 Elien Eeckhout1,2
Elien Eeckhout1,2 Tomoko Asaoka1,2
Tomoko Asaoka1,2 Hanne Van Gorp1,2Dieter Demon1,2
Hanne Van Gorp1,2Dieter Demon1,2 Charlotte Girard-Guyonvarc’h3
Charlotte Girard-Guyonvarc’h3 Vanessa Andries1,2
Vanessa Andries1,2 Lars Vereecke1,2
Lars Vereecke1,2 Cem Gabay3Mohamed Lamkanfi1
Cem Gabay3Mohamed Lamkanfi1 Geert van Loo2,4
Geert van Loo2,4 Andy Wullaert1,2,5*
Andy Wullaert1,2,5*- 1Department of Internal Medicine and Pediatrics, Ghent University, Ghent, Belgium
- 2VIB-UGent Center for Inflammation Research, VIB, Ghent, Belgium
- 3Division of Rheumatology, Department of Medicine, University Hospital of Geneva, Department of Pathology and Immunology, University of Geneva Faculty of Medicine, Geneva, Switzerland
- 4Department of Biomedical Molecular Biology, Ghent University, Ghent, Belgium
- 5Laboratory of Proteinscience, Proteomics and Epigenetic Signalling (PPES), Department of Biomedical Sciences, University of Antwerp, Antwerp, Belgium
Background: Autoinflammation with infantile enterocolitis (AIFEC) is an often fatal disease caused by gain-of-function mutations in the NLRC4 inflammasome. This inflammasomopathy is characterized by macrophage activation syndrome (MAS)-like episodes as well as neonatal-onset enterocolitis. Although elevated IL-18 levels were suggested to take part in driving AIFEC pathology, the triggers for IL-18 production and its ensuing pathogenic effects in these patients are incompletely understood.
Methods: Here, we developed and characterized a novel genetic mouse model expressing a murine version of the AIFEC-associated NLRC4V341A mutation from its endogenous Nlrc4 genomic locus.
Results: NLRC4V341A expression in mice recapitulated increased circulating IL-18 levels as observed in AIFEC patients. Housing NLRC4V341A-expressing mice in germfree (GF) conditions showed that these systemic IL-18 levels were independent of the microbiota, and unmasked an additional IL-18-inducing effect of NLRC4V341A expression in the intestines. Remarkably, elevated IL-18 levels did not provoke detectable intestinal pathologies in NLRC4V341A-expressing mice, even not upon genetically ablating IL-18 binding protein (IL-18BP), which is an endogenous IL-18 inhibitor that has been used therapeutically in AIFEC. In addition, NLRC4V341A expression did not alter susceptibility to the NLRC4-activating gastrointestinal pathogens Salmonella Typhimurium and Citrobacter rodentium.
Conclusion: As observed in AIFEC patients, mice expressing a murine NLRC4V341A mutant show elevated systemic IL-18 levels, suggesting that the molecular mechanisms by which this NLRC4V341A mutant induces excessive IL-18 production are conserved between humans and mice. However, while our GF and infection experiments argue against a role for commensal or pathogenic bacteria, identifying the triggers and mechanisms that synergize with IL-18 to drive NLRC4V341A-associated pathologies will require further research in this NLRC4V341A mouse model.
1 Introduction
Inflammasomes are multiprotein complexes that serve as a platform for inflammatory caspase activation. During canonical inflammasome activation, detection of pathogen-associated or danger-associated molecular patterns (PAMPs/DAMPs) by cytosolic sensor proteins leads to recruitment and proteolytic activation of caspase-1. Subsequently, activated caspase-1 proteolytically matures the proinflammatory cytokines pro-IL-1β and pro-IL-18, and cleaves Gasdermin D (GSDMD). The resulting N-terminal fragment of GSDMD is a pore forming protein that facilitates pyroptosis, a lytic form of cell death that allows the release of the mature IL-1β and IL-18 cytokines (1). Among various sensor proteins capable of inducing this inflammasome response, NLRC4 is activated by cytosolic flagellin or by components of the Type 3 secretion system (T3SS), which endows it with a pivotal role in antimicrobial defense against a wide range of bacterial pathogens such as Pseudomonas aeruginosa and Salmonella Typhimurium (2).
Besides its critical role in host defense against bacterial infections, NLRC4 was found to drive the development of a spectrum of autoinflammatory diseases (AIDs), collectively referred to as NLRC4-associated AIDs (NLRC4-AIDs) or NLRC4 inflammasomopathies (3). Several gain-of-function mutations in human NLRC4 such as the V341A (4, 5), T337S (6), T337N (7) and S171F (8) substitutions were described to cause such NLRC4-AIDs. Each of these NLRC4 mutations cause severe/recurrent macrophage activation syndrome (MAS)-like episodes characterized by elevated IL-18 serum levels (3). In addition, the NLRC4-AIDs caused by V341A (4, 5), T337S (6), T337N (7) or S171F (8) mutations exhibit neonatal-onset enterocolitis and are therefore also referred to as autoinflammation with infantile enterocolitis (AIFEC) (3). The first onset of AIFEC commonly occurs during infancy and leads to a potentially lethal disease. Based on NLRC4 crystal structure analysis, AIFEC-associated mutations are proposed to destabilize an auto-inhibitory mechanism of NLRC4, which might lead to ligand-independent spontaneous NLRC4 inflammasome activation (3, 4, 6, 8–11). However, possible triggers of AIFEC flares and the pathogenic mechanisms at work in AIFEC patients remain incompletely understood.
Here, we generated and characterized a knock-in mouse model of the AIFEC-associated NLRC4V341A mutation during both homeostasis and infection. Our data show that murine NLRC4V341A expression increases IL-18 levels in circulation as well as in the colon. However, elevated IL-18 levels in NLRC4V341A-expressing mice did not induce intestinal pathology and did not impact on gastrointestinal infections with NLRC4-activating pathogens. Interestingly, NLRC4V341A induced IL-18 expression independently of the gut microbiota. Together, these observations argue against a role for commensal or pathogenic bacteria in driving NLRC4V341A-associated intestinal pathology, but further research will be needed to identify the actual triggers mediating NLRC4V341A-associated AIFEC.
2 Materials and methods
2.1 Mice
NLRC4V341A/V341A mice were generated by Cyagen Biosciences by CRISPR/Cas9-mediated genome editing of the V341-encoding GTG codon to an A341-encoding GCC codon in exon 4 of the endogenous NLRC4 gene of C57BL/6 mice (see also Figure 1). Briefly, C57BL/6 zygotes were co-injected with a vector encoding Cas9 as well as the gRNA 5’-CAGGTGATCACCACGAAGAG-3’ and with the 5’-tgtgggcccaaatccaggagtccaggtgcctgagaaatctgatgaagacccctctcttcGCCgtgatcacctgtgcaattcagatgggcagacaggaattccaagctcacacccaaaccatg-3’oligo donor for homology-directed repair. Pups were genotyped for the mutated GCC codon by sequencing a 361bp PCR amplicon around the mutated codon. Two founder animals carrying the correct GCC mutation and no other mutations within the amplicon were crossed to C57BL/6J mice and then intercrossed to obtain homozygous Nlrc4V341A/V341A mice. Upon observing that Nlrc4V341A/V341A offspring from both founders were born in Mendelian ratios and appeared healthy, all further analyses were performed on progeny from one founder animal. Il18bptm1.1(KOMP)Vlcg (IL-18BP-/-) mice were generated by the Knockout Mouse Project Repository (University of California, Davis, Davis, CA) on C57BL/6N background by the Velocigene approach, in which the entire coding region of the Il18bp gene was replaced by a LacZ gene and a LoxP-flanked Neomycin cassette (12). Specific pathogen free (SPF) mice were housed in individually ventilated cages in the animal facility of the IRC-VIB. Germfree (GF) Nlrc4V341A/V341A mice were generated by transfer of Nlrc4V341A/+ embryo’s in GF foster mothers that had been mated with GF vasectomized males, after which the resulting GF Nlrc4V341A/+ mice were intercrossed to generate GF Nlrc4+/+ and Nlrc4V341A/V341A littermates. GF mice were generated at the Axenic/Gnoto Facility of the Instituto Gulbenkian de Ciência (Lisbon, Portugal) in the context of an INFRAFRONTIER2020 project, and were then transfered to a GF facility at Ghent University where they were housed in positive-pressure flexible film isolators (North Kent Plastics). Food and water were provided ad libitum. All animal experiments were performed according to institutionally approved protocols according to national (Belgian Laws 14/08/1986 and 22/12/2003, Belgian Royal Decree 06/04/2010) and European (EU Directives 2010/63/EU, 86/609/EEG) animal regulations. Animal protocols were reviewed and approved by the Ethical Committee Animal Experimentation VIB site Ghent - Ghent University - Faculty of Sciences (permit number LA1400091) with approval IDs 2018-032 and 2019-072. All necessary efforts were made to minimize suffering of the animals.
