 Gaurang Jhala1,2
Gaurang Jhala1,2 Claudia Selck1†
Claudia Selck1† Jonathan Chee3,4
Jonathan Chee3,4 Chun-Ting J. Kwong1
Chun-Ting J. Kwong1 Evan G. Pappas1
Evan G. Pappas1 Helen E. Thomas1,2
Helen E. Thomas1,2 Thomas W.H. Kay1,2*‡
Thomas W.H. Kay1,2*‡ Balasubramanian Krishnamurthy1,2‡
Balasubramanian Krishnamurthy1,2‡- 1St. Vincent’s Institute, Fitzroy, VIC, Australia
- 2Department of Medicine, The University of Melbourne, St Vincent’s Hospital, Fitzroy, VIC, Australia
- 3National Centre for Asbestos Related Diseases, Institute of Respiratory Health, University of Western Australia, Perth, WA, Australia
- 4School of Biomedical Sciences, University of Western Australia, Perth, WA, Australia
T-cell responses to insulin and its precursor proinsulin are central to islet autoimmunity in humans and non-obese diabetic (NOD) mice that spontaneously develop autoimmune diabetes. Mice have two proinsulin genes proinsulin -1 and 2 that are differentially expressed, with predominant proinsulin-2 expression in the thymus and proinsulin-1 in islet beta-cells. In contrast to proinsulin-2, proinsulin-1 knockout NOD mice are protected from autoimmune diabetes. This indicates that proinsulin-1 epitopes in beta-cells maybe preferentially targeted by autoreactive T cells. To study the contribution of proinsulin-1 reactive T cells in autoimmune diabetes, we generated transgenic NOD mice with tetracycline-regulated expression of proinsulin-1 in antigen presenting cells (TIP-1 mice) with an aim to induce immune tolerance. TIP-1 mice displayed a significantly reduced incidence of spontaneous diabetes, which was associated with reduced severity of insulitis and insulin autoantibody development. Antigen experienced proinsulin specific T cells were significantly reduced in in TIP-1 mice indicating immune tolerance. Moreover, T cells from TIP-1 mice expressing proinsulin-1 transferred diabetes at a significantly reduced frequency. However, proinsulin-1 expression in APCs had minimal impact on the immune responses to the downstream antigen islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) and did not prevent diabetes in NOD 8.3 mice with a pre-existing repertoire of IGRP reactive T cells. Thus, boosting immune tolerance to proinsulin-1 partially prevents islet-autoimmunity. This study further extends the previously established role of proinsulin-1 epitopes in autoimmune diabetes in NOD mice.
Introduction
Recognition of proinsulin by the immune system is a major determinant in the pathogenesis of autoimmune diabetes in both humans and non-obese diabetic (NOD) mice (1, 2). A polymorphic variable number of tandem repeats (VNTR) located in the promoter region of the insulin locus controls the transcription level of the Ins gene and is strongly associated with susceptibility to type 1 diabetes (T1D) in humans (3–5). Mice do not have a VNTR upstream of the insulin locus; however, they have two Insulin genes, Ins1 and Ins2 encoding proteins that are highly homologous with 92% identity at the amino acid level. Proinsulin 1 and 2 proteins have identical A chains but differ by two amino acids in the B chain, three amino acids in the connecting peptide (C-peptide) and six amino acids in the leader peptide (6). The two proinsulin isoforms are differentially expressed with proinsulin 1 (PIns1) predominantly expressed in the pancreatic beta-cells and proinsulin 2 (PIns2) being the predominant isoform detected in the thymus (7–9).
Immune responses to native insulin peptides, in particular the B chain amino acids 9-23 (Ins B:9-23), are essential for autoimmune diabetes in NOD mice (10, 11). The two proinsulin isoforms differ by a single amino acid in the B: 9-23 region (PIns1: B9 proline, PIns2: B9 serine) and strong cross-reactivity of T cells for the Ins B: 9-23 epitope in both proinsulin molecules has also been reported (12). Despite the high degree of homology in the B:9-23 epitope and cross-reactivity of T cells for the Ins B: 9-23 epitope, a divergent immune response was observed when NOD mice were immunized with either Ins1 B:9-23 or Ins2 B:9-23 peptides, with Ins2 peptide conferring protection from diabetes onset, whereas Ins1 peptide did not prevent disease (13, 14). Further differences in cellular and humoral immune responses to both proinsulin isoforms have been highlighted by individual gene knockouts. NOD mice lacking Ins2 gene develop accelerated diabetes, ascribed to loss of central tolerance to insulin peptides; however, development of insulin autoantibodies (IAA) in Ins2 -/- mice suggests that immune responses against PIns1 epitopes are intact (15). In contrast, genetic deletion of Ins1 or replacement of murine Ins1 with human insulin gene (INS) in NOD mice provides significant protection from diabetes (16, 17). Protection from diabetes in NOD mice lacking Ins1 is likely due to the absence of cognate antigen in the target tissue, indicating that PIns1 peptides may be primarily targeted by insulin reactive T cells. Immunogenic epitopes in the PIns1 molecule have been reported (18), and T cells recognizing PIns1 amino acids 47-64 in the C-peptide region induce diabetes in NOD.SCID recipients (19). Thus, epitopes in PIns1 molecule may contribute to islet autoimmunity.
In contrast to NOD mice, non-autoimmune strains lacking Ins2 globally (20), or in medullary thymic epithelial cells (mTECs) did not develop pathological islet destruction however, when C57Bl/6 mice lacking Ins2 in mTECs were crossed to Ins1 knockout mice, the progeny developed spontaneous autoimmune diabetes within 3 weeks after birth (21). These studies suggest that thymic expression of PIns1 may add to the effect of PIns2 in eliminating insulin-specific autoreactive T cells. Constitutive or temporal expression of PIns2 in APCs induces recessive tolerance to PIns2 as it provides lasting protection from autoimmune diabetes in NOD mice (22). These mice were also thought to be tolerant to PIns1 epitopes because of cross-reactivity of the T cells to the conserved Ins B: 9-23 epitope. However, the role of PIns1 specific immune responses in pathogenesis of islet autoimmunity in NOD mice remains unclear, given the differential immune response observed upon immunization with Ins1 B:9-23 or Ins2 B:9-23 peptide. To resolve this, we investigated the impact of induced PIns1 expression in APCs on the development of antigen-specific T cells as well as insulitis and diabetes in NOD mice.
Materials and Methods
TetO-Ins1 Mice
To generate the TetO-Ins1 construct, a 411 bp cDNA fragment spanning the coding region of murine PIns1 was amplified by PCR using NOD pancreatic islet cDNA as a template and cloned into HindIII and EcoRV sites of the pTRE-tight plasmid (Clontech). A 1100 bp transgene cassette comprising of the TetO-minimal CMV promoter, followed by the PIns1 gene and a polyA signal was excised between Xho I sites and purified for injection into fertilized NOD/Lt ova using standard techniques. Founders and transgene positive offspring were screened by PCR using primers spanning the PIns1 gene (5’-TTAAGATATCTTCATTCATTATAGAACTC -3’) and the tetO-CMV promoter (5’-TCAGTGATAGAGAACGTATGTCG -3’).
Other Mice
NOD/Lt mice were bred and housed at the bioresources center St. Vincent’s Hospital, Fitzroy. The NOD-IEα-tTA mice that drive the expression of tetracycline transactivator (tTA) under the control of MHC class II IEα promoter have been previously described (23) and were obtained from Prof. C. Benoist and Prof. D. Mathis (Dept of Pathology, Harvard Medical School, Boston, Massachusetts, USA). Generation of NOD8.3 mice, which express the TCRαβ rearrangements of the H-2Kd-restricted, β cell-reactive, CD8+ T cell clone NY8.3, was previously described in detail (24). TIP-1/8.3 mice were generated by crossing NOD-IEα-tTA-TetO-Ins1double transgenic TIP-1 mice with TCR transgenic NOD8.3 mice. All mice were bred, maintained and used under specific pathogen free conditions at St Vincent’s Institute (Melbourne, Australia). All experimental procedures followed the guidelines approved by the institutional animal ethics committee.
