 Beatriz Beamud
Beatriz Beamud María Alma Bracho
María Alma Bracho Fernando González-Candelas
Fernando González-Candelas- 1Instituto de Biología Integrativa de Sistemas, Consejo Superior de Investigaciones Científicas, Universitat de València, Valencia, Spain
- 2Unidad Mixta de Investigación Infección y Salud Pública, Fundación para el Fomento de la Investigación Sanitaria y Biomédica de la Comunitat Valenciana, Universitat de València, Valencia, Spain
- 3Área de Genómica y Salud, Fundación para el Fomento de la Investigación Sanitaria y Biomédica de la Comunitat Valenciana, Valencia, Spain
- 4Centro de Investigación Biomédica en Red de Epidemiología y Salud Pública, Valencia, Spain
Recombination is one of the main processes shaping the evolution of HIV-1, with relevant consequences for its epidemiology. In fact, Circulating and Unique Recombinant Forms (CRFs and URFs) cause 23% of current infections. The routine analyses of antiretroviral resistance yield partial pol gene sequences that can be exploited for molecular epidemiology surveillance but also to study viral diversity and to detect potential recombinant samples. Among the pol sequences derived from a large sample dataset from the Comunitat Valenciana (Spain), we identified nine putative recombinant samples. We aimed at fully characterizing these samples and performing a detailed analysis of the corresponding recombination events. We obtained nearly full-genome sequences and used jpHMM and RDP4 to detect and characterize recombinant fragments. We assessed the confidence of these inferences by likelihood mapping and phylogenetic placement with topology congruence tests. Next, we performed a phylogenetic analysis of each putative recombinant fragment to determine its relationships to previously described recombinant forms. We found that two samples related to CRF44_BF whereas the rest corresponded to new URFs (two URF_AD, one URF_BG that can constitute a new CRF resulting from subtype B and CRF24_BG, and two URF_cpx composed of A, G, K, H, and J subtypes). These URFs have a complex recombination pattern that cannot be determined accurately. They seem to have arisen by successive recombination events among lineages, including other CRFs. Our results highlight the usefulness of routine surveillance analysis for the detection of new HIV-1 recombination forms and, at the same time, the need for full-genome sequencing and recombination detection guidelines to properly characterize this complex process.
Introduction
The high genetic variability of HIV-1 is one of the main hurdles to control the current epidemic of this virus. This diversity is the result of high mutation (Abram et al., 2010), replication (Rodrigo et al., 1999), and recombination rates. The recombinogenic nature of HIV-1 is well-known, as this virus has one of the highest rates known (Rodrigo et al., 1999; Shriner et al., 2004). Recombination has important evolutionary consequences for viruses and has been associated with the expansion of viral host ranges, the emergence of new variants, increases in pathogenesis and virulence, the alteration of tropisms, the immune escape, and resistance to antivirals (Martin et al., 2011; Simon-Loriere and Holmes, 2011). In addition, recombination also plays an important role in HIV-1 epidemiology. Up to date, 91 circulating recombinant forms (CRFs) have been described (Los Alamos National Laboratory1). CRFs are recombinant HIV-1 genomes that share the same recombination breakpoints (BPs) between the same parentals and have been found in at least three non-epidemiologically related individuals. If a recombinant form does not fulfill the requirements to be considered a CRF, it is called a unique recombinant form (URF). RFs can also be classified according to their mosaic structure: they are denoted as complex (cpx) if they are composed by three or more subtypes or second-generation recombinants (SGRs) if they result from a recombination event in which pre-existing RFs are involved. RFs account for 23% of the current infections and are responsible for almost half of total infections in areas such as Central Africa (Hemelaar et al., 2019).
HIV-1 subtyping is still dominated by the partial sequencing of the pol gene used in routine analyses of antiretroviral resistance mutations. However, the development of whole genome sequencing (WGS) and its reducing costs have led to a substantial increase in the availability of complete HIV-1 genomes. This is partially responsible for the increased reporting of RFs world-wide. This increase also results from the mixing of newly formed HIV-1 variants, resulting in a complex and dynamic epidemiology (Hemelaar et al., 2019). Spain is one of the western European countries with the highest prevalence of HIV-12. Subtype B is responsible for 82–88% of the infections in this country followed by RFs, which cause about 9–10% of cases (Yebra et al., 2012). In fact, the three CRFs described from Spanish samples so far, CRF14_BG, CRF47_BF, and CRF73_BG (Delgado et al., 2002; Fernández-García et al., 2010, 2016), involve the B subtype. Apart from these, only a few studies with complete HIV-1 genome sequences have been done in this country using non-B subtypes (Casado et al., 2003; Sierra et al., 2005; Holguin et al., 2008; Cuevas et al., 2010). Hence, the prevalence of recombinant forms in Spain, as in many other countries, might be underestimated (Yebra et al., 2012).
The pol sequences obtained in the analyses of resistance mutations can be used to gain insight into the population dynamics and evolution of the corresponding lineages. These analyses can also identify sequences that are not neatly included in any of the major subtypes or sub-subtypes of HIV-1. They may represent new variants or RFs, relevant for the epidemiological characterization of the viral population. There is no standard procedure to detect recombination in HIV-1 and different methods have been used so far (a comprehensive list of recombination analysis software3). Due to this lack of standardization and the intrinsic difficulties in identifying recombinant events, some authors have postulated that several methods should be used to obtain a precise picture of recombination (Posada, 2002). The hallmark of recombination is the presence of fragments with different ancestries in the same genome. Therefore, the ultimate test for recombination should be based on revealing a statistically significant lack of phylogenetic congruence among portions of the recombinant genomes (Pérez-Losada et al., 2015). In order to test the statistical significance of alternate topologies, the corresponding multiple alignments must incorporate adequate levels of genetic variation. Consequently, analyses of the phylogenetic signal should also be included in this procedure. This is most relevant for small recombinant regions in which an insufficient phylogenetic signal may lead to wrong inferences of phylogenies (Jia et al., 2014).
During routine analyses of resistance mutations in HIV-1 samples from the Comunitat Valenciana (Spain) (Patiño-Galindo et al., 2017), we detected several samples whose partial pol sequences did not cluster with high support in any sub-subtype. Our aim here is to describe their full genomic characterization and to perform an in-depth analysis of recombination using different methods and confirm them by means of phylogenetic congruence tests.
Materials and Methods
Samples
From 2010 to 2013, about 1,800 serum samples of individuals infected with HIV-1 were screened for antiretroviral resistance mutations in our laboratory (Patiño-Galindo et al., 2017). For this purpose, a partial region (1,302 nt) of the pol gene was sequenced. The phylogenetic analysis of this region revealed nine samples with a suspected recombinant nature because they did not cluster with high enough bootstrap support with reference sequences of pure subtypes and representative CRFs (data not shown). These nine samples, all from Spanish patients that attended hospitals of the Comunitat Valenciana (Spain), were subjected to nearly-WGS.
Extraction, Amplification, and Sequencing
RNA was extracted from sera using the NucliSENS® EasyMag® automated platform (bioMérieux, Marcy, L’Étoile, France). Reverse transcription (RT) of RNA was performed in two adjacent genome regions to improve yield. The first hemigenome was obtained by priming with IN-T-A2 (Thomson et al., 2002) in the vif gene (nts: 5192–5218, HXB2, GenBank Accession No. K03455). The second hemigenome was obtained with UNINEF7′ (Nadai et al., 2008), close to the 3′ end of the viral RNA (nts: 9605–9632, HXB2). The conditions for RT and PCR were slightly modified from Nadai et al. (2008). RNA (4 μL) was reverse transcribed in a total volume of 20 μL with 500 uM dNTP, 2.5 uM primer, 1x RT buffer, 40 U RNasin® (Promega, Madison, WI, United States), 10 mM DTT and 200 U of SuperScriptTM III reverse transcriptase (InvitrogenTM, Carlsbad, CA, United States). Reagents and RNA were incubated for cDNA synthesis at 50°C for 2 h, followed by 85°C for 5 min.
Next, four overlapping regions were amplified using first-round PCRs and, if needed, nested-PCRs (Figure 1). The first amplicon consisted of about 3 kb from 5′ LTR U5 to the middle of pol (nts: 571–3554, HXB2). The second amplicon, of 2.1 kb, covered from the second half of pol to the start of vif (nts: 2930–5042, HXB2). The third amplicon, of approximately 1.4 kb, covered until env gp160. Lastly, the fourth amplicon consisted of 3.9 kb from env gp160 to the end of nef (nts: 5582–9511, HXB2). The reaction volume was 25 μL, containing 1x PCR buffer, 350 uM dNTP mixture, 0.4 uM of each primer and 2.5 U TaKaRa Ex Taq® (Takara Bio, Shiga, Japan). Cycling conditions were 94°C for 2 min and then 10 cycles of (94°C 10 s, 60°C 30 s, 68°C 3 min), and 20 cycles of (94°C 10 s, 55°C 30 s, 68°C 3 min) followed by 68°C for 10 min. Nested-PCRs were done with the same conditions as above but adding 1.5 μL of the previous PCR product with appropriate primers.
