 Thomas Sajda
Thomas Sajda Animesh A. Sinha
Animesh A. Sinha- Department of Dermatology, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, Buffalo, NY, United States
Pemphigus vulgaris (PV) is an autoimmune skin blistering disease effecting both cutaneous and mucosal epithelia. Blister formation in PV is known to result from the binding of autoantibodies (autoAbs) to keratinocyte antigens. The primary antigenic targets of pathogenic autoAbs are known to be desmoglein 3, and to a lesser extent, desmoglein 1, cadherin family proteins that partially comprise the desmosome, a protein structure responsible for maintaining cell adhesion, although additional autoAbs, whose role in blister formation is still unclear, are also known to be present in PV patients. Nevertheless, there remain large gaps in knowledge concerning the precise mechanisms through which autoAb binding induces blister formation. Consequently, the primary therapeutic interventions for PV focus on systemic immunosuppression, whose side effects represent a significant health risk to patients. In an effort to identify novel, disease-specific therapeutic targets, a multitude of studies attempting to elucidate the pathogenic mechanisms downstream of autoAb binding, have led to significant advancements in the understanding of autoAb-mediated blister formation. Despite this enhanced characterization of disease processes, a satisfactory explanation of autoAb-induced acantholysis still does not exist. Here, we carefully review the literature investigating the pathogenic disease mechanisms in PV and, taking into account the full scope of results from these studies, provide a novel, comprehensive theory of blister formation in PV.
Introduction
Pemphigus vulgaris (PV) is an autoimmune skin blistering disease characterized by the presence of autoantibodies (autoAbs) directed against keratinocyte surface antigens. It has been well established that autoAbs alone are capable of driving blister formation in PV. Early studies identified the primary target of pathogenic autoAbs as desmoglein 3 (Dsg3) and to a lesser extent, desmoglein 1 (Dsg1). More recently, additional autoAbs specificities have been identified in PV patients that could potentially also contribute to disease pathogenicity (1, 2). Despite extensive research, the exact mechanisms through which autoAbs induce a loss of cell–cell adhesion (also termed acantholysis) are not well understood.
Since the primary target of PV autoAbs was shown to be a desmosomal protein, most of the earliest theories of acantholysis suggested that the loss of cell adhesion was simply the result of autoAbs sterically hindering the homo- and heterophillic binding of desmosomal proteins between neighboring cells (3, 4). Studies showing that staphylococcal exfoliative toxins, which cleave Dsg1, could produce blisters similar to those seen in PF, indicated that disturbance of desmosomal proteins was capable of causing a loss of cell–cell adhesion (5, 6). The relationship between desmoglein expression and cell adhesion was further supported by the observation that Dsg3-deficient mice develop mucosal lesions similar to those seen in PV patients (7). Together, these data demonstrated that interference with desmoglein interactions was sufficient to drive a loss of cell–cell adhesion.
The observation by multiple groups that PVIgG was seen only to bind at desmosomal areas also supported the idea that autoAb interference of desmosomal interaction drives blister formation (8–10). In addition, in areas of acantholysis, PVIgG was seen to bind to both desmosomal plaques and split desmosomes (11), indicating that PVIgG could access desmosome-associated desmogleins.
Further support for the steric hindrance theory came from later studies attempting to characterize the fine-epitope specificity of anti-Dsg3 autoAbs. It was shown that autoAbs primarily target the amino terminal of Dsg3 which, based on the crystal structure of other classical cadherins, was predicted to facilitate trans-interaction (12–17). Furthermore, only anti-Dsg3 autoAbs targeting the amino terminal EC1-2 of Dsg3 could illicit blister formation when passively transferred to neonatal mice, not anti-Dsg3 autoAbs targeting the more carboxyl domains EC3-5. Another study demonstrated that anti-Dsg3 autoAbs preferentially recognize the mature form of Dsg3, but not an immature form, which requires additional proteolytic processing to participate in adhesion (18, 19). It was also shown that autoAb binding was dependent on the proper calcium stabilized formation of Dsg3, which is known to be required for proper adhesive functioning (13, 20, 21). Later experiments using atomic force microscopy (AFM) were able to demonstrate that PVIgG could directly interfere with Dsg3 trans-interaction (22).
One potential liability of the steric hindrance theory was the possibility that blistering effects were mediated by the constant regions of the Abs. However, experiments showing that Fab and F(ab2) fragments, as well as single-chain variable fragments which lack the Fc domain, were able to induce blister formation in vitro proved that Fc-dependent mechanisms were not necessary for blister formation (23–26). Additional experiments demonstrating the pathogenicity of PVIgG in C5a-deficient mice indicated that compliment activation was not required for acantholysis (23).
Over time, evidence has accumulated suggesting steric hindrance may not be the primary or sole pathogenic mechanism operative in PV. One of the earliest indications that alternative mechanisms may drive pathogenesis was the observation that IgG from PF patients could induce disease in mice without interfering with trans-adhesion of Dsg1 (27). It was noted in multiple studies that PVIgG was seen to bind extra-desmosomal spaces on the surface of keratinocytes, allowing for the possibility that binding of autoAbs outside of desmosomes may affect disease (3, 28). It was also shown that PVIgG binding induced cytoskeletal changes and the retraction of keratin intermediate filaments before any visible changes in desmosomes (29–33). It was also noted that in early PV lesions keratinocytes first separate at inter-desmosomal areas and desmosomes are still intact and interacting with neighboring desmosomes (29, 34–36). Together, these findings suggested that desmosomal separation may be downstream of other processes induced by the binding of autoAbs. Recently, one research group used AFM to demonstrate that the loss of Dsg3 binding alone was not sufficient to cause a loss of cell adhesion, strongly indicating that steric hindrance by itself cannot sufficiently explain acantholysis in PV (37).
An early alternative to the steric hindrance theory was suggested by results showing that the binding of autoAbs initiated the activation of proteases which in turn degraded Dsg3 and inhibited cell–cell adhesion. Specifically, plasminogen activator was thought to play a role in disease (38). PVIgG was shown to induce signaling that led to increased production of plasminogen activator (39, 40). Furthermore, PVIgG induced keratinocyte expression of plasminogen activator receptor (38, 41). However, inhibition of plasminogen via dexamethasone did not prevent PVIgG-induced acantholysis (42). The role of other proteases was also shown not to be essential in disease by the failure of protease inhibitors and gene ablation to prevent blister formation (43, 44).
One of the earliest studies that indicated that autoAbs may exert their pathogenic effect through the activation of intracellular cascades demonstrated that plakoglobin (Pkg)-deficient mice were protected from PVIgG-induced blister formation (45). Pkg, an armadillo family protein, is well established as a major signaling molecule involved in the regulation of cell adhesion (46, 47). The inability of PVIgG to induce blisters in the absence of Pkg strongly suggests that alteration of Pkg signaling is a primary pathogenic mechanism of PVIgG. In addition, keratinocytes incubated at 4°C did not show any effects of PVIgG on cell adhesion, suggesting that the mechanisms underlying blister formation are energy dependent (48).
Identification and characterization of the precise signaling pathways driving autoAb-induced acantholysis has been a significant focus for PV research. As a result, large amounts of (often conflicting) information concerning the signaling alterations downstream of anti-Dsg and PVIgG binding have been characterized. Moreover, studies showing that autoAbs in PV sera directed at non-desmoglein antigens can also elicit intracellular signaling have further complicated efforts to elicit the precise mechanisms driving disease (49, 50). The primary signaling pathways and the evidence that supports their role in PV pathogenesis are reviewed below (see Table 1 for evidence supporting steric hindrance vs. intracellular signaling).

Table 1. Evidence supporting steric hindrance vs. intracellular signaling.
Signaling Pathways Implicated in PV
p38MAPK
p38 is one of the three major families of mitogen-activated protein kinases (MAPK), which are known to play a prominent role in a wide range of cellular pathways (51). In general, p38MAPK proteins can be activated by environmental stress and regulate the transcription of inflammatory cytokines (52). All MAPKs require dual phosphorylation for enzymatic activity, and each contains a characteristic dual phosphorylation sequence which affect both the substrate specificity and ability to auto-phosphorylate (53, 54). There are four types of p38MAPK: α, β, γ, and δ, each displaying unique patterns of tissue expression (52). Only p38MAPK α, β, and δ are known to be expressed in keratinocytes (52, 54–58) and have been primarily associated with differentiation and apoptosis (59).
The significance of p38MAPK signaling in PV pathogenesis was first suggested by a study which observed that PVIgG induced significant increases in the phosphorylation of p38MAPK, MAPKAP2 (MK2), and heat-shock protein (Hsp)27 (60). The degree of phosphorylation was shown to increase when cells were treated with higher concentrations of PVIgG. In addition, treatment of cells with p38MAPK inhibitors was able to prevent PVIgG-induced acantholysis as well as changes in the actin cytoskeleton and the retraction of KIFs from desmosomal attachments, both of which are hallmarks of acantholysis in PV. Inhibition of p38MAPK also prevented PVIgG-induced phosphorylation of MK2, Hsp27, and p38MAPK (60).
Other studies assessing the role of PVIgG on p38MAPK activation identified that PVIgG causes keratin retraction and p38MAPK activation within 30 min, and another peak at 6–10 h in cultured human keratinocytes. Only inhibition at the earlier time point was associated with prevention of blister formation and keratin retraction (61). Another study showed that AK23, a mouse-derived monoclonal anti-Dsg3 antibody, could also activate p38MAPK, demonstrating that autoAb binding to Dsg3 specifically can lead to p38MAPK activation (62).