Doxycycline Treatment
Untreated TIP-1 mice constitutively express proinsulin-1 in antigen presenting cells (APCs). To turn-off proinsulin-1 expression, doxycycline hyclate (Dox) (Sigma-Aldrich) was administered via drinking water at concentration of 2mg/ml. Water bottles were changed thrice weekly.
RT-PCR
For total RNA extraction, whole spleen and thymus were harvested in cold Phosphate Buffered Saline (PBS). Tissue homogenates were prepared in RNA lysis buffer RA1 (Macherey-Nagel) from a 15mg slice of tissue using a tissue homogenizer. RNA was isolated using Nucleospin RNA II-isolate kits (Macherey-Nagel), and first strand cDNA was generated from 500ng RNA using High Capacity cDNA Reverse Transcription kits (Applied Biosystem) according to the manufacturers’ instructions. cDNA was diluted (1:20) and Real-time PCR analysis was performed using Rotor-Gene-RG-3000 cycler (Corbett Research, Sydney, Australia). Taqman gene expression primers murine insulin 1 (Ins1; Mm01950294_s1), murine β-actin (Actb; Mm00607939_s1) and murine Glyceraldehyde 3-phosphate dehydrogenase (Gapdh; Mm99999915_g1) were purchased from Applied Biosystems. To determine relative expression, Ct values of Insulin were subtracted from Ct values of reference genes for each sample and the difference (dCt) was plotted to determine the abundance of the gene of interest.
Histology and Immunohistochemistry
For immunohistochemistry, pancreata were snap-frozen in optimal cutting temperature compound (OCT Compound; Sakura Finetek, Torrance, CA) and stored at −80°C. For histological analysis 5-µm frozen sections of pancreas were prepared from three levels (200 µm apart), acetone fixed, stained with guinea pig anti-insulin followed by horseradish peroxidase–conjugated anti–guinea pig Ig (Dako Cytomation, Carpenteria, CA) and counterstained with hematoxylin. Insulitis was graded using the following scale: 0 = no infiltrate, 1 = peri-islet infiltrate, 2 = extensive (>50%) peri-islet infiltrate, 3 = intraislet infiltrate, and 4 = extensive intraislet infiltrate (>80%) or total β-cell loss. The percentage of islets with each grade per pancreas was calculated by addition of the grades for the three sections. Individual insulitis scoring for each mouse was performed as previously described (22).
Incidence of Diabetes and Insulitis
Diabetes onset was monitored by weekly measurement of urine glucose levels using Diastix (Bayer Diagnostics). Blood glucose levels were measured in mice with glycosuria using Advantage II Glucose strips (Roche). Animals displaying two consecutive blood glucose measurements of ≥ 15mmol/L were considered diabetic. For adoptive transfer of diabetes, 2 x107 splenocytes from 13-17 week old pre-diabetic TIP-1 mice or control NOD mice were transferred (i.v.) into 9-12week old NOD Rag-/- recipients and diabetes development was monitored as above.
Flow Cytometry
Antibodies used were anti-CD4 (RM4-5) conjugated to PerCpCy5.5, anti-CD3 (145-2C11) conjugated to FITC or anti-CD3 (500 A2) V500, anti-CD44 (1M7) conjugated to AlexaFlour700 (all BD Biosciences), anti-CD11c (N418), anti-B220 (RA3-6B2), anti-CD11b (M1/70), anti-F4/80 (BM8) conjugated to eFlour450 and anti-FoxP3 (FJK-16S) conjugated to APC (all eBiosciences), anti-CD8a (5H10) conjugated to Pacific Orange (Invitrogen) or anti-CD8a (53-6.7) conjugated to PE-Cy7, anti-CD62L (MEL-14) conjugated to APC-Cy7 (BD Biosciences). FoxP3 was stained intracellularly using FoxP3/Transcription Factor Fixation/Permeabilization kit (eBiosciences). Data were collected on an LSR Fortessa flow-cytometer (BD) and analyzed using FlowJo (Treestar) software.
Tetramer and Magnetic Bead-Based Enrichment
The tetramer and magnetic bead-based enrichment method was previously described (25). I-Ag7 tetramers were obtained from NIH tetramer core facility (Emory University, Georgia, USA), Kd-tetramers were obtained from ImmunoID (Parkville, Victoria, Australia). To enrich insulin-specific CD4+ T cells single cell suspensions from peripheral lymphoid organs (PLO), (pooled spleen and non-draining lymph nodes), were stained with phycoerythrin (PE)-conjugated I-Ag7-INSB10-23 (HLVERLYLVCGGEG) tetramer for 1 hour at room temperature. The Ins B10-23 peptide in the I-Ag7-INSB10-23 tetramer has been mutated (Glutamic acid to Glycine (E-G) at position 20 and Arginine to Glycine (R-G) at position 21) to improve its binding to the I-Ag7 molecule, which allows for better detection of insulin-specific CD4+ T cells (26). Insulin-specific CD8+ T cells were enriched from pooled PLO by staining the cell suspensions with APC-conjugated H-2Kd- INSB15-23 (LYLVCGGEG) tetramer for 1 hour on ice. Hen Egg Lysozyme I-Ag7-HEL (AMKRHGLDNYRGYSL) tetramer or H-2Kd-TUM (KYQAVTTTL) were used as controls. Cells were then washed and stained with anti-PE or anti-APC microbeads (Miltenyi Biotec) followed by magnetic separation using an AutoMACSpro (Miltenyi Biotec) according to manufacturer’s instructions. IGRP206-214 specific CD8+ T cells (H2-Kd, VYLKTNVFL) were stained and enriched as previously described (27). The separated fractions were stained and analyzed by flow cytometry. Gating strategy for tetramer enrichment was as follows: single cells were gated on forward and side scatter, and dead cells excluded using propidium iodide. From the live cell population, CD11c-CD11b-B220-F4/80-CD3+ cells were gated as the T cell population for analysis. Further selection of CD4+ T cells or CD8+ T cells was followed by analysis of the insulin or IGRP tetramer positive population respectively.
Insulin Autoantibody (IAA) Assay
A non-competitive IAA assay was performed in a 96 well ELISA format as previously described (28, 29). Briefly, an ELISA plate (Costar) was coated with or without human insulin (10 µg/ml, Actrapid, Novo Nordisk) overnight at 4°C. Wells were blocked with PBS containing 2% BSA for 2 hours and room-temperature and then probed with sera from 12-15 weeks old TIP mice, NOD or C57BL/6 mice (1:10 dilution) for 2 hours. Wells were washed 4 times and a biotinylated anti-mouse IgG1 (AbCam, 1:10000 dilution) antibody was added for 30 minutes. After washing, horse-radish-peroxidase conjugated streptavidin (BioLegend) was added for 15 minutes. The plate was washed five times, TMB substrate solution (BioLegend) was added and absorbance was measured at 450 nm using a Polarstar (BMG labtech) microplate reader. Each sample was run in duplicate and absorbance (450 nm) of test sample without plate bound insulin was subtracted from absorbance of test sample with plate bound insulin to calculate the actual absorbance value for each sample.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 8 Software (GraphPad, San Diego, CA, USA). Pooled data are shown as dot-plots with individual mice and the mean ± SEM. Data were tested for normal distribution using D’Agostino-Pearson omnibus normality test or Shapiro-Wilks test. Comparisons between two groups were performed using two-tailed unpaired student t-tests. Multiple comparisons were performed using One-way ANOVA with Sidak’s post-hoc test. Survival curves were compared using Log-Rank (Mantel-Cox) test. Statistical significance was defined as P < 0.05.
