 Qiutong Huang
Qiutong Huang Cyril Seillet
Cyril Seillet Gabrielle T. Belz
Gabrielle T. Belz- 1Walter and Eliza Hall Institute of Medical Research, Melbourne, VIC, Australia
- 2Department of Medical Biology, University of Melbourne, Melbourne, VIC, Australia
Innate lymphoid cells (ILCs) are a key cell type that are enriched at mucosal surfaces and within tissues. Our understanding of these cells is growing rapidly. Paradoxically, these cells play a role in maintaining tissue integrity but they also function as key drivers of allergy and inflammation. We present here the most recent understanding of how genomics has provided significant insight into how ILCs are generated and the enormous heterogeneity present within the canonical subsets. This has allowed the generation of a detailed blueprint for ILCs to become highly sensitive and adaptive sensors of environmental changes and therefore exquisitely equipped to protect immune surfaces.
Introduction
The importance of innate immunity has been recognized for many years. These cells play a key role in rapid responses to pathogens to protect the mucosal and external surfaces of the body. Natural killer (NK) cells and lymphoid tissue-inducer (LTi) cells are the founding members of the innate lymphoid family with the former identified more than 40 years ago. Over the past 10 years, a number of new family members have been discovered revealing an entire network of innate cells that complement the adaptive immune network. These cells went largely unrecognized for several decades which begs the question as to how they were overlooked. In this review, we summarize the current knowledge on innate lymphoid cell (ILC) differentiation and critically discuss the key challenges in the field in understanding ILC homeostatic regulation.
Canonical ILC Subsets
The ILC family is divided into three major groups: group 1 ILCs (ILC1s) which includes NK cells and ILC1s that produce interferon-γ (IFN-γ) and depend on the transcription factors Eomesodermin (Eomes) and T-bet; group 2 ILCs (ILC2s) that secrete IL-5 and IL-13 and are characterized by Gata3 expression; and group 3 ILCs (ILC3s) that express the RAR-related orphan receptor, Rorγt, and includes LTi cells and multiple subsets of ILC3s capable of producing IL-17 and/or IL-22 (Figure 1A). The ILCs are distinguished from adaptive immune cells by their lack of germline rearranged antigen-specific receptors and generalized lack of lineage-specific markers normally used to distinguish B and T cells. ILCs are not thought to traffic through tissues and are often referred to as “tissue-resident” (1) but their precursors can be isolated in humans from blood (2) indicating that these cells are not completely sessile throughout their life cycle. At the very least, they transit around the body to achieve their strategic positioning close to barrier surfaces to allow them to respond rapidly to local environmental changes.
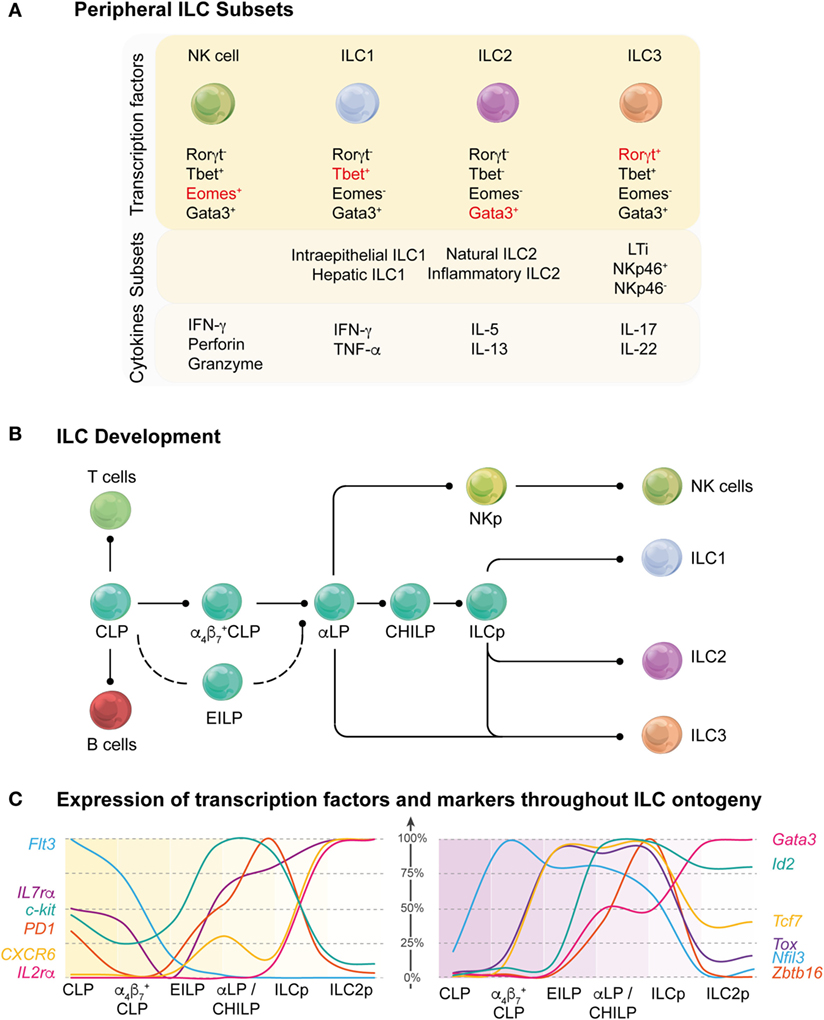
Figure 1. Overview of stages of innate lymphoid cell (ILC) development. (A) Current understanding of the regulation of peripheral ILC subsets. A variety of different transcription factors are required for the development of peripheral ILC subsets. Key transcription factors (red) are responsible for the lineage determination of the canonical ILC subsets (top panel). ILC subsets can also be further differentiated and categorized based on the organ in which they reside, functional differences, or the expression of different receptors and surface markers (middle panel). In response to activation signals, ILCs are able to produce effector molecules and cytokines to mediate an appropriate immune response (bottom panel). (B) Schematic showing the current understanding of ILC development from the common lymphoid progenitor (CLP) through multiple intermediary stages on their way to becoming mature ILC subsets ILC1, 2, and 3. CLP has multi-lineage potential, including T and B cell fate, but this potential is gradually lost as the progenitors differentiate into the more lineage restricted αLP. This occurs through the intermediate α4β7+ CLP and αLP/common helper-like ILC precursor (CHILP) progenitors or through an alternative pathway via the early innate lymphoid progenitors (EILPs). Within the αLP population, the natural killer (NK) cell lineage diverges from the ILC lineage and the ILC precursor (ILCp) exclusively develops into the remaining mature ILCs in the periphery. (C) Dynamic regulation of the surface markers (left panel) and transcription factors (right panel) throughout the ILC ontogeny. The graphs show the relative RNA expression among the different ILC progenitor stages (100% represents the highest expression for each gene detected across the six different populations).