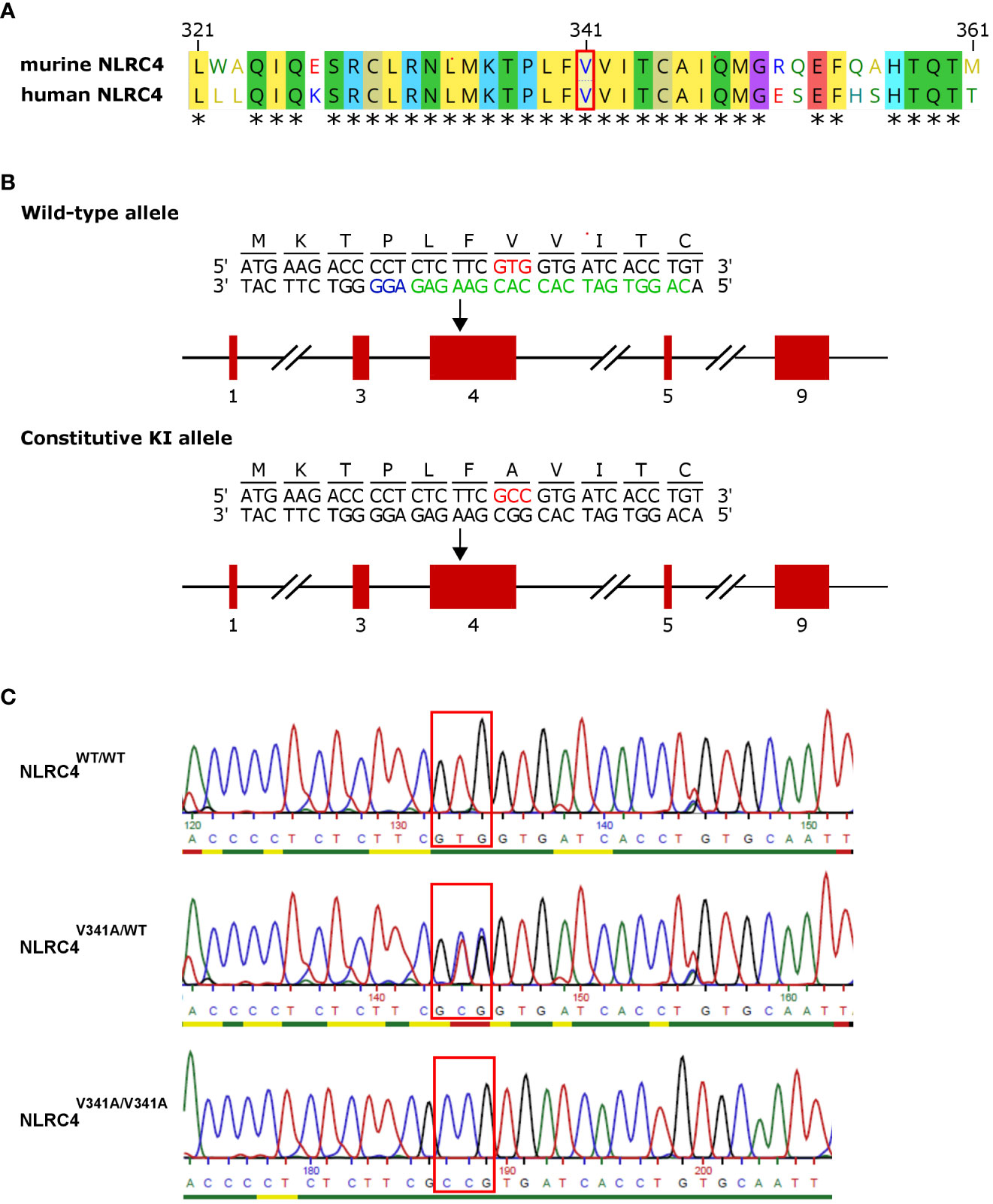
Figure 1 Generation of NLRC4V341A-expressing mice. (A) Alignment of amino acids 321-361 of human and murine NLRC4. (B) Nlrc4 targeting scheme for introducing the V341A mutation. Red boxes are exons; the red codon is the V341A substitution; and in green and blue are the gRNA and the NGG target used for CrisprCas9 editing. (C) Genotyping sequencing example of resulting mice. * = amino acid residues conserved in humans and mice.
2.2 Oral S. Typhimurium infection
Age- and sex-matched mice were infected by oral gavage with 104 or 105 CFU of the SL1344 S. Typhimurium strain administered in a 100 μl inoculum in the logarithmic phase of proliferation after starvation of 4h. Overall susceptibility to the infection was evaluated by survival.
2.3 Gastrointestinal Citrobacter rodentium infection
Age- and sex-matched mice were infected by oral gavage with 5 x 109 CFU of the nalidixic acid (NAL) resistant ICC169 C. rodentium strain administered in a 200 μl inoculum in the logarithmic phase of proliferation, as described (13). Enumeration of C. rodentium loads was performed by plating stool samples and colon tissues collected at the indicated time points after infection, on selective Luria-Bertani (LB) agar containing 50 μg/ml NAL. Colony forming units (CFUs) were normalized to the weight of the sample. Mice that did not surpass the threshold of 1 x 105 CFU/g feces at 7 days post-infection (dpi) were considered not successfully infected and were removed from the experiment.
2.4 Stimulation of bone marrow derived macrophages
Primary macrophages were generated from bone marrow cells flushed from femur and tibia. Cells were differentiated to bone marrow derived macrophages (BMDMs) by culturing them in Iscove’s modified Dulbecco’s medium (IMDM, Lonza) with 10% (v/v) heat-inactivated FBS, 30% (v/v) L929 cell-conditioned medium, 1% Gibco non-essential amino acids, penicillin (100 U/ml) and streptomycin (100 mg/ml) for 6 days in 37°C in the presence of 5% CO2. After differentiation, medium was aspirated and the cells were scraped in IMDM supplemented with 10% FBS and 1% Gibco non-essential amino acids. For specific experiments, 106 cells were seeded in 0.5 ml culture medium per well in 12-well plates and were incubated overnight at 37˚ C and 5% CO2. On day 7, FlaTox stimulations (1 µg/ml, 2h) and S. Typhimurium infections (MOI1, 3h) were performed on LPS-primed (1 µg/ml, 5h) or unprimed BMDMs. At 2h post S. Typhimurium infection 100 µg/ml gentamycin was added followed by 1h incubation. After incubation at 37°C and 5% CO2 samples were collected.
2.5 Cell death analysis
Lytic cell death was evaluated by detection of LDH released in the cell culture supernatant by LDH assay (Promega) according to manufacturer’s instructions. The data were plotted as percentage of total cell death as determined by a 100% cell death control from Triton X-100 treated wells.
2.6 Cytokine measurements
Tissue samples were weighed and were homogenized in PBS with protease inhibitors, after which lysis was completed by addition of lysis buffer (20 mM Tris HCl (pH 7.4), 200 mM NaCl, 1% Nonidet P-40) and incubation for 20 minutes on ice. Full speed centrifugation for 30 minutes cleared the homogenate and supernatant was used for further analysis. Mouse cytokines in cell culture supernatants, serum and tissue homogenates were determined by magnetic bead-based multiplex assay using Luminex technology (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. Cytokines from tissue homogenates were normalized to weight of tissue, while cytokines from cell culture supernatants and serum were expressed as concentration per ml of cell culture medium or serum.
2.7 Western blot analyses
Cells and culture supernatants, colon or ileum intestinal epithelial cells (IECs), or whole colon or ileum homogenates, were incubated with cell lysis buffer (20 mM Tris HCl (pH 7.4), 200 mM NaCl, 1% Nonidet P-40) and denatured in Laemlli buffer by boiling for 10 min. Proteins were separated by SDS-PAGE electrophoresis (Thermo Scientific) after which proteins were transferred to membranes using turbo (7 min) blotting. Blocking and antibody incubation were performed in PBS supplemented with 0.05% or 0.2% Tween20 (vol/vol) and 3% non-fat dry milk. The membranes were incubated overnight at 4˚ C with primary antibodies against caspase-1 (1:1000; Adipogen, AG-20B-0042-C100), IL-1β (1:2000; GeneTex, GTX74034), GSDMD (1:1000, Abcam, ab209845) and IL-18 (1:1000, Biovision, 5180R-100). After washing, membranes were incubated with HRP-conjugated anti-mouse, anti-rabbit or anti-goat antibodies (1:5000; Jackson ImmunoResearch Laboratories, 115-035-146, 111-035-144 and 305-035-003) or were incubated with the directly labeled primary antibody β-Actin-HRP (1:10000; Santa Cruz) for up to 3h. Proteins of interest were detected by the enhanced SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
2.8 Histology
Colon and ileum tissues were fixed in 4% paraformaldehyde, embedded in paraffin, and cut in 4 µm sections. For histopathological analysis hematoxylin and eosin staining were performed according to standard protocols. Histological crypt length quantifications were performed in a blinded fashion using Image-J-win4. Cell death was evaluated on paraffin sections by TUNEL staining (in situ cell death detection kit, TMR red, Roche) performed according to the manufacturer’s instructions. For immunohistochemical Ki67 staining paraffin sections were rehydrated and heat-induced antigen retrieval was performed in Antigen Unmasking Solution, Citric Acid Based (Vector Laboratories). Endogenous peroxidase activity was blocked by incubating the slides in methanol containing 3% H2O2. Sections were then incubated overnight with primary antibody for Ki67 (1/1000, Cell Signalling, D3B5) in PBS containing 10% goat serum. Biotinylated secondary antibody was purchased from Dako (E0432). Stainings were visualized with Vectastain ELITE ABC Kit and DAB substrate (ImmPACT DAB Substrate kit, Peroxidase, Vector Laboratories), after which sections were counterstained with hematoxylin. Incubation times with DAB substrate were equal for all samples. For AB-PAS staining paraffin sections were rehydrated, stained for 20 min with Alcian blue. This was followed by 15 min treatment with 1% periodic acid and 15 min treatment with Shiff’s reagent. After washing with running tap water for 10 min, nuclei were stained with Mayer’s hematoxylin for 20 sec. Finally, sections were dehydrated and mounted with depex. For immunofluorescent Lysozyme staining paraffin sections were rehydrated and heat-induced antigen retrieval was performed in Antigen Unmasking Solution, Citric Acid Based (Vector Laboratories). Endogenous peroxidase activity was blocked by incubating the slides in methanol containing 3% H2O2. Sections were incubated overnight with anti-lysozyme primary antibody (1/700, DAKO, A0099) in PBS containing 0.2% goat serum, 0.5% fish skin gelatin and 2% BSA, followed by one-hour incubation with a secondary antibody labeled with Dylight-488 (1/500, Thermofisher, 35553) and nuclei staining with DAPI (1/1000, Thermofisher, D21490) for 10min in Prolong Gold anti-fade. All pictures were taken with a high content screening microscope (Zeiss AxioScan) at the same exposure and intensity settings.
2.9 Isolation of IECs
Ileum and colon were dissected and flushed with PBS to remove fecal content. The tissue was turned inside-out, washed with PBS and incubated in HBSS containing 2 mM EDTA for 30 min at 37°C shaking. Afterwards tissue was removed and cells were collected by centrifugation at 3000 rpm for 5 min, followed by washing of the cells with PBS by centrifugation at 5000 rpm for 5 min. Cells were lysed in cell lysis buffer (20 mM Tris HCl (pH 7.4), 200 mM NaCl, 1% Nonidet P-40) for Western Blot analysis or kept in TRIsure (Bioline) until further isolation of RNA.
