 Shen-ping Tang1†
Shen-ping Tang1† Shao-wei Li
Shao-wei Li- 1Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, China
- 2Key Laboratory of Minimally Invasive Techniques & Rapid Rehabilitation of Digestive System Tumor of Zhejiang Province, Taizhou Hospital Affiliated to Wenzhou Medical University, Linhai, China
- 3Department of Gastroenterology, Taizhou Hospital of Zhejiang Province affiliated to Wenzhou Medical University, Linhai, China
- 4Institute of Digestive Disease, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, China
- 5Health Management Center, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, China
Liver transplantation is the ultimate method for treating end-stage liver disease. With the increasing prevalence of obesity, the number of patients with non-alcoholic fatty liver, a common cause of chronic liver disease, is on the rise and may become the main cause of liver transplantation in the future. With the increasing gap between the number of donor livers and patients waiting for liver transplantation and the increasing prevalence of non-alcoholic fatty liver, the proportion of steatosis livers among non-standard donor organs is also increasing. Ischemia-reperfusion injury has historically been the focus of attention in the liver transplantation process, and severe ischemia-reperfusion injury leads to adverse outcomes of liver transplantation. Studies have shown that the production of reactive oxygen species and subsequent oxidative stress play a key role in the pathogenesis of hepatic ischemia and reperfusion injury and non-alcoholic fatty liver. Furthermore, the sensitivity of fatty liver transplantation to ischemia-reperfusion injury has been suggested to be related to the production of reactive oxygen species (ROS) and oxidative stress. In ischemia-reperfusion injury, Kupffer cell and macrophage activation along with mitochondrial damage and the xanthine/xanthine oxidase system promote marked reactive oxygen species production and the inflammatory response and apoptosis, resulting in liver tissue injury. The increased levels of ROS and lipid peroxidation products, vicious circle of ROS and oxidative stress along with mitochondrial dysfunction promoted the progress of non-alcoholic fatty liver. In contrast to the non-fatty liver, a non-alcoholic fatty liver produces more reactive oxygen species and suffers more serious oxidative stress when subjected to ischemia-reperfusion injury. We herein review the effects of reactive oxygen species on ischemia-reperfusion injury and non-alcoholic fatty liver injury as well as highlight several treatment approaches.
Introduction
When the liver disease reaches terminal failure, the last resort is liver transplant. With the increasing global prevalence of obesity, type 2 diabetes, and metabolic syndrome, more patients than ever before are being diagnosed with non-alcoholic fatty liver disease (NAFLD) and reaching the terminal stage of their disease, it is reported that the global prevalence of NAFLD is about 25% (1). Patients with NAFLD may thus account for the greatest proportion of liver transplant candidates in the future (2). Over the past 10 years, the gap between the number of patients awaiting liver transplantation and the number of donor livers has increased, and the possibility of receiving a non-standard donor organ, including a fatty liver, has increased (3).Steatosis is considered when evaluating donor livers,which may affect transplant outcomes and is approximately 30% in deceased organ contributors and approximately 20% in living donors. In previous studies, the detection rate of NAFLD in living donor livers was between 14.5-53% (4). An evaluation of potential donors for living donor liver transplantation (LDLT) programs in Malaysia showed that NAFLD and obesity were significant reasons for low donor utilization (5). Severe steatosis liver (>60%) excludes donor liver range, while the use of moderate steatosis (30-60%) remains controversial (6, 7).
Ischemia and reperfusion (IR) injury is inevitable in cases of liver transplantation. IR injury involves various mechanisms, including macrophage polarization, necroptosis, reactive oxygen species (ROS) production, and oxidative stress (8). During liver reperfusion, the activation of Kupffer cells (KCs) and macrophages, mitochondrial damage, and large amounts of xanthine and hypoxanthine due to increased ATP consumption during ischemia promote the production of a large quantity of ROS (9). Excessive ROS and the consumption of endogenous antioxidants lead to redox imbalance, which causes oxidative stress (10). ROS combine with intracellular macromolecules to damage organelles, which eventually leads to cell damage and even apoptosis (11). Active oxygen damages lipids, resulting in the production of lipid peroxides, destruction of cell membrane permeability, apoptosis, the production of inflammatory factors, and the induction of inflammatory reactions (12). Reactive oxygen can also cooperate with Ca2+ to promote the opening of mitochondrial membrane permeability transition pore (MPTP), damage to mitochondria, release of cytochrome C, formation of apoptotic bodies, and promotion of cell apoptosis (13). Furthermore, cells that are damaged by ROS release damage-related model molecules (DAMPs), which combine with TLR4 or TLR9 on KCs to activate the NF-κB signaling pathway and thus generate more ROS to amplify the inflammatory response (14, 15).
When fatty livers are subjected to IR, they generate more ROS than non-fatty livers, the lipid peroxidation reaction is heavier, the antioxidant capacity is lower, and the mitochondria are more vulnerable to damage, so the resulting tissue damage is more serious. Such increased sensitivity and reduced tolerance of NAFLD to IR have increased the difficulty of treatment.
We herein review the role of ROS and its influence on fatty liver transplantation.
The Production of ROS in IR
During IR, ROS are produced by a variety of pathways, causing oxidative stress and damage. ROS in IR come are generated by a number of different sources, including the mitochondrial electron transport chain, xanthine oxidase, NADPH oxidase, and uncoupling nitric oxide synthase (NOS) (16) (Figure 1).
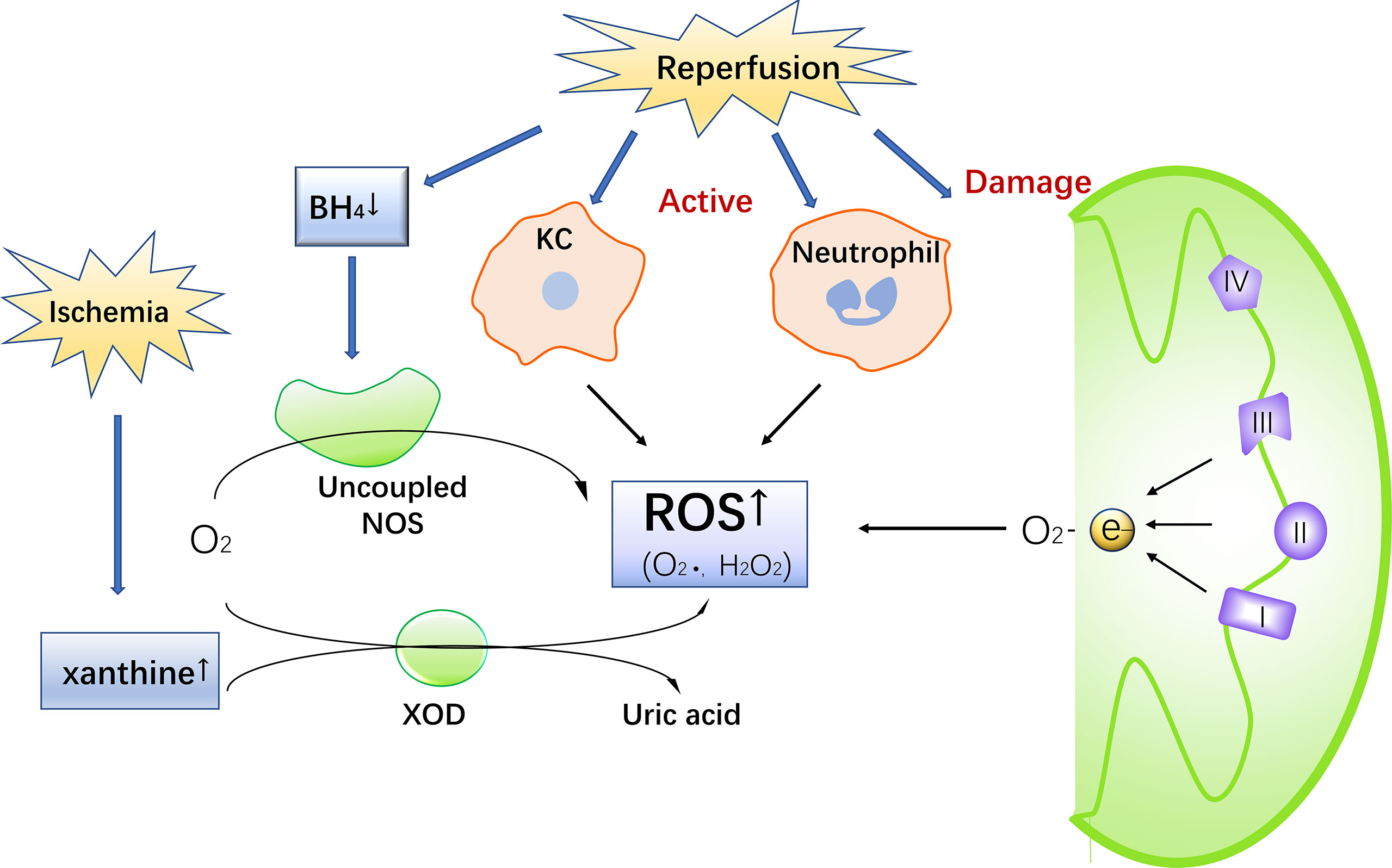
Figure 1 The Production of ROS during IR injury. Ischemia leads to an increase in xanthine. Reperfusion damages mitochondria, activates Kupffer cells and neutrophils, consumes BH4 and uncouples NOS, promotes ROS production, and simultaneous increase of xanthine in the catalysis of xanthine oxidase to produce ROS.
Mitochondrial Electron Transport Chain
ROS are mainly produced in the respiratory chain complex on the mitochondrial inner membrane during mitochondrial metabolism (9, 11). Under normal circumstances, mitochondria produce a small amount of ROS (17). However, when subjected to ischemia, a large quantity is produced in order to maintain the redox balance, leading to the consumption of endogenous antioxidants and the expression of antioxidant enzymes. When the damage is severe or sustained, the ROS-scavenging capacity of the antioxidant system is not sufficient to remove the increased amount of ROS, leading to oxidative stress, inflammation, cell death, and organ failure (10, 18). In the ischemic state, prolonged tissue hypoxia and the consequent increase in ATP depletion often lead to cell damage. Therefore, the cell metabolism pattern becomes anaerobic, leading to the accumulation of lactic acid, a decrease in intracellular pH, and disorders of cytoplasmic ions (19, 20). Taken together, these events can damage mitochondrial macromolecules, such as DNA and protein, and the damaged mitochondria gradually produce more ROS, creating a vicious cycle of ROS production and mitochondrial damage (11).
Xanthine Oxidase
Xanthine oxidizing reductase (XOR) can catalyze the conversion of hypoxanthine to xanthine, which is then converted to uric acid. It exists in the form of XO and XDH, and these two subtypes can be converted to each other (21).The xanthine/xanthine oxidase (XOD) system is an important source of ROS in hepatocytes. During the ischemic period, xanthine dehydrogenase (XDH) is transformed into XOD, which produces oxygen-free radicals. At the same time, xanthine, the substrate of XOD, is also accumulating. During the reperfusion period, XOD is transformed to ROS (22). The use of allopurinol, a type of XOD inhibitor, can inhibit ROS production and reduce liver IR injury (23).
NADPH Oxidase
The Nox/Duox family in NADHP oxidase, including NOX1-5, DUOX1 and DUOX2, is involved in the production of ROS (24). The increase in ROS after reperfusion is an important part of IR injury (25). During reperfusion, the depletion of superoxide dismutase (SOD) and glutathione (GSH) peroxidase and reduced GSH promote oxidative stress (26). Reperfusion can be divided into two stages. The initial stage is 0.5-4 h after reperfusion, and the late stage is 6-24 h after reperfusion (18). In the initial stage of reperfusion, KCs are activated to express Nox2 and its subunits, forming complexes on the cell membrane, catalyzing the reduction of oxygen and producing ROS, thus causing oxidative stress (24, 27, 28). At the same time, T lymphocytes secrete interferon-α (IFN-α) to promote the activation of KCs (9). In the late stage of reperfusion, oxidants derived from extrahepatic cells mediate inflammatory damage (29). Under constant stimulation, neutrophils continuously activate the NADPH oxidase complex, producing a large number of ROS (30). Meanwhile, neutrophils infiltrate, secreting pro-inflammatory factors, such as TNF and platelet-activating factor. TNF induces the expression of adhesion molecules in vascular endothelial cells to recruit neutrophils. Platelet activators are involved in the activation of ROS production by neutrophils (9). When neutrophils attack liver cells, they produce hydrogen peroxide and hypochlorous acid, which are cytotoxic and damage liver cells (31).