Innate lymphoid cell subsets were initially categorized based on their phenotype, function, and the key transcriptional regulators that drive their development. In many aspects, these subsets mirror CD4+ T cell subsets although some populations such as ILC1s have been quite difficult to position due to their lack of specific distinguishing markers (3, 4). The current classification model has served as an important framework to focus our thinking around canonical subset classifications. However, recent analyses of elegant reporters and genomic probing of individual cells has revealed that ILCs are dynamically tuned resulting in enormous heterogeneity (5–8). Potentially, this property would enable ILCs to respond to diverse stimuli in “real time.” It is widely accepted that CD4+ T cell subsets display extraordinary plasticity allowing them adapt to a broad spectrum of inflammatory signals, but such a program among ILCs has not been appreciated until recently. Indeed, the capacity for ILCs to exhibit a highly flexible program may be an essential element for tuning ILCs to ensure responsiveness to continuous changes in signals encountered at mucosal barriers.
Recent findings in the field have identified conceptually new ideas about how the immune system is regulated and how the innate arm might contribute to this process. For example, the ILC network forms an extensive interface between the external environment and the adaptive immune system. Their regulation is highly dynamic and relies on a highly integrated molecular signaling network, resulting in heterogeneity and plasticity. Finally, it appears to be highly complementary to the adaptive immune system providing a fail-safe mechanism for ensuring immune protection and repair processes. Excitingly, we are only just beginning to understand how this network of cells might work.
Core Transcription Factors Establish the ILC Differentiation Framework
Innate lymphoid cells arise from the common lymphoid progenitor (CLP) through multiple intermediary stages with changes in surface expression of key surface molecules and the temporal regulation of transcriptional regulators to become mature ILC1, 2, and 3 subsets (Figure 1B). Induction of the downstream molecular program involves the induction of α4β7 which identify the α4β7+ CLP (9) [also called αLP1 (10)] follow by the downmodulation of Flt3 expression leading to the emergence of the α lymphoid progenitor (αLP, also called αLP2) (10, 11). While the αLP can generate all ILC subset, a subpopulation seems to have lost the ability to generate NK cells and named common helper-like ILC precursor (CHILP) (12). The distinction between the αLP and CHILP is not clear as they appear to be very highly similar in their surface marker or transcription factor expression. Finally, the induction of promyelocytic leukemia zinc finger (PLZF) in the ILC precursor (ILCp) mark the bifurcation between LTi and NK cells with the other ILC1, 2, and 3 subsets (13). Their fate is guided by lineage-determining transcription factors that are also involved in specifying different subsets of T cells. Transcription factors control multiple aspects of the development of immune cell lineages including proliferation, migration, metabolism, and effector function (Table 1). Some transcription factors play unique roles in defining the fate of early progenitors, but increasingly it is emerging that overlapping and synergistic contributions by transcription factors may be critical in setting the threshold for fate decisions and the function of an individual cell. A major challenge for the field now is to understand the combinatorial interactions between transcription factors and how they define ILC developmental choices.
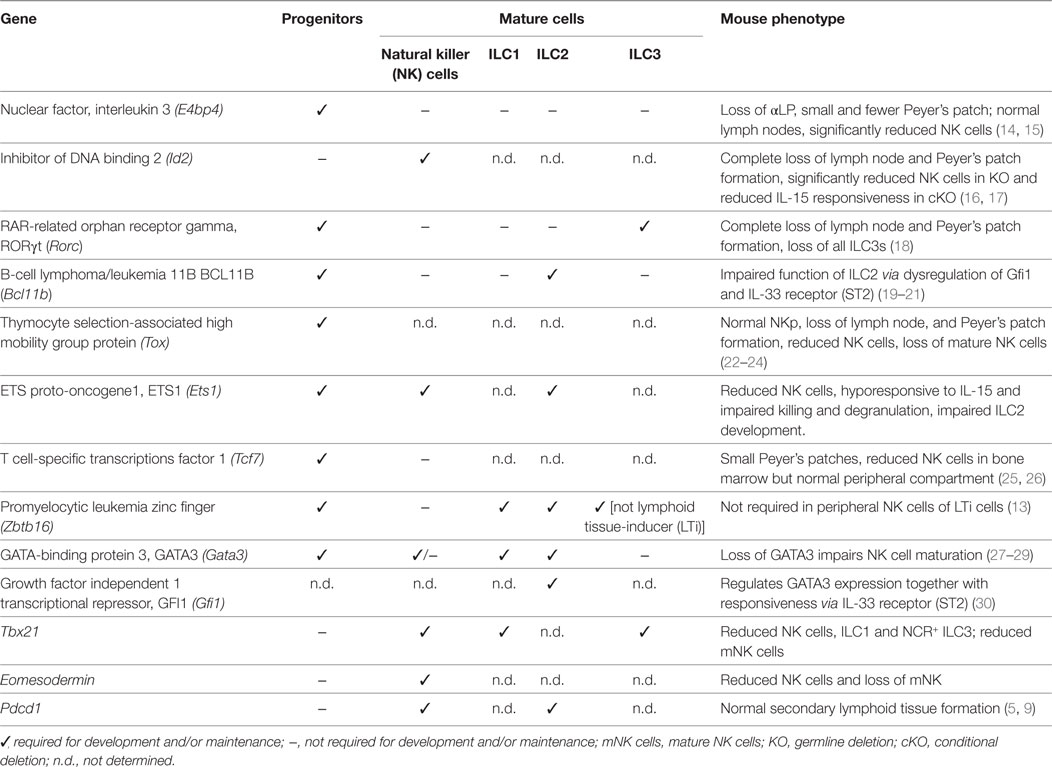
Table 1. Requirement for different transcription factors during innate lymphoid cell (ILC) development.