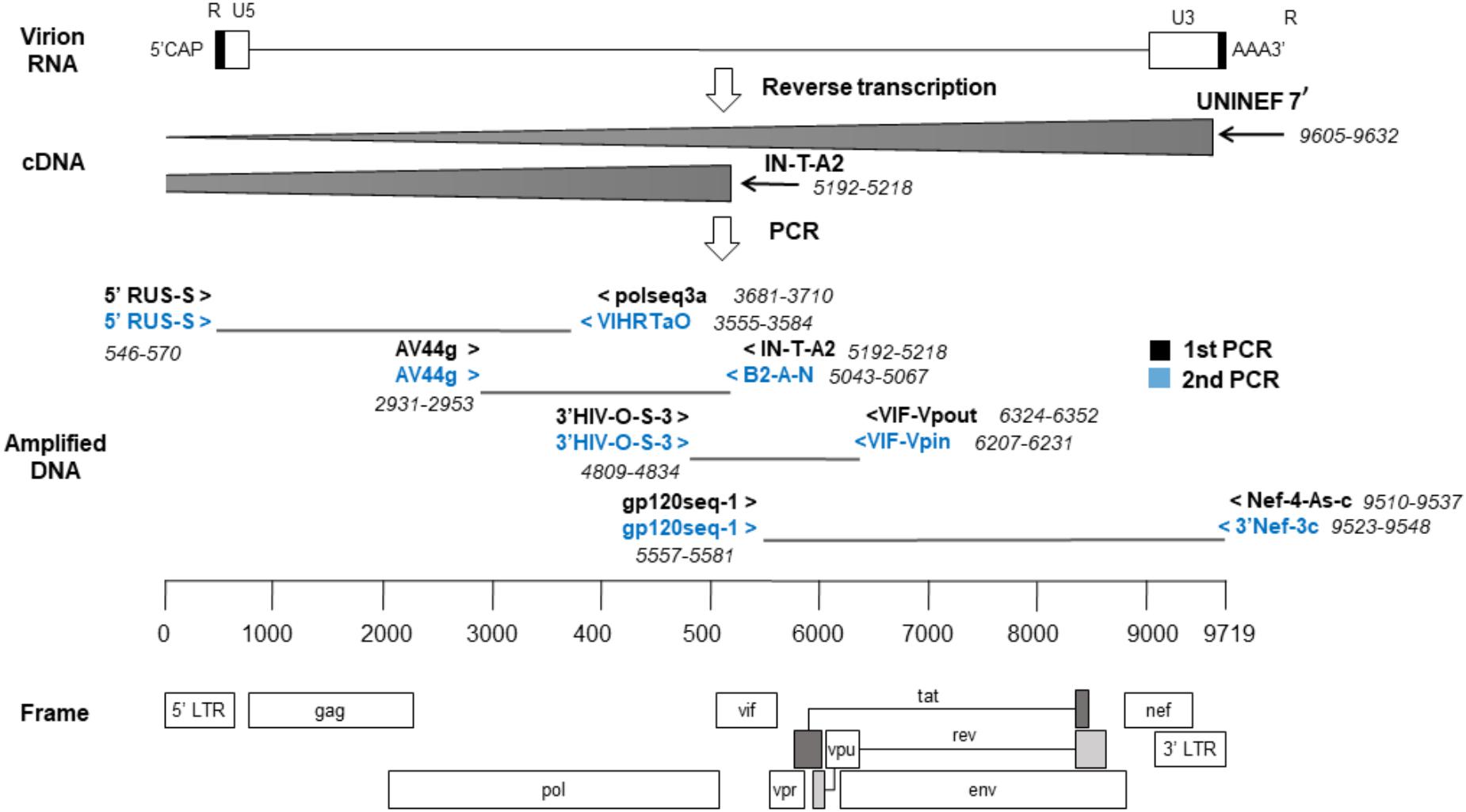
Figure 1. Sequencing strategy by the nearly full-length RT-PCR method. The locations of the primers used are relative to the reference genome, HXB2. cDNA synthesis was done in two reactions and a strategy of four overlapping amplicons was followed by PCR.
Positive PCR products were purified with a High Pure PCR Product Purification Kit (Roche Applied Science, Indianapolis, IN, United States) and sequenced using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, United States) in an ABI 3730xl DNA sequencer (Roche, Nutley, NJ, United States). The set of primers used in this study is available upon request. We obtained the consensus nearly full-length genome (8.9 kb) by assembling the sequence readings that passed the default quality filter of Pregap4 (Bonfield et al., 1995; Nadai et al., 2008) along with visual inspection of polymorphic regions. For genomes with more than one contig, scaffolding was done after comparison with the closest reference genome available at the Los Alamos National Laboratory (LANL) database. The final sequences have been deposited in the GenBank with Accession Nos. MK095228–MK095236.
Recombination Analysis
The mosaic recombinant structure of the samples was screened using two different strategies. Firstly, breakpoints (BPs) were determined with the default jpHMM options (Schultz et al., 2010). Secondly, we obtained recombination events and BPs that were supported by at least three of the seven selected tests implemented in RDP4: RDP (Martin and Rybicki, 2000), GENECONV (Padidam et al., 1999), BootScan (Martin et al., 2005), MaxChi (Smith, 1992), Chimaera (Posada and Crandall, 2001), SiScan (Gibbs et al., 2000) and 3Seq (Boni et al., 2007). Default parameter values were used except for window size that was increased to 60 nt in RDP, to 120 in MaxChi and Chimaera, and to 500 in BootScan and SiScan, as suggested (Lemey et al., 2009).
For jpHMM we used the nine sequences obtained above. To avoid confounding effects between recombinant samples, we used a separate alignment for each sample in RDP4. These alignments consisted of one sequence of each ‘pure’ subtype and sub-subtypes A1, A2, F1, and F2 (from RIP Alignment4) and each putative recombinant sample using the option -add of MAFFT v.7 (Katoh and Standley, 2013).
After detecting recombination, we wanted to further contrast the confidence of each recombination event independently. For this, we used a slightly modified phylogenetic congruence testing pipeline. The corresponding BPs of each detection strategy delimited sequence regions that were extracted and independently aligned using MAFFT v.7 (Katoh and Standley, 2013) (option –add) with the RIP reference alignment detailed above. This partial alignment was used to reconstruct a reference maximum likelihood (ML) tree using IQ-TREE v.1.6.5 (Nguyen et al., 2015) with the best evolutionary model (GTR+F+I+G4) determined by the same software (Kalyaanamoorthy et al., 2017). To test the phylogenetic congruence of the recombinant fragments in each genome, we constructed all possible alternative trees by clustering each sample with all the (sub-)subtypes in the reference alignment. For this, we used the R packages ape (Paradis and Schliep, 2018) and phytools (Revell, 2011). The 11 alternative trees for each sample were used for Expected Likelihood Weights (ELWs) (Strimmer and Rambaut, 2002) topological congruence test with the partial alignment corresponding to each fragment. This test assigns relative support values to the alternative topologies based on the likelihood values of the corresponding alignments. These tests were performed with IQ-TREE v.1.6.5 using 10,000 RELL replicates. Additionally, the level of phylogenetic signal of each partial alignment was also assessed by means of likelihood mapping with the evaluation of 1,000 random quartets using IQ-TREE v.1.6.5. A proportion higher than 0.25 of unresolved quartets was considered to represent a lack of phylogenetic signal (Strimmer and Rambaut, 2002; Lemey et al., 2009).
Next, each fragment of the recombinant samples was further characterized by phylogenetic analysis with representative sequences of HIV-1, including RFs, as above. The alignment was obtained from LANL database (RIP v.2017, 145 sequences) but adding, with MAFFT v.7, all the URFs identified as such (resulting in a total of 226 sequences). Then, new ML trees were built with 1,000 bootstrap replicates. The closest relatives (CRs) of each fragment were determined considering as CRs the minimum set of sequences that grouped with the fragment of interest with a bootstrap support value higher than 70%.
Finally, the inferred mosaic structure from the above analyses was drawn using the ggplot2 library in R (Wickham, 2016). These analyses, except the execution of RDP4, have been integrated into a publicly available pipeline accessible at https://github.com/BBeamud/PhyloRecomb/.
Results
We obtained nearly complete genome sequences of seven samples, but our sequencing strategy failed to yield enough information in two cases. Between 86 and 92% of the reference genome (HXB2) length was covered in our samples. The per base coverage ranged from 2.44 to 4.53. In four of these genomes, it was not possible to assemble all the sequencing reads into a single contig due to hypervariability in the env gene. This is reflected in the proportion of undetermined bases of each sequence that were used for scaffolding (Table 1). For the incomplete genomes, we obtained 41% (sample 678) and 49% (sample 3164) of the reference genome length with coverage depths of 1.64 and 1.66, respectively. Additional sequencing of these two samples was not possible due to lack of biological material.