NOS
In addition, during IR, the uncoupling of NOS also produces ROS (32). Coupled NOS oxidized L-arginine to nitric oxide (NO). NO attenuates IR injury through antioxidation and the inhibition of inflammatory cell migration, as well as to reductive superoxide (33, 34). IR reduces the content of BH4, a cofactor of nitric oxide synthase, leading to the uncoupling of NOS, thereby resulting in the production of ROS (33).
ROS Causes Damage to Liver During Ischemia-Reperfusion
Under normal circumstances, the production and removal of ROS in the body are in a dynamic equilibrium state, and there is no harmful effect on the body. However, when a cell encounters IR, in order to adapt to or resist the stimulus, the cell produce a large amount of highly active ROS. On one hand, ROS activate the antioxidant system, but on the other hand, they attack and damage endogenous antioxidants (35). The excessive production of ROS and weakened antioxidant capacity in cells lead to an imbalance between the production and removal of ROS, resulting in oxidative stress and thereby promoting inflammation, cell apoptosis and mitochondrial damage (36, 37) (Figure 2).
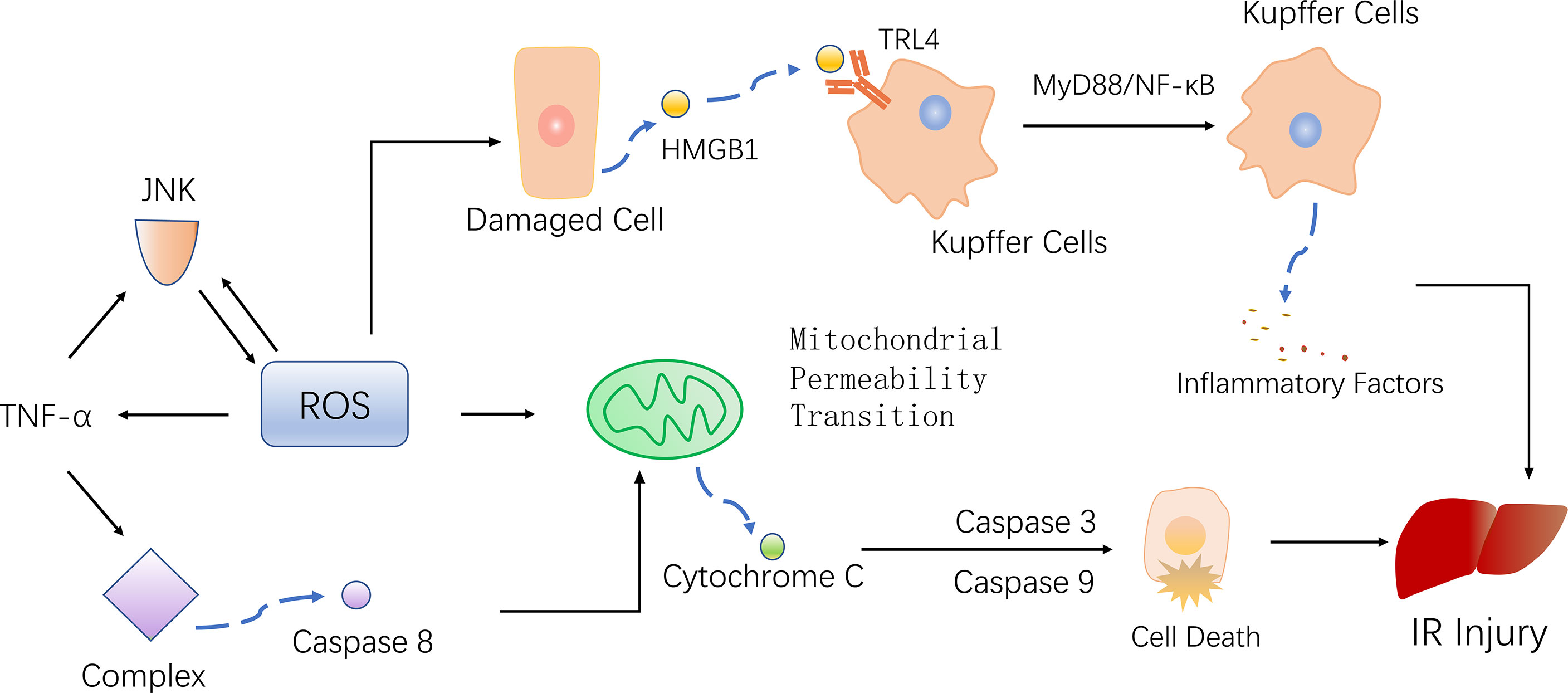
Figure 2 ROS mediates inflammation, mitochondrial damage, and cell death, ultimately promoting liver damage. KCs recognize HMGB1, promote the inflammatory response through the TRL4-MyD88-NF-κB pathway. At the same time, TNF-α activated by ROS, phosphorylating JNK and further producing ROS. Activated TNF-α also form complexes that activate caspase 8, leading to mitochondrial permeability transition (MTP) and finally apoptosis.
ROS Promote Inflammation
During reperfusion, a large amount of ROS is produced, which causes oxidative stress and cell and even tissue damage. ROS activates KCs and macrophages, subsequently the activated cells release a large number of pro-inflammatory cytokines, including TNFa, IL-1b, etc., which promote the inflammatory response to damage hepatocyte damage and accelerate the progression of IR damage (38). In the initial stage of reperfusion, KCs recognize and activate endogenous damage-related model molecules (DAMPs) released by damaged cells, which recruit monocytes and neutrophils to synergistically induce inflammation. Common DAMPs include high mobility group box-1 protein (HMGB1), DNA, and ATP (14). HMGB1 is a DNA-binding protein. Normally, HMGB1 is expressed in organs and tissues, but when IR occurs, HMGB1 is released from the damaged nucleus and into the blood under the promotion of ROS, leading to cell death (39). HMGB1 binds to toll-like receptor 4 (TLR4) on KCs and activates the NF-κB signaling pathway, leading to the massive release of proinflammatory factors (IL-1, IL-2, IL-3, IL-6, IL-8, TNF-α) and subsequent promotion of inflammatory reactions (14, 39, 40). DAMPs can also activate TLR9 on KCs, enhancing the production of TLR9-dependent ROS and inflammatory mediators and promoting inflammation. TLR9-knockout mice show a reduced inflammatory cytokine production and liver damage compared with wild-type mice (15, 41).
ROS Induce Mitochondrial Damage
ROS attacks mitochondria, leading to mitochondrial damage. The mitochondrial apoptosis pathway is one of the main apoptotic pathways, and the continuous opening of the mitochondrial membrane permeability transition pore (MPTP) is an important mechanism of mitochondrial function damage (42). The massive generation of ROS and Ca2+ overload caused by IR regulates the opening of the MPTP, resulting in the irreversible transformation of mitochondrial permeability transition (MPT). As a result, the electron transport chain is uncoupled, damaging the mitochondrial membrane. Cytochrome C is released through the channel formed by Bax to produce apoptotic bodies, which mediate endogenous cell apoptosis (13, 43). Cytochrome C released into the cell forms a complex with apoptosis activator 1, activating caspase-9 and caspase-3. This activated caspase-3 damages DNA repair enzymes, aggravating cell DNA damage and leading to cell apoptosis (44). Sirtuin1 (SIRT1) is an NAD+-dependent class III protein deacetylase that regulates hepatic lipid metabolism, systemic inflammatory status and autophagy through mitofusin2 (MFN2) (45). In Overexpression of STRT1 and (MFN2) in mice promotes autophagy and prevents mitochondrial dysfunction (46).
IR Induce Apoptosis
During IR, multiple types of cells such as Kupffer cells and lymphocytes release TNF-α, which induces NOS and chemotaxis of leukocytes (47). When TNF-α binds to a specific receptor, the death signaling pathway, which depends on c-Jun N-terminal kinase (JNK) phosphorylation, is activated, inducing the production of ROS in mitochondria. Aggravated oxidative stress further stabilizes the JNK phosphorylation level, resulting in MPT and consequent cell death (16). Under the continuous stimulation of TNF-α, TNF-α receptor-associated protein with death domain(TRADD) bind to the Fas-associated protein with death domain(FADD), summon and activate Caspase 8, which leads to changes in mitochondrial permeability and initiates the process of apoptosis (48–50). TNF-α can also increase the expression of NF-κB, thereby promoting the production of ROS and causing damage as well as gathering CD4+ T cells, promoting the secretion of colony-stimulating factors (IFN-γ and TNF-β), and accelerating cell apoptosis during IR (9).
In IR, autophagy has both damaging and protective effects. Proper mitochondrial autophagy during the ischemic stage can clear away damaged mitochondria and reduce subsequent injury. However, during the reperfusion stage, ROS levels are significantly increased, the MPTP remains open, and the PINK1/Parkin pathway is activated, mediating mitochondrial autophagy and concentrated injury (51).
ROS and Fatty Liver Transplantation
The histological features of NASH are vesicular steatosis and lobular hepatitis with necrosis or balloon degeneration and fibrosis (52, 53). According to the percentage of fat content in the liver, hepatic steatosis can be divided into mild, moderate and severe steatosis, and moderate to severe steatosis may lead to adverse outcomes after liver transplantation. However, views on the utilization of donor livers with moderate steatosis are not consistent (7). Steatosis hepatocytes are more sensitive to IR damage, possible reasons are the production of ROS, and more severe oxidative stress, inflammation, mitochondrial damage (Figure 3)
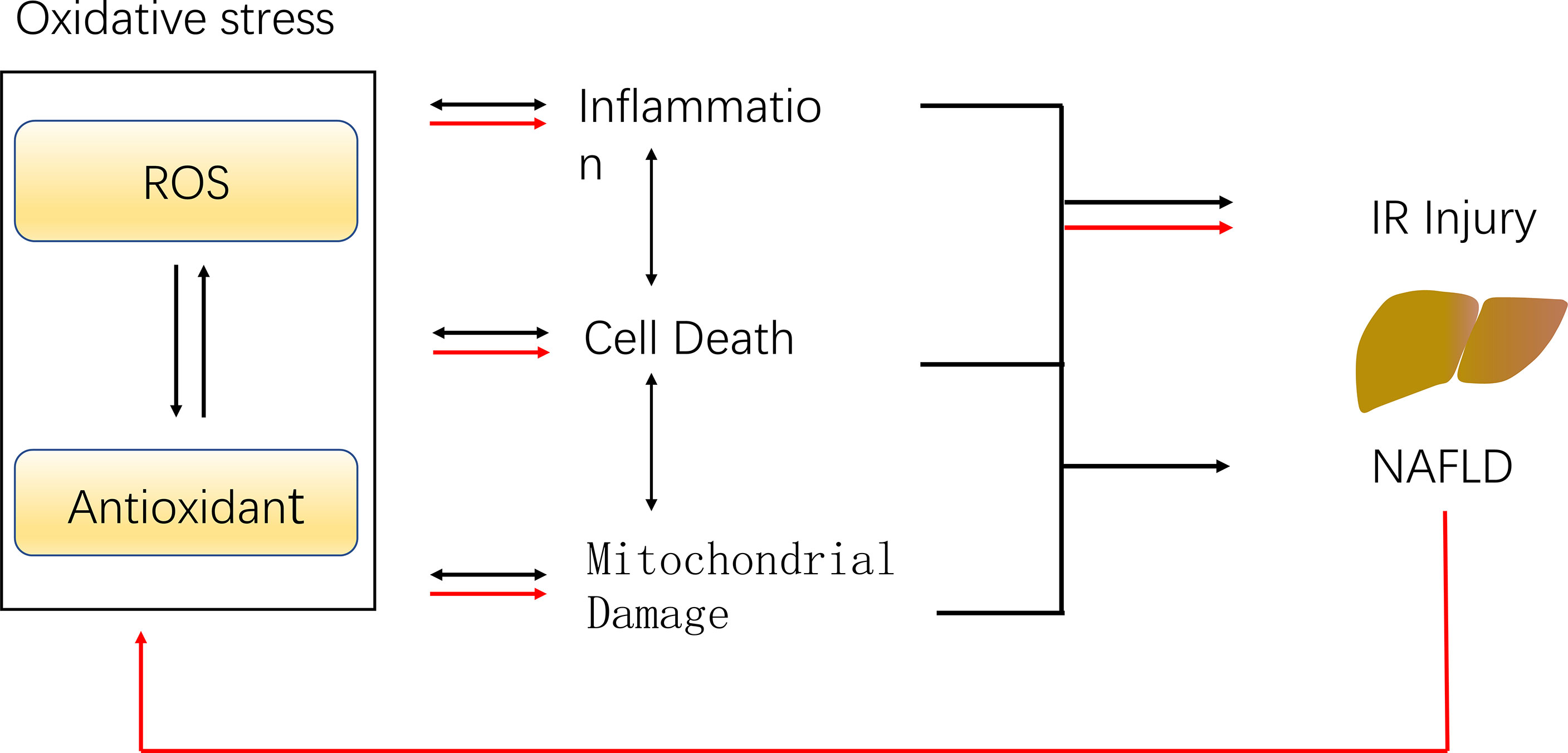
Figure 3 The potential mechanism of IR injury and NAFLD caused by ROS. ROS leads to the inactivation of antioxidants, which reduce the scavenging of ROS, and the imbalance of the ROS and antioxidant results in oxidative stress. Oxidative stress promotes inflammation, mitochondrial damage as well as cell death, leading to hepatic ischemia-reperfusion injury and NAFLD (black arrows). The presence of NAFLD resulted in excessive ROS generation during IR, weaker antioxidant capacity, aggravated oxidative stress and enhanced IR injury (red arrows).
