 Wenfeng Liu1
Wenfeng Liu1 Tatiana Rochat2
Tatiana Rochat2 Claire Toffano-Nioche1
Claire Toffano-Nioche1 Thao Nguyen Le Lam1
Thao Nguyen Le Lam1 Philippe Bouloc1*
Philippe Bouloc1* Claire Morvan1*
Claire Morvan1*- 1Institute for Integrative Biology of the Cell (I2BC), CEA, Centre National de la Recherche Scientifique, Université Paris-Sud, Université Paris-Saclay, Gif-sur-Yvette, France
- 2VIM, Institut National de la Recherche Agronomique, Université Paris-Saclay, Institut National de la Recherche Agronomique Centre Jouy-en-Josas, Jouy-en-Josas, France
Bacterial regulatory RNAs have been extensively studied for over a decade, and are progressively being integrated into the complex genetic regulatory network. Transcriptomic arrays, recent deep-sequencing data and bioinformatics suggest that bacterial genomes produce hundreds of regulatory RNAs. However, while some have been authenticated, the existence of the others varies according to strains and growth conditions, and their detection fluctuates with the methodologies used for data acquisition and interpretation. For example, several small RNA (sRNA) candidates are now known to be parts of UTR transcripts. Accurate annotation of regulatory RNAs is a complex task essential for molecular and functional studies. We defined bona fide sRNAs as those that (i) likely act in trans and (ii) are not expressed from the opposite strand of a coding gene. Using published data and our own RNA-seq data, we reviewed hundreds of Staphylococcus aureus putative regulatory RNAs using the DETR'PROK computational pipeline and visual inspection of expression data, addressing the question of which transcriptional signals correspond to sRNAs. We conclude that the model strain HG003, a NCTC8325 derivative commonly used for S. aureus genetic regulation studies, has only about 50 bona fide sRNAs, indicating that these RNAs are less numerous than commonly stated. Among them, about half are associated to the S. aureus sp. core genome and a quarter are possibly expressed in other Staphylococci. We hypothesize on their features and regulation using bioinformatic approaches.
Introduction
Bacterial regulatory RNAs are essential elements of complex genetic networks that tune gene expression according to growth conditions (Wagner and Romby, 2015). Most of them associate by base pairing to target sequences, and affect stability, structure and translation efficiency of target RNAs. Regulatory RNAs are divided into two categories, cis- and trans-acting RNAs. Cis-acting RNAs regulate expression of adjacent genes without reaching their substrate by diffusion (Mellin and Cossart, 2015). In contrast, trans-acting RNAs are expressed from loci not necessarily genetically linked to their target. RNAs and in some cases proteins are targets of trans-acting RNAs. When a trans-acting RNA is expressed from a complementary strand of another gene, it is often called antisense RNA (asRNA) (Georg and Hess, 2011); the predicted target of these asRNAs is the RNA transcribed from the complementary sequence. Trans-acting RNAs that are not asRNAs are often referred to as sRNAs because most of them are of small size. In bacteria, they are usually 50–300 nucleotides long, non-coding and conditionally expressed (i.e., depending upon specific stress and/or growth phase), although several sRNAs do not fit this description. Of interest, RNAIII, which is over 500 nucleotides long and encodes delta haemolysin, is an “exceptional” staphylococcal sRNA paradigm (Novick et al., 1993). This example alone underlines the difficulty of giving a straightforward definition of a bona fide sRNA.
Since 2005, S. aureus non-coding RNAs have been searched by bioinformatics (Pichon and Felden, 2005; Geissmann et al., 2009; Marchais et al., 2009), DNA-arrays (Anderson et al., 2006; Roberts et al., 2006; Mäder et al., 2016), cDNA sequencing (Abu-Qatouseh et al., 2007, 2010), and RNA-seq methods (Beaume et al., 2010; Bohn et al., 2010; Howden et al., 2013; Broach et al., 2016; Carroll et al., 2016). The data are difficult to compare because of the different strains, growth conditions and experimental procedures used. In addition, many regulatory RNAs were renamed and in some cases, previously published work was overlooked. Data from different studies suggest that S. aureus may have hundreds sRNAs, but <10 have thus far been functionally characterized.
Despite recent releases of compilation and cross-comparison of available data in different S. aureus strains, (Felden et al., 2011; Sassi et al., 2015; Carroll et al., 2016; Mäder et al., 2016), it is still difficult to determine bona fide sRNAs from transcriptional background noise, asRNAs, and untranslated region (UTR) derived RNAs. We applied rigorous criteria to define sRNAs, and then used visual curation and bioinformatic approaches on compiled experimental data to assess bona fide sRNAs in S. aureus. S. aureus HG003 (Herbert et al., 2010), an NCTC8325 derivative, was used as the model strain to list bona fide sRNAs. Our main objectives were to identify sRNAs likely to act in trans and to clarify redundancies in the literature due to the use of different nomenclature. We then performed in silico analysis on these sRNAs to determine their phylogenetic conservation and to predict their putative regulators. The reassessment of the number of expressed sRNAs in S. aureus provided by this study may be applicable to other bacteria.
Bona Fide sRNA Definition
Bacterial genomes have complex organization with condensed information and flexible gene expression driven by multiple promoters with some internal to ORFs, operon organization, alternative premature termination, leader-less translation, and translational coupling (e.g., Mäder et al., 2016). The extent to which antisense RNA impacts gene expression in S. aureus is debatable, with reports of both high (Lasa et al., 2011) or more marginal (Mäder et al., 2016) effects. For these reasons, RNA boundaries are difficult to predict and may vary with strains and growth conditions.
We consider that a theoretical bona fide sRNA is (i) a gene not overlapping any other genes from the opposite strand, a definition excluding asRNAs, (ii) not a putative processed UTR and (iii) not a transcript derived from premature termination (i.e., riboswitch). It would therefore have its own promoter and a transcriptional terminator detected by computational predictions (Figure 1A), or interpreted as such because of clear expression up- and down-shifts (Figure 1B). This restrictive definition excludes processed UTRs and short transcripts from premature transcription termination that could also act in trans (Loh et al., 2009); riboswitches and long UTRs are thus excluded as putative sRNAs (Figure 1C). Type I toxin-antitoxin systems comprising a small open reading frame post-transcriptionally controlled by an antisense RNA are also excluded; this concerns several spr genes located within pathogenicity islands (Pichon and Felden, 2005).
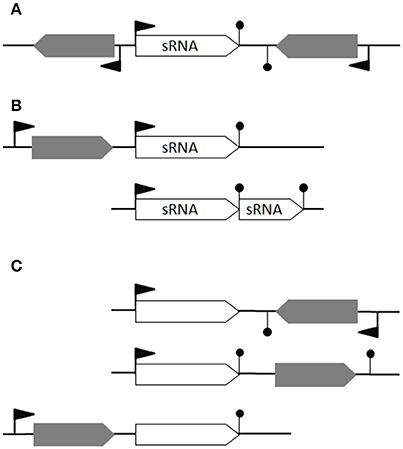
Figure 1. Defining bona fide sRNAs. (A) An ideal bona fide sRNA gene has its own promotor and transcriptional terminator. Its transcription does not overlap any antisense transcription. (B) In the first case, transcription from the second promoter leads to a bona fide sRNA while the transcription from the first promoter would likely generate a transcript with a long 3′UTR (e.g., RsaG). In the second case, a transcriptional termination read-through generates an alternative longer sRNA (e.g., RsaE-S390). (C) Three examples of non-coding RNAs not considered as bona fide sRNAs: a putative asRNA, a cis-regulatory element and a long 3′-UTR, respectively. Flag: promotor; hairpin loop: terminator; gray arrow: open reading frames; empty arrow: non-coding RNA.
The first RNA-seq studies were performed with low read densities. Reads distant from coding sequences and not homologous to known non-coding RNAs (e.g., tRNAs, rRNAs, known UTRs) were first interpreted as putative bona fide sRNAs, and consequently, compiling results from different publications on S. aureus overestimated the number of sRNAs per bacterial strain. Indeed, recent high-density RNA-seq and tiling-array data reveal that many sequences previously considered as sRNAs are UTRs or premature termination products from longer transcripts.
Another consequence of sRNA identification with low read coverage was ambiguous identification of transcription start and termination sites. Even for well-studied sRNAs (e.g., RsaE), transcript boundaries differ according to studies, possibly because of strains, and growth conditions used for experiments. The recent high density transcriptome information is used here to define sRNA boundaries.