2.10 Quantitative real-time PCR
Total RNA was isolated using TRIsure reagent (Bioline) and the RNeasy® Mini Kit (QIAGEN) according to manufacturer’s instructions. cDNA was synthesized using the iScript gDNA clear cDNA Synthesis kit (Biorad) according to the manufacturer’s instructions. cDNA was amplified on quantitative PCR in a total volume of 5 µl with SensiFAST SYBR® No-ROX Kit (Bioline) and specific IL-18 primers (Fwd ACTTTGGCCGACTTCACTGTA, Rev CTTCACAGAGAGGGTCACAGC) on a LightCycler 480 (Roche). The reactions were performed in triplicates. Primer sequences reference genes: Gapdh (Fwd TGAAGCAGGCATCTGAGGG, Rev CGAAGGTGGAAGAGTGGGAG), Hprt1 (Fwd AGTGTTGGATACAGGCCAGAC, Rev CGTGATTCAAATCCCTGAAGT), Ubc (Fwd AGGTCAAACAGGAAGACAGACGTA, Rev TCACACCCAAGAACAAGCACA).
2.11 Statistics
All statistical analyses were performed using GraphPad Prism version 9.0. For mouse survival curves, statistical significance was determined by log-rank Mantel-Cox test. Other data were analyzed by applying either unpaired two-sided student t-tests or unpaired two-sided Mann-Whitney tests in case of not normal distribution of the values. Data are shown as means of biological replicates with SD as indicated in figure legends. Statistical results are indicated as ns not significant; *p < 0.05; **p < 0.01 or ***p < 0.001.
3 Results
3.1 NLRC4V341A expression elevates IL-18 in circulation but does not provoke intestinal inflammation in mice
The V341A mutation of human NLRC4 was reported to cause AIFEC characterized by neonatal-onset enterocolitis, periodic fevers, and fatal or near-fatal episodes of autoinflammation in four individuals from two unrelated pedigrees (4, 5). To better understand the pathogenic mechanisms involved in these NLRC4V341A-expressing AIFEC patients, we created mice expressing a V341A variant of the murine NLRC4 protein, as the region around the V341 residue is conserved in humans and mice (Figure 1A). For this purpose, we introduced a codon encoding the V341A substitution in the endogenous Nlrc4 gene of C57BL/6 mice by CRISPR/Cas9-mediated genome editing (Figures 1B, C). Both NLRC4V341A/WT and NLRC4V341A/V341A mice were born in Mendelian ratios and aged indistinguishably from their NLRC4WT/WT littermates. Therefore, to maximize potential pathologic effects of NLRC4V341A expression, we proceeded with analyzing inflammatory parameters in homozygous NLRC4V341A/V341A mice as compared to their NLRC4WT/WT littermates.
AIFEC patients show chronically elevated serum IL-18 levels that are thought to originate from increased NLRC4 inflammasome activity (4–7). Therefore, to determine whether NLRC4V341A-expressing mice displayed a similar IL-18-dominant serum signature, we measured the inflammasome-dependent IL-18 and IL-1β as well as the inflammasome-independent IL-6 and TNF cytokines in serum of NLRC4V341A/V341A and NLRC4WT/WT mice. NLRC4V341A/V341A mice showed significantly higher circulating IL-18 levels than NLRC4WT/WT littermates (Figure 2A), while IL-1β, IL-6 and TNF serum levels did not differ between these genotypes (Figures 2B–D). Thus, as observed in AIFEC patients (4–7), NLRC4V341A expression in mice induces a specific increase in circulating IL-18 levels.
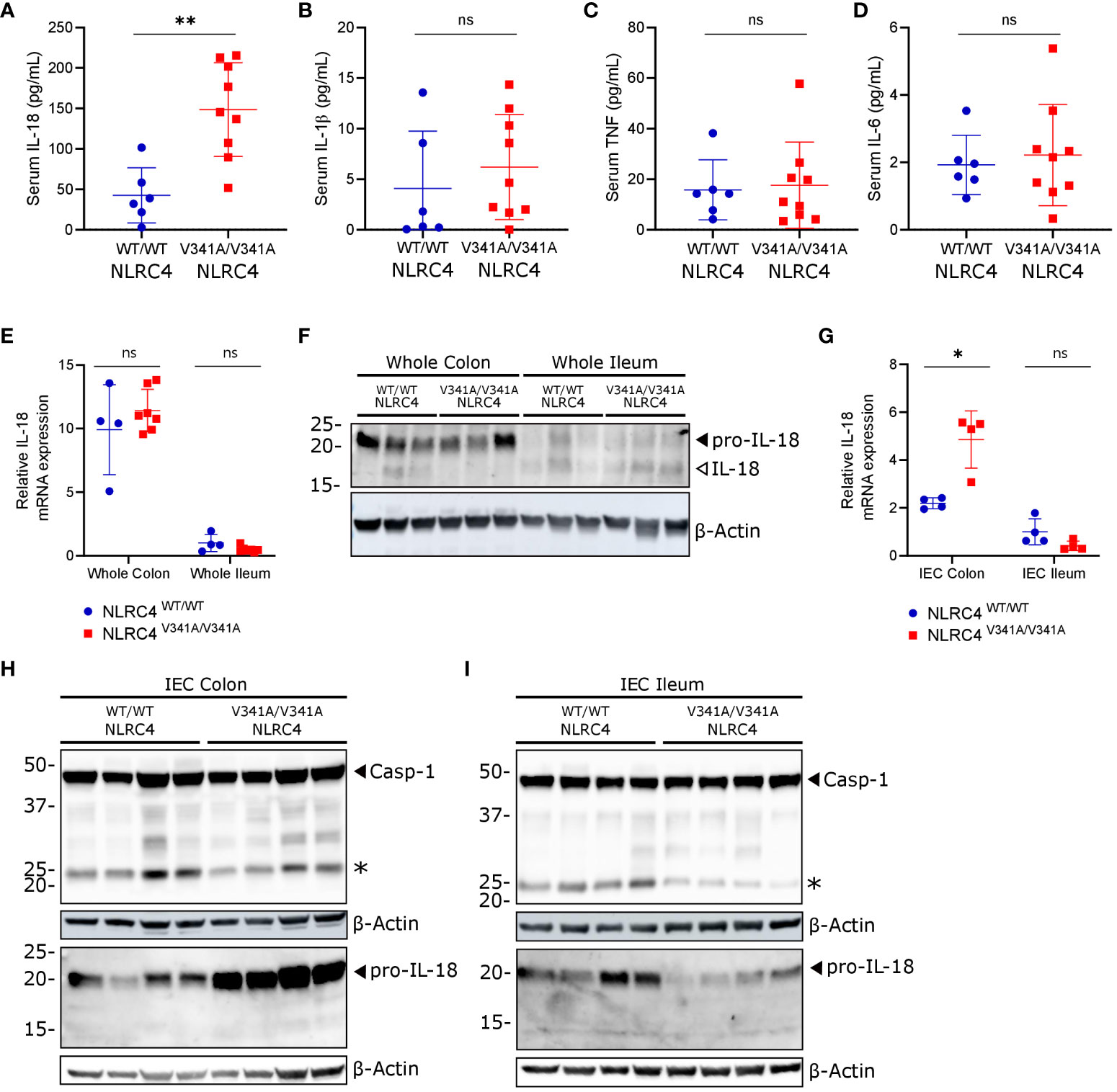
Figure 2 NLRC4V341A expression provokes elevated IL-18 circulation and increased pro-IL-18 expression in colon IECs. (A–D) Serum (A) IL-18, (B) IL-1β, (C) TNF and (D) IL-6 levels in age- and sex-matched NLRC4V341A/V341A and NLRC4WT/WT littermates. (E–I) IL-18 (E, G) mRNA and (F, H, I) protein expression in (E, F) whole colon and ileum or in (G–I) colon and ileum IECs of NLRC4V341A/V341A and NLRC4WT/WT littermates. Data in (A–E, G) represent individual mice and their means +/- SD. In (F, H, I) every lane represents a lysate from a different mouse, and * indicates an unspecific signal in (H, I). * p<0.05, ** p<0.01, ns, not significant.
A previously described genetic mouse model expressing an NLRC4T337S mutant showed that IL-18 production in these mice mainly originated from IECs (14). We therefore investigated whether the gastrointestinal tract was the source of increased circulating IL-18 levels in NLRC4V341A/V341A mice. Whole tissue qPCR analyses showed no differences in pro-IL-18 mRNA expression in ileum or colon of NLRC4V341A/V341A mice, while these analyses did show that IL-18 mRNA expression was higher in colon compared to ileum in both genotypes (Figure 2E). In accordance, pro-IL-18 protein was mainly detectable in colon lysates, but NLRC4V341A/V341A mice did not show different pro-IL-18 levels compared to NLRC4WT/WT mice when analyzing complete ileum or colon lysates (Figure 2F). In addition, these protein analyses on complete tissue lysates only detected low levels of mature IL-18 in either ileum or colon, and did not reveal clear effects of NLRC4V341A expression on IL-18 maturation (Figure 2F). Next, given their high IL-18 expression levels (14–16), we analyzed IL-18 production specifically in IECs of ileum and colon of NLRC4V341A/V341A mice. Interestingly, whereas IECs isolated from ileum showed no differences in pro-IL-18 mRNA expression, colon IECs from NLRC4V341A/V341A mice showed higher pro-IL-18 mRNA expression than colon IECs from NLRC4WT/WT mice (Figure 2G). Accordingly, immunoblotting analyses showed that NLRC4V341A expression increased pro-IL-18 protein levels in colon but not in ileum IECs (Figures 2H, I). However, these immunoblotting analyses did not allow detecting mature IL-18 in IECs. In addition, cleavage of caspase-1 to its active p20 subunit was undetectable in colon or ileum IECs of naïve NLRC4V341A/V341A mice (Figures 2H, I). Overall, these observations indicate that NLRC4V341A expression increases pro-IL-18 mRNA as well as protein levels specifically in colonic IECs, but inflammasome activation leading to IL-18 maturation could not be detected in these cells under homeostatic conditions. Therefore, although our observations suggest that NLRC4V341A-expressing colon IECs have a higher capacity to produce mature IL-18, it remains unclear whether the elevated circulating IL-18 levels in these mice derive from the intestines.
