Glomerular Endothelial Cell Crosstalk With Podocytes in Diabetic Kidney Disease
 Nassim Mahtal
Nassim Mahtal  Olivia Lenoir
Olivia Lenoir Pierre-Louis Tharaux
Pierre-Louis Tharaux- Université de Paris, Paris Cardiovascular Center, Inserm, Paris, France
Diabetes is the main cause of renal failure worldwide. Complications of the kidney micro-and macro-circulation are common in diabetic patients, leading to proteinuria and can progress to end-stage renal disease. Across the complex interplays aggravating diabetes kidney disease progression, lesions of the glomerular filtration barrier appear crucial. Among its components, glomerular endothelial cells are known to be central safeguards of plasma filtration. An array of evidence has recently pinpointed its intricate relations with podocytes, highly specialized pericytes surrounding glomerular capillaries. During diabetic nephropathy, endothelial cells and podocytes are stressed and damaged. Besides, each can communicate with the other, directly affecting the progression of glomerular injury. Here, we review recent studies showing how in vitro and in vivo studies help to understand pathological endothelial cells-podocytes crosstalk in diabetic kidney disease.
Introduction
Diabetes is a multifactorial disease and encompasses multi-organ complications, including kidney lesions leading to diabetic kidney disease (DKD). DKD is characterized by elevated urinary albumin excretion rate (UAER), increase in blood pressure, and decline in renal function leading to end-stage renal disease (ESRD). In addition, these patients have a high risk of cardiovascular disease, which further increases with deteriorating renal function. Although the methodologies to assess diabetes complications and consequences lack accuracy, possibly underestimating its burden, diabetes is recognized as major public health and economic plague (1). In parallel, the prevalence of diabetes in ESRD has been increasing constantly and diabetes is now the main cause of ESRD worldwide (2) and a rapidly increasing problem in the developing countries with the epidemic of type 2 diabetes. Albuminuria is an indicator of glomerular injury during diabetes, and a first step through ESRD (3).
The glomerular filtration barrier (GFB) is altered in DKD, a consequence of the combination of long-term hyperglycemia, advanced glycation end products through glycation reaction between reducing sugars, such as glucose, and proteins, lipids or nucleic acids; dysregulated insulinemia (with alternated hypo- and hyperinsulinemia), and frequently associated endothelial dysfunction and hypertension. Alterations of the GFB involve glomerular endothelial cells (ECs) and podocyte lesions. Endothelial dysfunction, increased extracellular matrix deposition, loss of podocyte permselectivity and progressive podocyte apoptosis occur along the time-course of DKD and contribute to the GFB dysfunction and progressive demise. Podocytes and ECs are physically close and isolated from each other by the glomerular basal membrane (GBM). ECs form a fenestrated endothelium delimiting the vascular compartment, whereas on the other side of the GBM podocytes board the urinary pole in the glomerulus. Communications between ECs and podocytes are physiological and occur from development to adult. During DKD however, pathological mechanisms such as hyperglycemia and hypertension impact the GFB, and lesions of the glomerular endothelium and the podocyte monolayer are common in DKD patients (4). EC are central players of the GFB, and damages of ECs participate to glomerulosclerosis and albuminuria in various pathological contexts, including diabetes (5–7). Evidence of a negative loop taking place between podocytes and ECs has been reported and reviewed, where stressed ECs impair podocytes and vice versa (8, 9). In this review, we focus on recent in vitro and in vivo data illustrating ECs-podocytes crosstalk in diabetic conditions.
Endothelin-1 (ET-1) Is Detrimental for Podocytes and ECs in DKD
Endothelin-1 (ET-1) is a powerful vasoconstrictor and mitogen that has emerged as an interesting novel target for the treatment of DKD (10). ET-1 (EDN1) expression is increased in diabetic kidneys and higher plasmatic ET-1 levels are found in patients with diabetes as well as in animal models of DKD (11–14). ET-1 receptor blockers have renoprotective properties in several DKD (10, 15–17). ET-1 has a key role in regulating renal hemodynamics, salt and water homeostasis, and acid-base balance and in modulating cell proliferation, extracellular matrix accumulation, inflammation, and fibrosis. Consequently, any abnormality in the intrarenal ET system may result in renal dysfunction (e.g., salt sensitivity) and/or injury. Notably, the ET system is present in the renal areas targeted by diabetes, including the microvasculature, mesangial cells, and podocytes. To decipher whether the ET-1 system is a disease modifier beyond its role in the glomerular hemodynamics and sclerosis processes, our group investigated the roles of the ET receptors in podocytes in mice wherein podocyte-specific, double deletion of the ETA (EDNRA), and ETB (EDNRB) receptors was induced. These mice were protected against diabetes-induced podocyte loss and glomerulosclerosis but also provide evidence that the ETB receptor may play as important a role as does the ETA receptor. ETB receptor activation increased intracellular calcium and triggered the NF-κB and β-catenin signaling pathways, analogous to activation of the ETA receptor (Figure 1). The quantitative contribution of the ETB receptor may be substantial, as suggested by the fact that it is upregulated to a larger extent than the ETA receptors in the podocytes of diabetic mice. This study suggests an important role for it in mediating podocyte injury upon stimulation by ET-1, presumably produced by ECs during diabetes (18).
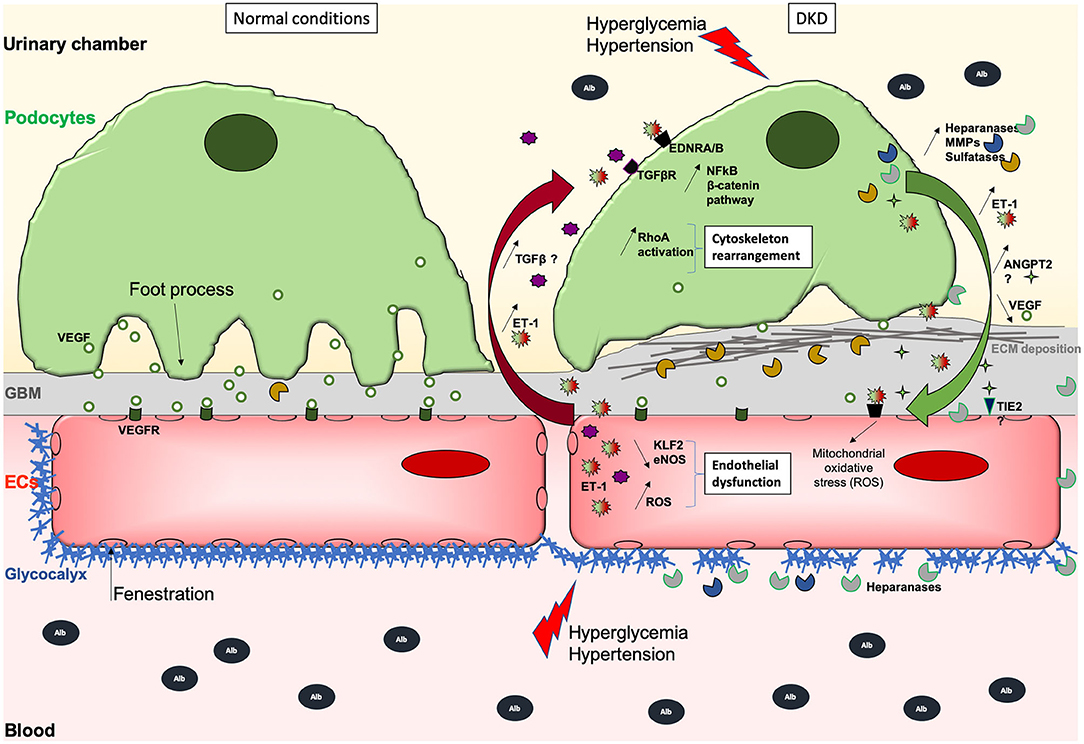
Figure 1. Schematic illustration of the GFB during normal (left) and DKD (right) conditions. During DKD, podocytes lose their foot processes, dedifferentiate with transiently increased ANGPT2 and VEGF production, and then detach or die, which then leads to fewer VEGF secretion. Initial and sustained loss of endothelial permselectivity is also fostered by ANGPT2/ANGPT1 imbalance fostering EC lesions through binding to its receptor TIE2. This initially reduces ECs quiescence and impairs permselectivity then ECs viability, and stimulates ET-1 synthesis that in turn acts on the other side of the GFB, activating podocyte Wnt/beta-catenin and NFkB pathways, heparanase release, inappropriate cytoskeletal remodeling, and abnormal extracellular matrix synthesis causing GBM thickening. ET-1 produced by EC but also by podocytes may also contribute to increased oxidative stress and secretion of proteases, which can degrade the glycocalyx, creating a vicious loop. Hence, ECs are subjected to oxidative and mitochondrial stress, lose their fenestrations, do not exert their functions properly, and may die. Deleterious effects of (ECs-derived?) -TGFβ-mediated signaling on podocytes are suggested. Moreover, the overproduction of sulfatases in the GBM could decrease the bioavailability of growth factors needed by ECs. The GBM thickens due to extracellular matrix (ECM) protein deposits, such as collagen fibers and fibronectin which augments the distance between rarefied podocytes and ECs, potentially altering podocyte to EC bidirectional signal cross-talk.