Staphylococcus aureus Regulatory RNA Data
Staphylococcus aureus genomes differ by the presence of variable elements (e.g., pathogenicity islands, SCCmec elements, prophages, transposable elements, insertion sequences, and plasmids). A recent study based on 64 S. aureus strains from different ecological niches reveals a core genome of 1,441 genes (not counting sRNAs genes, except tmRNA) and a pangenome of more than 7,400 genes, indicating a wide genetic diversity between strains (Bosi et al., 2016). Transcriptional patterns are influenced by these variable elements, which may affect virulence and antibiotic susceptibility. In addition, strain-specific elements have sRNA genes (Pichon and Felden, 2005) that can directly modulate core genome gene expression (Chabelskaya et al., 2010). sRNA expression has been experimentally studied by global approaches in different S. aureus strains, including NCTC8325 derivatives and methicillin resistant strains isolated in Japan, the USA and Europe (N315, USA300, MRSA252, respectively) (Felden et al., 2011; Sassi et al., 2015; Carroll et al., 2016; Mäder et al., 2016).
NCTC8325 (aka RN1) is a S. aureus strain isolated from a sepsis patient in 1960 widely used for genetic and physiological studies (Novick and Richmond, 1965). This strain is defective for two main regulators encoded by rsbU and tcaR: a positive activator of the general stress response regulator σB and a transcriptional activator of protein A-encoding gene, respectively. HG001, the strain used in the impressive transcriptional landscape study of 44 growth conditions published by Mäder et al. (2016), is a NCTC8325 derivative repaired solely for rsbU, whereas HG003, the strain we focus on in this analysis and that is now used as a model strain for S. aureus regulation studies (Herbert et al., 2010), is repaired for both rsbU and tcaR genes.
According to the compilation of data from several publications, S. aureus N315, NCTC8325, and Newman strains each could have over 500 putative regulatory RNAs (Sassi et al., 2015), the precise figure changing according to strains and sources (Carroll et al., 2016; Mäder et al., 2016). Most sRNAs were rediscovered in each independent analysis often under a different name. In order to compile an accurate list of bona fide sRNAs, we visually analyzed high-density coverage published data plus our own RNA-seq data (deep sequencing of pooled RNA extracts from cultures of HG003 strain grown in 16 different growth conditions; GEO GSE104971) as reported in Methods, and performed in-depth curation according to the rules defined in the previous chapter.
Methods
RNA-seq for sRNA Detection
Experiments were performed with the S. aureus HG003 strain grown in different conditions: (i) eight samples in rich medium (BHI) at OD600nm 0.6, 1.8, 3.3, 4.5, 7.2, 9.8, and 12.8, and stationary phase (24 h), (ii) seven samples under stress conditions (cold shock, heat shock, oxygen limitation, alkaline stress, oxidative stress, disulfide stress, iron-depleted condition and (iii) one sample from colonies on BHI-agar plates (also see GSE10497 in GEO database). Total RNAs were extracted from these 16 growth conditions, pooled together and processed using the MICROBExpress kit (Ambion, AM1905) as recommended by the suppliers, to remove rRNAs. They were then sequenced using an Illumina Genome Analyzer IIx generating single-end 40-nt reads. After a FastQC (v0.10.1) quality control, reads from the stranded and single-end sequencing were mapped onto the reference genome (S. aureus subsp. aureus NCTC8325, CP000253.1 version) using Bowtie 2 with default parameters and an overlapping rate of 69%. DETR'PROK_2.1.2.sh pipeline was run to detect sRNAs (Toffano-Nioche et al., 2012, 2013); parameters are supplied as supplementary materials. The logarithm of the read coverage was computed for multiple or unique mapping reads on each strand; values were used to identify RNAs containing repeated regions (home-made shell scripts) and visualized with the Artemis genome viewer (Rutherford et al., 2000).
Literature and Experimental Data Integration
S. aureus global studies available in literature are summed up in Table S1. sRNA annotations (coordinates and strand) were collected from the following whole transcriptome analyses: 255 “indep” (transcripts with a promoter determined independently of annotated features) or “inter” (between two annotated regions transcribed from independent promoters) (Mäder et al., 2016), 286 sRNAs (Carroll et al., 2016), 352 NCTC8325 automatically annotated sRNAs (Sassi et al., 2015) using HG003 RNA-seq data (this work; GEO GSE104971), and 53 sRNAs (Beaume et al., 2010). These sRNA annotations were pooled together as a GFF file for the present expert analysis. sRNA expression profiles and reported annotations from different strains were compared with HG001 transcription profiles of S. aureus expression data browser (http://genome.jouy.inra.fr/cgi-bin/aeb/index.py). This manual expertise led us to draw up a bona fide sRNA list with their most probable positions as described in Tables 1, 2.
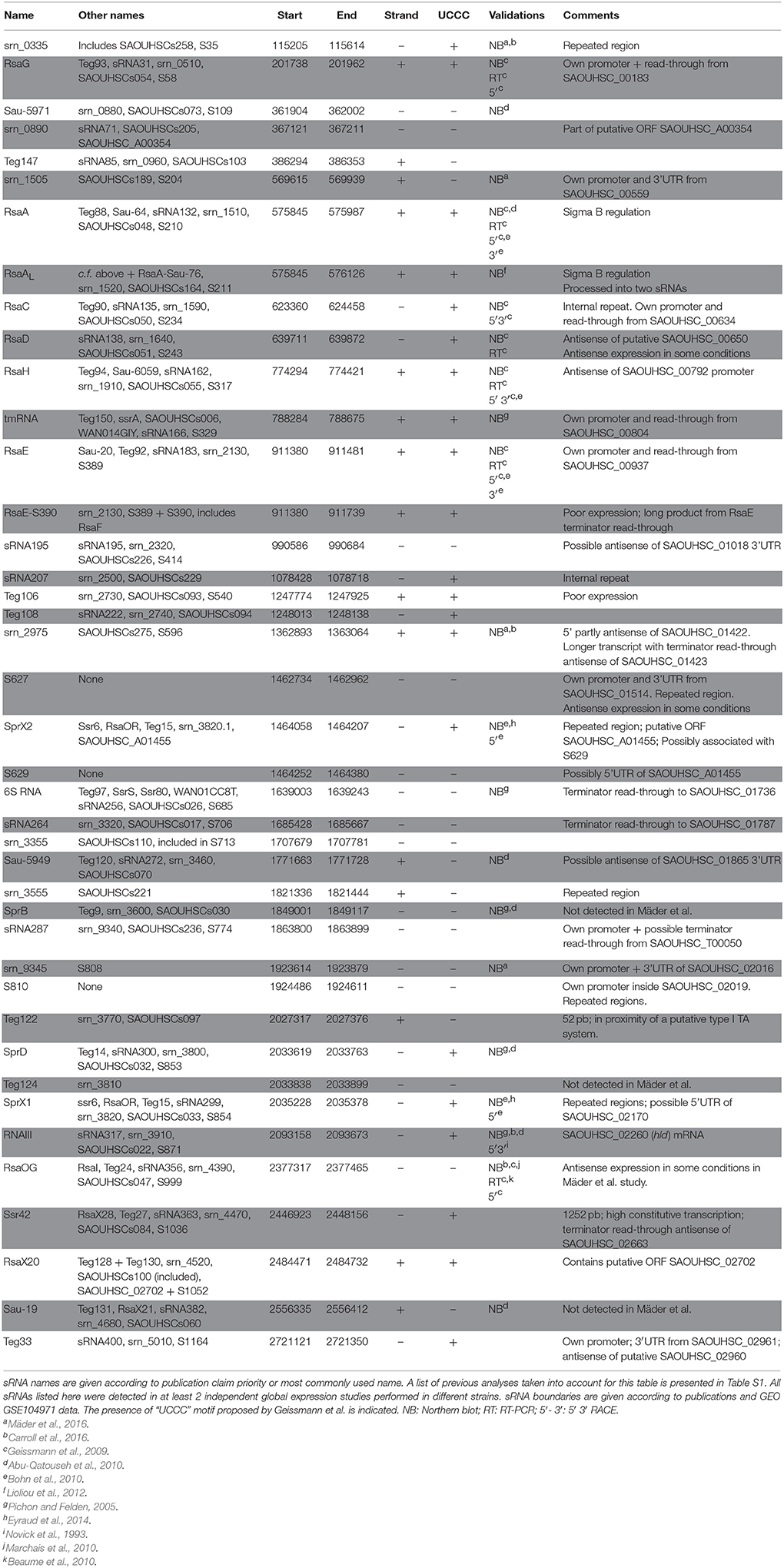
Table 1. Bona fide sRNAs expressed in HG003.
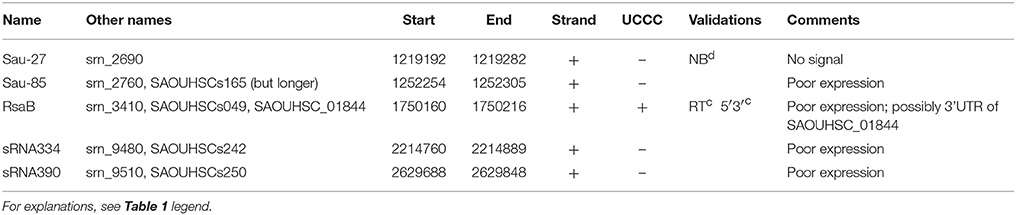
Table 2. Bona fide sRNAs in HG003 with poor expression in the tested conditions of RNA-seq (this study) and tiling arrays (Mäder et al., 2016) datasets.