Results
Conditional Expression of Proinsulin-1 in NOD Mice
To test whether inducing immune tolerance to proinsulin-1 (PIns1) influenced autoimmune diabetes we generated transgenic NOD mice to facilitate conditional expression of PIns1 in the antigen presenting cells (APCs). Reporter NOD mice expressing PIns1 under the control of the tetracycline-responsive CMV promoter (TetO-Ins1 mice) were bred with previously described driver NOD mice expressing TetR-VP16 tetracycline transactivator protein (tTA) under the control of IEα-MHC-II promoter referred to as TA-NOD mice (23). Bi-transgenic progeny referred to as TIP-1 (Tet Inducible PIns1) mice (Figure 1A) express Pins1 in the APCs, which can be turned-off upon doxycycline (Dox) treatment. Analysis of PIns1 expression in TIP-1 mice revealed that PIns1 transgene was expressed in the thymus and spleen as measured by RT-PCR (Figure 1B). After one week of Dox treatment, PIns1 expression dropped to baseline levels (Figure S1). Thus, PIns1 expression in TIP-1 mice was conditional, and tightly regulated.
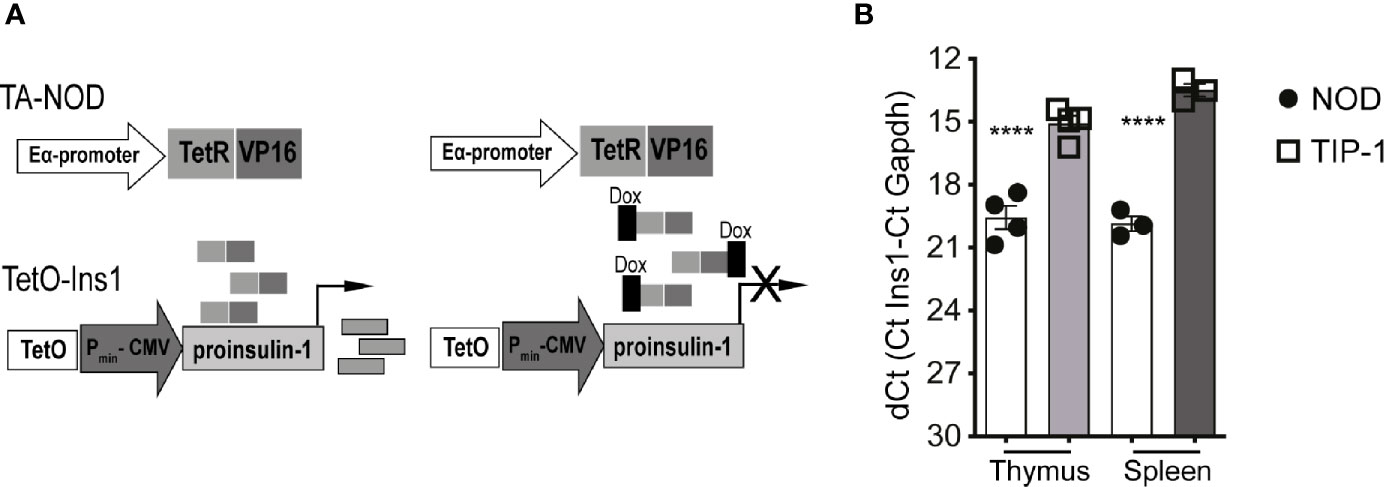
Figure 1 Conditional proinsulin 1 expression in TIP-1 mice (A) Scheme of generation of tetracycline regulated NOD.IEα-tTA (TA-NOD) and tetO-Ins1 dual transgenic mice referred to herein as TIP-1 mice. TA-NOD mice were crossed with tetO-Ins1 mice. Bi-transgenic animals constitutively express PIns1 in APCs (B) Quantitative RT-PCR was performed using Taqman probes for Ins1 and Gapdh in thymic and splenic lysates of WT-NOD mice TIP-1 mice. Data represent dCT values (Mean ± SEM) from 2-3 independent experiments run in duplicate for each probe. ****P < 0.0001. Data compared using One-way ANOVA with Sidak’s multiple comparisons test.
TIP-1 Mice Have Reduced Insulitis and Insulin Autoantibody Expression
We recently reported that constitutive or temporal expression of PIns2 (PIns2) in the APCs limited to the perinatal period prevented insulitis and diabetes in NOD mice (22). To test whether PIns1 expression in the APCs influenced the progression of islet autoimmunity, we examined the immune infiltrate (insulitis) in the pancreata of TIP-1 mice expressing PIns1 continuously. At 12-14 weeks of age, insulitis was significantly reduced in TIP-1 mice expressing PIns1 compared to age matched NOD mice or TIP-1 mice fed dox to suppress PIns1 expression (Figures 2A–C). Analysis of pancreas histology from TIP-1 mice at 20-25 weeks of age revealed that approximately 50% of the islets examined were free of insulitis, whereas more than 80% of the islets examined from non-transgenic littermates were infiltrated (Figures 2D, E and Table S1), indicating that PIns1 expression in the APCs decreased but did not completely abolish development of insulitis, which progressed over time. Production of insulin autoantibodies (IAA) indicates spontaneous anti-insulin autoimmunity and IAA are frequently detected prior to diabetes onset in both humans and NOD mice (30, 31). We examined whether induced PIns1 expression in TIP-1 mice influenced B cell mediated humoral responses against insulin by measuring IAA in TIP-1 mice. IAA was significantly reduced in 12-15 weeks old TIP-1 mice as compared to age matched non-transgenic NOD mice (Figure 2F). Previously described PIns2 tolerant NOD-PI mice that are protected from diabetes and non-autoimmune prone C57BL/6 mice were used to set the baseline. Collectively, these results indicate that immune tolerance to PIns1 influenced progression of insulitis and reduced the development of IAA.
