 Hua Wang
Hua Wang Jiatong Liu1
Jiatong Liu1- 1Department of Pediatric Neurology, Shengjing Hospital of China Medical University, Shenyang, China
- 2Chigene (Beijing) Translational Medical Research Center Co., Ltd., Beijing, China
Objective: Neurodevelopmental disorder with or without seizure and gait abnormalities (NEDSGA, MIM * 617864) is a newly described autosomal dominant inherited disease caused by a heterozygous variant in the GRIA4 gene. GRIA4 plays an essential role in excitatory synaptic transmission. In this study, we presented the clinical and genetic features of a female patient carrying a novel de novo variant in GRIA4 and further reviewed the previously reported five different patients.
Methods: Evaluation of the patient included a detailed history and clinical examination. Trio-whole exome sequencing (WES) was performed to identify pathogenic variants in NEDSGA. Sanger sequencing was further used to validate the variants.
Results: We described the clinical features of an infant diagnosed with NEDSGA caused by a GRIA4 variant, who presented with severe developmental delay, limb hypertonia, generalized seizure, retinal hypoplasia, and chorioretinal hyperpigmentation. The patient developed tricuspid regurgitation, and imaging examination revealed a patent foramen ovale. Trio-WES identified a novel de novo heterozygous missense variant c.1918G>T, p.Ala640Ser in the GRIA4 gene. Multiple in silico tools predicted deleterious effects of p.Ala640Ser.
Conclusion: A novel heterozygous missense variant in the GRIA4 gene (c.1918G>T) identified in the proband expanded the genotypic and phenotypic spectrum of disorders associated with GRIA4 variants. This is the first NEDSGA case reported in China. Our findings provide valuable information for the differential diagnosis of neonatal onset neurodevelopmental disorders.
Introduction
Neurodevelopmental disorder with or without seizure and gait abnormalities (NEDSGA, MIM * 617864) is an early onset of neurodevelopmental disorder associated with global developmental delay and variable intellectual disability. Most patients presented with irritability, stiffness, seizure, and hypertonia early in life, followed by spasticity and impaired gait. NEDSGA is caused by the gene GRIA4 (MIM * 138246) located on chromosomes 11q22 and is inherited in an autosomal dominant manner. GRIA4 encodes glutamate ionotropic receptor AMPA type subunit 4 and plays an essential role in excitatory synaptic transmission. GRIA4 is ubiquitous in the central nervous system and is highly expressed in the thalamus, especially in the thalamic reticular nucleus (Beyer et al., 2008). GRIA4 in the rat brain is relatively high in CA1 pyramidal cells, hippocampal dentate gyrus, cerebral cortex, and cerebellar granule cells (Keinänen et al., 1990). Since Martin first reported NEDSGA in 2017 (Martin et al., 2017), only five NEDSGA patients with GRIA4 variants have been reported, ranging in age from 4 to 21 years. All GRIA4 variants described are de novo heterozygous missense variants that cluster in the transmembrane and ligand-binding domains of the GRIA4 protein.
In this study, we reported a 9-month-old girl from a nonconsanguineous family with healthy parents. The patient presented with severe global developmental delay (HP:0011344), limb hypertonia (HP:0002509), partial seizure (HP:0007359), retinal hypoplasia (HP:0007770), chorioretinal hyperpigmentation (HP:0040031), tricuspid regurgitation (HP:0005180), and patent foramen ovale (HP:0001655). Using trio-whole exome sequencing (WES), we identified a novel de novo heterozygous missense variant in GRIA4, c.1918G>T, p.Ala640Ser. To the best of our knowledge, this is the first NEDSGA case reported in China. Our finding expands the genotypic and phenotypic spectrum associated with GRIA4.
Patients and Methods
Patients
The Ethics Committee of the Shengjing Hospital of China Medical University approved this study. The patient’s legal guardians signed the informed consent for the study.