The emergence of the innate cells from the CLP and the divergence of this pathway away from the adaptive lineages is an extremely controlled process that involves the coordinated actions of several transcription factors. Detailed analysis of the transcriptional landscape of the ILC development from the earliest precursor to the committed cells has revealed that regulation of the different developmental stages is highly dynamic. The sequential expression of nuclear factor interleukin 3 (NFIL3), inhibitor of DNA binding 2 (ID2), thymocyte selection-associated high mobility group box protein (TOX), and GATA-binding protein 3 (GATA3) establishes the framework for ILC differentiation (7, 9) (Figure 1C). ID2 counterbalances the effects of E proteins to direct cell choices away from T and B cell outcomes (31). Other transcription factors such as EOMES, PLZF, transcriptions factor 1 (TCF-1), and RUNX influence subset divergence. We now have new insight to the key factors that determine the fate outcome of progenitor cells under steady-state conditions. However, it still remains unclear how higher order genomic architecture establishes and maintains the differentiation program.
The Early Regulators: A Quartet
Two major transcription factors, NFIL3 and TOX, have emerged as critical initiators of development of early αLP. Induction of NFIL3 appears to be the critical initiating step in driving the αLP toward the ILC lineage (7, 9). NFIL3 is induced in α4β7+ CLP, but the factors responsible for this induction have yet to be elucidated (Figures 1B,C). Concomitantly, TOX and ID2 are only expressed at low levels in the CLP but TOX expression rapidly increases in the early innate lymphoid progenitors (EILPs) while ID2 levels remains low until the late αLP and common helper ILC (CHILP) stages (12, 32–34). Nevertheless, the expression of ID2 has two patterns in ILCps; the first phase in which ID2 is expressed at low levels (and the E protein E2A is concomitantly high) and does not appear to be required for ILC development, and the second in which ID2 is strongly upregulated with concurrent downregulation of E2A and is essential for ILC lineage progression (9, 31). Indeed, while the deletion of NFIL3 blocks the development of the α4β7+CLP, ID2 deletion has does not appear to affect the development of the αLP. However, all cells derived from the ILC progenitor are absent in ID2−/− mice suggesting that ID2 is more important in late differentiation and in maintaining the long-term identity of ILCs (16, 35). Loss of ID2 in ILCs has also been shown to repress genes belonging to the stem cell program such as Gfi1b, Tal1, Lmo2, Gata2, and Hhex (9).
Precisely how NFIL3 and TOX regulate the development of the ILC progenitors remains unclear. TOX-deficient progenitors appear to lack the expression of key factors thought to be essential for ILC development including Gata3, Rora, Rorc, Tcf7, and Zbtb16 (22). Despite this, Nfil3 expression, which could be regulated by TOX, was not found to be different from that of wild-type cells. Thus, more work will be necessary to ascertain whether NFIL3 is a direct target of TOX or not. NFIL3 has been shown to directly bind to ID2; however, it is not clear that this binding is actually responsible for the induction of ID2 (36). In ILC progenitors, ID2 expression has been found to be reduced when NFIL3 is deleted in the hematopoietic compartment. However, in mature cells, ablation of NFIL3 did not alter ID2 expression suggesting that the developmental stage of the cell influenced the interactions (37, 38). Furthermore, overexpression of either ID2 or TOX in NFIL3-deficient CLPs revealed that both transcription factors could at least partially rescue ILC development independent of NFIL3. Therefore, it is likely that the key role of NFIL3 is to promote the emergence of ILCs by induction of the expression of these two key transcription factors.
Expression Patterning of NFIL3 and ID2 Establishes the Landscape for ILCs
Although several details of the fine tuning of NFIL3 remain unanswered, the timing and action of NFIL3 is very interesting. Nfil3flox/flox mice crossed to the Id2ERT2Cre strain generated a model in which deletion of NFIL3 could be timed relative to ID2 (9). This approach demonstrated that NFIL3 expression preceded that of ID2 but that surprisingly, deletion of NFIL3 in ID2+ cells either in vivo or in vitro did not affect the subsequent development of any of the ILC subsets. These findings are consistent with earlier work showing that NFIL3 was not required for the maintenance of mature NK cells (14). What was particularly unexpected, however, in the study, was the very transient nature of the NFIL3 expression which was both necessary and sufficient to promote ILC development. Indeed, only a short pulse of NFIL3 expression in the progenitors was required and it subsequently rapidly decreased as ILC progressed through each developmental stage (7, 9) and is repressed in mature cells in the periphery (15).
This pattern of short-lived expression found in NFIL3 may represent a more generalized pattern for orchestrating the complex integration of different transcriptional signals. This expression pattern has also been reported for Zbtb16 (that encodes PLZF), another transcription factor important for ILC development but which was originally implicated in NKT cell development (39). Mature ILCs do not express PLZF; however, lineage tracing experiments revealed that 60–75% of ILCs exhibited a fluorescent imprint marking their previous expression of PLZF during development (13). In this setting, only ~5% of LTi cells and ~20% of NK cells were labeled indicating that bifurcation of these lineages from other ILC subsets occurred before the induction of PLZF. The role of the transient expression of PLZF in ILCp is not known. Using the PLZF-reporter mice, Constantinides et al. (13) also showed that PLZF is expressed only transiently. This allowed the identification of the ILCp, but PLZF was subsequently downregulated after this stage. PLZF does not appear to be absolutely required for ILCs as deletion of PLZF results in ~4-fold reduction in the number of ILCs that develop in contrast to ablation of NFIL3 which results in more than a 10-fold reduction. PLZF is expressed after NFIL3 coordinately with E2A and ID2 resulting in a stepwise progression through the early progenitor stage to generate E2AhiPLZF−ID2lo, E2AhiPLZF+ID2lo, E2AloPLZFhiID2hi, and E2AloPLZF−ID2hi expressing cells (31). In competitive situation, PLZF-deficiency appears selective, mainly affected ILC2s in the lamina propria and ILC1s in the liver (13). It was notable that ~40% of the ILC2s in this study were not labeled in these tracing experiments, so it remains possible that they could be derived from an alternative pathway that is independent of PLZF.
