 Lan Wang
Lan Wang Zheng Cao
Zheng Cao Zi Wang
Zi Wang Jimin Guo
Jimin Guo Jing Wen
Jing Wen- 1Department of Microbiology, Immunology and Molecular Genetics, David Geffen School of Medicine, University of California Los Angeles (UCLA), Los Angeles, CA, United States
- 2UCLA Acquired Immune Deficiency Syndrome (AIDS) Institute, University of California Los Angeles (UCLA), Los Angeles, CA, United States
- 3Department of Chemical and Biomolecular Engineering, University of California Los Angeles (UCLA), Los Angeles, CA, United States
An appropriate level of reactive oxygen species (ROS) is necessary for cell proliferation, signaling transduction, and apoptosis due to their highly reactive character. ROS are generated through multiple metabolic pathways under a fine-tuned control between oxidant and antioxidant signaling. A growing number of evidence has proved their highly relevant role in modulating inflammation during influenza virus infection. As a network of biological process for protecting organism from invasion of pathogens, immune system can react and fight back through either innate immune system or adaptive immune system, or both. Herein, we provide a review about the mechanisms of ROS generation when encounter influenza virus infection, and how the imbalanced level of ROS influences the replication of virus. We also summarize the pathways used by both the innate and adaptive immune system to sense and attack the invaded virus and abnormal levels of ROS. We further review the limitation of current strategies and discuss the direction of future work.
Introduction
Reactive oxygen species (ROS) are a class of partially reduced metabolites of oxygen (O2). There are two categories, radical and non-radical, of ROS based on the number of unpaired electrons in their outmost shell (1). ROS is highly reactive and can oxidize intracellular macromolecules in response to endogenous and/or exogenous stimuli, which makes them crucial elements for cellular activities (2). The production of ROS is fine-tuned by oxidant and antioxidant mechanisms under physiological metabolism. An appropriate level of ROS is necessary for cellular processes, such as cell growth, signaling transduction, and apoptosis (3). It is believed that excessive accumulation of ROS leads to aggravation of inflammation, augment of protease secretion, and accumulation of ROS intermediates, which ultimately resulting in inflammation response, apoptosis, and tissue injury (4). Evidence has proved that ROS have served as a crucial contributor to viral disease. This review focuses on the impact of ROS on viral replication and immune response during influenza virus (IV) infection.
ROS production after IAV infection
IV is a family of negative-sense single-stranded RNA virus which belongs to Orthomyxoviridae. It can be classified into four major genera, including Alphainfluenzavirus (Influenza A virus, IAV), Betainfluenzavirus (Influenza B virus, IBV), Gammainfluenzavirus (Influenza C virus, ICV), and Deltainfluenzavirus (Influenza D virus, IDV). Since IAV is responsible for the majority of seasonal epidemic or pandemic threat to human (5, 6), this review focuses on the effect of ROS induced by IAV infection. Overall, dysfunctional mitochondria, activated nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase), and inhibited antioxidant signaling pathway and enzymes, are three main mechanisms involved in the multi-coordinated process of ROS production post IAV infection (7) (Figure 1).
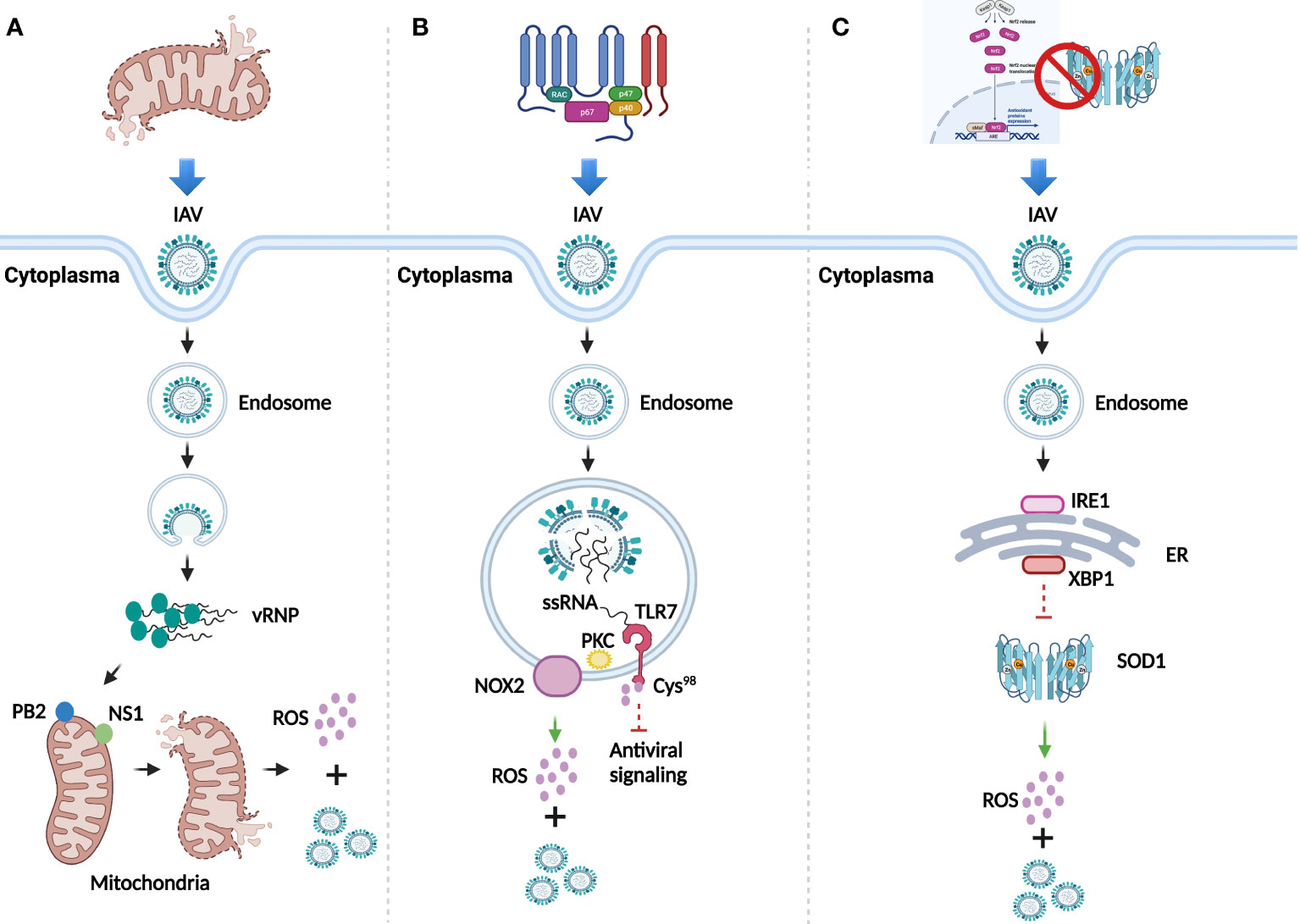
Figure 1 ROS generation after IAV infection. (A) Dysfunction of mitochondria mediated by IAV infection for the generation of ROS and viral replication. (B) ROS generation and viral replication mediated by activation of NADPH oxidase. (C) Inhibition of antioxidant signaling pathway and antioxidant enzymes for ROS generation and viral replication.
