. B., D’Souza, M., Rausche, M., et al. (2001). Hermansky-Pudlak Syndrome Type 3 in Ashkenazi Jews and Other Non-Puerto Rican Patients with Hypopigmentation and Platelet Storage-Pool Deficiency. Am. J. Hum. Genet. 69 (5), 1022–1032. doi:10.1086/324168
Hujová, P., Souček, P., Grodecká, L., Grombiříková, H., Ravčuková, B., Kuklínek, P., et al. (2020). Deep Intronic Mutation in SERPING1 Caused Hereditary Angioedema through Pseudoexon Activation. J. Clin. Immunol. 40 (3), 435–446. doi:10.1007/s10875-020-00753-2
Ikeda, H., Matsubara, Y., Mikami, H., Kure, S., Owada, M., Gough, T., et al. (1997). Molecular Analysis of Dihydropteridine Reductase Deficiency: Identification of Two Novel Mutations in Japanese Patients. Hum. Genet. 100 (5-6), 637–642. doi:10.1007/s004390050566
Ikezawa, M., Minami, N., Takahashi, M., Goto, Y.-i., Miike, T., and Nonaka, I. (1998). Dystrophin Gene Analysis on 130 Patients with Duchenne Muscular Dystrophy with a Special Reference to Muscle mRNA Analysis. Brain Dev. 20 (3), 165–168. doi:10.1016/s0387-7604(98)00012-6
Inaba, H., Koyama, T., Shinozawa, K., Amano, K., and Fukutake, K. (2013). Identification and Characterization of an Adenine to Guanine Transition within Intron 10 of the Factor VIII Gene as a Causative Mutation in a Patient with Mild Haemophilia A. Haemophilia 19 (1), 100–105. doi:10.1111/j.1365-2516.2012.02906.x
Ing, A., Wlodaver, A., Kirschmann, D., Toledo, E., McCabe, C., Kadri, S., et al. (2021). Transcript Analysis for Variant Classification Resolution in a Child with Primary Ciliary Dyskinesia. Cold Spring Harb Mol. Case Stud. 7 (1), 1. doi:10.1101/mcs.a005363
Ishibashi, K., Takeshima, Y., Yagi, M., Nishiyama, A., and Matsuo, M. (2006). Novel Cryptic Exons Identified in Introns 2 and 3 of the Human Dystrophin Gene with Duplication of Exons 8-11. Kobe J. Med. Sci. 52 (3-4), 61–75.
Ishigaki, K., Nicolle, D., Krejci, E., Leroy, J.-P., Koenig, J., Fardeau, M., et al. (2003). Two Novel Mutations in the COLQ Gene Cause Endplate Acetylcholinesterase Deficiency. Neuromuscul. Disord. 13 (3), 236–244. doi:10.1016/s0960-8966(02)00243-2
Ishigaki, S., Masuda, A., Fujioka, Y., Iguchi, Y., Katsuno, M., Shibata, A., et al. (2012). Position-dependent FUS-RNA Interactions Regulate Alternative Splicing Events and Transcriptions. Sci. Rep. 2, 529. doi:10.1038/srep00529
Ishii, S., Nakao, S., Minamikawa-Tachino, R., Desnick, R. J., and Fan, J.-Q. (2002). Alternative Splicing in the α-Galactosidase A Gene: Increased Exon Inclusion Results in the Fabry Cardiac Phenotype. Am. J. Hum. Genet. 70 (4), 994–1002. doi:10.1086/339431
Ishmukhametova, A., Van Kien, P. K., Méchin, D., Thorel, D., Vincent, M.-C., Rivier, F., et al. (2012). Comprehensive Oligonucleotide Array-Comparative Genomic Hybridization Analysis: New Insights into the Molecular Pathology of the DMD Gene. Eur. J. Hum. Genet. 20 (10), 1096–1100. doi:10.1038/ejhg.2012.51
Isler, J., Rüfenacht, V., Gemperle, C., Allegri, G., and Häberle, J. (2020). Improvement of Diagnostic Yield in Carbamoylphosphate Synthetase 1 ( CPS1 ) Molecular Genetic Investigation by RNA Sequencing. JIMD Rep. 52 (1), 28–34. doi:10.1002/jmd2.12091
Jamshidi, F., Place, E. M., Mehrotra, S., Navarro-Gomez, D., Maher, M., Branham, K. E., et al. (2019). Contribution of Noncoding Pathogenic Variants to RPGRIP1-Mediated Inherited Retinal Degeneration. Genet. Med. 21 (3), 694–704. doi:10.1038/s41436-018-0104-7
Jang, M.-A., Kim, Y.-E., Kim, S. K., Lee, M.-K., Kim, J.-W., and Ki, C.-S. (2016). Identification and Characterization of NF1 Splicing Mutations in Korean Patients with Neurofibromatosis Type 1. J. Hum. Genet. 61 (8), 705–709. doi:10.1038/jhg.2016.33
Janin, A., Chanavat, V., Rollat‐Farnier, P. A., Bardel, C., Nguyen, K., Chevalier, P., et al. (2020). Whole MYBPC3 NGS Sequencing as a Molecular Strategy to Improve the Efficiency of Molecular Diagnosis of Patients with Hypertrophic Cardiomyopathy. Hum. Mutat. 41 (2), 465–475. doi:10.1002/humu.23944
Jiang, C., and Zhao, Z. (2006). Mutational Spectrum in the Recent Human Genome Inferred by Single Nucleotide Polymorphisms. Genomics 88 (5), 527–534. doi:10.1016/j.ygeno.2006.06.003
Jin, M., Li, J.-J., Xu, G.-R., Wang, N., and Wang, Z.-Q. (2020). Cryptic Exon Activation Causes Dystrophinopathy in Two Chinese Families. Eur. J. Hum. Genet. 28 (7), 947–955. doi:10.1038/s41431-020-0578-z
Jin, X., Yan, Y., Zhang, C., Tai, Y., An, L., Yu, X., et al. (2021). Identification of Novel Deep Intronic PAH Gene Variants in Patients Diagnosed with Phenylketonuria. Hum Mutat.. doi:10.1002/humu.24292
Jo, B.-S., and Choi, S. S. (2015). Introns: The Functional Benefits of Introns in Genomes. Genomics Inform. 13 (4), 112–118. doi:10.5808/GI.2015.13.4.112
Jourdy, Y., Janin, A., Fretigny, M., Lienhart, A., Négrier, C., Bozon, D., et al. (2018). Reccurrent F8 Intronic Deletion Found in Mild Hemophilia A Causes Alu Exonization. Am. J. Hum. Genet. 102 (2), 199–206. doi:10.1016/j.ajhg.2017.12.010
Juan-Mateu, J., González-Quereda, L., Rodríguez, M. J., Verdura, E., Lázaro, K., Jou, C., et al. (2013). Interplay between DMD point Mutations and Splicing Signals in Dystrophinopathy Phenotypes. PLoS One 8 (3), e59916. doi:10.1371/journal.pone.0059916
Kaida, D. (2016). The Reciprocal Regulation between Splicing and 3′‐end Processing. WIREs RNA 7 (4), 499–511. doi:10.1002/wrna.1348
Kaimori, J.-y., Ichimaru, N., Isaka, Y., Hashimoto, F., Fu, X., Hashimura, Y., et al. (2013). Renal Transplantations from Parents to Siblings with Autosomal Recessive Alport Syndrome Caused by a Rearrangement in an Intronic Antisense Alu Element in the COL4A3 Gene Led to Different Outcomes. CEN Case Rep. 2 (1), 98–101. doi:10.1007/s13730-012-0049-7
Kalscheuer, V. M., Musante, L., Fang, C., Hoffmann, K., Fuchs, C., Carta, E., et al. (2009). A Balanced Chromosomal Translocation disruptingARHGEF9is Associated with Epilepsy, Anxiety, Aggression, and Mental Retardation. Hum. Mutat. 30 (1), 61–68. doi:10.1002/humu.20814
Kannu, P., Nour, M., Irving, M., Xie, J., Loder, D., Lai, J., et al. (2013). Paraspinal Ganglioneuroma in the Proband of a Large Family with Mild Cutaneous Manifestations of NF1, Carrying a deepNF1intronic Mutation. Clin. Genet. 83 (2), 191–194. doi:10.1111/j.1399-0004.2012.01882.x
Känsäkoski, J., Jääskeläinen, J., Jääskeläinen, T., Tommiska, J., Saarinen, L., Lehtonen, R., et al. (2016). Complete Androgen Insensitivity Syndrome Caused by a Deep Intronic Pseudoexon-Activating Mutation in the Androgen Receptor Gene. Sci. Rep. 6, 32819. doi:10.1038/srep32819
Kazakov, D. V., Thoma-Uszynski, S., Vanecek, T., Kacerovska, D., Grossmann, P., and Michal, M. (2009). A Case of Brooke-Spiegler Syndrome with a Novel Germline Deep Intronic Mutation in the CYLD Gene Leading to Intronic Exonization, Diverse Somatic Mutations, and Unusual Histology. Am. J. Dermatopathol 31 (7), 664–673. doi:10.1097/DAD.0b013e3181a05dad
Ke, S., Shang, S., Kalachikov, S. M., Morozova, I., Yu, L., Russo, J. J., et al. (2011). Quantitative Evaluation of All Hexamers as Exonic Splicing Elements. Genome Res. 21 (8), 1360–1374. doi:10.1101/gr.119628.110
Keeratichamroen, S., Cairns, J. R., Wattanasirichaigoon, D., Wasant, P., Ngiwsara, L., Suwannarat, P., et al. (2008). Molecular Analysis of the Iduronate-2-Sulfatase Gene in Thai Patients with Hunter Syndrome. J. Inherit. Metab. Dis. 31 (Suppl. 2), S303–S311. doi:10.1007/s10545-008-0876-z
Kelly, S., Georgomanolis, T., Zirkel, A., Diermeier, S., O'Reilly, D., Murphy, S., et al. (2015). Splicing of many Human Genes Involves Sites Embedded within Introns. Nucleic Acids Res. 43 (9), 4721–4732. doi:10.1093/nar/gkv386
Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M., Pringle, T. H., Zahler, A. M., et al. (2002). The Human Genome Browser at UCSC. Genome Res. 12 (6), 996–1006. doi:10.1101/gr.229102