The proband was a 9-month-old girl, the first child of healthy Chinese parents. The mother was 34 years old, and the father was 40 years old. The patient was transferred to our hospital due to the occurrence of breathing difficulties, limb hypertonia, and seizure. Electroencephalogram (EEG) and brain magnetic resonance imaging (MRI) were performed on her clinical presentation.
Variation Analysis
DNA was obtained from the peripheral venous blood of the girl and the parents and submitted to the Chigene Translational Medicine Research Center Co., Ltd., Beijing, for trio (parents and proband)-WES. Whole-exome capture was xGen Exome Research Panel v2.0 (IDT, Iowa, United States). The sequencing operation flow was standardized on the DNBSEQ-T7 (BGI, China) platform. Raw-sequencing reads were processed by fastp (https://github.com/OpenGene/fastp) for adapter removal and low-quality read filtering. High-quality sequencing data were generated and performed on the Ensemble GRCh37/hg19 reference genome using the Burrows-Wheeler Aligner (BWA, https://github.com/lh3/bwa). GATK (http://www.broadinstitute.org/gatk/) was used for base quality score recalibration and SNP and INDEL calling. Trio-WES had a mean depth of coverage of at least 172× per sample, with 98% of the exome covered 20× or greater. The sequencing depth ranged from 192×−413× coverage of the GRIA4 gene, with 100% target region coverage >10× sequencing depth. Pathogenicity of the genetic variants were predicted by bioinformatics tools such as PolyPhen (http://www.bork.embl-heidelberg.de/PolyPhen/), Mutation Taster (http://www.mutationtaster.org), REVEL (https://sites.google.com/site/revelgenomics/), and CADD (http://cadd.gs.washington.edu/). Finally, the variants were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015).
Pathogenic variants were detected using trio-WES, followed by Sanger sequencing validation. Primer 5.0 primer software was used to design the GRIA4 primers: forward primer (5′- GCTAAAGCCCATGGTATAATTGTTG-3′) and reverse primer (5′-GCATGGTGAATTGACGGTATTTCTT-3′). Sanger sequencing was further performed using the 3730xl DNA Analyzer (Applied Biosystems, United States). The identified rare variants have been submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) (Accession number: SCV002055968).
The GRIA4 protein is highly conserved among vertebrates, showing 86% sequence identity among 10 different species, including primates, rodents, laurasiatheria, placental mammals, sauropsida, and fish (http://asia.ensembl.org/index.html). Multiple protein sequence alignments were conducted on MEGA X (Kumar et al., 2018). 3D modeling of structural effects was performed using the GRIA4 protein structure (AlphaFold, AF-P48058-F1) (Varadi et al., 2022). The models were visualized using Pymol (www.pymol.org) (Schrodinger, 2015).
Result
Clinical Features
The girl was born by an uncomplicated delivery at 38 weeks’ gestation, with a normal birth weight of 3,200 g and a body length of 50 cm. Neonatally she presented with irritability, breathing difficulties, limb hypertonia, and partial seizure. Cardiac ultrasonography showed tricuspid regurgitation and patent foramen ovale. Fundus examinations revealed hypoplasia of the retina and chorioretinal hyperpigmentation. Brain magnetic resonance imaging (MRI) was normal. Her electroencephalogram (EEG) demonstrated multifocal sharp waves and low waves during sleep (Figures 1A–C). Laboratory tests showed hyperlactemia and hyperammonemia.