The relevance of p38MAPK in disease was emphasized by findings that both p38MAPK and Hsp27 are phosphorylated in the lesional skin of PV patients (63). Further studies demonstrated that p38MAPK inhibitor blocks acantholysis in vivo, as well as p38MAPK activation, suggesting auto-phosphorylation of p38MAPK (64). p38MAPK activation was also shown to cause Dsg3 internalization, and p38MAPK inhibition can prevent this phenomenon in vivo (65). A more detailed assessment of the effects of p38MAPK showed that p38 depletes extra-desmosomal Dsg3 early as 30 min, and also is responsible for later depletion (2–24 h) of other desmosomal cadherins as well as DP (66–70).
The regulation of cytoskeletal changes by p38MAPK is especially relevant in understanding how p38MAPK plays a role in PV pathogenesis. In epithelial cells, cell detachment has been shown to induce p38MAPK activation (71), indicating a close relationship between p38MAPK and cellular adhesion. Furthermore, p38MAPK activation can lead to phosphorylation, and subsequent destabilization of keratin intermediate filaments (72), which could be one explanation for the characteristic retraction of KIFs seen in PV. p38MAPK is known to regulate actin filaments as well (73). Since extra-desmosomal Dsg3 complexes with actin cytoskeleton and is required to bring DP to desmosomal plaques (74, 75), it is possible that PVIgG-induced dysregulation of p38MAPK could interfere with proper desmosome assembly.
MAPKAP2 is phosphorylated and activated by p38MAPK (76, 77). The activation of MK2 has been associated with cell cycle control, cytokine production, and regulation of the keratin and actin cytoskeletons (78–80). The inhibition of MK2 has been shown to prevent PVIgG-induced spontaneous blister formation in mice, but not blistering solicited via the application of mechanical stress (81). This suggests that while MK2 may mediate some of the pathogenic effects of PVIgG, additional pathways downstream of p38MAK are likely also contributing to acantholysis.
Heat-shock protein 27 is another signaling molecule activated by MK2 (82). Hsp27 regulates both actin (83–85) and keratin cytoskeleton (86, 87). Both p38MAPK and MK2 regulate the effect of Hsp27 on the cytoskeleton via phosphorylation (88–90). Taken together, these findings demonstrate that one possible pathogenic mechanism of PVIgG-induced p38MAPK activation could be the perversion of typical cytoskeletal regulation, resulting in impaired cell adhesion.
The degree of evidence supporting a role for p38MAPK activation in the pathogenesis of PV has led researchers to investigate the utility of p38MAPK inhibition for the clinical treatment of PV. In a small clinical trial, 15 PV patients were treated with KC-706, a small molecule allosteric inhibitor of p38MAPK. Unfortunately, the trial was terminated before completion due to severe side effects of the drug. At the time of cessation, half of the patients were seen to exhibit at least a partial response to treatment, whereas the other half showed no improvement or a worsening of symptoms (91). Hopefully, the development of newer, more specific inhibitors of p38MAPK and other downstream targets will allow for effective pathway inhibition while avoiding serious side effects (92).
In addition to the effects listed above, p38MAPK can affect epidermal growth factor receptor (EGFR) signaling (93), RhoA activation (60, 68, 94, 95), and various apoptotic pathways (96). All of these pathways have been implicated in PV pathogenesis and are discussed in greater detail below.
Calcium/Protein Kinase C (PKC)/Phospholipase C (PLC)
The role of calcium signaling in keratinocyte differentiation and adhesion is well established. Increasing the Ca2+ concentration in keratinocyte culture medium increases intracellular calcium which in turn induces cell–cell contact (mainly adherens junctions) within 5 min, and formation of desmosomes within 2 h (97–104). PLC, an isoenzyme that is responsible for the cleavage of phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG), plays a significant role in calcium-induced keratinocyte differentiation (103, 105–108). PKC is a downstream target of PLC and is activated by calcium and DAG. Of the five isoforms known to be expressed in keratinocytes, PKC-alpha has been shown to play a major role in epidermal differentiation and proliferation (109–112).
Early studies showing that PVIgG leads to a rapid increase in intracellular calcium in keratinocytes were the first to implicate calcium signaling as a pathogenic signaling pathway in PV (113, 114). Additional studies demonstrated that PKC was activated within 30 s of treatment of PVIgG (115). A significant role for calcium signaling in PV pathogenesis was strengthened by studies which showed inhibition of PKC could prevent acantholysis both in vitro and in vivo (116, 117). It was also shown that inhibition of PLC prevented PVIgG-induced acantholysis, as well as increases in intracellular calcium and PKC activation (118). These results suggest that PVIgGs may exert their pathogenic effect by eliciting an increase in intracellular calcium, which leads to the activation of downstream signaling pathways.
The identification of PKC as a potential driver of PVIgG-induced pathogenesis is especially interesting due to the well-established role of PKC in cell adhesion. It has been shown that PKC activation leads to weakened cell–cell adhesion, whereas PKC inhibition results in increased adhesion (119, 120). In addition, PKC is known to be required for desmosome assembly and disassembly (41, 97, 121–126). The association of PKC with cell adhesion and desmosomal regulation may provide insight to help identify exactly how PVIgG induced PKC activation results in a loss of cell adhesion. One study showed that PVIgG-induced activation of PKC leads to its dissociation from KIFs and subsequent phosphorylation of DP, which resulted in desmosomal instability (127, 128). Another mechanism through which PKC activation may contribute to disease pathogenesis is by its effect on KIF turnover. It has been shown that PKC directly phosphorylates keratin molecules, leading to a turnover in KIFs (129, 130). This phenomenon may also explain the mechanistic background for the detachment of KIFs which is the hallmark of acantholysis.
Epidermal Growth Factor Receptor
Epidermal growth factor receptor is a well-studied signaling pathway that impacts a multitude of cellular processes either through the direct binding of its ligand, epidermal growth factor, or by cross-activation from a number of other signaling pathways (131–134). EGFR signaling has been shown to impact cell adhesion via both adherens junctions and desmosomes (135–137). Specifically, association with Dsg1 has been shown to suppress EGFR extracellular signal-regulated kinase 1/2 signaling in skin (138). In addition, it has been shown that EGFR activation can induce the phosphorylation of Pkg and decrease the association of desmoplakin with the desmosome, resulting in weakened cellular adhesion (139). In general, these studies associate an activation of EGFR signaling with destabilization of desmosomal adhesion.
The association between EGFR signaling and cell adhesion provided a rationale for researchers to investigate if EGFR signaling played a role in acantholysis, and it was eventually shown that PVIgG led to the activation of EGFR in keratinocytes (49, 93). Further studies determined that the activation of EGFR could be detected as early as 30 min after exposure to PVIgG, but the activation occurred downstream of p38MAPK activation (49, 66, 67, 140). Another group was able to show that anti-Dsg3 autoAbs could also lead to EGFR activation (14). Multiple studies then showed that the inhibition of EGFR could prevent PVIgG-induced skin blistering both in vivo and in human skin explants (66, 93, 141).
In addition, EGFR is implicated in PV pathogenesis via a second signaling axis independent of p38MAPK. PVIgG binding induces secretion of EGF and related mediators from basal keratinocytes, which in turn activate Src, focal adhesion kinase, and mammalian target of rapamycin (mTOR) via EGFR and nitric oxide synthase. The end result is the activation of caspases 3 and 9, which have been proposed to contribute to bister formation (142–145). The role for traditional apoptosis in PV pathogenesis is currently unclear; our group showed that the induction of apoptosis-related mechanisms after anti-Dsg antibody binding is reversible and independent of the Fas/FasL axis (146). However, studies showing that inhibition of the low level caspase-3 induction caused by PVIgG prevented acantholysis in vitro and in vivo suggest that caspase-3 activation does indeed play a role in disease (147).
Rho Family GTPases
Rho and Rac are both members of the Rho small GTPases family, which are known to play a role cytoskeletal reorganization, cell polarity, morphogenesis, and cell migration (61). These Rho family GTPases are known to affect the turnover of adherens junctions through multiple pathways (148). Both Rho and Rac are required for the establishment of adherens junctions (149, 150), and the activation of Rac has also been shown inhibit desmosomal adhesion in human keratinocytes (151).
The first association of Rho GTPases with PV pathogenesis was seen in experiments which demonstrated that the activation of Rho GTPases could prevent PVIgG-induced blister formation in human skin. Additional studies were able to show that RhoA activation was able to block the PVIgG-induced retraction of KIFs as well as loss of cell adhesion in HaCaT cells (41). Also, it was observed that HaCaT cells treated with PVIgG demonstrated a reduction in RhoA activity. In addition, p38MAPK inhibitors were shown to block the PVIgG-induced reduction of RhoA activity (94). These results suggest that PVIgG-induced activation of p38MAPK may induce blister formation, at least in part, by inhibiting the activity of RhoA. Given that the formation of adherens junctions has been shown to be necessary for proper assembly and disassembly of desmosomes (47, 152, 153), the loss of desmosomal adhesion seen in PV may be secondary to the inhibition of adherens junctions caused by RhoA inhibition.
For a summary of evidence in support of PV-associated signaling pathways, see Table 2.