Khan, A. O., Becirovic, E., Betz, C., Neuhaus, C., Altmüller, J., Maria Riedmayr, L., et al. (2017). A Deep Intronic CLRN1 (USH3A) Founder Mutation Generates an Aberrant Exon and Underlies Severe Usher Syndrome on the Arabian Peninsula. Sci. Rep. 7 (1), 1411. doi:10.1038/s41598-017-01577-8
Khan, M., Arno, G., Fakin, A., Parfitt, D. A., Dhooge, P. P. A., Albert, S., et al. (2020a). Detailed Phenotyping and Therapeutic Strategies for Intronic ABCA4 Variants in Stargardt Disease. Mol. Ther. - Nucleic Acids 21, 412–427. doi:10.1016/j.omtn.2020.06.007
Khan, M., Cornelis, S. S., De Pozo-Valero, M. l., Whelan, L., Runhart, E. H., Mishra, K., et al. (2020b). Resolving the Dark Matter of ABCA4 for 1054 Stargardt Disease Probands through Integrated Genomics and Transcriptomics. Genet. Med. 22 (7), 1235–1246. doi:10.1038/s41436-020-0787-4
Khan, M., Cornelis, S. S., Khan, M. I., Elmelik, D., Manders, E., Bakker, S., et al. (2019). Cost‐effective Molecular Inversion Probe‐based ABCA4 Sequencing Reveals Deep‐intronic Variants in Stargardt Disease. Hum. Mutat. 40 (10), 1749–1759. doi:10.1002/humu.23787
Khelifi, M. M., Ishmukhametova, A., Van Kien, P. K., Thorel, D., Méchin, D., Perelman, S., et al. (2011). Pure Intronic Rearrangements Leading to Aberrant Pseudoexon Inclusion in Dystrophinopathy: a New Class of Mutations? Hum. Mutat. 32 (4), 467–475. doi:10.1002/humu.21471
Khourieh, J., Rao, G., Habib, T., Avery, D. T., Lefèvre-Utile, A., Chandesris, M.-O., et al. (2019). A Deep Intronic Splice Mutation of STAT3 Underlies Hyper IgE Syndrome by Negative Dominance. Proc. Natl. Acad. Sci. USA 116 (33), 16463–16472. doi:10.1073/pnas.1901409116
Kim, S. W., Taggart, A. J., Heintzelman, C., Cygan, K. J., Hull, C. G., Wang, J., et al. (2017). Widespread Intra-dependencies in the Removal of Introns from Human Transcripts. Nucleic Acids Res. 45 (16), 9503–9513. doi:10.1093/nar/gkx661
King, K., Flinter, F., Nihalani, V., and Green, P. (2002). Unusual Deep Intronic Mutations in the COL4A5 Gene Cause X Linked Alport Syndrome. Hum. Genet. 111 (6), 548–554. doi:10.1007/s00439-002-0830-3
Knebelmann, B., Forestier, L., Drouot, L., Quinones, S., Chuet, C., Benessy, F., et al. (1995). Splice-mediated Insertion of an Alu Sequence in the COL4A3 mRNA Causing Autosomal Recessive Alport Syndrome. Hum. Mol. Genet. 4 (4), 675–679. doi:10.1093/hmg/4.4.675
Knight, S., Vulliamy, T., Morgan, B., Devriendt, K., Mason, P., and Dokal, I. (2001). Identification of Novel DKC1 Mutations in Patients with Dyskeratosis Congenita: Implications for Pathophysiology and Diagnosis. Hum. Genet. 108 (4), 299–303. doi:10.1007/s004390100494
Kollberg, G., Tulinius, M., Melberg, A., Darin, N., Andersen, O., Holmgren, D., et al. (2009). Clinical Manifestation and a New ISCU Mutation in Iron-sulphur Cluster Deficiency Myopathy. Brain 132 (Pt 8), 2170–2179. doi:10.1093/brain/awp152
Kossack, N., Simoni, M., Richter-Unruh, A., Themmen, A. P. N., and Gromoll, J. (2008). Mutations in a Novel, Cryptic Exon of the Luteinizing Hormone/chorionic Gonadotropin Receptor Gene Cause Male Pseudohermaphroditism. Plos Med. 5 (4), e88. doi:10.1371/journal.pmed.0050088
Koster, R., Brandão, R. D., Tserpelis, D., van Roozendaal, C. E. P., van Oosterhoud, C. N., Claes, K. B. M., et al. (2021). Pathogenic Neurofibromatosis Type 1 (NF1) RNA Splicing Resolved by Targeted RNAseq. NPJ Genom Med 6, 95. doi:10.1038/s41525-021-00258-w
Královičová, J., Hwang, G., Asplund, A. C., Churbanov, A., Smith, C. I. E., and Vořechovský, I. (2011). Compensatory Signals Associated with the Activation of Human GC 5′ Splice Sites. Nucleic Acids Res. 39 (16), 7077–7091. doi:10.1093/nar/gkr306
Královičová, J., Knut, M., Cross, N. C. P., and Vořechovský, I. (2016). Exon-centric Regulation of ATM Expression Is Population-dependent and Amenable to Antisense Modification by Pseudoexon Targeting. Sci. Rep. 6, 18741. doi:10.1038/srep18741
Královičová, J., and Vořechovský, I. (2007). Global Control of Aberrant Splice-Site Activation by Auxiliary Splicing Sequences: Evidence for a Gradient in Exon and Intron Definition. Nucleic Acids Res. 35 (19), 6399–6413. doi:10.1093/nar/gkm680
Krawczak, M., Thomas, N. S. T., Hundrieser, B., Mort, M., Wittig, M., Hampe, J., et al. (2007). Single Base-Pair Substitutions in Exon-Intron Junctions of Human Genes: Nature, Distribution, and Consequences for mRNA Splicing. Hum. Mutat. 28 (2), 150–158. doi:10.1002/humu.20400
Kremer, L. S., Bader, D. M., Mertes, C., Kopajtich, R., Pichler, G., Iuso, A., et al. (2017). Genetic Diagnosis of Mendelian Disorders via RNA Sequencing. Nat. Commun. 8, 15824. doi:10.1038/ncomms15824
Kröll-Hermi, A., Ebstein, F., Stoetzel, C., Geoffroy, V., Schaefer, E., Scheidecker, S., et al. (2020). Proteasome Subunit PSMC3 Variants Cause Neurosensory Syndrome Combining Deafness and Cataract Due to Proteotoxic Stress. EMBO Mol. Med. 12 (7), e11861. doi:10.15252/emmm.201911861
Kuehl, P., Zhang, J., Lin, Y., Lamba, J., Assem, M., Schuetz, J., et al. (2001). Sequence Diversity in CYP3A Promoters and Characterization of the Genetic Basis of Polymorphic CYP3A5 Expression. Nat. Genet. 27 (4), 383–391. doi:10.1038/86882
Lai, C. Y., Tsai, I. J., Chiu, P. C., Ascher, D. B., Chien, Y. H., Huang, Y. H., et al. (2021). A Novel Deep Intronic Variant Strongly Associates with Alkaptonuria. NPJ Genom Med. 6 (89), 89. doi:10.1038/s41525-021-00252-2
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., et al. (2001). Initial Sequencing and Analysis of the Human Genome. Nature 409 (6822), 860–921. doi:10.1038/35057062
Landrith, T., Li, B., Cass, A. A., Conner, B. R., LaDuca, H., McKenna, D. B., et al. (2020). Splicing Profile by Capture RNA-Seq Identifies Pathogenic Germline Variants in Tumor Suppressor Genes. Npj Precis. Onc. 4, 4. doi:10.1038/s41698-020-0109-y
Larrue, R., Chamley, P., Bardyn, T., Lionet, A., Gnemmi, V., Cauffiez, C., et al. (2020). Diagnostic Utility of Whole-Genome Sequencing for Nephronophthisis. NPJ Genom Med. 5 (1), 38. doi:10.1038/s41525-020-00147-8
Lassalle, F., Jourdy, Y., Jouan, L., Swystun, L., Gauthier, J., Zawadzki, C., et al. (2020). The challenge of Genetically Unresolved Haemophilia A Patients: Interest of the Combination of Whole F8 Gene Sequencing and Functional Assays. Haemophilia 26 (6), 1056–1063. doi:10.1111/hae.14179
Latchman, K., Brown, J., Sineni, C. J., Ragin‐Dames, L., Guo, S., Huang, J., et al. (2020). A Founder Noncoding GALT Variant Interfering with Splicing Causes Galactosemia. Jrnl Inher Metab. Disea 43 (6), 1199–1204. doi:10.1002/jimd.12293
Laugel, V., Dalloz, C., Durand, M., Sauvanaud, F., Kristensen, U., Vincent, M. C., et al. (2010). Mutation Update for theCSB/ERCC6andCSA/ERCC8genes Involved in Cockayne Syndrome. Hum. Mutat. 31 (2), 113–126. doi:10.1002/humu.21154
Lebon, S., Minai, L., Chretien, D., Corcos, J., Serre, V., Kadhom, N., et al. (2007). A Novel Mutation of the NDUFS7 Gene Leads to Activation of a Cryptic Exon and Impaired Assembly of Mitochondrial Complex I in a Patient with Leigh Syndrome. Mol. Genet. Metab. 92 (1-2), 104–108. doi:10.1016/j.ymgme.2007.05.010
Lee, H., Huang, A. Y., Wang, L.-k., Yoon, A. J., Renteria, G., Eskin, A., et al. (2020). Diagnostic Utility of Transcriptome Sequencing for Rare Mendelian Diseases. Genet. Med. 22 (3), 490–499. doi:10.1038/s41436-019-0672-1
Lee, M., Roos, P., Sharma, N., Atalar, M., Evans, T. A., Pellicore, M. J., et al. (2017). Systematic Computational Identification of Variants that Activate Exonic and Intronic Cryptic Splice Sites. Am. J. Hum. Genet. 100 (5), 751–765. doi:10.1016/j.ajhg.2017.04.001
Lee, W.-I., Torgerson, T. R., Schumacher, M. J., Yel, L., Zhu, Q., and Ochs, H. D. (2005). Molecular Analysis of a Large Cohort of Patients with the Hyper Immunoglobulin M (IgM) Syndrome. Blood 105 (5), 1881–1890. doi:10.1182/blood-2003-12-4420
Levitus, M., Waisfisz, Q., Godthelp, B. C., Vries, Y. d., Hussain, S., Wiegant, W. W., et al. (2005). The DNA Helicase BRIP1 Is Defective in Fanconi Anemia Complementation Group J. Nat. Genet. 37 (9), 934–935. doi:10.1038/ng1625
