 Litao Guo
Litao Guo Chao Tang1†
Chao Tang1† Chunsheng Gao
Chunsheng Gao Yi Cheng
Yi Cheng Tuhong Wang
Tuhong Wang Jianping Xu
Jianping Xu- 1Institute of Bast Fiber Crops, Chinese Academy of Agricultural Sciences, Changsha, China
- 2Department of Biology, McMaster University, Hamilton, ON, Canada
The hemp flea beetle Psylliodes attenuata (Coleoptera: Chrysomelidae: Psylliodes) is a common pest of Cannabis sativa, including cultivars of both medicinal marijuana and industrial hemp. Both the larval and adult stages of this beetle can cause significant damages to C. sativa, resulting in substantial crop losses. At present, little is known about the bacterial and fungal community diversity among populations of this pest insect. In the present study, we obtained P. attenuata samples from nine field sites representing broad industrial hemp productions in China and analyzed their microbial communities using DNA metabarcoding. Bacterial sequences of all the samples were assigned to 3728 OTUs, which belonged to 45 phyla, 1058 genera and 1960 known species. The most common genera were Rickettsia, Wolbachia, and Candidatus_Brownia. Fungal sequences of all the samples were assigned to 910 OTUs, which belonged to 9 phyla, 308 genera and 464 known species. The most common fungal genera were Cladosporium, Cutaneotrichosporon, and Aspergillus. Principal coordinate analysis revealed a significant difference in the bacterial and fungal community structure among the nine P. attenuata populations. Understanding the microbial symbionts may provide clues to help develop potential biocontrol techniques against this pest.
Introduction
Being one of the world’s oldest crops with significant economic and medicinal importance, Cannabis sativa L. (cannabis) has attracted an enormous amount of attention and become one of the most commonly cultivated plants worldwide (Deguchi et al., 2020; Gao et al., 2020). Based on the statistics from Organization for Economic Co-operation and Development (OECD), industrial hemp is grown in more than 130 countries with 94 certified hemp varieties for large-scale productions of food, textiles and building materials. Similarly, C. sativa is of significant medical and recreational interests due to its productions of the psychoactive compound D-9-tetrahydrocannabinol (THC) and/or other cannabinoids such as cannabidiol (CBD). CBD and THC have shown pharmacological properties in the treatment of mood disorders, pain, cancer, diabetes, and inflammatory and neurodegenerative diseases (Andre et al., 2016; Irakli et al., 2019).
The hemp flea beetle, Psylliodes attenuata (Koch) 1803 (Coleoptera: Chrysomelidae: Psylliodes), is among the most common pests that can cause severe crop losses in Cannabis production areas (Goncalves et al., 2019). P. attenuata is geographically broadly distributed across Eurasia, from the United Kingdom to France, the Middle East, eastern Siberia, Japan, and China (Goncalves et al., 2019). The larvae and adult of the hemp flea beetle can damage hemp crops, with adults causing more damages than grubs. The larvae feed on hemp roots, and the adult feed on apical leaves, flowers, and seeds of hemp (McParland et al., 2000). Many flea beetles are polyphagous, but the hemp flea beetle is mostly an oligophagous insect, feeding on hemps (Zhao et al., 2018; Deguchi et al., 2020). Indeed, hemp flea beetle has been suggested as a candidate biocontrol agent for eradicating illegal marijuana growth due to its obligatory feeding on hemp (Hall, 1999).
Insect microbiota have attracted a lot of attention because they have shown to provide a diverse range of effects on insects, from nutrition, physiology, to behavior. For example, insect microbiota are known to detoxify noxious secondary metabolites and xenobiotics, such as terpenes (Berasategui et al., 2017), caffeine (Ceja-Navarro et al., 2015; Summers et al., 2015), nicotine (Brandsch, 2006), and insecticides; defending against parasites and other pathogens; generating and relaying signals among hosts; improving behavior and immunity; and producing nutrients to supplement host’s diet and to enhance their digestion (Dillon and Dillon, 2004; Ezemwa et al., 2012; Wu et al., 2016). In recent years, a growing number of studies have cataloged and characterized microbial communities, particularly in bees (Moran et al., 2012), fruit flies (Erkosar et al., 2017; Bing et al., 2018), termites (Rossmassler et al., 2015), silkworm (Chen et al., 2018), whiteflies (Santos-Garcia et al., 2020), and beetles (Kudo et al., 2018). For example, Zhang et al. (2022) found that a normal gut microbiota is required for olfactory learning and memory abilities, mainly by regulating tryptophan metabolism, with host-specific Lactobacillus strains enhancing memory by transforming tryptophan to indole derivatives that activate the host aryl hydrocarbon receptor in honeybee (Zhang et al., 2022).
In a previous study, we analyzed the genetic diversity and geographic structure of P. attenuata in hemp fields in China based on mitochondrial COI gene sequences. Our analyses revealed that geographic separation among the regional populations accounted for 58% of the total genetic variance, while within-local populations accounted for 42% of the total observed genetic variance (Guo et al., 2020). However, the significance of the observed genetic diversity and geographic differences remains unknown. In this study, we analyzed the bacterial and fungal communities associated P. attenuata to investigate whether the different geographic populations of P. attenuata contain distinct bacterial and fungal communities. In addition, we were interested in whether there were common shared bacterial and fungal species among the diverse P. attenuata populations. Commonly shared microbes among geographic diverse hosts would suggest their potential key functions to these pests. In contrast, geography-specific patterns would suggest unique microbial communities for individual geographic regions. Such discoveries could potentially help us design novel strategies to control and prevent the damaging effects of these pests on hemp crops.
To achieve our objectives, we broadly sampled hemp flea beetles in four hemp-growing regions in China where nine P. attenuata populations were collected. We then obtained data on microbial communities inside these flea beetles using high-throughput next-generation DNA sequencing. Finally, we analyzed the alpha diversity, the beta diversity, and predicted functions of the microbial community. The observed data were discussed in the context of general microbial diversity in insects, their potential roles in the biology of the hemp flea beetles, and implications for developing future control and management strategies against these pests.
Materials and methods
Sample collection and identification of Psylliodes attenuata
For this study, we broadly sampled hemp flea beetles in four hemp-growing regions in China: northeastern China (including Heilongjiang and Jilin provinces), central-east China (Shandong and Anhui provinces), south-central China (Hunan province), and southwestern China (Yunnan province). Nine sites within the four regions were sampled and the site information is presented in Figure 1 and Table 1 (Guo et al., 2020). The collected adult beetles were soaked in 99.7% anhydrous alcohol, and then surface-cleaned with PBS buffer for three times, placed in a sterile centrifuge tube, frozen with liquid nitrogen for more than 1h and stored at −80°C. These beetles were identified as hemp flea beetles based on their morphological features and mitochondrial COI sequences (McParland et al., 2000; Guo et al., 2020). At each of the nine sites, we obtained 20 flea beetles for analyses, with each flea beetle collected from a different hemp plant. The 20 flea beetles from each site were randomly grouped into four samples as biological repeats, with each sample containing five beetles to ensure that sufficient microbial DNA could be extracted for analyses from each sample. A total of 36 samples were collected and analyzed in this study.
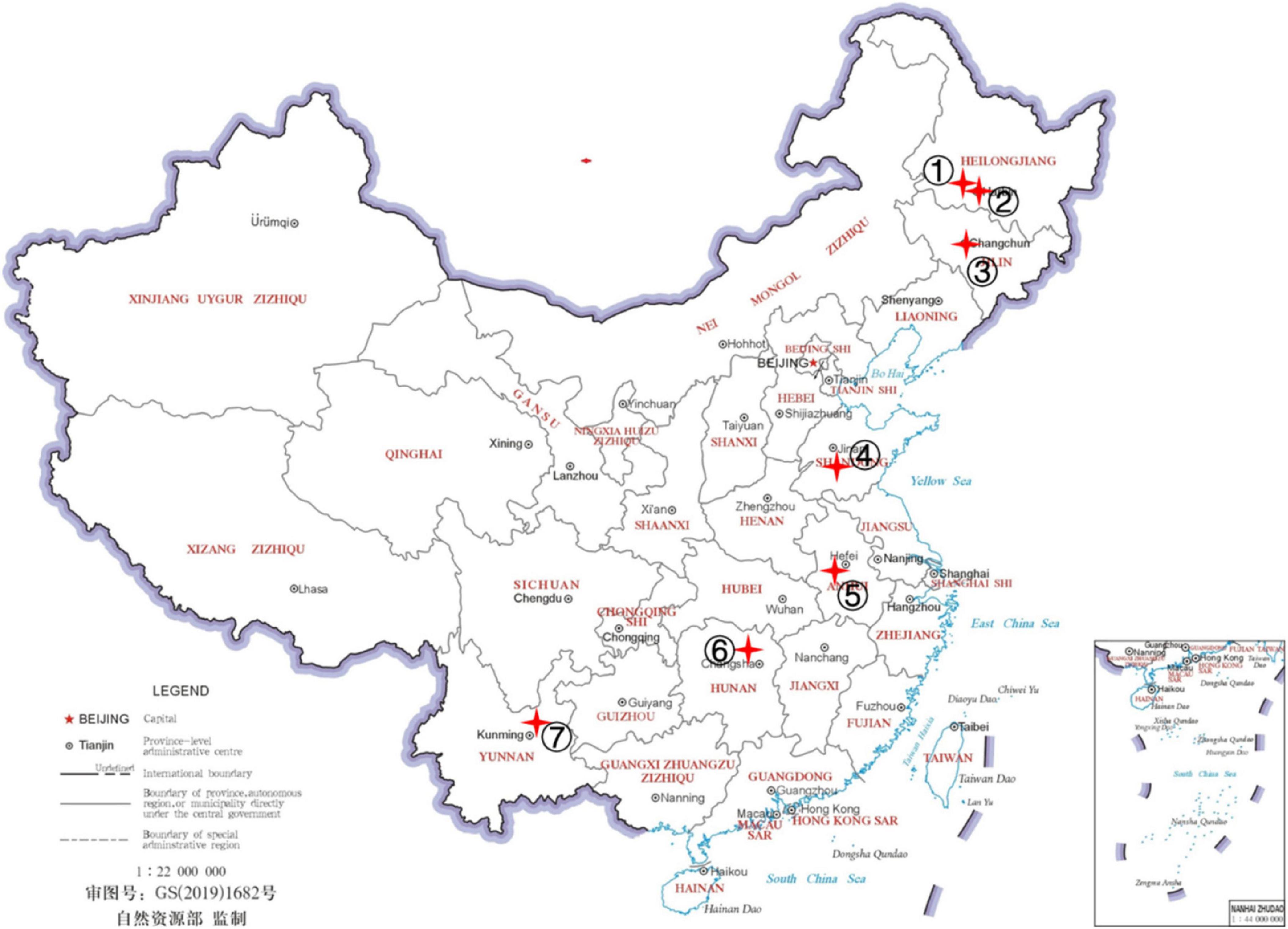
Figure 1. Collection localities for Psylliodes attenuata. The map was downloaded from http://bzdt.ch.mnr.gov.cn. The locations of sampling sites are noted with numbers and red cross on the map. The details of P. attenuata collections were shown in Table 1.