Dysfunction of mitochondria
Mitochondria, the powerhouse of the cells, can generate ROS through respiration. Viral polymerase is responsible for viral genome replication and transcription in the nuclei of host cells (8). IAV-dependent RNA polymerase complex contains a subunit, the polymerase basic 2 (PB2) protein, which can import IAV into the matrix space of mitochondria due to its N-terminal mitochondrial targeting sequence. Accumulation of IAV in the mitochondria breaks the equilibrium of mitochondrial fusion and fission, causing their fragmentation (9) and leading to the leak of superoxide ion into the cytoplasm (10). Besides PB2, the nonstructural protein 1 (NS1) can also induce fragmentation of mitochondria by altering its dynamics (11), which indicates the stimulation role of NS1 in ROS production. An increased level of ROS promotes the replication of IAV to enhance its pathogenesis (12) (Figure 1A).
Activation of NADPH oxidase
NADPH oxidase also known as NOX, is a membrane-bound complex present in cells and phagosomes. Human NOX isoforms comprise NOX1 to NOX5, dual oxidase 1 (DUOX1), and DUOX2 (13). Evidence has proved that ROS significantly increase in lung epithelial cell lines and primary murine cells in a NOX4-dependent manner after IAV infection (14). Besides, observed in primary murine macrophages, endosomal NOX2-induced ROS generation was mediated by the toll-like receptor 7 (TLR7) and subsequent activation of protein kinase C (PKC), while the generated ROS further suppressed the antiviral signaling through the modification of cysteine residue Cys98 of TLR7 to support viral proliferation (15) (Figure 1B). Inhibition of NOX2 significantly attenuated neutrophil influx, alveolitis, and ROS generation from inflammatory cells in IAV infected C57Bl/6 mice, which confirmed the role of NOX2 oxidase for promoting ROS generation (16). Additionally, the presence of IAV promotes the translocation of p67-phox, a cytosolic protein of NOX, to the cell membrane for ROS production at the early infection stage (17).
Inhibition of antioxidant signaling pathway and antioxidant enzymes
The Nrf2/Keap1 pathway is the principal antioxidant signaling cascade for protecting against ROS stress. Evidence has showed that in isolated human alveolar type II (ATII) cells and alveolar macrophages (AM), IAV infection increases the level of ROS and translocation of Nrf2 to the nuclei, whereas overexpression of Nrf2 decreases viral replication and oxidative stress (18). In addition, downregulated Nrf2 and upregulated ROS were observed in both IAV infected primary normal human bronchial epithelial (pNHBE) cells and BALB/c mice with IAV infection (19). Besides antioxidant signaling pathway, ROS can also be neutralized by antioxidant enzymes, such as superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx). The investigation of IAV infection in human alveolar cells has showed that the loss of copper-zinc SOD1 contributed to the increased level of superoxide anion and viral replication (20). A study has further disclosed the underlying mechanism that endoplasmic reticulum (ER)-associated degradation (ERAD) regulates the redox state to potentiate IAV infection (21). The presence of IAV first stimulated inositol-requiring 1(IRE1), a core sensor of ER stress signaling pathway, then IRE1 activated downstream factor X-box binding protein-1 (XBP1) through mRNA splicing, which could further promote ERAD. The whole process is likely to be responsible for the reduction of SOD1, thus leading to the increase of ROS and viral replication (Figure 1C).
Other mechanisms
Apart from the three main mechanisms, other mechanisms of IAV to produce ROS and favor its replication have been proposed. In human lung cancer A549 cells, decreased sirtuin 2(SIRT2) reduces the expression and activity of glucose-6-phosphate dehydrogenase (G6PD) by acetylation, resulting in enhanced production of ROS and IAV replication (22). The binding between aryl hydrocarbon receptor (AhR) and its ligand, quinone 1 (NQO1) is also reported to involve in the production of ROS (5, 23, 24). Additionally, neutrophil is another major source of ROS either via a neutrophil elastase (NE) mediated mechanism (25) or myeloperoxidase (MPO) enzyme (26) to assist pathogen clearance.
The immune response mediated by ROS responding to IAV infection
The innate and adaptive immune systems are critical for pathogen-specific defense. Substantial evidence has revealed that ROS are essential messengers in immune cells. The balanced production and elimination of ROS maintains a healthy immune system in physiological situation. The presence of virus induces increased levels and disturbs the balance of ROS within immune cells, resulting in activation of both innate and adaptive immune response. Additionally, ROS not only work as critical components of the host to fight against the invading virus, but also possess significant role to transmit signals from multiple signaling pathways to regulate the phenotype and function of immune cells (27). However, accumulated ROS in pathological condition may persistently stimulate the immune system and induce hyperactivation of inflammatory responses, resulting in tissue damage and pathology (28, 29).
Innate immune response
Inflammasome
Inflammasome pathway enables the detection of pathogens, release of cytokines, and recruitment of effector cell to the infection site (30). It shows that stimulation of NF-κB activates NLRP3 inflammasome (31). IAV virulence protein, PB1-F2, can activate NLRP3 inflammasome as well, but also impaired its activation via various mechanisms (31). Besides, mitochondrial-derived ROS are proved to promote the activity of NLRP3 inflammasome (32, 33). This activation could further drive the expression of interleukin-1β (IL-1β) (33). A novel MxA inflammasome has also been reported to be involved in IAV mediated immune response (34). In respiratory epithelial cells, MxA recognizes the nucleoprotein of IAV, then interacts with ASC to release IL-1β through the activation of caspase 1 (35).
Neutrophils
Neutrophils are first responders to be recruited to the site of infection and recognize IAV through TLR7/8 (36). A mode used by neutrophils for their recruitment in lung microenvironment is due to the induction of chemokine receptors (37). By using single-cell RNA sequencing assay, a study shows that PD-L1+ neutrophils are the major contributor for releasing pro-inflammatory factors in the first 1-3 days post infection (38). The function of neutrophils during IAV infection is controversial (39). A protective role is found to inhibit viral spread based on mice data that, early recruitment of neutrophils and their derived chemokine CXCL12 were essential for the migration of CD8+ T-cells to infected trachea (40). Another report proved that CD11b/CD18 integrin (MAC-1) helped neutrophil to suppress IAV-induced, T-cell mediated pathology probably by restraining the proliferation of T-cells (41). Neutrophil-derived secretion of IL-1β is thought to protect against virus. By releasing peptide mCRAMP, NLRP3 inflammasomes in alveolar macrophages were activated by neutrophils, which led to the release of IL-1β to combat infection (42). However, hyperactivated neutrophils with excessive recruitment, robust inflammatory reaction, and neutrophil extracellular traps (NETs) (39), can induce sever lethal effect in the acute respiratory distress syndrome (ARDS) caused by IAV infection (43). NETs contain histone and granule proteins, their formation relies on neutrophil produced ROS, NE and MPO (44). Evidence has demonstrated that high levels of NETs are correlated with poor prognosis of severe IAV infection and fatality in patients (45, 46).