Recent publications from the Ilse Daehn group have also highlighted the role of ET-1 signaling in EC-podocyte crosstalk but with different mechanisms. They showed that when the forced expression of ET-1 by podocytes (and likely, in ECs) was induced through podocyte-specific activation of TGF-β signaling in transgenic mice and BALB/c mice with adriamycin-induced glomerulosclerosis, activation of ET-1 receptor type A in ECs induced mitochondrial oxidative stress and dysfunction, which in turn lead to release of yet unidentified factors mediating injury and depletion of podocytes in such experimental focal and segmental glomerulosclerosis (19). Glomerular endothelial mitochondrial dysfunction was also associated with increased glomerular ET-1 receptor type A expression and increased circulating ET-1 in experimental DKD. Moreover, pharmacological prevention of EC mitochondrial stress in this diabetes model prevented podocyte loss (20). Secreted factors from dysfunctional ECs were sufficient to cause podocyte apoptosis in supernatant transfer experiments or co-culture but this did not occur when ECs had been previously treated with mitoTEMPO, a mitochondrial antioxidant (21). Thus, ET-1 seems to be a key mediator in podocytes-to-ECs and ECs-to-podocytes communications promoting cell injury in several renal pathologies including DKD. In line with these experimental studies, the SONAR trial suggested that the ETA receptor antagonist atrasentan decreases albuminuria and the risk of major kidney outcomes when given to adults with type 2 diabetes, estimated glomerular filtration rate (eGFR) 25–75 mL/min per 1.73 m2, and a urine albumin-to-creatinine ratio (UACR) of 300–5,000 mg/g who had received maximum labeled or tolerated renin–angiotensin system inhibition for at least 4 weeks (10). Interestingly, albuminuria decrease with atrasentan was consistent irrespective of sodium glucose cotransporter 2 inhibitor (SGLT2i) use before enrolment in the SONAR trial, suggesting that the effects of atrasentan are additive to SGLT2i (22).
Glomerular Glycocalyx Degradation in DKD
In glomeruli, the glycocalyx surrounding endothelial cells creates a space between the blood and the endothelium, controlling vessel permeability, restricting leukocyte and platelet adhesion, and allowing an appropriate endothelial response through mechanosensing. The negative charge of the glycocalyx on podocytes also repulses proteins, contributing to permselectivity towards negatively charged plasma proteins that limit their leakage in the urine. A study of Pima Indians with type 2 diabetes found that both podocyte damage and glomerular endothelial injury were commonly present in a cohort with macroalbuminuria. Interestingly, compared with podocyte injury, endothelial abnormalities were more closely associated with increased urine albumin excretion, suggesting that endothelial cell injury may be more critical to glomerular alterations in DKD compared with the commonly viewed importance of podocyte injury. Glycocalyx composition includes proteoglycans, glycoproteins, glycolipids, and glycosaminoglycans. Increased expression of proteolytic enzymes such as MMP9 (23–25), hyaluronidase (26), or heparanase [reviewed in van der Vlag and Buijsers (27)] was observed in diabetic patients and could participate in glycocalyx degradation in such pathological context, thus promoting proteinuria in diabetic patients (28, 29). MMP9 is mainly produced by podocytes and parietal epithelial cells in DKD where it participates in podocyte injury and promotes extracellular matrix (ECM) synthesis (23). Interestingly, MMP9 also promotes syndecan-4 shedding at ECs cell surface (30) and MMPs inhibition in a mouse diabetic model prevents syndecan-4 degradation and glycocalyx disruption in glomeruli (31). Whether it is the production by podocytes or by other cell types that induce ECs glycocalyx degradation in this context remains to be explored.
Heparanase is strongly up-regulated in podocytes exposed to high glucose (32, 33) (Figure 1). The increased heparanase expression by podocytes in kidneys has been demonstrated in DN (32, 34), and is essential for the development of albuminuria DN in both animal models and likely, in human (35, 36). Mice that lack heparanase develop less proteinuria or structural injury in diabetes induced with streptozotocin. Notably, loss of glycocalyx has been suggested in patients with type I diabetes 1 (37). Further, the development of microalbuminuria in diabetic patients results in further reductions of the systemic glycocalyx, leading to systemic vascular dysfunction (38). In Pima Indians with type 2 diabetes, podocyte foot processes in microalbuminuric participants were not different from those in control subjects and although microalbuminuria in type 2 diabetic Pima Indians often heralds progressive glomerular injury, it is not the result of defects in the size permselectivity of the glomerular barrier but rather of changes in either glomerular charge selectivity or tubular handling of filtered proteins or of a combination of these two factors (39). Another interesting study confirmed more directly that glycocalyx is perturbed in individuals with type 2 diabetes mellitus, and oral glycocalyx precursor treatment improved glycocalyx properties (28).
Garsen et al. demonstrated that heparanase production by podocytes promotes heparan sulfate degradation and glycocalyx disruption at podocyte and the endothelial cell surface in diabetic context by using in vivo mouse models and in vitro podocyte-to-EC supernatant transfer approaches (40) (Figure 1). Interestingly, in this latter article, the authors demonstrated that diabetes-mediated heparanase production in podocytes is mediated by endothelin pathway activation in podocytes in response to ET-1 production by ECs. Eberfors et al. also found that ET-1 signaling mediates degradation of the glomerular endothelial glycocalyx in non-diabetic kidney disease via pathological crosstalk between activated podocytes and glomerular endothelial cells but with a different mechanism. Indeed, here the authors found increased heparanase and hyaluronoglucosaminidase gene expression in glomerular ECs in response to podocyte-released factors and to ET-1 (41). Boels et al. further confirmed the crucial role of the endothelin pathway on heparanase expression and glycocalyx injury. Atrasentan, an antagonist of the endothelin receptor ETA, prevented glycocalyx degradation in DKD through reduction of glomerular and endothelial heparanase expression, although the production of heparanase by podocytes was not specifically explored in this context (42).