Intestinal biopsies and autopsy specimens from NLRC4V341A-expressing AIFEC patients showed mixed immune cell infiltration, villous blunting with tissue edema, and tissue autolysis (4, 5). Given this remarkable intestinal pathology and the increased pro-IL-18 production by colonic IECs in NLRC4V341A/V341A mice, we performed histopathological analyses on ileum and colon of these mice. H&E staining revealed no abnormal immune cell infiltration or structural differences in both ileum or colon of NLRC4V341A/V341A mice (Figure 3A). Furthermore, normal numbers of goblet cells and Paneth cells were observed in NLRC4V341A/V341A mice as assessed by Alcian Blue-Periodic acid-Schiff (AB-PAS) staining and Lysozyme immunostaining, respectively (Figures 3B, C). Finally, NLRC4V341A/V341A mice showed normal rates of intestinal epithelial proliferation (Figure 3D), and a TUNEL staining revealed no increased cell death in ileum or colon of NLRC4V341A/V341A mice (Figure 3E). Taken together, these analyses suggest that the increased pro-IL-18 levels in colon IECs do not give rise to sufficient IL-18 activity to induce histopathological changes in the intestine of NLRC4V341A/V341A mice under homeostatic conditions.
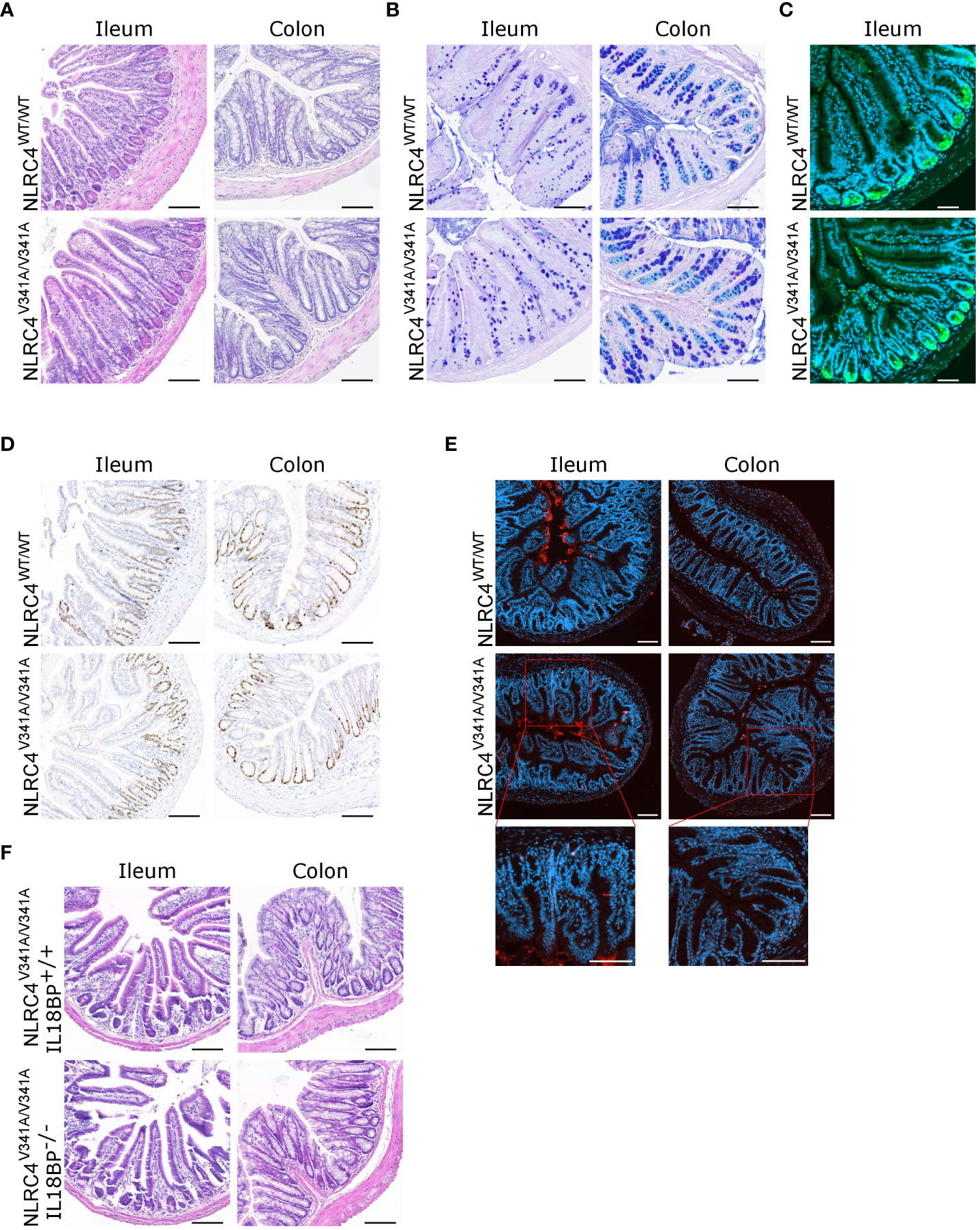
Figure 3 Nlrc4V341A expression does not affect intestinal homeostasis even in the absence of IL-18BP. (A–E) Representative (A) H&E staining, (B) AB-PAS staining, (C) Lysozyme staining, (D) Ki67 staining and (E) TUNEL staining on colon and/or ileum of NLRC4V341A/V341A and NLRC4WT/WT littermates. (F) Representative ileum and colon H&E staining of age- and sex-matched NLRC4V341A/V341AIL-18BP-/- and NLRC4V341A/V341AIL-18BP+/+ littermates. Scale bars 100 µm.
IL-18 Binding Protein (IL-18BP)-mediated sequestering of IL-18 prevents the mature cytokine from binding with its receptor (17–19). To investigate whether this might provide a possible explanation for the lack of intestinal inflammation in NLRC4V341A/V341A mice, we generated NLRC4V341A/V341A mice in an IL-18BP-deficient background. However, the absence of IL-18BP did not provoke histopathology in ileum or colon of these NLRC4V341A/V341AIL-18BP-/- mice (Figure 3F). This indicates that the absence of intestinal pathology in NLRC4V341A/V341A mice did not result from IL-18 neutralization by IL-18BP. Overall, the above analyses show that despite their increased IL-18 circulation, naïve NLRC4V341A/V341A mice do not display gastrointestinal pathologies even in the absence of the endogenous IL-18 antagonist IL-18BP.
3.2 NLRC4V341A expression does not alter host defense against infections with NLRC4-triggering gastrointestinal pathogens
As our above analyses showed that elevated IL-18 circulation in naïve NLRC4V341A/V341A mice was not associated with gastrointestinal pathologies, we next evaluated the response of these mice to gastrointestinal infections. Indeed, NLRC4 plays important roles in host defense against a wide range of NLRC4-activating bacterial pathogens, for which the enteropathogen Salmonella Typhimurium is a prototypical example (20). To assess whether NLRC4V341A intrinsically alters NLRC4-mediated inflammasome activation, we first evaluated NLRC4V341A responses in bone-marrow-derived macrophages (BMDMs). For this purpose, we treated BMDMs with FlaTox to deliver flagellin to the cytosol (21, 22), and we infected BMDMs with S. Typhimurium. Upon both of these NLRC4-activating triggers, NLRC4V341A/V341A and NLRC4WT/WT BMDMs showed similar caspase-1, GSDMD and IL-18 cleavage (Figure 4A), and showed no significant differences in IL-18 secretion (Figure 4B). In addition, although LPS-primed NLRC4V341A/V341A BMDMs showed a slight reduction in IL-1β cleavage compared to NLRC4WT/WT BMDMs upon FlaTox stimulation (Figure 4A), FlaTox- as well as S. Typhimurium-induced IL-1β secretion was equal between these genotypes (Figure 4C). In line with these cytokine observations, NLRC4V341A/V341A and NLRC4WT/WT BMDMs died to a similar extent upon FlaTox stimulation or S. Typhimurium infection (Figure 4D). Notably, in each of these inflammasome activation, cytokine and cell death analyses, untreated NLRC4V341A/V341A and NLRC4WT/WT BMDMs showed no baseline differences (Figures 4A–D). Taken together, these data indicate that NLRC4V341A expression does not alter either baseline or stimulated NLRC4 inflammasome activity in BMDMs. Furthermore, baseline as well as LPS- and S. Typhimurium-induced IL-6 production was similar between NLRC4V341A/V341A and NLRC4WT/WT BMDMs (Figure 4E), suggesting that also NF-κB transcriptional effects in BMDMs were not affected by NLRC4V341A expression. Overall, these data show that NLRC4V341A expression does not alter S. Typhimurium responses in BMDMs. Next, to extend these analyses to a physiological infectious context, we infected NLRC4V341A/V341A and NLRC4WT/WT mice orally with S. Typhimurium as a model of lethal typhoid fever caused by systemic pathogen dissemination from the gastrointestinal tract (23, 24). However, neither female nor male NLRC4V341A/V341A mice showed differences in survival upon S. Typhimurium infection as compared to their NLRC4WT/WT littermates (Figures 4F, G). This observation shows that NLRC4V341A expression does not influence the overall host response against S. Typhimurium induced typhoid fever-like disease, which together with our BMDM observations argues against NLRC4V341A critically altering NLRC4 inflammasome responses during S. Typhimurium infection.