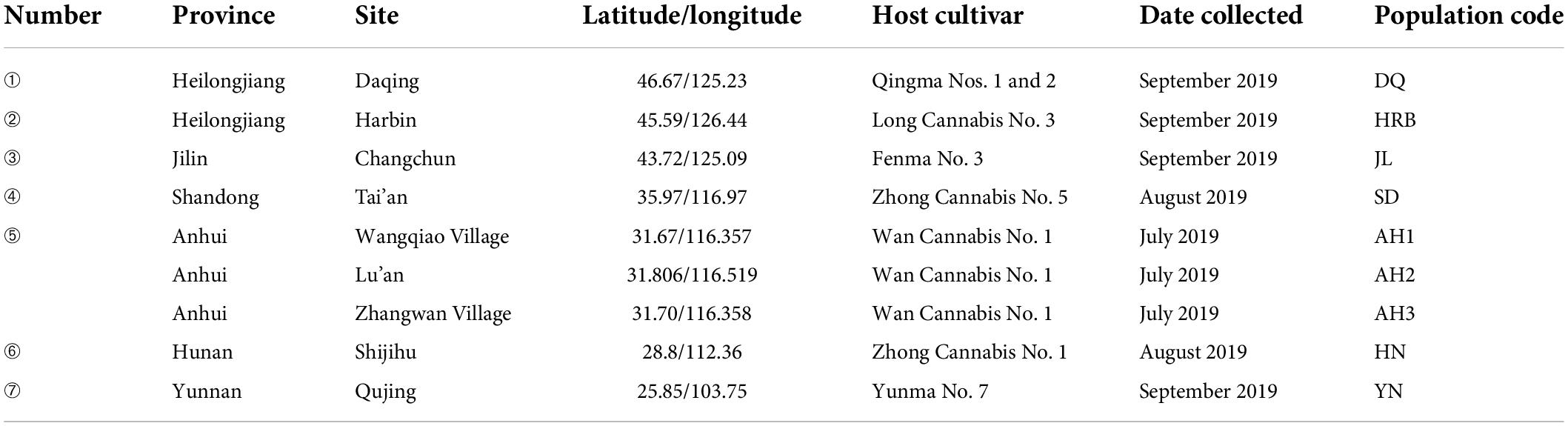
Table 1. The details of Psylliodes attenuata collections.
DNA extraction and PCR Amplification
Microbial community genomic DNA was extracted from each of four samples at each of the nine sites using the E.Z.N.A.® soil DNA Kit (Omega Bio-Tek, Norcross, GA, United States) according to manufacturer’s instructions. The DNA extract was checked on 1% agarose gel, and DNA concentration and purity were determined with NanoDrop 2000 UV-vis spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, United States). The 16S ribosomal RNA (rRNA) gene spanning hypervariable regions (V3−V4) and fungal internal transcribed spacer 1 (ITS1) region were respectively amplified using the following primer pairs: 338F/806R (5′-ACTCCTACGGGAGGCAGCAG-3′ and 5′-GGACTACHVGGGTWTCTAAT-3′) and ITS1F/ITS2R (5′-CTTGGTCATTTAGAGGAAGTAA-3′ and 5′-GCTGCGTTCTTCATCGATGC-3′) by an ABI GeneAmp 9700 PCR thermocycler (ABI, CA, United States). The PCR amplification of 16S rRNA/ITS1 DNA fragments was performed as follows: initial denaturation at 95°C for 3 min, followed by 28/35 cycles of denaturing at 95°C for 30 s, annealing at 55°C for 30 s and extension at 72°C for 45 s, and single extension at 72°C for 10 min, and end at 10°C. Each PCR mixture contained 5 × TransStart FastPfu buffer 4 μL, 2.5 mM dNTPs 2 μL, forward primer (5 μM) 0.8 μL, reverse primer (5 μM) 0.8 μL, TransStart FastPfu DNA Polymerase 0.4 μL, BSA 0.2 μL, template DNA 10 ng, and finally ddH2O up to 20 μL. PCR reactions were performed in triplicate. The PCR products were extracted from 2% agarose gel and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, United States) according to manufacturer′s instructions and quantified using Quantus™ Fluorometer (Promega, United States).
Illumina MiSeq sequencing
For both the amplified 16S rRNA and ITS1 sequences, the purified amplicons from each of the 36 samples were separately barcoded and the 36 barcoded samples were pooled in equimolar and paired-end sequenced on an Illumina MiSeq PE300 platform/NovaSeq PE250 platform (Illumina, San Diego, CA, United States) according to the standard protocols by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China). The raw reads were deposited into the NCBI Sequence Read Archive (SRA) database (BioProject Number: PRJNA843905; sequence accession numbers SRR19449852-SRR19449887).
Processing of sequencing data and statistical analysis
The raw 16S rRNA and ITS1 sequencing reads were demultiplexed, quality-filtered by fastp version 0.20.0 (Chen et al., 2015) and merged by FLASH version 1.2.7 (Magoč and Salzberg, 2011) with the following criteria. First, the 300 bp reads were truncated at any site receiving an average quality score of <20 over a 50 bp sliding window, and the truncated reads shorter than 50 bp were discarded, reads containing ambiguous nucleotide bases were also discarded; (ii) only overlapping sequences longer than 10 bp were assembled according to their overlapped sequence. The maximum mismatch ratio of overlap region was set at 0.2. Reads that could not be assembled were discarded; (iii) Samples were distinguished and sorted according to their barcode and primers.
The data were analyzed on the free online platform of Majorbio Cloud Platform1. Operational taxonomic units (OTUs) with 97% similarity cutoff (Stackebrandt and Goebel, 1994; Edgar, 2013; Xu, 2016, 2020) were identified and clustered using UPARSE version 7.1 (Edgar, 2013), and chimeric sequences were identified and removed. The taxonomy of each OTU representative sequence was analyzed by RDP Classifier version 2.11 (Wang et al., 2007) against the 16S rRNA database (Silva v138) and ITS database (Unite v8.0) (Kõljalg et al., 2013) using confidence threshold of 0.7. The curated OTU data in QIIME were used to calculate the alpha diversity indices by Mothur version 1.30.2, including Sobs, Chao, Ace estimators, Shannon and Simpson indices, respectively. Values were compared using one-way ANOVA Tukey test among samples, P < 0.05 was considered statistically significant. These analyses were performed with the Statistical Package for the Social Sciences, version 20.0 (SPSS, Chicago, United States). Bray–Curtis distance matrices were constructed using rarefied OTU abundance table and visualized in principal coordinate analysis (PCoA). Taxonomic heat maps were clustered by the average linkage method with the Bray–Curtis distance and generated by the R package. Both the total OTU count data and rarefied data were used for comparisons among samples. The functional of the bacterial and fungal community was respectively predicted by PICRUSt2 and FUNGuild.
Results
Comparison of bacterial communities across all samples
The composition of the bacterial communities within and among the nine geographic populations of the hemp flea beetle P. attenuata in China was investigated by high-throughput metabarcode DNA sequencing. Our sequencing generated a total of 1,887,854 reads of bacterial 16S rRNA genes with an average length of 412 bp. In total, 3728 bacterial OTUs were identified among the nine geographic populations. These bacterial OTUs belonged to 45 phyla, 1058 genera, and 1960 species (Table 2 and Supplementary Table 1). After rarefaction according to the lowest sample sequence number, 2933 OTUs were clustered, attributed to 42 phyla and 970 genera and 1751 species for the nine P. attenuata populations (Table 2 and Supplementary Table 2). Rarefaction curves from both the original sequencing data and rarefied data sets showed that the curves of all 36 samples approached asymptote, indicating that the amount of sequencing data was sufficiently representative of the microbial diversity within each of the 36 samples (Supplementary Figure 1).
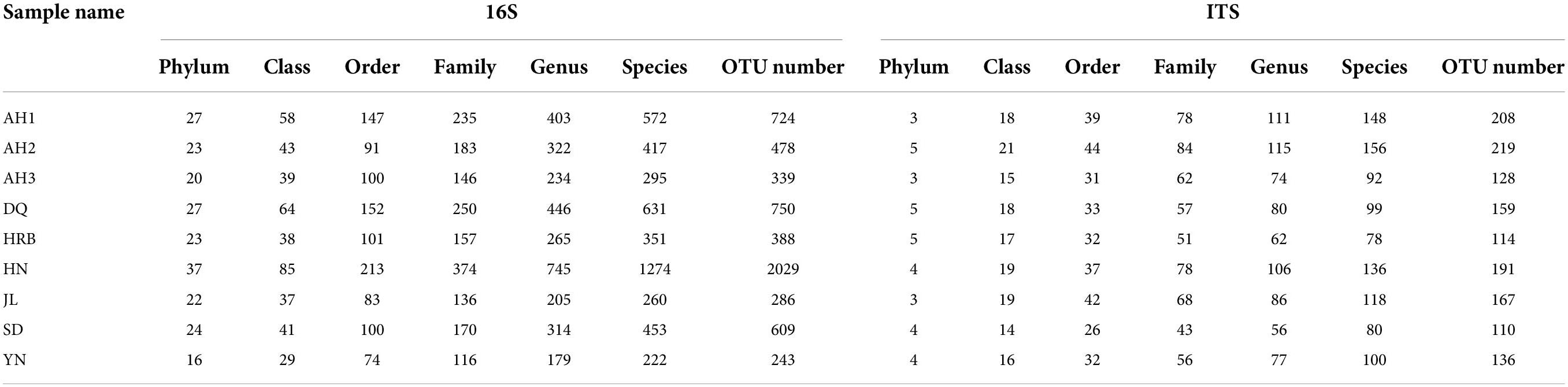
Table 2. Summary of the 16S ribosomal RNA (rRNA) and internal transcribed spacer (ITS) read counts of bacteria across nine geographic populations of the hemp flea beetle P. attenuata in China.
There were significant differences in bacterial species richness (observed OTUs) and community diversity (Shannon) with Alpha-diversity analyses between the HN and the other eight geographic populations (including AH1, AH2, AH3, DQ, HRB, JL, SD, and YN) (Figures 2A,B and Tables 2, 3). In addition, species richness of the HN population was the highest among the nine geographic populations (Figure 2A). At the phylum level, Proteobacteria and Bacteroidetes dominated the bacterial community across all samples but HN (Supplementary Figure 2). At the genus level, OTUs of the AH1 population were mainly annotated to Rickettsia (82.2%) and Candidatus_Brownia (7.04%); OTUs of AH2 were mainly annotated to Rickettsia (76.42%), Cutibacterium (4.67%), and Rhodococcus (4.5%); OTUs of AH3 were mainly annotated to Rickettsia (83.86%) and Candidatus_Brownia (9.81%); OTUs of SD were mainly annotated to Rickettsia (36.2%), Pantoea (33.21%), and Prevotella_9 (13.11%); OTUs of DQ were mainly annotated to Wolbachia (35.73%), Candidatus_Brownia (30.48%), and Enterobacter (13.45%); OTUs of HRB were mainly annotated to Wolbachia (53.12%), Candidatus_Brownia (33.76%), and Rickettsia (6.69%); OTUs of JL were mainly annotated to Rickettsia (21.55%), Enterobacter (20.69%), Lactococcus (20.64%), Wolbachia (18.56%), and Candidatus_Brownia (9.34%); OTUs of YN were mainly annotated to Wolbachia (62.55%), Rickettsia (21.41%), and Candidatus_Brownia (12.81%) (Figure 2C). The similarities and differences in composition and structure of the bacterial communities among all geographic samples were revealed by Principal components analysis (PCoA). PCoA separated most field populations (ADONIS test with 999 permutations, P < 0.05) except the SD, JL, and DQ samples (Figure 2D).