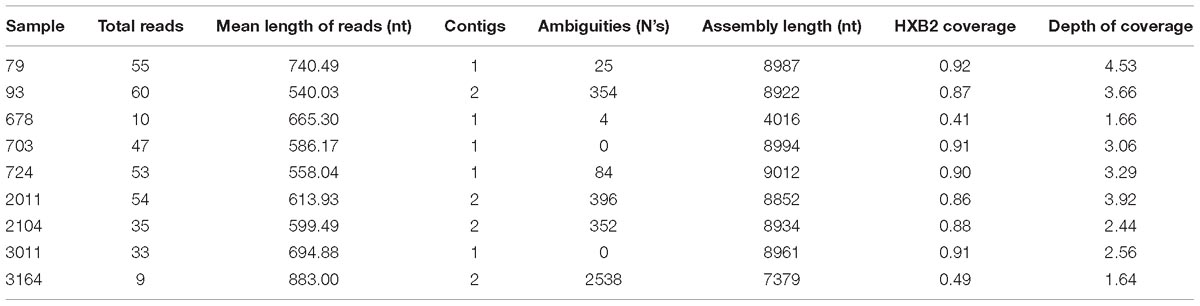
Table 1. Summary statistics of the near complete genomes obtained for the 9 HIV-1 samples studied.
Recombinant mosaic structures were obtained by two strategies. Firstly, we considered the recombination events and their defining BPs supported by at least three of the methods tested in RDP4. Additionally, we considered the BPs reported by jpHMM. These two programs assign a subtype for each fragment. All the strategies revealed putative recombination events in all the samples, but the total number of such events differed among them. To facilitate the comparison between detection strategies, we retained as major parental (MP) the subtype with most genomic nucleotides assigned. In addition, we considered the same recombination event if the minor assigned subtype and the inferred BPs were similar ( ± 200 nt) in both sides for both strategies. A total of 42 recombination events were detected but only eight were shared by the two strategies. Of the 34 remaining events, 21 were detected exclusively by jpHMM and 13 by RDP4. There were substantial differences in the performance of the methods implemented in RDP4, with a minimum of 3 (GENECONV) and a maximum of 19 (MaxChi and 3Seq) detected events (Table 2). Shared events had an average length of 1454.9 nt with a difference in the start and end positions of 14.9 and 59.0 nt, respectively. The shared events detected by RDP4 were on average 11.9 nt shorter than those detected by jpHMM. On the contrary, unique events by jpHMM and RDP4 had lengths of 513.6 and 1399.4 nt. Surprisingly, some large events detected by RDP4 were not shared with jpHMM. This was due to RDP4 failing to detect several events of the minor subtypes and reporting them as unique, larger recombination events (Table 3, samples 2011, 2104, 3011, and 3164).
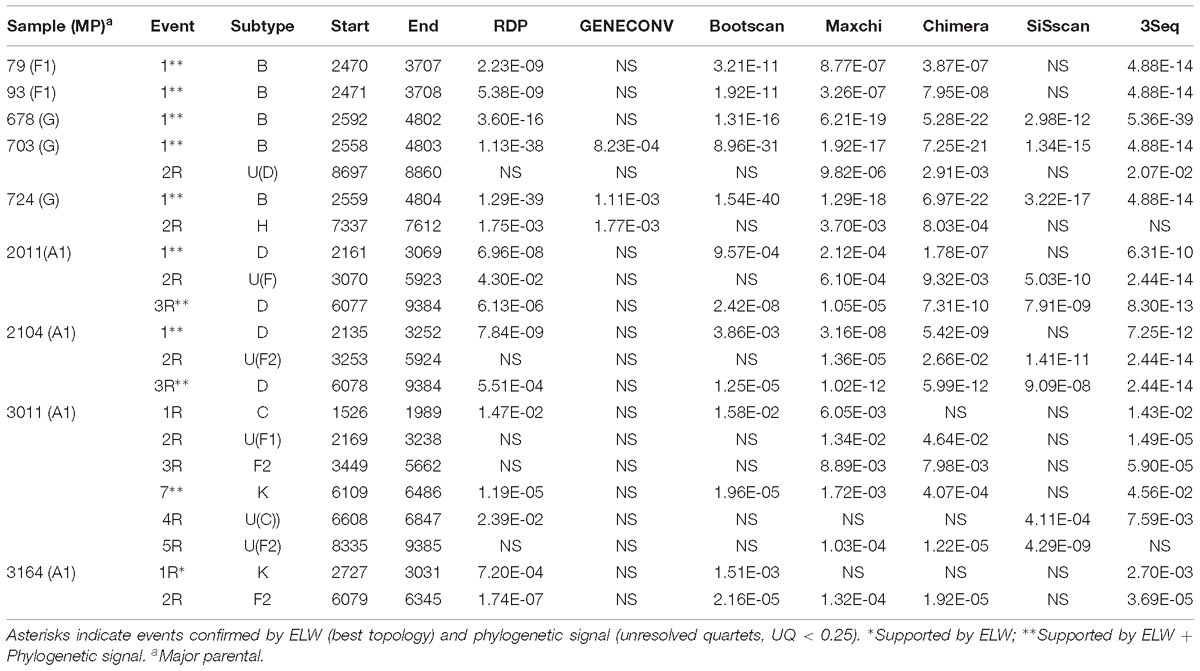
Table 2. Support of each putative recombination event detected by the different methods implemented in RDP4.
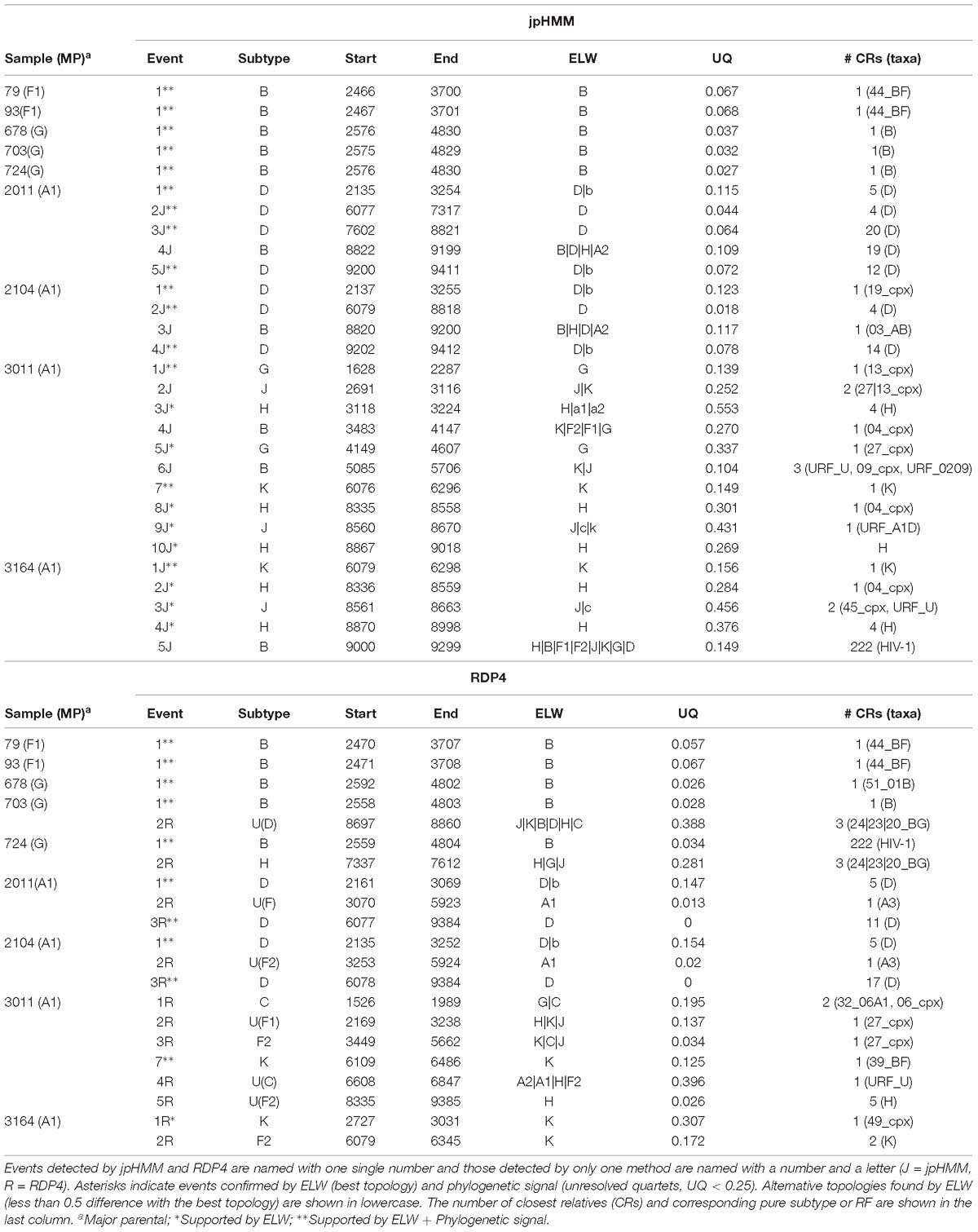
Table 3. Recombination events detected by jpHMM and RPD4 and phylogenetic analysis of the studied samples.