Loss of IL-7R Expression: A New Progenitor or an Alternative Pathway?
The identification of the early ILC progenitor in the bone marrow, or EILP, noted for its high expression of TCF-1, has raised some questions around the linear model of ILC development (25). This EILP lacks B or T cell potential but can generate all ILC subsets including the NK cells similar to the αLP progenitor capacity, but it differs from the other precursors described as it lacks IL7Rα expression (25). One possibility is that IL-7Rα expression is lost between the αLP and the ILCp stages. Such changes could be regulated via posttranslational modifications though the biological relevance for such downregulation is unclear. A second possibility is that the EILP is a precursor for an alternative pathway for ILC development. EILPs express high levels of NFIL3, TOX, and TCF-1, low levels of ID2 and PLZF is almost undetectable. Interestingly, the EILPs are not affected by ID2 deletion, thus the EILPs appear to be very similar to the IL-7R-expressing αLP. A comparison between these two cell types may help to better understand the relationship between the IL7Rα+ and IL7Rα− ILCps and define the factors that regulate IL-7R signaling in ILCs that is essential for their development. To date, TCF-1 and ID2 are known to be upregulated in EILP but additional transcriptional requirements of this progenitor have not been investigated. Therefore, whether EILP represents an intermediate stage of the ILCp, or an alternative precursor that does not fit in the current linear model of ILC development, remains an open question.
Heterogeneity and Plasticity of ILCs are Key to Maintain Homeostasis
Initial categorization of ILC subsets relied on cytokines and effector molecules they produced combined with signature transcription factors that appeared to be central regulators of the different subsets. The patterns found in ILCs were aligned to the categorization of CD4+ T cell subsets. Unlike CD4+ T cells, however, it quickly emerged that several ILC phenotypes did not necessarily neatly fit into the unified nomenclature coined in 2013 with subsets described as being “like” other subsets (40). The full spectrum of ILC heterogeneity has recently been unveiled using comprehensive single-cell sequencing combined with mass cytometry approaches which has allowed the field to bridge between our understanding of ILCs in mouse and man (2, 6, 41, 42) (Figure 2A). This approach revealed that potentially as many as 15 transcriptionally distinct identities could be delineated in the small intestine and that crosstalk orchestrated via cytokines such as IL-12 and IL-23 and microbial signals were substantially responsible for mediating this subset plasticity (6). It is undoubtable that these approaches are transforming our understanding of diversity in immune cell subsets but these data also throw up new technical and intellectual challenges in understanding how diversity arises. Historically, various surface markers have been considered to represent “lineage specificity” but we now recognize that this is seldom the case, challenging that how we interpret complex data and underlying that subsets cannot be defined exclusively by particular markers or transcription factors (43). Nevertheless, it provides a rich landscape for understanding how different stimuli affect the homeostatic balance of ILC subsets. It has been revealed that ILCs exhibit tissue-specific characteristics and this in part reflects alterations in phenotype that can be significantly regulated by responses to inflammation and infection.
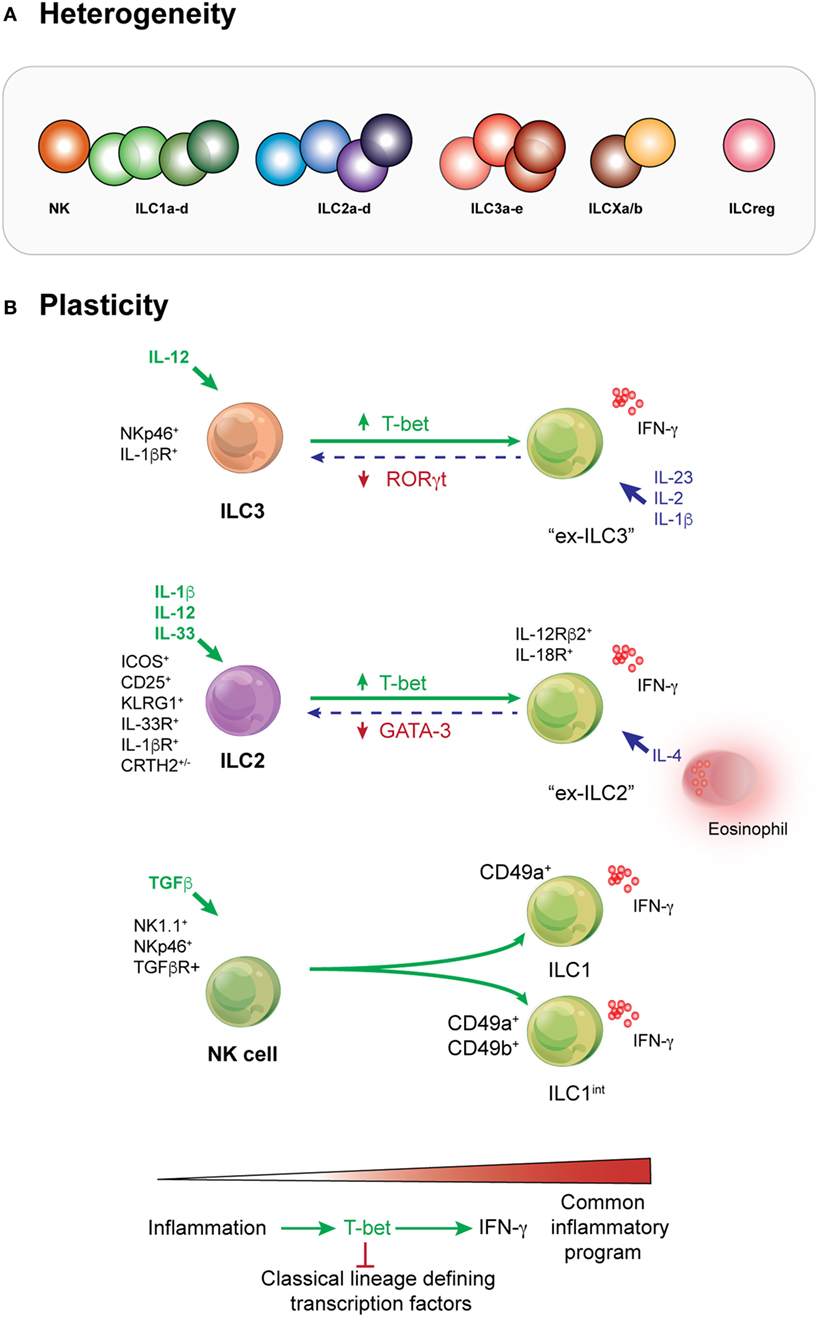
Figure 2. Heterogeneity and plasticity are prominent features of innate lymphoid cell (ILC) behavior across all subsets. (A) Deep analysis into the transcriptome of ILCs demonstrates that heterogeneity occurs within every subset. Depending on the type or intensity of the stimuli received by these cells, different molecular pathways may be activated by cells of the same subset. This results in phenotypic or functional variation and a subsequent spectrum of ILCs within each compartment. Whether additional subsets such as the proposed regulatory subset exist is yet to be fully determined. (B) ILC3s can adopt an ILC1-like phenotype when activated by IL-12. They are known as “ex-ILC3s.” In human cells, this pathway can be reversed by the action of IL-23, IL-2, and IL-1β. ILC2s are activated by IL-2 and IL-33. Stimulation with IL-1β primes the responsiveness of ILC2 by enhancing the expression of cytokine receptors such as IL-25R, IL-33R, and TSLP to potentiate ILC2 responsiveness and induce a significant increase in the population. Critically, however, IL-12 is essential to effect remodeling of the chromatin landscape in ILC2 allowing them to induce phenotypic changes and become more like ILC1s (ILC1-like or “ex-ILC2s”) that produce IFN-γ. Natural killer (NK) cells respond to TGF-β to form “intermediate ILC1” reflecting their acquisition of CD49a and bone fide ILC1. In many situations, it appears that the transcription factor T-bet is key to augmentation of the inflammatory program and concurrently represses signature transcription factors that typically define individual lineages.