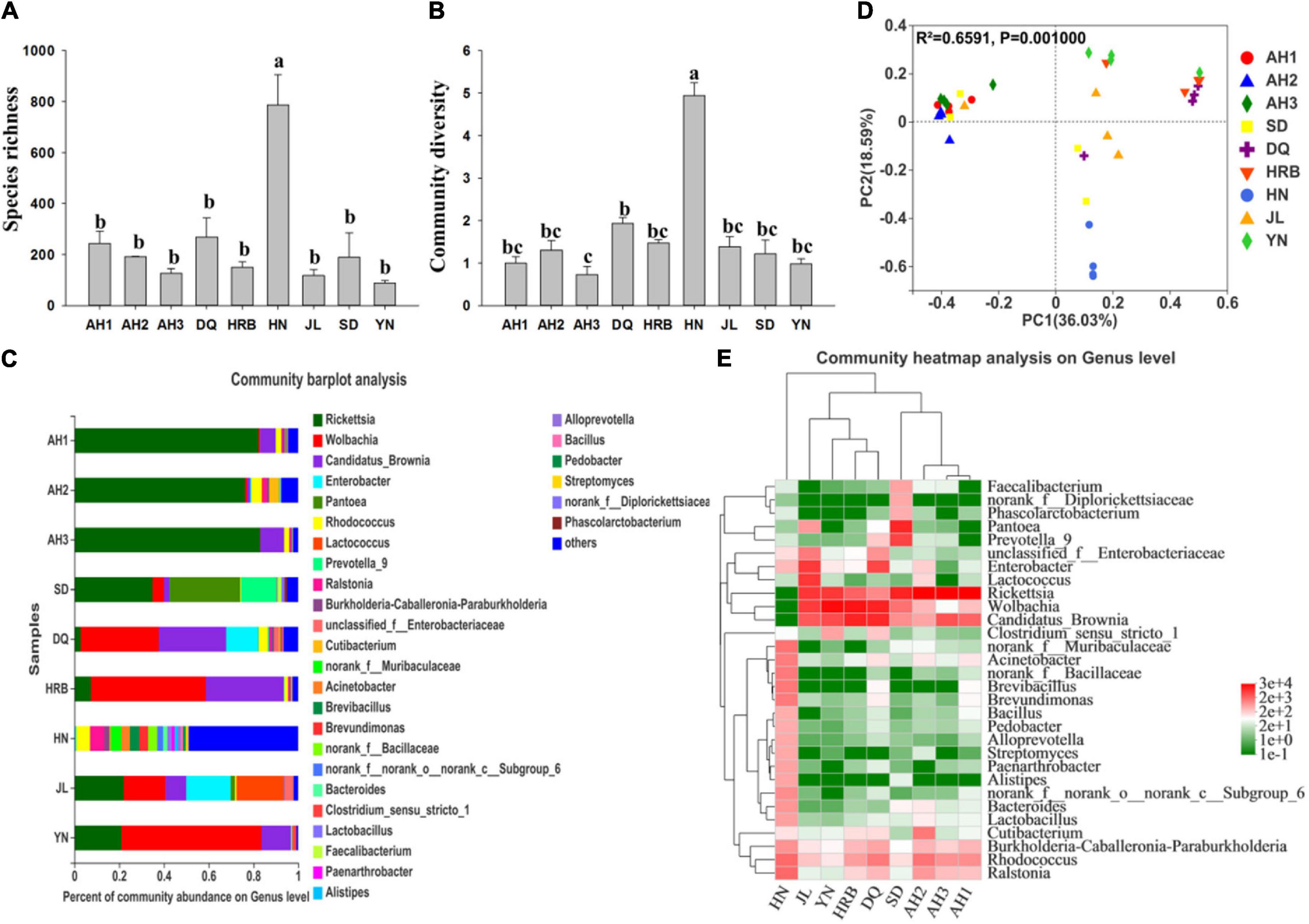
Figure 2. Bacterial community composition within and among nine geographic populations of the hemp flea beetle P. attenuata in China. (A) Histogram of species richness (number of OTUs). (B) Histogram of community diversity measured by Shannon index. Letters above each population indicate significant differences (one-way ANOVA, Tukey test, P < 0.05, see Table 3) in the mean values. (C) Relative abundance of bacterial genera in different populations. (D) Principal components analysis (PCoA) plots of bacterial communities based on the Bray-Curtis distance. Each symbol or color represents a sample. The significant differences in beta diversities were analyzed using Adonis analysis with 999 permutations, P < 0.05. (E) Heatmap showing the relative abundance of top thirty bacterial genera. Hierarchical cluster analysis was based on the Bray-Curtis distance with average method.
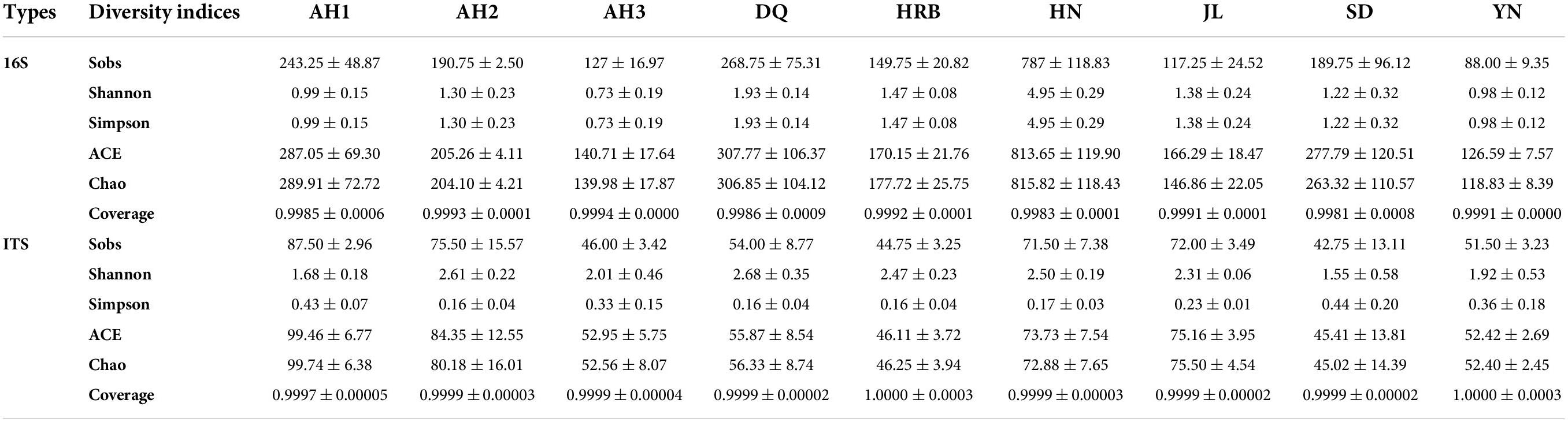
Table 3. The diversity indices of bacteria and fungi based on 16S rRNA and ITS sequences respectively across nine geographic populations of the hemp flea beetle P. attenuata in China.
We selected the bacterial genera with the top thirty abundance ratios and drew heat maps based on their relative abundances in different geographic populations (Figure 2E). From the overall distribution of the microbial community at different populations, Rickettsia, Wolbachia, and Candidatus_Brownia were the dominant genera (Figure 2E and Supplementary Figure 3) across most of the samples except the HN samples. Pantoea (33.21%) was abundant in SD, but the numbers were relatively small in the other populations. The abundance ratio of Lactococcus in JL was significantly higher than those in the other populations (Figure 2E). Overall, the bacterial communities from the three AH populations (AH1, AH2, and AH3 samples) were clustered together, with the SD sample being most similar to them among the non-AH samples. HRB, DQ, YN and JL populations clustered into a different group, while the HN populations were distinctly different. There was no genus in the HN population exceeding 6.30%.
Abundance of bacteria across Samples
Figure 2 shows the bacterial community overlap among the nine geographic regions of the hemp flea beetle P. attenuata in China at different taxonomic levels. At all taxonomic levels, the HN population had the highest richness (Figure 3). At the OTU level, there were 54 shared OTUs among the nine geographic regions, and the unique OTUs of AH1, AH2, AH3, SD, DQ, HRB, HN, JL, and YN were respectively 127, 109, 55, 155, 115, 74, 1040, 30, and 29 (Figure 3). At other taxonomic levels, there were 13 shared Phyla, 19 shared Classes, 39 shared Orders, 56 shared Families, 57 shared Genera, 56 shared known species, and 54 shared OTUs among the nine geographic regions (Figure 3).
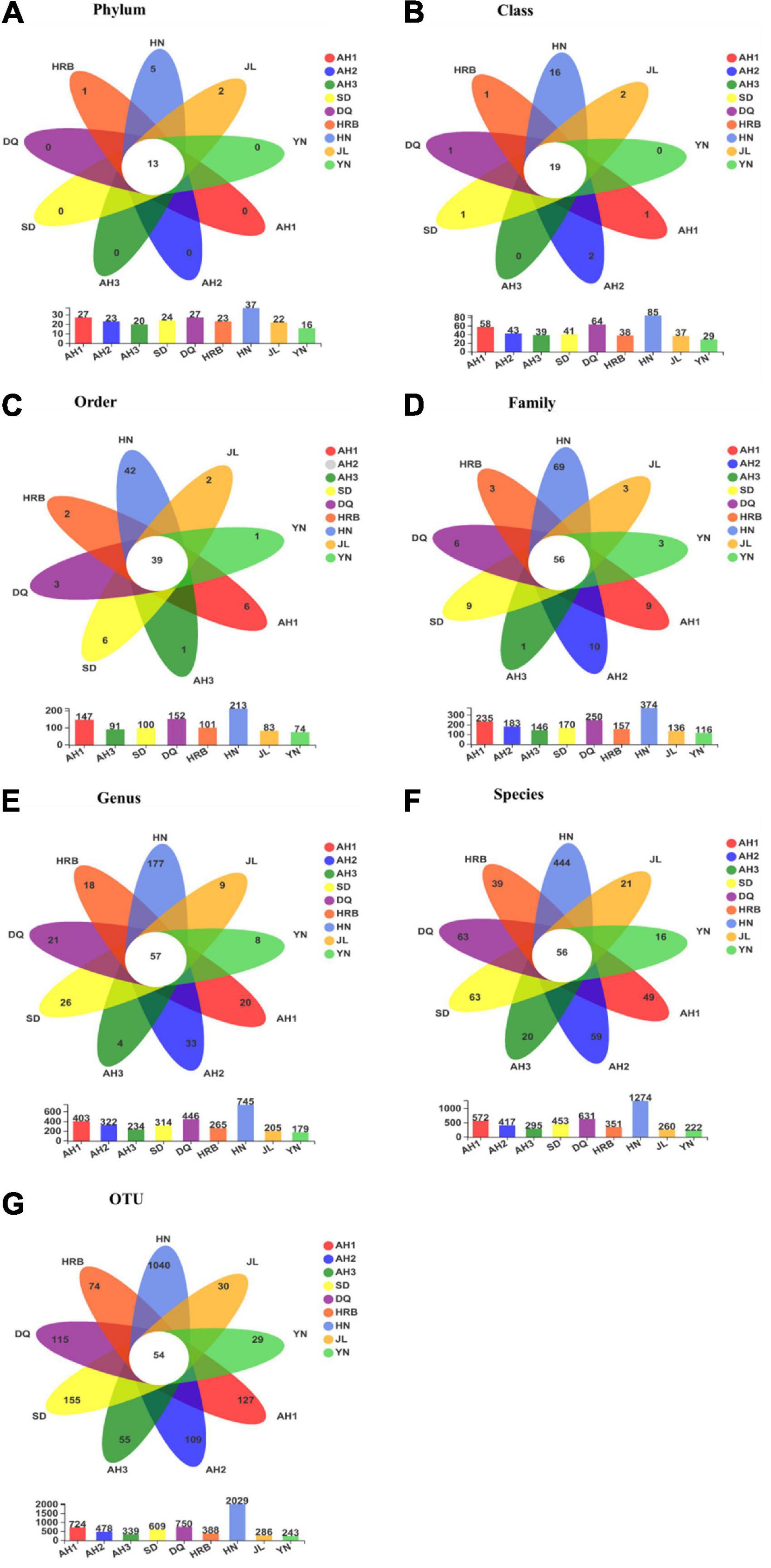
Figure 3. (A–G) Overlap among bacterial communities at different taxonomic levels among the nine geographic populations of the hemp flea beetle P. attenuata in China, with the number of species unique to each population in the petal and the number of species common to all populations in the center. Below is a bar chart of the total number of species in each population at selected taxonomic level.