To further corroborate these events, we performed ELW congruence tests and likelihood mapping analyses for each fragment. A recombination event was considered verified only when the tree with the sample fragment grouped with the minor subtype identified had a relative support value, as determined by ELW, larger than 0.5 than that of the second-best tree. Additionally, we considered fragments with less than 0.25 unresolved quartets reliable for phylogenetic inferences. All the events jointly detected by jpHMM and RPD4 were verified by ELW tests and phylogenetic signal. However, 15 out of the 21 events detected only by jpHMM were corroborated, regardless of their phylogenetic signal, whereas only 3 of the 13 events detected only by RDP4 were verified. For the latter, the main discrepancies were observed in the assigned subtype whereas for jpHMM a high proportion of cases with low phylogenetic signal was observed (Table 3). A summary of true positive, false positive and false negative recombination events, detected by the different methods implemented in RDP4 and jpHMM, is shown in Figure 2. The overall best performance corresponds to jpHMM, with 23 corroborated events of the 29 detected with this method. The worst performing method was GENECONV with only two corroborated events of the three detected.
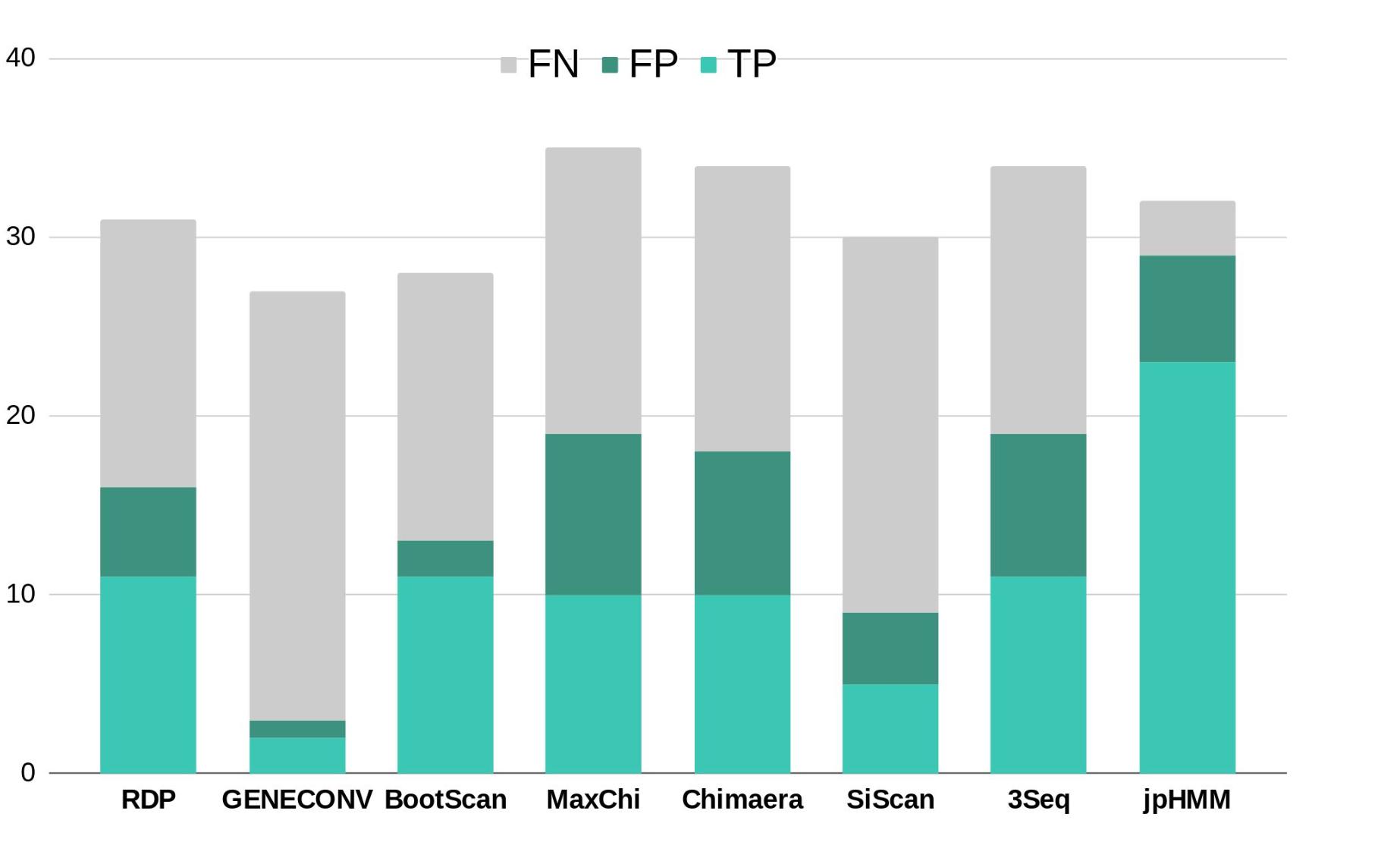
Figure 2. Summary of true positive (TP), false positive (FP) and false negative (FN) recombination events detected by the different methods used in this study according to the results of ELW tests. RDP, GENECONV, BootScan, Maxchi, Chimaera, SiScan, and 3Seq were analyzed with RDP4.
Different mosaic structures were observed in the samples considering verified events. In particular, we observed two BF recombinants, two AD recombinants, three BG recombinants, and two complex recombinant forms. These mosaic structures were refined with the reconstruction of ML trees for each fragment based on the analysis of closest relatives (CRs) with 226 representative sequences of HIV-1. We performed these analyses for all the major and minor parental fragments although the results of the former analyses are not shown for simplification (Table 3).
The two BF recombinants corresponded to samples 79 and 93. They showed the same pattern and similar BPs by both jpHMM and RDP4. A single recombination event of subtype B was detected and verified in the region located between nucleotides 2,466 and 3,708 (all the positions reported are referred to HXB2). This structure resembles that of CRF38_BF (Ruchansky et al., 2009) although the CRs analyses of the recombinant fragments of samples 79 and 93 showed that they are related to CRF44_BF (Delgado et al., 2010) (Table 3). The latter has three recombination events (all of subtype B) that were not detected by jpHMM nor RDP4. We re-analyzed samples 79 and 93 by performing congruence analyses with the reference breakpoints of CRF44_BF5 to confirm it relatedness. The results of the ELW tests showed a lack of congruence for the three recombination events with their grouping with subtype B, thus corroborating them (Figure 3). Hence, the CRs analyses allowed us to classify samples 79 and 93 as CRF44_BF, despite their initial identification as CRF38_BF.
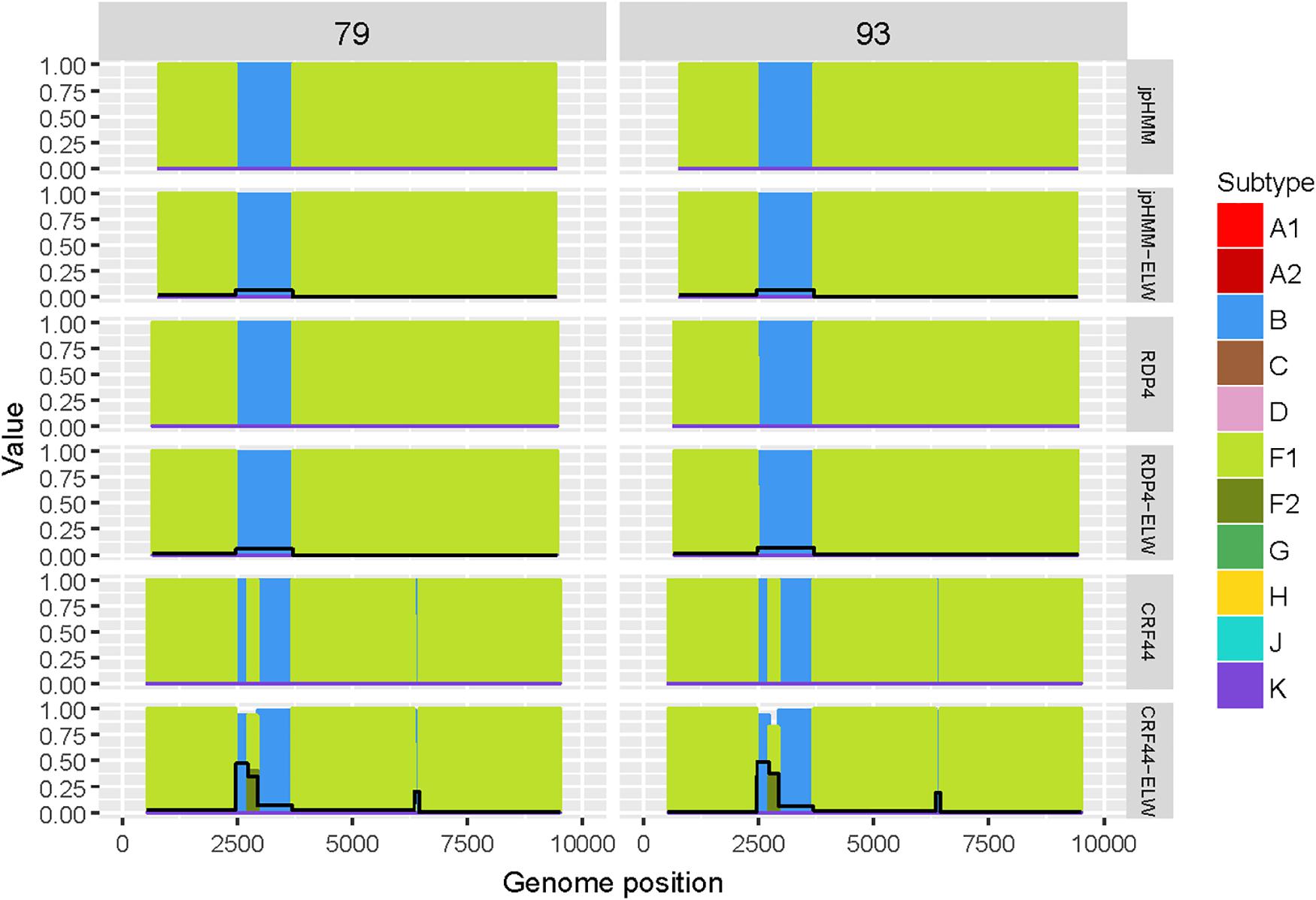
Figure 3. Recombination analyses of samples 79 and 93 with recombination BPs determined by different sources (jpHMM, RDP4 and CRF44_BF reference BPs) and phylogenetic congruence (ELW) tests. The black line represents the proportion of unresolved quartets, which correspond to the inverse of the phylogenetic signal.