Macrophages
Alveolar macrophages (AMs) are the most relevant macrophages for initiating inflammatory and immune responses to IAV infection in the lung. An animal study shows that a group of platelet factor 4-positive (Pf4+)-macrophages, probably the precursors of AMs, can generate pro-inflammatory factors 7 days after IAV infection (38). AMs contribute to protect alveolar epithelial cells (AECs) from IAV infection., The cysteinyl leukotriene (CysLT) trigged pathway is suppressed by AMs, which is considered to prevent AECs damage from the virus (47). The protective role of AMs during IAV infection might be due to their peroxisome proliferator-activated receptor gamma (PPAR-γ). Activation of PPAR-γ ameliorates virus-associated inflammation and increases the level of MMP7 and MMP9, tissue remodeling factors, and EGF and VEGF, epi-endothelial growth factor, for repair of damaged sites (48, 49). The impact of ROS on macrophages against IAV infection, as well as ROS mediated signaling pathways in infected macrophages, are indirect (50). Evidence has showed that the polarization of M1 macrophages depends on ROS mediated pathway (51); in M2 macrophages, ROS either promote their polarization via NF-κB (52), or induce autophagy to inhibit their polarization via MAPK pathway (53). Besides, ROS destroy exogenous materials in the antigen presenting route to reduce the antigen presentation of macrophages (54). Furthermore, ROS mediated autophagy strengthens the formation of MHC class II for macrophages (55). These data indicates that ROS can impact the function of macrophages, but further investigation is needed to fully understand the effects of IAV infection on ROS-influenced macrophages.
Natural killer cells
NK cells are recruited in the lung within the first few days following IAV infection in mice (56). The presence of IAV activates NK cells through STAT4 to secrete IFN-γ and release granzymes, as well as perforin to remove infected cells and strengthen CD8+ T-cell response (57, 58). Accumulation of NK is found in the lung and airways dependent on the cell surface chemokine receptors, such as CC-chemokine receptor 5 (CCR5) and CXC-chemokine receptor 3 (CXCR3) (59). Activation of natural cytotoxicity receptors (NCRs) expressed on NK cells relied on the binding between NKp46 and NKp44 to viral hemagglutinin (HA) (60, 61). This kind of activation results in the lysis of infected cells mediated by NK cells through degranulation, perforin and granzyme release, and IFNγ secretion (62, 63). The function of NK cells can be influenced by ROS. It has been proved that ROS derived from monocytes alters the signaling transduction of NK by reducing CD16ζ chain to inhibit their function (64). ROS signaling is necessary for pro-inflammatory cytokine release, including type I IFN produced by NK cells. Therefore, ROS may participate in the regulation of cytokine production of NK cells responding to IAV infection, which needs to be proved by further studies.
Adaptive immune response
The adaptive immune response is the second line of defense against pathogens. Unlike fast response by the innate immune system, the development of adaptive immune response is a highly specific and long-lasting process, which is necessary for the clearance of virus and protection against future invasion through the establishment of long-term memory (65).
Dendritic cells
Evidence has showed that acute infection of IAV resulted in reduced number of cDCs (CD11c+ conventional DCs) and pDCs (plasmacytoid DCs) in peripheral circulation, but a sustained enhancement in respiratory tract (62). The murine CD103+ DCs traffic viral antigen to lymph nodes and present to CD8+ T cells to control replication of virus (66–68); however, CD11b+ cDCs activated by IAV fail to prime CD8+ T cells (69). Massive accumulation of IFN-α produced by pDCs may contribute to uncontrolled inflammation and pathology (70). Accumulation of pDCs in lymph nodes under lethal infection upregulate the expression of Fas ligand, which recognize its receptor Fas expressed on IAV-infected CD8+ T cells, to promote elimination by Fas-dependent apoptosis (71, 72). Altered levels of ROS impact the antigen presenting capacity of DCs. It has reported that inhibited levels of ROS significantly decrease antigen uptake of DCs (73, 74); reduced mitochondrial ROS, to be specific, decreases antigen presentation of pDCs to CD8+ T cells (75); nevertheless, enhanced level of ROS suppressed antigen presentation of DCs as well by induction of mitochondrial disorder (76). DCs themselves produce ROS at a slow but prolonged manner. It was revealed that ROS could be produced by DCs within minutes and sustained for at least 10h with an average rate around 0.5mM/sec per phagosome, which was about 10-fold lower than that in neutrophils (77). ROS, on the other hand, has ability to increase the intracellular Ca2+ concentration by activating the transient receptor potential melastatin 2 (TRPM2) channels, which are essential for Ca2+-permeable and preferentially present in the lysosomal membranes in DCs (78). The release of Ca2+ from activated TRPM2 channel provides vital signal for the maturation and chemotaxis of DCs (78, 79). However, further investigation on the ROS-mediated function of DCs responding to IAV infection is still needed.
T cells
CD4 T cells are found to be correlated with lower IAV shedding and less severe outcome in infected patients (80). They can migrate to the infection sites by contacting with virus-specific antigens to activate CD8 T cells and modulate immune response mediated by virus-specific CD8 T cells (81, 82). Release of IFN-γ by CD4 Th1 cells is necessary for the development and preservation of CD8 T memory (83, 84). The function of CD4 T cells during the initial priming phase of infection limits exhaustion of CD8 T cells, which can rapidly recall the viral memory in future infection (85). CD4 CTL utilizes a perforin/granzyme-mediated mechanism to perform cytotoxicity role for elimination (86, 87). IL-2 induced Jak3 and STAT5 are required for optimal formation of CD4 CTL. A recent study revealed that STAT1 protected CD4 CTL against NK during IAV infection, and STAT4 enhanced the promotion of Th1 identity to improve their anti-viral impact (88).
CD8 T cells contribute to defense immunity against IAV infection by releasing cytotoxic granules and cytokines and inducing direct apoptosis of infected cells (89). IAV-specific CD8 T cells have been mostly enriched in the lung of patients (90) and reach the peaks of frequency at approximately day 10 after infection (91). Following infection, CD8 T cells recognize highly conserved epitopes derived from internal influenza components (92). CD8 T cells then get activated and acquire the effector ability to secret inflammatory cytokines and effector molecules. During the pandemic in 2009 caused by IAV, development of severe diseases is correlated with less frequency of virus-specific CD8 T cells (93). A subset of virus-specific CD8 T cells keep in the host to protect against further infection by forming long-lasting memory population (91). Memory subtypes can remain in human up to several months, an over 13 years presence in peripheral blood have also been found (94). The memory response to IAV in human is stable and can trigger rapid expansion in the lung towards the secondary infection (95, 96). Their expansion in the lung and airways correlates with increased CXCR3- and CCR5-binding chemokines (97). After expansion, memory subtypes produce cytokines, like IFN-γ and TNF, in a rapid manner (98, 99); meanwhile, high expression of CD11a helps them to produce cytolytic molecules to clear and protect against the virus (100). CD8 T cells express Fas ligand (CD95L) and TNF receptor apoptosis-inducing ligand (TRAIL), which can interact with their receptor Fas (CD95) and DR4 and/or DR5, respectively, to mediate apoptosis of infected cells (91).
Activation of both CD4 and CD8 T cells triggers the respiratory burst either by direct contact with phagocytes or by cytokines, meanwhile, phagocytes-generated ROS in turn impacts T cells and leads to oxidative stress. NOX-2 produced ROS have been found involved in the differentiation of T cells (101). The susceptibility to ROS strongly depends on the subtype of T cells, CD45+RA T naive cells are more resistant to ROS-induced apoptosis than CD45+RO T memory cells (102). Effector T cells are largely protected from ROS-mediated cell death (103), which may partly due to the glutathione precursor cysteine and the thiol-reducing enzyme thioredoxin released by macrophages and DCs (101). The generation of ROS plays an essential role for massive expansion of CD8 T cells responding to IAV infection (104). The metabolism of mitochondria is also critical for T-cell activation that mitochondrial ROS activated nuclear factor of activated T cells (NFAT) are necessary for IL-2 secretion (105). Meanwhile, mitochondrial ROS can be converted to hydrogen peroxide signal, which induces CD95L expression to regulate activation-induced T-cell death (AICD) (106). As modulators, ROS play an indispensable role in T-cell receptor-induced transcription. The generation of ROS constitutes an intriguing issue with multiple implications for both T-cell-activated bioenergetics and T-cell-mediated pathologies (107).