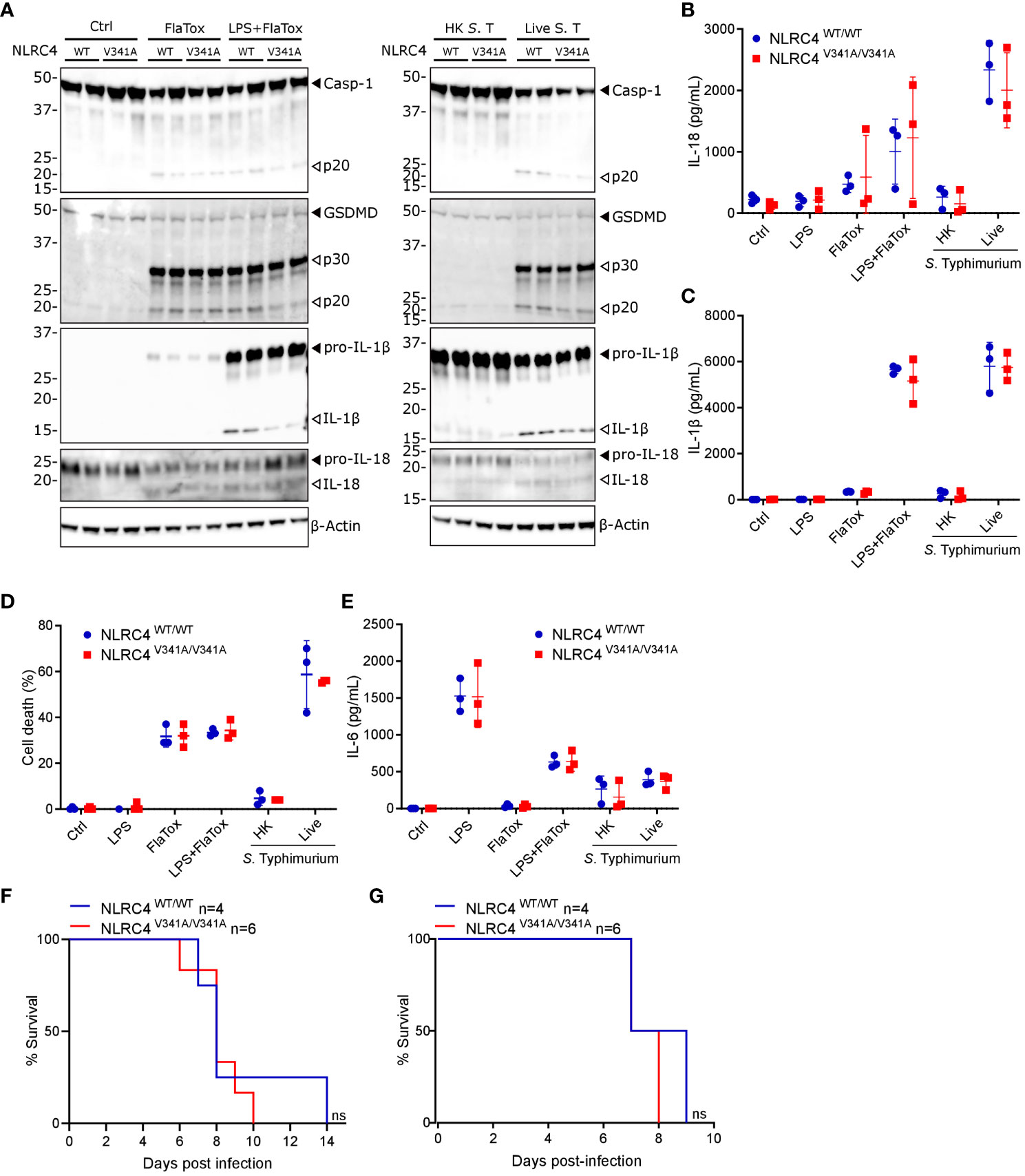
Figure 4 Nlrc4V341A expression does not impact on inflammasome activation by NLRC4 stimuli in BMDMs or on S. Typhimurium susceptibility. (A–E) NLRC4WT/WT and NLRC4V341A/V341A BMDMs were left untreated (Ctrl), were primed or not for 5h with LPS, were treated with FlaTox for 2h, or were infected with heat-killed (HK) or live S. Typhimurium (S. T) at MOI 1 for 3h, as indicated. (A) Western blot analysis of caspase-1, GSDMD, IL-1β and IL-18. (B–E) Culture supernatant levels of (B) IL-18, (C) IL-1β, (D) Lactate Dehydrogenase (LDH), and (E) IL-6. (F, G) Survival analyses of age-matched (F) female or (G) male NLRC4V341A/V341A and NLRC4WT/WT littermates infected by oral gavage with (F) 105 CFU or (G) 104 CFU S. Typhimurium. In (A) every lane represents BMDMs from a different mouse. Data in (B–E) are means +/- SD of biological triplicates. ns, not significant.
As a second gastrointestinal infection model we inoculated NLRC4V341A/V341A mice by oral gavage with C. rodentium, a murine enteropathogen used to mimic enteropathogenic and enterohaemorrhagic Escherichia coli infections in humans (25). Notably, NLRC4 inflammasome activation was demonstrated to participate in host defense against C. rodentium by acting in non-hematopoietic cells (26), thereby providing an opportunity to assess potential in vivo effects of epithelial NLRC4V341A expression. During C. rodentium infection, NLRC4V341A/V341A mice did not display consistent body weight changes compared to NLRC4WT/WT littermates (Figure 5A), and these mice cleared the infection with similar kinetics as NLRC4WT/WT littermates (Figure 5B). Furthermore, NLRC4V341A/V341A mice did not show differences in colonic C. rodentium loads at the peak of infection [10 days post-infection (dpi)] (Figure 5C). In addition, whereas transmissible colonic crypt hyperplasia is a characteristic histopathological feature of C. rodentium colitis, NLRC4V341A/V341A and NLRC4WT/WT mice showed comparable crypt lengths at 10 dpi (Figure 5D). Overall, these observations indicate no obvious influence of NLRC4V341A expression on the host defense against a gastrointestinal C. rodentium infection. However, C. rodentium further increased serum IL-18 levels in NLRC4V341A/V341A mice and, as observed in uninfected conditions, C. rodentium-infected NLRC4V341A/V341A mice showed higher IL-18 levels in the serum compared to their NLRC4WT/WT littermates (Figure 5E). Moreover, in contrast to our observations in uninfected mice, whole colon lysates of C. rodentium-infected mice showed the presence of mature IL-18 as well as cleaved caspase-1 and GSDMD, indicating that this infection induced inflammasome activation in the colon (Figure 5F). Nevertheless, these Western blotting analyses showed similar levels of colonic inflammasome activation in C. rodentium-infected NLRC4V341A/V341A and NLRC4WT/WT mice (Figure 5F). An IL-18 ELISA approach to more specifically quantify mature IL-18 levels confirmed this semi-quantitative Western blotting observation, as whole colon lysates of NLRC4V341A/V341A and NLRC4WT/WT mice showed equal IL-18 upregulation (Figure 5G). Together, these observations suggest that NLRC4V341A expression does not induce excessive inflammasome responses during a C. rodentium infection and thereby does not impact on the host response against this enteropathogen. Considering also our observations with S. Typhimurium, these C. rodentium observations argue that NLRC4V341A expression does not influence the host response to NLRC4-triggering pathogens.
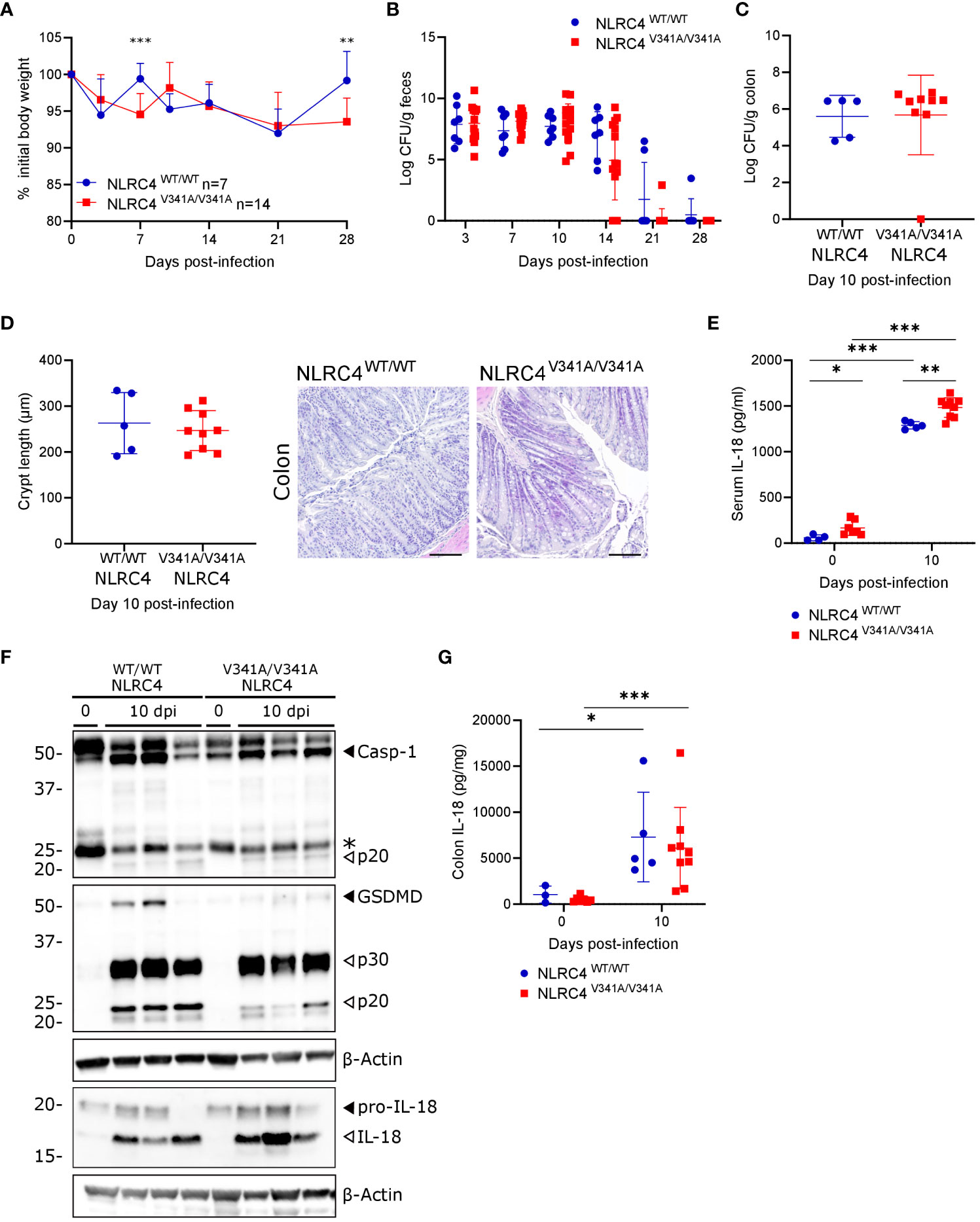
Figure 5 Nlrc4V341A expression does not impact on the host defense against a gastrointestinal C. rodentium infection. (A–G) Age- and sex-matched NLRC4V341A/V341A and NLRC4WT/WT littermates were not infected or infected by oral gavage with 5x109 CFU C. rodentium and analyzed at indicated days post-infection (dpi). (A) Weight change, (B) fecal C. rodentium loads, (C) colonic C. rodentium loads, (D) representative colon H&E staining and colon crypt length measurements, (E) serum IL-18 levels, (F) Western blot analyses on whole colon lysates, and (G) whole colon IL-18 levels. Data in (A) represent means + SD; data in (B–E, G) represent individual mice and their means +/- SD. In (F) every lane represents a whole colon lysate from a different mouse, and * indicates an unspecific signal. Scale bars (D) 100 µm. * p<0.05, ** p<0.01, *** p<0.001.