VEGF Family Pathway Dysregulation in Glomeruli During DKD
Podocytes act as pericyte-like cells to support ECs differentiation and notably produce VEGF which is crucial to maintain glomerular ECs differentiation. In a pioneer work published in 2008, Hirschberg et al. showed that VEGF is upregulated in podocytes after high glucose (HG) treatment (43). VEGF is sufficient to promote proliferation and tube formation of blood outgrowth EC (BOEC) through Flk-1 in vitro, and co-culture of podocytes and BOEC also enhances proliferation of the latter, hence emphasizing the role of podocyte-derived VEGF. These data suggested a podocyte role on angiogenesis during the early onset of diabetic nephropathy in vivo (44, 45). The role of the VEGF family in vivo has already been extensively reviewed elsewhere (46–51), and the growing evidence point to the role of VEGFA and VEGFC during DKD. VEGFA is mainly expressed by podocytes, can be alternatively spliced in different isoforms such as VEGFA165b, and bind the endothelial receptors VEGFR1 and VEGFR2 (Figure 1). Besides, other members of the family include VEGFB, VEGFC, VEGFD, and PlGF. VEGFC can act both on lymphatic and blood vessels through VEGFR3 and VEGFR2, respectively. In vitro, VEGFC protects glomerular ECs from the negative influence of VEGFA reducing their permeability, and podocyte VEGFC overexpression protects ECs during diabetes in vivo (52). Besides, VEGFA and diabetic conditions (in db/db mice) increase glomerular albumin permeability ex vivo, which is rescued by VEGFC treatment. VEGFA is increased in HG-cultured podocytes, and a direct axis TGFβ1/VEGFA/AP-1, terminating in Bcl2 reduction and podocyte apoptosis, has been described (53). In the same study, inhibition of VEGFA or AP-1 was beneficial for diabetic rats. However, VEGF-A165b improved the permeability of isolated diabetic human glomeruli, and diabetic mice treated with VEGF-A165b or having a specific podocyte-overexpression of it develop a less severe phenotype (54). Dysregulation of the VEGF pathway during DKD would be probably more complicated than just dysregulation of VEGF synthesis by podocytes (Figure 1). The Semaphorin 3-Neuropilin axis is an important regulator of podocyte-to-endothelial cells during development (55) and plasmatic and urinary Semaphorin 3 expressions are positively associated with DKD (56, 57). Furthermore, advanced glycation end-products suppress Neuropilin 1 expression in podocytes, thus promoting their migration in vitro (58). Podocyte-selective Semaphorin 3A overexpression exacerbates DKD in mice by remodeling podocyte cytoskeleton and ECM synthesis (59). In addition to Semaphorin 3A signaling, Neuropilin 1 plays an important role in endothelial cells via its binding to VEGFA. Nevertheless, Neuropilin 1 expression in glomeruli seems restrained to podocyte and decreased during diabetes in mice (58) and to our knowledge, no one has modulated Neuropilin 1 expression specifically in endothelial cells during DKD to explore its function in such pathological context. Neuropilin 1 receptor could function as an extracellular scaffold protein generating podocyte-endothelial cell cross-signaling during DKD. Together, these data illustrate the intertwined and multiple effects of the VEGF family during diabetes.
Other Influences of Glomerular ECs on Podocyte Injury During DKD
Physiologically, loops of glomerular capillaries are subjected to mean laminar shear stress (LSS) estimated at 10–20 dyne/cm2 (60). Slater et al. have shown that glomerular ECs submitted to LSS have increased ERK5 pathway activation leading to high KLF2 expression, which promotes ET-1, NO, and eNOS secretion (61). High KLF2 expression has also been observed in human glomeruli ex vivo. However, using conditioned media transfer from ECs to podocytes, and a co-culture strategy, they also show that LSS-exposed ECs secrete factors reducing the resistance of a podocyte monolayer. Of note, KLF2 is reduced in ECs exposed to high glucose but increased by insulin (62). In the same work, the authors showed that mice deficient for KLF2 in ECs have an aggravated phenotype during diabetes, and more pronounced podocyte lesions associated with higher glomerular mRNA levels of Vegfa, Flk1, and angiopoietin 2, and lower Flt1, Tie2, and angiopoietin 1 levels. These data highlight crosstalk between ECs and podocytes relying on KLF2 endothelial expression level, which promotes in basal conditions an anti-inflammatory phenotype and appears required for ECs-podocytes homeostasis (Figure 1).
One of the first works identifying EC-to-podocyte crosstalk in DKD came from Isermann et al. Indeed, they demonstrated that activated protein C (APC), which is regulated by endothelial thrombomodulin, is downregulated in DKD. They used gain-of-function and loss-of-function complementary approaches in diabetic mice to show that APC inhibits hyperglycemia-induced endothelial and podocyte mitochondrial-dependent apoptosis (63).
Yuen et al. unraveled the role of eNOS in the EC-podocyte crosstalk (64). Mice deficient in eNOS develop podocytopathy, although eNOS is expressed in endothelial cells, but not in podocytes (65, 66). Authors have observed a marked cytoskeleton rearrangement of podocytes treated with the serum of diabetic eNOS-deficient mice, which suggests a modulation of the RhoA family that controls cytoskeleton dynamics. Moreover, increased activation of RhoA in podocytes treated with supernatants from glomerular ECs exposed to high glucose and/or angiotensin II isolated from diabetic eNOS-deficient mice was observed, despite the reduction of RhoA activity when ECs were from control mice. Nevertheless, the modulation of RhoA in podocytes has been reported detrimental (67).
Transforming growth factor-beta (TGF-β) is a well-described mediator of renal fibrosis in DKD with pleiotropic effects on glomerular cells. It promotes mesangial cell hypertrophy and extracellular matrix deposition and induces endothelial and podocyte dedifferentiation or death in mouse diabetic models [Reviewed in Chang et al. (68) and Ghayur and Margetts (69)]. TGF-β1 and TGF-β2 may originate from several cell types in DKD (in particular mesangial cells and EC) and as a secreted molecule, it would not be surprising that TGF-β could promote glomerulosclerosis and GFB dysfunction in DKD through paracrine mechanisms and participate in cross-communication within the GFB. The mechanisms for such deleterious effects of TGF-β on the GFB cellular components, podocytes and EC, are still unclear, whereas this growth factor displays contrasting hypertrophic and survival actions on other cell types such as fibroblast, mesangial cells or vascular smooth muscle cells.
A recent publication from regret Detlef Schlondorff's group highlighted the role of BAMBI a negative modulator of TGF-β1 in DKD with specific roles in ECs and podocytes (70). Interestingly the authors demonstrated that in diabetes, selective EC-Bambi deletion induced podocyte injury similarly to a selective podocyte-Bambi deletion. Diabetes-induced podocyte loss was even more pronounced in the EC-Bambi KO than in the podocyte-Bambi KO mice. Similarly, endothelial-selective autophagy inhibition also promotes podocyte injury in DN, supporting the concept that ECs injury in DKD may be a crucial mediator of podocyte injury and underscoring the importance of glomerular crosstalk in DKD (71).
ECs communicate with podocytes through secreted proteins, but also via exosomes during diabetes. To our knowledge, only one work from Wu et al. demonstrates that during diabetes, ECs could negatively affect podocytes by releasing exosomes (72). In HG conditions, ECs undergo endothelial-to-mesenchymal transition, secrete more exosomes, and the latter are internalized by podocytes which increased the TGF-β1/Wnt/β-catenin pathway. Consistently, podocytes treated with HG-cultured EC-derived exosomes are more permeable to albumin in vitro. Activation of the Wnt/β-catenin pathway in podocytes during diabetes and other proteinuric kidney diseases is known to be detrimental (73–75), and leads to oxidative stress (76).