B cells
B cells contribute to the production of pathogen-specific antibodies to inactivate pathogens and to eliminate the infected cells (108). They also present viral antigens for downstream stimulation (109) and secrete anti- and pro-inflammatory cytokines during viral clearance (110). Following IAV exposure, naïve B cells are activated by viral antigens and differentiate either into short-lived antibody-producing plasmablasts (PBs) or germinal center (GC) B cells (111). GC is the place for affinity maturation and clonal selection of GC B cells (111, 112). Within GC, successfully mutated GC B cel clones present antigen peptides to T follicular helper (Tfh) cells via MHC-II (111). With the help of Tfh, the affinity of GC B cells decides their differentiate that the highest affinity is selected for long-lived plasma cells (LLPCs), while lower ones for memory B cells (MBCs) (113–115). Encountered the secondary infection, MBCs are recalled and differentiate rapidly into PBs, or re-enter into GCs for affinity mature (116–118). Simultaneously, naïve B cells are recruited again to generate a de novo B cell response to variate the antibody response against shared and drifted epitopes (117, 118). As a result, newly educated and generated LLPCs and MBCs fight together to protect host against new viral variant.
ROS production is involved in B cell receptor (BCR) stimulation. BCR stimulation of primary resting B cells induces a rapid generation of ROS for at least 24h in mice, in where early production of ROS (0-2h) relies on Nox2, while the later ROS (6-24h) depends on mitochondrial respiration (119). Produced ROS further stimulate BCR downstream for effective B cell response through PI3K pathway (119). A further study confirms the role of prolonged ROS in B cell proliferation (120). In mouse splenic B cells, ROS generated by BCR ligation are produced in two phases. The first one happened immediately and ceased in 1h, while the second one started at 2h and lasted for 4-6h. ROS, produced by NOX3 not NOX2 in the late phase, enhance the activation of essential pathways for B cell proliferation, including NF-κB and PI3K pathways (120).
Discussion
Human respiratory virus infections lead to a spectrum of respiratory symptoms and disease severity, contributing to substantial morbidity, mortality and economic losses worldwide, as seen in the COVID-19 pandemic. Respiratory virus, mainly including IV, respiratory syncytial virus (RSV), MERS-related coronavirus (MERS-CoV), and severe acute respiratory syndrome coronavirus (SARS and SARS-CoV-2), show comparable symptoms in patients. In this review, we discuss the ROS involvement of immune responses to IAV infection, a representative of respiratory virus, including mitochondria function, oxidant and antioxidant enzymes, and signaling pathways in various immune cells. Extensive research has suggested that hyperinflammation induced by IAV is heavily involved with oxidative stress. Therefore, the elevated oxidative stress is commonly observed in IAV infected cases with severe disease progress (121–123). Consistently, several factors that are known to be associated with increased ROS levels and oxidative stress, such as chronic diseases, morbid obesity, smoking, and older age, are listed as risk factors for a more severe course on IAV infected patients (124–126). Severe hyperinflammation caused by high levels of ROS is spurred by an exuberant but dysregulated immune response (127). The current treatment in the clinic for hyperinflammation induced by mainly relies on immunomodulatory therapy, including broadly immunosuppressive approaches (such as glucocorticoids) (128, 129) and targeted immunomodulatory therapies (such as cytokine blockades) (130). The understanding of ROS impact on immune response will provide novel therapeutic targets to effectively treat IAV infection, and other respiratory virus infection in general.
Author contributions
LW, ZC, ZW, JG performed the literature review. LW wrote the manuscript. JW revised the manuscript, JW and LW organized and provided the frame of this manuscript. All authors read and approved the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by R01 CA253215 (JW), the UCLA AIDS Institute, the James B. Pendleton Charitable Trust and the McCarthy Family Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
JW has a financial interest in Vivibaba and The Regents have licensed intellectual property invented by JW to Vivibaba. No funding was provided by these companies to support this work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yang Y, Bazhin AV, Werner J, Karakhanova S. Reactive oxygen species in the immune system. Int Rev Immunol (2013) 32(3):249–70. doi: 10.3109/08830185.2012.755176
2. Li Z, Xu X, Leng X, He M, Wang J, Cheng S, et al. Roles of reactive oxygen species in cell signaling pathways and immune responses to viral infections. Arch Virol (2017) 162(3):603–10. doi: 10.1007/s00705-016-3130-2
3. Banerjee S, Ghosh S, Mandal A, Ghosh N, Sil PC. ROS-associated immune response and metabolism: a mechanistic approach with implication of various diseases. Arch Toxicol (2020) 94(7):2293–317. doi: 10.1007/s00204-020-02801-7
4. Liu M, Chen F, Liu T, Chen F, Liu S, Yang J. The role of oxidative stress in influenza virus infection. Microbes Infect (2017) 19(12):580–6. doi: 10.1016/j.micinf.2017.08.008
5. Chen KK, Minakuchi M, Wuputra K, Ku CC, Pan JB, Kuo KK, et al. Redox control in the pathophysiology of influenza virus infection. BMC Microbiol (2020) 20(1):214. doi: 10.1186/s12866-020-01890-9
6. Flerlage T, Boyd DF, Meliopoulos V, Thomas PG, Schultz-Cherry S. Influenza virus and SARS-CoV-2: pathogenesis and host responses in the respiratory tract. Nat Rev Microbiol (2021) 19(7):425–41. doi: 10.1038/s41579-021-00542-7
7. Sander WJ, Fourie C, Sabiu S, O'Neill FH, Pohl CH, O'Neill HG. Reactive oxygen species as potential antiviral targets. Rev Med Virol (2022) 32(1):e2240. doi: 10.1002/rmv.2240
8. Hutchinson EC, Fodor E. Transport of the influenza virus genome from nucleus to nucleus. Viruses (2013) 5(10):2424–46. doi: 10.3390/v5102424
9. Long JC, Fodor E. The PB2 subunit of the influenza a virus RNA polymerase is imported into the mitochondrial matrix. J Virol (2016) 90(19):8729–38. doi: 10.1128/JVI.01384-16
10. Malik G, Zhou Y. Innate immune sensing of influenza a virus. Viruses (2020) 12(7):755. doi: 10.3390/v12070755