3.3 NLRC4V341A increases systemic and intestinal IL-18 levels independently of the microbiota
While our above observations argued against a role for pathogenic bacteria in exacerbating NLRC4V341A-induced IL-18 production, we next aimed to investigate whether commensal bacteria were responsible for the observed IL-18 production in NLRC4V341A/V341A mice. For this purpose, we generated germfree (GF) NLRC4V341A/V341A mice that are devoid of microbiota. Remarkably, GF NLRC4V341A/V341A mice showed significantly higher IL-18 levels in circulation compared to GF NLRC4WT/WT mice, similar as observed under specific pathogen free (SPF) conditions (Figure 6A). This showed that systemic IL-18 elevation in NLRC4V341A/V341A mice was independent of the microbiota. Moreover, analyzing IL-18 levels in ileum and colon of GF NLRC4V341A/V341A mice revealed that the absence of microbiota unmasked an IL-18-inducing effect of NLRC4V341A in the intestine. Consistent with a role for the gut microbiota in inducing basal IL-18 production (27–30), ileal and colonic IL-18 production were abolished in GF NLRC4WT/WT mice (Figures 6B, C). However, GF NLRC4V341A/V341A mice showed increased IL-18 levels in both ileum and colon (Figures 6B, C), thereby demonstrating that NLRC4V341A/V341A mice produce intestinal IL-18 independently of the microbiota. Consistent with these ELISA-based mature IL-18 measurements, western blot analyses showed increased IL-18 cleavage in both the colon and ileum of GF NLRC4V341A/V341A mice compared to GF NLRC4WT/WT mice (Figure 6D). This suggests that the microbiota-independent IL-18 production in GF NLRC4V341A/V341A mice could be associated with NLRC4V341A-mediated inflammasome activation. However, despite the detectable elevations in intestinal mature IL-18 production in GF mice, like SPF NLRC4V341A/V341A mice also GF NLRC4V341A/V341A mice showed no obvious histopathology in both ileum or colon (Figure 6E). Taken together, these observations show that NLRC4V341A expression increases intestinal as well as systemic IL-18 levels independently of the microbiota, while these elevated IL-18 levels do not suffice to provoke gastrointestinal pathology.
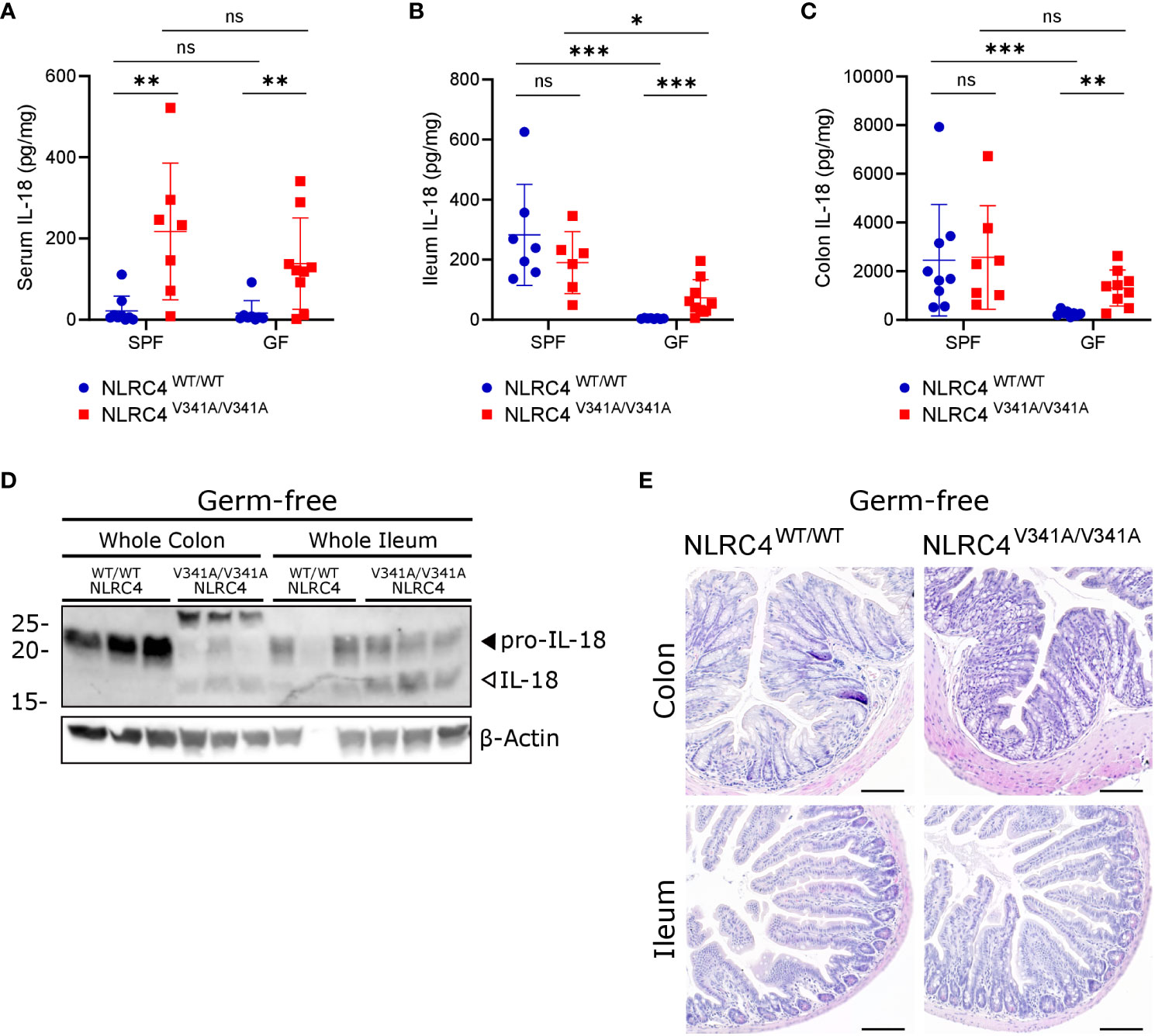
Figure 6 NLRC4V341A expression increases systemic and intestinal IL-18 levels independently of the microbiota. (A–E) Age- and sex-matched NLRC4V341A/V341A and NLRC4WT/WT littermates, housed in either SPF or GF conditions as indicated, were analyzed for (A) IL-18 serum levels, (B) ileum IL-18 levels, (C) colon IL-18 levels, (D) IL-18 maturation in whole colon and ileum lysates, and (E) ileum and colon histopathology by H&E staining. Data in (A–C) represent individual mice and their means +/- SD. Every lane in (D) represents a whole ileum or colon lysate from a different mouse. Scale bars (E) 100 µm. * p<0.05, ** p<0.01, *** p<0.001, ns, not significant.
4 Discussion
Inflammasomopathies are autoinflammatory disorders caused by gain-of-function mutations in inflammasome sensor proteins. Among the various known inflammasomopathies, NLRC4-AIDs distinguish themselves from NLRP3-associated Cyropyrin-associated periodic syndromes (CAPS) and Pyrin-related Familial Mediterranean Fever (FMF) by the occurrence of MAS, elevated IL-18 production and the manifestation of potentially lethal infantile enterocolitis in the AIFEC subset of NLRC4-AID patients (3, 31, 32). In addition, while CAPS and FMF patients are treated effectively with IL-1-antagonizing therapeutics (33), an NLRC4V341A-expressing AIFEC patient was found to be refractory to anakinra treatment. However, supplementing anakinra treatment with recombinant human IL-18BP, which is a circulating decoy protein for IL-18, was able to resolve MAS parameters as well as enterocolitis in this one NLRC4V341A-expressing AIFEC patient (5). While this demonstrated a potential pathogenic contribution for IL-18 in some individuals expressing an NLRC4V341A variant, the mechanisms underlying NLRC4V341A-associated IL-18 production and subsequent colitis development are not known.
In this study, we generated a genetic mouse model expressing a murine NLRC4V341A variant from its endogenous genomic locus. Interestingly, homozygous NLRC4V341A/V341A mice recapitulated elevated serum IL-18 levels as in AIFEC patients (4–7), although the IL-18 serum levels observed in NLRC4V341A/V341A mice were much lower than observed in NLRC4V341A-expressing AIFEC patients (4, 5). This species difference could relate either to an intrinsically lower caspase-1 activating capacity of the murine NLRC4V341A mutant, to less pro-IL-18 expression in mouse IECs as compared to human IECs, or to insufficient levels of NLRC4-activating stimuli in SPF-housed mice. However, regarding the latter possibility it should be noted that even C. rodentium-infected NLRC4V341A/V341A mice showed tenfold less serum IL-18 than AIFEC patients, indicating that a pathogenic NLRC4 activator in the mouse colon was not sufficient to induce these human IL-18 levels. The reasons why NLRC4V341A/V341A mice produce less IL-18 than AIFEC patients thus remains unknown but are likely multifactorial. In line with their only moderately elevated IL-18 production, NLRC4V341A/V341A mice showed no signs of spontaneous inflammation and did not develop detectable intestinal pathology. This suggested that IL-18 levels in NLRC4V341A/V341A mice were not sufficiently high, or alternatively that endogenous antagonists prevented IL-18-mediated pathogenic effects. In this respect, considering the effective treatment of a critically ill neonatal AIFEC patient with recombinant IL-18BP (5), it is quite remarkable that additional IL-18BP deletion did not allow IL-18 to provoke spontaneous colitis in NLRC4V341A/V341A mice. This shows that the absence of clinical signs in NLRC4V341A/V341A mice does not result from the inability of their IL-18 levels to surpass endogenous inhibition by IL-18BP. Instead, additional yet to be identified triggers may be needed to unleash the pathogenic actions of IL-18 in NLRC4V341A/V341A mice. In line with this hypothesis, also previously described NLRC4T337S-expressing mice that displayed much higher IL-18 serum levels than NLRC4V341A/V341A mice did not develop spontaneous autoinflammatory pathology (14). Moreover, since AIFEC patients show chronically elevated IL-18 levels also in between flares, elevated IL-18 levels appear to require additional cues to drive disease in AIFEC patients as well (4–7). Therefore, it is likely that the absence of pathology in NLRC4V341A/V341A mice is not merely due to their low IL-18 levels but also involves insufficient induction of additional factors that could cooperate with IL-18 to induce inflammation. For instance, evaluating IL-18-mediated induction of IFNγ in NLRC4V341A/V341A mice could be informative to better understand the reasons why these mice did not develop autoinflammation.