Bacterial community dissimilarity across samples
According to the UPGMA analyses, there were three geography-specific bacterial community clusters. The bacterial communities in AH1, AH2, and AH3 samples from Anhui were very similar to each other (Figure 4, group A). Similarly, the four samples from Yunnan (Figure 4, group C) and four samples from Hunan (Figure 4, group D) were respectively clustered together based on their geographic locations. However, for samples from the northeast (Heilongjiang and Jilin) and eastern China (Shandong), their samples were inter-mixed. For example, three samples each from HRB and DQ were clustered in group B, but the remaining sample from HRB and DQ (one each from these two regions) were clustered with those from other geographic areas (Figure 4). Heatmap analyses showed similar results. Groups A, B, C, D, and E described in the UPGMA graph were similarly found in the heat map. Within groups A, B, and C, the sample distances at the OTU level were close except DQ_3. In contrast, within group D (i.e., four samples within the HN region), the distances among the four biological replicates at the OTU level were far apart from each other (Figure 4).
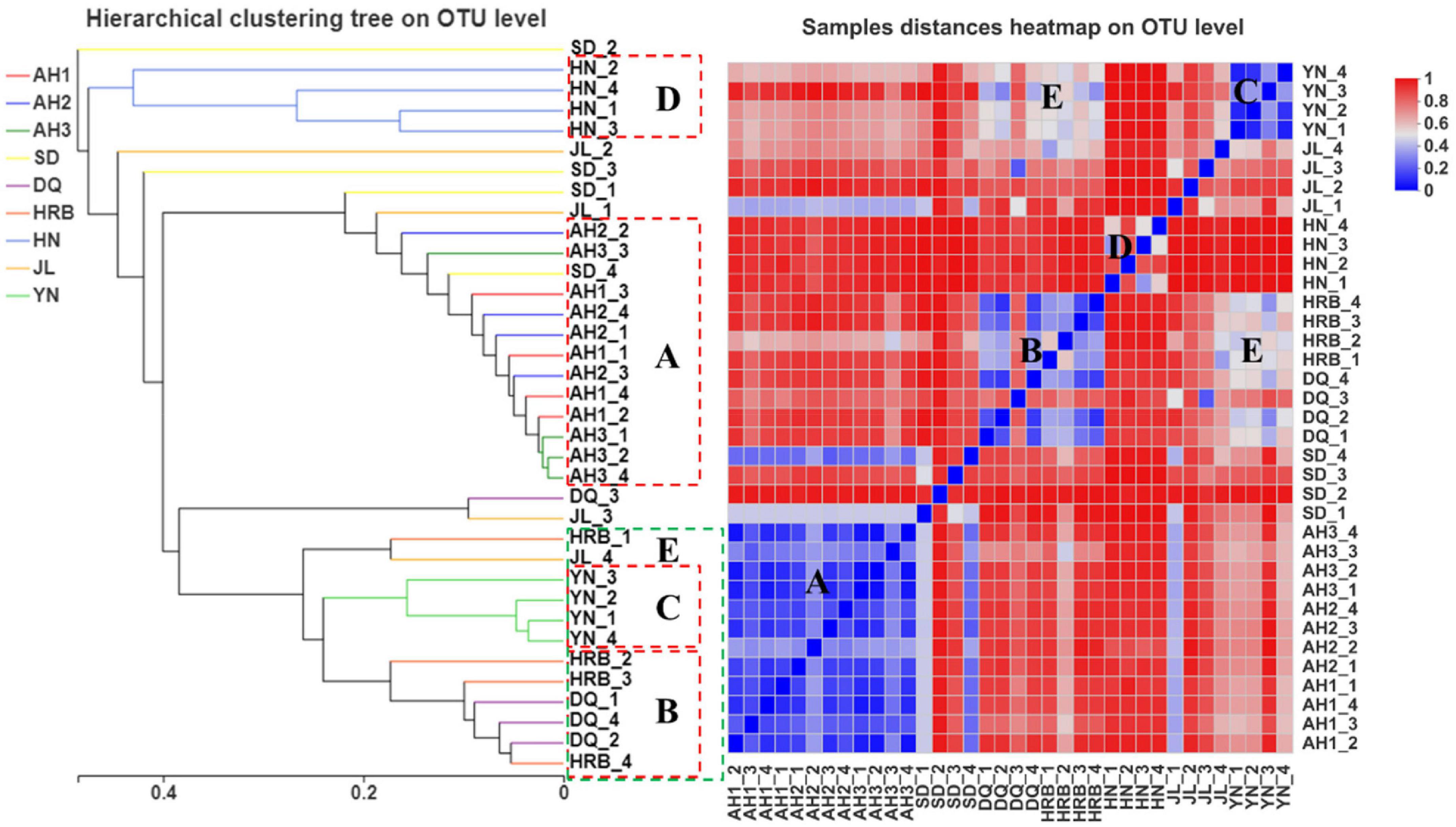
Figure 4. Bacterial community dissimilarity among samples from nine geographic populations of the hemp flea beetle P. attenuata in China. The picture on the left: analysis of unweighted pair-group method with arithmetic mean (UPGMA) based on hierarchical clustering at the OTU level. The picture on the right: sample distance heatmap analysis at the OTU level.
Functional prediction analysis of bacterial communities
To understand the potential roles of the bacterial community at the nine local populations of the hemp flea beetle P. attenuata in China, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and the Cluster of Orthologous Groups (COG) functions were predicted (Figure 5). We found that the highest relative abundance of KEGG pathways at level 3 prediction was “metabolic pathways,” “biosynthesis of secondary metabolites,” and “microbial metabolism in diverse environments” (Figure 5A). The relative abundances of “xenobiotics biodegradation and metabolism,” “metabolism of terpenoids,” and “polyketides metabolic pathways” at level 2 pathway predictions, which belong to the “general metabolism” in level 1 pathway prediction were overall high (Figure 5B). The relative abundance of predicted COG functions was shown in Figure 5C. The functional category with the highest abundance was “function unknown.” The main metabolism functional features included translation, ribosomal structure and biogenesis, energy production and conversion, cell wall/membrane/envelope biogenesis, replication, recombination and repair.
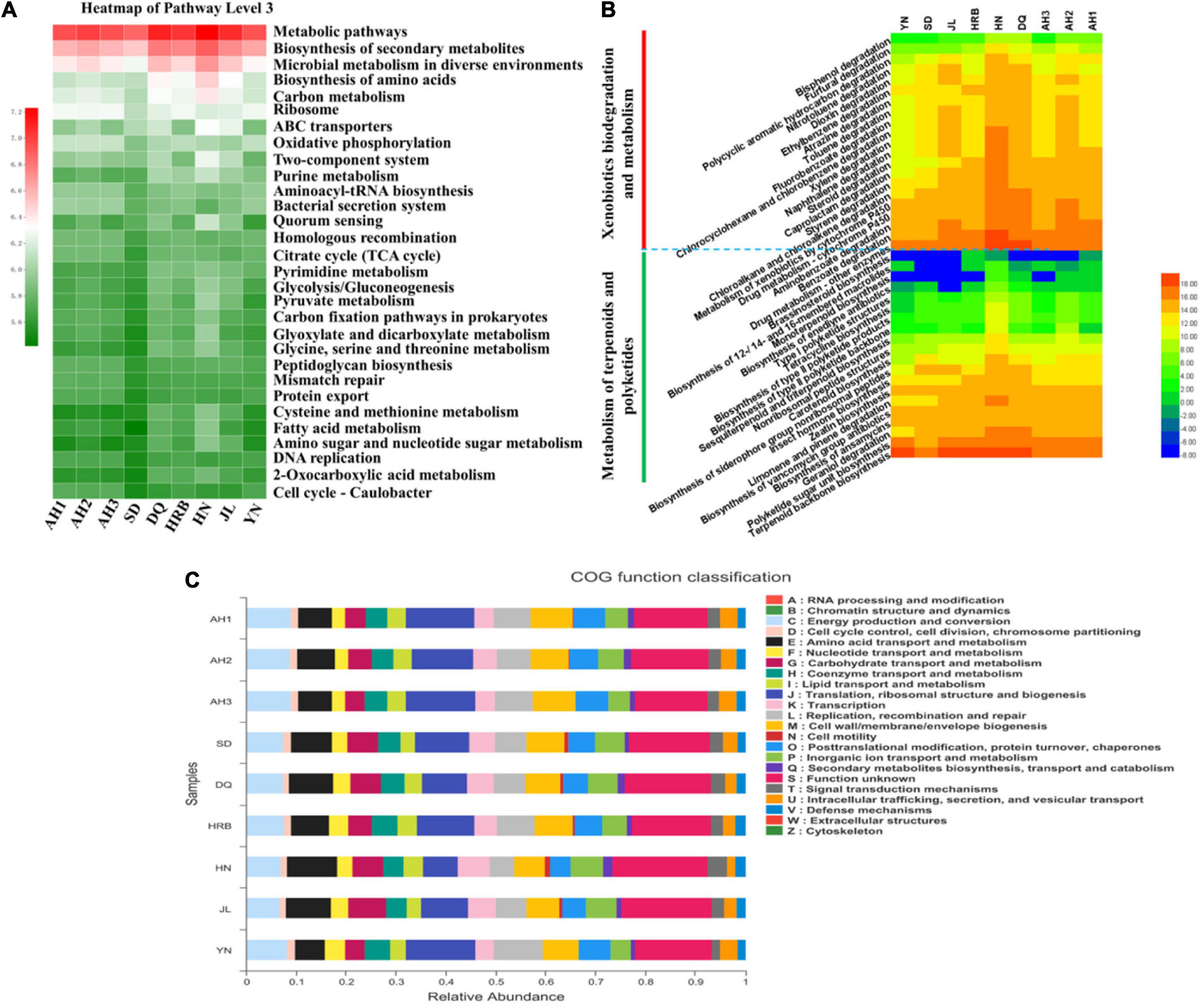
Figure 5. Comparison of predicted KEGG pathways and COG functions among nine geographic populations of the hemp flea beetle P. attenuata in China by PICRUSt2. (A) The relative abundance of KEGG pathways prediction based on level 3. (B) The relative abundance of metabolism pathways. The inferred metabolic pathways are shown with xenobiotics biodegradation and metabolism and metabolism of terpenoids and polyketides in the pathway level 2, which belong to the metabolism in pathway level 1. (C) The relative abundance of predicted COG functions.