Transcription Factor Binding Sites in sRNA Promoter Regions
The 49 predictions of N315 Transcription Factors Binding Sites (TFBS) were downloaded (“reference regulons,” version 4.0, Fasta format) from the RegPrecise web site (Novichkov et al., 2013). Equivalences were searched for strain NCTC8325 as follows: When TFBS predictions are supported by only one promoter sequence in N315, we collected the predicted TFBS from other Staphylococcus species (using curl facilities of the RegPrecise web site). For each TFBS, a Position-Specific Scoring Matrix (PSSM) was computed with the MEME tool (Bailey and Elkan, 1994) using a background model built on the NCTC8325 genome sequence (fasta-get-markov in MEME suite, k-mer size of 3, -oops, -dna). The S. aureus NCTC8325 chromosome was scanned with the corresponding PSSM for each of the 49 TFBS with MAST (4.12.0) (Bailey and Gribskov, 1998). Only results on sRNA promoter regions (ranging from −100 nt to +50 nt from the 5′ sRNA end) and with a statistical E-value < 0.01 were conserved in order to report only the most probable predictions. However, this high stringency may discard effective TFBSs.
sRNA Coregulation and Identification of Putative sRNA Targets
Genes coregulated with the sRNAs from Table 3 were identified using the web browser from Mäder et al. showing S. aureus expression data (Mäder et al., 2016). Relevant pages are indicated in Table S3. The RNApredator website (Eggenhofer et al., 2011) was used to predict sRNA-mRNA interactions between the sRNAs from Table 3 and the NCTC8325 genome (accession # NC_007795). In the absence of conservation data, RNAplex program used by RNA predator is among the best predictor (Pain et al., 2015). Results are presented in Data Sheet 2.
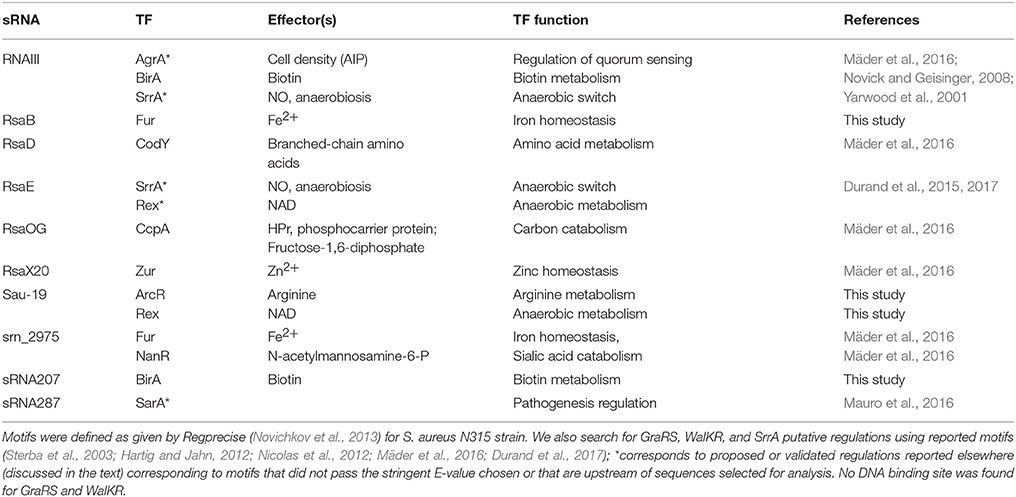
Table 3. List of transcription factor motifs found by MAST analysis (E-value < 0.01) in the putative promoter region (−100 to +50 nts from transcription start sites) of bona fide sRNAs.
sRNA Conservation
sRNA sequence similarities were searched against a nucleotide database (see Table S2 for the list of strains). Complete genomic sequences were downloaded from the NCBI database. Similarity search parameters (blastall 2.2.26) were defined to report a maximum of hits (-e 1000 -W7) with specific scoring criteria (-r2 -G5 -E2) designed for sRNA identification (Ott et al., 2012). For each genome, only the blast hit with the best score was kept and divided by the score obtained in S. aureus NCTC8325. The resulting score ratios are represented by a color scale: the more the sequence of the hit is similar to the sRNA sequence, the darker the pixel is (R script). A 50% similarity ratio threshold was applied to define conserved sRNA genes.
Results and Hypothesis
HG003 Bona Fide sRNAs
Based on a computational analysis of our HG003 RNA-seq data (GEO GSE104971), 88 UTRs, 22 antisense RNAs, 24 CDSs, 11 T-boxes, and riboswitches, and 3 toxin-antitoxin systems were annotated among the 527 putative regulatory RNAs found and indexed in the SRD database (Sassi et al., 2015). According to the definition given above, we considered a restricted list of 352 putative sRNA candidates to which we adjoined those of Carroll et al.'s and Mäder et al.'s sRNA lists. A gene-finding format (GFF) file including these putative sRNAs was generated (Data Sheet 1) and visually analyzed and compared to HG003 RNA-seq profiles using Artemis genome browser (Rutherford et al., 2000). In addition, HG001 tiling array profiles were scrutinized for each putative sRNA using the S. aureus expression data browser (http://genome.jouy.inra.fr/cgi-bin/aeb/index.py). From these inspections, we applied the bona fide criteria to compile a curated list of 41 bona fide sRNAs expressed in at least one biological condition in the HG003 strain (Table 1). We also added 5 bona fide sRNAs described in other strains but poorly expressed in HG003 and HG001 in the tested conditions. For instance, no expression was detected for Sau-27 in our HG003 RNA-seq or in HG001 tiling arrays data. As conditions might exist in which these sRNAs are expressed, we retained them in a separate table (Table 2).
Most of the rejected sRNAs were found to be part of UTRs, or displayed a strong antisense-transcription signal. We discarded from the bona fide sRNA list, most RNAs with antisense expression and those likely part of type I toxin-anti-toxin systems [e.g., Teg13, RsaOI, srn_2335, SprC and S929 (Figure S1)]. However, we retained sRNA genes transcribed in antisense of putative small ORFs with no reported expression [e.g., Teg33, S596 and RsaD (Figure 2)]. Either the peptide does not exist or the antisense decay activity on the mRNA is efficient and completely turns off peptide expression.
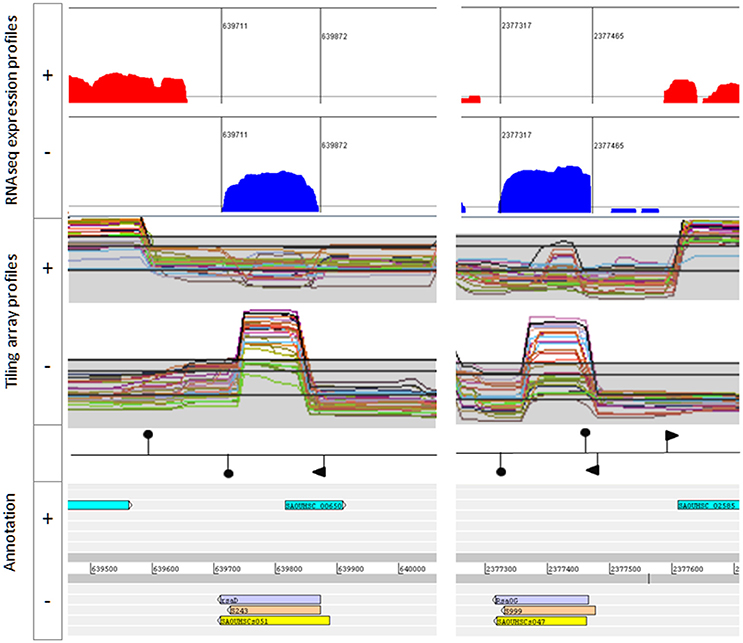
Figure 2. Example of bona fide sRNAs. RsaD (Left) and RsaOG (Right). Upper: Artemis viewer window showing read log-coverages from pooled RNA samples extracted from HG003 grown in 16 growth conditions. Middle panel: screen snapshots of tiling array data from HG001 grown in different conditions (http://genome.jouy.inra.fr/cgi-bin/aeb/index.py, Mäder et al., 2016). Lower: annotations including genomic coordinates and sRNA names from Carroll et al. (yellow), Mäder et al. (light orange), and this study (mauve). Promoters (flags) and transcription terminators (hairpin loops) are placed according to Mäder et al. and/or TranstermHP software terminator predictions (Kingsford et al., 2007).