11. Lee JH, Oh SJ, Yun J, Shin OS. Nonstructural protein NS1 of influenza virus disrupts mitochondrial dynamics and enhances mitophagy via ULK1 and BNIP3. Viruses (2021) 13(9):1845. doi: 10.3390/v13091845
12. Peterhans E. Oxidants and antioxidants in viral diseases: disease mechanisms and metabolic regulation. J Nutr (1997) 127(5 Suppl):962S–5S. doi: 10.1093/jn/127.5.962S
13. Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol (2015) 12(1):5–23. doi: 10.1038/cmi.2014.89
14. Amatore D, Sgarbanti R, Aquilano K, Baldelli S, Limongi D, Civitelli L, et al. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by NOX4-derived ROS. Cell Microbiol (2015) 17(1):131–45. doi: 10.1111/cmi.12343
15. To EE, Vlahos R, Luong R, Halls ML, Reading PC, King PT, et al. Endosomal NOX2 oxidase exacerbates virus pathogenicity and is a target for antiviral therapy. Nat Commun (2017) 8(1):69. doi: 10.1038/s41467-017-00057-x
16. To EE, Luong R, Diao J, JJ OL, Brooks DA, Vlahos R, et al. Novel endosomal NOX2 oxidase inhibitor ameliorates pandemic influenza a virus-induced lung inflammation in mice. Respirology (2019) 24(10):1011–7. doi: 10.1111/resp.13524
17. Lejal N, Truchet S, Bechor E, Bouguyon E, Khedkar V, Bertho N, et al. Turning off NADPH oxidase-2 by impeding p67(phox) activation in infected mouse macrophages reduced viral entry and inflammation. Biochim Biophys Acta Gen Subj (2018) 1862(6):1263–75. doi: 10.1016/j.bbagen.2018.03.004
18. Kosmider B, Messier EM, Janssen WJ, Nahreini P, Wang J, Hartshorn KL, et al. Nrf2 protects human alveolar epithelial cells against injury induced by influenza a virus. Respir Res (2012) 13:43. doi: 10.1186/1465-9921-13-43
19. Guo Y, Tu YH, Wu X, Ji S, Shen JL, Wu HM, et al. ResolvinD1 protects the airway barrier against injury induced by influenza a virus through the Nrf2 pathway. Front Cell Infect Microbiol (2020) 10:616475. doi: 10.3389/fcimb.2020.616475
20. Pyo CW, Shin N, Jung KI, Choi JH, Choi SY. Alteration of copper-zinc superoxide dismutase 1 expression by influenza a virus is correlated with virus replication. Biochem Biophys Res Commun (2014) 450(1):711–6. doi: 10.1016/j.bbrc.2014.06.037
21. Jung KI, Ko DH, Shin N, Pyo CW, Choi SY. Endoplasmic reticulum-associated degradation potentiates the infectivity of influenza a virus by regulating the host redox state. Free Radic Biol Med (2019) 135:293–305. doi: 10.1016/j.freeradbiomed.2019.03.021
22. De Angelis M, Amatore D, Checconi P, Zevini A, Fraternale A, Magnani M, et al. Influenza virus down-modulates G6PD expression and activity to induce oxidative stress and promote its replication. Front Cell Infect Microbiol (2021) 11:804976. doi: 10.3389/fcimb.2021.804976
23. Wheeler JL, Martin KC, Resseguie E, Lawrence BP. Differential consequences of two distinct AhR ligands on innate and adaptive immune responses to influenza a virus. Toxicol Sci (2014) 137(2):324–34. doi: 10.1093/toxsci/kft255
24. Lee C. Therapeutic modulation of virus-induced oxidative stress via the Nrf2-dependent antioxidative pathway. Oxid Med Cell Longev (2018) 2018:6208067. doi: 10.1155/2018/6208067
25. Yipp BG, Kubes P. NETosis: how vital is it? Blood (2013) 122(16):2784–94. doi: 10.1182/blood-2013-04-457671
26. Dahlgren C, Karlsson A, Bylund J. Intracellular neutrophil oxidants: From laboratory curiosity to clinical reality. J Immunol (2019) 202(11):3127–34. doi: 10.4049/jimmunol.1900235
27. Kim HJ, Kim CH, Ryu JH, Kim MJ, Park CY, Lee JM, et al. Reactive oxygen species induce antiviral innate immune response through IFN-lambda regulation in human nasal epithelial cells. Am J Respir Cell Mol Biol (2013) 49(5):855–65. doi: 10.1165/rcmb.2013-0003OC
28. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal (2014) 20(7):1126–67. doi: 10.1089/ars.2012.5149
29. Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol (2014) 24(10):R453–62. doi: 10.1016/j.cub.2014.03.034
30. Sarvestani ST, McAuley JL. The role of the NLRP3 inflammasome in regulation of antiviral responses to influenza a virus infection. Antiviral Res (2017) 148:32–42. doi: 10.1016/j.antiviral.2017.10.020
31. Zhao C, Zhao W. NLRP3 inflammasome-a key player in antiviral responses. Front Immunol (2020) 11:211. doi: 10.3389/fimmu.2020.00211
32. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature (2011) 472(7344):476–80. doi: 10.1038/nature09973
33. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469(7329):221–5. doi: 10.1038/nature09663
34. Carty M, Guy C, Bowie AG. Detection of viral infections by innate immunity. Biochem Pharmacol (2021) 183:114316. doi: 10.1016/j.bcp.2020.114316
35. Lee S, Ishitsuka A, Noguchi M, Hirohama M, Fujiyasu Y, Petric PP, et al. Influenza restriction factor MxA functions as inflammasome sensor in the respiratory epithelium. Sci Immunol (2019) 4(40):eaau4643. doi: 10.1126/sciimmunol.aau4643
36. Cole SL, Ho LP. Contribution of innate immune cells to pathogenesis of severe influenza virus infection. Clin Sci (Lond) (2017) 131(4):269–83. doi: 10.1042/CS20160484
37. Rudd JM, Pulavendran S, Ashar HK, Ritchey JW, Snider TA, Malayer JR, et al. Neutrophils induce a novel chemokine receptors repertoire during influenza pneumonia. Front Cell Infect Microbiol (2019) 9:108. doi: 10.3389/fcimb.2019.00108
38. Zhang J, Liu J, Yuan Y, Huang F, Ma R, Luo B, et al. Two waves of pro-inflammatory factors are released during the influenza a virus (IAV)-driven pulmonary immunopathogenesis. PloS Pathog (2020) 16(2):e1008334. doi: 10.1371/journal.ppat.1008334
39. George ST, Lai J, Ma J, Stacey HD, Miller MS, Mullarkey CE. Neutrophils and influenza: A thin line between helpful and harmful. Vaccines (Basel) (2021) 9(6):597. doi: 10.3390/vaccines9060597
40. Lim K, Hyun YM, Lambert-Emo K, Capece T, Bae S, Miller R, et al. Neutrophil trails guide influenza-specific CD8(+) T cells in the airways. Science (2015) 349(6252):aaa4352. doi: 10.1126/science.aaa4352
41. Tak T, Rygiel TP, Karnam G, Bastian OW, Boon L, Viveen M, et al. Neutrophil-mediated suppression of influenza-induced pathology requires CD11b/CD18 (MAC-1). Am J Respir Cell Mol Biol (2018) 58(4):492–9. doi: 10.1165/rcmb.2017-0021OC
42. Peiro T, Patel DF, Akthar S, Gregory LG, Pyle CJ, Harker JA, et al. Neutrophils drive alveolar macrophage IL-1beta release during respiratory viral infection. Thorax (2018) 73(6):546–56. doi: 10.1136/thoraxjnl-2017-210010