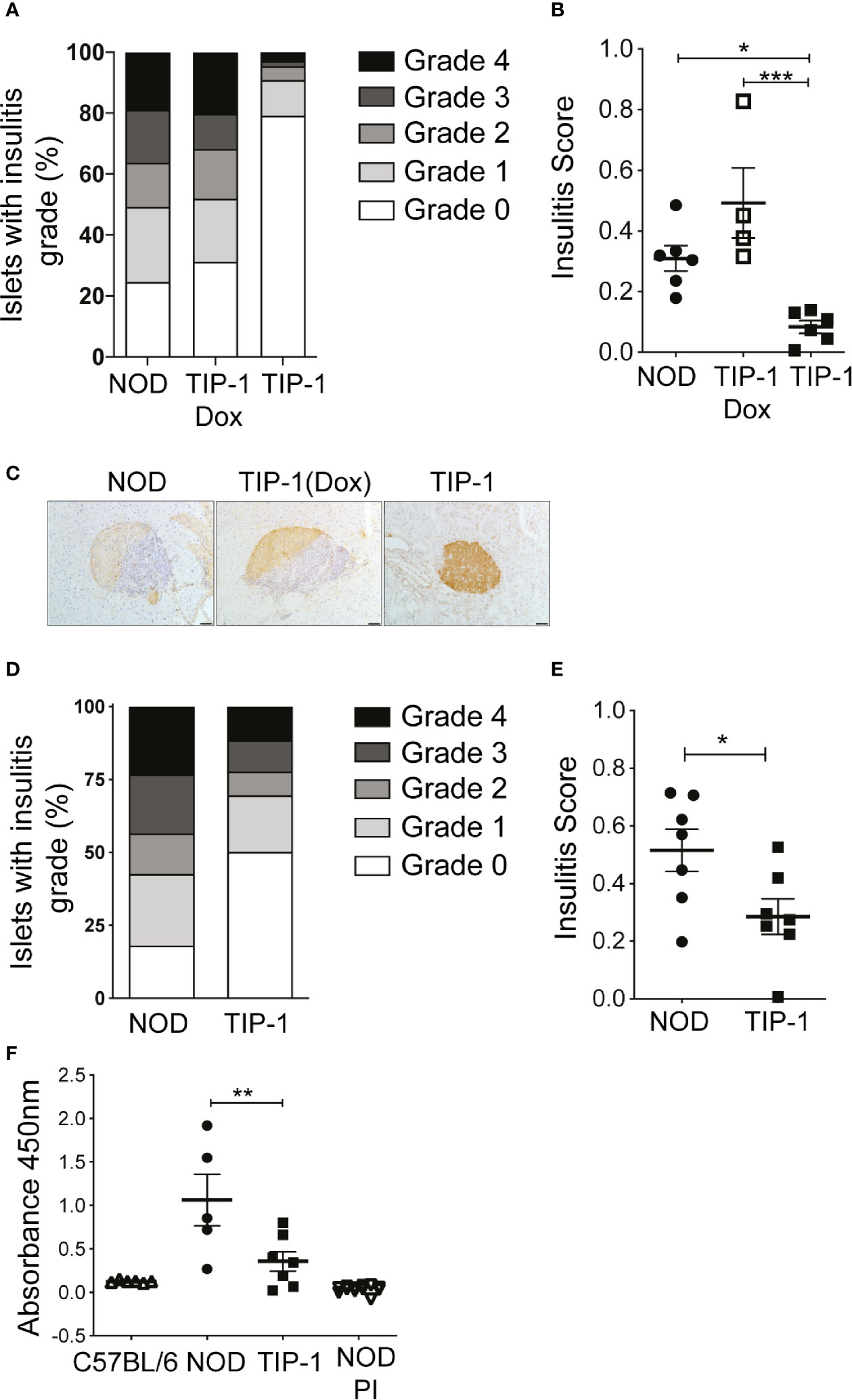
Figure 2 Insulitis and insulin autoantibodies (IAA) in TIP-1 mice (A, B) Histological grading and individual insulitis scores in NOD mice, Doxycycline treated and untreated TIP-1 mice at 12-14 weeks of age. (C) Representative images of islet histology from 12-14 week old NOD, TIP-1 and Dox treated TIP-1 mice, 200x magnification, scale bar 50 μm. (D, E) Histological grading and individual insulitis scores at 20-25 weeks of age in NOD mice and TIP-1 mice (n=4-7, > 60 islets scored per mouse). (F) Sera from 12-16 weeks old C57BL/6 mice, NOD mice, TIP-1 and NOD-PI mice were tested for the presence of insulin autoantibodies (IAA) by ELISA assay. Absorbance values at 450nm are plotted. Each symbol in the scatter plot represents data from individual animals. Data plotted as Mean ± SEM, ***P < 0.001,**P < 0.01, *P < 0.05. Data compared using One-way ANOVA with Sidak’s multiple comparisons test (B, F) and 2-tailed unpaired t-test (E).
PIns1 Overexpression Partially Suppresses Spontaneous Diabetes in NOD Mice
Reduced insulitis and IAA suggest that diabetes development may be altered in TIP-1 mice. A cohort of female TIP-1 mice expressing PIns1 continuously and control NOD mice were observed for incidence of spontaneous diabetes. TIP-1 mice developed diabetes but at a significantly reduced incidence compared to non-transgenic control NOD mice. By 300 days of age 40% of TIP-1 mice and 65% of the control mice developed diabetes (Figure 3A). In addition, we investigated whether PIns1 expression in TIP-1 mice influenced the pathogenic potential of effector T cells. Splenocytes from 15-18 weeks old TIP-1 mice with ongoing expression of PIns1 and age matched control NOD mice were transferred into NOD.Rag1 -/- recipients. All recipient mice receiving control splenocytes developed diabetes between 50-70 days post-transfer, whereas only 2 out of 6 (33%) animals that received splenocytes from TIP-1 mice developed diabetes 70-90 days post-transfer (Figure 3B). Taken together these results suggest that overexpression of PIns1 in APCs is able to partially dampen immune responses against insulin and reduce diabetes incidence in NOD mice.
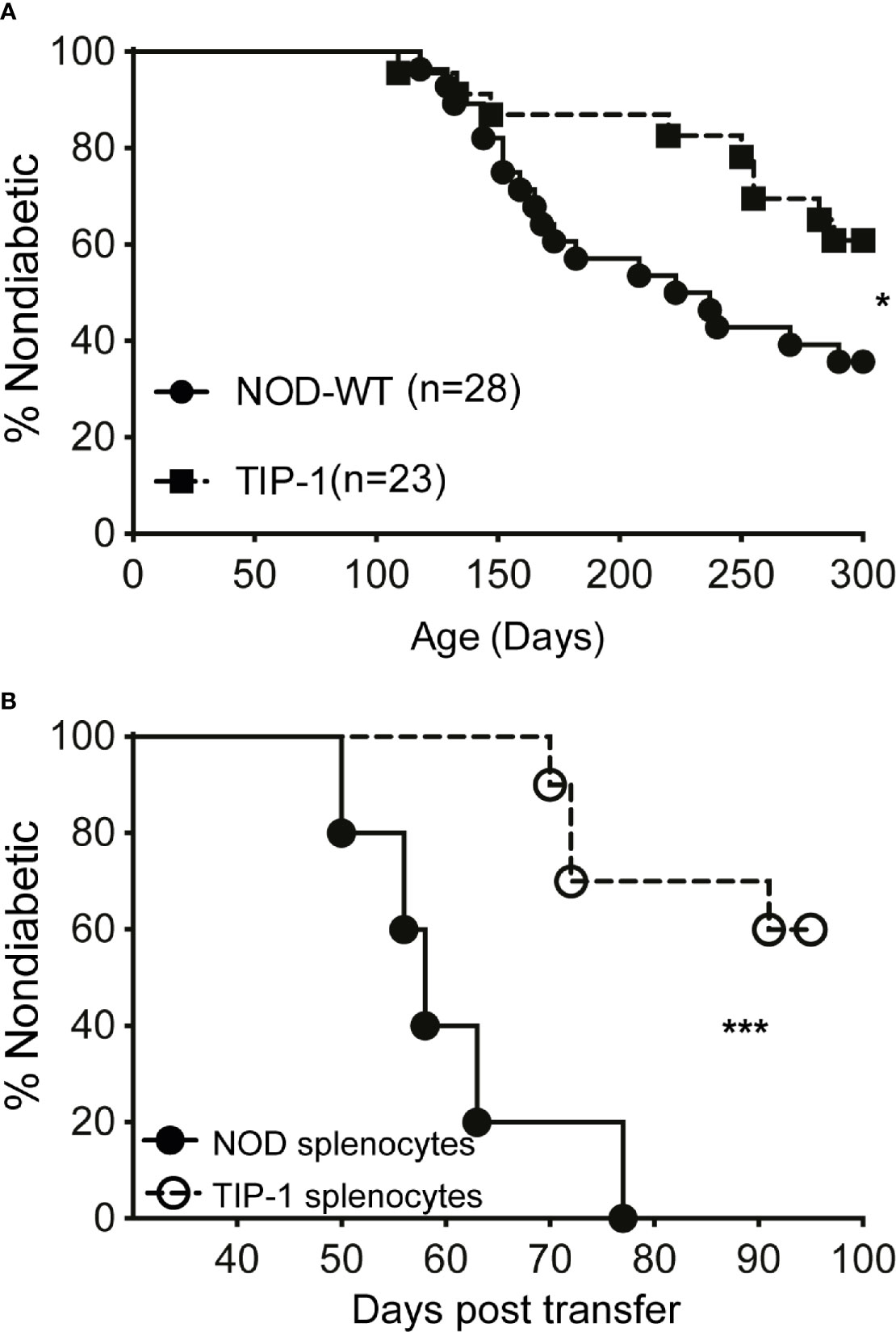
Figure 3 Spontaneous diabetes incidence in TIP-1 mice (A) Incidence of spontaneous diabetes in female TIP-1 mice and non-transgenic littermates until 300 days of age. Numbers in parentheses indicate the number of mice analyzed. (B) Incidence of diabetes following transfer of splenocytes (2 x 107 cells/recipient) from 13-17 weeks old non-diabetic TIP-1 mice or NOD mice into 8-9 weeks old NOD.Rag1-/- recipients (n > 5 each). *P < 0.05; ***P < 0.0001. Survival curves were compared using log-rank (Mantel-Cox) test.