Ligtenberg, M. J. L., Kuiper, R. P., Chan, T. L., Goossens, M., Hebeda, K. M., Voorendt, M., et al. (2009). Heritable Somatic Methylation and Inactivation of MSH2 in Families with Lynch Syndrome Due to Deletion of the 3′ Exons of TACSTD1. Nat. Genet. 41 (1), 112–117. doi:10.1038/ng.283
Lim, B. C., Ki, C.-S., Kim, J.-W., Cho, A., Kim, M. J., Hwang, H., et al. (2010). Fukutin Mutations in Congenital Muscular Dystrophies with Defective Glycosylation of Dystroglycan in Korea. Neuromuscul. Disord. 20 (8), 524–530. doi:10.1016/j.nmd.2010.06.005
Liquori, A., Vaché, C., Baux, D., Blanchet, C., Hamel, C., Malcolm, S., et al. (2016). WholeUSH2AGene Sequencing Identifies Several New Deep Intronic Mutations. Hum. Mutat. 37 (2), 184–193. doi:10.1002/humu.22926
Liu, W., Han, B., Zhu, W., Cheng, T., Fan, M., Wu, J., et al. (2017). Polymorphism in the Alternative Donor Site of the Cryptic Exon of LHCGR: Functional Consequences and Associations with Testosterone Level. Sci. Rep. 7, 45699. doi:10.1038/srep45699
Lo, Y.-F., Nozu, K., Iijima, K., Morishita, T., Huang, C.-C., Yang, S.-S., et al. (2011). Recurrent Deep Intronic Mutations in theSLC12A3Gene Responsible for Gitelman's Syndrome. Cjasn 6 (3), 630–639. doi:10.2215/CJN.06730810
Lornage, X., Schartner, V., Balbueno, I., Biancalana, V., Willis, T., Echaniz-Laguna, A., et al. (2019). Clinical, Histological, and Genetic Characterization of PYROXD1-Related Myopathy. Acta Neuropathol. Commun. 7 (1), 138. doi:10.1186/s40478-019-0781-8
Lou, H., Helfman, D. M., Gagel, R. F., and Berget, S. M. (1999). Polypyrimidine Tract-Binding Protein Positively Regulates Inclusion of an Alternative 3′-Terminal Exon. Mol. Cel Biol 19 (1), 78–85. doi:10.1128/MCB.19.1.78
Lu, X., Han, C., Mai, J., Jiang, X., Liao, J., Hou, Y., et al. (2021). Novel Intronic Mutations Introduce Pseudoexons in DMD that Cause Muscular Dystrophy in Patients. Front. Genet. 12, 657040. doi:10.3389/fgene.2021.657040
Lualdi, S., Pittis, M. G., Regis, S., Parini, R., Allegri, A. E., Furlan, F., et al. (2006). Multiple Cryptic Splice Sites Can Be Activated by IDS point Mutations Generating Misspliced Transcripts. J. Mol. Med. 84 (8), 692–700. doi:10.1007/s00109-006-0057-1
Lucien, N., Chiaroni, J., Cartron, J.-P., and Bailly, P. (2002). Partial Deletion in the JK Locus Causing a Jknull Phenotype. Blood 99 (3), 1079–1081. doi:10.1182/blood.v99.3.1079
Ma, S. L., Vega-Warner, V., Gillies, C., Sampson, M. G., Kher, V., Sethi, S. K., et al. (2015). Whole Exome Sequencing Reveals Novel PHEX Splice Site Mutations in Patients with Hypophosphatemic Rickets. PLoS One 10 (6), e0130729. doi:10.1371/journal.pone.0130729
Madan, V., Kanojia, D., Li, J., Okamoto, R., Sato-Otsubo, A., Kohlmann, A., et al. (2015). Aberrant Splicing of U12-type Introns Is the Hallmark of ZRSR2 Mutant Myelodysplastic Syndrome. Nat. Commun. 6, 6042. doi:10.1038/ncomms7042
Madden, H. R., Fletcher, S., Davis, M. R., and Wilton, S. D. (2009). Characterization of a Complex Duchenne Muscular Dystrophy-Causing Dystrophin Gene Inversion and Restoration of the reading Frame by Induced Exon Skipping. Hum. Mutat. 30 (1), 22–28. doi:10.1002/humu.20806
Magri, F., Del Bo, R., D'Angelo, M. G., Govoni, A., Ghezzi, S., Gandossini, S., et al. (2011). Clinical and Molecular Characterization of a Cohort of Patients with Novel Nucleotide Alterations of the Dystrophin Gene Detected by Direct Sequencing. BMC Med. Genet. 12, 37. doi:10.1186/1471-2350-12-37
Malueka, R. G., Takaoka, Y., Yagi, M., Awano, H., Lee, T., Dwianingsih, E. K., et al. (2012). Categorization of 77 Dystrophinexons into 5 Groups by a Decision Tree Using Indexes of Splicing Regulatory Factors as Decision Markers. BMC Genet. 13, 23. doi:10.1186/1471-2156-13-23
Mameli, E., Lepori, M. B., Chiappe, F., Ranucci, G., Di Dato, F., Iorio, R., et al. (2015). Wilson's Disease Caused by Alternative Splicing and Alu Exonization Due to a Homozygous 3039-bp Deletion Spanning from Intron 1 to Exon 2 of the ATP7B Gene. Gene 569 (2), 276–279. doi:10.1016/j.gene.2015.05.067
Marek-Yagel, D., Eliyahu, A., Veber, A., Shalva, N., Philosoph, A. M., Barel, O., et al. (2021). Deep Intronic Variant in the ARSB Gene as the Genetic Cause for Maroteaux-Lamy Syndrome (MPS VI). Am. J. Med. Genet. A. 185 (12), 3804–3809. doi:10.1002/ajmg.a.62453
Martinez, F. J., Lee, J. H., Lee, J. E., Blanco, S., Nickerson, E., Gabriel, S., et al. (2012). Whole Exome Sequencing Identifies a Splicing Mutation in NSUN2 as a Cause of a Dubowitz-like Syndrome. J. Med. Genet. 49 (6), 380–385. doi:10.1136/jmedgenet-2011-100686
Martínez, M. A., Rincón, A., Desviat, L. R., Merinero, B., Ugarte, M., and Pérez, B. (2005). Genetic Analysis of Three Genes Causing Isolated Methylmalonic Acidemia: Identification of 21 Novel Allelic Variants. Mol. Genet. Metab. 84 (4), 317–325. doi:10.1016/j.ymgme.2004.11.011
Martínez-Pizarro, A., Dembic, M., Pérez, B., Andresen, B. S., and Desviat, L. R. (2018). Intronic PAH Gene Mutations Cause a Splicing Defect by a Novel Mechanism Involving U1snRNP Binding Downstream of the 5' Splice Site. Plos Genet. 14 (4), e1007360. doi:10.1371/journal.pgen.1007360
Masala, M. V., Scapaticci, S., Olivieri, C., Pirodda, C., Montesu, M. A., Cuccuru, M. A., et al. (2007). Epidemiology and Clinical Aspects of Werner's Syndrome in North Sardinia: Description of a Cluster. Eur. J. Dermatol. 17 (3), 213–216. doi:10.1684/ejd.2007.0155
Mayer, A. K., Rohrschneider, K., Strom, T. M., Glöckle, N., Kohl, S., Wissinger, B., et al. (2016). Homozygosity Mapping and Whole-Genome Sequencing Reveals a Deep Intronic PROM1 Mutation Causing Cone-Rod Dystrophy by Pseudoexon Activation. Eur. J. Hum. Genet. 24 (3), 459–462. doi:10.1038/ejhg.2015.144
Mayer, K., Ballhausen, W., Leistner, W., and Rott, H.-D. (2000). Three Novel Types of Splicing Aberrations in the Tuberous Sclerosis TSC2 Gene Caused by Mutations Apart from Splice Consensus Sequences. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1502 (3), 495–507. doi:10.1016/s0925-4439(00)00072-7
McConville, C. M., Stankovic, T., Byrd, P. J., McGuire, G. M., Yao, Q. Y., Lennox, G. G., et al. (1996). Mutations Associated with Variant Phenotypes in Ataxia-Telangiectasia. Am. J. Hum. Genet. 59 (2), 320–330.
McDonell, L. M., Mirzaa, G. M., Alcantara, D., Schwartzentruber, J., Carter, M. T., Lee, L. J., et al. (2013). Mutations in STAMBP, Encoding a Deubiquitinating Enzyme, Cause Microcephaly-Capillary Malformation Syndrome. Nat. Genet. 45 (5), 556–562. doi:10.1038/ng.2602
Medina, M. W., Theusch, E., Naidoo, D., Bauzon, F., Stevens, K., Mangravite, L. M., et al. (2012). RHOA Is a Modulator of the Cholesterol-Lowering Effects of Statin. Plos Genet. 8 (11), e1003058. doi:10.1371/journal.pgen.1003058
Meili, D., Královičová, J., Zagalak, J., Bonafé, L., Fiori, L., Blau, N., et al. (2009). Disease-causing Mutations Improving the branch Site and Polypyrimidine Tract: Pseudoexon Activation of LINE-2 and antisenseAlulacking the Poly(T)-tail. Hum. Mutat. 30 (5), 823–831. doi:10.1002/humu.20969
Meischl, C., de Boer, M., Åhlin, A., and Roos, D. (2000). A New Exon Created by Intronic Insertion of a Rearranged LINE-1 Element as the Cause of Chronic Granulomatous Disease. Eur. J. Hum. Genet. 8 (9), 697–703. doi:10.1038/sj.ejhg.5200523
Mele, C., Lemaire, M., Iatropoulos, P., Piras, R., Bresin, E., Bettoni, S., et al. (2015). Characterization of a New DGKE Intronic Mutation in Genetically Unsolved Cases of Familial Atypical Hemolytic Uremic Syndrome. Cjasn 10 (6), 1011–1019. doi:10.2215/CJN.08520814
Messiaen, L. M., and Wimmer, K. (2008). “NF1 Mutational Spectrum,” in Monographs in Human Genetics 16. Editor D. Kaufmann (Basel: Karger), 63–77. doi:10.1159/000126545
Messiaen, L., and Wimmer, K. (2012). “Mutation Analysis of the NF1 Gene by cDNA-Based Sequencing of the Coding Region,” in Advances in Neurofibromatosis Research. Editors K. S. G. Cunha, and M. Geller (Hauppauge, NY: Nova Science Publishers, Inc.), 89–101.