The mechanism underlying the pathogenesis and progression of NAFLD remains unclear, although the hypothesis of “multiple hits” is currently proposed. Insulin resistance, oxidative damage, stellate cell activation, fibrosis pathway activation, changes in the expression of adipokines, and many other sources of damage lead to the occurrence of NASH and liver cirrhosis (54). Excessive ROS generation and oxidative stress play an important role in the pathogenesis and development of NAFLD. In addition, mitochondrial oxidative stress is the main factor involved in the increased sensitivity of fatty liver to IR injury (55). Compared with healthy subjects, NAFLD patients have higher levels of ROS and lipid peroxidation products, lower levels of antioxidant enzymes [e.g. SOD and catalase (CAT)], and decreased levels of antioxidant compounds (e.g. GSH). The imbalance between the scavenging ability of antioxidants and ROS production led to oxidative stress (56, 57). In the pathogenesis of NAFLD, in order to eliminate excessive free fatty acids in the liver, mitochondria enhance β-oxidation and promote the production of ROS in the respiratory chain, leading to oxidative stress. Oxidative stress also aggravates lipid accumulation in hepatocytes, further promotes the production of ROS, and damages organelles, proteins, DNA, and lipids, resulting in a vicious cycle (58, 59). At the same time, the reaction of unfolded proteins in endoplasmic reticulum is enhanced, which also produces ROS (60).
Some studies have found that mitochondrial dysfunction in liver tissue during NAFLD affects the liver lipid balance, promotes ROS production, lipid peroxidation as well as cytokine release, ultimately leading to cell death (61, 62). Damage to mitochondrial function in NAFLD makes mitochondria more susceptible to MPT and subsequently causing cell death when suffer from IR (25). Phospholipids on the mitochondrial membrane are oxidized, reducing fluidity and hindering the entry of GSH into the mitochondria, causing an imbalance between antioxidants and ROS, inducing oxidative stress, which makes increased expression of the uncoupling protein 2 (UCP2). UCP2 induces electron transport chains(ETC) uncoupling and thus reduces mitochondrial ATP synthesis (63). As a result, patients with NAFLD have lower reserves of ATP, and when the liver undergoing ischemia and hypoxia injury, ATP depletes faster, making cells more susceptible to necrosis and triggering an inflammatory response (25) (Figure 4).
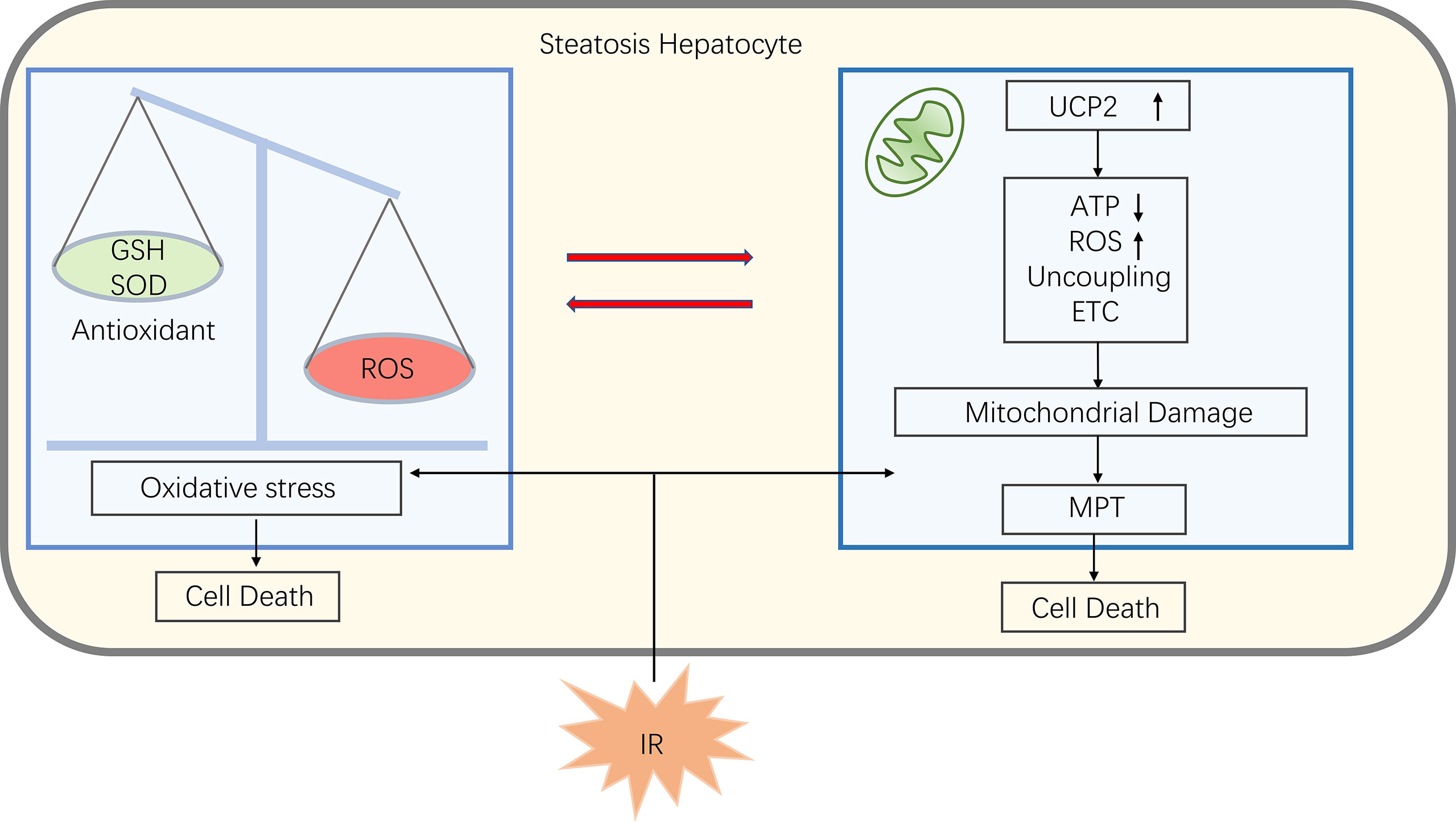
Figure 4 In steatosis hepatocyte, an imbalance of ROS and antioxidants leads to oxidative stress that damages mitochondria. Also oxidative stress promotes uncoupling protein 2 (UCP2) expression, which can uncouple electron transport chains (ETC), thereby reducing ATP synthesis and induce ROS production, further promoting oxidative stress. When fatty liver encounter IR, the mitochondria at this statute are more susceptible to mitochondrial permeability transitions (MPT), leading to cell death.
ROS binds to polyunsaturated fatty acids (PUFAs) to produce lipid peroxides. Unstable lipid peroxides are easily decomposed to active 4-hydroxy-2-nonenal (4-HNE) and Malondialdehyde (MDA), causing damage to cells (56, 64). In fatty liver IR, lipids are most vulnerable to ROS attack. However, NAFLD itself contains the accumulation of fatty acids and extensive lipid peroxidation, which makes the fatty liver IR injury more serious (54). In the pathogenesis of NALFD, ROS also attack proteins, especially antioxidant enzymes, and the antioxidant capacity is weakened after oxidation. Furthermore, in NAFLD, GSH is chronically consumed, and the combination of lipid peroxidation products with GSH also consumes GSH (65) (Figure 5).
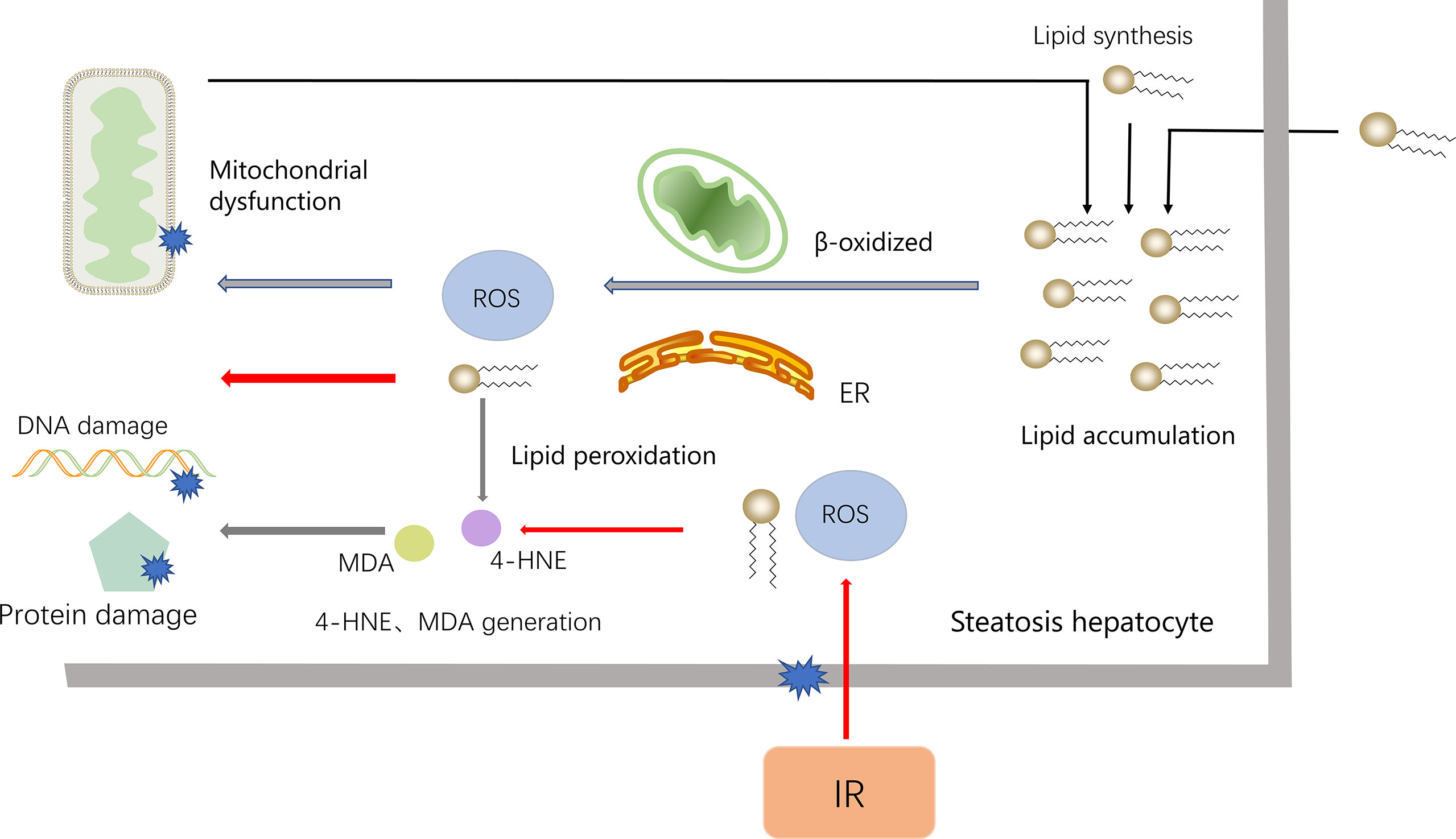
Figure 5 The accumulation of lipids is an important reason for the NAFLD. To process the accumulated lipids, hepatocytes increase mitochondrial β-oxidation and the activity of ER, which lead to the synthesis of ROS. ROS, in turn, damages the mitochondria, further leading to the accumulation of fatty acids (black arrows). When NAFLD encounters IR, ROS increased (red arrow), combined to accumulated fatty acids and release 4-HNE and MDA, which cause DNA and protein damage and worsen liver damage.