Table 2. Evidence in support of pemphigus vulgaris (PV)-associated signaling pathways.
Integrated Model of PV autoAb-Induced Signaling
The pathogenic processes following the binding of autoAbs that eventually drive the loss of cell adhesion are diverse and complex. Taken together, the above data suggest that the disease mechanisms underlying PV are the result of both the direct steric interference of adhesion molecule interaction by autoAb binding and the activation of intracellular signaling pathways elicited via autoAb binding. A viewpoint that could tie both these mechanisms together is to see the desmosome/keratin intermediate filament complex as a signaling complex which participates in mechanosensing in addition to providing structural stability. Although desmosomal proteins have not yet been shown to function in this manner, a wealth of data exists which describes a similar function in adherens junctions (154). If this was shown to be the case with desmosomal proteins as well, it would be reasonable to assume that, in addition to physically interfering with desmosomal adhesion, the binding of autoAbs in PV may alter signaling pathways associated with the desmosomal complex, such as p38MAPK, EGFR, and PKC that ultimately interfere with the structural stability of keratinocytes.
Taking into consideration the breadth of experimental data detailing autoAb-induced activation of intracellular signaling pathways, we propose that blister formation in PV may result from the following mechanism: (1) binding of autoAbs to target desmosomal antigens (either desmosomal or extra-desmosomal) induces the activation of PLC, leading to the activation of PKC via Ca2+ and DAG, which in turn activates p38MAPK via MAP3ks [such as Ask1 (155, 156)]; (2) activated PKC (either through direct phosphorylation of keratin filaments or DP) and p38MAPK (via MK2 and Hsp27 phosphorylation) then induce the retraction of the KIFs as well as the turnover of the actin cytoskeleton; (3) the retraction of KIFs from the desmosomal plaques, as well as cytoskeletal rearrangements, then cause a destabilization of desmosomes, and a weakening of cell adhesion; (4) finally, the weakening of cell adhesion, coupled with the mechanical stress induced by cytoskeletal rearrangements, induces a cellular stress response, resulting in the activation of Src, EGFR, and Rac1 (as well as other pathways), and re-initiation/perpetuation of the pathological cycle (Figure 1).
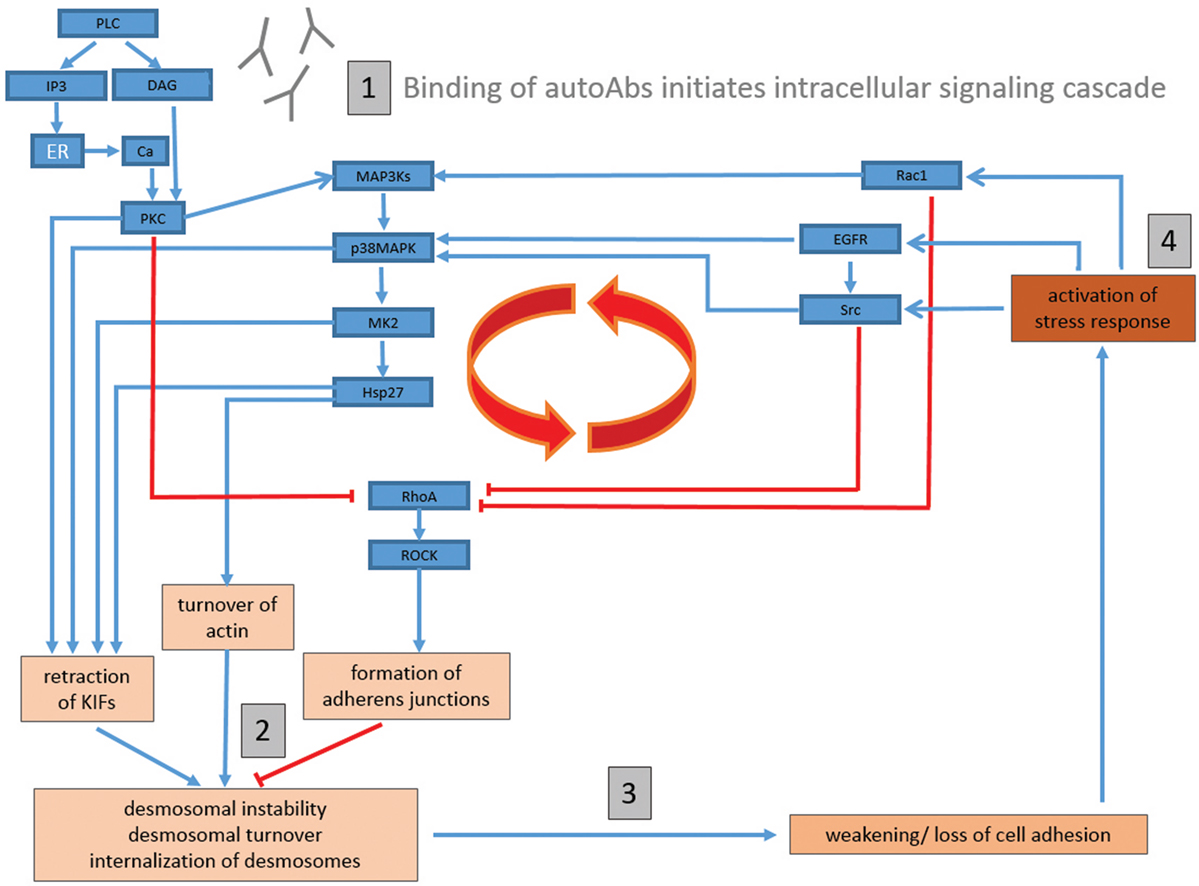
Figure 1. Proposed comprehensive model of signaling mechanisms underlying blister formation in pemphigus vulgaris. (1) Binding of autoantibodies (autoAbs) to target keratinocyte antigens (either desmosomal or extra-desmosomal) induces the activation of phospholipase C (PLC), and subsequently the activation of protein kinase C (PKC) via Ca2+ and diacylglycerol (DAG), which then activates p38MAPK via MAP3ks (such as Ask1); (2) activated PKC {either through direct phosphorylation of keratin filaments or DP} and p38MAPK {via MAPKAP2 (MK2) and heat-shock protein (Hsp)27 phosphorylation} then induce the retraction of the KIFs as well as the turnover of the actin cytoskeleton; (3) the retraction of KIFs from the desmosomal plaques, as well as cytoskeletal rearrangements, then cause a destabilization of desmosomes, and a weakening of cell adhesion; (4) finally, the weakening of cell adhesion, coupled with the mechanical stress induced by cytoskeletal rearrangements, induces a cellular stress response, resulting in the activation of Src, epidermal growth factor receptor (EGFR), and Rac1 (as well as other pathways), and re-initiation/perpetuation of the pathological cycle. RhoA is inhibited by multiple pathways (such as PKC, EGFR/Src, and Rac1). As RhoA activation has been shown to inhibit PVIgG-induced acantholysis, an inhibition of RhoA would promote cell dissociation. Blue arrow-headed lines indicate activation, while red bar-headed lines indicate inhibition of selected molecules/pathways.
A primary advantage of this model is the ability to provide a mechanistic framework for understanding how the widely variegated set of factors that have been implicated across multiple studies may be contributing to PV pathogenesis. For example, multiple studies have linked increased levels of reactive oxygen species (ROS) to PV (157–161), but a potential mechanistic contribution to disease has not been well defined. Using our model, however, the known ability of ROS to affect KIF and actin cytoskeletons as well as PKC, Src, p38MAPK, and RhoA signaling (162–167) potentially demonstrates how ROS may contribute to blister formation in a number of ways. As a result, our model can also explain how anti-mitochondrial autoAbs, which have been suggested to play a role in PV and shown to increase ROS production, can directly mediate disease pathogenesis (50, 168–172). Furthermore, the activation of pro-apoptotic pathways by autoAb binding has been shown to modulate p38MAPK (50) and may further contribute to the signaling pathologies that drive acantholysis.
Although this model lays a comprehensive framework for the mechanism of blister formation incorporating all of the major signaling pathways implicated in pathogenesis by the literature, future experiments are required to test the validity of this model and more precisely define the series of signaling events which occur downstream of autoAb binding. Determining the degree to which both desmosomal and extra-desmosomal Dsg proteins associate with signaling molecules and whether or not the binding of autoAbs to these proteins is sufficient to elicit intracellular signaling is a high priority to ascertain if these molecules could function as signal transducers and if so, through which pathways. Immunofluorescent studies of keratinocyte monolayers before and after PVIgG treatment may be one way to determine which molecules are physically associated with desmosomal proteins and determine if autoAb binding leads to their activation/translocation. Another important step would be to more precisely define the upstream MAPK cascade that leads to p38MAPK activation. There are multiple upstream signaling molecules which are known to activate p38MAPK and the characterization of the molecules involved in this process could allow for a better understanding of how autoAbs, or additional factors such as hormones or cytokines (which are known to utilize specific cascades) could activate p38MAPK (173–175). Protein microarray analysis, which allows for rapid, highly specific, multiplexed analysis of the signal transduction pathways (176, 177), would be well suited for this task.