The first example of ILC ability to adopt a different phenotype was the identification of ILC3s that downregulated RORγt but expressed T-bet, NK1.1, and produced IFN-γ (also known as ex-ILC3s) (12, 44, 45) under chronic stimulation and RORγt+ ILC3 that produced IL-17 which were found in the large intestine (46) (Figure 2B). IL-12 was found to be a key driver of this pathway. Similarly, IL-1β, together with combinations of IL-4, IL-12, and IL-33, can drive ILC2s to adopt an IFN-γ-producing phenotype called ex-ILC2 (47–49). ILC2s can be divided into two subsets: homeostatic or natural ILC2s which typically reside in barrier tissues and respond to IL-33; and inflammatory ILC2s which are generally not found in peripheral tissues at steady-state but respond to IL-25 which induces multipotency (50–53). In this setting, T-bet (encoded by Tbx21) was induced, whereas GATA3 was diminished. These studies highlight the variability that can occur in ILCs, particularly in vitro as demonstrated for ILC2s (47–49). Indeed, even NK cells can undergo this sort of transformation (54). Signaling through the TGF-β pathway can convert NK cells (CD49a−CD49b+Eomes+) into intermediate ILC1 (CD49a+CD49b+Eomes+) and bone fide ILC1 (CD49a+CD49b+Eomes+/−) within a tumor microenvironment. Strikingly, these latter ILC1 were disabled in their capacity to control local tumor growth and to prevent metastasis, while NK cells retained their ability to undertake immune surveillance (54). It is striking that once activated through these different pathways, each subset converges on an “IFN-γ-producing ILC1-like” phenotype. This suggests that this fate may represent a common outcome for multiple ILC subsets, even perhaps an essential adaptive program for all ILCs. It remains a challenge, however, to understand how these IFN-γ-producing cells arise and whether it represents a protective response or the first steps to loss of immune control (Figure 2B). Teasing this apart will require much more extensive study particularly in the context of pathogen infection, inflammation, and tumor development.
Are More Subsets Possible?
How ILCs populate tissues after birth or maintain their presence in peripheral tissue is not clear. Our current understanding indicates that ILCs are almost exclusively tissue resident and that they do not routinely circulate throughout the body. This perception is predicated on the findings that during engraftment following irradiation, donor ILCs largely fail to replace ILCs originally found in the host (1). Instead, they depend on local proliferation to expand and replace ILCs, and it is only late in an infection or physiological disturbance that replenishment from blood-borne precursors restores the integrity of the ILC compartment. But have we got this right? These findings depend on a number of assumptions. For example, it is presumed that the transcriptional regulators, surface molecules, maturity, and frequency of relatively “mature” ILCs are also the most useful for pinpointing circulating ILCs. In addition, the high similarity of markers in murine ILCs that are comparable in man may have obscured our ability to identify circulating progenitors, or more mature cells, that are critical to maintain tissue homeostasis.
Circulating Precursors
The first clue that programming might potentially differ between mouse and man came from Scoville et al. (55) who observed that RORγt was expressed in all human ILCs. This was in striking contrast to murine ILCs where RORγt expression was highly restricted to the ILC3 subset (18). Interestingly, CD34+ progenitors expressing c-kit and RORγt could generate all ILC subsets, including NK cells. These progenitors selectively resided in secondary lymphoid tissues. This helped to identify the pathway of ILC development in man; however, the involvement of these precursors remained unclear. Later, an extensive analysis of the c-kit+ ILCs from the blood and tissues revealed that circulating ILCp in humans exist and do not typically express many of the markers associated with mature cells but do express low levels of RORγt (2). Such cells are maintained in RORC-deficient patients and retain the potential to produce different populations of ILC except ILC3. Thus, it appears that high expression of RORγt is necessary to generate an ILC3 in both man and mouse, but these observations raise the question of whether in mice similar progenitors have been simply discounted due to their low expression of this transcription factor. However, while the role of RORγt in the development of human ILCps is unclear, transcriptomic and epigenomic analysis of circulating human ILCps revealed the upregulation of many transcription factors known to be critical for ILC development in mice such as NFIL3, ID2, TOX, TCF7, ZBTB16, and GATA3 (2). This suggests that transcriptional regulation of ILC development share common factors in human and mouse.