Samples 678, 703, and 724 are BG recombinants, with G being the major parental with a single B recombination event. A similar situation to that in BF recombinants was found in this group, except for two recombination events detected only by RDP4, 703_2R (U|D) and 724_2R(H), which were not verified by ELW and phylogenetic signal tests. CRs analyses showed that the G fragment was related to CRF24_BG whereas the B fragment grouped consistently with pure B sequences or CRF51_01B (Table 3). To confirm that samples 678, 703, and 724 were indeed CRF24_BG we proceeded as above, with the BPs defined at the LANL database for this CRF. In this case, the results of the ELW tests did not reveal four different B recombination events, as in CRF24_BG, but recognized only the single B event detected jointly by RDP4 and jpHMM (Figure 4). It is important to note that in the case of sequence 678, the comparisons were limited because only a partial genome was obtained.
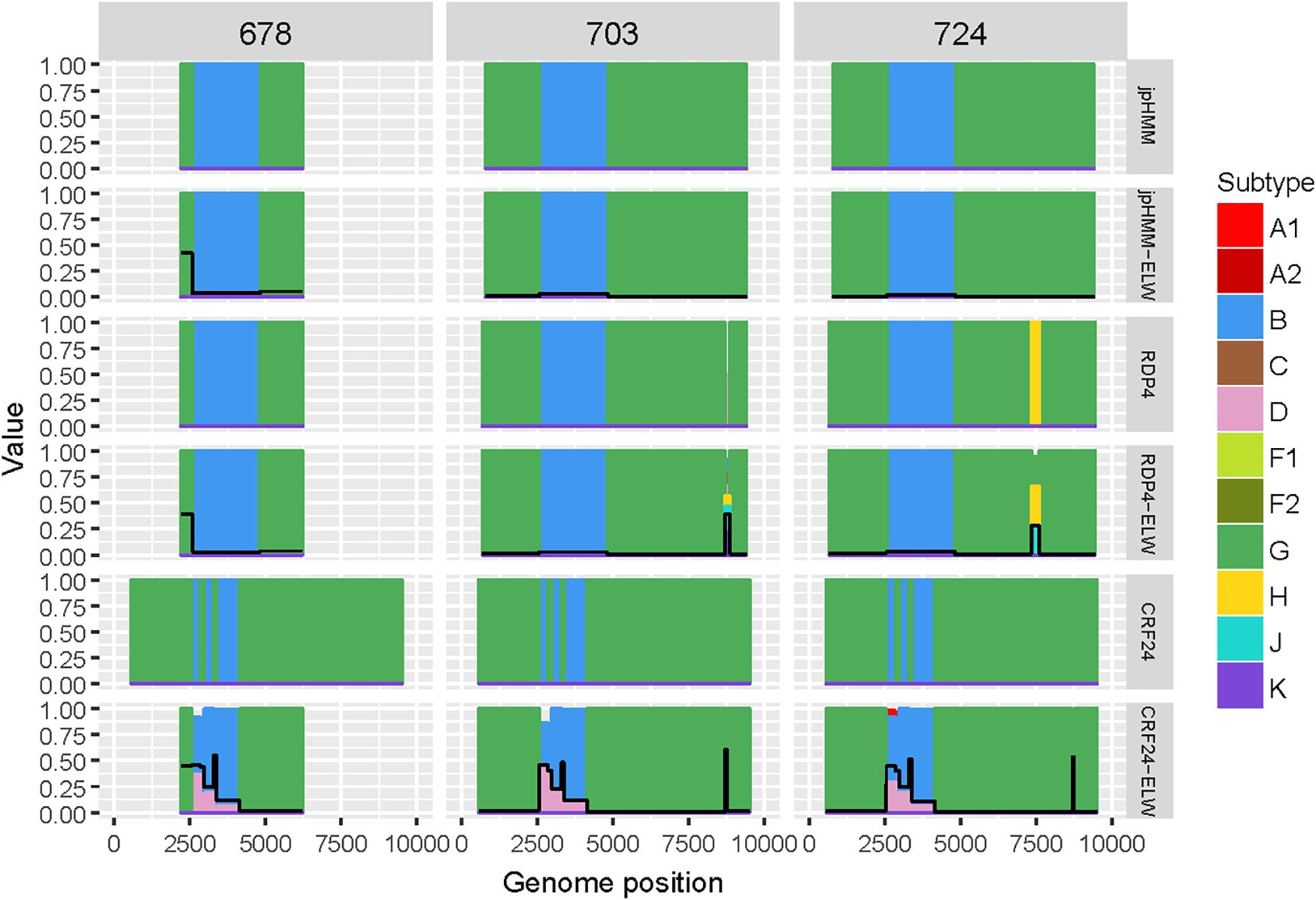
Figure 4. Recombination analyses of samples 678, 703, and 724 with recombination BPs determined by different sources (jpHMM, RDP4, and CRF24_BG reference BPs) and phylogenetic congruence (ELW) tests. The black line represents the proportion of unresolved quartets, which correspond to the inverse of the phylogenetic signal.
The two AD recombinants corresponded to samples 2011 and 2014 with A subtype being the major parental and with four and three fragments of subtype D, respectively. Initially, more recombination events of other subtypes were identified by jpHMM and RDP4. However, they were discarded based on the results of the ELW congruence tests. RDP4 failed to detect three events that were detected by jpHMM and further verified by tree-topology and phylogenetic signal (Figure 5). CRs analyses showed that the A portion corresponded to sub-subtype A3 whereas the D fragments clustered with D ‘pure’ or CRF19_cpx sequences (Table 3). Despite small discrepancies, 2011 and 2014 present a similar recombination pattern but, to our knowledge, no similar pattern has been reported to the databases yet.
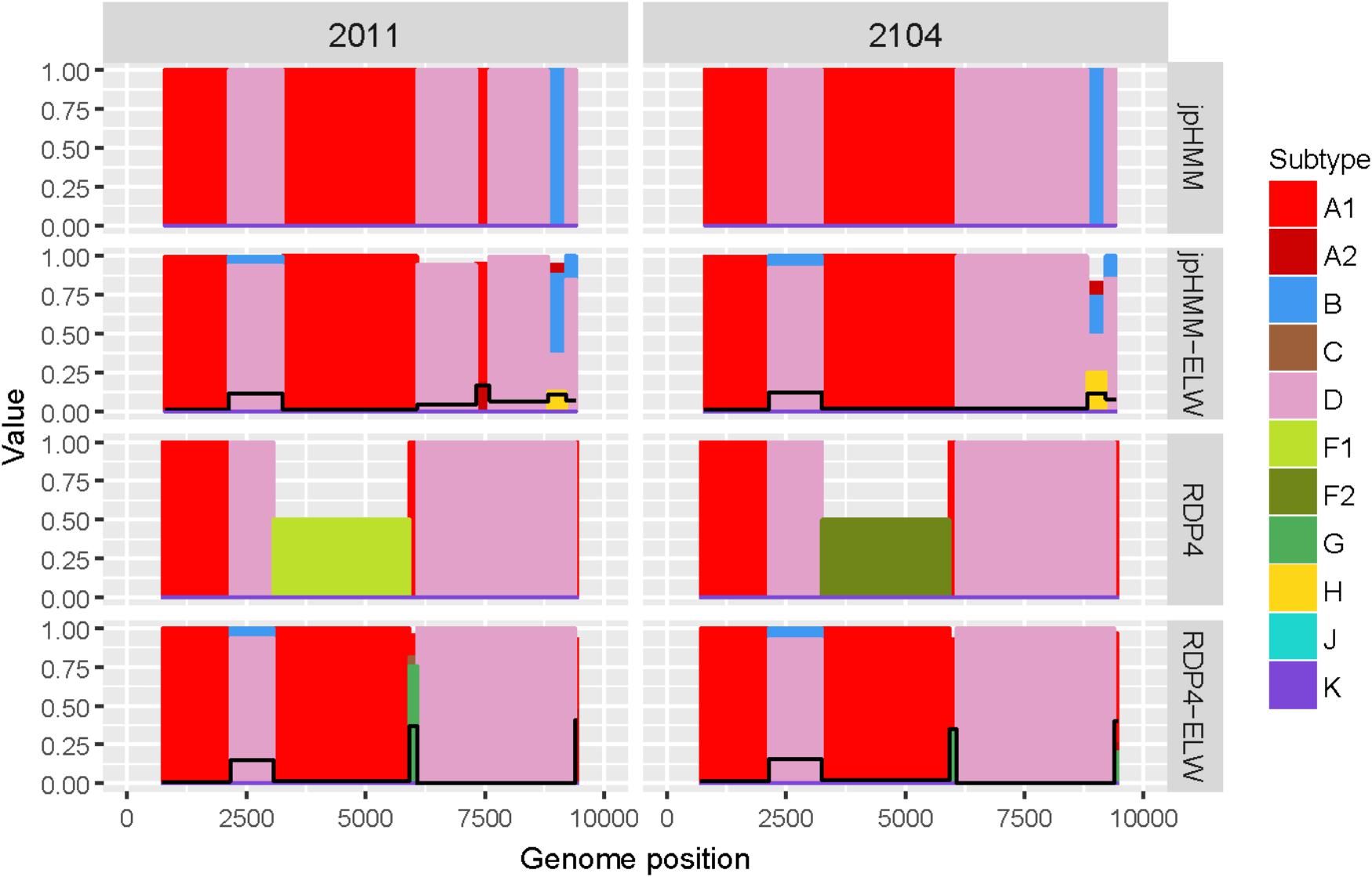
Figure 5. Recombination analyses of samples 2011 and 2104 with recombination BPs determined by different sources (jpHMM and RDP4) and phylogenetic congruence (ELW) tests. Values for inconclusive results by RDP4 are arbitrarily represented as 0.5. The black line represents the proportion of unresolved quartets, which correspond to the inverse of the phylogenetic signal.