Michel-Calemard, L., Dijoud, F., Till, M., Lambert, J. C., Vercherat, M., Tardy, V., et al. (2009). Pseudoexon Activation in the PKHD1 Gene: a French Founder Intronic Mutation IVS46+653A>G Causing Severe Autosomal Recessive Polycystic Kidney Disease. Clin. Genet. 75 (2), 203–206. doi:10.1111/j.1399-0004.2008.01106.x
Milh, M., Pop, A., Kanhai, W., Villeneuve, N., Cano, A., Struys, E. A., et al. (2012). Atypical Pyridoxine-dependent Epilepsy Due to a Pseudoexon in ALDH7A1. Mol. Genet. Metab. 105 (4), 684–686. doi:10.1016/j.ymgme.2012.01.011
Mitchell, G. A., Labuda, D., Fontaine, G., Saudubray, J. M., Bonnefont, J. P., Lyonnet, S., et al. (1991). Splice-mediated Insertion of an Alu Sequence Inactivates Ornithine delta-aminotransferase: a Role for Alu Elements in Human Mutation. Proc. Natl. Acad. Sci. 88 (3), 815–819. doi:10.1073/pnas.88.3.815
Mochel, F., Knight, M. A., Tong, W.-H., Hernandez, D., Ayyad, K., Taivassalo, T., et al. (2008). Splice Mutation in the Iron-Sulfur Cluster Scaffold Protein ISCU Causes Myopathy with Exercise Intolerance. Am. J. Hum. Genet. 82 (3), 652–660. doi:10.1016/j.ajhg.2007.12.012
Moles-Fernández, A., Domènech-Vivó, J., Tenés, A., Balmaña, J., Diez, O., and Gutiérrez-Enríquez, S. (2021). “Role of Splicing Regulatory Elements and,” in Silico Tools Usage in the Identification of Deep Intronic Splicing Variants in Hereditary Breast/Ovarian Cancer Genes (Basel: Cancers), 13. doi:10.3390/cancers13133341
Mollin, M., Beaumel, S., Vigne, B., Brault, J., Roux-Buisson, N., Rendu, J., et al. (2021). Clinical, Functional and Genetic Characterization of 16 Patients Suffering from Chronic Granulomatous Disease Variants - Identification of 11 Novel Mutations in CYBB. Clin. Exp. Immunol. 203 (2), 247–266. doi:10.1111/cei.13520
Monnier, N., Gout, J. P., Pin, I., Gauthier, G., and Lunardi, J. (2001). A Novel 3600+11.5 Kb C>G Homozygous Splicing Mutation in a Black African, Consanguineous CF Family. J. Med. Genet. 38 (1), E4. doi:10.1136/jmg.38.1.e4
Monnier, N., Ferreiro, A., Marty, I., Labarre-Vila, A., Mezin, P., and Lunardi, J. (2003). A Homozygous Splicing Mutation Causing a Depletion of Skeletal Muscle RYR1 Is Associated with Multi-Minicore Disease Congenital Myopathy with Ophthalmoplegia. Hum. Mol. Genet. 12 (10), 1171–1178. doi:10.1093/hmg/ddg121
Montalban, G., Bonache, S., Moles-Fernández, A., Gisbert-Beamud, A., Tenés, A., Bach, V., et al. (2019). Screening of BRCA1/2 Deep Intronic Regions by Targeted Gene Sequencing Identifies the First Germline BRCA1 Variant Causing Pseudoexon Activation in a Patient with Breast/ovarian Cancer. J. Med. Genet. 56 (2), 63–74. doi:10.1136/jmedgenet-2018-105606
Morello, R., Bertin, T. K., Chen, Y., Hicks, J., Tonachini, L., Monticone, M., et al. (2006). CRTAP Is Required for Prolyl 3- Hydroxylation and Mutations Cause Recessive Osteogenesis Imperfecta. Cell 127 (2), 291–304. doi:10.1016/j.cell.2006.08.039
Morrison, F. S., Locke, J. M., Wood, A. R., Tuke, M., Pasko, D., Murray, A., et al. (2013). The Splice Site Variant Rs11078928 May Be Associated with a Genotype-dependent Alteration in Expression of GSDMB Transcripts. BMC Genomics 14, 627. doi:10.1186/1471-2164-14-627
Movassat, M., Forouzmand, E., Reese, F., and Hertel, K. J. (2019). Exon Size and Sequence Conservation Improves Identification of Splice-Altering Nucleotides. RNA 25 (12), 1793–1805. doi:10.1261/rna.070987.119
Murdock, D. R., Dai, H., Burrage, L. C., Rosenfeld, J. A., Ketkar, S., Müller, M. F., et al. (2021). Transcriptome-directed Analysis for Mendelian Disease Diagnosis Overcomes Limitations of Conventional Genomic Testing. J. Clin. Invest. 131 (1), 1. doi:10.1172/JCI141500
Nakamura, K., Du, L., Tunuguntla, R., Fike, F., Cavalieri, S., Morio, T., et al. (2012). Functional Characterization and Targeted Correction of ATM Mutations Identified in Japanese Patients with Ataxia-Telangiectasia. Hum. Mutat. 33 (1), 198–208. doi:10.1002/humu.21632
Naruto, T., Okamoto, N., Masuda, K., Endo, T., Hatsukawa, Y., Kohmoto, T., et al. (2015). Deep Intronic GPR143 Mutation in a Japanese Family with Ocular Albinism. Sci. Rep. 5, 11334. doi:10.1038/srep11334
Nasim, M. T., Chernova, T. K., Chowdhury, H. M., Yue, B. G., and Eperon, I. C. (2003). HnRNP G and Tra2beta: Opposite Effects on Splicing Matched by Antagonism in RNA Binding. Hum. Mol. Genet. 12 (11), 1337–1348. doi:10.1093/hmg/ddg136
Navarrete, R., Leal, F., Vega, A. I., Morais-López, A., Garcia-Silva, M. T., Martín-Hernández, E., et al. (2019). Value of Genetic Analysis for Confirming Inborn Errors of Metabolism Detected through the Spanish Neonatal Screening Program. Eur. J. Hum. Genet. 27 (4), 556–562. doi:10.1038/s41431-018-0330-0
Neidhardt, J., Glaus, E., Barthelmes, D., Zeitz, C., Fleischhauer, J., and Berger, W. (2007). Identification and Characterization of a Novel RPGR Isoform in Human Retina. Hum. Mutat. 28 (8), 797–807. doi:10.1002/humu.20521
Nieminen, T. T., Pavicic, W., Porkka, N., Kankainen, M., Järvinen, H. J., Lepistö, A., et al. (2016). Pseudoexons Provide a Mechanism for Allele-specific Expression of APC in Familial Adenomatous Polyposis. Oncotarget 7 (43), 70685–70698. doi:10.18632/oncotarget.12206
Njålsson, R., Carlsson, K., Winkler, A., Larsson, A., and Norgren, S. (2003). Diagnostics in Patients with Glutathione Synthetase Deficiency but without Mutations in the Exons of the GSS Gene. Hum. Mutat. 22 (6), 497. doi:10.1002/humu.9199
Noack, D., Heyworth, P. G., Curnutte, J. T., Rae, J., and Cross, A. R. (1999). A Novel Mutation in the CYBB Gene Resulting in an Unexpected Pattern of Exon Skipping and Chronic Granulomatous Disease. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1454 (3), 270–274. doi:10.1016/s0925-4439(99)00044-7
Noack, D., Heyworth, P. G., Newburger, P. E., and Cross, A. R. (2001). An Unusual Intronic Mutation in the CYBB Gene Giving Rise to Chronic Granulomatous Disease. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1537 (2), 125–131. doi:10.1016/s0925-4439(01)00065-5
Nogueira, C., Silva, L., Marcão, A., Sousa, C., Fonseca, H., Rocha, H., et al. (2021). Role of RNA in Molecular Diagnosis of MADD Patients. Biomedicines 9 (5), 1. doi:10.3390/biomedicines9050507
Novoselova, T. V., Rath, S. R., Carpenter, K., Pachter, N., Dickinson, J. E., Price, G., et al. (2015). NNTPseudoexon Activation as a Novel Mechanism for Disease in Two Siblings with Familial Glucocorticoid Deficiency. J. Clin. Endocrinol. Metab. 100 (2), E350–E354. doi:10.1210/jc.2014-3641
Nozu, K., Iijima, K., Igarashi, T., Yamada, S., Kralovicova, J., Nozu, Y., et al. (2017a). A Birth of Bipartite Exon by Intragenic Deletion. Mol. Genet. Genomic Med. 5 (3), 287–294. doi:10.1002/mgg3.277
Nozu, K., Iijima, K., Nozu, Y., Ikegami, E., Imai, T., Fu, X. J., et al. (2009). A Deep Intronic Mutation in the SLC12A3 Gene Leads to Gitelman Syndrome. Pediatr. Res. 66 (5), 590–593. doi:10.1203/PDR.0b013e3181b9b4d3
Nozu, K., Nozu, Y., Nakanishi, K., Konomoto, T., Horinouchi, T., Shono, A., et al. (2017b). Cryptic Exon Activation in SLC12A3 in Gitelman Syndrome. J. Hum. Genet. 62 (2), 335–337. doi:10.1038/jhg.2016.129
Nozu, K., Vorechovsky, I., Kaito, H., Fu, X. J., Nakanishi, K., Hashimura, Y., et al. (2014). X-linked Alport Syndrome Caused by Splicing Mutations in COL4A5. Cjasn 9 (11), 1958–1964. doi:10.2215/CJN.04140414
Ogino, W., Takeshima, Y., Nishiyama, A., Okizuka, Y., Yagi, M., Tsuneishi, S., et al. (2007). Mutation Analysis of the Ornithine Transcarbamylase (OTC) Gene in Five Japanese OTC Deficiency Patients Revealed Two Known and Three Novel Mutations Including a Deep Intronic Mutation. Kobe J. Med. Sci. 53 (5), 229–240.