Regulatory ILCs
Innate lymphoid cell subsets appear to largely mimic those defined for CD4+ T cells. Recent evidence suggests that this extends to the presence of regulatory ILCs. Among the ILCs, NK cells have been described to produce IL-10 (56, 57) that acts to depress B cell (58) and dendritic cell immune responsiveness (59, 60) and suppress activation. The Ohashi group (61) strengthens the notion that a regulatory subset might exist. During their evaluation of a tumor-infiltrating cell-based adoptive immunotherapy for ovarian cancer, it was noticed that a high frequency of CD56+CD3− cells was strongly correlated with suppression of tumor-infiltrating cell outgrowth and proposed that these cells played a regulatory role. While this effect has only been tested in vitro, they ascertained that regulatory and conventional CD56+CD3− ILCs exhibited high levels of the transcription factors ID2, ZBTB16 (PLZF), RUNX3, and TOX, but similar amounts of EOMES, TBX21, GATA3, RORA, and AHR, factors also shared with NK cells, ILC2s, and ILC3s. Very recently, however, Wang et al. (62) provide the first description of regulatory ILCs in mice and humans which were shown to be important in response to gut inflammation such as Citrobacter rodentium. This subset arises from an ID2-expressing progenitor and depends on a second inhibitor of DNA-binding protein, ID3 but not PLZF or RORγt. Intriguingly, this subset lacks expression of any of the classical transcription factors required for the early steps in development by other ILCs including NFIL3, TOX, TCF-1, GATA3, or PLZF. Thus, it remains unclear whether the early progenitor is the ILCp, or alternatively a distinct progenitor gives rise to this subset. However, like other ILC subsets they lack typical lineage markers but do express CD25 and CD90 while autocrine expression of TGF-β1 drives expansion of this IL-10+ subset during inflammation and results in suppression of activation of ILC1 and ILC3.
Combined, these important studies point toward the existence of a regulatory subset, but important questions still remain. For example, how and when do regulatory ILCs emerge, what other transcription factors drive this process, and do they express receptors that can be targeted to restore immune homeostasis during chronic and autoimmune diseases. If they do, this proves another avenue to unleash the protective power of ILCs either within tumors or during inflammation and in maintaining normal homeostasis to prevent autoimmunity.
Concluding Remarks
With increasing understanding of the regulation of the ILC, we realize how extensive is their ability to adapt their microenvironment. While ILC subsets are often seen as innate counterpart of T helper cells, it may be interesting to imagine the ILCp as an innate counterpart of naive T cells. ILC subsets also to appear to be an extremely plastic population that can profoundly change their predicted responses in reaction of extracellular mediators. One important challenge will be to identify the large variety of environmental and host-derived signals they can integrate to understand the role of ILCs in the homeostasis of the tissue and during inflammation.
Author Contributions
All authors contributed to the manuscript and read, edited, and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors apologize to all investigators whose works were not cited in this article due to space limitations.
Funding
This work was supported by grants and fellowships from the National Health and Medical Research Council (NHMRC) of Australia (GB and CS; 1047903 and 1054925), Australian Research Council Future Fellowship (GB); The Rebecca L. Cooper Foundation Medical Research Foundation (GB), and a Page Betheras Award (QH). This work was made possible through Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIIS.
References
1. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science (2015) 350:981–5. doi:10.1126/science.aac9593
2. Lim AI, Li Y, Lopez-Lastra S, Stadhouders R, Paul F, Casrouge A, et al. Systemic human ILC precursors provide a substrate for tissue ILC differentiation. Cell (2017) 168:1086–100. doi:10.1016/j.cell.2017.02.021
3. Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol (2016) 17:758–64. doi:10.1038/ni.3482
4. Jiao Y, Huntington ND, Belz GT, Seillet C. Type 1 innate lymphoid cell biology: lessons learnt from natural killer cells. Front Immunol (2016) 7:426. doi:10.3389/fimmu.2016.00426
5. Yu Y, Tsang JC, Wang C, Clare S, Wang J, Chen X, et al. Single-cell RNA-seq identifies a PD-1hi ILC progenitor and defines its development pathway. Nature (2016) 539:102–6. doi:10.1038/nature20105
6. Gury-BenAri M, Thaiss CA, Serafini N, Winter DR, Giladi A, Lara-Astiaso D, et al. The spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell (2016) 166:1231–46. doi:10.1016/j.cell.2016.07.043
7. Ishizuka IE, Chea S, Gudjonson H, Constantinides MG, Dinner AR, Bendelac A, et al. Single-cell analysis defines the divergence between the innate lymphoid cell lineage and lymphoid tissue-inducer cell lineage. Nat Immunol (2016) 17:269–76. doi:10.1038/ni.3344
8. Viant C, Rankin LC, Girard-Madoux MJ, Seillet C, Shi W, Smyth MJ, et al. Transforming growth factor-beta and Notch ligands act as opposing environmental cues in regulating the plasticity of type 3 innate lymphoid cells. Sci Signal (2016) 9:ra46. doi:10.1126/scisignal.aaf2176