Peroxisome proliferator-activated receptor-α (PPAR-α) and Peroxisome proliferator-activated receptor-γ (PPAR-γ) are important regulators of IR damage in steatotic liver (55). The PPAR-γ agonist WY-14643 reduces steatosis and the IR injury in mouse liver (66). In addition, studies have shown that pioglitazone-activated PPAR-γ attenuates liver injury induced by IR. In contrast, mice with a significantly decreased expression of PPAR-γ showed relatively severe liver injury (67, 68). PGC-1α regulates antioxidants in vivo, and its overexpression can significantly reduce ROS in IR, whereas knockout of PGC-1α leads to a further increase in the intracellular ROS level (69). ROS is produced during NAFLD, which consumes antioxidants and damages mitochondria. On this basis, when IR occurs, more ROS are produced, more antioxidants are exhausted, and oxidative stress and damage are exacerbated.
Possible Strategies for Treating IR
Given the current high proportion of donor hepatic steatosis and the increase in the number of fatty liver patients on the liver transplant waiting list, improving the availability of grafts and reducing the risk of post-transplant dysfunction has become a hot topic. Numerous studies have been conducted, including the treatment of potential donors for transplantation, as well as treatment during transplantation, thereby improving the success rate of transplantation. Here, we summarize some of the treatments for some antioxidant therapies during transplantation (Table 1) and for potential donors diagnosed with NAFLD/NASH.
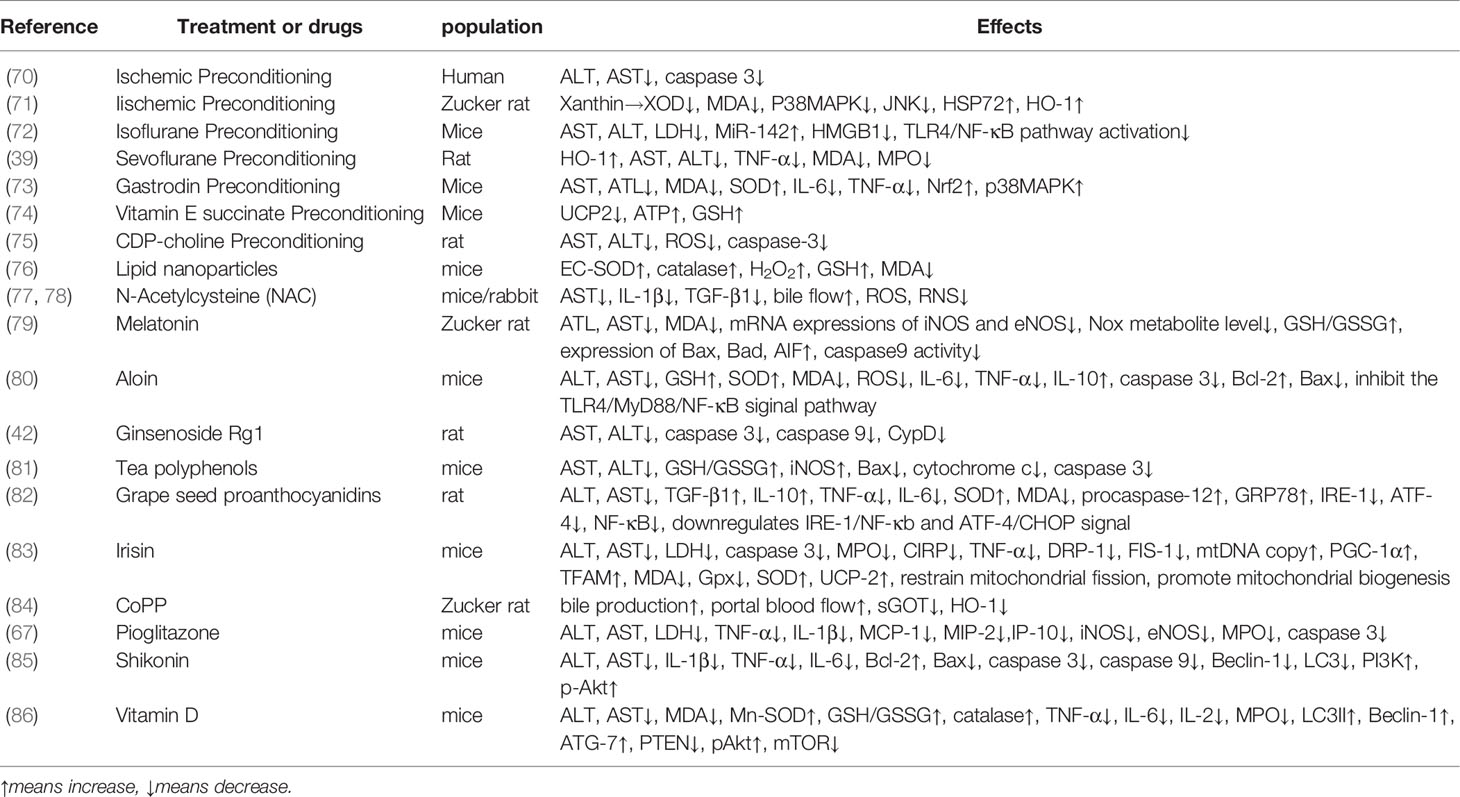
Table 1 Possible treatment or drugs on alleviating IR injury and mechanism.
Preconditioning
A common anti-IR approach is ischemic preconditioning (IPC), the process of transient IR before a long period of ischemia. An experimental model showed that liver IPC can protect the liver from the adverse effects of IR, and clinical practice has also demonstrated the protective effect of IPC on steatotic liver (70, 87). IPC reduce the production of ROS by inducing the production of NO, reducing the accumulation of xanthine during ischemia, and preventing the conversion of XDH to XOD (88). IPC also regulates inflammatory factors, activates the PPAR-α signaling pathway, and reduces the production of ROS, thereby reducing oxidative stress, mitochondrial dysfunction, and the activation of neutrophils and KCs (9). IPC over-induced heme oxygenase-1 (HO-1) can protect fatty liver from IR injury, and the protective effect of IPC is weakened after using HO-1 inhibitors (71). Studies have shown that the protective effect of IPC on steatotic liver IR injury is related to the activation of AMPK. AMPK activation reduces the further consumption of ATP, promotes NO synthesis, inhibits the NF-κB signaling pathway, and reduces lipid peroxidation and liver cell damage (55).
In addition, simulating the effect of IPC by regulating the internal and external components of cells via drugs is called pharmacological preconditioning (31). Pretreatment with volatile anesthesia, such as isoflurane and sevoflurane, reduces IR injury in mice (39, 72). Sevoflurane reportedly inhibits the expression of HMGB1 by up-regulating the expression of miR-142 in IR mice and inhibits the activation of the TLR4/NF-κB inflammatory pathway, thereby reducing oxidative stress and liver damage (39). Preconditioning with gastrodin, vitamin E succinate, CDP-choline, and other drugs protects the liver from IR injury (73–75).
Anti-Oxidants
In fatty liver, the SOD activity and GSH levels decrease. When such livers are exposed to IR, these parameters decrease even further, leading to a sharp increase in ROS and thereby promoting an imbalance between oxidation and reduction, leading to oxidative stress. Therefore, antioxidants play an important role in reducing fatty liver IR (72). Common endogenous antioxidant enzymes include SOD, CAT, GSH, and vitamin C. SOD catalyzes the superoxide anion and water to form H2O2 and remove ROS (89). There are three isozymes of SOD: copper/zinc SOD (Cu/Zn-SOD), manganese SOD (Mn-SOD), and extracellular SOD (EC-SOD). In IR, the inactivation of the MnSOD function weakens the antioxidant capacity of NAFLD and affects the viability of hepatocytes (90). CAT is a H2O2 scavenger that plays an additional scavenging role when SOD is insufficient to remove ROS. Lipid nanoparticle-mediated antioxidant gene delivery significantly increases the expression of human EC-SOD and CAT genes in the liver, thus reducing IR injury (76).
The application of antioxidants also reduces the oxidative stress of liver IR. At present, the studies of antioxidants to alleviate ischemia and reperfusion damage in animal models is intense with fewer studies of antioxidants in human ischemia and reperfusion damage. GSH is a substrate of Gpx and decomposes the precursor molecule of H2O2. N-Acetylcysteine (NAC) has been shown to increase the activity of reduced GTH-related enzymes, reduce ROS, and improve IR injury in fatty liver (77, 78). The administration of postoperative or peri-operative NAC in patients undergoing hepatic resection did not achieve great complication prevention results (91, 92). In another study, the use of NAC during donor liver collection significantly improves graft survival and post-transplant outcomes (93). Existing antioxidant studies of human ischemia-reperfusion injury are rarely assessed for antioxidant capacity. Melatonin alleviates IR injury in rats with fatty liver degeneration by strengthening the clearance of ROS and NOS, restoring the mitochondrial function, reducing the expression of pro-apoptotic genes, and reducing oxidative stress (79). In a randomized controlled double-blind pilot clinical trial, patients received preoperative care of high-dose (50 mg/Kg) melatonin, exhibiting less non-infectious complications ICU and total hospital stay, the authors speculate that antioxidant capacity is a possible cause (94). The increase in the ratio of ω6/ω3 plays an important role in NAFLD, and the NAFLD patients suffer from less damage when the ratio decreases. ω3 polyunsaturated fatty acids (PUFA) plays good anti-inflammatory activity, while reducing the fat content of the liver (95, 96). In animal renal ischemia-reperfusion models, ω3PUFA has been reported to reduce damage through antioxidant action (97). Mice consuming a omega-3PUFA-containing diet were less sensitive to liver ischemia reperfusion injury (98). Previous studies have shown that postoperative use of omega-3 PUFA parenteral nutrition has some benefit in postoperative liver patients (95, 99). In contrast to previous animal experiments reported to reduce liver damage and improve liver regeneration a recent study showed that perioperatively intravenous omega-3PUFA achieve no improvement on complications (100).
In addition, some antioxidants have been shown in animal models to reduce oxidative stress. Aloin increases the SOD activity and GSH level, reduces oxidative stress and inflammation, and alleviates liver IR injury (80). Ginsenoside Rg1 plays a protective role by inhibiting the expression of apoptosis-related proteins and down-regulating inflammatory mediators and antioxidation (42, 101). Tea polyphenols have strong antioxidant properties. The use of tea polyphenols increases the level of GSH, inhibits oxidative stress, and alleviates IR in mice (81). Grape seed proanthocyanidins protects against IR injury through antioxidant, anti-inflammatory, and anti-apoptotic effects (82). Irisin can relieve oxidative stress by mediating the expression of coupling protein 2 and ameliorate IR through its anti-apoptotic effect (83). Curcumin increases animal survival after transplantation by playing a protective role against inflammation and oxidative stress (102, 103).
Some endogenous genes or gene products have been proven to exert protective effects in IR injury. Nrf-2 is a transcription factor that is sensitive to oxidative stress and positively regulates the basic and induced expression of a large number of cytoprotective genes (104). In liver IR, the regulation of the keap1-Nrf2-ARE pathway promotes the gene expression of antioxidant enzymes, such as SOD, GTH peroxidase, CAT, and HO-1, thereby playing a protective role (105, 106). Under oxidative stress, Nrf2 is activated, and the expression of HO-1 is up-regulated. HO-1, with NADPH as a cofactor, catalyzes the oxidative degradation of heme to the antioxidants biliverdin, carbon monoxide, and iron, and exerts the effect of scavenging ROS and anti-inflammation (107, 108). The up-regulation of HO-1 expression by HO-1 inducers, such as cobalt protoporphyrin and adenovirus HO-1, alleviates steatotic liver IR injury in rats (84). ROS activates the PI3K/Akt pathway, and the activation of Akt inactivates p53 and other pro-apoptotic proteins, thereby increasing the expression of anti-apoptotic proteins and inhibiting cell apoptosis. Shikonin treatment reduce IR injury by activating PI3K/Akt (85). Vitamin D treatment of mice was shown to reduce ROS and down-regulate the PTEN-activated PI3K/Akt pathway, thus regulating autophagy and reducing injury (86).