Comparison of fungal communities
The composition of the fungal communities within and among the nine geographic populations of the hemp flea beetle P. attenuata in China was investigated by high-throughput metabarcoding ITS1 sequencing. The finalized ITS sequencing data of the total sample contained 2,458,552 reads with an average length of 228 bp. In total, 910 OTUs were identified. These OTUs belonged to 9 phyla, 308 genera and 464 species (Table 2 and Supplementary Table 3). After rarefaction to the sample with the lowest number of sequence reads, 746 OTUs were clustered, attributed to 9 phyla and 277 genera and 410 known species for the nine P. attenuata populations (Table 2 and Supplementary Table 4). Rarefaction curves showed that all 36 samples approached asymptote, consistent with sufficient sequencing depths for each of the 36 samples analyzed here for fungal diversity (Supplementary Figure 4).
There were significant differences in fungal species richness (observed OTUs) between AH1 and AH3, HRB, SD (Figure 6A and Table 2). There was no significant difference in fungal community diversity (Shannon) based on Alpha-diversity analyses among the nine local populations of the hemp flea beetle P. attenuata in China (Figure 6B and Table 3). At the phylum level, Ascomycota and Basidiomycota were found to constitute the main members of the fungal community (Supplementary Figure 5). At the genus level, OTUs of AH1 were mainly annotated to Cutaneotrichosporon (64.86%) and Fusarium (9.80%); OTUs of AH2 were mainly annotated to Cutaneotrichosporon (16.54%), Aspergillus (15.57%), and Nigrospora (5.36%); OTUs of AH3 were mainly annotated to Aspergillus (23.88%) and Alternaria (9.65%); OTUs of SD were mainly annotated to unclassified_f__Ophiocordycipitaceae (41.79%) and Latorua (9.42%); OTUs of DQ were mainly annotated to Cladosporium (16.35%), Candida (14.31%), and Cutaneotrichosporon (9.18%); OTUs of HRB were mainly annotated to Cladosporium (12.86%), Cutaneotrichosporon (12.52%), unclassified_f__Didymellaceae (11.26%), unclassified_o__Hypocreales (16.52%), and Filobasidium (12.42%); OTUs of JL were mainly annotated to Cladosporium (44.79%) and Metarhizium (9.09%); and OTUs of YN were mainly annotated to Cladosporium (27.93%), Purpureocillium (28.65%), and Filobasidium (11.9%) (Figure 6C). The similarities and differences in composition and structure of the fungal communities of all samples were investigated by Principal components analysis (PCoA). PCoA separated most field populations (ADONIS test with 999 permutations, P < 0.05) but not the DQ, HN, HRB, and YN (Figure 6D). We selected the fungal genera with the top thirty abundance ratios and drew heat maps based on their relative abundance within different populations (Figure 6E). From the overall distribution of the fungal community at different populations, Cladosporium, Cutaneotrichosporon and Aspergillus were the dominant genera (Figure 6E and Supplementary Figure 6) across most of the samples but not the AH3 and SD populations. Unclassified_f__Ophiocordycipitaceae (41.79%) was abundant in SD, relatively rare in other populations. The abundance ratio of Purpureocillium in YN was significantly higher than those of the other populations (Figure 6E and Supplementary Figure 6).
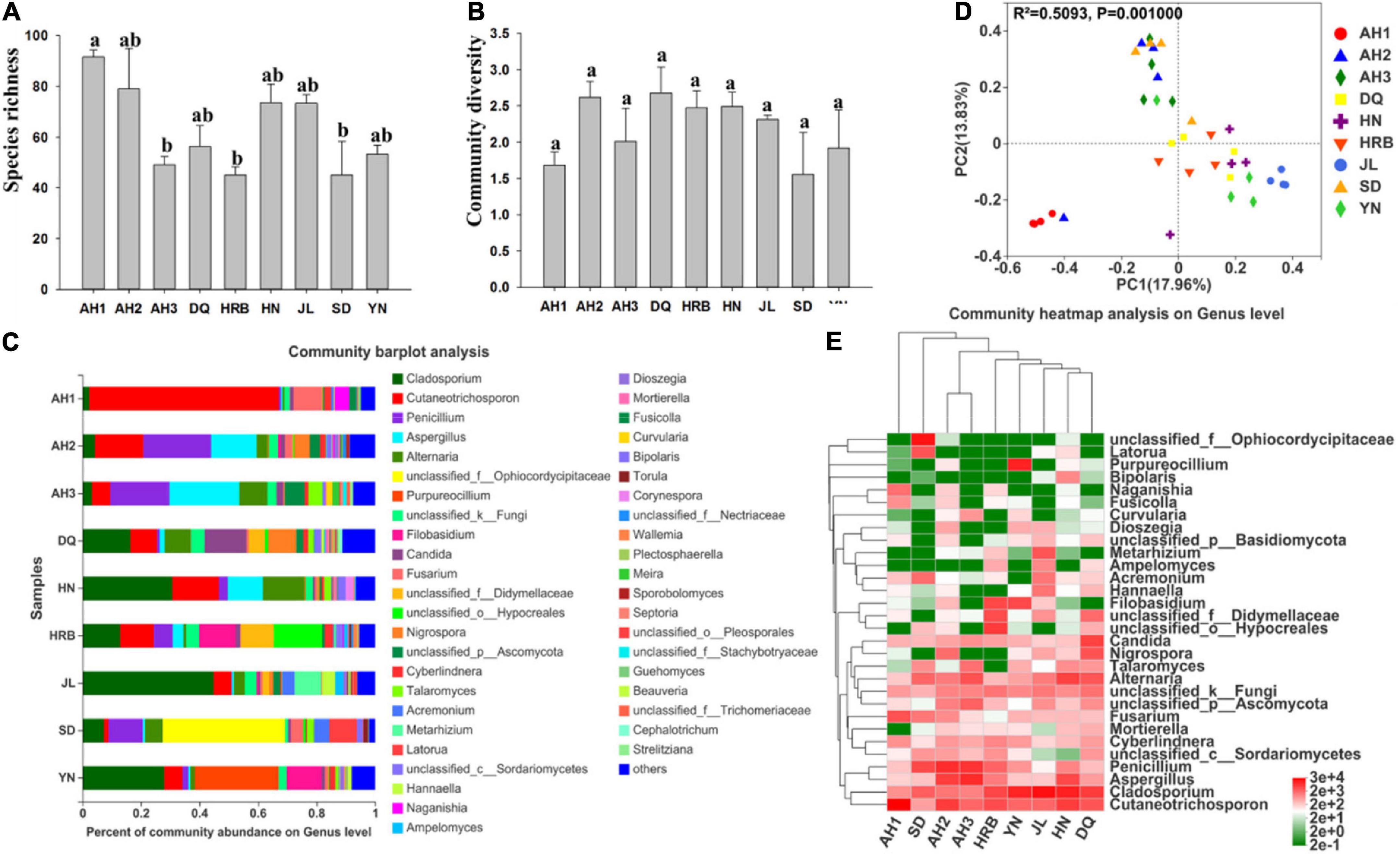
Figure 6. Fungal community composition within and among the nine geographic populations of the hemp flea beetle P. attenuata in China. (A) Histogram of species richness (number of OTUs). (B) Histogram of community diversity measured by Shannon index. Letters above each population indicate significant differences (one-way ANOVA, Tukey test, P < 0.05, see Table 3) in the mean values. (C) Relative abundance of fungal genera in different populations. (D) Principal components analysis (PCoA) plots of fungal communities based on the Bray-Curtis distance. Each symbol or color represents a sample. The significant differences in beta diversities were analyzed using Adonis analysis with 999 permutations, P < 0.05. (E) Heatmap showing the relative abundance of top thirty genera. Hierarchical cluster analysis was based on the Bray – Curtis distance with average method.
Abundance of fungi across samples
Fungal community overlap at different taxonomic levels among the nine local populations of the hemp flea beetle P. attenuata in China is shown in Figure 6. At the OTU level, the AH2 population has the highest fungal species richness (Figure 7). Overall, there were 13 shared fungal OTUs among all geographic samples, and the unique number of OTUs of the AH1, AH2, AH3, DQ, HN, HRB, JL, SD, and YN were respectively 67, 73, 46, 59, 63, 42, 54, 39, and 50 (Figure 7). At the other taxonomic levels, there were 10 shared Classes, 12 shared Orders, 14 shared Families, 12 shared Genera, and 16 shared known species among the nine geographic samples (Figure 7).
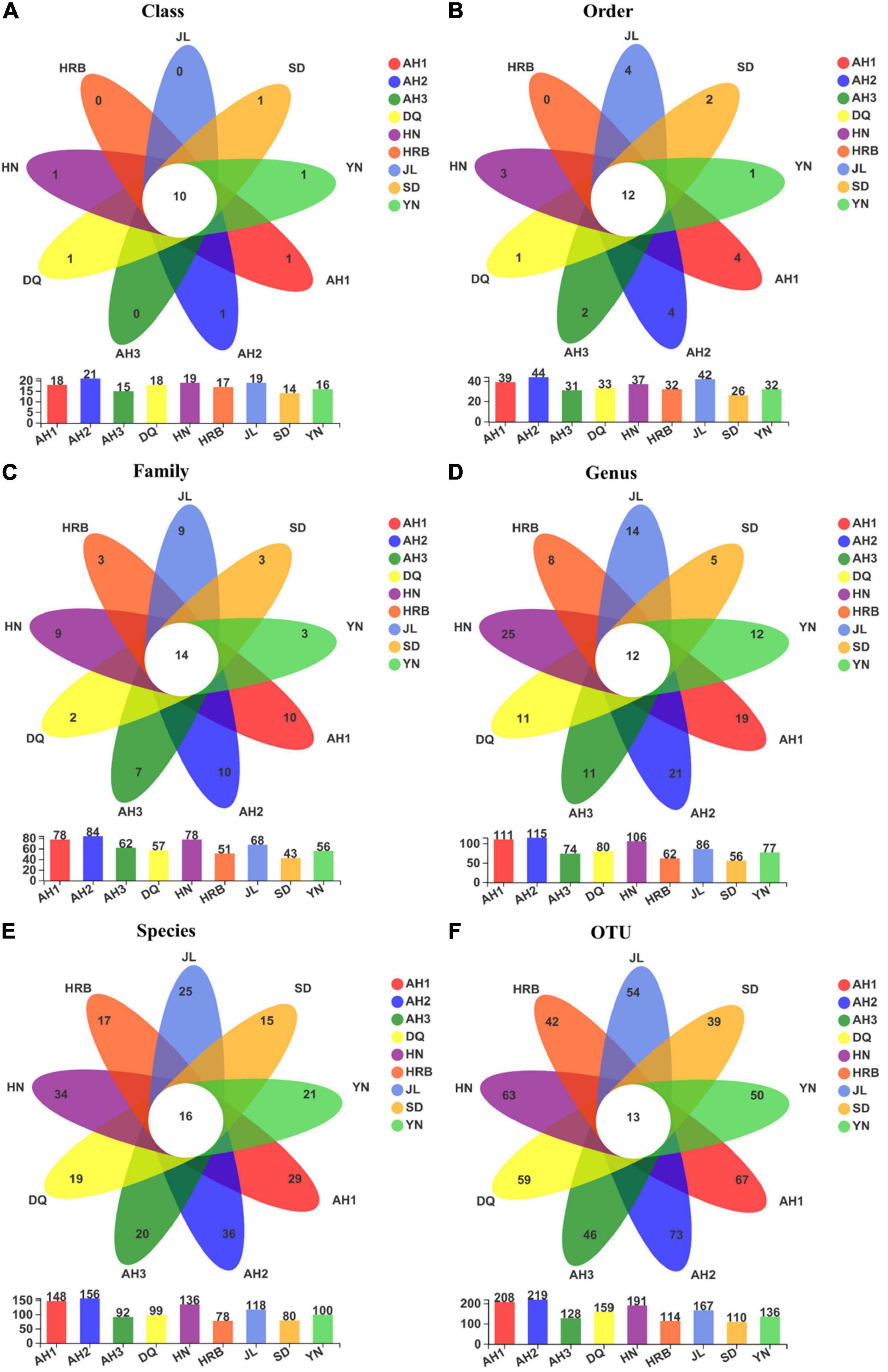
Figure 7. (A–F) Overlap in fungal community composition at different taxonomic levels among the nine geographic populations of the hemp flea beetle P. attenuata in China, with the number of fungi unique to each population in the petal and the number of fungi common to all populations in the center. Below is a bar chart of the total number of species in each population at selected taxonomic level.