Many short transcripts may encode small ORFs (sORF); however, for most of them, their expression is not confirmed. To avoid considering sORF genes as bona fide sRNA genes, we discarded those with either high conservation or with a hydrophobic domain. Four sRNAs (srn_0890, SprX2, RsaB and RsaX20) with putative sORFs were retained as their translation was uncertain. We also kept RNAIII expressing the delta hemolysin as its main function and structural part are associated with trans-acting regulation (Novick et al., 1993). Small peptides are also predicted by the microbial gene annotation platform MicroScope (Vallenet et al., 2017) for RsaA, Sau-76, 6S RNA, and sRNA264. Moreover, a ribosome profiling study suggests new ORFs corresponding to sRNA genes (e.g., RsaAL, SprB, 6S RNA, and tmRNA) (Davis et al., 2014); however, a ribosome binding on RNAs is not sufficient to confirm protein expression. In the absence of further biological validation and because sRNAs can have a regulatory activity both through RNA targeting and via the expression of small peptides, we retained all of them but their status may change in the future.
As the number and the depth of deep-sequencing analyses increase, separated adjacent sRNA transcription units can be merged. Here, we consider that Teg128 and Teg130 likely do not exist per se and annotation should be merged to correspond to RsaX20 (Figure S2). In another example, RsaA and Sau-76 share the same promoter, and RNase III-dependent processing generates shorter transcripts (Lioliou et al., 2012); in Table 1, we considered, as previously published, the two transcriptional entities, the short transcript RsaA, and the longer form RsaAL (Figure S3).
The transcriptional study of HG001 in multiple growth conditions indicates a transcript named S390 downstream of the rsaE transcriptional terminator (Mäder et al., 2016). S390 has a putative terminator but no obvious promoter. Its expression is low compared to that of rsaE, possibly suggesting that S390 may result from a transcriptional terminator read-through of RsaE. Weak conservation of S390 beyond S. aureus, as opposed to high conservation of RsaE, questions its functional importance. RsaF is a 105 nucleotide sRNA. rsaF transcription was proposed to initiate from a promoter embedded in the rsaE gene, with expression resulting from transcriptional terminator read-through (Geissmann et al., 2009). As RsaF and its promoter were not detected in the transcriptome databases, we chose to consider just two transcripts, RsaE and the RsaE/S390 fusion.
Also, many previously reported sRNAs are now known to be part of UTRs. One example is Teg49: initially characterized as a bona fide sRNA (Beaume et al., 2010), it is also within the 5′UTR of sarA mRNA, yet Teg49 plays a trans-acting role by modulating sarA expression (Kim et al., 2014; Manna et al., 2017). For two recently proposed sRNAs, S1077 (Figure S4) and S736, which have their own terminators, authors showed that they are both part of longer transcripts that extend downstream of their terminators (Mäder et al., 2016) and are probably cis-acting elements. Alternatively, transcriptional terminator read-through from sRNA genes could generate longer regulatory RNAs.
The 4.5S RNA, which is the RNA component of the signal recognition particle ribonucleoprotein complex and is not a regulatory RNA was removed from the bona fide sRNA list. Two other sRNAs that interact with proteins, 6S RNA and tmRNA, were kept in the sRNA list as they may have regulatory functions (Makhlin et al., 2007; Cavanagh and Wassarman, 2014).
Our RNA-seq transcriptome data are similar to those produced by tiling arrays and presented in the S. aureus expression data browser (http://genome.jouy.inra.fr/cgi-bin/aeb/index.py). Indeed, many bona fide sRNAs listed in Tables 1, 2 are independently detected using these two methodologies. Our transcriptome analysis contains 27 bona fide sRNAs not annotated as such in the Mäder et al. study, although reported elsewhere. Six are located in repeat regions not evaluated by the tiling array method (e.g., S627 and S629 Figure S5). Others have either no expression or an expression level that does not fulfill the cut-off selection imposed by the authors (Figure S6). The slight expression differences observed [namely for, srn_0890, Teg147, Teg108 (Figure S6), Sau-85, RsaB, Sau-5949, SprB, Teg122, Teg124, sRNA334, Sau-19 (Figure S6), and sRNA390] could be due to allelic variation of the tcaR regulator between the two sister strains HG001 and HG003 or to specific expression of sRNAs in at least one of the conditions tested only in our dataset (e.g., heat shock).
sRNA Features
S. aureus is a low guanine-cytosine (33% GC) content member among Firmicutes. Local variation within the genome of this percentage may reflect DNA acquisition by horizontal transfer (Garcia-Vallve et al., 2000). This could be the case for Teg122, tmRNA, Teg147, srn_9345 and 6S RNA, whose GC content is above 40%. However, for tmRNA and 6S RNA, the composition is likely constrained by their interaction with proteins.
Base-pair associations between RNA molecules initiate with unpaired nucleotides; the pairing may then extend beyond these seed motifs. Using the MEME suite (Bailey et al., 2009), we searched for over-represented motifs within the 46 selected sRNAs, which may serve as seed of sRNA/RNA interactions. A conserved C-rich motif (UCCC) in unpaired regions was reported for several S. aureus non-coding RNAs (Geissmann et al., 2009). Impressively, this motif is present in 48% of HG003 bona fide sRNAs, often in multi-copy (from 1 to 5 motifs in HG003 RsaC; Tables 1, 2). sRNAs lacking this motif are often of small size. A stretch of C and/or G is possibly an efficient discriminating element since S. aureus is only 33% GC. As suggested by the authors, it also may indicate that sRNAs with GC-rich unpaired patches may share a mode of action (Geissmann et al., 2009). We have also looked for an alternative motif in sRNAs not featuring UCCC but found none, suggesting that each of these sRNAs would find their target with specific sequences.
HG003 sRNA Conservation
Among 46 HG003 bona fide sRNA genes, 54% are conserved in all tested S. aureus strains (Figure 3) and may be part of the core genome. 24% of the 46 bona fide sRNA genes are conserved among other species of the Staphylococcaceae family. However, most HG003 bona fide sRNAs are species specific. sRNA genes present on pathogenicity islands such as sprX1, sprX2, and sprD are de facto present solely in strains bearing these elements. S629, S810, and srn_9345 genes are poorly conserved among the 43 S. aureus strains included in the analysis, and S627 was found in only three of these strains, M1, CA347 and NCTC8325. Of note, srn_3555, while absent in many aureus strains, is conserved in non-aureus staphylococci such as S. lugdunensis, S. haemolyticus, and S. epidermidis suggesting its acquisition by horizontal transfer. The phylogenetic study suggests rsaC is poorly conserved in S. aureus. However, its conservation is probably underestimated due to the presence of repeat sequences, whose number varies according to strains (Figure S7).
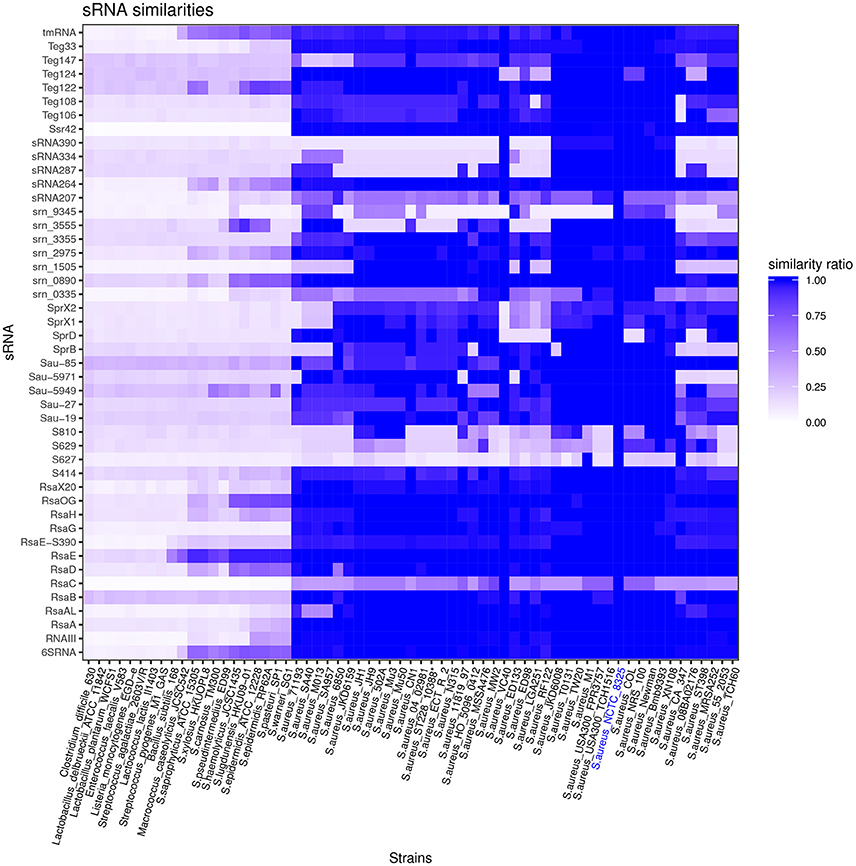
Figure 3. Staphylococcus aureus sRNAs conservation across the Firmicutes phylum. Vertical axe: list of bona fide sRNAs. Horizontal axe: list of S. aureus strains and other Firmicutes (for accession numbers, see Table S2). Similarity ratios between sRNAs of indicated strains and NCTC8325 sRNAs (reference strain) are represented by the indicated color code.