The genome sequences derived from samples 3011 and 3164 were identified as complex forms, composed by more than two ‘pure’ subtypes: A, G, H, J, and K for 3011 and A, H, J, and K for 3164. For both samples, the A subtype was considered as the major parental although, in this case, there is little difference between the portion of the genome covered by the different subtypes. As before, the description of 3164 is limited because about only half of its genome sequence was obtained. These recombinant forms were very complex both in the detection and verification results (Figure 6). A total of 16 and seven different recombination events were detected for samples 3011 and 3164, respectively. JpHMM detected 10 events in sample 3011, which were different from the five events detected by RDP4. Only one event, 3011_7(K), was detected simultaneously by both methods (Table 3). None of the seven events detected in sample 3164, five by jpHMM and two by RDP4, was shared by the two methods.
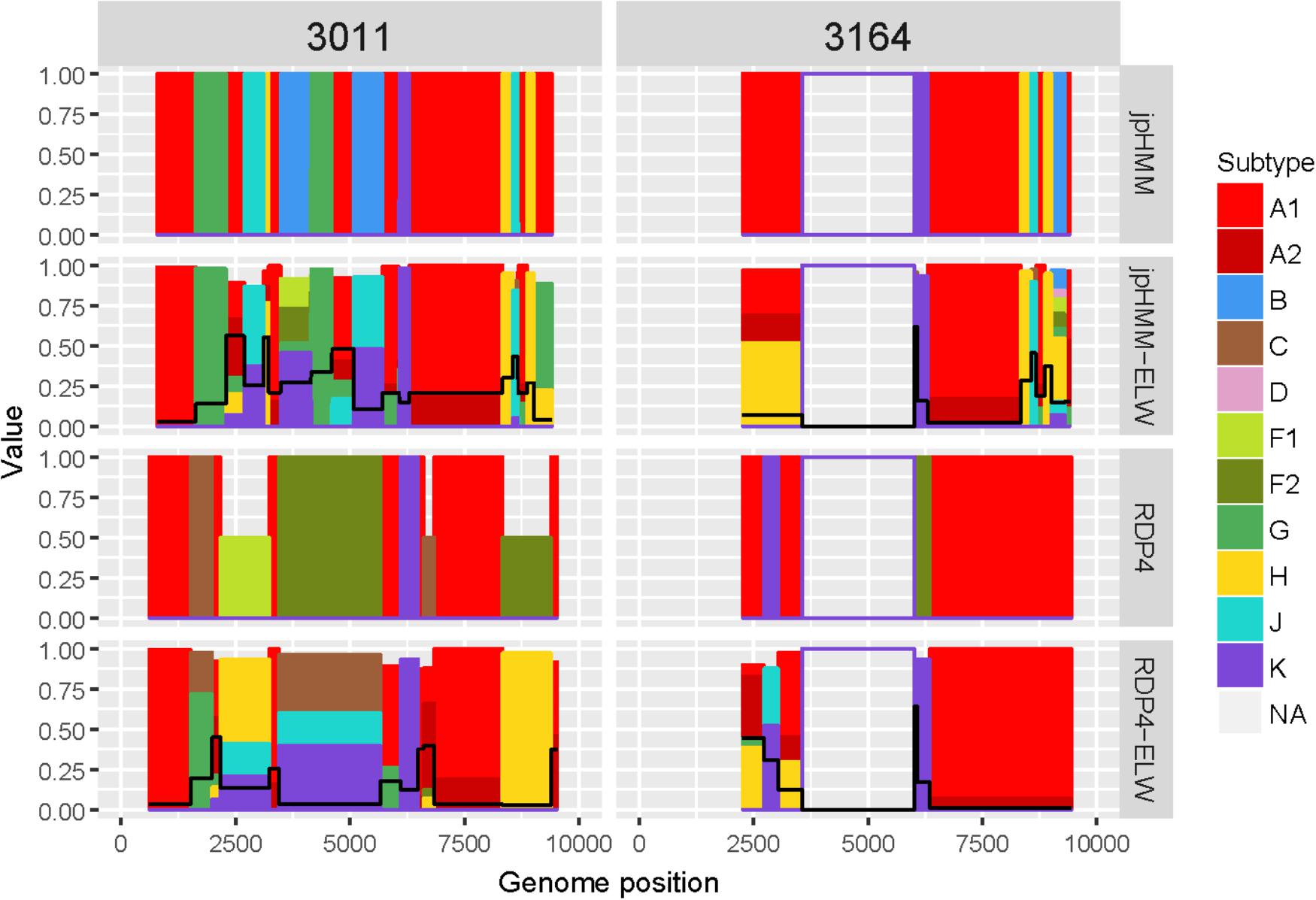
Figure 6. Recombination analyses of samples 3011 and 3164 with recombination BPs determined by different sources (jpHMM and RDP4) and phylogenetic congruence (ELW) tests. Values for inconclusive results by RDP4 are arbitrarily represented as 0.5. The black line represents the proportion of unresolved quartets, which correspond to the inverse of the phylogenetic signal.
Most of the events detected by jpHMM in sample 3011 (7/11) showed a low phylogenetic signal, with more than 25% of unresolved quartets (Table 3) whereas only one event detected by RDP4 in this sample failed this test. A similar result was observed for sample 3164, with three of the five events detected by jpHMM presenting low phylogenetic signal and with one of the two events detected by RDP4 also failing this test. This low signal might also affect subtyping inferences, with several discrepancies between the subtypes identified by jpHMM or RDP4 and those inferred as most likely by the phylogenetic congruence analyses (Table 3). Finally, the results of CRs analyses presented several cases of unclear relatedness. For instance, fragments of the major subtype of these two samples grouped within clade A but also with distinct URFs and complex forms, such as CRF45_cpx. The G and J fragments grouped with other complex forms, such as CRF13_cpx and CRF27_cpx. Finally, recombinant fragments of subtype H clustered with ‘pure’ H sequences or CRF04_cpx and those of subtype K were related to K ‘pure’ sequences (Table 3). Altogether, the mosaic structure of these genomes could not be determined unambiguously. Samples 3011 and 3164 showed important discrepancies between them and at present, no CRFs_cpx with these patterns has been reported to the databases.
Discussion
In this study, we have described seven nearly-complete and two partial genomes of different HIV-1 recombinant forms, detected in infected individuals from Comunitat Valenciana (Spain). We have performed detailed analyses to characterize their recombination events. Firstly, we identified putative recombinant fragments using different methods, jpHMM and seven of those included in RDP4. In a second phase, we corroborated the recombinant fragments by evaluating their phylogenetic signal, testing their topological congruence and analyzing their closest relatives using a comprehensive dataset of HIV-1 sequences including representatives of all the subtypes, CRF and URFs.