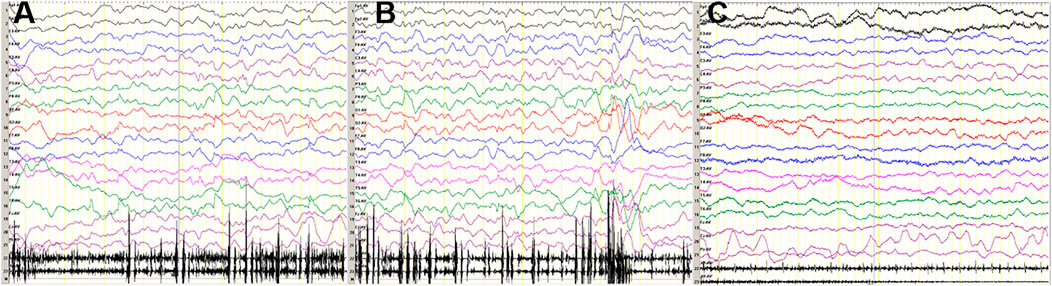
FIGURE 1. (A, B) At the age of 3 months, sleep EEG of the patient revealed multifocal sharp waves and low waves. (C) Wake EEG was normal.
In the follow-up, she suffered from severe developmental delay. At 4 months, she started taking lysine, inosite and vitamin B12 oral solution, GABA compound nutritious solid drink, and cerebroprotein hydrolysate oral solution. Limb hypertonia persisted, but seizures were well controlled.
To the best of our knowledge, only one publication (Table1) (Martin et al., 2017) reported a total of five cases with GRIA4 variants. Our patient is the sixth case of NEDSGA, in which neurodevelopmental disorders with seizures and abnormal gait were the most common phenotypes caused by pathogenic variants in GRIA4.
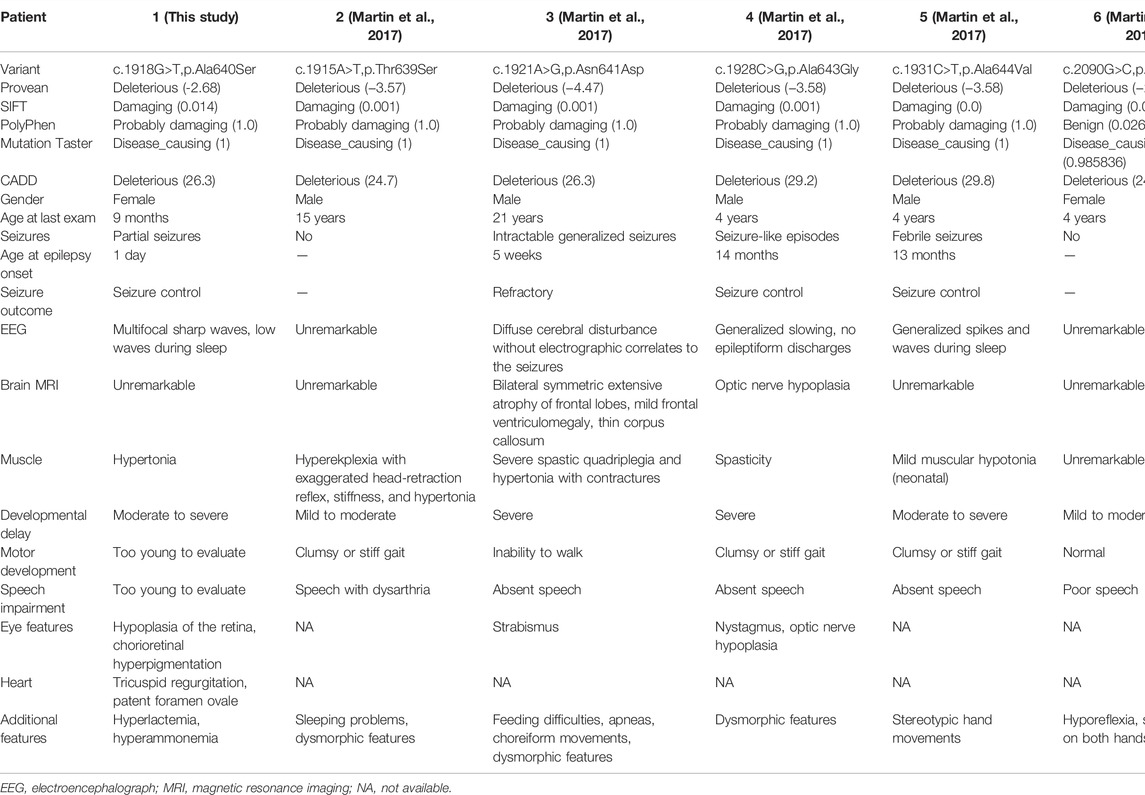
TABLE 1. Clinical features of individuals with de novo GRIA4 variants.