Proinsulin-Specific Tolerance in TIP-1 Mice
The partial protection from insulitis and diabetes in TIP-1 mice expressing PIns1 in the APCs could be due to immune tolerance to PIns1 epitopes. To demonstrate tolerance to PIns1, we enumerated the frequency of Insulin B 9-23 reactive CD4+ T cells and Insulin B 15-23 reactive CD8+ T cells in the peripheral lymphoid organs (PLO) (pooled spleen and non-draining lymph nodes) of 20-25 weeks old non-diabetic TIP-1 mice and age matched control mice using respective I-A (g7) and Kd tetramers. There was a significant reduction in the absolute number of insulin B:9-23 specific CD4+ T cells binding to insulin B:10-23/I-A (g7) tetramer (26) and the antigen-experienced CD44hi subset of insulin B:9-23 specific CD4+ T cells in TIP-1 mice (Figures 4A–D). The absolute number of CD8+T cells recognizing insulin B:15-23 epitope (32) as well as the number of antigen-experienced CD44hi subset of insulin B:15-23 specific CD8+ T cells were comparable in both TIP-1 mice and controls (Figures 4E–H). While the significant reduction of insulin-specific CD4+ and CD8+ T cells in TIP-1 is suggestive of deletional tolerance, it is possible that transgenic antigen expression in APCs may induce regulatory T cells (Tregs) that confer dominant tolerance and prevent diabetes in TIP-1 mice. We examined the expression of Foxp3 on insulin B:9-23 specific CD4+ T cells in PLO of TIP-1 mice and non-transgenic controls and did not observe any significant differences (Figure S2A, B), In addition we examined the frequency of Foxp3+ CD4+ Tregs in the thymus and pancreatic lymph node (PLN). The proportion of Tregs was similar in both TIP-1 and control mice (Figure S2C, D).Taken together our data indicate that ectopic PIns1 expression induces deletion of cognate CD4+ T cells, but does not induce antigen specific Tregs. The few remaining insulin reactive CD4+T cells could not be activated by the expressed antigen, whereas the low-affinity insulin B:15-23 reactive CD8+ T cells (33) are not influenced by transgenic PIns1 expression.
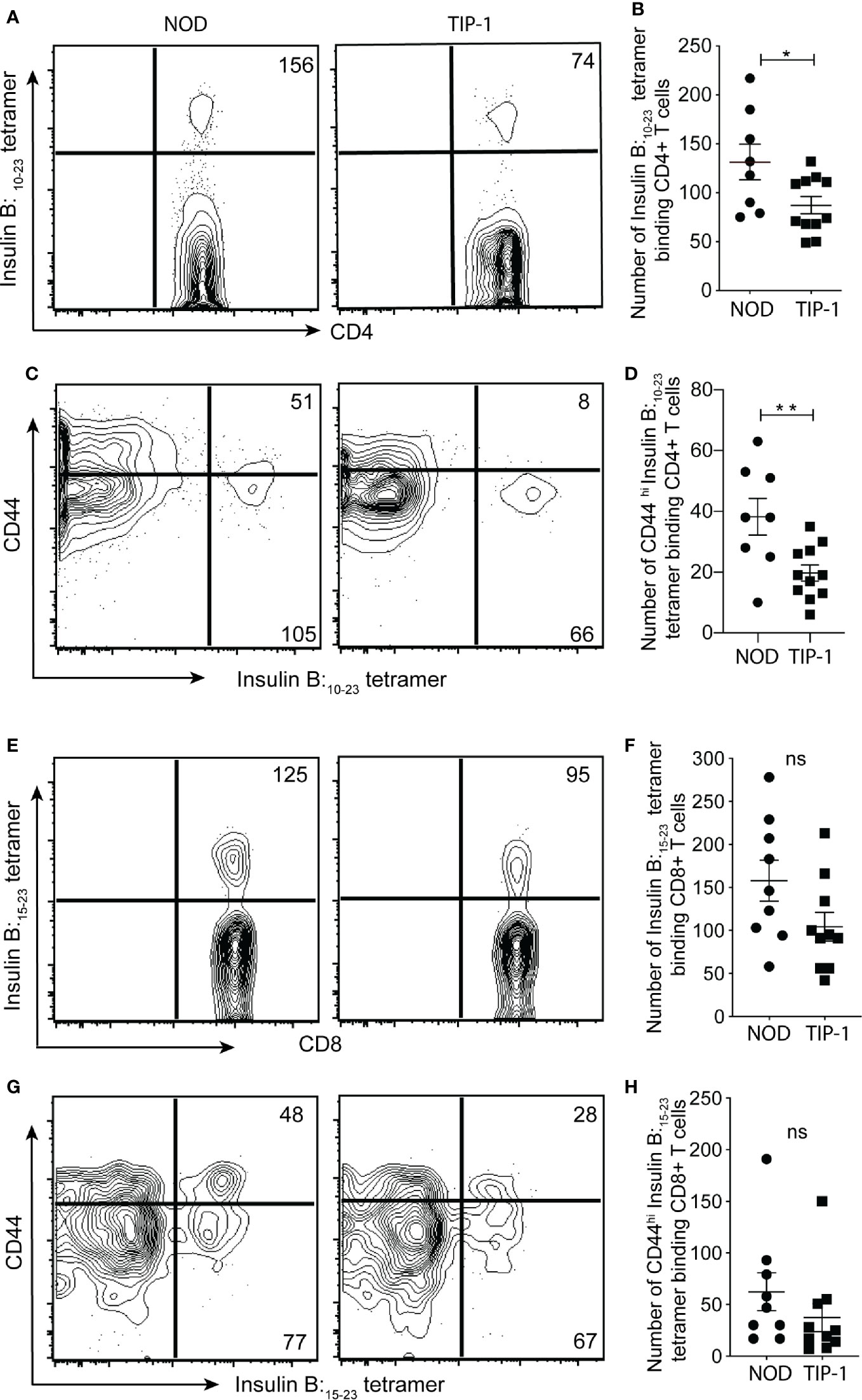
Figure 4 Immune tolerance to insulin specific T cells in TIP-1 mice Insulin B:10-23-specific CD4+ T cells or Insulin B:15-23-specific CD8+ T cells were stained with respective tetramers and enriched from pooled peripheral lymphoid organs (PLO) of 20-25 weeks old TIP-1 mice and NOD mice using magnetic beads and enumerated by flow-cytometry. Representative FACS plots (A, C, E, G) and enumeration of insulin tetramer+ CD4+ T (B) cells, insulin tetramer+ CD8+ T cells (F), CD44hi Insulin tetramer + CD4+ T cells (D) and CD44hi Insulin tetramer + CD8+ T cells (H) in TIP-1 and NOD mice. Values in the FACS plots indicate absolute number of tetramer binding cells. Each symbol in the scatter plots (Mean ± SEM) represents data from an individual mouse. **P < 0.01, *P < 0.05, ns= not significant. Data compared using 2-tailed unpaired t-test.