Transfer-messenger RNA (tmRNA) and 6S RNA, which interact with SmpB protein and RNA polymerase, respectively, are widely conserved in bacteria. Apart from these sRNAs, RsaE is the only HG003 bona fide sRNA conserved in distantly-related Firmicutes. It differs from Bacillus subtilis RsaE almost exclusively by its terminator region. This unusual conservation reveals an unexpected selective pressure to preserve RsaE sequence integrity; we hypothesize that in addition to its numerous mRNA targets (Geissmann et al., 2009; Bohn et al., 2010), RsaE may interact with a protein constraining the RNA sequence to ensure it regulatory activity.
HG003 sRNA Transcriptional Regulation
Transcription in S. aureus depends on four sigma (σ) factors: σA, the primary σ factor responsible for the transcription of most genes, σB, involved in the general stress response, σH, implicated in the competence state but cryptic in NCTC8325 (Morikawa et al., 2012), and σS, the extracytoplasmic function sigma factor (Burda et al., 2014).
In S. aureus, the σB regulon comprises about 249 coding genes expressed from 145 promoters (Bischoff et al., 2004; Mäder et al., 2016). The σB consensus recognition site was used to find small σB regulated genes called sbr (for σB-dependent small RNA) (Nielsen et al., 2011). Three were found in several strains including SH1000, a NCTC8325 σB+ derivative. However, sbrA and sbrB encode putative small basic peptides and are not regulatory RNA genes. The 3' end of sbrC overlaps with the 3′ end of mntC, that codes a metal binding lipoprotein, and their corresponding RNAs interact in vitro, indicating that SbrC should be categorized as an asRNA. σB-mediated regulation has been proposed for RsaA, RsaF and RsaD, as their expression was enhanced in σB proficient strains, and a characteristic σB promoter was found upstream of rsaA (Geissmann et al., 2009). However, so far, σB regulation was confirmed only for rsaA and its derivative rsaAL (including sau-76) (Mäder et al., 2016). No σB promoter was found upstream of rsaD despite activation of the σB regulon by several growth conditions (Mäder et al., 2016). Hence, only one out of 46 bona fide sRNAs, rsaA, appears to be transcribed by σB. Remarkably, while often associated with adaptation and stress responses, almost all sRNAs have σA promoters. Since they are usually modulated by specific growth conditions, their expression likely relies on additional regulatory factors.
Transcription factors (TFs) bind specific DNA sites that can be detected by biocomputing tools when consensus sequences are already described. We performed such analyses for the bona fide sRNAs using MAST (Bailey et al., 2009) and predicted TF regulation for 8 sRNA genes (Table 3). The putative regulatory targets, and those previously reported, are discussed below.
RNAIII activates virulence genes either directly or indirectly at high S. aureus cell density. It is positively regulated by the quorum sensing regulator AgrA (Novick and Geisinger, 2008). AgrA also activates its own operon and psm (phenol-soluble modulins) genes that encode toxins (Queck et al., 2008), sometimes inadvertently annotated as sRNA genes. A putative BirA binding motif is detected upstream of the RNAIII gene (Mäder et al., 2016). BirA is a biotin-dependent repressor that downregulates genes implicated in biotin synthesis and transport (Henke and Cronan, 2016). In addition, RNAIII is reportedly repressed by SrrAB, a two-component system involved in aerobic to anaerobic adaptation and energy metabolism similar to B. subtilis ResDE (Yarwood et al., 2001). SrrAB-dependent RNAIII repression may result from a direct interaction of SrrA with the agr P3 promoter (Pragman et al., 2004).
SrrAB has an opposite effect on RsaE expression compared to that on RNAIII. The absence of SrrAB results in a drastic reduction of RsaE and an SrrA binding motif is detected 125 nucleotides upstream rsaE transcriptional start site (Durand et al., 2015). In B. subtilis, expression of roxS, the rsaE ortholog, is submitted to a double regulation by the activator ResDE, the SrrAB functional homolog, and the redox sensing repressor of anaerobic metabolism Rex (Pagels et al., 2010; Durand et al., 2017). As an identical Rex binding motif is also present within the RsaE promoter region, this double regulation is likely conserved for rsaE in S. aureus. RNAIII and RsaE would both exert a role in response to impaired respiration and indeed, in B. subtilis the absence of RoxS, results in the modulation of genes related to redox homeostasis (Durand et al., 2015).
In anaerobiosis, ArcR, a Crp/Fnr family transcriptional activator, stimulates arginine utilization as an energy source (Makhlin et al., 2007). We found that sau-19, an sRNA gene poorly expressed in conditions thus far tested, has ArcR and Rex binding motifs; these motifs resemble each other and concern the same sequence. Full activation of Sau-19 may need growth conditions in which Rex is inactive and ArcR is active, as observed for the arginine deiminase pathway (Makhlin et al., 2007).
S. aureus adapts to nutrient shifts with dedicated TFs. CcpA is a master regulator of carbon utilization in Gram-positive bacteria (Halsey et al., 2017). It binds to catabolite-response elements (cre) DNA sequences, and may act as an activator or a repressor. A cre box is detected within the promoter region of rsaOG (alias rsaI). RsaOG regulation by CcpA is supported by its coregulation with other CcpA regulated genes such as lip, putA, fadXEDB, and rocA (Mäder et al., 2016) (Table S3). This sRNA with a predicted pseudoknot (Marchais et al., 2010) is strongly modulated by growth conditions, and is increased in oxidative stress, during stationary phase and in human serum (Howden et al., 2013; Carroll et al., 2016). Fructose-1,6-bisphosphate is an allosteric effector of CcpA function (Schumacher et al., 2007). Interestingly, in addition to sugar transporters (i.e., SAOUHSC_02520, SAOUHSC_02815), the fructose-1,6-bisphosphate aldolase (SAOUHSC_02926) is a putative RsaOG target (Data Sheet 2), which in turn may contribute to CcpA regulation.
In S. aureus, CodY is a pleiotropic regulator affecting expression of numerous metabolic and virulence genes in response to branched amino acid and GTP availability (Geiger and Wolz, 2014; Waters et al., 2016). The presence of a CodY box in the promoter region of rsaD suggests that this sRNA belongs to its regulon. This proposal is strongly supported by the observation that rsaD is expressed in the same condition as CodY-regulated genes such as SAOUHSC_00962, mtnE-ddh, and oppBCDFA (Mäder et al., 2016) (Table S3).
Iron starvation is known to limit bacterial development during infection, but at the same time, an excess of iron generates deleterious reactive oxygen radicals. Consequently, intracellular iron homeostasis is tightly controlled and in many bacteria, the iron-sensing regulator Fur is involved. RhyB is an important Fur-regulated sRNA conserved in many Gram-negative bacteria that represses numerous genes and contributes to virulence (Oglesby-Sherrouse and Murphy, 2013). Iron-responsive sRNAs with a similar function are also present in Gram-positive bacteria (e.g., Gaballa et al., 2008). From the HG003 bona fide sRNA list, Fur boxes were detected in front of rsaB and srn_2975 (S596), suggesting their implication in iron homeostasis. Regulation of srn_2975 by iron is supported by (i) its co-expression with isd and sbn genes related to heme/hemin and iron uptake and utilization, respectively and (ii) predicted targets that are related to iron metabolism (Mäder et al., 2016). Srn_2975 would be the S. aureus functional ortholog of RhyB. Two NanR binding motifs are also found upstream of srn_2975. NanR is a repressor controlling sialic acid (N-acetylneuraminic acid) catabolism enzymes that may play an important role during growth in the host (Olson et al., 2013). Like iron, the metal ion zinc is essential. Zur, a zinc-sensing Fur-like protein (Lindsay and Foster, 2001) regulates zinc intracellular concentration. One sRNA gene, rsaX20, is preceded by a Zur binding motif, and interestingly RsaX20 is co-expressed with genes from the Zur regulon (Mäder et al., 2016) (Table S3). Consequently, RsaX20 is possibly associated with metal homeostasis.
sRNA regulation can be directly linked to virulence and pathogenicity factors. Besides the Agr system, as an example, SarA is a transcriptional factor belonging to the core genome, which is implicated in infectivity and biofilm formation. SarA represses sRNA287 and SprC, two sRNAs located on the same pathogenicity island (Mauro et al., 2016).
The sRNA regulators discussed here are associated with quorum sensing, aerobic to anaerobic transition, carbon source availability, metal metabolism or infectivity. All these processes crucial for virulence and survival within the host indicate that functional studies of S. aureus sRNAs are essential for understanding the global regulatory network governing bacterial pathogenicity.