Other Influences of Podocytes on Glomerular ECs
In recent work, Ngo et al. collected plasma samples to assess renal arteriovenous gradients (77). A positive correlation between testican-2 and eGFR, and an association between higher baseline testican-2 levels and slower decline of eGFR, were observed in cohorts of patients including diabetics. Interestingly, testican-2 is expressed by podocytes and glomerular basal membrane, and can increase glomerular EC tube formation and motility, but not proliferation, in vitro. Hence, secreted testican-2 from podocytes seems beneficial for glomerular ECs in a variety of chronic kidney diseases including diabetes. Of note, testican-2 is not expressed in mice (78), illustrating the need for in vitro models to study podocyte-EC crosstalks. More complex culture models have been investigated, being 3D tri-partite cell cultures or “glomerulus-on-chip” systems. Waters et al. used glomerular EC, mesangial cells, and podocyte to reproduce the glomerular filtration barrier in 3D cultures (79). They showed that TGFβ-induced glomerulosclerosis, as seen in DKD, was prevented in 3D tri-cultures by conjoint inhibition of ALK5 and CTGF, and differential effects of TGFβ on mesangial cells and glomerular ECs. TGFβ led to nodule formation and loss of ECs arborization in 3D tri-cultures, and in mono-cultures, it increased the mediator CTGF expression in podocytes and increased ALK5 expression in mesangial cells, which favors an upregulation of TGFβ pathway activation through SMAD2/3. Moreover, BMP7 appeared to modulate the effects of TGFβ on ECs but not in mesangial cells.
Another well-known angiogenic signaling is likely to be involved in podocyte-to-endothelial cell cross-communication in DKD, whereas direct evidence is missing. Angiopoietin 1 (ANGPT1) produced by podocytes promotes maturation and stabilization of glomerular capillaries via TIE2 receptor on endothelial cells (80), playing an important role in the regulation of angiogenesis, endothelial cell survival, proliferation, migration, adhesion, and spreading, but also maintenance of vascular quiescence. Angiopoietin 2 (ANGPT2) has opposite effects to ANGPT1 by destabilizing blood vessels and effects of ANGPT2 are dependent on VEGFA levels (81, 82). ANGPT2 levels are associated with indexes of endothelial dysfunction in clinical diabetes mellitus (83–85). A decreased circulating ANGPT1/ANGPT-2 ratio may contribute to the development of DKD after administration of STZ in mice (86) and STZ-induced DKD rats (87). Meanwhile, plasma ANGPT2, like VEGF, was found to be raised in human diabetes regardless of vascular disease. Whereas, both growth factors correlated with HbA1c and with each other, ANGPT2 levels did not correlate with carotid atherosclerosis, plasma von Willebrand factor (vWf), and urine albumin to creatinine ratio in humans with type 2 diabetes after multiple adjustment (88). This is not surprising as the ANGPT system is regulated locally in the microvessels. Regulation of glomerular angiopoietin levels is key in animal models of DKD. To further investigate the role of ANGPT1 in diabetes, Jeansson et al. compared diabetic controls and Angpt1-deleted mice induced with STZ. The Angpt1 knockout kidney showed accelerated diabetes-mediated glomerular damage, suggesting that ANGPT1 could potentially protect the glomerular microvasculature from diabetes-induced injury (89). Mice with podocyte-specific inducible ANGPT-1 overexpression in the early stage of DKD led to a 70% reduction of albuminuria and prevented diabetes-induced GEC proliferation via increased TIE-2 phosphorylation suggesting a critical role of ANGPT1-1/ANGPT-2 in early DKD. Meanwhile, hyperfiltration and renal morphology were unchanged, indicating a still limited role (90). The role of angiopoietins in DKD has been nicely reviewed elsewhere (91, 92). Overall, the authors consider that angiopoietins are produced by podocyte and signal via TIE2 in endothelial cells only, which is most likely the mechanism but has not been entirely demonstrated.
Finally, dysregulation of the GBM composition by podocytes could also impair ECs in DKD. Indeed, heparan sulfate, the major component of glomerular ECM, modulates growth factor signaling, notably modulating VEGFA availability to the surrounding ECs. Schumacher et al. showed that Wilms' Tumor 1 changes VEGFA and FGF2 signaling by increasing the expression of the 6-O-endosulfatases Sulf1 and Sulf2, which remodel the heparan sulfate 6-O-sulfation pattern in the GBM (93). Mice deficient in both Sulf1 and Sulf2 developed age-dependent proteinuria as a result of ultrastructural abnormalities in podocytes and endothelial cells. Sulf1 and Sulf2 double-knockout (DKO) mice also showed glomerular hypercellularity, matrix accumulation, and GBM irregularity. Platelet-derived growth factor (PDGF)-B and PDGF receptor-β were upregulated in Sulf1 and Sulf2 DKO mice. Diabetic mice showed an upregulation of glomerular Sulf1 and Sulf2 expression and diabetic Sulf1 and Sulf2 DKO mice showed an acceleration of the glomerular pathology without glomerular hypertrophy (94). Thus, Sulf1 and Sulf2 may play protective roles in DKD, probably by modulating growth factor availability to podocytes and ECs.
Discussion
Across the years, researchers tried to model pathological conditions happening during DKD, by culturing EC and/or podocytes with HG. More recently, this EC-podocyte crosstalk has been explored with conditioned media transfers. This shed light on the crucial role of VEGF, produced by podocytes for EC survival and dysregulated during DKD (43, 50, 95), but also to signaling pathways modulated in EC or podocytes during the excess of glucose. In parallel, rodent DKD models were developed, confirmed both of these observations, and even more. Transgenic animals gave substances to the former hypothesis and opened new horizons, as integrated systems (96). Nevertheless, cell cultures remain powerful tools, they tend to naturally evolve from monolayered and mono-typed to multi-typed, organoids and even glomerulus-on-chip, to represent an alternative, or at least an intermediate, to in vivo studies. Recently, Wang et al. developed a tri-partite glomerular filtration barrier in a 3D culture from rat glomeruli and showed that high glucose conditions in the endothelial side increased barrier permeability and podocyte migration in this model (97). Zhou et al. developed a device composed of ECs and podocytes cultured together but physically separated by a porous membrane (98). Hypertensive conditions on the endothelial compartment led to increased barrier permeability and damages of both EC and podocytes. Hence, in vitro models constitute an efficient way to study glomerular ECs – podocytes crosstalk during DKD. More importantly, they are useful to dissect molecular pathways involved in pathophysiology. Recent advances in 3D cell cultures and microfluidics tend to combine the comfort and high throughput of in vitro assays with partial biological relevance of in vivo studies. A significant limitation of such 3D systems being obviously to poorly mimic the pathophysiological environment of glomerular cells as would occur in chronic diabetic condition.
ECs - podocytes crosstalk is crucial during DKD development, where ECs also dialogue with other renal cell types. As an example, the elevation of EC-secreted ET-1 directly enhances mesangial expansion and extracellular matrix deposits, characteristics of DKD (99). ECs injury could also participate in parietal epithelial cell activation, a condition seen in focal segmental glomerulosclerosis (FSGS), a rare-to-common renal complication of diabetic patients. Indeed, Luque et al. demonstrated that Hif2α pathway inhibition in endothelial cells only sensitized mice to the development of hypertension-induced FSGS, suggesting that signals from the ECs could be transferred to PEC (100). Finally, renal tubular cell injury in diabetes modulates ECs [reviewed in Chen et al. (101)].
Together, these studies showed through different methodologies, from cell cultures to human samples, that glomerular ECs are crucial actors of DKD pathophysiology, and cross-communications with podocytes constitute major events for diabetic renal disease progression. Intensive control of glucose and blood pressure along with RAS inhibition and SGLT2i (for patients with type 2 DM) remain the clinical gold standards to deter the progression of DKD. The armament may be complemented in the near future with glucagon-like peptide 1 (GLP-1) receptor agonists, non-steroidal selective mineralocorticoid receptor antagonists (MRAs) or ETRAs. In fact, metformin, RAAS and ET-1 inhibitors were shown to prevent endothelial dysfunction beyond their effect on insulin resistance. Recent anti-diabetic drugs also display clear effects on the microcirculation in animal models and patients (102–107). Additional work is needed to understand the mechanisms involved, and new treatments that aim to prevent microvascular injury or restore microvascular function could be an effective strategy for preventing; or even reversing DKD. Such strategies may consider crosstalk within the glomerular system. Fine tuning of angiogenic systems and cellular energetics, promotion of autophagy, of glycocalyx protection, alleviation of chronic sterile inflammation and senescence may offer promising perspectives. The variety of molecules involved represents as many potential therapeutic targets to better take charge of the DKD burden and improve patient lives. Future studies need to consolidate the concept of the glomerulus as an integrated functional unit.