Downstream Responses to IGRP Are Delayed in TIP-1 Mice
Previous work from our group has demonstrated that autoreactive responses to islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) are dependent upon immune response to PIns2 (1). To investigate if tolerance to PIns1 influenced the immune response to IGRP we examined the frequency of pathogenic IGRP 206-214 reactive CD8+ T cells in TIP-1 mice. The number of IGRP 206-214 specific CD8+ T cells was significantly reduced in 12-14 weeks old TIP-1 mice expressing PIns1 as compared to age matched controls. However, the frequency of IGRP 206-214 specific CD8+ T cells in TIP-1 mice expressing PIns1 did not differ from age-matched controls at 20-25 weeks of age (Figures 5A, B). This indicates that tolerance to PIns1 delays but does not prevent the spreading of immune responses to downstream antigen IGRP.
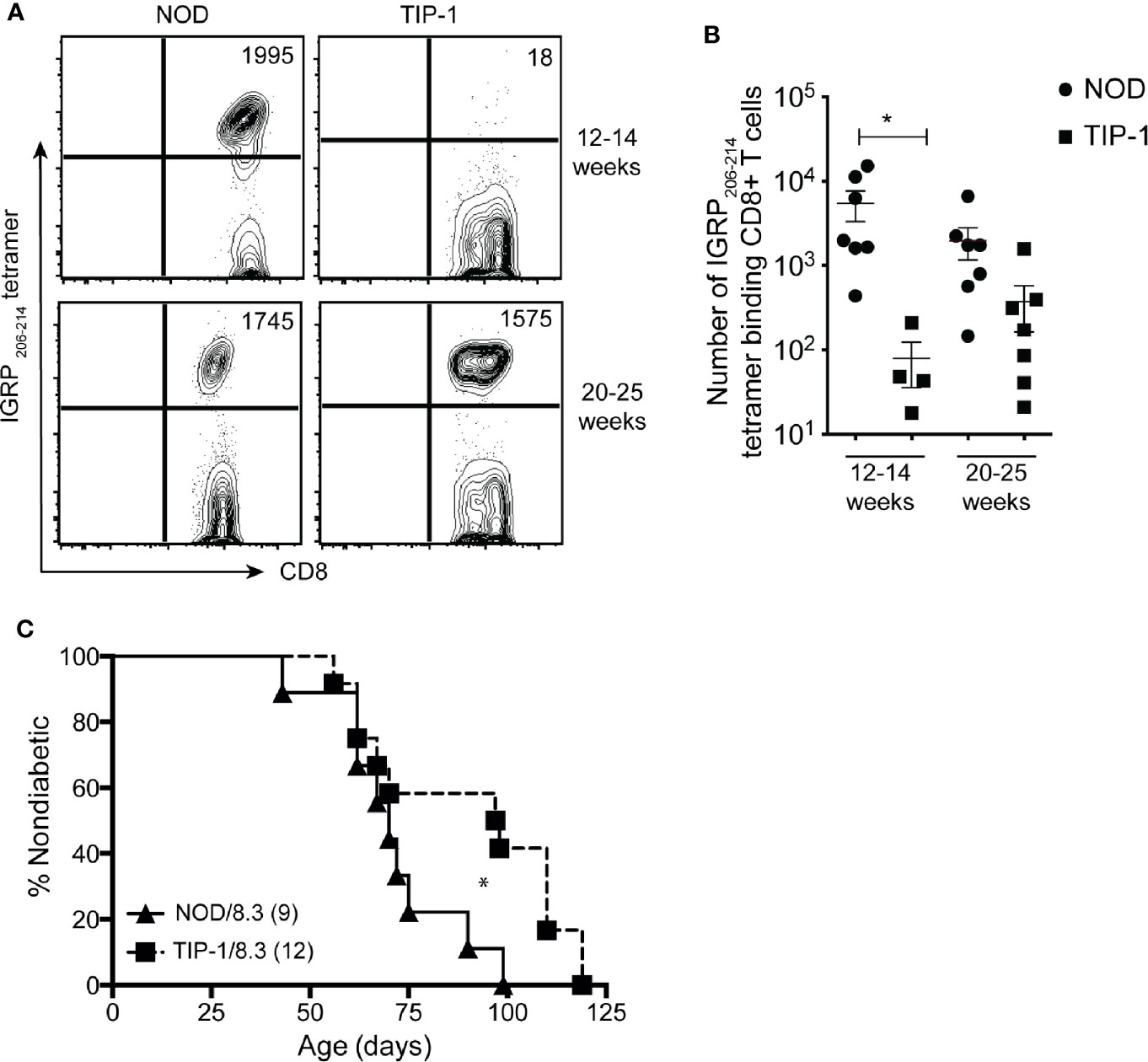
Figure 5 Enumeration of IGRP specific CD8+ T cells in TIP-1 mice IGRP 206-214-specific CD8+ T cells were stained with Kd- IGRP tetramer and enriched from pooled peripheral lymphoid organs (PLO) of 12-14 weeks and 20-25 weeks old NOD mice and TIP-1 mice. Representative FACS plots (A) and quantification (B) of absolute number of IGRP 206-214 tetramer + CD8+ T cells at indicated ages. Values in the FACS plots indicate absolute number of tetramer binding cells. Each symbol in the scatter plots (Mean ± SEM) represents data from an individual mouse. *P<0.05, data in (B) compared using one-way ANOVA with Sidak’s multiple comparisons test. (C) Incidence of spontaneous diabetes in female TIP-1/8.3 mice and NOD 8.3 littermates. Numbers in parentheses indicate the number of mice analyzed. *P < 0.05. Survival curves were compared using log-rank (Mantel-Cox) test.
Immune Response to Proinsulin-1 Is Not Required for Diabetes in NOD 8.3 Mice
Autoreactivity to PIns2 is required for diabetes development in NOD 8.3 mice that have a pre-existing repertoire of IGRP specific T cells (34). Since we observed reduced frequency of IGRP reactive CD8+ T cells in 12-14 weeks old TIP-1 mice, we wished to know if immune responses to PIns1 were necessary for diabetes development in NOD 8.3 mice. TIP-1 mice were crossed with NOD 8.3 mice to generate offspring that were TIP-1/NOD8.3 double transgenic or NOD 8.3 transgenic alone. TIP-1/8.3 mice developed diabetes with significantly delayed kinetics (median survival 97 days) compared to NOD8.3 mice (median survival 70 days) but all mice eventually developed disease (Figure 5C). The frequency of insulin specific T cells is very low even in NOD mice and with a skewed T cell repertoire in NOD 8.3 transgenic mice it is not possible to detect any insulin specific T cells. We were unable to detect insulin-specific T cells in TIP-1/8.3 mice (data not shown). Therefore, tolerance to PIns1 significantly delays but does not prevent diabetes development in NOD 8.3 mice.
Discussion
In this study we generated transgenic NOD mice to induce PIns1 expression in the APCs and examined the impact of antigen specific tolerance on autoimmune diabetes. The main findings of this study are 1) TIP-1 mice expressing PIns1 in the APCs show significantly reduced incidence of diabetes, which is associated with reduced insulitis and insulin autoantibody (IAA) expression. 2) Proinsulin specific CD4+ T cells are detectable in TIP-1 mice at a reduced frequency and are not activated. 3) Immune responses to downstream antigen IGRP are delayed but not absent in TIP-1 mice.
Given the high degree of homology between proinsulin 1 and 2 proteins, especially in the immunodominant insulin B chain epitope Ins B:9-23 we expected to achieve robust protection from diabetes onset in TIP-1 mice, similar to previously described proinsulin-2 tolerant NOD mice (35); however, the partial protection from insulitis and diabetes observed in TIP-1 mice points to the existence of distinct pathogenic peptide epitopes in the PIns2 protein that can precipitate autoimmunity in NOD mice. A previous study characterizing immunogenic epitopes in NOD mice reported existence of multiple epitopes on both PIns1 and PIns2 molecules recognized by CD4+T cells (18). Importantly, epitopes outside of the highly homologous Ins B:9-23 peptide were identified in the leader and A chain sequences of PIns2 molecule. Thus, it is likely that PIns2 reactive T-cells recognizing these unique epitopes may induce islet destruction and subsequent diabetes onset in TIP-1 mice.