Desmosomal proteins as a mechanosensing signaling complex would provide a logical intersection between the two primary theories of autoAb-induced blister formation in PV. Although future studies are still needed, there exists a great potential to significantly and directly affect the development of future treatments in PV. Identification of the signals transduced by autoAb binding could lead to the identification of potentially novel drug targets, or, at the very least allow researchers to focus on disease-specific signaling pathologies, resulting in small molecule inhibitors with a lower chance of harmful side effects and the ability to minimize systemic suppression of the immune system. In addition, large scale profiling of the signal transduction pathways upstream of MAPK may represent an especially beneficial endeavor as such studies could identify cytokines or hormones which may be contributing to disease process. In addition to facilitating the use of disease specific anti-cytokine or hormonal therapies which may already be in use for other disease modalities, such information could be individualized to the specific pathologies driving disease in a given patient, informing more tailored and effective treatment strategies and greatly enhancing clinical management of disease.
Author Contributions
AS and TS contributed to the conceptualization and writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank K. Seiffert-Sinha for critical review of the manuscript. We thank B.K.S. and M.S. for their continued guidance and support.
References
1. Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell (1991) 67:869–77. doi:10.1016/0092-8674(91)90360-B
2. Ahmed AR, Carrozzo M, Caux F, Cirillo N, Dmochowski M, Alonso AE, et al. Monopathogenic vs multipathogenic explanations of pemphigus pathophysiology. Exp Dermatol (2016) 25:839–46. doi:10.1111/exd.13106
3. Patel HP, Diaz LA, Anhalt GJ, Labib RS, Takahashi Y. Demonstration of pemphigus antibodies on the cell surface of murine epidermal cell monolayers and their internalization. J Invest Dermatol (1984) 83:409–15. doi:10.1111/1523-1747.ep12273480
4. Jones JC, Yokoo KM, Goldman RD. Further analysis of pemphigus autoantibodies and their use in studies on the heterogeneity, structure, and function of desmosomes. J Cell Biol (1986) 102:1109–17. doi:10.1083/jcb.102.3.1109
5. Amagai M, Matsuyoshi N, Wang ZH, Andl C, Stanley JR. Toxin in bullous impetigo and staphylococcal scalded-skin syndrome targets desmoglein 1. Nat Med (2000) 6:1275–7. doi:10.1038/81385
6. Hanakawa Y, Schechter NM, Lin C, Nishifuji K, Amagai M, Stanley JR. Enzymatic and molecular characteristics of the efficiency and specificity of exfoliative toxin cleavage of desmoglein 1. J Biol Chem (2004) 279:5268–77. doi:10.1074/jbc.M311087200
7. Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, et al. Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol (1997) 137:1091–102. doi:10.1083/jcb.137.5.1091
8. Shimizu H, Masunaga T, Ishiko A, Kikuchi A, Hashimoto T, Nishikawa T. Pemphigus vulgaris and pemphigus foliaceus sera show an inversely graded binding pattern to extracellular regions of desmosomes in different layers of human epidermis. J Invest Dermatol (1995) 105:153–9. doi:10.1111/1523-1747.ep12316695
9. Akiyama M, Hashimoto T, Sugiura M, Nishikawa T. Ultrastructural localization of pemphigus vulgaris and pemphigus foliaceus antigens in cultured human squamous carcinoma cells. Br J Dermatol (1991) 125:233–7. doi:10.1111/j.1365-2133.1991.tb14746.x
10. Karpati S, Amagai M, Prussick R, Cehrs K, Stanley JR. Pemphigus vulgaris antigen, a desmoglein type of cadherin, is localized within keratinocyte desmosomes. J Cell Biol (1993) 122:409–15. doi:10.1083/jcb.122.2.409
11. Shimizu A, Ishiko A, Ota T, Tsunoda K, Amagai M, Nishikawa T. IgG binds to desmoglein 3 in desmosomes and causes a desmosomal split without keratin retraction in a pemphigus mouse model. J Invest Dermatol (2004) 122:1145–53. doi:10.1111/j.0022-202X.2004.22426.x
12. Li N, Aoki V, Hans-Filho G, Rivitti EA, Diaz LA. The role of intramolecular epitope spreading in the pathogenesis of endemic pemphigus foliaceus (fogo selvagem). J Exp Med (2003) 197:1501–10. doi:10.1084/jem.20022031
13. Sekiguchi M, Futei Y, Fujii Y, Iwasaki T, Nishikawa T, Amagai M. Dominant autoimmune epitopes recognized by pemphigus antibodies map to the N-terminal adhesive region of desmogleins. J Immunol (2001) 167:5439–48. doi:10.4049/jimmunol.167.9.5439
14. Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, et al. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol (2003) 170:2170–8. doi:10.4049/jimmunol.170.4.2170
15. Shapiro L, Fannon AM, Kwong PD, Thompson A, Lehmann MS, Grubel G, et al. Structural basis of cell-cell adhesion by cadherins. Nature (1995) 374:327–37. doi:10.1038/374327a0
16. Nagar B, Overduin M, Ikura M, Rini JM. Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature (1996) 380:360–4. doi:10.1038/380360a0
17. Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science (2002) 296:1308–13. doi:10.1126/science.1071559
18. Yokouchi M, Saleh MA, Kuroda K, Hachiya T, Stanley JR, Amagai M, et al. Pathogenic epitopes of autoantibodies in pemphigus reside in the amino-terminal adhesive region of desmogleins which are unmasked by proteolytic processing of prosequence. J Invest Dermatol (2009) 129:2156–66. doi:10.1038/jid.2009.61
19. Ozawa M, Kemler R. Correct proteolytic cleavage is required for the cell adhesive function of uvomorulin. J Cell Biol (1990) 111:1645–50. doi:10.1083/jcb.111.4.1645
20. Eyre RW, Stanley JR. Human autoantibodies against a desmosomal protein complex with a calcium-sensitive epitope are characteristic of pemphigus foliaceus patients. J Exp Med (1987) 165:1719–24. doi:10.1084/jem.165.6.1719
21. Kamiya K, Aoyama Y, Shirafuji Y, Hamada T, Morizane S, Fujii K, et al. A higher correlation of the antibody activities against the calcium-dependent epitopes of desmoglein 3 quantified by ethylenediaminetetraacetic acid-treated enzyme-linked immunosorbent assay with clinical disease activities of pemphigus vulgaris. J Dermatol Sci (2013) 70:190–5. doi:10.1016/j.jdermsci.2013.02.011
22. Heupel WM, Zillikens D, Drenckhahn D, Waschke J. Pemphigus vulgaris IgG directly inhibit desmoglein 3-mediated transinteraction. J Immunol (2008) 181:1825–34. doi:10.4049/jimmunol.181.3.1825
23. Anhalt GJ, Till GO, Diaz LA, Labib RS, Patel HP, Eaglstein NF. Defining the role of complement in experimental pemphigus vulgaris in mice. J Immunol (1986) 137:2835–40.
24. Mascaro JM Jr, Espana A, Liu Z, Ding X, Swartz SJ, Fairley JA, et al. Mechanisms of acantholysis in pemphigus vulgaris: role of IgG valence. Clin Immunol Immunopathol (1997) 85:90–6. doi:10.1006/clin.1997.4408
25. Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, et al. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest (2005) 115:888–99. doi:10.1172/JCI24185
26. Rock B, Labib RS, Diaz LA. Monovalent Fab’ immunoglobulin fragments from endemic pemphigus foliaceus autoantibodies reproduce the human disease in neonatal Balb/c mice. J Clin Invest (1990) 85:296–9. doi:10.1172/JCI114426
27. Waschke J, Bruggeman P, Baumgartner W, Zillikens D, Drenckhahn D. Pemphigus foliaceus IgG causes dissociation of desmoglein 1-containing junctions without blocking desmoglein 1 transinteraction. J Clin Invest (2005) 115:3157–65. doi:10.1172/JCI23475
28. Bedane C, Prost C, Thomine E, Intrator L, Joly P, Caux F, et al. Binding of autoantibodies is not restricted to desmosomes in pemphigus vulgaris: comparison of 14 cases of pemphigus vulgaris and 10 cases of pemphigus foliaceus studied by western immunoblot and immunoelectron microscopy. Arch Dermatol Res (1996) 288:343–52.