9. Seillet C, Mielke LA, Amann-Zalcenstein DB, Su S, Gao J, Almeida FF, et al. Deciphering the innate lymphoid cell transcriptional program. Cell Rep (2016) 17:436–47. doi:10.1016/j.celrep.2016.09.025
10. Chea S, Schmutz S, Berthault C, Perchet T, Petit M, Burlen-Defranoux O, et al. Single-cell gene expression analyses reveal heterogeneous responsiveness of fetal innate lymphoid progenitors to notch signaling. Cell Rep (2016) 14:1500–16. doi:10.1016/j.celrep.2016.01.015
11. Yu X, Wang Y, Deng M, Li Y, Ruhn KA, Zhang CC, et al. The basic leucine zipper transcription factor NFIL3 directs the development of a common innate lymphoid cell precursor. Elife (2014) 3:e04406. doi:10.7554/eLife.04406
12. Klose CS, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell (2014) 157:340–56. doi:10.1016/j.cell.2014.03.030
13. Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature (2014) 508:397–401. doi:10.1038/nature13047
14. Firth MA, Madera S, Beaulieu AM, Gasteiger G, Castillo EF, Schluns KS, et al. Nfil3-independent lineage maintenance and antiviral response of natural killer cells. J Exp Med (2013) 210:2981–90. doi:10.1084/jem.20130417
15. Seillet C, Rankin LC, Groom JR, Mielke LA, Tellier J, Chopin M, et al. Nfil3 is required for the development of all innate lymphoid cell subsets. J Exp Med (2014) 211:1733–40. doi:10.1084/jem.20140145
16. Yokota Y, Mansouri A, Mori S, Sugawara S, Adachi S, Nishikawa S, et al. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature (1999) 397:702–6. doi:10.1038/17812
17. Delconte RB, Shi W, Sathe P, Ushiki T, Seillet C, Minnich M, et al. The helix-loop-helix protein ID2 governs NK cell fate by tuning their sensitivity to interleukin-15. Immunity (2016) 44:103–15. doi:10.1016/j.immuni.2015.12.007
18. Eberl G, Marmon S, Sunshine M-J, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor Rorγt in the generation of fetal lymphoid tissue inducer cells. Nat Immunol (2004) 5:64–72. doi:10.1038/ni1022
19. Califano D, Cho JJ, Uddin MN, Lorentsen KJ, Yang Q, Bhandoola A, et al. Transcription factor Bcl11b controls identity and function of mature type 2 innate lymphoid cells. Immunity (2015) 43:354–68. doi:10.1016/j.immuni.2015.07.005
20. Yu Y, Wang C, Clare S, Wang J, Lee SC, Brandt C, et al. The transcription factor Bcl11b is specifically expressed in group 2 innate lymphoid cells and is essential for their development. J Exp Med (2015) 212:865–74. doi:10.1084/jem.20142318
21. Walker JA, Oliphant CJ, Englezakis A, Yu Y, Clare S, Rodewald HR, et al. Bcl11b is essential for group 2 innate lymphoid cell development. J Exp Med (2015) 212:875–82. doi:10.1084/jem.20142224
22. Seehus CR, Aliahmad P, de la Torre B, Iliev ID, Spurka L, Funari VA, et al. The development of innate lymphoid cells requires TOX-dependent generation of a common innate lymphoid cell progenitor. Nat Immunol (2015) 16:599–608. doi:10.1038/ni.3168
23. Aliahmad P, Seksenyan A, Kaye J. The many roles of TOX in the immune system. Curr Opin Immunol (2012) 24:173–7. doi:10.1016/j.coi.2011.12.001
24. Yun S, Lee SH, Yoon S-R, Kim MS, Piao Z-H, Myung P-K, et al. TOX regulates the differentiation of human natural killer cells from hematopoietic stem cells in vitro. Immunol Lett (2011) 136:29–36. doi:10.1016/j.imlet.2010.11.008
25. Yang Q, Li F, Harly C, Xing S, Ye L, Xia X, et al. TCF-1 upregulation identifies early innate lymphoid progenitors in the bone marrow. Nat Immunol (2015) 16:1044–50. doi:10.1038/ni.3248
26. Mielke LA, Groom JR, Rankin LC, Seillet C, Masson F, Putoczki T, et al. TCF-1 controls ILC2 and NKp46+RORgammat+ innate lymphocyte differentiation and protection in intestinal inflammation. J Immunol (2013) 191:4383–91. doi:10.4049/jimmunol.1301228
27. Mjosberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, et al. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity (2012) 37:649–59. doi:10.1016/j.immuni.2012.08.015
28. Yagi R, Zhong C, Northrup DL, Yu F, Bouladoux N, Spencer S, et al. The transcription factor GATA3 is critical for the development of all IL-7Rα-expressing innate lymphoid cells. Immunity (2014) 40:378–88. doi:10.1016/j.immuni.2014.01.012
29. Serafini N, Klein Wolterink RG, Satoh-Takayama N, Xu W, Vosshenrich CA, Hendriks RW, et al. Gata3 drives development of RORgammat+ group 3 innate lymphoid cells. J Exp Med (2014) 211:199–208. doi:10.1084/jem.20131038
30. Spooner CJ, Lesch J, Yan D, Khan AA, Abbas A, Ramirez-Carrozzi V, et al. Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nat Immunol (2013) 14:1229–36. doi:10.1038/ni.2743
31. Miyazaki M, Miyazaki K, Chen K, Jin Y, Turner J, Moore AJ, et al. The E-Id protein axis specifies adaptive lymphoid cell identity and suppresses thymic innate lymphoid cell development. Immunity (2017) 46:818–34.e4. doi:10.1016/j.immuni.2017.04.022
32. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature (2010) 463:540–4. doi:10.1038/nature08636
33. Boos MD, Yokota Y, Eberl G, Kee BL. Mature natural killer cell and lymphoid tissue-inducing cell development requires Id2-mediated suppression of E protein activity. J Exp Med (2007) 204:1119–30. doi:10.1084/jem.20061959
34. Zook EC, Ramirez K, Guo X, van der Voort G, Sigvardsson M, Svensson EC, et al. The ETS1 transcription factor is required for the development and cytokine-induced expansion of ILC2. J Exp Med (2016) 213:687–96. doi:10.1084/jem.20150851
35. Ikawa T, Fujimoto S, Kawamoto H, Katsura Y, Yokota Y. Commitment to natural killer cells requires the helix-loop-helix inhibitor Id2. Proc Natl Acad Sci U S A (2001) 98:5164–9. doi:10.1073/pnas.091537598