43. Short KR, Kroeze E, Fouchier RAM, Kuiken T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect Dis (2014) 14(1):57–69. doi: 10.1016/S1473-3099(13)70286-X
44. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol (2010) 191(3):677–91. doi: 10.1083/jcb.201006052
45. Zhu L, Liu L, Zhang Y, Pu L, Liu J, Li X, et al. High level of neutrophil extracellular traps correlates with poor prognosis of severe influenza a infection. J Infect Dis (2018) 217(3):428–37. doi: 10.1093/infdis/jix475
46. Tang BM, Shojaei M, Teoh S, Meyers A, Ho J, Ball TB, et al. Neutrophils-related host factors associated with severe disease and fatality in patients with influenza infection. Nat Commun (2019) 10(1):3422. doi: 10.1038/s41467-019-11249-y
47. Cardani A, Boulton A, Kim TS, Braciale TJ. Alveolar macrophages prevent lethal influenza pneumonia by inhibiting infection of type-1 alveolar epithelial cells. PloS Pathog (2017) 13(1):e1006140. doi: 10.1371/journal.ppat.1006140
48. Moseley CE, Webster RG, Aldridge JR. Peroxisome proliferator-activated receptor and AMP-activated protein kinase agonists protect against lethal influenza virus challenge in mice. Influenza Other Respir Viruses (2010) 4(5):307–11. doi: 10.1111/j.1750-2659.2010.00155.x
49. Huang S, Zhu B, Cheon IS, Goplen NP, Jiang L, Zhang R, et al. PPAR-gamma in macrophages limits pulmonary inflammation and promotes host recovery following respiratory viral infection. J Virol (2019) 93(9):e00030-19. doi: 10.1128/JVI.00030-19
50. Herb M, Schramm M. Functions of ROS in macrophages and antimicrobial immunity. Antioxidants (Basel) (2021) 10(2):313. doi: 10.3390/antiox10020313
51. Zhao Q, Chen H, Yang T, Rui W, Liu F, Zhang F, et al. Direct effects of airborne PM2.5 exposure on macrophage polarizations. Biochim Biophys Acta (2016) 1860(12):2835–43. doi: 10.1016/j.bbagen.2016.03.033
52. Formentini L, Santacatterina F, Nunez de Arenas C, Stamatakis K, Lopez-Martinez D, Logan A, et al. Mitochondrial ROS production protects the intestine from inflammation through functional M2 macrophage polarization. Cell Rep (2017) 19(6):1202–13. doi: 10.1016/j.celrep.2017.04.036
53. Shan M, Qin J, Jin F, Han X, Guan H, Li X, et al. Autophagy suppresses isoprenaline-induced M2 macrophage polarization via the ROS/ERK and mTOR signaling pathway. Free Radic Biol Med (2017) 110:432–43. doi: 10.1016/j.freeradbiomed.2017.05.021
54. Halleen JM, Raisanen SR, Alatalo SL, Vaananen HK. Potential function for the ROS-generating activity of TRACP. J Bone Miner Res (2003) 18(10):1908–11. doi: 10.1359/jbmr.2003.18.10.1908
55. Choi SH, Gonen A, Diehl CJ, Kim J, Almazan F, Witztum JL, et al. SYK regulates macrophage MHC-II expression via activation of autophagy in response to oxidized LDL. Autophagy (2015) 11(5):785–95. doi: 10.1080/15548627.2015.1037061
56. Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol (2006) 7(5):517–23. doi: 10.1038/ni1322
57. Ge MQ, Ho AW, Tang Y, Wong KH, Chua BY, Gasser S, et al. NK cells regulate CD8+ T cell priming and dendritic cell migration during influenza a infection by IFN-gamma and perforin-dependent mechanisms. J Immunol (2012) 189(5):2099–109. doi: 10.4049/jimmunol.1103474
58. Hwang I, Scott JM, Kakarla T, Duriancik DM, Choi S, Cho C, et al. Activation mechanisms of natural killer cells during influenza virus infection. PloS One (2012) 7(12):e51858. doi: 10.1371/journal.pone.0051858
59. Carlin LE, Hemann EA, Zacharias ZR, Heusel JW, Legge KL. Natural killer cell recruitment to the lung during influenza a virus infection is dependent on CXCR3, CCR5, and virus exposure dose. Front Immunol (2018) 9:781. doi: 10.3389/fimmu.2018.00781
60. Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, et al. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature (2001) 409(6823):1055–60. doi: 10.1038/35059110
61. Arnon TI, Lev M, Katz G, Chernobrov Y, Porgador A, Mandelboim O. Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur J Immunol (2001) 31(9):2680–9. doi: 10.1002/1521-4141(200109)31:9<2680::AID-IMMU2680>3.0.CO;2-A
62. Lamichhane PP, Samarasinghe AE. The role of innate leukocytes during influenza virus infection. J Immunol Res (2019) 2019:8028725. doi: 10.1155/2019/8028725
63. Bjorkstrom NK, Strunz B, Ljunggren HG. Natural killer cells in antiviral immunity. Nat Rev Immunol (2022) 22(2):112–23. doi: 10.1038/s41577-021-00558-3
64. Hansson M, Asea A, Ersson U, Hermodsson S, Hellstrand K. Induction of apoptosis in NK cells by monocyte-derived reactive oxygen metabolites. J Immunol (1996) 156(1):42–7.
65. Stambas J, Lu C, Tripp RA. Innate and adaptive immune responses in respiratory virus infection: implications for the clinic. Expert Rev Respir Med (2020) 14(11):1141–7. doi: 10.1080/17476348.2020.1807945
66. McGill J, Van Rooijen N, Legge KL. Protective influenza-specific CD8 T cell responses require interactions with dendritic cells in the lungs. J Exp Med (2008) 205(7):1635–46. doi: 10.1084/jem.20080314
67. Ho AW, Prabhu N, Betts RJ, Ge MQ, Dai X, Hutchinson PE, et al. Lung CD103+ dendritic cells efficiently transport influenza virus to the lymph node and load viral antigen onto MHC class I for presentation to CD8 T cells. J Immunol (2011) 187(11):6011–21. doi: 10.4049/jimmunol.1100987
68. Desch AN, Randolph GJ, Murphy K, Gautier EL, Kedl RM, Lahoud MH, et al. CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J Exp Med (2011) 208(9):1789–97. doi: 10.1084/jem.20110538
69. GeurtsvanKessel CH, Willart MA, van Rijt LS, Muskens F, Kool M, Baas C, et al. Clearance of influenza virus from the lung depends on migratory langerin+CD11b- but not plasmacytoid dendritic cells. J Exp Med (2008) 205(7):1621–34. doi: 10.1084/jem.20071365
70. Davidson S, Crotta S, McCabe TM, Wack A. Pathogenic potential of interferon alphabeta in acute influenza infection. Nat Commun (2014) 5:3864. doi: 10.1038/ncomms4864
71. Boonnak K, Vogel L, Feldmann F, Feldmann H, Legge KL, Subbarao K. Lymphopenia associated with highly virulent H5N1 virus infection due to plasmacytoid dendritic cell-mediated apoptosis of T cells. J Immunol (2014) 192(12):5906–12. doi: 10.4049/jimmunol.1302992
72. Langlois RA, Legge KL. Plasmacytoid dendritic cells enhance mortality during lethal influenza infections by eliminating virus-specific CD8 T cells. J Immunol (2010) 184(8):4440–6. doi: 10.4049/jimmunol.0902984
73. Huang CH, Huang CY, Huang MH. Unsaturated squalene content in emulsion vaccine adjuvants plays a crucial role in ROS-mediated antigen uptake and cellular immunity. Mol Pharm (2018) 15(2):420–9. doi: 10.1021/acs.molpharmaceut.7b00800