Ohura, T., Narisawa, K., Tada, K., and Iinuma, K. (1999). An 84bp Insertion Found in a Propionic Acidaemia Patient Is Not a Disease-Causing Mutation but a Product of Cryptic mRNA. J. Inherit. Metab. Dis. 22 (5), 676–677. doi:10.1023/a:1005506819699
Olsson, A., Lind, L., Thornell, L.-E., and Holmberg, M. (2008). Myopathy with Lactic Acidosis Is Linked to Chromosome 12q23.3-24.11 and Caused by an Intron Mutation in the ISCU Gene Resulting in a Splicing Defect. Hum. Mol. Genet. 17 (11), 1666–1672. doi:10.1093/hmg/ddn057
Olthof, A. M., Hyatt, K. C., and Kanadia, R. N. (2019). Minor Intron Splicing Revisited: Identification of New Minor Intron-Containing Genes and Tissue-dependent Retention and Alternative Splicing of Minor Introns. BMC Genomics 20 (1), 686. doi:10.1186/s12864-019-6046-x
Osborn, M. J., and Upadhyaya, M. (1999). Evaluation of the Protein Truncation Test and Mutation Detection in the NF1 Gene: Mutational Analysis of 15 Known and 40 Unknown Mutations. Hum. Genet. 105 (4), 327–332. doi:10.1007/s004399900135
Oshima, J., Yu, C. E., Piussan, C., Klein, G., Jabkowski, J., Balci, S., et al. (1996). Homozygous and Compound Heterozygous Mutations at the Werner Syndrome Locus. Hum. Mol. Genet. 5 (12), 1909–1913. doi:10.1093/hmg/5.12.1909
Pagani, F., Buratti, E., Stuani, C., Bendix, R., Dörk, T., and Baralle, F. E. (2002). A New Type of Mutation Causes a Splicing Defect in ATM. Nat. Genet. 30 (4), 426–429. doi:10.1038/ng858
Pagliarini, V., Jolly, A., Bielli, P., Di Rosa, V., De la Grange, P., and Sette, C. (2020). De la Grange, P., and Sette, CSam68 binds Alu-rich introns in SMN and promotes pre-mRNA circularization. Nucleic Acids Res. 48 (2), 633–645. doi:10.1093/nar/gkz1117
Parada, G. E., Munita, R., Cerda, C. A., and Gysling, K. (2014). A Comprehensive Survey of Non-canonical Splice Sites in the Human Transcriptome. Nucleic Acids Res. 42 (16), 10564–10578. doi:10.1093/nar/gku744
Paradis, C., Cloutier, P., Shkreta, L., Toutant, J., Klarskov, K., and Chabot, B. (2007). hnRNP I/PTB Can Antagonize the Splicing Repressor Activity of SRp30c. RNA 13 (8), 1287–1300. doi:10.1261/rna.403607
Park, V. M., and Pivnick, E. K. (1998). Neurofibromatosis Type 1 (NF1): a Protein Truncation Assay Yielding Identification of Mutations in 73% of Patients. J. Med. Genet. 35 (10), 813–820. doi:10.1136/jmg.35.10.813
Paz, I., Kosti, I., Ares, M., Cline, M., and Mandel-Gutfreund, Y. (2014). RBPmap: a Web Server for Mapping Binding Sites of RNA-Binding Proteins. Nucleic Acids Res. 42 (Web Server issue), W361–W367. doi:10.1093/nar/gku406
Pérez, B., Rincón, A., Jorge-Finnigan, A., Richard, E., Merinero, B., Ugarte, M., et al. (2009). Pseudoexon Exclusion by Antisense Therapy in Methylmalonic Aciduria (MMAuria). Hum. Mutat. 30 (12), 1676–1682. doi:10.1002/humu.21118
Perrault, I., Hanein, S., Gérard, X., Mounguengue, N., Bouyakoub, R., Zarhrate, M., et al. (2021). Whole Locus Sequencing Identifies a Prevalent Founder Deep Intronic RPGRIP1 Pathologic Variant in the French Leber Congenital Amaurosis Cohort. Basel: Genes, 12. doi:10.3390/genes12020287
Perrin, G., Morris, M. A., Antonarakis, S. E., Boltshauser, E., and Hutter, P. (1996). Two Novel Mutations Affecting mRNA Splicing of the Neurofibromatosis Type 1 (NF1) Gene. Hum. Mutat. 7 (2), 172–175. doi:10.1002/(SICI)1098-1004(1996)7:2<172::AID-HUMU15>3.0.CO;2-#
Pezeshkpoor, B., Zimmer, N., Marquardt, N., Nanda, I., Haaf, T., Budde, U., et al. (2013). Deep Intronic 'mutations' Cause Hemophilia A: Application of Next Generation Sequencing in Patients without Detectable Mutation in F8 cDNA. J. Thromb. Haemost. 11 (9), 1679–1687. doi:10.1111/jth.12339
Piovesan, A., Antonaros, F., Vitale, L., Strippoli, P., Pelleri, M. C., and Caracausi, M. (2019). Human Protein-Coding Genes and Gene Feature Statistics in 2019. BMC Res. Notes 12 (1), 315. doi:10.1186/s13104-019-4343-8
Piva, F., Giulietti, M., Burini, A. B., and Principato, G. (2012). SpliceAid 2: a Database of Human Splicing Factors Expression Data and RNA Target Motifs. Hum. Mutat. 33 (1), 81–85. doi:10.1002/humu.21609
Pros, E., Fernández-Rodríguez, J., Canet, B., Benito, L., Sánchez, A., Benavides, A., et al. (2009). Antisense Therapeutics for Neurofibromatosis Type 1 Caused by Deep Intronic Mutations. Hum. Mutat. 30 (3), 454–462. doi:10.1002/humu.20933
Pros, E., Gómez, C., Martín, T., Fábregas, P., Serra, E., and Lázaro, C. (2008). Nature and mRNA Effect of 282 differentNF1point Mutations: Focus on Splicing Alterations. Hum. Mutat. 29 (9), E173–E193. doi:10.1002/humu.20826
Purevsuren, J., Fukao, T., Hasegawa, Y., Fukuda, S., Kobayashi, H., and Yamaguchi, S. (2008). Study of Deep Intronic Sequence Exonization in a Japanese Neonate with a Mitochondrial Trifunctional Protein Deficiency. Mol. Genet. Metab. 95 (1-2), 46–51. doi:10.1016/j.ymgme.2008.06.013
Qu, W., Cingolani, P., Zeeberg, B. R., and Ruden, D. M. (2017). A Bioinformatics-Based Alternative mRNA Splicing Code that May Explain Some Disease Mutations Is Conserved in Animals. Front. Genet. 8, 38. doi:10.3389/fgene.2017.00038
Raponi, M., Upadhyaya, M., and Baralle, D. (2006). Functional Splicing Assay Shows a Pathogenic Intronic Mutation in Neurofibromatosis Type 1 (NF1) Due to Intronic Sequence Exonization. Hum. Mutat. 27 (3), 294–295. doi:10.1002/humu.9412
Rathmann, M., Bunge, S., Beck, M., Kresse, H., Tylki-Szymanska, A., and Gal, A. (1996). Mucopolysaccharidosis Type II (Hunter Syndrome): Mutation "hot Spots" in the Iduronate-2-Sulfatase Gene. Am. J. Hum. Genet. 59 (6), 1202–1209.
Ray, T. A., Cochran, K., Kozlowski, C., Wang, J., Alexander, G., Cady, M. A., et al. (2020). Comprehensive Identification of mRNA Isoforms Reveals the Diversity of Neural Cell-Surface Molecules with Roles in Retinal Development and Disease. Nat. Commun. 11 (1), 3328. doi:10.1038/s41467-020-17009-7
Reeskamp, L. F., Hartgers, M. L., Peter, J., Dallinga-Thie, G. M., Zuurbier, L., Defesche, J. C., et al. (2018). A Deep Intronic Variant in LDLR in Familial Hypercholesterolemia. Circ. Genom Precis Med. 11 (12), e002385. doi:10.1161/CIRCGEN.118.002385
Reeskamp, L. F., Balvers, M., Peter, J., van de Kerkhof, L., Klaaijsen, L. N., Motazacker, M. M., et al. (2021). Intronic Variant Screening with Targeted Next-Generation Sequencing Reveals First Pseudoexon in LDLR in Familial Hypercholesterolemia. Atherosclerosis 321, 14–20. doi:10.1016/j.atherosclerosis.2021.02.003
Rendu, J., Montjean, R., Coutton, C., Suri, M., Chicanne, G., Petiot, A., et al. (2017). Functional Characterization and Rescue of a Deep Intronic Mutation inOCRLGene Responsible for Lowe Syndrome. Hum. Mutat. 38 (2), 152–159. doi:10.1002/humu.23139
Rentas, S., Rathi, K. S., Kaur, M., Raman, P., Krantz, I. D., Sarmady, M., et al. (2020). Diagnosing Cornelia de Lange syndrome and related neurodevelopmental disorders using RNA sequencing. Genet. Med. 22 (5), 927–936. doi:10.1038/s41436-019-0741-5
Riant, F., Odent, S., Cecillon, M., Pasquier, L., de Baracé, C., Carney, M. P., et al. (2014). Deep Intronic KRIT1 Mutation in a Family with Clinically Silent Multiple Cerebral Cavernous Malformations. Clin. Genet. 86 (6), 585–588. doi:10.1111/cge.12322
Richard, N., Abeguilé, G., Coudray, N., Mittre, H., Gruchy, N., Andrieux, J., et al. (2012). A New Deletion Ablating NESP55 Causes Loss of Maternal Imprint of A/BGNASand Autosomal Dominant Pseudohypoparathyroidism Type Ib. J. Clin. Endocrinol. Metab. 97 (5), E863–E867. doi:10.1210/jc.2011-2804
Rimessi, P., Fabris, M., Bovolenta, M., Bassi, E., Falzarano, S., Gualandi, F., et al. (2010). Antisense Modulation of Both Exonic and Intronic Splicing Motifs Induces Skipping of a DMD Pseudo-exon Responsible for X-Linked Dilated Cardiomyopathy. Hum. Gene Ther. 21 (9), 1137–1146. doi:10.1089/hum.2010.010
Rincón, A., Aguado, C., Desviat, L. R., Sánchez-Alcudia, R., Ugarte, M., and Pérez, B. (2007). Propionic and Methylmalonic Acidemia: Antisense Therapeutics for Intronic Variations Causing Aberrantly Spliced Messenger RNA. Am. J. Hum. Genet. 81 (6), 1262–1270. doi:10.1086/522376
Rius, R., Riley, L. G., Guo, Y., Menezes, M., Compton, A. G., Van Bergen, N. J., et al. (2019). Cryptic Intronic NBAS Variant Reveals the Genetic Basis of Recurrent Liver Failure in a Child. Mol. Genet. Metab. 126 (1), 77–82. doi:10.1016/j.ymgme.2018.12.002
Rodríguez-Martín, C., Cidre, F., Fernández-Teijeiro, A., Gómez-Mariano, G., de la Vega, L., Ramos, P., et al. (2016). Familial Retinoblastoma Due to Intronic LINE-1 Insertion Causes Aberrant and Noncanonical mRNA Splicing of the RB1 Gene. J. Hum. Genet. 61 (5), 463–466. doi:10.1038/jhg.2015.173
Rodríguez-Pascau, L., Coll, M. J., Vilageliu, L., and Grinberg, D. (2009). Antisense Oligonucleotide Treatment for a Pseudoexon-Generating Mutation in theNPC1gene Causing Niemann-Pick Type C Disease. Hum. Mutat. 30 (11), E993–E1001. doi:10.1002/humu.21119
Romano, M., Buratti, E., and Baralle, D. (2013). Role of Pseudoexons and Pseudointrons in Human Cancer. Int. J. Cel Biol 2013, 810572. doi:10.1155/2013/810572