HG003 sRNA Transcriptional Termination
Bacterial transcription terminates either at secondary structures formed by nascent RNAs (intrinsic termination) (Ray-Soni et al., 2016) or via the activity of a termination factor such as Rho (Grylak-Mielnicka et al., 2016). Notably, while essential in several bacteria including Escherichia coli, Rho is dispensable in S. aureus (Washburn et al., 2001). Transcriptome data of HG001 rho in three different conditions is available (Mäder et al., 2016). Most intrinsic terminators are detected by bio-computing analysis. The presence of terminators within intergenic regions was initially used as an indication of the existence of sRNA genes (Wassarman et al., 2001) with the general belief that sRNA genes have Rho-independent terminators. Using TransTermHP with default parameters (http://transterm.cbcb.umd.edu) (Kingsford et al., 2007), intrinsic terminators were detected for 38 sRNAs among the 46 retained for HG003. By analyzing HG001 rho transcriptomic data for the nine sRNAs with no detected intrinsic terminator (Mäder et al., 2016), we conclude that they had no apparent Rho-dependent termination.
Several sRNAs have their own promoter but are also expressed because of a terminator read-through from upstream gene resulting in a longer RNA. In several cases, the expression of the sRNA gene and its upstream gene (or operon) is remarkably co-regulated (e.g., rsaG, srn_1505) suggesting that both genes are associated with the same function. In this case, the sRNA promoter would be present to boost sRNA expression.
Conclusion
Most reported small transcripts correspond to UTRs and asRNAs. Our curated analysis led to a number of bona fide sRNAs in S. aureus that is smaller than what would be expected from the compilation of all sRNA studies. Even among this restricted list, the regulation, targets and functions of these sRNAs are still mostly unknown. Studying bona fide sRNA genes present the advantage that their deletion, in principal, has no polar effect on adjacent genes, thus facilitating genetic approaches to search for phenotypes (Le Lam et al., 2017). Putative proposed sRNA regulators (Table 3) are starting points to elucidate their function. Indeed, sRNAs often act as effectors of the transcription factors controlling their expression. They are the polishing regulators that would fine tune genetic regulation and refine bacterial adaptability. Described sRNAs are mostly negative regulators and often act as invertors of regulatory responses: induction of an sRNA by a given activator may lead to gene down-regulation. The same reasoning applies conversely for a repressed sRNA. For the above-discussed regulators, exploring genes repressed by inactivation of a repressor or induced by the absence of an activator is a good hint to discover sRNA-targets.
The number of bona fide sRNAs is lower than initially proposed, although we expect that new candidates will be added to this group. Publications based on high-throughput sequencing data indicate dense transcription with numerous so far uncharacterized transcripts that are putative regulatory elements. This pervasive transcription is hidden and probably not selected in a natural environment; and mutations such as rnc and rho are required to unmask it (Lasa et al., 2011; Lioliou et al., 2012; Mäder et al., 2016). Active S. aureus RNA processing generates numerous alternative RNA species (Lioliou et al., 2013; Bonnin and Bouloc, 2015) and many transcripts have long UTRs with a regulatory role demonstrated only in a few cases (e.g., de Los Mozos et al., 2013; Bouloc and Repoila, 2016). It is likely that besides bona fide sRNAs, S. aureus has a plethora of RNA-based regulations nesting within these non-translated RNAs.
Author Contributions
TR, PB, and CM: Conception, design of the work. WL, TR, and TL: Data acquisition. CT-N, PB, and CM: Data analysis. TR, PB, and CM: Drafting of the work and revision.
Funding
This work was funded by the Agence Nationale pour la Recherche (ANR) (grant # ANR-15-CE12-0003-01 “sRNA-Fit”) and by the Fondation pour la Recherche Médicale (FRM) (grant # DBF20160635724 “Bactéries et champignons face aux antibiotiques et antifongiques”). WL and CM are the recipients of Chinese scholarship council, and fellowships from the ANR, respectively.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has benefited from the facilities and expertise of the high-throughput sequencing platform of I2BC (http://www.i2bc.paris-saclay.fr/). We thank the MIGALE platform, Pierre Nicolas (Jouy-en-Josas, France) and Ulrike Mäder (Greifswald University) for providing useful analyze tools and permission to print website captures; Alban Ott for help with Figure 3; Sandy Gruss for critical reading of the manuscript, Annick Jacq, and Mared Sous for helpful discussions and warm support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00228/full#supplementary-material
References
Abu-Qatouseh, L. F., Chinni, S. V., Seggewiss, J., Proctor, R. A., Brosius, J., Rozhdestvensky, T. S., et al. (2010). Identification of differentially expressed small non-protein-coding RNAs in Staphylococcus aureus displaying both the normal and the small-colony variant phenotype. J. Mol. Med. 88, 565–575. doi: 10.1007/s00109-010-0597-2
Abu-Qatouseh, L. F., Seggewiss, J., Chinni, S. V., Brosius, J., Rozhdestvensky, T. S., Peters, G., et al. (2007). RNomics: experimental identification of novel small nonprotein-coding RNAs in Staphylococcus aureus. Int. J. Med. Microbiol. 297, 106–106. doi: 10.1093/nar/gkj469
Anderson, K. L., Roberts, C., Disz, T., Vonstein, V., Hwang, K., Overbeek, R., et al. (2006). Characterization of the Staphylococcus aureus heat shock, cold shock, stringent, and SOS responses and their effects on log-phase mRNA turnover. J. Bacteriol. 188, 6739–6756. doi: 10.1128/JB.00609-06
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Bailey, T. L., and Elkan, C. (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36.
Bailey, T. L., and Gribskov, M. (1998). Combining evidence using p-values: application to sequence homology searches. Bioinformatics 14, 48–54. doi: 10.1093/bioinformatics/14.1.48
Beaume, M., Hernandez, D., Farinelli, L., Deluen, C., Linder, P., Gaspin, C., et al. (2010). Cartography of methicillin-resistant, S. aureus transcripts: detection, orientation and temporal expression during growth phase and stress conditions. PLoS ONE 5:e10725. doi: 10.1371/journal.pone.0010725
Bischoff, M., Dunman, P., Kormanec, J., Macapagal, D., Murphy, E., Mounts, W., et al. (2004). Microarray-based analysis of the Staphylococcus aureus sigmaB regulon. J. Bacteriol. 186, 4085–4099. doi: 10.1128/JB.186.13.4085-4099.2004
Bohn, C., Rigoulay, C., Chabelskaya, S., Sharma, C. M., Marchais, A., Skorski, P., et al. (2010). Experimental discovery of small RNAs in Staphylococcus aureus reveals a riboregulator of central metabolism. Nucleic Acids Res. 38, 6620–6636. doi: 10.1093/nar/gkq462
Bonnin, R. A., and Bouloc, P. (2015). RNA degradation in Staphylococcus aureus: diversity of ribonucleases and their impact. Int. J. Genomics 2015:395753. doi: 10.1155/2015/395753
Bosi, E., Monk, J. M., Aziz, R. K., Fondi, M., Nizet, V., and Palsson, B. O. (2016). Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc. Natl. Acad. Sci. U.S.A. 113, E3801–E3809. doi: 10.1073/pnas.1523199113
Bouloc, P., and Repoila, F. (2016). Fresh layers of RNA-mediated regulation in gram-positive bacteria. Curr. Opin. Microbiol. 30, 30–35. doi: 10.1016/j.mib.2015.12.008
Broach, W. H., Weiss, A., and Shaw, L. N. (2016). Transcriptomic analysis of staphylococcal sRNAs: insights into species-specific adaption and the evolution of pathogenesis. Microb Genom 2:e000065. doi: 10.1099/mgen.0.000065
Burda, W. N., Miller, H. K., Krute, C. N., Leighton, S. L., Carroll, R. K., and Shaw, L. N. (2014). Investigating the genetic regulation of the ECF sigma factor sigmaS in Staphylococcus aureus. BMC Microbiol. 14:280. doi: 10.1186/s12866-014-0280-9
Carroll, R. K., Weiss, A., Broach, W. H., Wiemels, R. E., Mogen, A. B., Rice, K. C., et al. (2016). Genome-wide annotation, identification, and global transcriptomic analysis of regulatory or small RNA gene expression in Staphylococcus aureus. MBio 7:e01990-15. doi: 10.1128/mBio.01990-15
Cavanagh, A. T., and Wassarman, K. M. (2014). 6S RNA, a global regulator of transcription in Escherichia coli, Bacillus subtilis, and beyond. Annu. Rev. Microbiol. 68, 45–60. doi: 10.1146/annurev-micro-092611-150135
Chabelskaya, S., Gaillot, O., and Felden, B. (2010). A Staphylococcus aureus small RNA is required for bacterial virulence and regulates the expression of an immune-evasion molecule. PLoS Pathog. 6:e1000927. doi: 10.1371/journal.ppat.1000927