All six patients (2 females and 4 males, aged 9 months to 21 years) had neurodevelopmental disorders, with mild (2/6; 33%) to severe (4/6; 67%) developmental delay. Four patients (4/6; 67%) had movement disorders, including clumsy or stiff gait and inability to walk. Three patients (3/6; 50%) presented with ocular anomalies, including strabismus, nystagmus, optic nerve hypoplasia, hypoplasia of the retina, and chorioretinal hyperpigmentation. Five patients (5/6, 83%) had poor speech or aphasia. Seizures occurred in four of six patients (67%). Patient 2 had sudden muscle cramps/seizures lasting up to 1 hour after trauma, while patient 6 had no seizures. The age of seizure onset ranged from 1 day to 14 months, with a median age of 7 months. Of note, seizure disorders were found in all patients with severe developmental delay. A broad spectrum, including generalized seizures, febrile seizures, and seizure-like episodes, was reported. Three patients (3/4; 75%) achieved seizure control by therapies. However, patient 3 developed refractory seizures and status epilepticus. All four patients with seizures exhibited abnormal EEG and two of them had abnormal MRI.
Our patient presented with severe developmental delay, limb hypertonia, partial seizure, retinal hypoplasia, chorioretinal hyperpigmentation, tricuspid regurgitation, and patent foramen ovale, whereas the girl had no signs of craniofacial or MRI abnormalities. In addition, our patient was too young to assess her ability to walk or speak. Together with our clinical findings, half of the affected patients had ocular abnormalities, so the patient’s ocular examination should not be ignored.
Variation Analysis
Trio-WES identified a de novo heterozygous variant in GRIA4 in the patient: chr11:105797537G>T(hg19), c.1918G>T transition. The c.1918G>T is predicted to result in the substitution of the alanine residue p.Ala640Ser. This variant has not been reported in public databases (gnomAD v2.1.1, http://gnomad.broadinstitute.org/ and the 1000 Genomes Project, http://www.internationalgenome.org). In addition, it was predicted as pathogenic by multiple bioinformatic tools (SIFT: damaging; PolyPhen: probably damaging; Mutation Taster: disease_causing; REVEL: deleterious; CADD: 26.3). According to the ACMG guidelines, we confirmed the variant to be pathogenic (PS2+PM1+PM2+PP2+PP3) (Richards et al., 2015; Harrison et al., 2019). Furthermore, we did not detect pathogenic or likely pathogenic variants in genes known to be associated with neurodevelopmental disorder in probands using Trio-WES. The variants of the GRIA4 gene were confirmed by Sanger sequencing (Figure 2A).