Rosenberg, J. B., Newman, P. J., Mosesson, M. W., Guillin, M. C., and Amrani, D. L. (1993). Paris I Dysfibrinogenemia: a point Mutation in Intron 8 Results in Insertion of a 15 Amino Acid Sequence in the Fibrinogen Gamma-Chain. Thromb. Haemost. 69 (3), 217–220. doi:10.1055/s-0038-1651583
Rump, A., Rösen-Wolff, A., Gahr, M., Seidenberg, J., Roos, C., Walter, L., et al. (2006). A Splice-Supporting Intronic Mutation in the Last Bp Position of a Cryptic Exon within Intron 6 of the CYBB Gene Induces its Incorporation into the mRNA Causing Chronic Granulomatous Disease (CGD). Gene 371 (2), 174–181. doi:10.1016/j.gene.2005.11.036
Rymen, D., Lindhout, M., Spanou, M., Ashrafzadeh, F., Benkel, I., Betzler, C., et al. (2020). Expanding the Clinical and Genetic Spectrum of CAD Deficiency: an Epileptic Encephalopathy Treatable with Uridine Supplementation. Genet. Med. 22 (10), 1589–1597. doi:10.1038/s41436-020-0933-z
Sabbagh, A., Pasmant, E., Imbard, A., Luscan, A., Soares, M., Blanché, H., et al. (2013). NF1 Molecular Characterization and Neurofibromatosis Type I Genotype-Phenotype Correlation: the French Experience. Hum. Mutat. 34 (11), 1510–1518. doi:10.1002/humu.22392
Saferali, A., Yun, J. H., Parker, M. M., Sakornsakolpat, P., Chase, R. P., Lamb, A., et al. (2019). Analysis of Genetically Driven Alternative Splicing Identifies FBXO38 as a Novel COPD Susceptibility Gene. Plos Genet. 15 (7), e1008229. doi:10.1371/journal.pgen.1008229
Sakaguchi, N., and Suyama, M. (2021). In Silico identification of Pseudo-exon Activation Events in Personal Genome and Transcriptome Data. RNA Biol. 18 (3), 382–390. doi:10.1080/15476286.2020.1809195
Sangermano, R., Garanto, A., Khan, M., Runhart, E. H., Bauwens, M., Bax, N. M., et al. (2019). Deep-intronic ABCA4 Variants Explain Missing Heritability in Stargardt Disease and Allow Correction of Splice Defects by Antisense Oligonucleotides. Genet. Med. 21 (8), 1751–1760. doi:10.1038/s41436-018-0414-9
Santoro, A., Cannella, S., Trizzino, A., Bruno, G., De Fusco, C., Notarangelo, L. D., et al. (2008). Mutations Affecting mRNA Splicing Are the Most Common Molecular Defect in Patients with Familial Hemophagocytic Lymphohistiocytosis Type 3. Haematologica 93 (7), 1086–1090. doi:10.3324/haematol.12622
Sargent, C. A., Kidd, A., Moore, S., Dean, J., Besley, G. T., and Affara, N. A. (2000). Five Cases of Isolated Glycerol Kinase Deficiency, Including Two Families: Failure to Find Genotype:phenotype Correlation. J. Med. Genet. 37 (6), 434–441. doi:10.1136/jmg.37.6.434
Sawyer, S. L., Hartley, T., Dyment, D. A., Beaulieu, C. L., Schwartzentruber, J., Smith, A., et al. (2016). Utility of Whole‐exome Sequencing for Those Near the End of the Diagnostic Odyssey: Time to Address Gaps in Care. Clin. Genet. 89 (3), 275–284. doi:10.1111/cge.12654
Schalk, A., Greff, G., Drouot, N., Obringer, C., Dollfus, H., Laugel, V., et al. (2018). Deep Intronic Variation in Splicing Regulatory Element of the ERCC8 Gene Associated with Severe but Long-Term Survival Cockayne Syndrome. Eur. J. Hum. Genet. 26 (4), 527–536. doi:10.1038/s41431-017-0009-y
Schneider, A., Maas, S. M., Hennekam, R. C. M., and Hanauer, A. (2013). Identification of the First Deep Intronic Mutation in the RPS6KA3 Gene in a Patient with a Severe Form of Coffin-Lowry Syndrome. Eur. J. Med. Genet. 56 (3), 150–152. doi:10.1016/j.ejmg.2012.11.007
Schollen, E., Keldermans, L., Foulquier, F., Briones, P., Chabas, A., Sánchez-Valverde, F., et al. (2007). Characterization of Two Unusual Truncating PMM2 Mutations in Two CDG-Ia Patients. Mol. Genet. Metab. 90 (4), 408–413. doi:10.1016/j.ymgme.2007.01.003
Schulz, H. L., Grassmann, F., Kellner, U., Spital, G., Rüther, K., Jägle, H., et al. (2017). Mutation Spectrum of the ABCA4 Gene in 335 Stargardt Disease Patients from a Multicenter German Cohort-Impact of Selected Deep Intronic Variants and Common SNPs. Invest. Ophthalmol. Vis. Sci. 58 (1), 394–403. doi:10.1167/iovs.16-19936
Schüssler, S., Gerhalter, T., Abicht, A., Müller-Felber, W., Nagel, A., and Trollmann, R. (2020). Rare Intronic Mutation between Exon 62 and 63 (c.9225-285A>G) of the Dystrophin Gene Associated with Atypical BMD Phenotype. Neuromuscul. Disord. 30 (8), 680–684. doi:10.1016/j.nmd.2020.06.003
Schwarze, U., Hata, R.-I., McKusick, V. A., Shinkai, H., Hoyme, H. E., Pyeritz, R. E., et al. (2004). Rare Autosomal Recessive Cardiac Valvular Form of Ehlers-Danlos Syndrome Results from Mutations in the COL1A2 Gene that Activate the Nonsense-Mediated RNA Decay Pathway. Am. J. Hum. Genet. 74 (5), 917–930. doi:10.1086/420794
Sedláčková, J., Vondráček, P., Hermanová, M., Zámečník, J., Hrubá, Z., Haberlová, J., et al. (2009). Point Mutations in Czech DMD/BMD Patients and Their Phenotypic Outcome. Neuromuscul. Disord. 19 (11), 749–753. doi:10.101610.1016/j.nmd.2009.08.011
Sege-Peterson, K., Chambers, J., Page, T., Jones, O. W., and Nyhan, W. L. (1992). Characterization of Mutations in Phenotypic Variants of Hypoxanthine Phosphoribosyltransferase Deficiency. Hum. Mol. Genet. 1 (6), 427–432. doi:10.1093/hmg/1.6.427
Seim, I., Lubik, A. A., Lehman, M. L., Tomlinson, N., Whiteside, E. J., Herington, A. C., et al. (2013). Cloning of a Novel Insulin-Regulated Ghrelin Transcript in Prostate Cancer. J. Mol. Endocrinol. 50 (2), 179–191. doi:10.1530/JME-12-0150
Sela, N., Mersch, B., Hotz-Wagenblatt, A., and Ast, G. (2010). Characteristics of Transposable Element Exonization within Human and Mouse. PLoS One 5 (6), e10907. doi:10.1371/journal.pone.0010907
Serra, T., Ars, E., Ravella, A., Sánchez, A., Puig, S., Rosenbaum, T., et al. (2001). Somatic NF1 Mutational Spectrum in Benign Neurofibromas: mRNA Splice Defects Are Common Among point Mutations. Hum. Genet. 108 (5), 416–429. doi:10.1007/s004390100514
Shi, F., Yao, Y., Bin, Y., Zheng, C. H., and Xia, J. (2019). Computational Identification of Deleterious Synonymous Variants in Human Genomes Using a Feature-Based Approach. BMC Med. Genomics 12 (Suppl. 1), 12. doi:10.1186/s12920-018-0455-6
Sibley, C. R., Emmett, W., Blázquez, L., Faro, A., Haberman, N., Briese, M., et al. (2015). Recursive Splicing in Long Vertebrate Genes. Nature 521 (7552), 371–375. doi:10.1038/nature14466
Simon, M. T., Eftekharian, S. S., Stover, A. E., Osborne, A. F., Braffman, B. H., Chang, R. C., et al. (2019). Novel Mutations in the Mitochondrial Complex I Assembly Gene NDUFAF5 Reveal Heterogeneous Phenotypes. Mol. Genet. Metab. 126 (1), 53–63. doi:10.1016/j.ymgme.2018.11.001
Sjarif, D. R., Hellerud, C., Amstel, J. K. P. v., Kleijer, W. J., Sperl, W., Lacombe, D., et al. (2004). Glycerol Kinase Deficiency: Residual Activity Explained by Reduced Transcription and Enzyme Conformation. Eur. J. Hum. Genet. 12 (6), 424–432. doi:10.1038/sj.ejhg.5201172
Smith, B. F., Kornegay, J. N., and Duan, D. (2007). Independent Canine Models of Duchenne Muscular Dystrophy Due to Intronic Insertions of Repetitive DNA. Mol. Ther. 15. doi:10.1016/s1525-0016(16)44336-4
Smith, M. J., Bowers, N. L., Banks, C., Coates‐Brown, R., Morris, K. A., Ewans, L., et al. (2020). A Deep Intronic SMARCB1 Variant Associated with Schwannomatosis. Clin. Genet. 97 (2), 376–377. doi:10.1111/cge.13637
Soliman, S. E., Racher, H., Lambourne, M., Matevski, D., MacDonald, H., and Gallie, B. (2018). A Novel Deep Intronic Low Penetrance RB1 Variant in a Retinoblastoma Family. Ophthalmic Genet. 39 (2), 288–290. doi:10.1080/13816810.2017.1393828
Spena, S., Asselta, R., Platé, M., Castaman, G., Duga, S., and Tenchini, M. L. (2007). Pseudo-exon Activation Caused by a Deep-Intronic Mutation in the Fibrinogen ?-chain Gene as a Novel Mechanism for Congenital Afibrinogenaemia. Br. J. Haematol. 139 (1), 128–132. doi:10.1111/j.1365-2141.2007.06758.x
Spier, I., Horpaopan, S., Vogt, S., Uhlhaas, S., Morak, M., Stienen, D., et al. (2012). Deep intronicAPCmutations Explain a Substantial Proportion of Patients with Familial or Early-Onset Adenomatous Polyposis. Hum. Mutat. 33 (7), 1045–1050. doi:10.1002/humu.22082
Spits, C., De Rycke, M., Van Ranst, N., Joris, H., Verpoest, W., Lissens, W., et al. (2005). Preimplantation Genetic Diagnosis for Neurofibromatosis Type 1. Mol. Hum. Reprod. 11 (5), 381–387. doi:10.1093/molehr/gah170
Stelzer, G., Rosen, N., Plaschkes, I., Zimmerman, S., Twik, M., Fishilevich, S., et al. (2016). The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinformatics 54, 1–33. doi:10.1002/cpbi.5
Stenson, P. D., Mort, M., Ball, E. V., Evans, K., Hayden, M., Heywood, S., et al. (2017). The Human Gene Mutation Database: towards a Comprehensive Repository of Inherited Mutation Data for Medical Research, Genetic Diagnosis and Next-Generation Sequencing Studies. Hum. Genet. 136 (6), 665–677. doi:10.1007/s00439-017-1779-6
Stucki, M., Suormala, T., Fowler, B., Valle, D., and Baumgartner, M. R. (2009). Cryptic Exon Activation by Disruption of Exon Splice Enhancer. J. Biol. Chem. 284 (42), 28953–28957. doi:10.1074/jbc.M109.050674
Stum, M., Davoine, C.-S., Vicart, S., Guillot-Noël, L., Topaloglu, H., Carod-Artal, F. J., et al. (2006). Spectrum ofHSPG2(Perlecan) Mutations in Patients with Schwartz-Jampel Syndrome. Hum. Mutat. 27 (11), 1082–1091. doi:10.1002/humu.20388