Our data complement the previous observations that reported detection of PIns1 reactive T cells (18, 19) in NOD mice. While the previous studies did not directly demonstrate the role of PIns1-reactive T cells in spontaneous disease, the significant reduction in diabetes incidence in TIP-1 mice suggests that PIns1 specific T cells participate in autoimmune destruction of beta cells. On the other hand, development of IAA and diabetes in TIP-1 mice may be related to ongoing immune responses to PIns2 epitopes.
A drawback of our study is that we have analyzed a single transgenic founder line expressing PIns1 in the APCs. Varying levels of transgenic insulin expression in the thymus may influence the diabetes progression in NOD mice. PIns2 levels were 7-fold higher in the spleen (~140 pmol/L) as compared to thymus (~20pmol/L) in the partially protected Pins2 tolerant mice previously described by Jaeckel et al. (12), whereas in the recently described TIP mice with robust protection from autoimmune diabetes upon conditional PIns2 expression in APCs, the level of thymic Pins2 expression (100pmol/L) was 5-fold more compared to peripheral tissues(20pmol/L) (22). TIP-1 mice may have relatively reduced transgenic expression of PIns1 in the thymic APCs as compared to transgenic PIns2 expression in the previously described TIP mice, thus imparting incomplete protection from autoimmune diabetes. Chentoufi and Polychronakos previously reported that Ins2 is expressed at a level more than 3-fold higher than Ins1 in the thymus of NOD mice (9). In TIP-1 mice analyzed here, induction of PIns1 results in approximately 5-fold higher expression as compared to non-transgenic NOD mice or uninduced TIP-1 mice. Moreover, protection from insulitis in TIP-1 mice is associated with the expression of PIns1 transgene, as TIP-1 mice fed doxycycline to suppress PIns1 expression develop islet infiltration comparable to non-transgenic controls indicating that ectopic PIns1 expression in APCs influences anti-islet immunity.
Does the reduction in the incidence of spontaneous diabetes in TIP-1 mice correlate with deletion of PIns1 specific T cells? Insulin B:10-23 and Insulin B:15-23 specific tetramers used in our study are likely to detect both PIns1 and 2 reactive CD4+ and CD8 +T cells, due to the invariant nature of the Insulin B:9-23 peptide between the two isoforms. Immune responses to Insulin B:9-23 epitope are required for both priming and effector phase of islet autoimmunity in NOD mice. Moreover, Insulin B:9-23 primed CD4+ T cells are able to induce islet autoimmunity evidenced by IAA production (36). The significant reduction in absolute number of Insulin B:9-23 tetramer binding CD4+T cells, and the antigen-experienced subset of tetramer binding CD4+ T cells, coupled with reduced IAA production in TIP-1 mice is suggestive of antigen-specific tolerance. While Tregs are an important tolerance mechanism, we did not find any evidence to suggest that the partial protection from diabetes in TIP-1mice is due to antigen-specific Tregs. We are currently unable to conclude whether central or peripheral tolerance mechanisms regulate the insulin specific T cells in TIP-1 mice; however, future studies with ectopic antigen expression induced after the exit of antigen-specific T cells from the thymus may resolve this question.
Autoimmunity to insulin determines immune responses to other downstream antigens such as IGRP (1). IGRP 206-214 reactive CD8+ T cells were reduced in TIP-1 mice at 12-14 weeks; but ongoing tolerance to PIns1 did not prevent development of diabetes onset in TIP-1/8.3 mice. The precursor frequency of IGRP reactive CD8+ T cells is low in NOD mice (27), and the residual immune response to PIns2 in TIP-1 mice may be reduced as compared to control mice. The reduced CD4+ T cell help possibly accounts for the delayed expansion of IGRP specific T cells seen in TIP-1 mice. However, the residual immune response to PIns2 in TIP-1/8.3 mice with a pre-existing repertoire of IGRP specific T cells may be sufficient to help IGRP specific CD8+ T cells to mediate beta-cell destruction.
In summary, we find that immune tolerance to PIns1, whilst partly protective, is not sufficient to prevent spontaneous diabetes in NOD mice. Our data clarifies the role of PIns1 in the pathogenesis of autoimmune diabetes in NOD mice and extends the previously established role of PIns1 in autoimmune diabetes. The experimental model we have presented here, with its conditional gene-expression system, has the potential to delineate whether antigen-specific interventions can induce immune tolerance after islet autoimmunity is well established. Understanding this is important for development of strategies to induce antigen-specific tolerance clinically in people with stage 1 or 2 type 1 diabetes (37).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by Animal Ethics Committee, St Vincents Hospital, Melbourne.
Author Contributions
GJ performed experiments, analyzed data, and wrote the manuscript. CS, JC, C-TK, and EP performed experiments and analyzed data. HT, BK, and TK designed the study, analyzed data, and wrote the manuscript. BK and TK supervised the study. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the National Health and Medical Research Council of Australia (GNT1037321 and GNT1150425) and fellowship from Juvenile Diabetes Research Foundation (BK). The St Vincent’s Institute receives support from the Operational Infrastructure Support Scheme of the Government of Victoria.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank S. Fynch, T. Catterall, I. Voelcker, V. Moshovakis, E. Tonkin, H. Barlow, and A. Cornelisz (St Vincent’s Institute) for excellent technical support and animal husbandry.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.645817/full#supplementary-material
References
1. Krishnamurthy B, Dudek NL, McKenzie MD, Purcell AW, Brooks AG, Gellert S, et al. Responses against islet antigens in NOD mice are prevented by tolerance to proinsulin but not IGRP. J Clin Invest (2006) 116(12):3258–65. doi: 10.1172/JCI29602
2. Mannering SI, Pathiraja V, Kay TW. The case for an autoimmune aetiology of type 1 diabetes. Clin Exp Immunol (2016) 183(1):8–15. doi: 10.1111/cei.12699
3. Lucassen AM, Julier C, Beressi J-P, Boitard C, Froguel P, Lathrop M, et al. Susceptibility to insulin dependent diabetes mellitus maps to a 4.1 kb segment of DNA spanning the insulin gene and associated VNTR. Nat Genet (1993) 4(3):305–10. doi: 10.1038/ng0793-305
4. Pugliese A, Zeller M, Fernandez A Jr., Zalcberg LJ, Bartlett RJ, Ricordi C, et al. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat Genet (1997) 15(3):293–7. doi: 10.1038/ng0397-293
5. Vafiadis P, Bennett ST, Todd JA, Nadeau J, Grabs R, Goodyer CG, et al. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet (1997) 15(3):289–92. doi: 10.1038/ng0397-289
6. Wentworth BM, Schaefer IM, Villa-Komaroff L, Chirgwin JM. Characterization of the two nonallelic genes encoding mouse preproinsulin. J Mol Evol (1986) 23(4):305–12. doi: 10.1007/BF02100639