Fungal community dissimilarity across samples
According to the UPGMA analyses, replicate samples within three of the nine geographic regions were clustered according to their geographic origins. Those included the AH1, JL, and HN samples respectively (Figure 8). For replicate samples from other six geographic locations, they were not uniformly clustered together based on their geographic locations. Instead, in most cases, 2−3 of the four samples from each location were clustered together while the remaining 1−2 showed more similar fungal assembly to samples from other geographic regions. Heatmap analyses showed a similar pattern as that shown on the UPGMA graph (Figure 8).
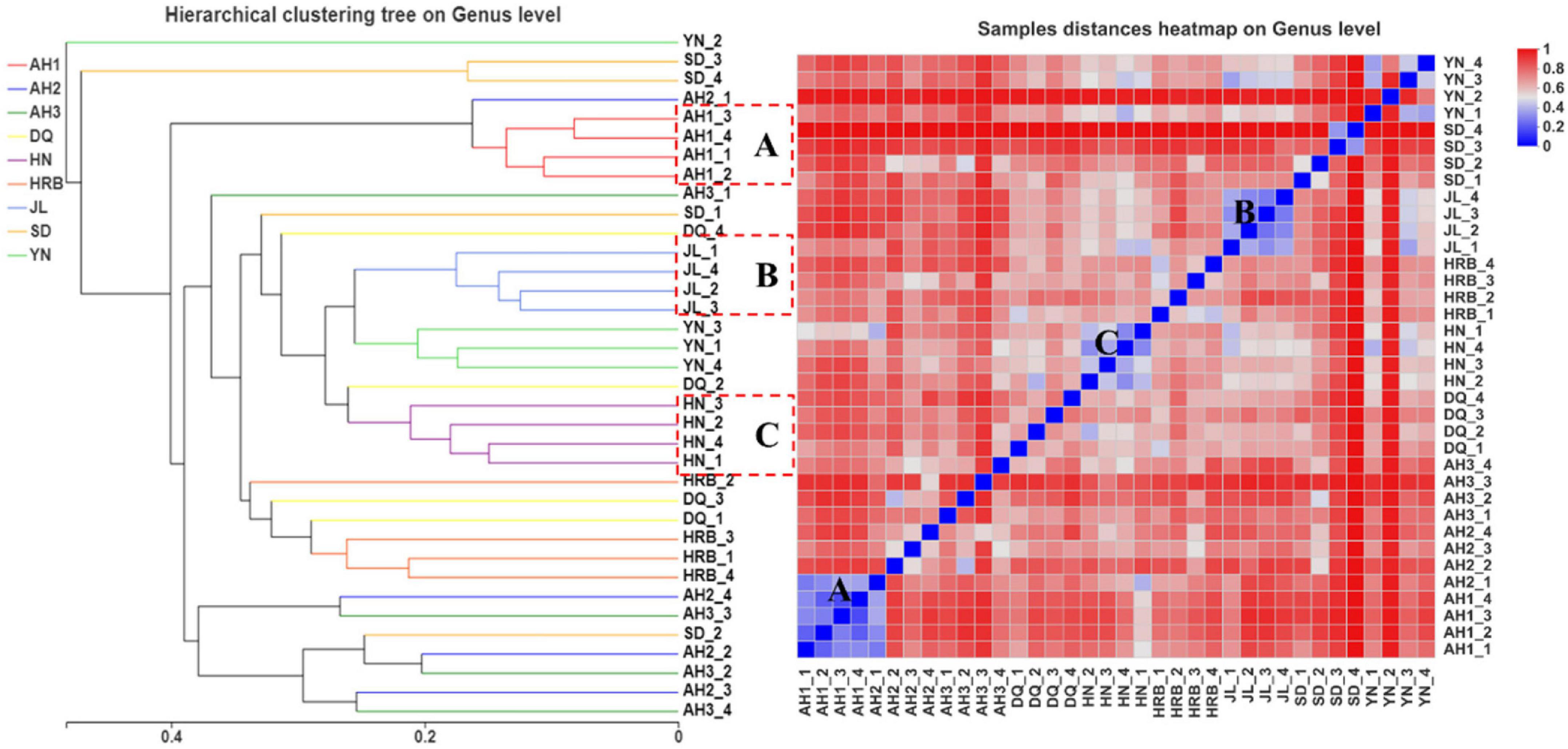
Figure 8. UPGMA (left) and heatmap (right) analyses of the fungal community in the nine geographic populations of the hemp flea beetle P. attenuata in China.
Functional predictive analysis of fungal communities
To understand the potential roles of the fungal community among the nine local populations of the hemp flea beetle P. attenuata in China, putative fungal functions were predicted based on FUNGuild (Figure 9). The main fungal functional features included (i) animal pathogen-endophyte-lichen parasite-plant pathogen-wood saprotroph, (ii) undefined saprotroph, (iii) animal pathogen, and (iv) unknown.
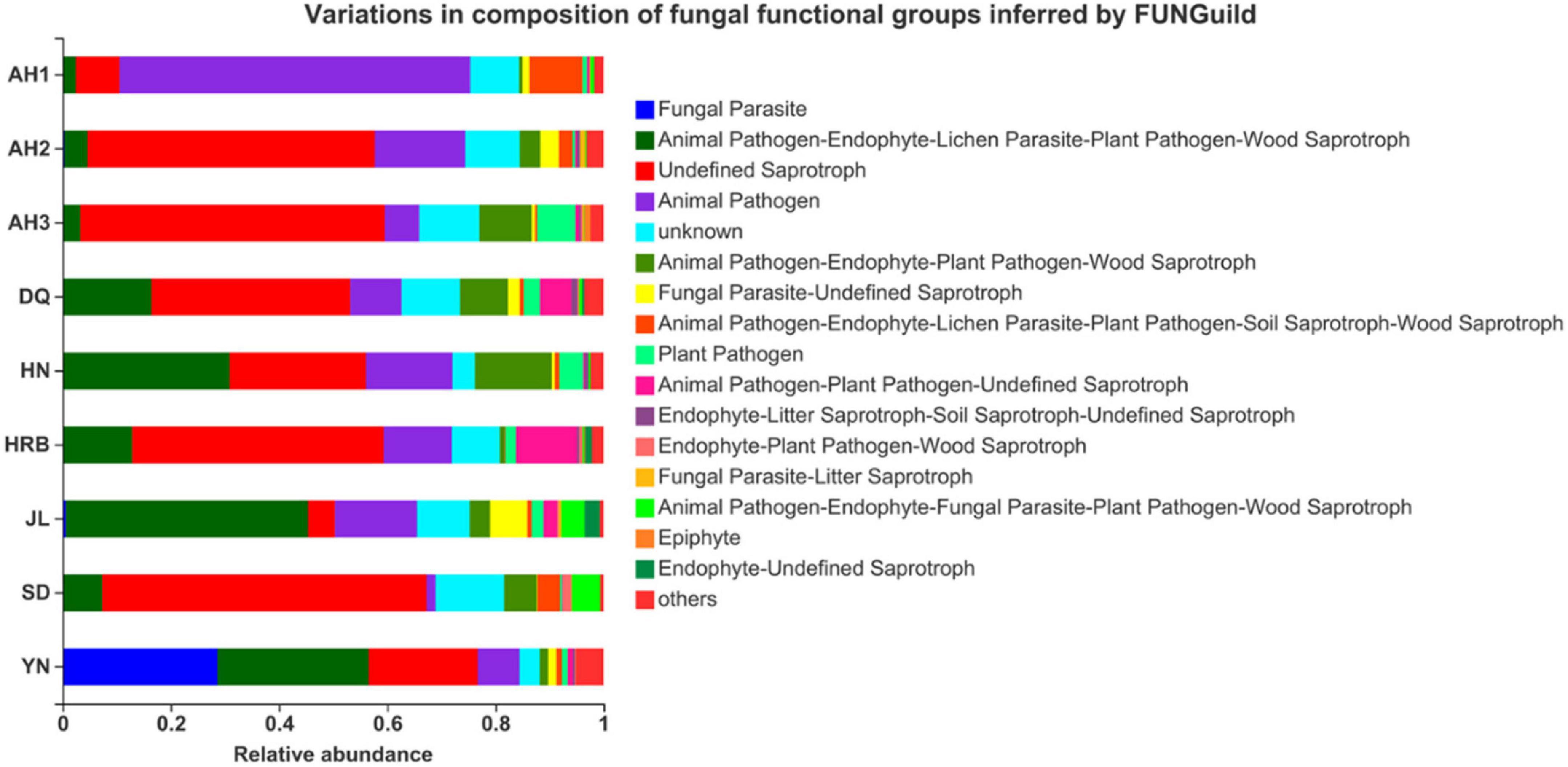
Figure 9. Comparison of predicted fungal functional groups among the nine geographic populations of the hemp flea beetle P. attenuata in China by FUNGuild.
Discussion
In contrast to many other agricultural pests, little is known about P. attenuata, including its internal microbial community. Here, the compositions and structures of the microbiota communities of P. attenuata from fields across the hemp growth regions in China were surveyed by high-throughput sequencing of 16S rDNA gene for bacteria and ITS1 DNA fragment for fungi. We found that Proteobacteria (59.57−86.88%) and Bacteroidetes (2.52−35.21%) dominated the bacterial community across eight of the nine geographic samples (HN was the exception). The observed bacterial distribution pattern showed both similarities and differences with those in other insect species (Supplementary Figure 2). For example, in sap-feeding insects, such as aphids (Gauthier et al., 2015; Zhao et al., 2016), Proteobacteria was also the most abundant bacteria phyla. In many insect species of Lepidoptera, Diptera, Coleoptera, Hymenoptera, Hemipter and mites, the most prevalent phyla were Proteobacteria and Firmicutes (Zhang et al., 2020). Species in phylum Proteobacteria are known to perform many important functions such as nitrogen fixation, metabolisms of critical nutritional components (including sugars and proteins), insecticide resistance, and protection against parasites and pathogens in fruit flies, aphids, and moths (Zhang et al., 2020). In our study, we found that the highest relative abundances of KEGG pathways prediction at “level 3” were “metabolic pathways,” “biosynthesis of secondary metabolites,” and “microbial metabolism in diverse environments” (Figure 5). Therefore, Proteobacteria species likely play an important role in the nutrition and metabolism of P. attenuata.
At the genus level, dominant bacterial genera were Rickettsia (Proteobacteria), Wolbachia (Proteobacteria), Candidatus_Brownia (Bacteroidetes), Enterobacter (Proteobacteria), Pantoea (Proteobacteria), and Rhodococcus (Actinobacteria) (Figure 2C). This observed pattern is different from those reported in several other insects. For example, in Dendroctonus beetles, the dominant bacterial genera were Erwinia, Serratia, Klebsiella, Rhodococcus, and Sphingomonas (Xu et al., 2019).