74. Gong J, Chen SS. Polyphenolic antioxidants inhibit peptide presentation by antigen-presenting cells. Int Immunopharmacol (2003) 3(13-14):1841–52. doi: 10.1016/j.intimp.2003.08.010
75. Oberkampf M, Guillerey C, Mouries J, Rosenbaum P, Fayolle C, Bobard A, et al. Mitochondrial reactive oxygen species regulate the induction of CD8(+) T cells by plasmacytoid dendritic cells. Nat Commun (2018) 9(1):2241. doi: 10.1038/s41467-018-04686-8
76. Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS, et al. Loss of phagocytic and antigen cross-presenting capacity in aging dendritic cells is associated with mitochondrial dysfunction. J Immunol (2015) 195(6):2624–32. doi: 10.4049/jimmunol.1501006
77. Paardekooper LM, Dingjan I, Linders PTA, Staal AHJ, Cristescu SM, Verberk W, et al. Human monocyte-derived dendritic cells produce millimolar concentrations of ROS in phagosomes per second. Front Immunol (2019) 10:1216. doi: 10.3389/fimmu.2019.01216
78. Sumoza-Toledo A, Lange I, Cortado H, Bhagat H, Mori Y, Fleig A, et al. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J (2011) 25(10):3529–42. doi: 10.1096/fj.10-178483
79. Syed Mortadza SA, Wang L, Li D, Jiang LH. TRPM2 channel-mediated ROS-sensitive Ca(2+) signaling mechanisms in immune cells. Front Immunol (2015) 6:407. doi: 10.3389/fimmu.2015.00407
80. Wilkinson TM, Li CK, Chui CS, Huang AK, Perkins M, Liebner JC, et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med (2012) 18(2):274–80. doi: 10.1038/nm.2612
81. Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science (2003) 300(5617):339–42. doi: 10.1126/science.1083317
82. Laidlaw BJ, Zhang N, Marshall HD, Staron MM, Guan T, Hu Y, et al. CD4+ T cell help guides formation of CD103+ lung-resident memory CD8+ T cells during influenza viral infection. Immunity (2014) 41(4):633–45. doi: 10.1016/j.immuni.2014.09.007
83. Bevan MJ. Helping the CD8(+) T-cell response. Nat Rev Immunol (2004) 4(8):595–602. doi: 10.1038/nri1413
84. Laidlaw BJ, Craft JE, Kaech SM. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat Rev Immunol (2016) 16(2):102–11. doi: 10.1038/nri.2015.10
85. Cullen JG, McQuilten HA, Quinn KM, Olshansky M, Russ BE, Morey A, et al. CD4(+) T help promotes influenza virus-specific CD8(+) T cell memory by limiting metabolic dysfunction. Proc Natl Acad Sci U S A (2019) 116(10):4481–8. doi: 10.1073/pnas.1808849116
86. Brown DM, Lampe AT, Workman AM. The differentiation and protective function of cytolytic CD4 T cells in influenza infection. Front Immunol (2016) 7:93. doi: 10.3389/fimmu.2016.00093
87. Juno JA, van Bockel D, Kent SJ, Kelleher AD, Zaunders JJ, Munier CM. Cytotoxic CD4 T cells-friend or foe during viral infection? Front Immunol (2017) 8:19. doi: 10.3389/fimmu.2017.00019
88. Finn CM, Dhume, McKinstry K. STAT1 protects CD4 T cells from deletion by NK cells during influenza infection and promotes Th1 identity that can be enhanced through STAT4 to improve their anti-viral impact. J Immunol (2021) 206(1 supplement):103.15.
89. Zhang Y, Xu Z, Cao Y. Host-virus interaction: How host cells defend against influenza a virus infection. Viruses (2020) 12(4):376. doi: 10.3390/v12040376
90. de Bree GJ, van Leeuwen EM, Out TA, Jansen HM, Jonkers RE, van Lier RA. Selective accumulation of differentiated CD8+ T cells specific for respiratory viruses in the human lung. J Exp Med (2005) 202(10):1433–42. doi: 10.1084/jem.20051365
91. Schmidt ME, Varga SM. The CD8 T cell response to respiratory virus infections. Front Immunol (2018) 9:678. doi: 10.3389/fimmu.2018.00678
92. Auladell M, Jia X, Hensen L, Chua B, Fox A, Nguyen THO, et al. Recalling the future: Immunological memory toward unpredictable influenza viruses. Front Immunol (2019) 10:1400. doi: 10.3389/fimmu.2019.01400
93. Sridhar S, Begom S, Bermingham A, Hoschler K, Adamson W, Carman W, et al. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat Med (2013) 19(10):1305–12. doi: 10.1038/nm.3350
94. van de Sandt CE, Hillaire ML, Geelhoed-Mieras MM, Osterhaus AD, Fouchier RA, Rimmelzwaan GF. Human influenza a virus-specific CD8+ T-cell response is long-lived. J Infect Dis (2015) 212(1):81–5. doi: 10.1093/infdis/jiv018
95. Heidema J, Rossen JW, Lukens MV, Ketel MS, Scheltens E, Kranendonk ME, et al. Dynamics of human respiratory virus-specific CD8+ T cell responses in blood and airways during episodes of common cold. J Immunol (2008) 181(8):5551–9. doi: 10.4049/jimmunol.181.8.5551
96. Crowe SR, Turner SJ, Miller SC, Roberts AD, Rappolo RA, Doherty PC, et al. Differential antigen presentation regulates the changing patterns of CD8+ T cell immunodominance in primary and secondary influenza virus infections. J Exp Med (2003) 198(3):399–410. doi: 10.1084/jem.20022151
97. Kohlmeier JE, Miller SC, Smith J, Lu B, Gerard C, Cookenham T, et al. The chemokine receptor CCR5 plays a key role in the early memory CD8+ T cell response to respiratory virus infections. Immunity (2008) 29(1):101–13. doi: 10.1016/j.immuni.2008.05.011
98. Schmidt ME, Knudson CJ, Hartwig SM, Pewe LL, Meyerholz DK, Langlois RA, et al. Memory CD8 T cells mediate severe immunopathology following respiratory syncytial virus infection. PloS Pathog (2018) 14(1):e1006810. doi: 10.1371/journal.ppat.1006810
99. McMaster SR, Wilson JJ, Wang H, Kohlmeier JE. Airway-resident memory CD8 T cells provide antigen-specific protection against respiratory virus challenge through rapid IFN-gamma production. J Immunol (2015) 195(1):203–9. doi: 10.4049/jimmunol.1402975
100. Ely KH, Roberts AD, Woodland DL. Cutting edge: effector memory CD8+ T cells in the lung airways retain the potential to mediate recall responses. J Immunol (2003) 171(7):3338–42. doi: 10.4049/jimmunol.171.7.3338
101. Belikov AV, Schraven B, Simeoni L. T Cells and reactive oxygen species. J BioMed Sci (2015) 22:85. doi: 10.1186/s12929-015-0194-3
102. Takahashi A, Hanson MG, Norell HR, Havelka AM, Kono K, Malmberg KJ, et al. Preferential cell death of CD8+ effector memory (CCR7-CD45RA-) T cells by hydrogen peroxide-induced oxidative stress. J Immunol (2005) 174(10):6080–7. doi: 10.4049/jimmunol.174.10.6080
103. Bogeski I, Kummerow C, Al-Ansary D, Schwarz EC, Koehler R, Kozai D, et al. Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci Signal (2010) 3(115)::ra24. doi: 10.1126/scisignal.2000672