29. Wilgram GF, Caulfield JB, Lever WF. An electron microscopic study of acantholysis in pemphigus vulgaris. J Invest Dermatol (1961) 36:373–82. doi:10.1038/jid.1961.58
30. Baroni A, Buommino E, Paoletti I, Orlando M, Ruocco E, Ruocco V. Pemphigus serum and captopril induce heat shock protein 70 and inducible nitric oxide synthase overexpression, triggering apoptosis in human keratinocytes. Br J Dermatol (2004) 150:1070–80. doi:10.1111/j.1365-2133.2004.05919.x
31. Jinbu Y, Kitajima Y, Koto S, Akasaka Y, Yaoita H. Different effects of pemphigus antibody and plasmin on the distribution of keratin intermediate filaments and desmoplakins between cultured oral and epidermal keratinocytes. J Dermatol Sci (1992) 3:6–12. doi:10.1016/0923-1811(92)90003-T
32. Kitajima Y, Inoue S, Yaoita H. Effects of pemphigus antibody on the organization of microtubules and keratin-intermediate filaments in cultured human keratinocytes. Br J Dermatol (1986) 114:171–9. doi:10.1111/j.1365-2133.1986.tb02795.x
34. Hashimoto K, Lever WF. An electron microscopic study on pemphigus vulgaris of the mouth and the skin with special reference to the intercellular cement. J Invest Dermatol (1967) 48:540–52. doi:10.1038/jid.1967.86
35. Hashimoto K, Lever WF. The intercellular cement in pemphigus vulgaris, an electron microscopic study. Dermatologica (1967) 135:27–34. doi:10.1159/000254157
36. Sams WM Jr, Gammon WR. Mechanism of lesion production in pemphigus and pemphigoid. J Am Acad Dermatol (1982) 6:431–52. doi:10.1016/S0190-9622(82)70036-2
37. Vielmuth F, Waschke J, Spindler V. Loss of desmoglein binding is not sufficient for keratinocyte dissociation in pemphigus. J Invest Dermatol (2015) 135:3068–77. doi:10.1038/jid.2015.324
38. Hashimoto K, Shafran KM, Webber PS, Lazarus GS, Singer KH. Anti-cell surface pemphigus autoantibody stimulates plasminogen activator activity of human epidermal cells. A mechanism for the loss of epidermal cohesion and blister formation. J Exp Med (1983) 157:259–72. doi:10.1084/jem.157.1.259
39. Schaefer BM, Jaeger CJ, Kramer MD. Plasminogen activator system in pemphigus vulgaris. Br J Dermatol (1996) 135:726–32. doi:10.1046/j.1365-2133.1996.d01-1070.x
40. Feliciani C, Toto P, Wang B, Sauder DN, Amerio P, Tulli A. Urokinase plasminogen activator mRNA is induced by IL-1alpha and TNF-alpha in in vitro acantholysis. Exp Dermatol (2003) 12:466–71. doi:10.1034/j.1600-0625.2002.120415.x
41. Seishima M, Satoh S, Nojiri M, Osada K, Kitajima Y. Pemphigus IgG induces expression of urokinase plasminogen activator receptor on the cell surface of cultured keratinocytes. J Invest Dermatol (1997) 109:650–5. doi:10.1111/1523-1747.ep12337662
42. Anhalt GJ, Patel HP, Labib RS, Diaz LA, Proud D. Dexamethasone inhibits plasminogen activator activity in experimental pemphigus in vivo but does not block acantholysis. J Immunol (1986) 136:113–7.
43. Schuh T, Besch R, Braungart E, Flaig MJ, Douwes K, Sander CA, et al. Protease inhibitors prevent plasminogen-mediated, but not pemphigus vulgaris-induced, acantholysis in human epidermis. Biol Chem (2003) 384:311–5. doi:10.1515/BC.2003.035
44. Mahoney MG, Wang ZH, Stanley JR. Pemphigus vulgaris and pemphigus foliaceus antibodies are pathogenic in plasminogen activator knockout mice. J Invest Dermatol (1999) 113:22–5. doi:10.1046/j.1523-1747.1999.00632.x
45. Caldelari R, de Bruin A, Baumann D, Suter MM, Bierkamp C, Balmer V, et al. A central role for the armadillo protein plakoglobin in the autoimmune disease pemphigus vulgaris. J Cell Biol (2001) 153:823–34. doi:10.1083/jcb.153.4.823
46. Zhurinsky J, Shtutman M, Ben-Ze’ev A. Plakoglobin and beta-catenin: protein interactions, regulation and biological roles. J Cell Sci (2000) 113(Pt 18):3127–39.
47. Getsios S, Huen AC, Green KJ. Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol (2004) 5:271–81. doi:10.1038/nrm1356
48. Calkins CC, Setzer SV, Jennings JM, Summers S, Tsunoda K, Amagai M, et al. Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J Biol Chem (2006) 281:7623–34. doi:10.1074/jbc.M512447200
49. Chernyavsky AI, Arredondo J, Kitajima Y, Sato-Nagai M, Grando SA. Desmoglein versus non-desmoglein signaling in pemphigus acantholysis: characterization of novel signaling pathways downstream of pemphigus vulgaris antigens. J Biol Chem (2007) 282:13804–12. doi:10.1074/jbc.M611365200
50. Marchenko S, Chernyavsky AI, Arredondo J, Gindi V, Grando SA. Antimitochondrial autoantibodies in pemphigus vulgaris: a missing link in disease pathophysiology. J Biol Chem (2010) 285:3695–704. doi:10.1074/jbc.M109.081570
51. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science (2002) 298:1911–2. doi:10.1126/science.1072682
52. Dashti SR, Efimova T, Eckert RL. MEK7-dependent activation of p38 MAP kinase in keratinocytes. J Biol Chem (2001) 276:8059–63. doi:10.1074/jbc.C000862200
53. Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J (1995) 9:576–96. doi:10.1096/fasebj.9.8.7768349
54. Han J, Lee JD, Jiang Y, Li Z, Feng L, Ulevitch RJ. Characterization of the structure and function of a novel MAP kinase kinase (MKK6). J Biol Chem (1996) 271:2886–91. doi:10.1074/jbc.271.6.2886
55. Lechner C, Zahalka MA, Giot JF, Moller NP, Ullrich A. ERK6, a mitogen-activated protein kinase involved in C2C12 myoblast differentiation. Proc Natl Acad Sci U S A (1996) 93:4355–9. doi:10.1073/pnas.93.9.4355
56. Li Z, Jiang Y, Ulevitch RJ, Han J. The primary structure of p38 gamma: a new member of p38 group of MAP kinases. Biochem Biophys Res Commun (1996) 228:334–40. doi:10.1006/bbrc.1996.1662
57. Court NW, dos Remedios CG, Cordell J, Bogoyevitch MA. Cardiac expression and subcellular localization of the p38 mitogen-activated protein kinase member, stress-activated protein kinase-3 (SAPK3). J Mol Cell Cardiol (2002) 34:413–26. doi:10.1006/jmcc.2001.1523
58. Kumar S, McDonnell PC, Gum RJ, Hand AT, Lee JC, Young PR. Novel homologues of CSBP/p38 MAP kinase: activation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem Biophys Res Commun (1997) 235:533–8. doi:10.1006/bbrc.1997.6849
59. Eckert RL, Efimova T, Dashti SR, Balasubramanian S, Deucher A, Crish JF, et al. Keratinocyte survival, differentiation, and death: many roads lead to mitogen-activated protein kinase. J Investig Dermatol Symp Proc (2002) 7:36–40. doi:10.1046/j.1523-1747.2002.19634.x
60. Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, Chua MP, et al. Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem (2005) 280:23778–84. doi:10.1074/jbc.M501365200
61. Lee HE, Berkowitz P, Jolly PS, Diaz LA, Chua MP, Rubenstein DS. Biphasic activation of p38MAPK suggests that apoptosis is a downstream event in pemphigus acantholysis. J Biol Chem (2009) 284:12524–32. doi:10.1074/jbc.M808204200
62. Kawasaki Y, Aoyama Y, Tsunoda K, Amagai M, Kitajima Y. Pathogenic monoclonal antibody against desmoglein 3 augments desmoglein 3 and p38 MAPK phosphorylation in human squamous carcinoma cell line. Autoimmunity (2006) 39:587–90. doi:10.1080/08916930600971943
63. Berkowitz P, Diaz LA, Hall RP, Rubenstein DS. Induction of p38MAPK and HSP27 phosphorylation in pemphigus patient skin. J Invest Dermatol (2008) 128:738–40. doi:10.1038/sj.jid.5701080
64. Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci U S A (2006) 103:12855–60. doi:10.1073/pnas.0602973103
65. Jolly PS, Berkowitz P, Bektas M, Lee HE, Chua M, Diaz LA, et al. p38MAPK signaling and desmoglein-3 internalization are linked events in pemphigus acantholysis. J Biol Chem (2010) 285:8936–41. doi:10.1074/jbc.M109.087999
66. Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, Payne AS, et al. Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PLoS One (2012) 7:e50696. doi:10.1371/journal.pone.0050696
67. Schulze K, Galichet A, Sayar BS, Scothern A, Howald D, Zymann H, et al. An adult passive transfer mouse model to study desmoglein 3 signaling in pemphigus vulgaris. J Invest Dermatol (2012) 132:346–55. doi:10.1038/jid.2011.299
68. Mao X, Choi EJ, Payne AS. Disruption of desmosome assembly by monovalent human pemphigus vulgaris monoclonal antibodies. J Invest Dermatol (2009) 129:908–18. doi:10.1038/jid.2008.339
69. Jennings JM, Tucker DK, Kottke MD, Saito M, Delva E, Hanakawa Y, et al. Desmosome disassembly in response to pemphigus vulgaris IgG occurs in distinct phases and can be reversed by expression of exogenous Dsg3. J Invest Dermatol (2011) 131:706–18. doi:10.1038/jid.2010.389
70. Oktarina DA, van der Wier G, Diercks GF, Jonkman MF, Pas HH. IgG-induced clustering of desmogleins 1 and 3 in skin of patients with pemphigus fits with the desmoglein nonassembly depletion hypothesis. Br J Dermatol (2011) 165:552–62. doi:10.1111/j.1365-2133.2011.10463.x
71. Rosen K, Shi W, Calabretta B, Filmus J. Cell detachment triggers p38 mitogen-activated protein kinase-dependent overexpression of Fas ligand. A novel mechanism of anoikis of intestinal epithelial cells. J Biol Chem (2002) 277:46123–30. doi:10.1074/jbc.M207883200
72. Ku NO, Azhar S, Omary MB. Keratin 8 phosphorylation by p38 kinase regulates cellular keratin filament reorganization: modulation by a keratin 1-like disease causing mutation. J Biol Chem (2002) 277:10775–82. doi:10.1074/jbc.M107623200
73. Gliem M, Heupel WM, Spindler V, Harms GS, Waschke J. Actin reorganization contributes to loss of cell adhesion in pemphigus vulgaris. Am J Physiol Cell Physiol (2010) 299:C606–13. doi:10.1152/ajpcell.00075.2010
74. Tsang SM, Brown L, Gadmor H, Gammon L, Fortune F, Wheeler A, et al. Desmoglein 3 acting as an upstream regulator of Rho GTPases, Rac-1/Cdc42 in the regulation of actin organisation and dynamics. Exp Cell Res (2012) 318:2269–83. doi:10.1016/j.yexcr.2012.07.002
75. Godsel LM, Dubash AD, Bass-Zubek AE, Amargo EV, Klessner JL, Hobbs RP, et al. Plakophilin 2 couples actomyosin remodeling to desmosomal plaque assembly via RhoA. Mol Biol Cell (2010) 21:2844–59. doi:10.1091/mbc.E10-02-0131
76. Freshney NW, Rawlinson L, Guesdon F, Jones E, Cowley S, Hsuan J, et al. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell (1994) 78:1039–49. doi:10.1016/0092-8674(94)90278-X
77. Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, et al. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell (1994) 78:1027–37. doi:10.1016/0092-8674(94)90277-1
78. Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science (1994) 265:808–11. doi:10.1126/science.7914033
79. Piotrowicz RS, Hickey E, Levin EG. Heat shock protein 27 kDa expression and phosphorylation regulates endothelial cell migration. FASEB J (1998) 12:1481–90. doi:10.1096/fasebj.12.14.1481
80. Rousseau S, Houle F, Landry J, Huot J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene (1997) 15:2169–77. doi:10.1038/sj.onc.1201380
81. Mao X, Li H, Sano Y, Gaestel M, Mo Park J, Payne AS. MAPKAP kinase 2 (MK2)-dependent and -independent models of blister formation in pemphigus vulgaris. J Invest Dermatol (2014) 134:68–76. doi:10.1038/jid.2013.224
82. Rogalla T, Ehrnsperger M, Preville X, Kotlyarov A, Lutsch G, Ducasse C, et al. Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor alpha by phosphorylation. J Biol Chem (1999) 274:18947–56. doi:10.1074/jbc.274.27.18947
83. Benndorf R, Hayess K, Ryazantsev S, Wieske M, Behlke J, Lutsch G. Phosphorylation and supramolecular organization of murine small heat shock protein HSP25 abolish its actin polymerization-inhibiting activity. J Biol Chem (1994) 269:20780–4.