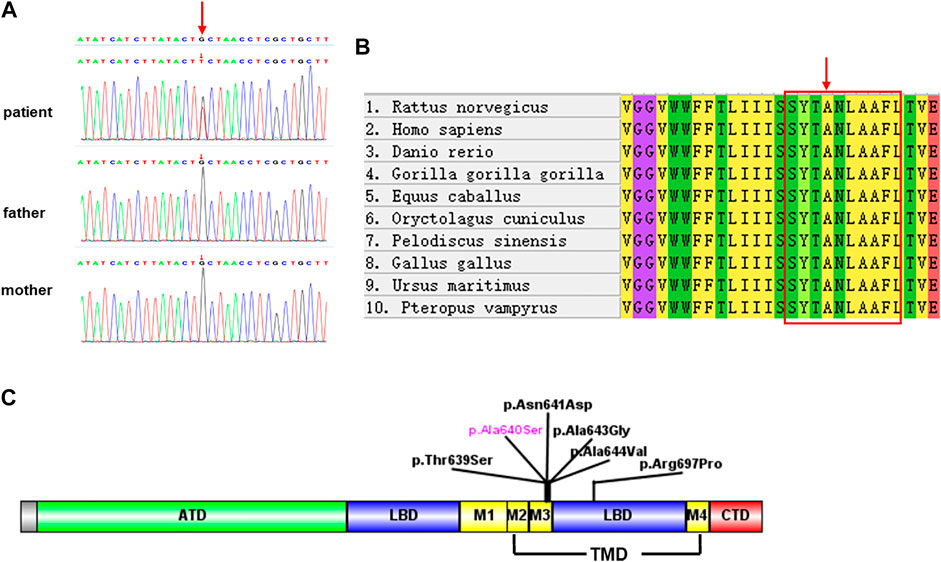
FIGURE 2. (A) Sanger sequencing validation of the variant c.1918G>T in the proband and parents. (B) Multiple species sequence alignment. The mutated alanine A640 (red arrow) falls within a highly conserved SYTANLAAF motif (red box). (C) Schematics depicting the location of GRIA4 variants. Amino-terminal domain (ATD, in green), ligand binding domain (LBD, in blue), transmembrane domain (TMD, in yellow), and carboxyl-terminal domain (CTD, in red). TMD contains three transmembrane domains (M1, M3, and M4) and re-entrant membrane loop M2.
Sequence alignment among multiple vertebrate species suggested that p.Ala640Ser was located at a highly conserved site (Figure 2B) and in the transmembrane domain (Figure 2C). 3D structural analysis of the GRIA4 protein showed that the mutated Ser640 residue formed a new hydrogen bond between Ser640 and the neighboring Ser636 compared to the WT model (Figures 3A–C). The mutation of Ala640Ser, located in an alpha-helix, causes the change of hydrogen bond between residues, and further affects protein folding.
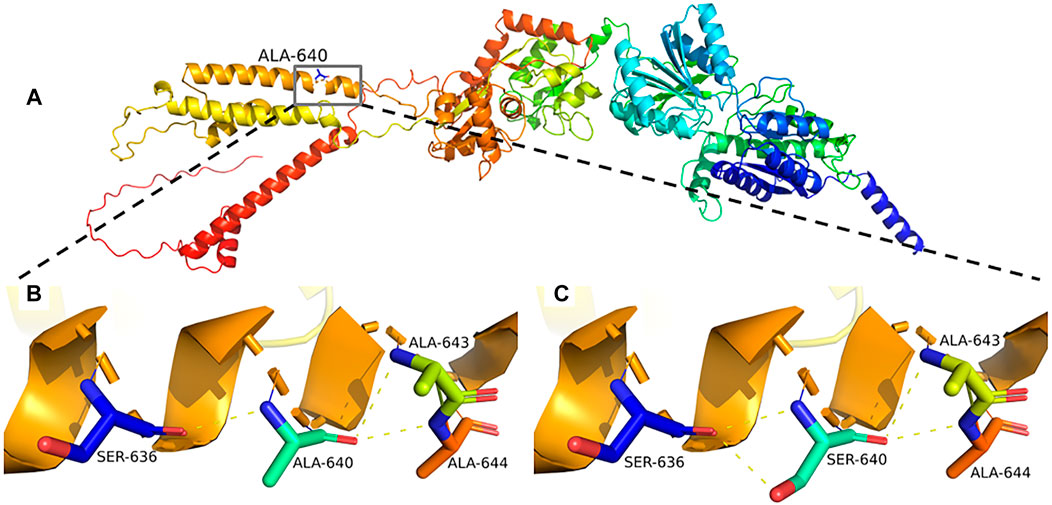
FIGURE 3. (A–C) Predicted mutational impact of p.Ala640Ser on the GRIA4 protein structure. Compared to the WT model (B), the new hydrogen bond formed between the mutant Ser640 (C) and the neighboring Ser636 hydrogen bonds, yellow dashed lines.