104. Laniewski NG, Grayson JM. Antioxidant treatment reduces expansion and contraction of antigen-specific CD8+ T cells during primary but not secondary viral infection. J Virol (2004) 78(20):11246–57. doi: 10.1128/JVI.78.20.11246-11257.2004
105. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38(2):225–36. doi: 10.1016/j.immuni.2012.10.020
106. Kaminski M, Kiessling M, Suss D, Krammer PH, Gulow K. Novel role for mitochondria: protein kinase ctheta-dependent oxidative signaling organelles in activation-induced T-cell death. Mol Cell Biol (2007) 27(10):3625–39. doi: 10.1128/MCB.02295-06
107. Kaminski MM, Roth D, Krammer PH, Gulow K. Mitochondria as oxidative signaling organelles in T-cell activation: physiological role and pathological implications. Arch Immunol Ther Exp (Warsz) (2013) 61(5):367–84. doi: 10.1007/s00005-013-0235-0
108. Newton AH, Cardani A, Braciale TJ. The host immune response in respiratory virus infection: balancing virus clearance and immunopathology. Semin Immunopathol (2016) 38(4):471–82. doi: 10.1007/s00281-016-0558-0
109. Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, et al. Dynamic signaling by T follicular helper cells during germinal center b cell selection. Science (2014) 345(6200):1058–62. doi: 10.1126/science.1257861
110. Upasani V, Rodenhuis-Zybert I, Cantaert T. Antibody-independent functions of b cells during viral infections. PloS Pathog (2021) 17(7):e1009708. doi: 10.1371/journal.ppat.1009708
111. Guthmiller JJ, Utset HA, Wilson PC. B cell responses against influenza viruses: Short-lived humoral immunity against a life-long threat. Viruses (2021) 13(6):965. doi: 10.3390/v13060965
112. Mesin L, Ersching J, Victora GD. Germinal center b cell dynamics. Immunity (2016) 45(3):471–82. doi: 10.1016/j.immuni.2016.09.001
113. Wong R, Belk JA, Govero J, Uhrlaub JL, Reinartz D, Zhao H, et al. Affinity-restricted memory b cells dominate recall responses to heterologous flaviviruses. Immunity (2020) 53(5):1078–94 e7. doi: 10.1016/j.immuni.2020.09.001
114. Purtha WE, Tedder TF, Johnson S, Bhattacharya D, Diamond MS. Memory b cells, but not long-lived plasma cells, possess antigen specificities for viral escape mutants. J Exp Med (2011) 208(13):2599–606. doi: 10.1084/jem.20110740
115. Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, et al. High affinity germinal center b cells are actively selected into the plasma cell compartment. J Exp Med (2006) 203(11):2419–24. doi: 10.1084/jem.20061254
116. Dugan HL, Guthmiller JJ, Arevalo P, Huang M, Chen YQ, Neu KE, et al. Preexisting immunity shapes distinct antibody landscapes after influenza virus infection and vaccination in humans. Sci Transl Med (2020) 12(573):eadb3601. doi: 10.1126/scitranslmed.abd3601
117. Turner JS, Zhou JQ, Han J, Schmitz AJ, Rizk AA, Alsoussi WB, et al. Human germinal centres engage memory and naive b cells after influenza vaccination. Nature (2020) 586(7827):127–32. doi: 10.1038/s41586-020-2711-0
118. Mesin L, Schiepers A, Ersching J, Barbulescu A, Cavazzoni CB, Angelini A, et al. Restricted clonality and limited germinal center reentry characterize memory b cell reactivation by boosting. Cell (2020) 180(1):92–106 e11. doi: 10.1016/j.cell.2019.11.032
119. Wheeler ML, Defranco AL. Prolonged production of reactive oxygen species in response to b cell receptor stimulation promotes b cell activation and proliferation. J Immunol (2012) 189(9):4405–16. doi: 10.4049/jimmunol.1201433
120. Feng YY, Tang M, Suzuki M, Gunasekara C, Anbe Y, Hiraoka Y, et al. Essential role of NADPH oxidase-dependent production of reactive oxygen species in maintenance of sustained b cell receptor signaling and b cell proliferation. J Immunol (2019) 202(9):2546–57. doi: 10.4049/jimmunol.1800443
121. Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, et al. Identification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell (2008) 133(2):235–49. doi: 10.1016/j.cell.2008.02.043
122. Lorente JA, Nin N, Villa P, Vasco D, Miguel-Coello AB, Rodriguez I, et al. Metabolomic diferences between COVID-19 and H1N1 influenza induced ARDS. Crit Care (2021) 25(1):390. doi: 10.1186/s13054-021-03810-3
123. Ng MP, Lee JC, Loke WM, Yeo LL, Quek AM, Lim EC, et al. Does influenza a infection increase oxidative damage? Antioxid Redox Signal (2014) 21(7):1025–31. doi: 10.1089/ars.2014.5907
124. Marshall RJ, Armart P, Hulme KD, Chew KY, Brown AC, Hansbro PM, et al. Glycemic variability in diabetes increases the severity of influenza. mBio (2020) 11(2):e02841-19. doi: 10.1128/mBio.02841-19
125. Chang DB, Cooper RL, Young AC, Martin CJ, Ancker-Johnson B. Restricted diffusion in biophysical systems: theory. J Theor Biol (1975) 50(2):285–308. doi: 10.1016/0022-5193(75)90082-X
126. Lawrence H, Hunter A, Murray R, Lim WS, McKeever T. Cigarette smoking and the occurrence of influenza - systematic review. J Infect (2019) 79(5):401–6. doi: 10.1016/j.jinf.2019.08.014
127. Ryabkova VA, Churilov LP, Shoenfeld Y. Influenza infection, SARS, MERS and COVID-19: Cytokine storm - the common denominator and the lessons to be learned. Clin Immunol (2021) 223:108652. doi: 10.1016/j.clim.2020.108652
128. Hardy RS, Raza K, Cooper MS. Therapeutic glucocorticoids: mechanisms of actions in rheumatic diseases. Nat Rev Rheumatol (2020) 16(3):133–44. doi: 10.1038/s41584-020-0371-y
129. Tinsley A, Navabi S, Williams ED, Liu G, Kong L, Coates MD, et al. Increased risk of influenza and influenza-related complications among 140,480 patients with inflammatory bowel disease. Inflammation Bowel Dis (2019) 25(2):369–76. doi: 10.1093/ibd/izy243
Keywords: reactive oxygen species, influenza virus, viral replication, innate immune response, adaptive immune response
Citation: Wang L, Cao Z, Wang Z, Guo J and Wen J (2022) Reactive oxygen species associated immunoregulation post influenza virus infection. Front. Immunol. 13:927593. doi: 10.3389/fimmu.2022.927593
Received: 24 April 2022; Accepted: 04 July 2022;
Published: 29 July 2022.
Edited by:
Kari Ann Shirey, University of Maryland, United StatesReviewed by:
Yung Jin Jeon, Seoul National University Hospital, South KoreaIrene Ramos, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2022 Wang, Cao, Wang, Guo and Wen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Wen, jingwen@mednet.ucla.edu
†Present address: Lan Wang, Hearing and Speech Rehabilitation Institute, College of Special Education, Binzhou Medical University, Yantai, China