Author Contributions
NM, OL, and P-LT wrote the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Zimmet P, Alberti KG, Magliano DJ, Bennett PH. Diabetes mellitus statistics on prevalence and mortality: facts and fallacies. Nat Rev Endocrinol. (2016) 12:616–22. doi: 10.1038/nrendo.2016.105
2. Cheng H-T, Xu X, Lim PS, Hung K-Y. Worldwide epidemiology of diabetes-related end-stage renal disease, 2000-2015. Diabetes Care. (2020) 44:89–97. doi: 10.2337/figshare.13105469
3. de Boer IH. Long-term renal outcomes of patients with type 1 diabetes mellitus and microalbuminuria an analysis of the diabetes control and complications trial/epidemiology of diabetes interventions and complications cohort microalbuminuria outcomes in type 1 diabetes. Arch Intern Med. (2011) 171:412. doi: 10.1001/archinternmed.2011.16
4. Toyoda M, Najafian B, Kim Y, Caramori ML, Mauer M. Podocyte detachment and reduced glomerular capillary endothelial fenestration in human type 1 diabetic nephropathy. Diabetes. (2007) 56:2155–60. doi: 10.2337/db07-0019
5. Sol M, Kamps JAAM, van den Born J, van den Heuvel MC, van der Vlag J, Krenning G, et al. Glomerular endothelial cells as instigators of glomerular sclerotic diseases. Front Pharmacol. (2020) 11:573557. doi: 10.3389/fphar.2020.573557
6. Ndisang JF. Glomerular endothelium and its impact on glomerular filtration barrier in diabetes: are the gaps still illusive? Curr Med Chem. (2018) 25:1525–9. doi: 10.2174/0929867324666170705124647
7. Fu J, Lee K, Chuang PY, Liu Z, He JC. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am J Physiol Renal Physiol. (2015) 308:F287–97. doi: 10.1152/ajprenal.00533.2014
8. Siddiqi FS, Advani A. Endothelial-podocyte crosstalk: the missing link between endothelial dysfunction and albuminuria in diabetes. Diabetes. (2013) 62:3647–55. doi: 10.2337/db13-0795
9. Daehn IS. Glomerular endothelial cell stress and cross-talk with podocytes in early [corrected] diabetic kidney disease. Front Med. (2018) 5:76. doi: 10.3389/fmed.2018.00076
10. Heerspink HJL, Parving H-H, Andress DL, Bakris G, Correa-Rotter R, Hou F-F, et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): a double-blind, randomised, placebo-controlled trial. Lancet Lond Engl. (2019) 393:1937–47. doi: 10.1016/S0140-6736(19)30772-X
11. Fukui M, Nakamura T, Ebihara I, Osada S, Tomino Y, Masaki T, et al. Gene expression for endothelins and their receptors in glomeruli of diabetic rats. J Lab Clin Med. (1993) 122:149–56.
12. Hargrove GM, Dufresne J, Whiteside C, Muruve DA, Wong NC. Diabetes mellitus increases endothelin-1 gene transcription in rat kidney. Kidney Int. (2000) 58:1534–45. doi: 10.1046/j.1523-1755.2000.00315.x
13. Bruno CM, Meli S, Marcinno M, Ierna D, Sciacca C, Neri S. Plasma endothelin-1 levels and albumin excretion rate in normotensive, microalbuminuric type 2 diabetic patients. J Biol Regul Homeost Agents. (2002) 16:114–7.
14. Zanatta CM, Veronese FV, Loreto MS, Sortica DA, Carpio VN, Eldeweiss MIA, et al. Endothelin-1 and endothelin a receptor immunoreactivity is increased in patients with diabetic nephropathy. Ren Fail. (2012) 34:308–15. doi: 10.3109/0886022X.2011.647301
15. Ding S-S, Qiu C, Hess P, Xi J-F, Zheng N, Clozel M. Chronic endothelin receptor blockade prevents both early hyperfiltration and late overt diabetic nephropathy in the rat. J Cardiovasc Pharmacol. (2003) 42:48–54. doi: 10.1097/00005344-200307000-00008
16. Cosenzi A, Bernobich E, Trevisan R, Milutinovic N, Borri A, Bellini G. Nephroprotective effect of bosentan in diabetic rats. J Cardiovasc Pharmacol. (2003) 42:752–6. doi: 10.1097/00005344-200312000-00009
17. Saleh MA, Boesen EI, Pollock JS, Savin VJ, Pollock DM. Endothelin receptor a-specific stimulation of glomerular inflammation and injury in a streptozotocin-induced rat model of diabetes. Diabetologia. (2011) 54:979–88. doi: 10.1007/s00125-010-2021-4
18. Lenoir O, Milon M, Virsolvy A, Hénique C, Schmitt A, Massé J-M, et al. Direct action of endothelin-1 on podocytes promotes diabetic glomerulosclerosis. J Am Soc Nephrol JASN. (2014) 25:1050–62. doi: 10.1681/ASN.2013020195
19. Daehn I, Casalena G, Zhang T, Shi S, Fenninger F, Barasch N, et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest. (2014) 124:1608–21. doi: 10.1172/JCI71195
20. Qi H, Casalena G, Shi S, Yu L, Ebefors K, Sun Y, et al. Glomerular endothelial mitochondrial dysfunction is essential and characteristic of diabetic kidney disease susceptibility. Diabetes. (2017) 66:763–78. doi: 10.2337/db16-0695
21. Casalena GA, Yu L, Gil R, Rodriguez S, Sosa S, Janssen W, et al. The diabetic microenvironment causes mitochondrial oxidative stress in glomerular endothelial cells and pathological crosstalk with podocytes. Cell Commun Signal CCS. (2020) 18:105. doi: 10.1186/s12964-020-00605-x
22. Heerspink HJL, Andress DL, Bakris G, Brennan JJ, Correa-Rotter R, Hou FF, et al. Baseline characteristics and enrichment results from the SONAR trial. Diabetes Obes Metab. (2018) 20:1829–35. doi: 10.1111/dom.13315
23. Li S-Y, Huang P-H, Yang A-H, Tarng D-C, Yang W-C, Lin C-C, et al. Matrix metalloproteinase-9 deficiency attenuates diabetic nephropathy by modulation of podocyte functions and dedifferentiation. Kidney Int. (2014) 86:358–69. doi: 10.1038/ki.2014.67
24. Rysz J, Banach M, Stolarek RA, Pasnik J, Cialkowska-Rysz A, Koktysz R, et al. Serum matrix metalloproteinases MMP-2 and MMP-9 and metalloproteinase tissue inhibitors TIMP-1 and TIMP-2 in diabetic nephropathy. J Nephrol. (2007) 20:444–52.