Therefore, we next evaluated the role of commensal as well as infectious gastrointestinal bacteria as potential triggers of IL-18-mediated pathogenic activities. Interestingly, neonatal-onset enterocolitis spontaneously normalizes after the first year of life in surviving AIFEC patients (3, 4). This led to the hypothesis that early gut colonization may promote enterocolitis development in AIFEC patients, while maturation of the gut microbiota could lead to spontaneous resolution. In accordance with a role for the gut microbiota composition regulating AIFEC severity, a recent study showed that a fecal microbiota transplant improved clinical symptoms in an NLRC4I343N-expressing NLRC4-AID patient (34). However, our observations in GF mice showed that NLRC4V341A expression elevated both systemic and intestinal IL-18 levels independently of the microbiota. In addition, we observed increased levels of cleaved IL-18 in whole colon and ileum of GF NLRC4V341A/V341A mice, while we could only observe increased levels of pro-IL-18 in colon IECs of NLRC4V341A/V341A mice housed in SPF conditions. These paradoxical observations likely relate to the higher IL-18 expression in SPF mice compared to GF mice, consistent with reports showing that the microbiota induce IL-18 in the gastrointestinal tract (27–30), making it more difficult to detect NLRC4V341A-induced IL-18 cleavage in SPF conditions as compared to GF conditions. Thus, intestinal inflammasome activation leading to IL-18 maturation in NLRC4V341A/V341A mice appears to be masked by the gut microbiota. Nevertheless, the observed intestinal IL-18 maturation in GF NLRC4V341A/V341A mice as well as the more specific ELISA-based detection of mature IL-18 in serum of GF and SPF NLRC4V341A/V341A mice suggests that the in vivo IL-18-inducing molecular mechanisms employed by both human and mouse NLRC4V341A at least partially rely on NLRC4 inflammasome activity. However, besides inflammasome-mediated IL-18 maturation, we also observed transcriptional upregulation of pro-IL-18 in colon IECs of SPF NLRC4V341A/V341A mice that could contribute to increased IL-18 production in these mice. Although transcriptional coactivator functions have been reported for other members of the NLR family such as CIITA and NLRC5 (35, 36), to our knowledge such roles have not been described for NLRC4. Since increased pro-IL-18 mRNA levels were observed specifically in colon IECs but not in ileum IECs nor in BMDMs of NLRC4V341A/V341A mice it seems rather unlikely that the V341A mutation confers a general transcriptional activator function to the NLRC4 protein. Instead, a low-grade inflammatory environment caused by IL-18 in the colon of NLRC4V341A/V341A mice might enhance pro-IL-18 expression from colon IECs. As IL-18 is known to induce NF-κB activation via MyD88 signaling (37), this could be created by autocrine IL-18 stimulation of colon IECs. Alternatively, indirect effects of IL-18 via the production of downstream inflammatory factors such as IFNγ could upregulate pro-IL-18 expression in colon IECs (38). However, future research will be needed to clarify how NLRC4V341A expression provokes transcriptional upregulation of pro-IL-18 and whether this may contribute to its effects in AIFEC patients.
While our observations in GF mice unequivocally showed that microbiota are not required to provoke production of mature IL-18 in NLRC4V341A/V341A mice, the lack of colitis in either GF or SPF NLRC4V341A/V341A mice suggested that additional bacterial triggers could be needed to allow IL-18 pathogenic activities. We therefore infected NLRC4V341A/V341A mice orally with S. Typhimurium. Although NLRC4V341A/V341A mice did not show altered susceptibility to this typhoid fever-like disease (23, 24), it should be noted that the NLRC4 inflammasome is not solely responsible for responding to this systemic infection. Indeed, S. Typhimurium dissemination was not altered in Nlrc4-/- mice but was increased in Nlrc4-/-Nlrp3-/- mice (39), raising the possibility that NLRC4V341A-induced inflammasome activation does not sufficiently augment Nlrp3 responses to alter the susceptibility of NLRC4V341A/V341A mice to S. Typhimurium. It would therefore be interesting to assess the effect of NLRC4V341A expression in S. Typhimurium-infected Nlrp3-/- mice in future research. To more specifically evaluate NLRC4V341A effects in a gastrointestinal infection model leading to IL-18 production from IECs, we infected NLRC4V341A/V341A mice with C. rodentium, as NLRC4 responses in non-hematopoietic cells were shown to restrict colitis induced by this enteropathogen (26). In addition, canonical inflammasome-induced IL-18 production was shown to correlate with survival of mice susceptible to a lethal C. rodentium infection (13). However, C. rodentium-infected NLRC4V341A/V341A mice did not display altered intestinal IL-18 levels nor altered host defense in comparison with C. rodentium-infected NLRC4WT/WT mice. Thus, NLRC4V341A expression does not alter the in vivo host defense against the NLRC4-activating C. rodentium and S. Typhimurium pathogens. However, it remains possible that other infectious triggers could be responsible for inducing disease in NLRC4V341A/V341A mice. In this respect, both IL-18tg and IL-18BP-/- mice that harbor increased IL-18 activities develop more severe MAS upon repeated TLR9 stimulation (12, 14), suggesting that perhaps CpG DNA could represent a relevant PAMP for triggering disease in NLRC4V341A/V341A mice. Future research in NLRC4V341A/V341AIL-18BP-/- mice could investigate this possibility.
Overall, our observations in NLRC4V341A/V341A mice are very reminiscent to a prior report on mice expressing the AIFEC-associated NLRC4T337S variant (14). Like NLRC4V341A/V341A mice, these NLRC4T337S mice showed elevated IL-18 levels without detectable inflammatory pathologies and were not more susceptible to various challenges such as endotoxemia, Dextran Sodium Sulfate colitis, and infection with abortive or chronic Lymphocytic Choriomeningitis Virus (14). Interestingly, this study convincingly showed that the increased systemic IL-18 levels in NLRC4T337S mice derived from the intestinal epithelium by specifically deleting IL-18 in IECs (14). Consistent herewith, we observed that colonic IECs from NLRC4V341A/V341A mice under SPF conditions showed increased pro-IL-18 production, while GF NLRC4V341A/V341A mice showed increased mature IL-18 levels in both colon and ileum. This suggests that the increased serum IL-18 levels seen in NLRC4V341A/V341A mice might also originate from IECs. Taken together, these two AIFEC mouse models suggest that IECs may be the cellular IL-18 source in AIFEC patients (4–7). However, it cannot completely be ruled out that other cells may also contribute to the elevated IL-18 levels in these patients. Indeed, peripheral blood derived macrophages from AIFEC patients showed enhanced spontaneous IL-18 secretion, possibly adding to the increased IL-18 levels in the circulation (4, 6). However, this macrophage inflammasome hyperactivity was not recapitulated in neither NLRC4V341A/V341A nor NLRC4T337S BMDMs (14). Interestingly, in contrast to these AIFEC-associated NLRC4 variants, murine macrophages expressing the FCAS-associated NLRC4H443P variant did display enhanced inflammasome activation as observed in human macrophages (40). Although further research is needed, these observations suggest that murine and human NLRC4 variants may employ very specific differences in the molecular signaling mechanisms leading to inflammasome activation.