84. Geum D, Son GH, Kim K. Phosphorylation-dependent cellular localization and thermoprotective role of heat shock protein 25 in hippocampal progenitor cells. J Biol Chem (2002) 277:19913–21. doi:10.1074/jbc.M104396200
85. Panasenko OO, Kim MV, Marston SB, Gusev NB. Interaction of the small heat shock protein with molecular mass 25 kDa (hsp25) with actin. Eur J Biochem (2003) 270:892–901. doi:10.1046/j.1432-1033.2003.03449.x
86. Perng MD, Cairns L, van den IP, Prescott A, Hutcheson AM, Quinlan RA. Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J Cell Sci (1999) 112(Pt 13):2099–112.
87. Evgrafov OV, Mersiyanova I, Irobi J, Van Den Bosch L, Dierick I, Leung CL, et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet (2004) 36:602–6. doi:10.1038/ng1354
88. Guay J, Lambert H, Gingras-Breton G, Lavoie JN, Huot J, Landry J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J Cell Sci (1997) 110(Pt 3):357–68.
89. Lavoie JN, Hickey E, Weber LA, Landry J. Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of heat shock protein 27. J Biol Chem (1993) 268:24210–4.
90. Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol Cell Biol (1995) 15:505–16. doi:10.1128/MCB.15.1.505
91. Schultz HY, Diaz LA, Sirois DA, Werth VP, Grando SA. Generating consensus research goals and treatment strategies for pemphigus and pemphigoid: the 2010 JC Bystryn Pemphigus and Pemphigoid Meeting. J Invest Dermatol (2011) 131:1395–9. doi:10.1038/jid.2011.120
92. Cohen P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr Opin Cell Biol (2009) 21:317–24. doi:10.1016/j.ceb.2009.01.015
93. Bektas M, Jolly PS, Berkowitz P, Amagai M, Rubenstein DS. A pathophysiologic role for epidermal growth factor receptor in pemphigus acantholysis. J Biol Chem (2013) 288:9447–56. doi:10.1074/jbc.M112.438010
94. Waschke J, Spindler V, Bruggeman P, Zillikens D, Schmidt G, Drenckhahn D. Inhibition of Rho A activity causes pemphigus skin blistering. J Cell Biol (2006) 175:721–7. doi:10.1083/jcb.200605125
95. Spindler V, Rotzer V, Dehner C, Kempf B, Gliem M, Radeva M, et al. Peptide-mediated desmoglein 3 crosslinking prevents pemphigus vulgaris autoantibody-induced skin blistering. J Clin Invest (2013) 123:800–11. doi:10.1172/JCI60139
96. Wu NL, Lee TA, Tsai TL, Lin WW. TRAIL-induced keratinocyte differentiation requires caspase activation and p63 expression. J Invest Dermatol (2011) 131:874–83. doi:10.1038/jid.2010.402
97. Sheu HM, Kitajima Y, Yaoita H. Involvement of protein kinase C in translocation of desmoplakins from cytosol to plasma membrane during desmosome formation in human squamous cell carcinoma cells grown in low to normal calcium concentration. Exp Cell Res (1989) 185:176–90. doi:10.1016/0014-4827(89)90047-5
98. Kitajima Y, Aoyama Y, Seishima M. Transmembrane signaling for adhesive regulation of desmosomes and hemidesmosomes, and for cell-cell datachment induced by pemphigus IgG in cultured keratinocytes: involvement of protein kinase C. J Investig Dermatol Symp Proc (1999) 4:137–44. doi:10.1038/sj.jidsp.5640197
99. Sharpe GR, Gillespie JI, Greenwell JR. An increase in intracellular free calcium is an early event during differentiation of cultured human keratinocytes. FEBS Lett (1989) 254:25–8. doi:10.1016/0014-5793(89)81002-6
100. Tu CL, Oda Y, Bikle DD. Effects of a calcium receptor activator on the cellular response to calcium in human keratinocytes. J Invest Dermatol (1999) 113:340–5. doi:10.1046/j.1523-1747.1999.00698.x
101. Oda Y, Tu CL, Chang W, Crumrine D, Komuves L, Mauro T, et al. The calcium sensing receptor and its alternatively spliced form in murine epidermal differentiation. J Biol Chem (2000) 275:1183–90. doi:10.1074/jbc.275.2.1183
102. Tu CL, Chang W, Bikle DD. The extracellular calcium-sensing receptor is required for calcium-induced differentiation in human keratinocytes. J Biol Chem (2001) 276:41079–85. doi:10.1074/jbc.M107122200
103. Xie Z, Bikle DD. The recruitment of phosphatidylinositol 3-kinase to the E-cadherin-catenin complex at the plasma membrane is required for calcium-induced phospholipase C-gamma1 activation and human keratinocyte differentiation. J Biol Chem (2007) 282:8695–703. doi:10.1074/jbc.M609135200
104. Berridge MJ, Irvine RF. Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature (1984) 312:315–21. doi:10.1038/312315a0
105. Punnonen K, Denning M, Lee E, Li L, Rhee SG, Yuspa SH. Keratinocyte differentiation is associated with changes in the expression and regulation of phospholipase C isoenzymes. J Invest Dermatol (1993) 101:719–26. doi:10.1111/1523-1747.ep12371682
106. Homma Y, Emori Y, Shibasaki F, Suzuki K, Takenawa T. Isolation and characterization of a gamma-type phosphoinositide-specific phospholipase C (PLC-gamma 2). Biochem J (1990) 269:13–8. doi:10.1042/bj2690013
107. Meldrum E, Parker PJ, Carozzi A. The PtdIns-PLC superfamily and signal transduction. Biochim Biophys Acta (1991) 1092:49–71. doi:10.1016/0167-4889(91)90177-Y
108. Suh PG, Ryu SH, Choi WC, Lee KY, Rhee SG. Monoclonal antibodies to three phospholipase C isozymes from bovine brain. J Biol Chem (1988) 263:14497–504.
109. Cataisson C, Joseloff E, Murillas R, Wang A, Atwell C, Torgerson S, et al. Activation of cutaneous protein kinase C alpha induces keratinocyte apoptosis and intraepidermal inflammation by independent signaling pathways. J Immunol (2003) 171:2703–13. doi:10.4049/jimmunol.171.5.2703
110. Hara T, Saito Y, Hirai T, Nakamura K, Nakao K, Katsuki M, et al. Deficiency of protein kinase Calpha in mice results in impairment of epidermal hyperplasia and enhancement of tumor formation in two-stage skin carcinogenesis. Cancer Res (2005) 65:7356–62. doi:10.1158/0008-5472.CAN-04-4241
111. Wang HQ, Smart RC. Overexpression of protein kinase C-alpha in the epidermis of transgenic mice results in striking alterations in phorbol ester-induced inflammation and COX-2, MIP-2 and TNF-alpha expression but not tumor promotion. J Cell Sci (1999) 112(Pt 20):3497–506.