25. García-Tejeda AU, Sampieri CL, Suárez-Torres I, Morales-Romero J, Demeneghi-Marini VP, Hernández-Hernández ME, et al. Association of urinary activity of MMP-9 with renal impairment in Mexican patients with type 2 diabetes mellitus. PeerJ. (2018) 6:e6067. doi: 10.7717/peerj.6067
26. Ikegami-Kawai M, Suzuki A, Karita I, Takahashi T. Increased hyaluronidase activity in the kidney of streptozotocin-induced diabetic rats. J Biochem. (2003) 134:875–80. doi: 10.1093/jb/mvg214
27. van der Vlag J, Buijsers B. Heparanase in kidney disease. Adv Exp Med Biol. (2020) 1221:647–67. doi: 10.1007/978-3-030-34521-1_26
28. Broekhuizen LN, Lemkes BA, Mooij HL, Meuwese MC, Verberne H, Holleman F, et al. Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia. (2010) 53:2646–55. doi: 10.1007/s00125-010-1910-x
29. Nieuwdorp M, van Haeften TW, Gouverneur MCLG, Mooij HL, van Lieshout MHP, Levi M, et al. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes. (2006) 55:480–6. doi: 10.2337/diabetes.55.02.06.db05-1103
30. Ramnath R, Foster RR, Qiu Y, Cope G, Butler MJ, Salmon AH, et al. Matrix metalloproteinase 9-mediated shedding of syndecan 4 in response to tumor necrosis factor α: a contributor to endothelial cell glycocalyx dysfunction. FASEB J Off Publ Fed Am Soc Exp Biol. (2014) 28:4686–99. doi: 10.1096/fj.14-252221
31. Ramnath RD, Butler MJ, Newman G, Desideri S, Russell A, Lay AC, et al. Blocking matrix metalloproteinase-mediated syndecan-4 shedding restores the endothelial glycocalyx and glomerular filtration barrier function in early diabetic kidney disease. Kidney Int. (2020) 97:951–65. doi: 10.1016/j.kint.2019.09.035
32. Maxhimer JB, Somenek M, Rao G, Pesce CE, Baldwin D, Gattuso P, et al. Heparanase-1 gene expression and regulation by high glucose in renal epithelial cells: a potential role in the pathogenesis of proteinuria in diabetic patients. Diabetes. (2005) 54:2172–8. doi: 10.2337/diabetes.54.7.2172
33. van den Hoven MJ, Rops AL, Bakker MA, Aten J, Rutjes N, Roestenberg P, et al. Increased expression of heparanase in overt diabetic nephropathy. Kidney Int. (2006) 70:2100–8. doi: 10.1038/sj.ki.5001985
34. Wijnhoven TJM, van den Hoven MJW, Ding H, van Kuppevelt TH, van der Vlag J, Berden JHM, et al. Heparanase induces a differential loss of heparan sulphate domains in overt diabetic nephropathy. Diabetologia. (2008) 51:372–82. doi: 10.1007/s00125-007-0879-6
35. Gil N, Goldberg R, Neuman T, Garsen M, Zcharia E, Rubinstein AM, et al. Heparanase is essential for the development of diabetic nephropathy in mice. Diabetes. (2012) 61:208–16. doi: 10.2337/db11-1024
36. Sidaway P. Diabetic nephropathy: Heparanase mediates renal injury. Nat Rev Nephrol. (2014) 10:483. doi: 10.1038/nrneph.2014.134
37. Deckert T, Kofoed-Enevoldsen A, Vidal P, Nørgaard K, Andreasen HB, Feldt-Rasmussen B. Size- and charge selectivity of glomerular filtration in Type 1 (insulin-dependent) diabetic patients with and without albuminuria. Diabetologia. (1993) 36:244–51. doi: 10.1007/BF00399958
38. Nieuwdorp M, Mooij HL, Kroon J, Atasever B, Spaan JAE, Ince C, et al. Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes. (2006) 55:1127–32. doi: 10.2337/diabetes.55.04.06.db05-1619
39. Lemley KV, Blouch K, Abdullah I, Boothroyd DB, Bennett PH, Myers BD, et al. Glomerular permselectivity at the onset of nephropathy in type 2 diabetes mellitus. J Am Soc Nephrol JASN. (2000) 11:2095–105.
40. Garsen M, Lenoir O, Rops ALWMM, Dijkman HB, Willemsen B, van Kuppevelt TH, et al. Endothelin-1 Induces Proteinuria by Heparanase-Mediated Disruption of the Glomerular Glycocalyx. J Am Soc Nephrol JASN. (2016) 27:3545–51. doi: 10.1681/ASN.2015091070
41. Ebefors K, Wiener RJ, Yu L, Azeloglu EU, Yi Z, Jia F, et al. Endothelin receptor-A mediates degradation of the glomerular endothelial surface layer via pathologic crosstalk between activated podocytes and glomerular endothelial cells. Kidney Int. (2019) 96:957–70. doi: 10.1016/j.kint.2019.05.007
42. Boels MGS, Avramut MC, Koudijs A, Dane MJC, Lee DH, van der Vlag J, et al. Atrasentan reduces albuminuria by restoring the glomerular endothelial glycocalyx barrier in diabetic nephropathy. Diabetes. (2016) 65:2429–39. doi: 10.2337/db15-1413
43. Hirschberg R, Wang S, Mitu GM. Functional symbiosis between endothelium and epithelial cells in glomeruli. Cell Tissue Res. (2008) 331:485–93. doi: 10.1007/s00441-007-0526-z
44. Nyengaard JR. Number and dimensions of rat glomerular capillaries in normal development and after nephrectomy. Kidney Int. (1993) 43:1049–57. doi: 10.1038/ki.1993.147
45. Nyengaard JR, Rasch R. The impact of experimental diabetes mellitus in rats on glomerular capillary number and sizes. Diabetologia. (1993) 36:189–94. doi: 10.1007/BF00399948
46. Chen S, Ziyadeh FN. Vascular endothelial growth factor and diabetic nephropathy. Curr Diab Rep. (2008) 8:470–6. doi: 10.1007/s11892-008-0081-3
47. Mironidou-Tzouveleki M, Tsartsalis S, Tomos C. Vascular endothelial growth factor (VEGF) in the pathogenesis of diabetic nephropathy of type 1 diabetes mellitus. Curr Drug Targets. (2011) 12:107–14. doi: 10.2174/138945011793591581
48. Tanabe K, Wada J, Sato Y. Targeting angiogenesis and lymphangiogenesis in kidney disease. Nat Rev Nephrol. (2020) 16:289–303. doi: 10.1038/s41581-020-0260-2
49. Majumder S, Advani A. VEGF and the diabetic kidney: More than too much of a good thing. J Diabetes Compl. (2017) 31:273–9. doi: 10.1016/j.jdiacomp.2016.10.020
50. Tufro A, Veron D. VEGF and podocytes in diabetic nephropathy. Semin Nephrol. (2012) 32:385–93. doi: 10.1016/j.semnephrol.2012.06.010
51. Domigan CK, Ziyad S, Iruela-Arispe ML. Canonical and non-canonical vascular endothelial growth factor pathways: new developments in biology and signal transduction. Arterioscler Thromb Vasc Biol. (2015) 35:30–9. doi: 10.1161/ATVBAHA.114.303215
52. Onions KL, Gamez M, Buckner NR, Baker SL, Betteridge KB, Desideri S, et al. VEGFC reduces glomerular albumin permeability and protects against alterations in VEGF receptor expression in diabetic nephropathy. Diabetes. (2019) 68:172–87. doi: 10.2337/db18-0045