Davis, A. R., Gohara, D. W., and Yap, M. N. (2014). Sequence selectivity of macrolide-induced translational attenuation. Proc. Natl. Acad. Sci. U.S.A. 111, 15379–15384. doi: 10.1073/pnas.1410356111
de Los Mozos, I. R., Vergara-Irigaray, M., Segura, V., Villanueva, M., Bitarte, N., Saramago, M., et al. (2013). Base pairing interaction between 5'- and 3'-UTRs controls icaR mRNA translation in Staphylococcus aureus. PLoS Genet 9:e1004001. doi: 10.1371/journal.pgen.1004001
Durand, S., Braun, F., Helfer, A. C., Romby, P., and Condon, C. (2017). sRNA-mediated activation of gene expression by inhibition of 5'-3' exonucleolytic mRNA degradation. Elife 6:e23602. doi: 10.7554/eLife.23602
Durand, S., Braun, F., Lioliou, E., Romilly, C., Helfer, A. C., Kuhn, L., et al. (2015). A nitric oxide regulated small RNA controls expression of genes involved in redox homeostasis in Bacillus subtilis. PLoS Genet. 11:e1004957. doi: 10.1371/journal.pgen.1004957
Eggenhofer, F., Tafer, H., Stadler, P. F., and Hofacker, I. L. (2011). RNApredator: fast accessibility-based prediction of sRNA targets. Nucleic Acids Res. 39, W149–W154. doi: 10.1093/nar/gkr467
Eyraud, A., Tattevin, P., Chabelskaya, S., and Felden, B. (2014). A small RNA controls a protein regulator involved in antibiotic resistance in Staphylococcus aureus. Nucleic Acids Res. 42, 4892–4905. doi: 10.1093/nar/gku149
Felden, B., Vandenesch, F., Bouloc, P., and Romby, P. (2011). The Staphylococcus aureus RNome and its commitment to virulence. PLoS Pathog. 7:e1002006. doi: 10.1371/journal.ppat.1002006
Gaballa, A., Antelmann, H., Aguilar, C., Khakh, S. K., Song, K. B., Smaldone, G. T., et al. (2008). The Bacillus subtilis iron-sparing response is mediated by a fur-regulated small RNA and three small, basic proteins. Proc. Natl. Acad. Sci. U.S.A. 105, 11927–11932. doi: 10.1073/pnas.0711752105
Garcia-Vallve, S., Romeu, A., and Palau, J. (2000). Horizontal gene transfer in bacterial and archaeal complete genomes. Genome Res. 10, 1719–1725. doi: 10.1101/gr.130000
Geiger, T., and Wolz, C. (2014). Intersection of the stringent response and the CodY regulon in low GC gram-positive bacteria. Int. J. Med. Microbiol. 304, 150–155. doi: 10.1016/j.ijmm.2013.11.013
Geissmann, T., Chevalier, C., Cros, M. J., Boisset, S., Fechter, P., Noirot, C., et al. (2009). A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res. 37, 7239–7257. doi: 10.1093/nar/gkp668
Georg, J., and Hess, W. R. (2011). cis-antisense RNA another level of gene regulation in bacteria. Microbiol. Mol. Biol. Rev. 75, 286–300. doi: 10.1128/MMBR.00032-10
Grylak-Mielnicka, A., Bidnenko, V., Bardowski, J., and Bidnenko, E. (2016). Transcription termination factor Rho: a hub linking diverse physiological processes in bacteria. Microbiology 162, 433–447. doi: 10.1099/mic.0.000244
Halsey, C. R., Lei, S., Wax, J. K., Lehman, M. K., Nuxoll, A. S., Steinke, L., et al. (2017). Amino acid catabolism in Staphylococcus aureus and the function of carbon catabolite repression. MBio 8:e01434-16. doi: 10.1128/mBio.01434-16
Hartig, E., and Jahn, D. (2012). Regulation of the anaerobic metabolism in Bacillus subtilis. Adv. Microb. Physiol. 61, 195–216. doi: 10.1016/B978-0-12-394423-8.00005-6
Henke, S. K., and Cronan, J. E. (2016). The Staphylococcus aureus group II biotin protein ligase BirA is an effective regulator of biotin operon transcription and requires the DNA binding domain for full enzymatic activity. Mol. Microbiol. 102, 417–429. doi: 10.1111/mmi.13470
Herbert, S., Ziebandt, A. K., Ohlsen, K., Schafer, T., Hecker, M., Albrecht, D., et al. (2010). Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect. Immun. 78, 2877–2889. doi: 10.1128/IAI.00088-10
Howden, B. P., Beaume, M., Harrison, P. F., Hernandez, D., Schrenzel, J., Seemann, T., et al. (2013). Analysis of the small RNA transcriptional response in multidrug-resistant Staphylococcus aureus after antimicrobial exposure. Antimicrob. Agents Chemother. 57, 3864–3874. doi: 10.1128/AAC.00263-13
Kim, S., Reyes, D., Beaume, M., Francois, P., and Cheung, A. (2014). Contribution of teg49 small RNA in the 5' upstream transcriptional region of sarA to virulence in Staphylococcus aureus. Infect. Immun. 82, 4369–4379. doi: 10.1128/IAI.02002-14
Kingsford, C. L., Ayanbule, K., and Salzberg, S. L. (2007). Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 8:R22. doi: 10.1186/gb-2007-8-2-r22
Lasa, I., Toledo-Arana, A., Dobin, A., Villanueva, M., De Los Mozos, I. R., Vergara-Irigaray, M., et al. (2011). Genome-wide antisense transcription drives mRNA processing in bacteria. Proc. Natl. Acad. Sci. U.S.A. 108, 20172–20177. doi: 10.1073/pnas.1113521108
Le Lam, T. N., Morvan, C., Liu, W., Bohn, C., Jaszczyszyn, Y., and Bouloc, P. (2017). Finding sRNA-associated phenotypes by competition assays: an example with Staphylococcus aureus. Methods 117, 21–27. doi: 10.1016/j.ymeth.2016.11.018
Lindsay, J. A., and Foster, S. J. (2001). zur: a Zn(2+)-responsive regulatory element of Staphylococcus aureus. Microbiology 147, 1259–1266. doi: 10.1099/00221287-147-5-1259
Lioliou, E., Sharma, C. M., Altuvia, Y., Caldelari, I., Romilly, C., Helfer, A. C., et al. (2013). In vivo mapping of RNA-RNA interactions in Staphylococcus aureus using the endoribonuclease III. Methods. 63, 135–143. doi: 10.1016/j.ymeth.2013.06.033
Lioliou, E., Sharma, C. M., Caldelari, I., Helfer, A. C., Fechter, P., Vandenesch, F., et al. (2012). Global regulatory functions of the Staphylococcus aureus endoribonuclease III in gene expression. PLoS Genet. 8:e1002782. doi: 10.1371/journal.pgen.1002782
Loh, E., Dussurget, O., Gripenland, J., Vaitkevicius, K., Tiensuu, T., Mandin, P., et al. (2009). A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell 139, 770–779. doi: 10.1016/j.cell.2009.08.046
Mäder, U., Nicolas, P., Depke, M., Pane-Farre, J., Debarbouille, M., Van Der Kooi-Pol, M. M., et al. (2016). Staphylococcus aureus transcriptome architecture: from laboratory to infection-mimicking conditions. PLoS Genet. 12:e1005962. doi: 10.1371/journal.pgen.1005962
Makhlin, J., Kofman, T., Borovok, I., Kohler, C., Engelmann, S., Cohen, G., et al. (2007). Staphylococcus aureus ArcR controls expression of the arginine deiminase operon. J. Bacteriol. 189, 5976–5986. doi: 10.1128/JB.00592-07
Manna, A. C., Kim, S., Cengher, L., Corvaglia, A., Leo, S., Francois, P., et al. (2017). Small RNA Teg49 is derived from a sarA transcript and regulates virulence genes independent of SarA in Staphylococcus aureus. Infect. Immun. 86:e00635-17. doi: 10.1128/IAI.00635-17
Marchais, A., Bohn, C., Bouloc, P., and Gautheret, D. (2010). RsaOG, a new staphylococcal family of highly transcribed non-coding RNA. RNA Biol. 7, 116–119. doi: 10.4161/rna.7.2.10925
Marchais, A., Naville, M., Bohn, C., Bouloc, P., and Gautheret, D. (2009). Single-pass classification of all noncoding sequences in a bacterial genome using phylogenetic profiles. Genome Res. 19, 1084–1092. doi: 10.1101/gr.089714.108
Mauro, T., Rouillon, A., and Felden, B. (2016). Insights into the regulation of small RNA expression: SarA represses the expression of two sRNAs in Staphylococcus aureus. Nucleic Acids Res. 44, 10186–10200. doi: 10.1093/nar/gkw777
Mellin, J. R., and Cossart, P. (2015). Unexpected versatility in bacterial riboswitches. Trends Genet. 31, 150–156. doi: 10.1016/j.tig.2015.01.005
Morikawa, K., Takemura, A. J., Inose, Y., Tsai, M., Nguyen Thi Le, T., Ohta, T., et al. (2012). Expression of a cryptic secondary sigma factor gene unveils natural competence for DNA transformation in Staphylococcus aureus. PLoS Pathog. 8:e1003003. doi: 10.1371/journal.ppat.1003003
Nicolas, P., Mader, U., Dervyn, E., Rochat, T., Leduc, A., Pigeonneau, N., et al. (2012). Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science 335, 1103–1106. doi: 10.1126/science.1206848
Nielsen, J. S., Christiansen, M. H., Bonde, M., Gottschalk, S., Frees, D., Thomsen, L. E., et al. (2011). Searching for small sigmaB-regulated genes in Staphylococcus aureus. Arch. Microbiol. 193, 23–34. doi: 10.1007/s00203-010-0641-1
Novichkov, P. S., Kazakov, A. E., Ravcheev, D. A., Leyn, S. A., Kovaleva, G. Y., Sutormin, R. A., et al. (2013). RegPrecise 3.0–a resource for genome-scale exploration of transcriptional regulation in bacteria. BMC Genomics 14:745. doi: 10.1186/1471-2164-14-745
Novick, R. P., and Geisinger, E. (2008). Quorum sensing in staphylococci. Annu. Rev. Genet. 42, 541–564. doi: 10.1146/annurev.genet.42.110807.091640
Novick, R. P., and Richmond, M. H. (1965). Nature and interactions of the genetic elements governing penicillinase synthesis in Staphylococcus aureus. J. Bacteriol. 90, 467–480.