Suga, Y., Tsuda, T., Nagai, M., Sakaguchi, Y., Jitsukawa, O., Yamamoto, M., et al. (2015). Lamellar Ichthyosis with Pseudoexon Activation in the Transglutaminase 1 Gene. J. Dermatol. 42 (6), 642–645. doi:10.1111/1346-8138.12846
Suminaga, R., Takeshima, Y., Adachi, K., Yagi, M., Nakamura, H., and Matsuo, M. (2002). A Novel Cryptic Exon in Intron 3 of the Dystrophin Gene Was Incorporated into Dystrophin mRNA with a Single Nucleotide Deletion in Exon 5. J. Hum. Genet. 47 (4), 196–201. doi:10.1007/s100380200023
Sutandy, F. X. R., Ebersberger, S., Huang, L., Busch, A., Bach, M., Kang, H.-S., et al. (2018). In Vitro iCLIP-based Modeling Uncovers How the Splicing Factor U2AF2 Relies on Regulation by Cofactors. Genome Res. 28 (5), 699–713. doi:10.1101/gr.229757.117
Sutton, I. J., Last, J. I. K., Ritchie, S. J., Harrington, H. J., Byrd, P. J., and Taylor, A. M. R. (2004). Adult-onset Ataxia Telangiectasia Due to ATM 5762ins137 Mutation Homozygosity. Ann. Neurol. 55 (6), 891–895. doi:10.1002/ana.20139
Suzuki, H., Aoki, Y., Kameyama, T., Saito, T., Masuda, S., Tanihata, J., et al. (2016). Endogenous Multiple Exon Skipping and Back-Splicing at the DMD Mutation Hotspot. Int. J. Mol. Sci. 17 (10), 1. doi:10.3390/ijms17101722
Svaasand, E. K. (2015). A Novel Deep Intronic Mutation Introducing a Cryptic Exon Causing Neurofibromatosis Type 1 in a Family with Highly Variable Phenotypes: A Case Study. Hereditary Genet. 04 (03), 1. doi:10.4172/2161-1041.1000152
Takahara, K., Schwarze, U., Imamura, Y., Hoffman, G. G., Toriello, H., Smith, L. T., et al. (2002). Order of Intron Removal Influences Multiple Splice Outcomes, Including a Two-Exon Skip, in a COL5A1 Acceptor-Site Mutation that Results in Abnormal Pro-α1(V) N-Propeptides and Ehlers-Danlos Syndrome Type I. Am. J. Hum. Genet. 71 (3), 451–465. doi:10.1086/342099
Takeshima, Y., Yagi, M., Okizuka, Y., Awano, H., Zhang, Z., Yamauchi, Y., et al. (2010). Mutation Spectrum of the Dystrophin Gene in 442 Duchenne/Becker Muscular Dystrophy Cases from One Japanese Referral center. J. Hum. Genet. 55 (6), 379–388. doi:10.1038/jhg.2010.49
Tosch, V., Vasli, N., Kretz, C., Nicot, A.-S., Gasnier, C., Dondaine, N., et al. (2010). Novel Molecular Diagnostic Approaches for X-Linked Centronuclear (Myotubular) Myopathy Reveal Intronic Mutations. Neuromuscul. Disord. 20 (6), 375–381. doi:10.1016/j.nmd.2010.03.015
Tozawa, Y., Abdrabou, S. S. M. A., Nogawa-Chida, N., Nishiuchi, R., Ishida, T., Suzuki, Y., et al. (2019). A Deep Intronic Mutation of c.1166-285 T > G in SLC46A1 Is Shared by Four Unrelated Japanese Patients with Hereditary Folate Malabsorption (HFM). Clin. Immunol. 208, 108256. doi:10.1016/j.clim.2019.108256
Trabelsi, M., Beugnet, C., Deburgrave, N., Commere, V., Orhant, L., Leturcq, F., et al. (2014). When a Mid-intronic Variation of DMD Gene Creates an ESE Site. Neuromuscul. Disord. 24 (12), 1111–1117. doi:10.1016/j.nmd.2014.07.003
Tran, V. K., Zhang, Z., Yagi, M., Nishiyama, A., Habara, Y., Takeshima, Y., et al. (2005). A Novel Cryptic Exon Identified in the 3′ Region of Intron 2 of the Human Dystrophin Gene. J. Hum. Genet. 50 (8), 425–433. doi:10.1007/s10038-005-0272-6
Treisman, R., Orkin, S. H., and Maniatis, T. (1983). Specific Transcription and RNA Splicing Defects in Five Cloned β-thalassaemia Genes. Nature 302 (5909), 591–596. doi:10.1038/302591a0
Tsuruta, M., Mitsubuchi, H., Mardy, S., Miura, Y., Hayashida, Y., Kinugasa, A., et al. (1998). Molecular Basis of Intermittent maple Syrup Urine Disease: Novel Mutations in the E2 Gene of the Branched-Chain α-keto Acid Dehydrogenase Complex. J. Hum. Genet. 43 (2), 91–100. doi:10.1007/s100380050047
Tubeuf, H., Charbonnier, C., Soukarieh, O., Blavier, A., Lefebvre, A., Dauchel, H., et al. (2020). Large‐scale Comparative Evaluation of User‐friendly Tools for Predicting Variant‐induced Alterations of Splicing Regulatory Elements. Hum. Mutat. 41 (10), 1811–1829. doi:10.1002/humu.24091
Tuffery-Giraud, S., Saquet, C. l., Chambert, S., and Claustres, M. (2003). Pseudoexon Activation in theDMD Gene as a Novel Mechanism for Becker Muscular Dystrophy. Hum. Mutat. 21 (6), 608–614. doi:10.1002/humu.10214
Tuffery-Giraud, S., Saquet, C., Thorel, D., Disset, A., Rivier, F., Malcolm, S., et al. (2005). Mutation Spectrum Leading to an Attenuated Phenotype in Dystrophinopathies. Eur. J. Hum. Genet. 13 (12), 1254–1260. doi:10.1038/sj.ejhg.5201478
Turunen, J. J., Niemelä, E. H., Verma, B., and Frilander, M. J. (2013). The Significant Other: Splicing by the Minor Spliceosome. Wiley Interdiscip Rev RNA 4 (1), 61–76. doi:10.1002/wrna.1141
Vaché, C., Besnard, T., le Berre, P., García-García, G., Baux, D., Larrieu, L., et al. (2012). Usher Syndrome Type 2 Caused by Activation of an USH2A Pseudoexon: Implications for Diagnosis and Therapy. Hum. Mutat. 33 (1), 104–108. doi:10.1002/humu.21634
Valcárcel, J., Gaur, R. K., Singh, R., and Green, M. R. (1996). Interaction of U2AF 65 RS Region with Pre-mRNA of Branch Point and Promotion Base Pairing with U2 snRNA. Science 273 (5282), 1706–1709. doi:10.1126/science.273.5282.1706
Valdmanis, P. N., Belzil, V. V., Lee, J., Dion, P. A., St-Onge, J., Hince, P., et al. (2009). A Mutation that Creates a Pseudoexon in SOD1 Causes Familial ALS. Ann. Hum. Genet. 73 (Pt 6), 652–657. doi:10.1111/j.1469-1809.2009.00546.x
Valero, M. C., Martín, Y., Hernández-Imaz, E., Marina Hernández, A., Meleán, G., Valero, A. M., et al. (2011). A Highly Sensitive Genetic Protocol to Detect NF1 Mutations. J. Mol. Diagn. 13 (2), 113–122. doi:10.1016/j.jmoldx.2010.09.002
van den Hurk, J. A. J. M., van de Pol, D. J. R., Wissinger, B., van Driel, M. A., Hoefsloot, L. H., de Wijs, I. J., et al. (2003). Novel Types of Mutation in the Choroideremia (CHM) Gene: a Full-Length L1 Insertion and an Intronic Mutation Activating a Cryptic Exon. Hum. Genet. 113 (3), 268–275. doi:10.1007/s00439-003-0970-0
van der Klift, H. M., Tops, C. M., Hes, F. J., Devilee, P., and Wijnen, J. T. (2012). Insertion of an SVA Element, a Nonautonomous Retrotransposon, inPMS2intron 7 as a Novel Cause of lynch Syndrome. Hum. Mutat. 33 (7), 1051–1055. doi:10.1002/humu.22092
van der Wal, E., Bergsma, A. J., van Gestel, T. J. M., in ‘t Groen, S. L. M., Zaehres, H., Araúzo-Bravo, M. J., et al. (2017). GAA Deficiency in Pompe Disease Is Alleviated by Exon Inclusion in iPSC-Derived Skeletal Muscle Cells. Mol. Ther. - Nucleic Acids 7, 101–115. doi:10.1016/j.omtn.2017.03.002
van Kuilenburg, A. B. P., Meijer, J., Mul, A. N. P. M., Meinsma, R., Schmid, V., Dobritzsch, D., et al. (2010). Intragenic Deletions and a Deep Intronic Mutation Affecting Pre-mRNA Splicing in the Dihydropyrimidine Dehydrogenase Gene as Novel Mechanisms Causing 5-fluorouracil Toxicity. Hum. Genet. 128 (5), 529–538. doi:10.1007/s00439-010-0879-3
Vandenbroucke, I., Callens, T., De Paepe, A., and Messiaen, L. (2002). Complex Splicing Pattern Generates Great Diversity in Human NF1 Transcripts. BMC Genomics 3, 13. doi:10.1186/1471-2164-3-13
Varon, R., Gooding, R., Steglich, C., Marns, L., Tang, H., Angelicheva, D., et al. (2003). Partial Deficiency of the C-Terminal-Domain Phosphatase of RNA Polymerase II Is Associated with Congenital Cataracts Facial Dysmorphism Neuropathy Syndrome. Nat. Genet. 35 (2), 185–189. doi:10.1038/ng1243
Vatanavicharn, N., Champattanachai, V., Liammongkolkul, S., Sawangareetrakul, P., Keeratichamroen, S., Ketudat Cairns, J. R., et al. (2012). Clinical and Molecular Findings in Thai Patients with Isolated Methylmalonic Acidemia. Mol. Genet. Metab. 106 (4), 424–429. doi:10.1016/j.ymgme.2012.05.012
Vaz-Drago, R., Custódio, N., and Carmo-Fonseca, M. (2017). Deep Intronic Mutations and Human Disease. Hum. Genet. 136 (9), 1093–1111. doi:10.1007/s00439-017-1809-4
Verdura, E., Schlüter, A., Fernández‐Eulate, G., Ramos‐Martín, R., Zulaica, M., Planas‐Serra, L., et al. (2020). A Deep Intronic Splice Variant Advises Reexamination of Presumably Dominant SPG7 Cases. Ann. Clin. Transl Neurol. 7 (1), 105–111. doi:10.1002/acn3.50967
Verrier, F., Dubois d'Enghien, C., Gauthier-Villars, M., Bonadona, V., Faure-Conter, C., Dijoud, F., et al. (2018). Mutiple DICER1 -related Lesions Associated with a Germline Deep Intronic Mutation. Pediatr. Blood Cancer 65 (6), e27005. doi:10.1002/pbc.27005
Vetrini, F., Tammaro, R., Bondanza, S., Surace, E. M., Auricchio, A., De Luca, M., et al. (2006). Aberrant Splicing in the Ocular Albinism Type 1 Gene (OA1/GPR143) Is Corrected In Vitro by Morpholino Antisense Oligonucleotides. Hum. Mutat. 27 (5), 420–426. doi:10.1002/humu.20303
Voith von Voithenberg, L., Sánchez-Rico, C., Kang, H.-S., Madl, T., Zanier, K., Barth, A., et al. (2016). Recognition of the 3′ Splice Site RNA by the U2AF Heterodimer Involves a Dynamic Population Shift. Proc. Natl. Acad. Sci. USA 113 (46), E7169–E7175. doi:10.1073/pnas.1605873113
Vorechovsky, I. (2010). Transposable Elements in Disease-Associated Cryptic Exons. Hum. Genet. 127 (2), 135–154. doi:10.1007/s00439-009-0752-4