Treatment for NAFLD
Treatment of potential donors prior to living donor transplantation (LDLT) could alleviate steatosis, increase donor availability to some extent, and reduce IR damage. The common methods include diet, exercise, and drugs (Figure 6). Physical activity and dietary restrictions are routine treatments for patients with NAFLD. It is reported that performing living liver transplantation with diet-treated donors is feasible (109). Short-term weight loss through diet combined with exercise can alleviate the degree of fat infiltration of fatty liver donors, and most donor who receive weight loss successfully complete LDLT (110–113).
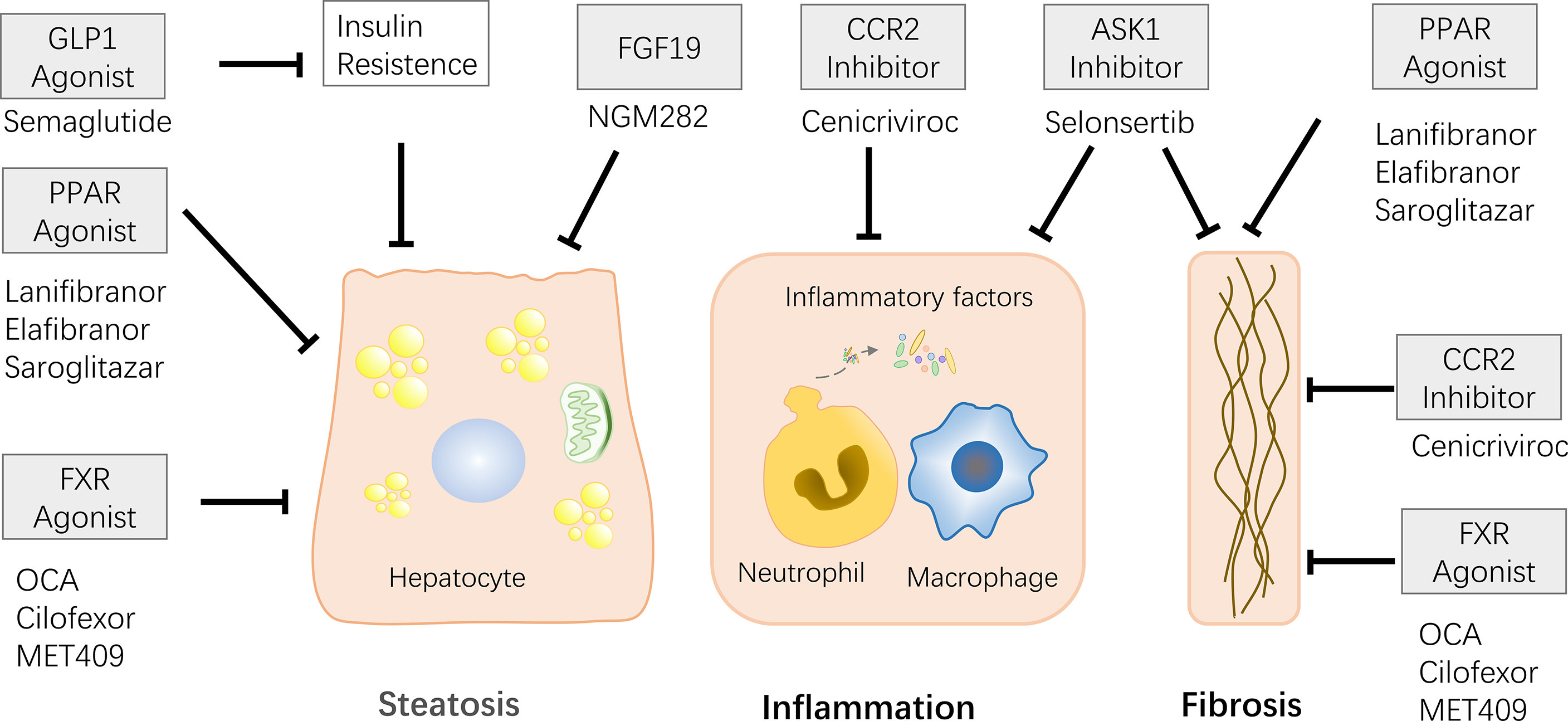
Figure 6 Some of the drugs currently in clinical trials and their effects are described. Effects include relieving steatosis, inflammatory response, and liver fibrosis. FXR agonists and PPAR agonists can alleviate steatosis and liver fibrosis. CCR2/5 and ASK1 inhibitors play role on anti-inflammatory and anti-fibrotic. FGF19 has the ability to relieve steatosis. FLP-I relieves steatosis by relieving insulin resistance.
The only current study of preoperative short-term drug therapy for donors was benzalabet (an activator of PPARα and PPARβ/δ). In a study of preoperative short-term treatment of donors, they were treated with a protein-rich (1000 kcal/day) diet, exercise (600 kcal/day) and benzabrate (400 mg/day) for 2-8 weeks before surgery, the authors observed a reduction in steatosis, no significant complications after surgery, and the transplanted grafts and donors showed good liver function (114).
PPAR is a hot therapeutic target, has been reported great capacity of reduce steatosis, inflammation and liver fibrosis (115–117). PPAR-γ has a protective effect on liver tissue in IR. IR injury can be alleviated by increasing the levels of SOD and hydrogen peroxide hydrolase and reducing IR injury using pioglitazone, a PPAR-γ agonist (67). Other PPAR agonists have also reported surprising results in clinical trials. Lanifibranor, a pan PPAR agonist that activates three PPARs, lowered the SAF-A score and relieved NASH in a recently published Phase 2b randomized, double-blind, placebo-controlled trial (115). Elafibranor (PPAR-α/δ agonist) is reported to alleviate histologic changes and improve metabolism in NASH patients (116). The phase 2 trial of Saroglitazar (a PPAR-α/γ agonist) reported the effect of remission of insulin resistance, atherosclerotic dyslipidemia, and ALT, LCF parameters in patients with NAFLD/NASH (117).
Farnesoid X receptor (FXR) is a member of the nuclear receptor superfamily that regulates bile acid metabolism, lipid metabolism, and glucose metabolism. Elevated bile acid levels could inhibit bile acid synthesis by activating FXR-SHP-SREBP1c (118, 119). FXR is now considered as new drug target, and clinical trials of some FXR agonists are underway. Obeticholic acid (OCA), a semi-synthetic derivative of primary human bile acid goose deoxycholic acid, works in animal models through reduce insulin resistance and hepatic steatosis. Previous studies have shown that after 6 weeks of treatment with 25/50 mg OCA, there was a decrease in liver inflammation and fibrosis markers in patients with type 2 diabetes and non-alcoholic fatty liver disease (120). Subsequent Phase 3 experiments on the treatment of NASH with OCA confirmed that it reduces NASH fibrosis and disease activity indicators (121). However, a large number of patients have experienced the side effect of itching after taking OCA, thus other FXR agonists without bile acids have been developed. MET409 is a novel FXR agonist without bile acid structure, has been reported to reduce liver fat content in NASH patients (122). Clinical trials of Cilofexor (nonsteroidal FXR agonist) have also reported similar results (123).
When FXR is activated by bile acids, fibroblast growth factor (FGF) 15 is induced, play the role of inhibiting the synthesis of bile acids through the down regulate the transcription of cholesterol 7α-hydroxylase (Cyp7a1). FGF19 is a human homolog of FGF15, is also regulated by the level of bile acids (124, 125). FGF19 analogues NGM282 and Aldafermin have been designed as novel therapeutics and are currently in clinical trials. The phase 2 trial of NGM282 confirmed that NGM282 induce a decrease in liver fat content in NASH patients (126). Similarly, phase 2 trials of Aldafermin confirmed its effects on reducing liver fat (127).
New drugs for other targets have also been designed, including targeting C-C motif chemokine receptor (CCR)2-CCR5, apoptosis signaling protease-1 (ASK-1), glucagon-like peptide (GLP)-1 and etc. CCR2/5 improve liver fibrosis by activating inflammatory signals and immune cell infiltration, causing liver damage (128). Cenicriviroc (CVC), an oral dual CCR2/CCR5 antagonist, the recent Phase 3 clinical trials surprisingly reported potent anti-inflammatory and antifibrotic activity (129). Oxidative stress activates the ASK1-Jun pathway, leading to increased liver inflammation and fibrosis. Selonsertib, a selective inhibitor of ASK1, was reported to reduce liver fibrosis in patients with NASH and stage 2-3 fibrosis in previously published phase 2 experiments. However, the recently published phase 3 experiment proved that there was no mitigating effect on fibrosis in patients with NASH-induced bridging fibrosis or compensated cirrhosis (130). Glucagon-like peptide (GLP)-1 stimulate insulin secretion and inhibit the secretion of glucagon, also has a certain effect of relieving NASH, authors observed that subcutaneous injection of Semaglutide in NASH patients could achieve NASH regression (131).
Conclusion and Future Directions
IR causes the adverse outcome of liver transplantation, and steatosis liver is more sensitive to IR, therefore, the steatosis liver transplantation is more likely to get the adverse outcome, which brings challenges to clinical work. The ROS production in non-alcoholic fatty liver is substantial, and IR further promotes ROS production, oxidative stress, and inflammation, thus exacerbating the condition. Preconditioning and certain antioxidants can reduce the IR injury seen in fatty liver in mice. Treatment of potential patients with fatty liver can increase transplant safety. However, the mechanisms underlying IR and the generation of non-alcoholic fatty liver itself are unclear. The rapid increase in fatty liver patients requires further research into the mechanism underlying fatty liver IR injury. At the same time, the drug treatment of NAFLD is also expected. In addition, most studies on antioxidant therapy are focused on IR, and clinical data are lacking. Therefore, further research on the treatment of fatty liver IR is needed.
Author Contributions
All of the authors contributed to the writing and editing of the manuscript and approved the submitted version.
Funding
This work was supported in part by the Medical Science and Technology Project of Zhejiang Province (2021PY083), Program of Taizhou Science and Technology Grant (20ywb29), Major Research Program of Taizhou Enze Medical Center Grant (19EZZDA2), Open Project Program of Key Laboratory of Minimally Invasive Techniques & Rapid Rehabilitation of Digestive System Tumor of Zhejiang Province (21SZDSYS01, 21SZDSYS09) and Key Technology Research and Development Program of Zhejiang Province (2019C03040).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Younossi ZM. Non-Alcoholic Fatty Liver Disease - A Global Public Health Perspective. J Hepatol (2019) 70(3):531–44. doi: 10.1016/j.jhep.2018.10.033
2. Zezos P, Renner EL. Liver Transplantation and Non-Alcoholic Fatty Liver Disease. World J Gastroenterol (2014) 1420(42):15532–8. doi: 10.3748/wjg.v20.i42.15532
3. McCormack L, Dutkowski P, El-Badry AM, Clavien P-A. Liver Transplantation Using Fatty Livers: Always Feasible? J Hepatol (2011) 54(5):1055–62. doi: 10.1016/j.jhep.2010.11.004
4. Minervini MI, Ruppert K, Fontes P, Volpes R, Vizzini G, de Vera ME, et al. Liver Biopsy Findings From Healthy Potential Living Liver Donors: Reasons for Disqualification, Silent Diseases and Correlation With Liver Injury Tests. J Hepatol (2009) 50(3):501–10. doi: 10.1016/j.jhep.2008.10.030
5. Rajaram RB, Jayaraman T, Yoong B-K, Koh P-S, Loh PS, Koong J-K, et al. Non-Alcoholic Fatty Liver Disease and Obesity Among Adult Donors Are Major Challenges to Living-Donor Liver Transplantation: A Single-Center Experience. Asian J Surg (2022) 45(1):441–7. doi: 10.1016/j.asjsur.2021.07.046
6. Majumdar A, Tsochatzis EA. Changing Trends of Liver Transplantation and Mortality From Non-Alcoholic Fatty Liver Disease. Metabolism (2020) 111S:154291. doi: 10.1016/j.metabol.2020.154291
7. Muhammad H, Zaffar D, Tehreem A, Ting P-S, Simsek C, Turan I, et al. An Update on Usage of High-Risk Donors in Liver Transplantation. J Clin Med (2021) 11(1):215. doi: 10.3390/jcm11010215
8. Li J, Li RJ, Lv GY, Liu HQ. The Mechanisms and Strategies to Protect From Hepatic Ischemia-Reperfusion Injury. Eur Rev Med Pharmacol Sci (2015) 19(11):2036–47.