For the first time, Rickettsia and Wolbachia infections were observed in P. attenuata. Wolbachia belongs to alpha-proteobacteria and strains in this genus are common intracellular parasites in arthropods and nematodes. It’s estimated that up to 70% of all insect species may contain Wolbachia (Kozek and Rao, 2007). These endosymbionts can infect many host tissues but the dominant tissues are those involved in sexual reproduction. Indeed, Wolbachia bacteria have been found to alter host insect biology in several ways, including nutrient provision, feminization, male-killing, parthenogenesis, and sperm-egg incompatibility or cytoplasmic incompatibility (Werren et al., 2008). Cytoplasmic incompatibility refers to the inability of con-specific sperm and egg to form viable offspring and it occurs when a Wolbachia infected male mates with a female that is either uninfected or infected by a different Wolbachia strain (Yen and Barr, 1971). Cytoplasmic incompatibility reduces the reproductive success of uninfected females and promotes the spread of Wolbachia strains infecting females. While the major mode of their transmission is vertical, they can also spread horizontally between host species (Werren et al., 2008). Indeed, due to their significant roles, Wolbachia bacteria have been explored as a potential agent in the control of mosquito-borne diseases in humans because of their unique ability to invade host mosquito populations rapidly (Pan et al., 2012). Similar suggestions have been made for controlling plant pathogens and pests. For example, Wolbachia endosymbionts in the Asian citrus pest psyllid Diaphorina citri can repress the holin promoter activity in the alpha-proteobacteria “Candidatus Liberibacter asiaticus,” a citrus disease agent, by killing the bacteria and preventing the disease from spreading (Jain et al., 2017).
The endosymbiotic Wolbachia is closely related to another intracellular bacterial genus Rickettsia (Perlman et al., 2006; Duron et al., 2008). While Rickettsia are most noted for causing human diseases, including Rocky Mountain spotted fever and epidemic typhus, they are also widely distributed in insects and play diverse roles (Perlman et al., 2006; Karkouri et al., 2022). For example, Rickettsia have shown to provide nutrition for host insects, improve their ability to resist high temperature and natural enemies, and significantly enhance the resistance of whitefly to infection by the bacterial pathogen Pseudomonas syringae (Zhang et al., 2021). Similar to those of Wolbachia, Rickettsia bacteria have shown to be capable of manipulating the reproductive behavior of host arthropods, including fertility, male-killing, and parthenogenesis (Perlman et al., 2006). In our study, the dominant bacterial genus in AH1, AH2, AH3, and SD were Rickettsia, the dominant bacterial genus in DQ, HRB, and YN were Wolbachia, and the dominant bacterial genera in JL was Rickettsia and Wolbachia. At present, the detailed roles of Wolbachia and Rickettsia to their host P. attenuata are not known. If they are found to play key roles in the survival and reproduction of P. attenuata in these regions, a potential novel control strategy against hemp flea beetles can be developed by targeting these endosymbiotic bacteria within these beetles.
Interestingly, the bacterial species richness and community diversity of the HN samples were significantly higher than those of other populations (Figures 2A,B). Furthermore, principal co-ordinate analysis showed that the community structures were similar among the four biological replicates within the HN region (Figure 2D). Therefore, the high bacterial species richness and community diversity of the HN samples were likely due to the uniqueness of the ecological niche at this site. Previous studies have shown that the microbial community in insects reflects the environmental microbial populations which in turn impact the insect host’s biology (Xu et al., 2019). The sampling site within Hunan province is rich in organic matter and has a climate different from other eight sites. Specifically, the site has had a long history of growing industrial hemp and at the site, the summers are hot, humid, and overcast while the winters are cold, humid, windy, and often cloudy. Over the course of the year, the temperature typically varies from 8°C to 33°C and is rarely below 4°C or above 36°C. At present, how these two features might have contributed to its distinct hemp beetle bacterial community in HN remain to be investigated. Additional sampling at sites with similar climates to those analyzed here as well as their corresponding soil samples are needed to determine the relative contributions of climate and soil niche to differences in the microbiome of hemp flea beetles.
The genus Lactococcus was abundant in the JL samples (20.64%) but rare in the other populations (<0.4%). Lactococcus is a genus of lactic acid-producing bacteria where only a single product lactic acid is produced during glucose fermentation. The broad distribution of Lactococcus bacteria in the Jilin samples suggested the potential importance of this fermentative pathway for this population of hemp flea beetles. In addition, this metabolic uniqueness of the Jilin samples may reflect other host phenotypic differences such as pesticide resistance. For instance, insecticide- resistant strains of the diamondback moth Plutella xylostella contained more abundant bacteria belonging to Lactobacillales, Pseudomonadales, and Xanthomonadales but fewer Enterobacteriales than an insecticide-susceptible strain (Xia et al., 2013). The genus Pantoea was abundant in the SD population (33.21%) and was rare in other populations (<2%). Pantoea is a highly diverse genus that can cause plant diseases and human diseases but also have functions in habitat restoration and pesticide degradation (Walterson and Stavrinides, 2015). It’s possible that Pantoea in the SD populations functioned similarly to those found in plants and humans. At present, how bacteria in genera Lactococcus and Pantoea might contribute to trait differences among P. attenuata populations and among the host plant Cannabis sativa are unknown.
Aside from comparing bacterial diversity, we also investigated the mycobiota of the P. attenuata samples. The dominant fungal phyla in our samples belonged to Ascomycota and Basidiomycota (Supplementary Figure 5). This result was not surprising because the great majority of known terrestrial fungi are in Ascomycota and Basidiomycota (Stajich et al., 2009), including molds, mushrooms, rusts, smuts, and yeasts. Species in Ascomycota and Basidiomycota play important ecological and economical roles, including making essential contributions to the bio-product industry, nutrient cycling, antibiotic production, and as animal, human and plant pathogens (Pei et al., 2019). In our study, the fungal communities of all samples of hemp flea beetles were dominated by common environmental fungi in Ascomycota and Basidiomycota. This result is consistent with the hypothesis that environmental fungal community is a key determinant of the fungal community within hemp flea beetles.
Our analyses revealed that the total number of known bacterial and fungal species was greater than the total number of bacterial and fungal OTUs. This result is not surprising because the OTUs were defined based on a 97% sequence similarity cutoff for both the bacterial and fungal datasets while many currently known closely related sister species had higher sequence similarities than 97% at these two barcode loci (Edgar, 2013; Xu, 2016, 2020). Given that there are unclassified novel taxa in both the bacterial and fungal datasets, the true numbers of bacterial and fungal species in our samples of P. attenuata are likely much higher than what we found here. High coverage metagenome sequencing coupled with culture-based surveys will help generate more robust estimates of bacterial and fungal diversities in these samples (Xu, 2020). The obtained microbial cultures will also allow direct tests of their potential roles in the survival and reproduction of hemp flea beetles.
We note that even though several geography-specific microbial community clusters were found in both the bacterial and fungal datasets, such clusters could be due to differences in other factors associated with the specific geographic regions, including climate, host cultivars, soil conditions, and host pest genotypes. Indeed, the four broad geographic regions (i.e., Northeast China, East-central China, Central-south China, and Southwestern China) analyzed here are known to differ from each other in at least one of the above factors. In addition, we observed samples in several geographic regions that were not clustered according to their geographic origins. At present, the reasons for such differences are not known. However, fine-scale heterogeneity in soil microflora, hemp clones/cultivars, health status (e.g., microbial infections) of sampled host hemp plants, and the host plant microbiome within crop fields could all have contributed to microbiome differences among hemp flea beetles sampled from different parts of a field. Fine-scale sampling of diverse microbiomes from the soil, host plants, and hemp flea beetles are needed in order to test their specific effects on the microbiome structures of hemp flea beetles.
Conclusion
The present study revealed the bacterial and fungal communities associated with hemp flea beetles across hemp production regions in China. Our analyses demonstrated that there were differences in microflora among field populations in different locations, which likely reflect their adaption to their local environments and to potential phenotypic differences among their host beetles. More research is required to explore the detailed roles of these microbes in P. attenuata, including how environmental factors may impact their interactions. One such approach is to isolate specific microbe and then test their effects on hemp flea beetles by inoculating them to hemp flea beetles from diverse locations. Similarly, the identification of widespread and dominant bacteria in genera Wolbachia and Rickettsia and several other bacteria suggests potentially key roles of these bacteria for hemp flea beetles, especially on the reproduction of these pest insects (Duron et al., 2008). Regarding the potential role of Wolbachia on cytoplasmic incompatibility in hemp flea beetles, additional research is needed on the distribution and genetic diversity of Wolbachia strains among hemp flea beetles from within and between geographic regions. Furthermore, genetic crosses between hemp flea beetles uninfected or infected with the same or different Wolbachia strains need to be analyzed. Such data will be crucial for evaluating whether these bacteria could be potentially used as biocontrol agents against these pest beetles in hemp fields and greenhouses.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
LG and JX conceived and designed the experiments and wrote the manuscript. LG and CT performed the experiments. LG analyzed the data. All authors contributed to the reagents, materials, and analysis tools.
Funding
This research was supported by the National Natural Science Foundation of China (31701818), Agricultural Science and Technology Innovation Program (No. ASTIP-IBFC-04), Key Laboratory of Biological and Processing for Bast Fiber Crops, MOAR.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.964735/full#supplementary-material
Supplementary Table 1 The original bacterial OTUs based on 16S rRNA sequences across nine geographic populations of the hemp flea beetle P. attenuata in China.
Supplementary Table 2 The bacterial OTUs based on 16S rRNA sequences after rarefaction to the lowest number of reads among the 36 samples across the nine geographic populations of the hemp flea beetle P. attenuata in China.
Supplementary Table 3 The original fungal OTUs based on ITS sequences across nine geographic populations of the hemp flea beetle P. attenuata in China.
Supplementary Table 4 The fungal OTUs based on ITS sequences after rarefaction to the lowest number of reads among the 36 samples across the nine geographic populations of the hemp flea beetle P. attenuata in China.
Supplementary Table 5 The raw data of the 16S rRNA read counts of bacteria across nine geographic populations of the hemp flea beetle P. attenuata in China.
Supplementary Table 6 The raw data of diversity indices of bacteria based on 16S rRNA sequences across nine geographic populations of the hemp flea beetle P. attenuata in China.
Supplementary Table 7 The raw data of the ITS read counts of fungi across nine geographic populations of the hemp flea beetle P. attenuata in China.
Supplementary Table 8 The raw data of the diversity indices of fungi based on ITS sequences across nine geographic populations of the hemp flea beetle P. attenuata in China.