36. Male V, Nisoli I, Kostrzewski T, Allan DS, Carlyle JR, Lord GM, et al. The transcription factor E4bp4/Nfil3 controls commitment to the NK lineage and directly regulates Eomes and Id2 expression. J Exp Med (2014) 211:635–42. doi:10.1084/jem.20132398
37. Xu W, Domingues RG, Fonseca-Pereira D, Ferreira M, Ribeiro H, Lopez-Lastra S, et al. NFIL3 orchestrates the emergence of common helper innate lymphoid cell precursors. Cell Rep (2015) 10:2043–54. doi:10.1016/j.celrep.2015.02.057
38. Seillet C, Huntington ND, Gangatirkar P, Axelsson E, Minnich M, Brady HJ, et al. Differential requirement for Nfil3 during NK cell development. J Immunol (2014) 192:2667–76. doi:10.4049/jimmunol.1302605
39. Savage AK, Constantinides MG, Han J, Picard D, Martin E, Li B, et al. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity (2008) 29:391–403. doi:10.1016/j.immuni.2008.07.011
40. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13:145–9. doi:10.1038/nri3365
41. Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol (2015) 16:306–17. doi:10.1038/ni.3094
42. Simoni Y, Fehlings M, Kloverpris HN, McGovern N, Koo SL, Loh CY, et al. Human innate lymphoid cell subsets possess tissue-type based heterogeneity in phenotype and frequency. Immunity (2017) 46:148–61. doi:10.1016/j.immuni.2016.11.005
43. Bernink JH, Mjosberg J, Spits H. Human ILC1: to be or not to be. Immunity (2017) 46:756–7. doi:10.1016/j.immuni.2017.05.001
44. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol (2013) 14:221–9. doi:10.1038/ni.2534
45. Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity (2010) 33:736–51. doi:10.1016/j.immuni.2010.10.017
46. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature (2010) 464:1371–5. doi:10.1038/nature08949
47. Ohne Y, Silver JS, Thompson-Snipes L, Collet MA, Blanck JP, Cantarel BL, et al. IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat Immunol (2016) 17:646–55. doi:10.1038/ni.3447
48. Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol (2016) 17:626–35. doi:10.1038/ni.3443
49. Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1beta, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol (2016) 17:636–45. doi:10.1038/ni.3444
50. Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat Immunol (2015) 16:161–9. doi:10.1038/ni.3078
51. Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, et al. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med (2006) 203:1105–16. doi:10.1084/jem.20051615
52. Voehringer D, Reese TA, Huang X, Shinkai K, Locksley RM. Type 2 immunity is controlled by IL-4/IL-13 expression in hematopoietic non-eosinophil cells of the innate immune system. J Exp Med (2006) 203:1435–46. doi:10.1084/jem.20052448
53. Hurst SD, Muchamuel T, Gorman DM, Gilbert JM, Clifford T, Kwan S, et al. New IL-17 family members promote Th1 or Th2 responses in the lung: in vivo function of the novel cytokine IL-25. J Immunol (2002) 169:443–53. doi:10.4049/jimmunol.169.1.443
54. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol (2017) 18:1004–15. doi:10.1038/ni.3800
55. Scoville SD, Mundy-Bosse BL, Zhang MH, Chen L, Zhang X, Keller KA, et al. Expressing transcription factor RORgammat generates all human innate lymphoid cell subsets. Immunity (2016) 44:1140–50. doi:10.1016/j.immuni.2016.04.007
56. Lee SH, Kim KS, Fodil-Cornu N, Vidal SM, Biron CA. Activating receptors promote NK cell expansion for maintenance, IL-10 production, and CD8 T cell regulation during viral infection. J Exp Med (2009) 206:2235–51. doi:10.1084/jem.20082387
57. Fodil-Cornu N, Lee SH, Belanger S, Makrigiannis AP, Biron CA, Buller RM, et al. Ly49h-deficient C57BL/6 mice: a new mouse cytomegalovirus-susceptible model remains resistant to unrelated pathogens controlled by the NK gene complex. J Immunol (2008) 181:6394–405. doi:10.4049/jimmunol.181.9.6394
58. Che S, Huston DP. Natural killer cell suppression of IgM production. Nat Immun (1994) 13:258–69.
59. Piccioli D, Sbrana S, Melandri E, Valiante NM. Contact-dependent stimulation and inhibition of dendritic cells by natural killer cells. J Exp Med (2002) 195:335–41. doi:10.1084/jem.20010934
60. Spaggiari GM, Carosio R, Pende D, Marcenaro S, Rivera P, Zocchi MR, et al. NK cell-mediated lysis of autologous antigen-presenting cells is triggered by the engagement of the phosphatidylinositol 3-kinase upon ligation of the natural cytotoxicity receptors NKp30 and NKp46. Eur J Immunol (2001) 31:1656–65. doi:10.1002/1521-4141(200106)31:6<1656::AID-IMMU1656>3.0.CO;2-V
61. Crome SQ, Nguyen LT, Lopez-Verges S, Yang SY, Martin B, Yam JY, et al. A distinct innate lymphoid cell population regulates tumor-associated T cells. Nat Med (2017) 23:368–75. doi:10.1038/nm.4278
Keywords: innate immunity, differentiation, gene expression, immune protection, innate lymphoid cell
Citation: Huang Q, Seillet C and Belz GT (2017) Shaping Innate Lymphoid Cell Diversity. Front. Immunol. 8:1569. doi: 10.3389/fimmu.2017.01569
Received: 01 September 2017; Accepted: 01 November 2017;
Published: 16 November 2017
Edited by:
Chiara Romagnani, Deutsches Rheuma-Forschungszentrum (DRFZ), GermanyReviewed by:
Barbara L. Kee, University of Chicago, United StatesAlan M. Hanash, Memorial Sloan Kettering Cancer Center, United States
Copyright: © 2017 Huang, Seillet and Belz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabrielle T. Belz, belz@wehi.edu.au;
Cyril Seillet, seillet@wehi.edu.au