Two samples, 79 and 93, were initially identified as CRF38_BF based on the results of jpHMM and RDP4. However, our additional analyses of closest relatives and ELW congruence test revealed that both were related to CRF44_BF, a recombinant form first described in Chile in 2010 (Delgado et al., 2010), but already circulating in Madrid (Spain) in 2005 to 2007 (Gonzalez-Alba et al., 2011). These samples were obtained in 2012 and 2013, thus indicating that this recombinant form was actively circulating in Spain a few years later. BF recombinants might be a larger family than previously thought with differences in the location of breakpoints or length of the inserted fragments (Cevallos et al., 2017). This hypothesis is confirmed in this study, providing two additional BF genomes that were not properly characterized based on the breakpoints reported by two detection methodologies but identified with additional analyses. Subsequent mutations or differences in methodology might prevent the detection of the actual mosaic structure of additional recombinant forms in this family. Thorough phylogenetic analyses, including congruence tests, should always be performed to reliably infer the recombination structure of HIV-1 samples.
The three BG recombinants shared a similar mosaic structure, which was further verified in our subsequent analyses. They might constitute a SGR resulting from a new recombination event between CRF24_BG and subtype B. CRF24_BG was firstly identified in Cuba in 2007 (Sierra et al., 2007; Cevallos et al., 2017). However, it was found in several Spanish individuals in Madrid a few years ago (Gonzalez-Alba et al., 2011). This, along with the high prevalence of subtype B and the origin of two other BG recombinant forms, CRF14_BG and CRF73_BG (Delgado et al., 2002; Fernández-García et al., 2016), in the Iberian Peninsula, suggests a scenario that fits well with the previous hypothesis as well as with the subsequent formation of new RFs with these subtypes.
Samples 2011 and 2014 might represent a new URF_AD, originated from several recombination events of ‘pure’ A3 and D sequences. In Spain, A and D subtypes are not frequent as they represent less than 1% of total infections (García et al., 2011; Gonzalez-Alba et al., 2011; Yebra et al., 2012; Patiño-Galindo et al., 2017) and no CRF_AD has been reported in this country so far. Hence, it is likely that this URF_AD has emerged in an endemic area for these subtypes and arrived in Spain by immigration.
Lastly, samples 3011 and 3164 represent complex forms composed of A, G, K, H and J subtypes that could not be identified accurately. Most of these subtypes, except G and A, are extremely rare in Spain (García et al., 2011; Gonzalez-Alba et al., 2011; Yebra et al., 2012; Patiño-Galindo et al., 2017). More specifically, in the Comunitat Valenciana, a previous analysis of the partial pol sequences of the 1,800 samples at the origin of this study revealed an important number of non-B infections (16.2%) but they did not include subtypes K, H, or J (Patiño-Galindo et al., 2017). These new URFs are an example of the enormous complexity of the recombination process in HIV-1 as several CRF_cpx are involved. The presence of CRFs from West-Central Africa, such as CRF9_cpx, CRF13_cpx, CRF27_cpx or CRF45_cpx, previously reported in Spain (Niama et al., 2009), might reflect an increased prevalence and generation of subsequent unique mosaic viruses in diverse geographical areas.
There are substantial differences in the performance of the eight methods used to detect recombination (Figure 2). The combined use of jpHMM and some (in our case at least three) methods implemented in RDP4 improves the reliable detection of recombination events: all the common events were further corroborated by ELW and phylogenetic signal analyses. This result agrees with previous suggestions of using several, methodologically different approaches to detect recombination events in HIV-1 (Posada, 2002). For those cases in which an event is not detected by one methodology, but it is positive according to others, additional corroboration analyses should be performed. In this case, 18 events detected by only jpHMM or RDP4 were later corroborated by ELW and phylogenetic signal analyses.
In general, RDP4 and jpHMM performed well for samples with one single recombination event. However, more false positive and negative events as well as cases of subtype misidentification were found for both programs with increasing complexity of the recombinant structures. Both methods showed the largest number of discrepancies in the detection of complex forms. The difficulties for the precise identification of the origin of these fragments were also evidenced by unresolved results in the congruence tests. Because some of these fragments are very short, they may not have enough phylogenetic signal to differentiate between subtypes. This poses problems for the two methods used to detect them. In addition, the paucity of complete sequence information about ‘rare’ subtypes makes the detection and verification of recombinants, especially those involving complex forms, a difficult task.
Conclusion
We have identified nine recombinant forms circulating in the Comunitat Valenciana (Spain). Two of them are related to CRF44_BF but the rest correspond to new URFs. Some samples of this study revealed a complex recombination nature and origin. This might suggest that, apart from the endemic areas that exhibit the highest levels of genetic diversity and therefore represent recombination hotspots, other regions could be sources of recombinant progeny due to the increasing global gene flow (Foster et al., 2014). To better understand the viral diversity and dynamics, as well as the real impact of recombination on them, more efforts should be devoted to obtaining complete genome sequences and to perform in-depth analyses of recombination.
The present study highlights the utility of surveillance analyses of HIV-1 for additional goals to identify resistance mutations, such as studying genetic diversity and characterizing new recombinant forms. A thorough analysis of partial genome sequences obtained in the surveillance of antiretroviral resistance mutations may provide the best starting point for the discovery and characterization of new recombinant forms in HIV-1.
Ethics Statement
Approval of surveillance studies by an ethics committee is not required as per local legislation and host institution (FISABIO) guidelines.
Author Contributions
The idea for this study was conceived by BB, MAB, and FG-C, who was also in charge of supervising the whole project. Data collection and reference searching were done by BB and MAB. The data analyses and drafting the manuscript were done by BB. The final manuscript was written, curated, and confirmed by BB, MAB and FG-C.
Funding
This research was supported by projects BFU2014-58656R and BFU2017-89594R from MINECO and MICU (Spanish Government), PROMETEO2016-0122 (from Generalitat Valenciana) and Conselleria de Sanitat Universal i Salut Pública (Generalitat Valenciana). BB was the recipient of an FPU contract for early stage researchers from MEC (Spanish Government).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
- ^ http://www.hiv.lanl.gov
- ^ http://apps.who.int/gho/data/node.main.620?lang=en
- ^ http://bioinf.man.ac.uk/robertson/recombination/programs.shtml
- ^ https://www.hiv.lanl.gov/content/sequence/NEWALIGN/align.html#RIP
- ^ https://www.hiv.lanl.gov/content/sequence/HIV/CRFs/breakpoints.html#CRF44
References
Abram, M. E., Ferris, A. L., Shao, G., Alvord, W. G., and Hughes, S. H. (2010). Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J. Virol. 84, 9864–9878. doi: 10.1128/JVI.00915-10
Bonfield, J. K., Smith, K. F., and Staden, R. (1995). A new DNA sequence assembly program. Nucleic Acids Res. 23, 4992–4999. doi: 10.1093/nar/23.24.4992
Boni, M. F., Posada, D., and Feldman, M. W. (2007). An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 176, 1035–1047. doi: 10.1534/genetics.106.068874
Casado, G., Thomson, M. M., Delgado, E., Sierra, M., Vázquez-De Parga, E., Pérez-Alvarez, L., et al. (2003). Near full-length genome characterization of an HIV type 1 CRF05_DF virus from Spain. AIDS Res. Hum. Retroviruses 19, 719–725. doi: 10.1089/088922203322280955
Cevallos, C. G., Jones, L. R., Pando, M. A., Carr, J. K., Avila, M. M., and Quarleri, J. (2017). Genomic characterization and molecular evolution analysis of subtype b and bf recombinant HIV-1 strains among Argentinean men who have sex with men reveal a complex scenario. PLoS One 12:e0189705. doi: 10.1371/journal.pone.0189705
Cuevas, M. T., Fernández-García, A., Pinilla, M., García-Alvarez, V., Thomson, M., Delgado, E., et al. (2010). Short communication: biological and genetic characterization of HIV type 1 subtype B and nonsubtype B transmitted viruses: usefulness for vaccine candidate assessment. AIDS Res. Hum. Retroviruses 26, 1019–1025. doi: 10.1089/aid.2010.0018
Delgado, E., Ríos, M., Fernández, J., Pérez-Alvarez, L., Nájera, R., and Thomson, M. M. (2010). Identification of a new HIV type 1 BF intersubtype circulating recombinant form (CRF44_BF) in Chile. AIDS Res. Hum. Retroviruses 26, 821–826. doi: 10.1089/aid.2010.0006
Delgado, E., Thomson, M. M., Villahermosa, M. L., Sierra, M., Ocampo, A., Miralles, C., et al. (2002). Identification of a newly characterized HIV-1 BG intersubtype circulating recombinant form in Galicia, Spain, which exhibits a pseudotype-like virion structure. J. Acquir. Immune Defic. Syndr. 29, 536–543. doi: 10.1097/00042560-200204150-00016
Fernández-García, A., Delgado, E., Cuevas, M. T., Vega, Y., Montero, V., Sánchez, M., et al. (2016). Identification of an HIV-1 BG intersubtype recombinant form (CRF73_BG), partially related to CRF14_BG, which is circulating in Portugal and Spain. PLoS One 11:e0148549. doi: 10.1371/journal.pone.0148549
Fernández-García, A., Pérez-Alvarez, L., Cuevas, M. T., Delgado, E., Muñoz-Nieto, M., Cilla, G., et al. (2010). Identification of a new HIV type 1 circulating BF intersubtype recombinant form (CRF47_BF) in Spain. AIDS Res. Hum. Retroviruses 26, 827–832. doi: 10.1089/aid.2009.0311
Foster, G. M., Ambrose, J. C., Hué, S., Delpech, V. C., Fearnhill, E., Abecasis, A. B., et al. (2014). Novel HIV-1 recombinants spreading across multiple risk groups in the United Kingdom: the identification and phylogeography of circulating recombinant form (CRF) 50_A1D. PLoS One 9:e83337. doi: 10.1371/journal.pone.0083337
García, F., Pérez-Cachafeiro, S., Guillot, V., Alvarez, M., Pérez-Romero, P., Pérez-Elías, M. J., et al. (2011). Transmission of HIV drug resistance and non-B subtype distribution in the Spanish cohort of antiretroviral treatment naïve HIV-infected individuals (CoRIS). Antiviral Res. 91, 150–153. doi: 10.1016/j.antiviral.2011.05.010
Gibbs, M. J., Armstrong, J. S., and Gibbs, A. J. (2000). Sister-scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 16, 573–582. doi: 10.1093/bioinformatics/16.7.573
Gonzalez-Alba, J. M., Holguin, A., Garcia, R., Garcia-Bujalance, S., Alonso, R., Suarez, A., et al. (2011). Molecular surveillance of HIV-1 in Madrid, Spain: a phylogeographic analysis. J. Virol. 85, 10755–10763. doi: 10.1128/JVI.00454-11
Hemelaar, J., Elangovan, R., Yun, J., Dickson-Tetteh, L., Fleminger, I., Kirtley, S., et al. (2019). Global and regional molecular epidemiology of HIV-1, 1990-2015: a systematic review, global survey, and trend analysis. Lancet Infect. Dis. 19, 143–155. doi: 10.1016/S1473-3099(18)30647-9
Holguin, A., Mulder, M., Yebra, G., Lopez, M., and Soriano, V. (2008). Increase of Non-B subtypes and recombinants among newly diagnosed HIV-1 native Spaniards and immigrants in Spain. Curr. HIV Res. 6, 327–334. doi: 10.2174/157016208785132455
Jia, L., Li, L., Li, H., Liu, S., Wang, X., Bao, Z., et al. (2014). Recombination pattern reanalysis of some HIV-1 circulating recombination forms suggest the necessity and difficulty of revision. PLoS One 9:e107349. doi: 10.1371/journal.pone.0107349
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Meth. 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Lemey, P., Salemi, M., and Vandamme, A.-M. (2009). The Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing. Cambridge: Cambridge University Press.