53. Bai X, Geng J, Li X, Yang F, Tian J. VEGF-A inhibition ameliorates podocyte apoptosis via repression of activating protein 1 in diabetes. Am J Nephrol. (2014) 40:523–34. doi: 10.1159/000369942
54. Oltean S, Qiu Y, Ferguson JK, Stevens M, Neal C, Russell A, et al. Vascular endothelial growth factor-A165b is protective and restores endothelial glycocalyx in diabetic nephropathy. J Am Soc Nephrol JASN. (2015) 26:1889–904. doi: 10.1681/ASN.2014040350
55. Reidy KJ, Villegas G, Teichman J, Veron D, Shen W, Jimenez J, et al. Semaphorin3a regulates endothelial cell number and podocyte differentiation during glomerular development. Dev Camb Engl. (2009) 136:3979–89. doi: 10.1242/dev.037267
56. Kwon SH, Shin JP, Kim IT, Park DH. Association of plasma semaphorin 3A with phenotypes of diabetic retinopathy and nephropathy. Invest Ophthalmol Vis Sci. (2016) 57:2983–9. doi: 10.1167/iovs.16-19468
57. Mohamed R, Ranganathan P, Jayakumar C, Nauta FL, Gansevoort RT, Weintraub NL, et al. Urinary semaphorin 3A correlates with diabetic proteinuria and mediates diabetic nephropathy and associated inflammation in mice. J Mol Med Berl Ger. (2014) 92:1245–56. doi: 10.1007/s00109-014-1209-3
58. Bondeva T, Rüster C, Franke S, Hammerschmid E, Klagsbrun M, Cohen CD, et al. Advanced glycation end-products suppress neuropilin-1 expression in podocytes. Kidney Int. (2009) 75:605–16. doi: 10.1038/ki.2008.603
59. Aggarwal PK, Veron D, Thomas DB, Siegel D, Moeckel G, Kashgarian M, et al. Semaphorin3a promotes advanced diabetic nephropathy. Diabetes. (2015) 64:1743–59. doi: 10.2337/db14-0719
60. Ballermann BJ, Dardik A, Eng E, Liu A. Shear stress and the endothelium. Kidney Int Suppl. (1998) 67:S100–8. doi: 10.1046/j.1523-1755.1998.06720.x
61. Slater SC, Ramnath RD, Uttridge K, Saleem MA, Cahill PA, Mathieson PW, et al. Chronic exposure to laminar shear stress induces Kruppel-like factor 2 in glomerular endothelial cells and modulates interactions with co-cultured podocytes. Int J Biochem Cell Biol. (2012) 44:1482–90. doi: 10.1016/j.biocel.2012.05.020
62. Zhong F, Chen H, Wei C, Zhang W, Li Z, Jain MK, et al. Reduced Krüppel-like factor 2 expression may aggravate the endothelial injury of diabetic nephropathy. Kidney Int. (2015) 87:382–95. doi: 10.1038/ki.2014.286
63. Isermann B, Vinnikov IA, Madhusudhan T, Herzog S, Kashif M, Blautzik J, et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. (2007) 13:1349–58. doi: 10.1038/nm1667
64. Yuen DA, Stead BE, Zhang Y, White KE, Kabir MG, Thai K, et al. eNOS deficiency predisposes podocytes to injury in diabetes. J Am Soc Nephrol JASN. (2012) 23:1810–23. doi: 10.1681/ASN.2011121170
65. Sörensson J, Fierlbeck W, Heider T, Schwarz K, Park DS, Mundel P, et al. Glomerular endothelial fenestrae in vivo are not formed from caveolae. J Am Soc Nephrol JASN. (2002) 13:2639–47. doi: 10.1097/01.ASN.0000033277.32822.23
66. Bachmann S, Bosse HM, Mundel P. Topography of nitric oxide synthesis by localizing constitutive NO synthases in mammalian kidney. Am J Physiol. (1995) 268:F885–98. doi: 10.1152/ajprenal.1995.268.5.F885
67. Zhu L, Jiang R, Aoudjit L, Jones N, Takano T. Activation of RhoA in podocytes induces focal segmental glomerulosclerosis. J Am Soc Nephrol JASN. (2011) 22:1621–30. doi: 10.1681/ASN.2010111146
68. Chang AS, Hathaway CK, Smithies O, Kakoki M. Transforming growth factor-β1 and diabetic nephropathy. Am J Physiol Renal Physiol. (2016) 310:F689–96. doi: 10.1152/ajprenal.00502.2015
69. Ghayur A, Margetts PJ. Transforming growth factor-beta and the glomerular filtration barrier. Kidney Res Clin Pract. (2013) 32:3–10. doi: 10.1016/j.krcp.2013.01.003
70. Lai H, Chen A, Cai H, Fu J, Salem F, Li Y, et al. Podocyte and endothelial-specific elimination of BAMBI identifies differential transforming growth factor-β pathways contributing to diabetic glomerulopathy. Kidney Int. (2020) 98:601–14. doi: 10.1016/j.kint.2020.03.036
71. Lenoir O, Jasiek M, Hénique C, Guyonnet L, Hartleben B, Bork T, et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy. (2015) 11:1130–45. doi: 10.1080/15548627.2015.1049799
72. Wu X, Gao Y, Xu L, Dang W, Yan H, Zou D, et al. Exosomes from high glucose-treated glomerular endothelial cells trigger the epithelial-mesenchymal transition and dysfunction of podocytes. Sci Rep. (2017) 7:9371. doi: 10.1038/s41598-017-09907-6
73. Bose M, Almas S, Prabhakar S. Wnt signaling and podocyte dysfunction in diabetic nephropathy. J Investig Med Off Publ Am Fed Clin Res. (2017) 65:1093–101. doi: 10.1136/jim-2017-000456
74. Zhou L, Liu Y. Wnt/β-catenin signalling and podocyte dysfunction in proteinuric kidney disease. Nat Rev Nephrol. (2015) 11:535–45. doi: 10.1038/nrneph.2015.88
75. Wang D, Dai C, Li Y, Liu Y. Canonical Wnt/β-catenin signaling mediates transforming growth factor-β1-driven podocyte injury and proteinuria. Kidney Int. (2011) 80:1159–69. doi: 10.1038/ki.2011.255
76. Zhou L, Chen X, Lu M, Wu Q, Yuan Q, Hu C, et al. Wnt/β-catenin links oxidative stress to podocyte injury and proteinuria. Kidney Int. (2019) 95:830–45. doi: 10.1016/j.kint.2018.10.032
77. Ngo D, Wen D, Gao Y, Keyes MJ, Drury ER, Katz DH, et al. Circulating testican-2 is a podocyte-derived marker of kidney health. Proc Natl Acad Sci USA. (2020) 117:25026–35. doi: 10.1073/pnas.2009606117
78. Vannahme C, Schübel S, Herud M, Gösling S, Hülsmann H, Paulsson M, et al. Molecular cloning of testican-2: defining a novel calcium-binding proteoglycan family expressed in brain. J Neurochem. (1999) 73:12–20. doi: 10.1046/j.1471-4159.1999.0730012.x
79. Waters JP, Richards YC, Skepper JN, Southwood M, Upton PD, Morrell NW, et al. A 3D tri-culture system reveals that activin receptor-like kinase 5 and connective tissue growth factor drive human glomerulosclerosis. J Pathol. (2017) 243:390–400. doi: 10.1002/path.4960
80. Satchell SC, Harper SJ, Tooke JE, Kerjaschki D, Saleem MA, Mathieson PW. Human podocytes express angiopoietin 1, a potential regulator of glomerular vascular endothelial growth factor. J Am Soc Nephrol JASN. (2002) 13:544–50.
81. Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. (1997) 277:55–60. doi: 10.1126/science.277.5322.55
82. Findley CM, Cudmore MJ, Ahmed A, Kontos CD. VEGF induces Tie2 shedding via a phosphoinositide 3-kinase/Akt dependent pathway to modulate Tie2 signaling. Arterioscler Thromb Vasc Biol. (2007) 27:2619–26. doi: 10.1161/ATVBAHA.107.150482
83. Khairoun M, de Koning EJP, van den Berg BM, Lievers E, de Boer HC, Schaapherder AFM, et al. Microvascular damage in type 1 diabetic patients is reversed in the first year after simultaneous pancreas-kidney transplantation. Am J Transplant Off J Am Soc Transplant Am Soc Transpl Surg. (2013) 13:1272–81. doi: 10.1111/ajt.12182
84. Tsai Y-C, Lee C-S, Chiu Y-W, Lee J-J, Lee S-C, Hsu Y-L, et al. Angiopoietin-2, renal deterioration, major adverse cardiovascular events and all-cause mortality in patients with diabetic nephropathy. Kidney Blood Press Res. (2018) 43:545–54. doi: 10.1159/000488826
85. Lim HS, Blann AD, Chong AY, Freestone B, Lip GYH. Plasma vascular endothelial growth factor, angiopoietin-1, and angiopoietin-2 in diabetes: implications for cardiovascular risk and effects of multifactorial intervention. Diabetes Care. (2004) 27:2918–24. doi: 10.2337/diacare.27.12.2918
86. Yamamoto Y, Maeshima Y, Kitayama H, Kitamura S, Takazawa Y, Sugiyama H, et al. Tumstatin peptide, an inhibitor of angiogenesis, prevents glomerular hypertrophy in the early stage of diabetic nephropathy. Diabetes. (2004) 53:1831–40. doi: 10.2337/diabetes.53.7.1831
87. Rizkalla B, Forbes JM, Cao Z, Boner G, Cooper ME. Temporal renal expression of angiogenic growth factors and their receptors in experimental diabetes: role of the renin-angiotensin system. J Hypertens. (2005) 23:153–64. doi: 10.1097/00004872-200501000-00026
88. Lim HS, Lip GYH, Blann AD. Angiopoietin-1 and angiopoietin-2 in diabetes mellitus: relationship to VEGF, glycaemic control, endothelial damage/dysfunction and atherosclerosis. Atherosclerosis. (2005) 180:113–8. doi: 10.1016/j.atherosclerosis.2004.11.004
89. Jeansson M, Gawlik A, Anderson G, Li C, Kerjaschki D, Henkelman M, et al. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J Clin Invest. (2011) 121:2278–89. doi: 10.1172/JCI46322
90. Dessapt-Baradez C, Woolf AS, White KE, Pan J, Huang JL, Hayward AA, et al. Targeted glomerular angiopoietin-1 therapy for early diabetic kidney disease. J Am Soc Nephrol JASN. (2014) 25:33–42. doi: 10.1681/ASN.2012121218
91. Gnudi L. Angiopoietins and diabetic nephropathy. Diabetologia. (2016) 59:1616–20. doi: 10.1007/s00125-016-3995-3
92. He F-F, Zhang D, Chen Q, Zhao Y, Wu L, Li Z-Q, et al. Angiopoietin-Tie signaling in kidney diseases: an updated review. FEBS Lett. (2019) 593:2706–15. doi: 10.1002/1873-3468.13568
93. Schumacher VA, Schlötzer-Schrehardt U, Karumanchi SA, Shi X, Zaia J, Jeruschke S, et al. WT1-dependent sulfatase expression maintains the normal glomerular filtration barrier. J Am Soc Nephrol JASN. (2011) 22:1286–96. doi: 10.1681/ASN.2010080860
94. Takashima Y, Keino-Masu K, Yashiro H, Hara S, Suzuki T, van Kuppevelt TH, et al. Heparan sulfate 6-O-endosulfatases, Sulf1 and Sulf2, regulate glomerular integrity by modulating growth factor signaling. Am J Physiol Renal Physiol. (2016) 310:F395–408. doi: 10.1152/ajprenal.00445.2015
95. Khamaisi M, Schrijvers BF, De Vriese AS, Raz I, Flyvbjerg A. The emerging role of VEGF in diabetic kidney disease. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc - Eur Ren Assoc. (2003) 18:1427–30. doi: 10.1093/ndt/gfg242
96. Alpers CE, Hudkins KL. Mouse models of diabetic nephropathy. Curr Opin Nephrol Hypertens. (2011) 20:278–84. doi: 10.1097/MNH.0b013e3283451901
97. Wang L, Tao T, Su W, Yu H, Yu Y, Qin J. A disease model of diabetic nephropathy in a glomerulus-on-a-chip microdevice. Lab Chip. (2017) 17:1749–60. doi: 10.1039/C7LC00134G
98. Zhou M, Zhang X, Wen X, Wu T, Wang W, Yang M, et al. Development of a functional glomerulus at the organ level on a chip to mimic hypertensive nephropathy. Sci Rep. (2016) 6:31771. doi: 10.1038/srep31771
99. Zou H-H, Wang L, Zheng X-X, Xu G-S, Shen Y. Endothelial cells secreted endothelin-1 augments diabetic nephropathy via inducing extracellular matrix accumulation of mesangial cells in ETBR-/- mice. Aging. (2019) 11:1804–20. doi: 10.18632/aging.101875
100. Luque Y, Lenoir O, Bonnin P, Hardy L, Chipont A, Placier S, et al. Endothelial Epas1 deficiency is sufficient to promote parietal epithelial cell activation and FSGS in experimental hypertension. J Am Soc Nephrol JASN. (2017) 28:3563–78. doi: 10.1681/ASN.2016090960
101. Chen S-J, Lv L-L, Liu B-C, Tang R-N. Crosstalk between tubular epithelial cells and glomerular endothelial cells in diabetic kidney disease. Cell Prolif. (2020) 53:e12763. doi: 10.1111/cpr.12763
102. Salim HM, Fukuda D, Yagi S, Soeki T, Shimabukuro M, Sata M. Glycemic control with ipragliflozin, a novel selective SGLT2 inhibitor, ameliorated endothelial dysfunction in streptozotocin-induced diabetic mouse. Front Cardiovasc Med. (2016) 3:43. doi: 10.3389/fcvm.2016.00043
103. Scheerer MF, Rist R, Proske O, Meng A, Kostev K. Changes in HbA1c, body weight, and systolic blood pressure in type 2 diabetes patients initiating dapagliflozin therapy: a primary care database study. Diabetes Metab Syndr Obes Targets Ther. (2016) 9:337–45. doi: 10.2147/DMSO.S116243
104. Ott C, Jumar A, Striepe K, Friedrich S, Karg MV, Bramlage P, et al. A randomised study of the impact of the SGLT2 inhibitor dapagliflozin on microvascular and macrovascular circulation. Cardiovasc Diabetol. (2017) 16:26. doi: 10.1186/s12933-017-0510-1
105. Solini A, Giannini L, Seghieri M, Vitolo E, Taddei S, Ghiadoni L, et al. Dapagliflozin acutely improves endothelial dysfunction, reduces aortic stiffness and renal resistive index in type 2 diabetic patients: a pilot study. Cardiovasc Diabetol. (2017) 16:138. doi: 10.1186/s12933-017-0621-8
106. Shigiyama F, Kumashiro N, Miyagi M, Ikehara K, Kanda E, Uchino H, et al. Effectiveness of dapagliflozin on vascular endothelial function and glycemic control in patients with early-stage type 2 diabetes mellitus: DEFENCE study. Cardiovasc Diabetol. (2017) 16:84. doi: 10.1186/s12933-017-0564-0
107. Jax T, Stirban A, Terjung A, Esmaeili H, Berk A, Thiemann S, et al. A randomised, active- and placebo-controlled, three-period crossover trial to investigate short-term effects of the dipeptidyl peptidase-4 inhibitor linagliptin on macro- and microvascular endothelial function in type 2 diabetes. Cardiovasc Diabetol. (2017) 16:13. doi: 10.1186/s12933-016-0493-3
Keywords: podocyte, endothelium/physiopathology, diabetes, glomerulosclerosis, disease module identification, angiocrine factors, glycocalyx (glycocalix)
Citation: Mahtal N, Lenoir O and Tharaux P-L (2021) Glomerular Endothelial Cell Crosstalk With Podocytes in Diabetic Kidney Disease. Front. Med. 8:659013. doi: 10.3389/fmed.2021.659013
Received: 26 January 2021; Accepted: 03 March 2021;
Published: 24 March 2021.
Edited by:
Quan Hong, Chinese PLA General Hospital, ChinaReviewed by:
Ilse Sofia Daehn, Icahn School of Medicine at Mount Sinai, United StatesJia Fu, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2021 Mahtal, Lenoir and Tharaux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pierre-Louis Tharaux, pierre-louis.tharaux@inserm.fr; Olivia Lenoir, olivia.lenoir@inserm.fr