Waddell, L. B., Bryen, S. J., Cummings, B. B., Bournazos, A., Evesson, F. J., Joshi, H., et al. (2021). WGS and RNA Studies Diagnose Noncoding DMD Variants in Males with High Creatine Kinase. Neurol. Genet. 7 (1), e554. doi:10.1212/NXG.0000000000000554
Walker, S., Lamoureux, S., Khan, T., Joynt, A. C. M., Bradley, M., Branson, H. M., et al. (2021). Genome Sequencing for Detection of Pathogenic Deep Intronic Variation: A Clinical Case Report Illustrating Opportunities and Challenges. Am. J. Med. Genet. A. 185 (10), 3129–3135. doi:10.1002/ajmg.a.62389
Walsh, T., Casadei, S., Munson, K. M., Eng, M., Mandell, J. B., Gulsuner, S., et al. (2020). CRISPR-Cas9/long-read Sequencing Approach to Identify Cryptic Mutations in BRCA1 and Other Tumour Suppressor Genes. J. Med. Genet. 58 (12), 850–852. doi:10.1136/jmedgenet-2020-107320
Wan, Y., Anastasakis, D. G., Rodriguez, J., Palangat, M., Gudla, P., Zaki, G., et al. (2021). Dynamic Imaging of Nascent RNA Reveals General Principles of Transcription Dynamics and Stochastic Splice Site Selection. Cell 184 (11), 2878–2895. e2820. doi:10.1016/j.cell.2021.04.012
Wang, X., Zhang, Y., Ding, J., and Wang, F. (2021). mRNA Analysis Identifies Deep Intronic Variants Causing Alport Syndrome and Overcomes the Problem of Negative Results of Exome Sequencing. Sci. Rep. 11 (1), 18097. doi:10.1038/s41598-021-97414-0
Webb, T. R., Parfitt, D. A., Gardner, J. C., Martinez, A., Bevilacqua, D., Davidson, A. E., et al. (2012). Deep Intronic Mutation in OFD1, Identified by Targeted Genomic Next-Generation Sequencing, Causes a Severe Form of X-Linked Retinitis Pigmentosa (RP23). Hum. Mol. Genet. 21 (16), 3647–3654. doi:10.1093/hmg/dds194
Weisschuh, N., Mazzola, P., Bertrand, M., Haack, T. B., Wissinger, B., Kohl, S., et al. (2021). Clinical Characteristics of POC1B-Associated Retinopathy and Assignment of Pathogenicity to Novel Deep Intronic and Non-canonical Splice Site Variants. Int. J. Mol. Sci. 22 (10), 1. doi:10.3390/ijms22105396
Weisschuh, N., Sturm, M., Baumann, B., Audo, I., Ayuso, C., Bocquet, B., et al. (2020). Deep‐intronic Variants in CNGB3 Cause Achromatopsia by Pseudoexon Activation. Hum. Mutat. 41 (1), 255–264. doi:10.1002/humu.23920
Welander, J., Larsson, C., Bäckdahl, M., Hareni, N., Sivlér, T., Brauckhoff, M., et al. (2012). Integrative Genomics Reveals Frequent Somatic NF1 Mutations in Sporadic Pheochromocytomas. Hum. Mol. Genet. 21 (26), 5406–5416. doi:10.1093/hmg/dds402
Whatley, S. D., Mason, N. G., Rhodes, J. M., Stewart, M. F., Reed, P., Crowley, V., et al. (2013). Pseudoexon Activation in the HMBS Gene as a Cause of the Nonerythroid Form of Acute Intermittent Porphyria. Clin. Chem. 59 (7), 1123–1125. doi:10.1373/clinchem.2012.199117
Will, K., Dörk, T., Stuhrmann, M., Meitinger, T., Bertele-Harms, R., Tümmler, B., et al. (1994). A Novel Exon in the Cystic Fibrosis Transmembrane Conductance Regulator Gene Activated by the Nonsense Mutation E92X in Airway Epithelial Cells of Patients with Cystic Fibrosis. J. Clin. Invest. 93 (4), 1852–1859. doi:10.1172/JCI117172
Wilson, A., Leclerc, D., Rosenblatt, D. S., and Gravel, R. A. (1999). Molecular Basis for Methionine Synthase Reductase Deficiency in Patients Belonging to the cblE Complementation Group of Disorders in Folate/cobalamin Metabolism. Hum. Mol. Genet. 8 (11), 2009–2016. doi:10.1093/hmg/8.11.2009
Wilson, A., Leclerc, D., Saberi, F., Campeau, E., Hwang, H. Y., Shane, B., et al. (1998). Functionally Null Mutations in Patients with the cblG-Variant Form of Methionine Synthase Deficiency. Am. J. Hum. Genet. 63 (2), 409–414. doi:10.1086/301976
Wilund, K. R., Yi, M., Campagna, F., Arca, M., Zuliani, G., Fellin, R., et al. (2002). Molecular Mechanisms of Autosomal Recessive Hypercholesterolemia. Hum. Mol. Genet. 11 (24), 3019–3030. doi:10.1093/hmg/11.24.3019
Wimmer, K., Callens, T., Wernstedt, A., and Messiaen, L. (2011). The NF1 Gene Contains Hotspots for L1 Endonuclease-dependent De Novo Insertion. Plos Genet. 7 (11), e1002371. doi:10.1371/journal.pgen.1002371
Xie, Z., Sun, C., Liu, Y., Yu, M., Zheng, Y., Meng, L., et al. (2020). Practical Approach to the Genetic Diagnosis of Unsolved Dystrophinopathies: a Stepwise Strategy in the Genomic Era. J. Med. Genet. 58 (11), 743–751. doi:10.1136/jmedgenet-2020-107113
Xu, Y., Song, T., Li, Y., Guo, F., Jin, X., Cheng, L., et al. (2020). Identification of Two Novel Insertion Abnormal Transcripts in Two Chinese Families Affected with Dystrophinopathy. J. Clin. Lab. Anal. 34 (4), e23142. doi:10.1002/jcla.23142
Yagi, M., Takeshima, Y., Wada, H., Nakamura, H., and Matsuo, M. (2003). Two Alternative Exons Can Result from Activation of the Cryptic Splice Acceptor Site Deep within Intron 2 of the Dystrophin Gene in a Patient with as yet Asymptomatic Dystrophinopathy. Hum. Genet. 112 (2), 164–170. doi:10.1007/s00439-002-0854-8
Yamaguchi, H., Fujimoto, T., Nakamura, S., Ohmura, K., Mimori, T., Matsuda, F., et al. (2010). Aberrant Splicing of the Milk Fat Globule-EGF Factor 8 (MFG-E8) Gene in Human Systemic Lupus Erythematosus. Eur. J. Immunol. 40 (6), 1778–1785. doi:10.1002/eji.200940096
Yamano, S., Nhamburo, P. T., Aoyama, T., Meyer, U. A., Inaba, T., Kalow, W., et al. (1989). cDNA Cloning and Sequence and cDNA-Directed Expression of Human P450 IIB1: Identification of a normal and Two Variant cDNAs Derived from the CYP2B Locus on Chromosome 19 and Differential Expression of the IIB mRNAs in Human Liver. Biochemistry 28 (18), 7340–7348. doi:10.1021/bi00444a029
Yasmeen, S., Lund, K., De Paepe, A., De Bie, S., Heiberg, A., Silva, J., et al. (2014). Occipital Horn Syndrome and Classical Menkes Syndrome Caused by Deep Intronic Mutations, Leading to the Activation of ATP7A Pseudo-exon. Eur. J. Hum. Genet. 22 (4), 517–521. doi:10.1038/ejhg.2013.191
Yeo, G., and Burge, C. B. (2004). Maximum Entropy Modeling of Short Sequence Motifs with Applications to RNA Splicing Signals. J. Comput. Biol. 11 (2-3), 377–394. doi:10.1089/1066527041410418
Yuste-Checa, P., Medrano, C., Gámez, A., Desviat, L. R., Matthijs, G., Ugarte, M., et al. (2015). Antisense-mediated Therapeutic Pseudoexon Skipping in TMEM165-CDG. Clin. Genet. 87 (1), 42–48. doi:10.1111/cge.12402
Zaum, A.-K., Stüve, B., Gehrig, A., Kölbel, H., Schara, U., Kress, W., et al. (2017). Deep Intronic Variants Introduce DMD Pseudoexon in Patient with Muscular Dystrophy. Neuromuscul. Disord. 27 (7), 631–634. doi:10.1016/j.nmd.2017.04.003
Zenteno, J. C., García-Montaño, L. A., Cruz-Aguilar, M., Ronquillo, J., Rodas-Serrano, A., Aguilar-Castul, L., et al. (2020). Extensive Genic and Allelic Heterogeneity Underlying Inherited Retinal Dystrophies in Mexican Patients Molecularly Analyzed by Next-Generation Sequencing. Mol. Genet. Genomic Med. 8 (1), 1. doi:10.1002/mgg3.1044
Zhang, J., Sun, X., Qian, Y., LaDuca, J. P., and Maquat, L. E. (1998). At Least One Intron Is Required for the Nonsense-Mediated Decay of Triosephosphate Isomerase mRNA: a Possible Link between Nuclear Splicing and Cytoplasmic Translation. Mol. Cel Biol 18 (9), 5272–5283. doi:10.1128/MCB.18.9.5272
Zhang, K., Nowak, I., Rushlow, D., Gallie, B. L., and Lohmann, D. R. (2008). Patterns of Missplicing Caused byRB1gene Mutations in Patients with Retinoblastoma and Association with Phenotypic Expression. Hum. Mutat. 29 (4), 475–484. doi:10.1002/humu.20664
Zhang, X.-O., Fu, Y., Mou, H., Xue, W., and Weng, Z. (2018). The Temporal Landscape of Recursive Splicing during Pol II Transcription Elongation in Human Cells. Plos Genet. 14 (8), e1007579. doi:10.1371/journal.pgen.1007579
Zhang, Y., Qian, J., Gu, C., and Yang, Y. (2021). Alternative Splicing and Cancer: a Systematic Review. Sig Transduct Target. Ther. 6 (1), 78. doi:10.1038/s41392-021-00486-7
Zhang, Z., Habara, Y., Nishiyama, A., Oyazato, Y., Yagi, M., Takeshima, Y., et al. (2007). Identification of Seven Novel Cryptic Exons Embedded in the Dystrophin Gene and Characterization of 14 Cryptic Dystrophin Exons. J. Hum. Genet. 52 (7), 607–617. doi:10.1007/s10038-007-0163-0
Zhu, F., Zhang, F., Hu, L., Liu, H., and Li, Y. (2021). Integrated Genome and Transcriptome Sequencing to Solve a Neuromuscular Puzzle: Miyoshi Muscular Dystrophy and Early Onset Primary Dystonia in Siblings of the Same Family. Front. Genet. 12, 672906. doi:10.3389/fgene.2021.672906
Keywords: pseudoexons, cryptic splicing, splicing mutations, recursive splicing, poison exons, genetic disease
Citation: Keegan NP, Wilton SD and Fletcher S (2022) Analysis of Pathogenic Pseudoexons Reveals Novel Mechanisms Driving Cryptic Splicing. Front. Genet. 12:806946. doi: 10.3389/fgene.2021.806946
Received: 01 November 2021; Accepted: 09 December 2021;
Published: 24 January 2022.
Edited by:
Michael R. Ladomery, University of the West of England, United KingdomReviewed by:
John Conboy, Lawrence Berkeley National Laboratory, United StatesIan Eperon, University of Leicester, United Kingdom
Copyright © 2022 Keegan, Wilton and Fletcher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Niall P. Keegan, N.Keegan@murdoch.edu.au