112. Jansen AP, Dreckschmidt NE, Verwiebe EG, Wheeler DL, Oberley TD, Verma AK. Relation of the induction of epidermal ornithine decarboxylase and hyperplasia to the different skin tumor-promotion susceptibilities of protein kinase C alpha, -delta and -epsilon transgenic mice. Int J Cancer (2001) 93:635–43. doi:10.1002/ijc.1395
113. Lyubimov H, Goldshmit D, Michel B, Oron Y, Milner Y. Pemphigus – identifying the autoantigen and its possible induction of epidermal acantholysis via Ca2+ signalling. Isr J Med Sci (1995) 31:42–8.
114. Seishima M, Esaki C, Osada K, Mori S, Hashimoto T, Kitajima Y. Pemphigus IgG, but not bullous pemphigoid IgG, causes a transient increase in intracellular calcium and inositol 1,4,5-triphosphate in DJM-1 cells, a squamous cell carcinoma line. J Invest Dermatol (1995) 104:33–7. doi:10.1111/1523-1747.ep12613469
115. Kitajima Y. New insights into desmosome regulation and pemphigus blistering as a desmosome-remodeling disease. Kaohsiung J Med Sci (2013) 29:1–13. doi:10.1016/j.kjms.2012.08.001
116. Cirillo N, Lanza A, Prime SS. Induction of hyper-adhesion attenuates autoimmune-induced keratinocyte cell-cell detachment and processing of adhesion molecules via mechanisms that involve PKC. Exp Cell Res (2010) 316:580–92. doi:10.1016/j.yexcr.2009.10.005
117. Spindler V, Endlich A, Hartlieb E, Vielmuth F, Schmidt E, Waschke J. The extent of desmoglein 3 depletion in pemphigus vulgaris is dependent on Ca(2+)-induced differentiation: a role in suprabasal epidermal skin splitting? Am J Pathol (2011) 179:1905–16. doi:10.1016/j.ajpath.2011.06.043
118. Esaki C, Seishima M, Yamada T, Osada K, Kitajima Y. Pharmacologic evidence for involvement of phospholipase C in pemphigus IgG-induced inositol 1,4,5-trisphosphate generation, intracellular calcium increase, and plasminogen activator secretion in DJM-1 cells, a squamous cell carcinoma line. J Invest Dermatol (1995) 105:329–33. doi:10.1111/1523-1747.ep12319948
119. Kimura TE, Merritt AJ, Garrod DR. Calcium-independent desmosomes of keratinocytes are hyper-adhesive. J Invest Dermatol (2007) 127:775–81. doi:10.1038/sj.jid.5700643
120. Hobbs RP, Amargo EV, Somasundaram A, Simpson CL, Prakriya M, Denning MF, et al. The calcium ATPase SERCA2 regulates desmoplakin dynamics and intercellular adhesive strength through modulation of PKCα signaling. FASEB J (2011) 25:990–1001. doi:10.1096/fj.10-163261
121. Godsel LM, Hsieh SN, Amargo EV, Bass AE, Pascoe-McGillicuddy LT, Huen AC, et al. Desmoplakin assembly dynamics in four dimensions: multiple phases differentially regulated by intermediate filaments and actin. J Cell Biol (2005) 171:1045–59. doi:10.1083/jcb.200510038
122. Bass-Zubek AE, Hobbs RP, Amargo EV, Garcia NJ, Hsieh SN, Chen X, et al. Plakophilin 2: a critical scaffold for PKC alpha that regulates intercellular junction assembly. J Cell Biol (2008) 181:605–13. doi:10.1083/jcb.200712133
123. Thomason HA, Cooper NH, Ansell DM, Chiu M, Merrit AJ, Hardman MJ, et al. Direct evidence that PKCalpha positively regulates wound re-epithelialization: correlation with changes in desmosomal adhesiveness. J Pathol (2012) 227:346–56. doi:10.1002/path.4016
124. Wallis S, Lloyd S, Wise I, Ireland G, Fleming TP, Garrod D. The alpha isoform of protein kinase C is involved in signaling the response of desmosomes to wounding in cultured epithelial cells. Mol Biol Cell (2000) 11:1077–92. doi:10.1091/mbc.11.3.1077
125. Osada K, Seishima M, Kitajima Y. Pemphigus IgG activates and translocates protein kinase C from the cytosol to the particulate/cytoskeleton fractions in human keratinocytes. J Invest Dermatol (1997) 108:482–7. doi:10.1111/1523-1747.ep12289726
126. Aoyama Y, Yamamoto Y, Yamaguchi F, Kitajima Y. Low to high Ca2+-switch causes phosphorylation and association of desmocollin 3 with plakoglobin and desmoglein 3 in cultured keratinocytes. Exp Dermatol (2009) 18:404–8. doi:10.1111/j.1600-0625.2008.00814.x
127. Kroger C, Loschke F, Schwarz N, Windoffer R, Leube RE, Magin TM. Keratins control intercellular adhesion involving PKC-alpha-mediated desmoplakin phosphorylation. J Cell Biol (2013) 201:681–92. doi:10.1083/jcb.201208162
128. Hobbs RP, Green KJ. Desmoplakin regulates desmosome hyperadhesion. J Invest Dermatol (2012) 132:482–5. doi:10.1038/jid.2011.318
129. Sivaramakrishnan S, Schneider JL, Sitikov A, Goldman RD, Ridge KM. Shear stress induced reorganization of the keratin intermediate filament network requires phosphorylation by protein kinase C zeta. Mol Biol Cell (2009) 20:2755–65. doi:10.1091/mbc.E08-10-1028
130. Ridge KM, Linz L, Flitney FW, Kuczmarski ER, Chou YH, Omary MB, et al. Keratin 8 phosphorylation by protein kinase C delta regulates shear stress-mediated disassembly of keratin intermediate filaments in alveolar epithelial cells. J Biol Chem (2005) 280:30400–5. doi:10.1074/jbc.M504239200
131. Hackel PO, Zwick E, Prenzel N, Ullrich A. Epidermal growth factor receptors: critical mediators of multiple receptor pathways. Curr Opin Cell Biol (1999) 11:184–9. doi:10.1016/S0955-0674(99)80024-6
132. Keely SJ, Uribe JM, Barrett KE. Carbachol stimulates transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T84 cells. Implications for carbachol-stimulated chloride secretion. J Biol Chem (1998) 273:27111–7. doi:10.1074/jbc.273.42.27111
133. Li X, Lee JW, Graves LM, Earp HS. Angiotensin II stimulates ERK via two pathways in epithelial cells: protein kinase C suppresses a G-protein coupled receptor-EGF receptor transactivation pathway. EMBO J (1998) 17:2574–83. doi:10.1093/emboj/17.9.2574
134. Tsai W, Morielli AD, Peralta EG. The m1 muscarinic acetylcholine receptor transactivates the EGF receptor to modulate ion channel activity. EMBO J (1997) 16:4597–605. doi:10.1093/emboj/16.15.4597
135. Hoschuetzky H, Aberle H, Kemler R. Beta-catenin mediates the interaction of the cadherin-catenin complex with epidermal growth factor receptor. J Cell Biol (1994) 127:1375–80. doi:10.1083/jcb.127.5.1375
136. Yin T, Getsios S, Caldelari R, Godsel LM, Kowalczyk AP, Muller EJ, et al. Mechanisms of plakoglobin-dependent adhesion: desmosome-specific functions in assembly and regulation by epidermal growth factor receptor. J Biol Chem (2005) 280:40355–63. doi:10.1074/jbc.M506692200
137. Miravet S, Piedra J, Castano J, Raurell I, Franci C, Dunach M, et al. Tyrosine phosphorylation of plakoglobin causes contrary effects on its association with desmosomes and adherens junction components and modulates beta-catenin-mediated transcription. Mol Cell Biol (2003) 23:7391–402. doi:10.1128/MCB.23.20.7391-7402.2003
138. Getsios S, Simpson CL, Kojima S, Harmon R, Sheu LJ, Dusek RL, et al. Desmoglein 1-dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. J Cell Biol (2009) 185:1243–58. doi:10.1083/jcb.200809044
139. Gaudry CA, Palka HL, Dusek RL, Huen AC, Khandekar MJ, Hudson LG, et al. Tyrosine-phosphorylated plakoglobin is associated with desmogleins but not desmoplakin after epidermal growth factor receptor activation. J Biol Chem (2001) 276:24871–80. doi:10.1074/jbc.M102731200
140. Frusic-Zlotkin M, Raichenberg D, Wang X, David M, Michel B, Milner Y. Apoptotic mechanism in pemphigus autoimmunoglobulins-induced acantholysis – possible involvement of the EGF receptor. Autoimmunity (2006) 39:563–75. doi:10.1080/08916930600971836
141. Sanchez-Carpintero I, Espana A, Pelacho B, Lopez Moratalla N, Rubenstein DS, Diaz LA, et al. In vivo blockade of pemphigus vulgaris acantholysis by inhibition of intracellular signal transduction cascades. Br J Dermatol (2004) 151:565–70. doi:10.1111/j.1365-2133.2004.06147.x
142. Pretel M, Espana A, Marquina M, Pelacho B, Lopez-Picazo JM, Lopez-Zabalza MJ. An imbalance in Akt/mTOR is involved in the apoptotic and acantholytic processes in a mouse model of pemphigus vulgaris. Exp Dermatol (2009) 18:771–80. doi:10.1111/j.1600-0625.2009.00893.x