Discussion
NEDSGA is a newly recognized rare neurodevelopmental disorder caused by a heterozygous variant in the GRIA4 gene. All the reported GRIA4 variants were heterozygous missense, located in the transmembrane and ligand-binding domains (Figure 2C) (Martin et al., 2017).
AMPARs (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors) consist of four subunits GluR1-GluR4, mediate fast excitatory neurotransmission in the central nervous system, and play a critical role in learning, memory formation, and brain development. Each iGluR subunit comprises an amino-terminal domain (ATD), a ligand-binding domain (LBD), a transmembrane domain (TMD), and a carboxyl-terminal domain (CTD) (Figure 2C) (Traynelis et al., 2010; Sobolevsky, 2015). TMD contains three transmembrane domains (M1, M3, and M4) and re-entrant membrane loop M2 (Twomey et al., 2017). M3 transmembrane segment contains a nine amino acid sequence, SYTANLAAF motif, that plays a crucial role in channel activation and gating, and is highly sensitive to a conservative amino acid change (Salpietro et al., 2019). The variant p.Ala640Ser is located in the SYTANLAAF motif that is highly conserved throughout all members of the glutamate receptor family (Jones et al., 2002). Furthermore, multiple sequence alignment analysis revealed that p.Ala640 is highly conserved in different species (Figure 2B). For the variant p.Ala640Ser, the presence of the serine acid residue is predicted to lead to the formation of a new hydrogen bond with the p.Ala636 residue. p.Ala640Ser is in hydrophobic membrane-spanning helix M3, mutation of a critical hydrophobic residue (Ala) to a hydrophilic one (Ser) is predicted to destroy the hydrophobic interactions and further disrupt the gating mechanism (Sobolevsky et al., 2003; Sobolevsky, 2015). In the previous study, Salpietro et al. identified the variant p.Ala639Ser in GRIA2 in two unrelated infants with uncontrolled seizures from the first days of life (Salpietro et al.). And the variant p.Ala639Ser in GRIA2 is located in the same position (in the motif of SYTANLAAF) as the variant p.Ala640Ser in GRIA4 in our patient. An in vitro functional study showed that the variant p.Ala639Ser in GRIA2 reduced agonist-induced current amplitude, resulting in a significantly reduced cell-surface expression of GRIA2 (Salpietro et al.). Therefore, we speculated that the variant p.Ala640Ser in GRIA4 had a high probability of pathogenicity, as it was also located in the same important functional domain. Moreover, further investigation by the electrophysiology experiments and biotinylation assay will be necessary to determine whether p.Ala640Ser in GRIA4 affects the channel synthesis or trafficking and its effect on currents.
In conclusion, using trio-WES, we identified a novel de novo heterozygous missense variant in the GRIA4 gene and diagnosed the sixth NEDSGA patient with severe developmental delay, limb hypertonia, partial seizure, hypoplasia of the retina, chorioretinal hyperpigmentation, and other clinical characteristics. Our findings enriched the phenotypic spectrum of genetic disorders associated with GRIA4 variants. And genetic evidence further supports the association of rare and newly reported NEDSGA caused by the GRIA4 gene. Our data will be helpful in diagnosing NEDSGA, especially in the affected newborns.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, SCV002055968.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Shengjing Hospital of China Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
HW drafted the initial manuscript. FL, WG, and ML were responsible for genetic analysis. HW, JL, and ZT collected clinical information. HW designed the study, and critically reviewed the manuscript. All the authors read and approved the final manuscript.