9. Elias-Miró M, Jiménez-Castro MB, Rodés J, Peralta C. Current Knowledge on Oxidative Stress in Hepatic Ischemia/Reperfusion. Free Radic Res (2013) 47(8):555–68. doi: 10.3109/10715762.2013.811721
10. Mailloux RJ, Harper M-E. Uncoupling Proteins and the Control of Mitochondrial Reactive Oxygen Species Production. Free Radic Biol Med (2011) 51(6):1106–15. doi: 10.1016/j.freeradbiomed.2011.06.022
11. Bhat AH, Dar KB, Anees S, Zargar MA, Masood A, Sofi MA, et al. Oxidative Stress, Mitochondrial Dysfunction and Neurodegenerative Diseases; a Mechanistic Insight. BioMed Pharmacother (2015) 74:101–10. doi: 10.1016/j.biopha.2015.07.025
12. Chang Y-Y, Liu Y-C, Kuo Y-H, Lin Y-L, Wu Y-HS, Chen J-W, et al. Effects of Antrosterol From Antrodia Camphorata Submerged Whole Broth on Lipid Homeostasis, Antioxidation, Alcohol Clearance, and Anti-Inflammation in Livers of Chronic-Alcohol Fed Mice. J Ethnopharmacol (2017) 202:200–7. doi: 10.1016/j.jep.2017.03.003
13. Kim J-S, Wang J-H, Lemasters JJ. Mitochondrial Permeability Transition in Rat Hepatocytes After Anoxia/Reoxygenation: Role of Ca2+-Dependent Mitochondrial Formation of Reactive Oxygen Species. Am J Physiol Gastrointest Liver Physiol (2012) 302(7):G723–31. doi: 10.1152/ajpgi.00082.2011
14. Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, et al. HMGB1 Release Induced by Liver Ischemia Involves Toll-Like Receptor 4 Dependent Reactive Oxygen Species Production and Calcium-Mediated Signaling. J Exp Med (2007) 204(12):2913–23. doi: 10.1084/jem.20070247
15. Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. Toll-Like Receptor 9 Inhibition Confers Protection From Liver Ischemia-Reperfusion Injury. Hepatology (2010) 51(2):621–32. doi: 10.1002/hep.23365
16. Uehara T, Bennett B, Sakata ST, Satoh Y, Bilter GK, Westwick JK, et al. JNK Mediates Hepatic Ischemia Reperfusion Injury. J Hepatol (2005) 42(6):850–9. doi: 10.1016/j.jhep.2005.01.030
17. Murphy MP. How Mitochondria Produce Reactive Oxygen Species. Biochem J (2009) 417(1):1–13. doi: 10.1042/BJ20081386
18. Glantzounis GK, Salacinski HJ, Yang W, Davidson BR, Seifalian AM. The Contemporary Role of Antioxidant Therapy in Attenuating Liver Ischemia-Reperfusion Injury: A Review. Liver Transpl (2005) 11(9):1031–47. doi: 10.1002/lt.20504
19. Zhang W, Wang M, Xie HY, Zhou L, Meng XQ, Shi J, et al. Role of Reactive Oxygen Species in Mediating Hepatic Ischemia-Reperfusion Injury and its Therapeutic Applications in Liver Transplantation. Transplant Proc (2007) 39(5):1332–7. doi: 10.1016/j.transproceed.2006.11.021
20. Cannistra M, Ruggiero M, Zullo A, Gallelli G, Serafini S, Maria M, et al. Hepatic Ischemia Reperfusion Injury: A Systematic Review of Literature and the Role of Current Drugs and Biomarkers. Int J Surg (2016) 33(Suppl 1):S57–70. doi: 10.1016/j.ijsu.2016.05.050
21. Casas AI, Nogales C, Mucke HAM, Petraina A, Cuadrado A, Rojo AI, et al. On the Clinical Pharmacology of Reactive Oxygen Species. Pharmacol Rev (2020) 72(4):801–28. doi: 10.1124/pr.120.019422
22. Fernández L, Heredia N, Grande L, Gómez G, Rimola A, Marco A, et al. Preconditioning Protects Liver and Lung Damage in Rat Liver Transplantation: Role of Xanthine/Xanthine Oxidase. Hepatology (2002) 36(3):562–72. doi: 10.1053/jhep.2002.34616
23. Peglow S, Toledo AH, Anaya-Prado R, Lopez-Neblina F, Toledo-Pereyra LH. Allopurinol and Xanthine Oxidase Inhibition in Liver Ischemia Reperfusion. J Hepatobiliary Pancreat Sci (2011) 18(2):137–46. doi: 10.1007/s00534-010-0328-7
24. Bedard K, Krause K-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol Rev (2007) 87(1):245–313. doi: 10.1152/physrev.00044.2005
25. Reiniers MJ, van Golen RF, van Gulik TM, Heger M. Reactive Oxygen and Nitrogen Species in Steatotic Hepatocytes: A Molecular Perspective on the Pathophysiology of Ischemia-Reperfusion Injury in the Fatty Liver. Antioxid Redox Signal (2014) 21(7):1119–42. doi: 10.1089/ars.2013.5486
26. Dhalla NS, Elmoselhi AB, Hata T, Makino N. Status of Myocardial Antioxidants in Ischemia-Reperfusion Injury. Cardiovasc Res (2000) 47(3):446–56. doi: 10.1016/S0008-6363(00)00078-X
27. Van Houten B, Woshner V, Santos JH. Role of Mitochondrial DNA in Toxic Responses to Oxidative Stress. DNA Repair (Amst) (2006) 5(2):145–52. doi: 10.1016/j.dnarep.2005.03.002
28. Liu P-G, He S-Q, Zhang Y-H, Wu J. Protective Effects of Apocynin and Allopurinol on Ischemia/Reperfusion-Induced Liver Injury in Mice. World J Gastroenterol (2008) 14(18):2832–7. doi: 10.3748/wjg.14.2832
29. Cutrn JC, Perrelli MG, Cavalieri B, Peralta C, Rosell Catafau J, Poli G. Microvascular Dysfunction Induced by Reperfusion Injury and Protective Effect of Ischemic Preconditioning. Free Radic Biol Med (2002) 33(9):1200–8. doi: 10.1016/S0891-5849(02)01017-1
30. El-Benna J, Dang PM-C, Gougerot-Pocidalo M-A. Priming of the Neutrophil NADPH Oxidase Activation: Role of P47phox Phosphorylation and NOX2 Mobilization to the Plasma Membrane. Semin Immunopathol (2008) 30(3):279–89. doi: 10.1007/s00281-008-0118-3
31. Casillas-Ramirez A, Mosbah IB, Ramalho F, Rosello-Catafau J, Peralta C. Past and Future Approaches to Ischemia-Reperfusion Lesion Associated With Liver Transplantation. Life Sci (2006) 79(20):1881–94. doi: 10.1016/j.lfs.2006.06.024
32. De Pascali F, Hemann C, Samons K, Chen C-A, Zweier JL. Hypoxia and Reoxygenation Induce Endothelial Nitric Oxide Synthase Uncoupling in Endothelial Cells Through Tetrahydrobiopterin Depletion and S-Glutathionylation. Biochemistry (2014) 53(22):3679–88. doi: 10.1021/bi500076r
33. Granger DN, Kvietys PR. Reperfusion Injury and Reactive Oxygen Species: The Evolution of a Concept. Redox Biol (2015) 6:524–51. doi: 10.1016/j.redox.2015.08.020
34. Phillips L, Toledo AH, Lopez-Neblina F, Anaya-Prado R, Toledo-Pereyra LH. Nitric Oxide Mechanism of Protection in Ischemia and Reperfusion Injury. J Invest Surg (2009) 22(1):46–55. doi: 10.1080/08941930802709470
35. Prieto I, Monsalve M. ROS Homeostasis, a Key Determinant in Liver Ischemic-Preconditioning. Redox Biol (2017) 12:1020–5. doi: 10.1016/j.redox.2017.04.036
36. Cichoż-Lach H, Michalak A. Oxidative Stress as a Crucial Factor in Liver Diseases. World J Gastroenterol (2014) 20(25):8082–91. doi: 10.3748/wjg.v20.i25.8082
37. Chen H-H, Chen Y-T, Yang C-C, Chen K-H, Sung P-H, Chiang H-J, et al. Melatonin Pretreatment Enhances the Therapeutic Effects of Exogenous Mitochondria Against Hepatic Ischemia-Reperfusion Injury in Rats Through Suppression of Mitochondrial Permeability Transition. J Pineal Res (2016) 61(1):52–68. doi: 10.1111/jpi.12326
38. Teoh NC. Hepatic Ischemia Reperfusion Injury: Contemporary Perspectives on Pathogenic Mechanisms and Basis for Hepatoprotection-the Good, Bad and Deadly. J Gastroenterol Hepatol (2011) 26 Suppl 1:180–7. doi: 10.1111/j.1440-1746.2010.06584.x
39. Xu L, Ge F, Hu Y, Yu Y, Guo K, Miao C. Sevoflurane Postconditioning Attenuates Hepatic Ischemia-Reperfusion Injury by Limiting HMGB1/TLR4/NF-κb Pathway Modulating microRNA-142. Front Pharmacol (2021) 12:646307. doi: 10.3389/fphar.2021.646307
40. Xu Z, Yu J, Wu J, Qi F, Wang H, Wang Z, et al. The Effects of Two Anesthetics, Propofol and Sevoflurane, on Liver Ischemia/Reperfusion Injury. Cell Physiol Biochem (2016) 38(4):1631–42. doi: 10.1159/000443103
41. Shaker ME, Trawick BN, Mehal WZ. The Novel TLR9 Antagonist COV08-0064 Protects From Ischemia/Reperfusion Injury in non-Steatotic and Steatotic Mice Livers. Biochem Pharmacol (2016) 112:90–101. doi: 10.1016/j.bcp.2016.05.003
42. Lin J, Huang H-F, Yang S-K, Duan J, Qu S-M, Yuan B, et al. The Effect of Ginsenoside Rg1 in Hepatic Ischemia Reperfusion (I/R) Injury Ameliorates Ischemia-Reperfusion-Induced Liver Injury by Inhibiting Apoptosis. BioMed Pharmacother (2020) 129:110398. doi: 10.1016/j.biopha.2020.110398
43. Varela AT, Rolo AP, Palmeira CM. Fatty Liver and Ischemia/Reperfusion: Are There Drugs Able to Mitigate Injury? Curr Med Chem (2011) 18(32):4987–5002. doi: 10.2174/092986711797535164
44. Wang C, Chen K, Xia Y, Dai W, Wang F, Shen M, et al. N-Acetylcysteine Attenuates Ischemia-Reperfusion-Induced Apoptosis and Autophagy in Mouse Liver via Regulation of the ROS/JNK/Bcl-2 Pathway. PloS One (2014) 9(9):e108855. doi: 10.1371/journal.pone.0108855
45. Cho H-I, Seo M-J, Lee S-M. 2-Methoxyestradiol Protects Against Ischemia/Reperfusion Injury in Alcoholic Fatty Liver by Enhancing Sirtuin 1-Mediated Autophagy. Biochem Pharmacol (2017) 131:40–51. doi: 10.1016/j.bcp.2017.02.008
46. Chun SK, Lee S, Flores-Toro J, RY U, Yang M-J, Go KL, et al. Loss of Sirtuin 1 and Mitofusin 2 Contributes to Enhanced Ischemia/Reperfusion Injury in Aged Livers. Aging Cell (2018) 17(4):e12761. doi: 10.1111/acel.12761
47. Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding Cell Death Signals in Liver Inflammation. J Hepatol (2013) 59(3):583–94. doi: 10.1016/j.jhep.2013.03.033
48. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by Caspase 8 Mediates the Mitochondrial Damage in the Fas Pathway of Apoptosis. Cell (1998) 94(4):491–501. doi: 10.1016/S0092-8674(00)81590-1
49. Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, et al. Bid-Deficient Mice Are Resistant to Fas-Induced Hepatocellular Apoptosis. Nature (1999) 400(6747):886–91. doi: 10.1038/23730
50. Harper N, Hughes M, MacFarlane M, Cohen GM. Fas-Associated Death Domain Protein and Caspase-8 Are Not Recruited to the Tumor Necrosis Factor Receptor 1 Signaling Complex During Tumor Necrosis Factor-Induced Apoptosis. J Biol Chem (2003) 278(28):25534–41. doi: 10.1074/jbc.M303399200
51. Kang J-W, Hong J-M, Lee S-M. Melatonin Enhances Mitophagy and Mitochondrial Biogenesis in Rats With Carbon Tetrachloride-Induced Liver Fibrosis. J Pineal Res (2016) 60(4):383–93. doi: 10.1111/jpi.12319
52. Spahis S, Delvin E, Borys J-M, Levy E. Oxidative Stress as a Critical Factor in Nonalcoholic Fatty Liver Disease Pathogenesis. Antioxid Redox Signal (2017) 26(10):519–41. doi: 10.1089/ars.2016.6776
53. Taylor RS, Taylor RJ, Bayliss S, Hagstrom H, Nasr P, Schattenberg JM, et al. Association Between Fibrosis Stage and Outcomes of Patients With Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Gastroenterology (2020) 158(6):1611–25.e12. doi: 10.1053/j.gastro.2020.01.043
54. Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, et al. A Lipidomic Analysis of Nonalcoholic Fatty Liver Disease. Hepatology (2007) 46(4):1081–90. doi: 10.1002/hep.21763
55. Tashiro H, Kuroda S, Mikuriya Y, Ohdan H. Ischemia–reperfusion Injury in Patients With Fatty Liver and the Clinical Impact of Steatotic Liver on Hepatic Surgery. Surg Today (2014) 44(9):1611–25. doi: 10.1007/s00595-013-0736-9
56. Malaguarnera L, Madeddu R, Palio E, Arena N, Malaguarnera M. Heme Oxygenase-1 Levels and Oxidative Stress-Related Parameters in non-Alcoholic Fatty Liver Disease Patients. J Hepatol (2005) 42(4):585–91. doi: 10.1016/j.jhep.2004.11.040
57. Videla LA, Rodrigo R, Orellana M, Fernandez V, Tapia G, Quiñones L, et al. Oxidative Stress-Related Parameters in the Liver of non-Alcoholic Fatty Liver Disease Patients. Clin Sci (Lond) (2004) 106(3):261–8. doi: 10.1042/CS20030285
58. Serviddio G, Sastre J, Bellanti F, Vina J, Vendemiale G, Altomare E. Mitochondrial Involvement in non-Alcoholic Steatohepatitis. Mol Aspects Med (2008) 29(1-2):22–35. doi: 10.1016/j.mam.2007.09.014
59. Fromenty B, Robin MA, Igoudjil A, Mansouri A, Pessayre D. The Ins and Outs of Mitochondrial Dysfunction in NASH. Diabetes Metab (2004) 30(2):121–38. doi: 10.1016/S1262-3636(07)70098-8
60. Svegliati-Baroni G, Pierantonelli I, Torquato P, Marinelli R, Ferreri C, Chatgilialoglu C, et al. Lipidomic Biomarkers and Mechanisms of Lipotoxicity in Non-Alcoholic Fatty Liver Disease. Free Radic Biol Med (2019) 144:293–309. doi: 10.1016/j.freeradbiomed.2019.05.029
61. Nassir F, Ibdah JA. Role of Mitochondria in Nonalcoholic Fatty Liver Disease. Int J Mol Sci (2014) 15(5):8713–42. doi: 10.3390/ijms15058713
62. Bessone F, Razori MV, Roma MG. Molecular Pathways of Nonalcoholic Fatty Liver Disease Development and Progression. Cell Mol Life Sci (2019) 76(1):99–128. doi: 10.1007/s00018-018-2947-0
63. Llacuna L, Fernández A, Montfort CV, Matías N, Martínez L, Caballero F, et al. Targeting Cholesterol at Different Levels in the Mevalonate Pathway Protects Fatty Liver Against Ischemia-Reperfusion Injury. J Hepatol (2011) 54(5):1002–10. doi: 10.1016/j.jhep.2010.08.031
64. Poli G, Schaur RJ, Siems WG, Leonarduzzi G. 4-Hydroxynonenal: A Membrane Lipid Oxidation Product of Medicinal Interest. Med Res Rev (2008) 28(4):569–631. doi: 10.1002/med.20117
65. Hardwick RN, Fisher CD, Canet MJ, Lake AD, Cherrington NJ. Diversity in Antioxidant Response Enzymes in Progressive Stages of Human Nonalcoholic Fatty Liver Disease. Drug Metab Dispos (2010) 38(12):2293–301. doi: 10.1124/dmd.110.035006
66. Teoh NC, Williams J, Hartley J, Yu J, McCuskey RS, Farrell GC. Short-Term Therapy With Peroxisome Proliferation-Activator Receptor-Alpha Agonist Wy-14,643 Protects Murine Fatty Liver Against Ischemia-Reperfusion Injury. Hepatology (2010) 51(3):996–1006. doi: 10.1002/hep.23420
67. Akahori T, Sho M, Hamada K, Suzaki Y, Kuzumoto Y, Nomi T, et al. Importance of Peroxisome Proliferator-Activated Receptor-Gamma in Hepatic Ischemia/Reperfusion Injury in Mice. J Hepatol (2007) 47(6):784–92. doi: 10.1016/j.jhep.2007.07.030
68. Kuboki S, Shin T, Huber N, Eismann T, Galloway E, Schuster R, et al. Peroxisome Proliferator-Activated Receptor-Gamma Protects Against Hepatic Ischemia/Reperfusion Injury in Mice. Hepatology (2008) 47(1):215–24. doi: 10.1002/hep.21963
69. Wang C, Li Z, Zhao B, Wu Y, Fu Y, Kang K, et al. PGC-1 Protects Against Hepatic Ischemia Reperfusion Injury by Activating PPAR and PPAR and Regulating ROS Production. Oxid Med Cell Longev (2021) 2021:6677955. doi: 10.1155/2021/6677955
70. Clavien PA, Yadav S, Sindram D, Bentley RC. Protective Effects of Ischemic Preconditioning for Liver Resection Performed Under Inflow Occlusion in Humans. Ann Surg (2000) 232(2):155–62. doi: 10.1097/00000658-200008000-00001
71. Massip-Salcedo M, Casillas-Ramirez A, Franco-Gou R, Bartrons R, Ben Mosbah I, Serafin A, et al. Heat Shock Proteins and Mitogen-Activated Protein Kinases in Steatotic Livers Undergoing Ischemia-Reperfusion: Some Answers. Am J Pathol (2006) 168(5):1474–85. doi: 10.2353/ajpath.2006.050645
72. Lv X, Yang L, Tao K, Liu Y, Yang T, Chen G, et al. Isoflurane Preconditioning at Clinically Relevant Doses Induce Protective Effects of Heme Oxygenase-1 on Hepatic Ischemia Reperfusion in Rats. BMC Gastroenterol (2011) 11:31. doi: 10.1186/1471-230X-11-31
73. Yuan B, Huang H, Qu S, Zhang H, Lin J, Jin L, et al. Gastrodin Pretreatment Protects Liver Against Ischemia-Reperfusion Injury via Activation of the Nrf2/HO-1 Pathway. Am J Chin Med (2020) 48(5):1159–78. doi: 10.1142/S0192415X20500573
74. Evans ZP, Mandavilli BS, Ellett JD, Rodwell D, Fariss MW, Fiorini RN, et al. Vitamin E Succinate Enhances Steatotic Liver Energy Status and Prevents Oxidative Damage Following Ischemia/Reperfusion. Transplant Proc (2009) 41(10):4094–8. doi: 10.1016/j.transproceed.2009.09.055
75. Zazueta C, Buelna-Chontal M, Macías-López A, Román-Anguiano NG, González-Pacheco H, Pavón N, et al. Cytidine-5’-Diphosphocholine Protects the Liver From Ischemia/Reperfusion Injury Preserving Mitochondrial Function and Reducing Oxidative Stress. Liver Transpl (2018) 24(8):1070–83. doi: 10.1002/lt.25179
76. He S-Q, Zhang Y-H, Venugopal SK, Dicus CW, Perez RV, Ramsamooj R, et al. Delivery of Antioxidative Enzyme Genes Protects Against Ischemia/Reperfusion-Induced Liver Injury in Mice. Liver Transpl (2006) 12(12):1869–79. doi: 10.1002/lt.21001
77. Cayuela NC, Koike MK, Jacysyn JF, Rasslan R, Azevedo ARA, Costa SKP, et al. N-Acetylcysteine Reduced Ischemia and Reperfusion Damage Associated With Steatohepatitis in Mice. Int J Mol Sci (2020) 21(11):4106. doi: 10.3390/ijms21114106
78. Fusai G, Glantzounis GK, Hafez T, Yang W, Quaglia A, Sheth H, et al. N-Acetylcysteine Ameliorates the Late Phase of Liver Ischaemia/Reperfusion Injury in the Rabbit With Hepatic Steatosis. Clin Sci (Lond) (2005) 109(5):465–73. doi: 10.1042/CS20050081
79. Kireev R, Bitoun S, Cuesta S, Tejerina A, Ibarrola C, Moreno E, et al. Melatonin Treatment Protects Liver of Zucker Rats After Ischemia/Reperfusion by Diminishing Oxidative Stress and Apoptosis. Eur J Pharmacol (2013) 701(1-3):185–93. doi: 10.1016/j.ejphar.2012.11.038
80. Du Y, Qian B, Gao L, Tan P, Chen H, Wang A, et al. Aloin Preconditioning Attenuates Hepatic Ischemia/Reperfusion Injury via Inhibiting TLR4/MyD88/NF-B Signal Pathway and. Oxid Med Cell Longev (2019) 2019:3765898. doi: 10.1155/2019/3765898
81. Tao J, Shen X, Ai Y, Han X. Tea Polyphenols Protect Against Ischemia/Reperfusion-Induced Liver Injury in Mice Through Anti-Oxidative and Anti-Apoptotic Properties. Exp Ther Med (2016) 12(5):3433–9. doi: 10.3892/etm.2016.3789
82. Xu Z-C, Yin J, Zhou B, Liu Y-T, Yu Y, Li G-Q. Grape Seed Proanthocyanidin Protects Liver Against Ischemia/Reperfusion Injury by Attenuating Endoplasmic Reticulum Stress. World J Gastroenterol (2015) 21(24):7468–77. doi: 10.3748/wjg.v21.i24.7468
83. Bi J, Zhang J, Ren Y, Du Z, Li Q, Wang Y, et al. Irisin Alleviates Liver Ischemia-Reperfusion Injury by Inhibiting Excessive Mitochondrial Fission, Promoting Mitochondrial Biogenesis and Decreasing Oxidative Stress. Redox Biol (2019) 20:296–306. doi: 10.1016/j.redox.2018.10.019
84. Amersi F, Buelow R, Kato H, Ke B, Coito AJ, Shen XD, et al. Upregulation of Heme Oxygenase-1 Protects Genetically Fat Zucker Rat Livers From Ischemia/Reperfusion Injury. J Clin Invest (1999) 104(11):1631–9. doi: 10.1172/JCI7903
85. Liu T, Zhang Q, Mo W, Yu Q, Xu S, Li J, et al. The Protective Effects of Shikonin on Hepatic Ischemia/Reperfusion Injury are Mediated by the Activation of the PI3K/Akt Pathway. Sci Rep (2017) 7:44785. doi: 10.1038/srep44785
86. Yang J, Chen Q, Tian S, Song S, Liu F, Wang Q, et al. The Role of 1,25-Dyhydroxyvitamin D3 in Mouse Liver Ischemia Reperfusion Injury: Regulation of Autophagy Through Activation of MEK/ERK Signaling and PTEN/PI3K/Akt/mTORC1 Signaling. Am J Transl Res (2015) 7(12):2630–45.