Taken together, in this study we characterized a novel AIFEC mouse model expressing the NLRC4V341A variant, thereby supporting observations in previously described NLRC4T337S mice that AIFEC-associated NLRC4 variants elevate intestinal and systemic IL-18 levels. In addition, we show that NLRC4V341A expression induces IL-18 production independently of the microbiota, and that this variant does not induce pathologies even in the absence of IL-18BP. This novel NLRC4V341A mouse model sets the stage for further investigation of the triggers and pathogenic mechanisms involved during AIFEC development in humans.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Ethical Committee Animal Experimentation VIB site Ghent - Ghent University - Faculty of Sciences. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
EE: Conceptualization, Formal Analysis, Writing – original draft, Writing – review & editing, Investigation, Methodology. TA: Formal Analysis, Investigation, Methodology, Writing – review & editing. HG: Writing – review & editing, Investigation, Methodology, Resources. DD: Investigation, Methodology, Writing – review & editing, Resources. CG-G: Resources, Writing – review & editing, Methodology. VA: Resources, Writing – review & editing. LV: Resources, Writing – review & editing. CG: Resources, Writing – review & editing. ML: Resources, Writing – review & editing. GvL: Supervision, Writing – review & editing. AW: Conceptualization, Formal Analysis, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Research in the AW lab is supported by the research grants 3G044718, 3G044818 and G0A3422N from the Fund for Scientific Research-Flanders, the BOF UGent grant BOF.24Y.2019.0032.01, and for this project also by an INFRAFRONTIER2020 project funded by the European Union Research and Innovation programme Horizon 2020 (Grant Agreement Number 730879). EE is a Doctoral Research Fellow supported by the BOF UGent fellowship BOFDOC2018004302. GL acknowledges funding from VIB, Ghent University (BOF23/GOA/001) and from the Research Foundation - Flanders (EOS-G0H2522N-40007505). Research in the ML lab is supported by Ghent University grant BOF23/GOA/001, grants from the Fund for Scientific Research-Flanders (GOI5722N, G017121N, G014221N) and ERC (ERC-2022-PoC 101101075). CG is supported by a grant from the Swiss National Science Foundation (310030B-201269). Both CG and AW were funded by the ERARE2 Cure-AID Grant from the European Union ERA-Net for Research Programs on Rare Diseases. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgments
We are grateful for excellent technical support by Sze Men Choi, Amelie Fossoul and Maarten Verdonckt. All microscopy was performed using infrastructure from the VIB Bioimaging Core headed by Saskia Lippens. We thank all core personnel for extensive training and assistance. We thank the Animal House Facility staffs in the IRC SPF animalarium as well as in the Ghent Germ-Free and Gnotobiotic Mouse Facility for support with animal maintenance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Barnett KC, Li S, Liang K, Ting JPY. A 360° view of the inflammasome: Mechanisms of activation, cell death, and diseases. Cell (2023) 186(11):2288–312. doi: 10.1016/j.cell.2023.04.025
2. Egan MS, Zhang J, Shin S. Human and mouse NAIP/NLRC4 inflammasome responses to bacterial infection. Curr Opin Microbiol (2023) 73:102298. doi: 10.1016/j.mib.2023.102298
3. Romberg N, Vogel TP, Canna SW. NLRC4 inflammasomopathies. Curr Opin Allergy Clin Immunol (2017) 17(6):398–404. doi: 10.1097/ACI.0000000000000396
4. Romberg N, Al Moussawi K, Nelson-Williams C, Stiegler AL, Loring E, Choi M, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet (2014) 46(10):1135–9. doi: 10.1038/ng.3066
5. Canna SW, Girard C, Malle L, De Jesus A, Romberg N, Kelsen J, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol (2017) 139(5):1698–701. doi: 10.1016/j.jaci.2016.10.022
6. Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet (2014) 46(10):1140–6. doi: 10.1038/ng.3089
7. Bracaglia C, Gatto A, Pardeo M, Lapeyre G, Ferlin W, Nelson R, et al. Anti interferon-gamma (IFNγ) monoclonal antibody treatment in a patient carrying an NLRC4 mutation and severe hemophagocytic lymphohistiocytosis. Pediatr Rheumatol (2015) 13(S1):O68. doi: 10.1186/1546-0096-13-S1-O68
8. Liang J, Alfano DN, Squires JE, Riley MM, Parks WT, Kofler J, et al. Novel NLRC4 mutation causes a syndrome of perinatal autoinflammation with hemophagocytic lymphohistiocytosis, hepatosplenomegaly, fetal thrombotic vasculopathy, and congenital anemia and ascites. Pediatr Dev Pathol (2017) 20(6):498–505. doi: 10.1177/1093526616686890
9. Hu Z, Yan C, Liu P, Huang Z, Ma R, Zhang C, et al. Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science (2013) 341(6142):172–5. doi: 10.1126/science.1236381
10. Hu Z, Zhou Q, Zhang C, Fan S, Cheng W, Zhao Y, et al. Structural and biochemical basis for induced self-propagation of NLRC4. Science (2015) 350(6259):399–404. doi: 10.1126/science.aac5489
11. Zhang L, Chen S, Ruan J, Wu J, Tong AB, Yin Q, et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science (2015) 350(6259):404–9. doi: 10.1126/science.aac5789
12. Girard-Guyonvarc’h C, Palomo J, Martin P, Rodriguez E, Troccaz S, Palmer G, et al. Unopposed IL-18 signaling leads to severe TLR9-induced macrophage activation syndrome in mice. Blood (2018) 131(13):1430–41. doi: 10.1182/blood-2017-06-789552
13. Eeckhout E, Hamerlinck L, Jonckheere V, Van Damme P, Van Loo G, Wullaert A. Gasdermin D independent canonical inflammasome responses cooperate with caspase-8 to establish host defense against gastrointestinal Citrobacter rodentium infection. Cell Death Dis (2023) 14(4):282. doi: 10.1038/s41419-023-05801-4
14. Weiss ES, Girard-Guyonvarc’h C, Holzinger D, De Jesus AA, Tariq Z, Picarsic J, et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood (2018) 131(13):1442–55. doi: 10.1182/blood-2017-12-820852
15. Takeuchi M, Nishizaki Y, Sano O, Ohta T, Ikeda M, Kurimoto M. Immunohistochemical and immuno-electron-microscopic detection of interferon-γ-inducing factor (”interleukin-18") in mouse intestinal epithelial cells. Cell Tissue Res (1997) 289(3):499–503. doi: 10.1007/s004410050895
16. Nowarski R, Jackson R, Gagliani N, de Zoete MR, Palm NW, Bailis W, et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell (2015) 163(6):1444–56. doi: 10.1016/j.cell.2015.10.072
17. Harel M, Fauteux-Daniel S, Girard-Guyonvarc’h C, Gabay C. Balance between Interleukin-18 and Interleukin-18 binding protein in auto-inflammatory diseases. Cytokine (2022) 150:155781. doi: 10.1016/j.cyto.2021.155781
18. Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 binding protein: A novel modulator of the th1 cytokine response. Immunity (1999) 10(1):127–36. doi: 10.1016/S1074-7613(00)80013-8
19. Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol (2013) 4:289. doi: 10.3389/fimmu.2013.00289
20. Clare B. Inflammasome activation by salmonella. Curr Opin Microbiol (2021) 64:27–32. doi: 10.1016/j.mib.2021.09.004
21. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477(7366):596–600. doi: 10.1038/nature10510
22. Von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, Van Rooijen N, et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature (2012) 490(7418):107–11. doi: 10.1038/nature11351
23. Bauer R, Rauch I. The NAIP/NLRC4 inflammasome in infection and pathology. Mol Aspects Med (2020) 76:100863. doi: 10.1016/j.mam.2020.100863
24. Santos RL, Zhang S, Tsolis RM, Kingsley RA, Garry Adams L, Bäumler AJ. Animal models of Salmonella infections: enteritis versus typhoid fever. Microbes Infect (2001) 3(14–15):1335–44. doi: 10.1016/S1286-4579(01)01495-2
25. Mullineaux-Sanders C, Sanchez-Garrido J, Hopkins EGD, Shenoy AR, Barry R, Frankel G. Citrobacter rodentium–host–microbiota interactions: immunity, bioenergetics and metabolism. Nat Rev Microbiol (2019) 17(11):701–15. doi: 10.1038/s41579-019-0252-z
26. Nordlander S, Pott J, Maloy KJ. NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal Immunol (2014) 7(4):775–85. doi: 10.1038/mi.2013.95
27. Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun (2015) 6(1):6734. doi: 10.1038/ncomms7734
28. Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell (2015) 163(6):1428–43. doi: 10.1016/j.cell.2015.10.048
29. Van Der Kraak LA, Schneider C, Dang V, Burr AHP, Weiss ES, Varghese JA, et al. Genetic and commensal induction of IL-18 drive intestinal epithelial MHCII via IFNγ. Mucosal Immunol (2021) 14(5):1100–12. doi: 10.1038/s41385-021-00419-1
30. Chudnovskiy A, Mortha A, Kana V, Kennard A, Ramirez JD, Rahman A, et al. Host-protozoan interactions protect from mucosal infections through activation of the inflammasome. Cell (2016) 167(2):444–456.e14. doi: 10.1016/j.cell.2016.08.076
31. Sönmez HE, Özen S. A clinical update on inflammasomopathies. Int Immunol (2017) 29(9):393–400. doi: 10.1093/intimm/dxx020
32. Alehashemi S, Goldbach-Mansky R. Human autoinflammatory diseases mediated by NLRP3-, pyrin-, NLRP1-, and NLRC4-inflammasome dysregulation updates on diagnosis, treatment, and the respective roles of IL-1 and IL-18. Front Immunol (2020) 11:1840. doi: 10.3389/fimmu.2020.01840
33. Malcova H, Strizova Z, Milota T, Striz I, Sediva A, Cebecauerova D, et al. IL-1 inhibitors in the treatment of monogenic periodic fever syndromes: from the past to the future perspectives. Front Immunol (2021) 11:619257. doi: 10.3389/fimmu.2020.619257
34. Bracaglia C, Marucci G, Del Chierico F, Russo A, Pardeo M, Pires Marafon D, et al. Microbiota transplant to control inflammation in a patient with NLRC4 gain-of-function–induced disease. J Allergy Clin Immunol (2023) 152(1):302–3. doi: 10.1016/j.jaci.2023.03.031
35. Masternak K, Muhlethaler-Mottet A, Villard J, Zufferey M, Steimle V, Reith W. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev (2000) 14(9):1156–66. doi: 10.1101/gad.14.9.1156
36. Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci USA (2010) 107(31):13794–9. doi: 10.1073/pnas.1008684107
37. Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, et al. Targeted disruption of the myD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity (1998) 9(1):143–50. doi: 10.1016/S1074-7613(00)80596-8
38. Kim YM, Im JY, Han SH, Kang HS, Choi I. IFN-γ Up-regulates IL-18 gene expression via IFN consensus sequence-binding protein and activator protein-1 elements in macrophages. J Immunol (2000) 165(6):3198–205. doi: 10.4049/jimmunol.165.6.3198
39. Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med (2010) 207(8):1745–55. doi: 10.1084/jem.20100257
Keywords: NLRC4 inflammasome, inflammasomopathies, autoinflammation, microbiota, germfree mice, Citrobacter rodentium, Salmonella Typhimurium
Citation: Eeckhout E, Asaoka T, Van Gorp H, Demon D, Girard-Guyonvarc’h C, Andries V, Vereecke L, Gabay C, Lamkanfi M, van Loo G and Wullaert A (2023) The autoinflammation-associated NLRC4V341A mutation increases microbiota-independent IL-18 production but does not recapitulate human autoinflammatory symptoms in mice. Front. Immunol. 14:1272639. doi: 10.3389/fimmu.2023.1272639
Received: 04 August 2023; Accepted: 13 November 2023;
Published: 28 November 2023.
Edited by:
Daniela Novick, Weizmann Institute of Science, IsraelReviewed by:
Diana Boraschi, Chinese Academy of Science (CAS), ChinaJoost Frenkel, Utrecht University, Netherlands
Seth Lucian Masters, The University of Melbourne, Australia
Copyright © 2023 Eeckhout, Asaoka, Van Gorp, Demon, Girard-Guyonvarc’h, Andries, Vereecke, Gabay, Lamkanfi, van Loo and Wullaert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andy Wullaert, andy.wullaert@uantwerpen.be