143. Gil MP, Modol T, Espana A, Lopez-Zabalza MJ. Inhibition of FAK prevents blister formation in the neonatal mouse model of pemphigus vulgaris. Exp Dermatol (2012) 21:254–9. doi:10.1111/j.1600-0625.2012.01441.x
144. Espana A, Modol T, Gil MP, Lopez-Zabalza MJ. Neural nitric oxide synthase participates in pemphigus vulgaris acantholysis through upregulation of Rous sarcoma, mammalian target of rapamycin and focal adhesion kinase. Exp Dermatol (2013) 22:125–30. doi:10.1111/exd.12088
145. Marquina M, Espana A, Fernandez-Galar M, Lopez-Zabalza MJ. The role of nitric oxide synthases in pemphigus vulgaris in a mouse model. Br J Dermatol (2008) 159:68–76. doi:10.1111/j.1365-2133.2008.08582.x
146. Seiffert-Sinha K, Yang R, Fung CK, Lai KW, Patterson KC, Payne AS, et al. Nanorobotic investigation identifies novel visual, structural and functional correlates of autoimmune pathology in a blistering skin disease model. PLoS One (2014) 9:e106895. doi:10.1371/journal.pone.0106895
147. Luyet C, Schulze K, Sayar BS, Howald D, Muller EJ, Galichet A. Preclinical studies identify non-apoptotic low-level caspase-3 as therapeutic target in pemphigus vulgaris. PLoS One (2015) 10:e0119809. doi:10.1371/journal.pone.0119809
148. Braga VM. Cell-cell adhesion and signalling. Curr Opin Cell Biol (2002) 14:546–56. doi:10.1016/S0955-0674(02)00373-3
149. Braga VM, Machesky LM, Hall A, Hotchin NA. The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell-cell contacts. J Cell Biol (1997) 137:1421–31. doi:10.1083/jcb.137.6.1421
150. Ehrlich JS, Hansen MD, Nelson WJ. Spatio-temporal regulation of Rac1 localization and lamellipodia dynamics during epithelial cell-cell adhesion. Dev Cell (2002) 3:259–70. doi:10.1016/S1534-5807(02)00216-2
151. Braga VM, Betson M, Li X, Lamarche-Vane N. Activation of the small GTPase Rac is sufficient to disrupt cadherin-dependent cell-cell adhesion in normal human keratinocytes. Mol Biol Cell (2000) 11:3703–21. doi:10.1091/mbc.11.11.3703
152. Gumbiner B, Stevenson B, Grimaldi A. The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J Cell Biol (1988) 107:1575–87. doi:10.1083/jcb.107.4.1575
153. Vasioukhin V, Bauer C, Yin M, Fuchs E. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell (2000) 100:209–19. doi:10.1016/S0092-8674(00)81559-7
154. Hatzfeld M, Keil R, Magin TM. Desmosomes and intermediate filaments: their consequences for tissue mechanics. Cold Spring Harb Perspect Biol (2017) 9(6):a029157. doi:10.1101/cshperspect.a029157
155. Ammoun S, Lindholm D, Wootz H, Akerman KE, Kukkonen JP. G-protein-coupled OX1 orexin/hcrtr-1 hypocretin receptors induce caspase-dependent and -independent cell death through p38 mitogen-/stress-activated protein kinase. J Biol Chem (2006) 281:834–42. doi:10.1074/jbc.M508603200
156. Salvador JM, Mittelstadt PR, Guszczynski T, Copeland TD, Yamaguchi H, Appella E, et al. Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat Immunol (2005) 6:390–5. doi:10.1038/ni1177
157. Javanbakht MH, Djalali M, Daneshpazhooh M, Zarei M, Eshraghian MR, Derakhshanian H, et al. Evaluation of antioxidant enzyme activity and antioxidant capacity in patients with newly diagnosed pemphigus vulgaris. Clin Exp Dermatol (2015) 40:313–7. doi:10.1111/ced.12489
158. Shah AA, Dey-Rao R, Seiffert-Sinha K, Sinha AA. Increased oxidative stress in pemphigus vulgaris is related to disease activity and HLA-association. Autoimmunity (2016) 49:248–57. doi:10.3109/08916934.2016.1145675
159. Shah AA, Sinha AA. Oxidative stress and autoimmune skin disease. Eur J Dermatol (2013) 23:5–13. doi:10.1684/ejd.2012.1884
160. Yesilova Y, Ucmak D, Selek S, Dertlioglu SB, Sula B, Bozkus F, et al. Oxidative stress index may play a key role in patients with pemphigus vulgaris. J Eur Acad Dermatol Venereol (2013) 27:465–7. doi:10.1111/j.1468-3083.2012.04463.x
161. Naziroglu M, Kokcam I, Simsek H, Karakilcik AZ. Lipid peroxidation and antioxidants in plasma and red blood cells from patients with pemphigus vulgaris. J Basic Clin Physiol Pharmacol (2003) 14:31–42. doi:10.1515/JBCPP.2003.14.1.31
162. Zeller KS, Riaz A, Sarve H, Li J, Tengholm A, Johansson S. The role of mechanical force and ROS in integrin-dependent signals. PLoS One (2013) 8:e64897. doi:10.1371/journal.pone.0064897
163. Hancock JT, Desikan R, Neill SJ. Role of reactive oxygen species in cell signalling pathways. Biochem Soc Trans (2001) 29:345–50. doi:10.1042/bst0290345
164. Aslan M, Ozben T. Oxidants in receptor tyrosine kinase signal transduction pathways. Antioxid Redox Signal (2003) 5:781–8. doi:10.1089/152308603770380089
165. Dalle-Donne I, Rossi R, Milzani A, Di Simplicio P, Colombo R. The actin cytoskeleton response to oxidants: from small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic Biol Med (2001) 31:1624–32. doi:10.1016/S0891-5849(01)00749-3
166. Chiarugi P. Reactive oxygen species as mediators of cell adhesion. Ital J Biochem (2003) 52:28–32.
167. Huot J, Houle F, Marceau F, Landry J. Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circ Res (1997) 80:383–92. doi:10.1161/01.RES.80.3.383
168. Grando SA. The mitochondrion is a common target of disease pathophysiology in pemphigus and pemphigoid. Exp Dermatol (2015) 24:655–6. doi:10.1111/exd.12772
169. Chen Y, Chernyavsky A, Webber RJ, Grando SA, Wang PH. Critical role of the neonatal Fc receptor (FcRn) in the pathogenic action of antimitochondrial autoantibodies synergizing with anti-desmoglein autoantibodies in pemphigus vulgaris. J Biol Chem (2015) 290:23826–37. doi:10.1074/jbc.M115.668061
170. Chernyavsky A, Chen Y, Wang PH, Grando SA. Pemphigus vulgaris antibodies target the mitochondrial nicotinic acetylcholine receptors that protect keratinocytes from apoptolysis. Int Immunopharmacol (2015) 29:76–80. doi:10.1016/j.intimp.2015.04.046
171. Kalantari-Dehaghi M, Chen Y, Deng W, Chernyavsky A, Marchenko S, Wang PH, et al. Mechanisms of mitochondrial damage in keratinocytes by pemphigus vulgaris antibodies. J Biol Chem (2013) 288:16916–25. doi:10.1074/jbc.M113.472100
172. Saleh MA, Salem H, El Azizy H. Autoantibodies other than anti-desmogleins in pemphigus vulgaris patients. Indian J Dermatol (2017) 62:47–51. doi:10.4103/0019-5154.198032
173. Kaur R, Liu X, Gjoerup O, Zhang A, Yuan X, Balk SP, et al. Activation of p21-activated kinase 6 by MAP kinase kinase 6 and p38 MAP kinase. J Biol Chem (2005) 280:3323–30. doi:10.1074/jbc.M406701200
174. Vergarajauregui S, San Miguel A, Puertollano R. Activation of p38 mitogen-activated protein kinase promotes epidermal growth factor receptor internalization. Traffic (2006) 7:686–98. doi:10.1111/j.1600-0854.2006.00420.x
175. Mittelstadt PR, Salvador JM, Fornace AJ Jr, Ashwell JD. Activating p38 MAPK: new tricks for an old kinase. Cell Cycle (2005) 4:1189–92. doi:10.4161/cc.4.9.2043
176. Chruscinski AJ, Singh H, Chan SM, Utz PJ. Broad-scale phosphoprotein profiling of beta adrenergic receptor (beta-AR) signaling reveals novel phosphorylation and dephosphorylation events. PLoS One (2013) 8:e82164. doi:10.1371/journal.pone.0082164
Keywords: pemphigus vulgaris, autoantibodies, signaling pathways, p38MAPK, calcium, epidermal growth factor receptor, Rho GTPases
Citation: Sajda T and Sinha AA (2018) Autoantibody Signaling in Pemphigus Vulgaris: Development of an Integrated Model. Front. Immunol. 9:692. doi: 10.3389/fimmu.2018.00692
Received: 01 February 2018; Accepted: 21 March 2018;
Published: 19 April 2018
Edited by:
Ralf J. Ludwig, Universität zu Lübeck, GermanyReviewed by:
Robert Gniadecki, University of Alberta, CanadaSergei Grando, University of California, Irvine, United States
Copyright: © 2018 Sajda and Sinha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Animesh A. Sinha, aasinha@buffalo.edu