Martin, D., and Rybicki, E. (2000). RDP: detection of recombination amongst aligned sequences. Bioinformatics 16, 562–563. doi: 10.1093/bioinformatics/16.6.562
Martin, D. P., Biagini, P., Lefeuvre, P., Golden, M., Roumagnac, P., and Varsani, A. (2011). Recombination in eukaryotic single stranded DNA viruses. Viruses 3, 1699–1738. doi: 10.3390/v3091699
Martin, D. P., Posada, D., Crandall, K. A., and Williamson, C. (2005). A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retroviruses 21, 98–102. doi: 10.1089/aid.2005.21.98
Nadai, Y., Eyzaguirre, L. M., Constantine, N. T., Sill, A. M., Cleghorn, F., Blattner, W. A., et al. (2008). Protocol for nearly full-length sequencing of HIV-1 RNA from plasma. PLoS One 3:e1420. doi: 10.1371/journal.pone.0001420
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Niama, F. R., Vidal, N., Bazepeo, S. E., Mpoudi, E., Toure-Kane, C., Parra, H. J., et al. (2009). CRF45_AKU, a circulating recombinant from Central Africa, is probably the common ancestor of HIV Type 1 MAL and HIV type 1 NOGIL. AIDS Res. Hum. Retroviruses 25, 1345–1353. doi: 10.1089/aid.2009.0169
Padidam, M., Sawyer, S., and Fauquet, C. M. (1999). Possible emergence of new Geminiviruses by frequent recombination. Virology 265, 218–225. doi: 10.1006/viro.1999.0056
Paradis, E., and Schliep, K. (2018). Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Patiño-Galindo, J. Á, Torres-Puente, M., Bracho, M. A., Alastrué, I., Juan, A., Navarro, D., et al. (2017). The molecular epidemiology of HIV-1 in the Comunidad Valenciana (Spain): analysis of transmission clusters. Sci. Rep. 7:11584. doi: 10.1038/s41598-017-10286-1
Pérez-Losada, M., Arenas, M., Galán, J. C., Palero, F., and González-Candelas, F. (2015). Recombination in viruses: mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 30, 296–307. doi: 10.1016/j.meegid.2014.12.022
Posada, D. (2002). Evaluation of methods for detecting recombination from DNA sequences: empirical data. Mol. Biol. Evol. 19, 708–717. doi: 10.1093/oxfordjournals.molbev.a004129
Posada, D., and Crandall, K. A. (2001). Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc. Natl. Acad. Sci. U.S.A. 98, 13757–13762. doi: 10.1073/pnas.241370698
Revell, L. J. (2011). Phytools: an R package for phylogenetic comparative biology (and other things). Meth. Ecol. Evol. 3, 217–223. doi: 10.1111/j.2041-210x.2011.00169.x
Rodrigo, A. G., Shpaer, E. G., Delwart, E. L., Iversen, A. K. N., Gallo, M. V., Brojatsch, J., et al. (1999). Coalescent estimates of HIV-1 generation time in vivo. Proc. Natl. Acad. Sci. U.S.A. 96, 2187–2191. doi: 10.1073/pnas.96.5.2187
Ruchansky, D., Casado, C., Russi, J. C., Arbiza, J. R., and Lopez-Galindez, C. (2009). Identification of a new HIV type 1 circulating recombinant form (CRF38_BF1) in Uruguay. AIDS Res. Hum. Retroviruses 25, 351–356. doi: 10.1089/aid.2008.0248
Schultz, A.-K., Zhang, M., Bulla, I., Leitner, T., Korber, B., Morgenstern, B., et al. (2010). jpHMM: improving the reliability of recombination prediction in HIV-1. Nucleic Acids Res. 38, 1059–1059. doi: 10.1093/nar/gkp371
Shriner, D., Rodrigo, A. G., Nickle, D. C., and Mullins, J. I. (2004). Pervasive genomic recombination of HIV-1 in vivo. Genetics 167, 1573–1583. doi: 10.1534/genetics.103.023382
Sierra, M., Thomson, M. M., Posada, D., Pérez, L., Aragonés, C., González, Z., et al. (2007). Identification of 3 phylogenetically related HIV-1 BG intersubtype circulating recombinant forms in Cuba. J. Acquir. Immune Defic. Syndr. 45, 151–160. doi: 10.1097/qai.0b013e318046ea47
Sierra, M., Thomson, M. M., Ríos, M., Casado, G., Ojea-de Castro, R., Delgado, E., et al. (2005). The analysis of near full-length genome sequences of human immunodeficiency virus type 1 BF intersubtype recombinant viruses from Chile, Venezuela and Spain reveals their relationship to diverse lineages of recombinant viruses related to CRF12_BF. Infect. Genet. Evol. 5, 209–217. doi: 10.1016/j.meegid.2004.07.010
Simon-Loriere, E., and Holmes, E. C. (2011). Why do RNA viruses recombine? Nat. Rev. Microbiol. 9, 617–626. doi: 10.1038/nrmicro2614
Strimmer, K., and Rambaut, A. (2002). Inferring confidence sets of possibly misspecified gene trees. Proc. R. Soc. B Biol. Sci. 269, 137–142. doi: 10.1098/rspb.2001.1862
Thomson, M. M., Delgado, E., Herrero, I., Villahermosa, M. L., Vázquez-de Parga, E., Cuevas, M. T., et al. (2002). Diversity of mosaic structures and common ancestry of human immunodeficiency virus type 1 BF intersubtype recombinant viruses from Argentina revealed by analysis of near full-length genome sequences. J. Gen. Virol. 83, 107–119. doi: 10.1099/0022-1317-83-1-107
Yebra, G., de Mulder, M., Martín, L., Rodríguez, C., Labarga, P., Viciana, I., et al. (2012). Most HIV type 1 non-Binfections in the Spanish cohort of antiretroviral treatment-naïve HIV-infected patients (CoRIS) are due to recombinant viruses. J. Clin. Microbiol. 50, 407–413. doi: 10.1128/JCM.05798-11
Keywords: HIV-1, nearly-full genome, recombination, phylogeny, CRFs, URFs
Citation: Beamud B, Bracho MA and González-Candelas F (2019) Characterization of New Recombinant Forms of HIV-1 From the Comunitat Valenciana (Spain) by Phylogenetic Incongruence. Front. Microbiol. 10:1006. doi: 10.3389/fmicb.2019.01006
Received: 15 October 2018; Accepted: 18 April 2019;
Published: 22 May 2019.
Edited by:
Kok Keng Tee, University of Malaya, MalaysiaReviewed by:
Miguel Arenas, University of Vigo, SpainFederico Garcia, Hospital Universitario San Cecilio, Spain
Copyright © 2019 Beamud, Bracho and González-Candelas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fernando González-Candelas, fernando.gonzalez@uv.es