Conflict of Interest
FL and WG were employed by Chigene (Beijing) Translational Medical Research Center Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Beyer, B., Deleuze, C., Letts, V. A., Mahaffey, C. L., Boumil, R. M., Lew, T. A., et al. (2008). Absence Seizures in C3H/HeJ and Knockout Mice Caused by Mutation of the AMPA Receptor Subunit Gria4. Hum. Mol. Genet. 17 (12), 1738–1749. doi:10.1093/hmg/ddn064
Harrison, S. M., Biesecker, L. G., and Rehm, H. L. (2019). Overview of Specifications to the ACMG/AMP Variant Interpretation Guidelines. Curr. Protoc. Hum. Genet. 103 (1), 93. doi:10.1002/cphg.93
Jones, K. S., VanDongen, H. M. A., and VanDongen, A. M. J. (2002). The NMDA Receptor M3 Segment Is a Conserved Transduction Element Coupling Ligand Binding to Channel Opening. J. Neurosci. 22 (6), 2044–2053. doi:10.1523/jneurosci.22-06-02044.2002
Keinänen, K., Wisden, W., Sommer, B., Werner, P., Herb, A., Verdoorn, T. A., et al. (1990). A Family of AMPA-Selective Glutamate Receptors. Science 249 (4968), 556–560. doi:10.1126/science.2166337
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 35 (6), 1547–1549. doi:10.1093/molbev/msy096
Martin, S., Chamberlin, A., Shinde, D. N., Hempel, M., Strom, T. M., Schreiber, A., et al. (2017). De Novo Variants in GRIA4 Lead to Intellectual Disability with or without Seizures and Gait Abnormalities. Am. J. Hum. Genet. 101 (6), 1013–1020. doi:10.1016/j.ajhg.2017.11.004
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Salpietro, V., Dixon, C. L., Guo, H., Bello, O. D., Vandrovcova, J., Efthymiou, S., et al. (2019). AMPA Receptor GluA2 Subunit Defects Are a Cause of Neurodevelopmental Disorders. Nat. Commun. 10 (1), 3094–10910. doi:10.1038/s41467-019-10910-w
Schrodinger, L. L. C. (2015). The AxPyMOL Molecular Graphics Plugin for Microsoft PowerPoint. Version 1.8.
Sobolevsky, A. I. (2015). Structure and Gating of Tetrameric Glutamate Receptors. J. Physiol. 593 (1), 29–38. doi:10.1113/jphysiol.2013.264911
Sobolevsky, A. I., Yelshansky, M. V., and Wollmuth, L. P. (2003). Different Gating Mechanisms in Glutamate Receptor and K+Channels. J. Neurosci. 23 (20), 7559–7568. doi:10.1523/jneurosci.23-20-07559.2003
Traynelis, S. F., Wollmuth, L. P., McBain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., et al. (2010). Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 62 (3), 405–496. doi:10.1124/pr.109.002451
Twomey, E. C., Yelshanskaya, M. V., Grassucci, R. A., Frank, J., and Sobolevsky, A. I. (2017). Channel Opening and Gating Mechanism in AMPA-Subtype Glutamate Receptors. Nature 549 (7670), 60–65. doi:10.1038/nature23479
Keywords: GRIA4, NEDSGA, neurodevelopmental disorder, trio-whole exome sequencing, novel heterozygous missense variant
Citation: Wang H, Liu J, Li F, Teng Z, Liu M and Gu W (2022) Novel Heterozygous Missense Variant in GRIA4 Gene Associated With Neurodevelopmental Disorder With or Without Seizures and Gait Abnormalities. Front. Genet. 13:859140. doi: 10.3389/fgene.2022.859140
Received: 21 January 2022; Accepted: 21 March 2022;
Published: 20 April 2022.
Edited by:
Tianyun Wang, University of Washington, United StatesReviewed by:
Yuki Hitomi, Hoshi University, JapanStefano Castellana, Home for Relief of Suffering (IRCCS), Italy
Copyright © 2022 Wang, Liu, Li, Teng, Liu and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Wang, shengjingwangh1@163.com