87. Gomez D, Homer-Vanniasinkam S, Graham AM, Prasad KR. Role of Ischaemic Preconditioning in Liver Regeneration Following Major Liver Resection and Transplantation. World J Gastroenterol (2007) 13(5):657–70. doi: 10.3748/wjg.v13.i5.657
88. Fernández L, Carrasco-Chaumel E, Serafín A, Xaus C, Grande L, Rimola A, et al. Is Ischemic Preconditioning a Useful Strategy in Steatotic Liver Transplantation? Am J Transplant (2004) 4(6):888–99. doi: 10.1111/j.1600-6143.2004.00447.x
89. Zhang H, Yan Q, Wang X, Chen X, Chen Y, Du J, et al. The Role of Mitochondria in Liver Ischemia-Reperfusion Injury: From Aspects of Mitochondrial Oxidative Stress, Mitochondrial Fission, Mitochondrial Membrane Permeable Transport Pore Formation, Mitophagy, and Mitochondria-Related Protective Measures. Oxid Med Cell Longev (2021) 2021:6670579. doi: 10.1155/2021/6670579
90. Krautbauer S, Eisinger K, Lupke M, Wanninger J, Ruemmele P, Hader Y, et al. Manganese Superoxide Dismutase Is Reduced in the Liver of Male But Not Female Humans and Rodents With Non-Alcoholic Fatty Liver Disease. Exp Mol Pathol (2013) 95(3):330–5. doi: 10.1016/j.yexmp.2013.10.003
91. Grendar J, Ouellet JF, McKay A, Sutherland FR, Bathe OF, Ball CG, et al. Effect of N-Acetylcysteine on Liver Recovery After Resection: A Randomized Clinical Trial. J Surg Oncol (2016) 114(4):446–50. doi: 10.1002/jso.24312
92. Robinson SM, Saif R, Sen G, French JJ, Jaques BC, Charnley RM, et al. N-Acetylcysteine Administration Does Not Improve Patient Outcome After Liver Resection. HPB (Oxford) (2013) 15(6):457–62. doi: 10.1111/hpb.12005
93. D’Amico F, Vitale A, Piovan D, Bertacco A, Ramirez Morales R, Chiara Frigo A, et al. Use of N-Acetylcysteine During Liver Procurement: A Prospective Randomized Controlled Study. Liver Transpl (2013) 19(2):135–44. doi: 10.1002/lt.23527
94. Nickkholgh A, Schneider H, Sobirey M, Venetz WP, Hinz U, Pelzl LH, et al. The Use of High-Dose Melatonin in Liver Resection is Safe: First Clinical Experience. J Pineal Res (2011) 50(4):381–8. doi: 10.1111/j.1600-079X.2011.00854.x
95. Gong Y, Liu Z, Liao Y, Mai C, Chen T, Tang H, et al. Effectiveness of ω-3 Polyunsaturated Fatty Acids Based Lipid Emulsions for Treatment of Patients After Hepatectomy: A Prospective Clinical Trial. Nutrients (2016) 8(6):357. doi: 10.3390/nu8060357
96. Romero-Gómez M, Zelber-Sagi S, Trenell M. Treatment of NAFLD With Diet, Physical Activity and Exercise. J Hepatol (2017) 67(4):829–46. doi: 10.1016/j.jhep.2017.05.016
97. Ajami M, Davoodi SH, Habibey R, Namazi N, Soleimani M, Pazoki-Toroudi H. Effect of DHA+EPA on Oxidative Stress and Apoptosis Induced by Ischemia-Reperfusion in Rat Kidneys. Fundam Clin Pharmacol (2013) 27(6):593–602. doi: 10.1111/j.1472-8206.2012.01066.x
98. El-Badry AM, Moritz W, Contaldo C, Tian Y, Graf R, Clavien P-A. Prevention of Reperfusion Injury and Microcirculatory Failure in Macrosteatotic Mouse Liver by Omega-3 Fatty Acids. Hepatology (2007) 45(4):855–63. doi: 10.1002/hep.21625
99. Zhu X-H, Wu Y-F, Qiu Y-D, Jiang C-P, Ding Y-T. Liver-Protecting Effects of Omega-3 Fish Oil Lipid Emulsion in Liver Transplantation. World J Gastroenterol (2012) 18(42):6141–7. doi: 10.3748/wjg.v18.i42.6141
100. Linecker M, Botea F, Aristotele Raptis D, Nicolaescu D, Limani P, Alikhanov R, et al. Perioperative Omega-3 Fatty Acids Fail to Confer Protection in Liver Surgery: Results of a Multicentric, Double-Blind, Randomized Controlled Trial. J Hepatol (2020) 72(3):498–505. doi: 10.1016/j.jhep.2019.10.004
101. Gao Y, Chu S, Shao Q, Zhang M, Xia C, Wang Y, et al. Antioxidant Activities of Ginsenoside Rg1 Against Cisplatin-Induced Hepatic Injury Through Nrf2 Signaling Pathway in Mice. Free Radic Res (2017) 51(1):1–13. doi: 10.1080/10715762.2016.1234710
102. Liu Y, Zhang W, Cheng Y, Miao C, Gong J, Wang M. Activation of Pparγ by Curcumin Protects Mice From Ischemia/Reperfusion Injury Induced by Orthotopic Liver Transplantation via Modulating Polarization of Kupffer Cells. Int Immunopharmacol (2018) 62:270–6. doi: 10.1016/j.intimp.2018.07.013
103. Shen S-Q, Zhang Y, Xiang J-J, Xiong C-L. Protective Effect of Curcumin Against Liver Warm Ischemia/Reperfusion Injury in Rat Model is Associated With Regulation of Heat Shock Protein and Antioxidant Enzymes. World J Gastroenterol (2007) 13(13):1953–61. doi: 10.3748/wjg.v13.i13.1953
104. Klaassen CD, Reisman SA. Nrf2 the Rescue: Effects of the Antioxidative/Electrophilic Response on the Liver. Toxicol Appl Pharmacol (2010) 244(1):57–65. doi: 10.1016/j.taap.2010.01.013
105. Xu D, Xu M, Jeong S, Qian Y, Wu H, Xia Q, et al. The Role of Nrf2 in Liver Disease: Novel Molecular Mechanisms and Therapeutic Approaches. Front Pharmacol (2018) 9:1428. doi: 10.3389/fphar.2018.01428
106. Kudoh K, Uchinami H, Yoshioka M, Seki E, Yamamoto Y. Nrf2 Activation Protects the Liver From Ischemia/Reperfusion Injury in Mice. Ann Surg (2014) 260(1):118–27. doi: 10.1097/SLA.0000000000000287
107. Bauer M, Bauer I. Heme Oxygenase-1: Redox Regulation and Role in the Hepatic Response to Oxidative Stress. Antioxid Redox Signal (2002) 4(5):749–58. doi: 10.1089/152308602760598891
108. Yun N, Eum H-A, Lee S-M. Protective Role of Heme Oxygenase-1 Against Liver Damage Caused by Hepatic Ischemia and Reperfusion in Rats. Antioxid Redox Signal (2010) 13(10):1503–12. doi: 10.1089/ars.2009.2873
109. Oshita A, Tashiro H, Amano H, Kobayashi T, Onoe T, Ide K, et al. Safety and Feasibility of Diet-Treated Donors With Steatotic Livers at the Initial Consultation for Living-Donor Liver Transplantation. Transplantation (2012) 93(10):1024–30. doi: 10.1097/TP.0b013e31824c9e25
110. Fujii Y, Kawamura N, Zaitsu M, Watanabe M, Goto R, Kamiyama T, et al. Outcome of Living-Donor Liver Transplantation Using Grafts From Donors Treated for Fatty Liver. Ann Transplant (2020) 25:e920677. doi: 10.12659/AOT.920677
111. Hwang S, Lee S-G, Jang S-J, Cho S-H, Kim K-H, Ahn C-S, et al. The Effect of Donor Weight Reduction on Hepatic Steatosis for Living Donor Liver Transplantation. Liver Transpl (2004) 10(6):721–5. doi: 10.1002/lt.20172
112. Doyle A, Adeyi O, Khalili K, Fischer S, Dib M, Goldaracena N, et al. Treatment With Optifast Reduces Hepatic Steatosis and Increases Candidacy Rates for Living Donor Liver Transplantation. Liver Transpl (2016) 22(9):1295–300. doi: 10.1002/lt.24495
113. Choudhary NS, Saraf N, Saigal S, Gautam D, Lipi L, Rastogi A, et al. Rapid Reversal of Liver Steatosis With Life Style Modification in Highly Motivated Liver Donors. J Clin Exp Hepatol (2015) 5(2):123–6. doi: 10.1016/j.jceh.2015.04.002
114. Nakamuta M, Morizono S, Soejima Y, Yoshizumi T, Aishima S, Takasugi S-i, et al. Short-Term Intensive Treatment for Donors With Hepatic Steatosis in Living-Donor Liver Transplantation. Transplantation (2005) 80(5):608–12. doi: 10.1097/01.tp.0000166009.77444.f3
115. Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N Engl J Med (2021) 385(17):1547–58. doi: 10.1056/NEJMoa2036205
116. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology (2016) 150(5):1147–59.e5. doi: 10.1053/j.gastro.2016.01.038
117. Gawrieh S, Noureddin M, Loo N, Mohseni R, Awasty V, Cusi K, et al. Saroglitazar, a PPAR-α/γ Agonist, for Treatment of NAFLD: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology (2021) 74(4):1809–24. doi: 10.1002/hep.31843
118. de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic Roles of Bile Acids in Metabolism. Cell Metab (2013) 17(5):657–69. doi: 10.1016/j.cmet.2013.03.013
119. Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, et al. Bile Acids Lower Triglyceride Levels via a Pathway Involving FXR, SHP, and SREBP-1c. J Clin Invest (2004) 113(10):1408–18. doi: 10.1172/JCI21025
120. Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall H-U, Kipnes M, et al. Efficacy and Safety of the Farnesoid X Receptor Agonist Obeticholic Acid in Patients With Type 2 Diabetes and Nonalcoholic Fatty Liver Disease. Gastroenterology (2013) 145(3):574–82.e1. doi: 10.1053/j.gastro.2013.05.042
121. Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic Acid for the Treatment of non-Alcoholic Steatohepatitis: Interim Analysis From a Multicentre, Randomised, Placebo-Controlled Phase 3 Trial. Lancet (2019) 394(10215):2184–96. doi: 10.1016/S0140-6736(19)33041-7
122. Harrison SA, Bashir MR, Lee K-J, Shim-Lopez J, Lee J, Wagner B, et al. A Structurally Optimized FXR Agonist, MET409, Reduced Liver Fat Content Over 12 Weeks in Patients With non-Alcoholic Steatohepatitis. J Hepatol (2021) 75(1):25–33. doi: 10.1016/j.jhep.2021.01.047
123. Patel K, Harrison SA, Elkhashab M, Trotter JF, Herring R, Rojter SE, et al. Cilofexor, a Nonsteroidal FXR Agonist, in Patients With Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial. Hepatology (2020) 72(1):58–71. doi: 10.1002/hep.31205
124. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast Growth Factor 15 Functions as an Enterohepatic Signal to Regulate Bile Acid Homeostasis. Cell Metab (2005) 2(4):217–25. doi: 10.1016/j.cmet.2005.09.001
125. Kliewer SA, Mangelsdorf DJ. Bile Acids as Hormones: The FXR-FGF15/19 Pathway. Dig Dis (2015) 33(3):327–31. doi: 10.1159/000371670
126. Harrison SA, Rinella ME, Abdelmalek MF, Trotter JF, Paredes AH, Arnold HL, et al. NGM282 for Treatment of non-Alcoholic Steatohepatitis: A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet (2018) 391(10126):1174–85. doi: 10.1016/S0140-6736(18)30474-4
127. Harrison SA, Neff G, Guy CD, Bashir MR, Paredes AH, Frias JP, et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients With Nonalcoholic Steatohepatitis. Gastroenterology (2021) 160(1):219–31.e1. doi: 10.1053/j.gastro.2020.08.004
128. du Plessis J, van Pelt J, Korf H, Mathieu C, van der Schueren B, Lannoo M, et al. Association of Adipose Tissue Inflammation With Histologic Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology (2015) 149(3):635–48.e14. doi: 10.1053/j.gastro.2015.05.044
129. Ratziu V, Sanyal A, Harrison SA, Wong VW-S, Francque S, Goodman Z, et al. Cenicriviroc Treatment for Adults With Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology (2020) 72(3):892–905. doi: 10.1002/hep.31108
130. Harrison SA, Wong VW-S, Okanoue T, Bzowej N, Vuppalanchi R, Younes Z, et al. Selonsertib for Patients With Bridging Fibrosis or Compensated Cirrhosis Due to NASH: Results From Randomized Phase III STELLAR Trials. J Hepatol (2020) 73(1):26–39. doi: 10.1016/j.jhep.2020.02.027
Keywords: ROS, hepatic ischemia-reperfusion, fatty liver, apoptosis, inflammation
Citation: Tang S-p, Mao X-l, Chen Y-h, Yan L-l, Ye L-p and Li S-w (2022) Reactive Oxygen Species Induce Fatty Liver and Ischemia-Reperfusion Injury by Promoting Inflammation and Cell Death. Front. Immunol. 13:870239. doi: 10.3389/fimmu.2022.870239
Received: 09 February 2022; Accepted: 07 April 2022;
Published: 29 April 2022.
Edited by:
Samer Tohme, University of Pittsburgh, United StatesReviewed by:
Weitao Que, Shanghai General Hospital, ChinaJohn D. Imig, Medical College of Wisconsin, United States
Copyright © 2022 Tang, Mao, Chen, Yan, Ye and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li-ping Ye, yelp@enzemed.com; Shao-wei Li, li_shaowei81@hotmail.com
†These authors have contributed equally to this work