Novick, R. P., Ross, H. F., Projan, S. J., Kornblum, J., Kreiswirth, B., and Moghazeh, S. (1993). Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 12, 3967–3975.
Oglesby-Sherrouse, A. G., and Murphy, E. R. (2013). Iron-responsive bacterial small RNAs: variations on a theme. Metallomics 5, 276–286. doi: 10.1039/c3mt20224k
Olson, M. E., King, J. M., Yahr, T. L., and Horswill, A. R. (2013). Sialic acid catabolism in Staphylococcus aureus. J. Bacteriol. 195, 1779–1788. doi: 10.1128/JB.02294-12
Ott, A., Idali, A., Marchais, A., and Gautheret, D. (2012). NAPP: the nucleic acid phylogenetic profile database. Nucleic Acids Res. 40, D205–D209. doi: 10.1093/nar/gkr807
Pagels, M., Fuchs, S., Pane-Farre, J., Kohler, C., Menschner, L., Hecker, M., et al. (2010). Redox sensing by a Rex-family repressor is involved in the regulation of anaerobic gene expression in Staphylococcus aureus. Mol. Microbiol. 76, 1142–1161. doi: 10.1111/j.1365-2958.2010.07105.x
Pain, A., Ott, A., Amine, H., Rochat, T., Bouloc, P., and Gautheret, D. (2015). An assessment of bacterial small RNA target prediction programs. RNA Biol. 12, 509–513. doi: 10.1080/15476286.2015.1020269
Pichon, C., and Felden, B. (2005). Small RNA genes expressed from Staphylococcus aureus genomic and pathogenicity islands with specific expression among pathogenic strains. Proc. Natl. Acad. Sci. U.S.A. 102, 14249–14254. doi: 10.1073/pnas.0503838102
Pragman, A. A., Yarwood, J. M., Tripp, T. J., and Schlievert, P. M. (2004). Characterization of virulence factor regulation by SrrAB, a two-component system in Staphylococcus aureus. J. Bacteriol. 186, 2430–2438. doi: 10.1128/JB.186.8.2430-2438.2004
Queck, S. Y., Jameson-Lee, M., Villaruz, A. E., Bach, T. H., Khan, B. A., Sturdevant, D. E., et al. (2008). RNAIII-independent target gene control by the agr quorum-sensing system: insight into the evolution of virulence regulation in Staphylococcus aureus. Mol. Cell 32, 150–158. doi: 10.1016/j.molcel.2008.08.005
Ray-Soni, A., Bellecourt, M. J., and Landick, R. (2016). Mechanisms of bacterial transcription termination: all good things must end. Annu. Rev. Biochem. 85, 319–347. doi: 10.1146/annurev-biochem-060815-014844
Roberts, C., Anderson, K. L., Murphy, E., Projan, S. J., Mounts, W., Hurlburt, B., et al. (2006). Characterizing the effect of the Staphylococcus aureus virulence factor regulator, SarA, on log-phase mRNA half-lives. J. Bacteriol. 188, 2593–2603. doi: 10.1128/JB.188.7.2593-2603.2006
Rutherford, K., Parkhill, J., Crook, J., Horsnell, T., Rice, P., Rajandream, M. A., et al. (2000). Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945. doi: 10.1093/bioinformatics/16.10.944
Sassi, M., Augagneur, Y., Mauro, T., Ivain, L., Chabelskaya, S., Hallier, M., et al. (2015). SRD: a Staphylococcus regulatory RNA database. RNA 21, 1005–1017. doi: 10.1261/rna.049346.114
Schumacher, M. A., Seidel, G., Hillen, W., and Brennan, R. G. (2007). Structural mechanism for the fine-tuning of CcpA function by the small molecule effectors glucose 6-phosphate and fructose 1,6-bisphosphate. J. Mol. Biol. 368, 1042–1050. doi: 10.1016/j.jmb.2007.02.054
Sterba, K. M., Mackintosh, S. G., Blevins, J. S., Hurlburt, B. K., and Smeltzer, M. S. (2003). Characterization of Staphylococcus aureus SarA binding sites. J. Bacteriol. 185, 4410–4417. doi: 10.1128/JB.185.15.4410-4417.2003
Toffano-Nioche, C., Luo, Y., Kuchly, C., Wallon, C., Steinbach, D., Zytnicki, M., et al. (2013). Detection of non-coding RNA in bacteria and archaea using the DETR'PROK galaxy pipeline. Methods. 63, 60–65. doi: 10.1016/j.ymeth.2013.06.003
Toffano-Nioche, C., Nguyen, A. N., Kuchly, C., Ott, A., Gautheret, D., Bouloc, P., et al. (2012). Transcriptomic profiling of the oyster pathogen Vibrio splendidus opens a window on the evolutionary dynamics of the small RNA repertoire in the Vibrio genus. RNA 18, 2201–2219. doi: 10.1261/rna.033324.112
Vallenet, D., Calteau, A., Cruveiller, S., Gachet, M., Lajus, A., Josso, A., et al. (2017). MicroScope in 2017: an expanding and evolving integrated resource for community expertise of microbial genomes. Nucleic Acids Res. 45, D517–D528. doi: 10.1093/nar/gkw1101
Wagner, E. G. H., and Romby, P. (2015). Small RNAs in Bacteria and archaea: who they are, what they do, and how they do it. Adv. Genet. 90, 133–208. doi: 10.1016/bs.adgen.2015.05.001
Washburn, R. S., Marra, A., Bryant, A. P., Rosenberg, M., and Gentry, D. R. (2001). rho is not essential for viability or virulence in Staphylococcus aureus. Antimicrob. Agents Chemother. 45, 1099–1103. doi: 10.1128/AAC.45.4.1099-1103.2001
Wassarman, K. M., Repoila, F., Rosenow, C., Storz, G., and Gottesman, S. (2001). Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev. 15, 1637–1651. doi: 10.1101/gad.901001
Waters, N. R., Samuels, D. J., Behera, R. K., Livny, J., Rhee, K. Y., Sadykov, M. R., et al. (2016). A spectrum of CodY activities drives metabolic reorganization and virulence gene expression in Staphylococcus aureus. Mol. Microbiol. 101, 495–514. doi: 10.1111/mmi.13404
Keywords: bona fide sRNA, Staphylococcus aureus HG003, RNA-seq, transcription factors, gene regulation
Citation: Liu W, Rochat T, Toffano-Nioche C, Le Lam TN, Bouloc P and Morvan C (2018) Assessment of Bona Fide sRNAs in Staphylococcus aureus. Front. Microbiol. 9:228. doi: 10.3389/fmicb.2018.00228
Received: 11 October 2017; Accepted: 30 January 2018;
Published: 20 February 2018.
Edited by:
Haiwei Luo, School of Life Sciences, The Chinese University of Hong Kong, ChinaReviewed by:
Huan Wang, University of California, Riverside, United StatesFranz Narberhaus, Ruhr University Bochum, Germany
Copyright © 2018 Liu, Rochat, Toffano-Nioche, Le Lam, Bouloc and Morvan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philippe Bouloc, philippe.bouloc@i2bc.paris-saclay.fr
Claire Morvan, claire.morvan@i2bc.paris-saclay.fr