Footnotes
References
Andre, C. M., Hausman, J. F., and Guerriero, G. (2016). Cannabis sativa: The plant of the thousand and one molecules. Front. Plant Sci. 7:19. doi: 10.3389/fpls.2016.00019
Berasategui, A., Salem, H., Paetz, C., Santoro, M., Gershenzon, J., Kaltenpoth, M., et al. (2017). Gut microbiota of the pine weevil degrades conifer diterpenes and increases insect fitness. Mol. Ecol. 26, 4099–4110. doi: 10.1111/mec.14186
Bing, X., Gerlach, J., Loeb, G., and Buchon, N. (2018). Nutrient-dependent impact of microbes on Drosophila suzukii development. mBio 9, e2199–e2117. doi: 10.1128/mBio.02199-17
Brandsch, R. (2006). Microbiology and biochemistry of nicotine degradation. Appl. Microbiol. Biotechnol. 69, 493–498. doi: 10.1007/s00253-005-0226-0
Ceja-Navarro, J. A., Vega, F. E., Karaoz, U., Hao, Z., Jenkins, S., Lim, H. C., et al. (2015). Gut microbiota mediate caffeine detoxification in the primary insect pest of coffee. Nat. Commun. 6:7618. doi: 10.1038/ncomms8618
Chen, B., Du, K., Sun, C., Vimalanathan, A., Liang, X., Li, Y., et al. (2018). Gut bacterial and fungal communities of the domesticated silkworm (Bombyx mori) and wild mulberry-feeding relatives. ISME J. 12, 2252–2262. doi: 10.1038/S41396-018-0174-1
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2015). fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1101/274100
Deguchi, M., Bogush, D., Weeden, H., Spuhler, Z., Potlakayala, S., Kondo, T., et al. (2020). Establishment and optimization of a hemp (Cannabis sativa L.) agroinfiltration system for gene expression and silencing studies. Sci. Rep. 10:3504. doi: 10.1038/s41598-020-60323-9
Dillon, R. J., and Dillon, V. M. (2004). The gut bacteria of insects: Nonpathogenic interactions. Annu. Rev. Entomol. 49, 71–92. doi: 10.1146/annurev.ento.49.061802.123416
Duron, O., Bouchon, D., Boutin, S., Bellamy, L., Zhou, L., Engelstädter, J., et al. (2008). The diversity of reproductive parasites among arthropods: Wolbachia do not walk alone. BMC Biol. 6:27. doi: 10.1186/1741-7007-6-27
Edgar, R. C. (2013). UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/NMETH.2604
Erkosar, B., Kolly, S., van der Meer, J. R., and Kawecki, T. J. (2017). Adaptation to chronic nutritional stress leads to reduced dependence on microbiota in Drosophila melanogaster. mBio 8, e1496–e1417. doi: 10.1128/mBio.01496-17
Ezemwa, V. O., Gerardo, N. M., Inouye, D. W., Mónica, M., and Xavier, J. B. (2012). Aminal behaviour and the microbiome. Science 338:198. doi: 10.1126/science.1227412
Gao, S., Wang, B., Xie, S., Xu, X., Zhang, J., Pei, L., et al. (2020). A high-quality reference genome of wild Cannabis sativa. Hortic. Res. 7:73. doi: 10.1038/s41438-020-0295-3
Gauthier, J., Outreman, Y., Mieuzet, L., and Simon, J. (2015). Bacterial communities associated with host-adapted populations of pea aphids revealed by deep sequencing of 16S ribosomal DNA. PLoS One 10:e0120664. doi: 10.1371/journal.pone.0120664
Goncalves, J., Gonçalves, J., Rosado, T., Soares, S., Simão, A. Y., Caramelo, D., et al. (2019). Cannabis and Its Secondary Metabolites: Their Use as Therapeutic Drugs, Toxicological Aspects, and Analytical Determination. Medicines 6:1. doi: 10.3390/medicines6010031
Guo, L. T., Gao, F., Cheng, Y., Gao, C. S., Chen, J., Li, Z. M., et al. (2020). Mitochondrial COI Sequence Variations within and among Geographic Samples of the Hemp Pest Psylliodes attenuata from China. Insects 11:370. doi: 10.3390/insects11060370
Hall, T. A. (1999). BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 41, 95–98. doi: 10.1021/bk-1999-0734.ch008
Irakli, M., Tsaliki, E., Kalivas, A., Kleisiaris, F., Sarrou, E., and Cook, C. M. (2019). Effect of genotype and growing year on the nutritional, phytochemical, and antioxidant properties of industrial hemp (Cannabis sativa L.) Seeds. Antioxidants 8:491. doi: 10.3390/antiox8100491
Jain, M., Fleites, L. A., and Gabriel, D. W. (2017). A small Wolbachia protein directly represses phage lytic cycle genes in “Candidatus Liberibacter asiaticus within psyllids. mSphere 2, e171–e117. doi: 10.1128/mSphereDirect.00171-17
Karkouri, K. E., Ghigo, E., Raoult, D., and Fournier, P. E. (2022). Genomic evolution and adaptation of arthropod-associated Rickettsia. Sci. Rep. 12:3807. doi: 10.1038/s41598-022-07725-z
Kõljalg, U. Henrik Nilsson, R., Abarenkov, K., Tedersoo, L., Taylor, A. F. S., Bahram, M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Kozek, W. J., and Rao, R. U. (2007). The Discovery of Wolbachia in Arthropods and Nematodes – A Historical Perspective. Issues Infectious Diseases. 5, 1–14. doi: 10.1159/000104228
Kudo, R., Masuya, H., Endoh, R., Kikuchi, T., and Ikeda, H. (2018). Gut bacterial and fungal communities in ground-dwelling beetles are associated with host food habit and habitat. ISME J. 13, 676–685. doi: 10.1038/s41396-018-0298-3
Magoč, T., and Salzberg, S. L. (2011). FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
McParland, J. M., Clarke, R. C., and Watson, D. P. (2000). Hemp diseases and pest management and biological control. Australas Plant Path. 30:251. doi: 10.1079/9780851994543.0000
Moran, N. A., Hansen, A. K., Powell, J. E., and Sabree, Z. L. (2012). Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees. PLoS One 7:e36393. doi: 10.1371/journal.pone.0036393
Pan, X., Zhou, G., Wu, J., Bian, G., Lu, P., Raikhel, A. S., et al. (2012). Wolbachia induces reactive oxygen species (ROS)-dependent activation of the Toll pathway to control dengue virus in the mosquito Aedes aegypti. Proc. Natl. Acad. Sci. U.S.A. 109, E23–E31. doi: 10.1073/pnas.1116932108
Pei, G. L., Guo, J., Wang, Q., and Kang, Z. S. (2019). Comparative analysis of protein kinases and associated domains between Ascomycota and Basidiomycota. J. Integ. Agr. 18, 96–107. doi: 10.1016/S2095-3119(18)62022-2
Perlman, S. J., Hunter, M. S., and Zchori-Fein, E. (2006). The emerging diversity of Rickettsia. Proc. Biol. Sci. 273, 2097–2106. doi: 10.1098/rspb.2006.3541
Rossmassler, K., Dietrich, C., Thompson, C., Mikaelyan, A., Nonoh, J. O., Schef-frahn, R. H., et al. (2015). Metagenomic analysis of the microbiota in the highly compartmented hindguts of six wood- or soil-feeding higher termites. Microbiome 3:56. doi: 10.1186/s40168-015-0118-1
Santos-Garcia, D., Mestre-Rincon, N., Zchori-Fein, E., and Morin, S. (2020). Inside out: Microbiota dynamics during host-plant adaptation of whiteflies. ISME J. 14, 847–856. doi: 10.1038/s41396-019-0576-8
Stackebrandt, E., and Goebel, B. M. (1994). Taxonomic Note: A Place for DNA-DNA Reassociation and 16S rRNA Sequence Analysis in the Present Species Definition in Bacteriology. Int. J. Syst. Bacteriol. 44, 846–849. doi: 10.1099/00207713-44-4-846
Stajich, J. E., Berbee, M. L., Blackwell, M., Hibbett, D. S., James, T. Y., Spatafora, J. W., et al. (2009). The fungi. Curr. Biol. 19, R840–R845. doi: 10.1016/j.cub.2009.07.004
Summers, R. M., Mohanty, S. K., Gopishetty, S., and Subramanian, M. (2015). Genetic characterization of caffeine degradation by bacteria and its potential applications. J. Microbial. Biotechnol. 8, 369–378. doi: 10.1111/1751-7915.12262
Walterson, A. M., and Stavrinides, J. (2015). Pantoea: Insights into a highly versatile and diverse genus within the Enterobacteriaceae. FEMS Microbiol. Rev. 39, 968–984. doi: 10.1093/femsre/fuv027
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Werren, J. H., Baldo, L., and Clark, M. E. (2008). Wolbachia: Master manipulators of invertebrate biology. Nat. Rev. Microbiol. 6, 741–751. doi: 10.1038/nrmicro1969
Wu, K., Yang, B., Huang, W., Dobens, L., Song, H., and Ling, E. (2016). Gut immunity in Lepidopteran insects. Dev. Comp. Immunol. 64, 65–74. doi: 10.1016/j.dci.2016.02.010
Xia, X. F., Zheng, D. D., Zhong, H. Z., Qin, B. C., Gurr, G. M., Vasseur, L., et al. (2013). DNA sequencing reveals the midgut microbiota of diamondback moth, Plutella xylostella (L.) and a possible relationship with insecticide resistance. PLoS One 8:e68852. doi: 10.1371/journal.pone.0068852
Xu, J. (2020). Fungal species concepts in the genomics era. Genome 63, 459–468. doi: 10.1139/gen-2020-0022
Xu, L., Deng, J., Zhou, F., Cheng, C., Zhang, L., Zhang, J., et al. (2019). Gut microbiota in an invasive bark beetle infected by a pathogenic fungus accelerates beetle mortality. J. Pest. Sci. 92, 343–351. doi: 10.1007/s10340-018-0999-4
Yen, J. H., and Barr, A. R. (1971). New hypothesis of the cause of cytoplasmic incompatibility in Culex pipiens. Nature 232, 657–658. doi: 10.1038/232657a0
Zhang, Y. T., Huang, Z. X., Zhang, J. G., Zhou, M., and Zhou, L. Q. (2021). Diversity analysis of the intracellular bacterial community in Terminalinus Hopkins at different development ages. South China Fruits 50, 126–136. doi: 10.13938/j.issn.1007-1431.20200776
Zhang, Z., Ali, M. W., Saqib, H. S. A., Liu, S., Yang, X., Li, Q., et al. (2020). A Shift Pattern of Bacterial Communities Across the Life Stages of the Citrus Red Mite, Panonychus citri. Front. Microbiol. 11:1620. doi: 10.3389/fmicb.2020.01620
Zhang, Z. J., Mu, X. H., Cao, Q. N., Shi, Y., Hu, X. S., and Zheng, H. (2022). Honeybee gut Lactobacillus modulates host learning and memory behaviors via regulating tryptophan metabolism. Nat. Commun. 13:2037. doi: 10.1038/s41467-022-29760-0
Zhao, M., Gao, J., Meng, X., Fen, X., Lang, Y., and Kang, H. (2018). Study on Efficacy Test in the Prevention and Control of Psylliodes attenuata Koch. J. Shanxi. Agr. Sci. 46, 2082–2083.
Keywords: Psylliodes attenuata, microbial community diversity, 16S rRNA, ITS, hemp
Citation: Guo L, Tang C, Gao C, Li Z, Cheng Y, Chen J, Wang T and Xu J (2022) Bacterial and fungal communities within and among geographic samples of the hemp pest Psylliodes attenuata from China. Front. Microbiol. 13:964735. doi: 10.3389/fmicb.2022.964735
Received: 08 June 2022; Accepted: 29 July 2022;
Published: 06 September 2022.
Edited by:
Takema Fukatsu, National Institute of Advanced Industrial Science and Technology, JapanCopyright © 2022 Guo, Tang, Gao, Li, Cheng, Chen, Wang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Litao Guo, guolitao@caas.cn; Jianping Xu, jpxu@mcmaster.ca; 0000-0003-2915-2780
†These authors have contributed equally to this work