7. Deltour L, Leduque P, Blume N, Madsen O, Dubois P, Jami J, et al. Differential expression of the two nonallelic proinsulin genes in the developing mouse embryo. Proc Natl Acad Sci U S A (1993) 90(2):527–31. doi: 10.1073/pnas.90.2.527
8. Heath VL, Moore NC, Parnell SM, Mason DW. Intrathymic expression of genes involved in organ specific autoimmune disease. J Autoimmun (1998) 11(4):309–18. doi: 10.1006/jaut.1998.0210
9. Chentoufi AA, Polychronakos C. Insulin expression levels in the thymus modulate insulin-specific autoreactive T-cell tolerance: the mechanism by which the IDDM2 locus may predispose to diabetes. Diabetes (2002) 51(5):1383–90. doi: 10.2337/diabetes.51.5.1383
10. Wegmann DR, Norbury-Glaser M, Daniel D. Insulin-specific T cells are a predominant component of islet infiltrates in pre-diabetic NOD mice. Eur J Immunol (1994) 24(8):1853–7. doi: 10.1002/eji.1830240820
11. Nakayama M, Abiru N, Moriyama H, Babya N, Liu E, Miao D, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature (2005) 435(7039):220–3. doi: 10.1038/nature03523
12. Jaeckel E, Lipes MA, von Boehmer H. Recessive tolerance to preproinsulin 2 reduces but does not abolish type 1 diabetes. Nat Immunol (2004) 5(10):1028–35. doi: 10.1038/ni1120
13. Daniel D, Wegmann DR. Protection of nonobese diabetic mice from diabetes by intranasal or subcutaneous administration of insulin peptide B-(9-23). Proc Natl Acad Sci U S A (1996) 93(2):956–60. doi: 10.1073/pnas.93.2.956
14. Devendra D, Paronen J, Moriyama H, Miao D, Eisenbarth GS, Liu E, et al. Differential immune response to B:9-23 insulin 1 and insulin 2 peptides in animal models of type 1 diabetes. J Autoimmun (2004) 23(1):17–26. doi: 10.1016/j.jaut.2004.03.008
15. Thebault-Baumont K, Dubois-Laforge D, Krief P, Briand J-P, Halbout P, Vallon-Geoffroy K, et al. Acceleration of type 1 diabetes mellitus in proinsulin 2-deficient NOD mice. J Clin Invest (2003) 111(6):851–7. doi: 10.1172/JCI16584
16. Moriyama H, Abiru N, Paronen J, Sikora K, Liu E, Miao D, et al. Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis and diabetes in the nonobese diabetic mouse. Proc Natl Acad Sci U S A (2003) 100(18):10376–81. doi: 10.1073/pnas.1834450100
17. Elso CM, Scott NA, Mariana L, Masterman EI, Sutherland APR, Thomas HE, et al. Replacing murine insulin 1 with human insulin protects NOD mice from diabetes. PLoS One (2019) 14(12):e0225021. doi: 10.1371/journal.pone.0225021
18. Halbout P, Briand J-P, Becourt C, Muller S, Boitard C. T cell response to preproinsulin I and II in the nonobese diabetic mouse. J Immunol (2002) 169(5):2436–43. doi: 10.4049/jimmunol.169.5.2436
19. Levisetti MG, Lewis DM, Suri A, Unanue ER. Weak proinsulin peptide-major histocompatibility complexes are targeted in autoimmune diabetes in mice. Diabetes (2008) 57(7):1852–60. doi: 10.2337/db08-0068
20. Faideau B, Lotton C, Lucas B, Tardivel I, Elliott JF, Boitard C, et al. Tolerance to proinsulin-2 is due to radioresistant thymic cells. J Immunol (2006) 177(1):53–60. doi: 10.4049/jimmunol.177.1.53
21. Fan Y, Rudert WA, Grupillo M, He J, Sisino G, Trucco M. Thymus-specific deletion of insulin induces autoimmune diabetes. EMBO J (2009) 28(18):2812–24. doi: 10.1038/emboj.2009.212
22. Jhala G, Chee J, Trivedi PM, Selck C, Gurzov EN, Graham KL, et al. Perinatal tolerance to proinsulin is sufficient to prevent autoimmune diabetes. JCI Insight (2016) 1(10):e86065. doi: 10.1172/jci.insight.86065
23. Guerau-de-Arellano M, Martinic M, Benoist C, Mathis D. Neonatal tolerance revisited: a perinatal window for Aire control of autoimmunity. J Exp Med (2009) 206(6):1245–52. doi: 10.1084/jem.20090300
24. Verdaguer J, Schmidt D, Amrani A, Anderson B, Averill N, Santamaria P, et al. Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J Exp Med (1997) 186(10):1663–76. doi: 10.1084/jem.186.10.1663
25. Moon JJ, Chu HH, Hataye J, Pagan AJ, Pepper M, McLachlan JB, et al. Tracking epitope-specific T cells. Nat Protoc (2009) 4(4):565–81. doi: 10.1038/nprot.2009.9
26. Crawford F, Stadinski B, Jin N, Michels A, Nakayama M, Pratt P, et al. Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci U S A (2011) 108(40):16729–34. doi: 10.1073/pnas.1113954108
27. Chee J, Ko H-J, Skowera A, Jhala G, Catterall T, Graham KL, et al. Effector-memory T cells develop in islets and report islet pathology in type 1 diabetes. J Immunol (2014) 192(2):572–80. doi: 10.4049/jimmunol.1302100
28. Babaya N, Liu E, Miao D, Li M, Yu L, Eisenbarth GS, et al. Murine high specificity/sensitivity competitive europium insulin autoantibody assay. Diabetes Technol Ther (2009) 11(4):227–33. doi: 10.1089/dia.2008.0072
29. Daniel C, Weigmann B, Bronson R, von Boehmer H. Prevention of type 1 diabetes in mice by tolerogenic vaccination with a strong agonist insulin mimetope. J Exp Med (2011) 208(7):1501–10. doi: 10.1084/jem.20110574
30. Yu L, Robles DT, Abiru N, Kaur P, Rewers M, Kelemen GS. Early expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence for early determination of subsequent diabetes. Proc Natl Acad Sci U S A (2000) 97(4):1701–6. doi: 10.1073/pnas.040556697
31. Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA (2013) 309(23):2473–9. doi: 10.1001/jama.2013.6285
32. Wong FS, Karttunen J, Dumont C, Wen L, Visintin I, Pilip IM, et al. Identification of an MHC class I-restricted autoantigen in type 1 diabetes by screening an organ-specific cDNA library. Nat Med (1999) 5(9):1026–31. doi: 10.1038/12465
33. Wong FS, Siew LK, Scott G, Thomas IJ, Chapman S, Viret C, et al. Activation of insulin-reactive CD8 T-cells for development of autoimmune diabetes. Diabetes (2009) 58(5):1156–64. doi: 10.2337/db08-0800
34. Krishnamurthy B, Mariana L, Gellert SA, Colman PG, Harrison LC, Lew AM, et al. Autoimmunity to both proinsulin and IGRP is required for diabetes in nonobese diabetic 8.3 TCR transgenic mice. J Immunol (2008) 180(7):4458–64. doi: 10.4049/jimmunol.180.7.4458
35. French MB, Allison J, Cram DS, Thomas HE, Dempsey-Collier M, Silva A, et al. Transgenic expression of mouse proinsulin II prevents diabetes in nonobese diabetic mice. Diabetes (1997) 46(1):34–9. doi: 10.2337/diabetes.46.1.34
36. Nakayama M, Beilke JN, Jasinski JM, Kobayashi M, Miao D, Li M, et al. Priming and effector dependence on insulin B:9-23 peptide in NOD islet autoimmunity. J Clin Invest (2007) 117(7):1835–43. doi: 10.1172/JCI31368
Keywords: type 1 diabetes, proinsulin-1, CD4+ T cells, immune tolerance, NOD mice
Citation: Jhala G, Selck C, Chee J, Kwong C-TJ, Pappas EG, Thomas HE, Kay TWH and Krishnamurthy B (2021) Tolerance to Proinsulin-1 Reduces Autoimmune Diabetes in NOD Mice. Front. Immunol. 12:645817. doi: 10.3389/fimmu.2021.645817
Received: 24 December 2020; Accepted: 08 March 2021;
Published: 25 March 2021.
Edited by:
F. Susan Wong, Cardiff University, United KingdomReviewed by:
Harley Y. Tse, Wayne State University, United StatesSaeedeh Ghorbanalipoor, University of Lübeck, Germany
Clayton E. Mathews, University of Florida, United States
Copyright © 2021 Jhala, Selck, Chee, Kwong, Pappas, Thomas, Kay and Krishnamurthy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas W.H. Kay, tkay@svi.edu.au
†Present address: Imperial College London, United Kingdom
‡These authors have contributed equally to this work and share senior authorship